 Yunqing Cheng
Yunqing Cheng Siqi Jiang
Siqi Jiang Xingzheng Zhang
Xingzheng Zhang Hongli He
Hongli He Jianfeng Liu
Jianfeng Liu- Jilin Provincial Key Laboratory of Plant Resource Science and Green Production, Jilin Normal University, Siping, China
Yield loss in the economically important hazelnut (Corylus spp.) occurs through the frequent formation of blank nuts. Although the condition is associated with embryo abortion, we have not yet identified the regulatory genes involved. Therefore, this study aimed to determine the genes related to embryo abortion in hazel. We performed whole-genome re-sequencing and single-nucleotide polymorphism (SNP) analysis on four mutant hazelnut trees (Empty1 to Empty4, C. heterophylla) bearing blank nuts and four wild-type trees (Full1 to Full4, C. heterophylla). A paired comparison of Empty1 vs. Full1, Empty2 vs. Full2, Empty3 vs. Full3, and Empty4 vs. Full4, along with the intersection of Empty1 to Empty4, revealed 3 081 common SNPs in the four blank-nut mutants. Of these, 215 synonymous SNPs in exonic regions were distributed across 178 candidate genes. Heterozygosity analysis showed that average homozygous and heterozygous SNP ratios were respectively 0.409 and 0.591 in the samples. According to Gene Ontology classification, candidate genes were enriched in the categories of binding, catalysis, molecular transducer, transporter, and molecular function regulator. Among these, 18 of 178 genes had homozygous SNPs in Empty1–4. Cis elements in the promoter region of starch synthase 4 (SS4) contain the RY-element, implying seed-specific expression. Starch granules were absent from Empty1–4 cotyledon cells, but abundantly present in Full1–Full4 cotyledon cells. The blank-nut phenotype has heavier nut shells. Overall, we conclude that single-nucleotide variants of Acetyl-CoA carboxylase 1 (ACC1), intracellular sodium/hydrogen exchanger 2 (NHX2), UDP-glycosyltransferase 74E2 (UGT74E2), DEFECTIVE IN MERISTEM SILENCING 3 (DMS3), DETOXIFICATION 43 (FRD3), and SS4 may induce embryo abortion, leading to blank-nut formation. Our results will benefit future research on how the gain or loss of candidate genes influences seed development. Moreover, our study provides novel prospects for seedless cultivar development.
Introduction
Hazel (Corylus spp., Betulaceae, Fagales) is an important wind-pollinated species. Its nutritious and delicious nut is an important raw material for several food processing industries (Amaral et al., 2006). In recent years, the cultivation area and yield of hazelnut in China have increased rapidly. Hazel-related industries in mountainous areas of Northeast China, a major hazel cultivation region, have a significant effect on the development of local economy. Currently, Corylus heterophylla and hybrid hazel (C. heterophylla × C. avellana) are the most important species cultivated in China. Among these, the cultivation area of C. heterophylla is more than 1.0 million hm (Liu et al., 2014a; Liu et al., 2014b), more than that of hybrid hazel (Liu et al., 2014b; Cheng et al., 2018a; Cheng et al., 2018b). Thus, C. heterophylla contributes to most of the hazel products available in Chinese market.
Successful pollination, fertilization, and kernel development are prerequisites for good hazelnut yield. The failure of these important biological events during flower and fruit development might induce the drop of pistillate flowers, blank fruits, and shriveled kernels, causing varying degrees of yields loss. Frequent blank-fruit formation has been observed in C. avellana and C. heterophylla (Silva et al, 1996; Beyhan and Marangoz, 2007; Liu et al., 2012). Embryo abortion, rather than failed pollination or fertilization, has been implicated in blank-fruit formation (Liu et al., 2012). Seed formation is a pivotal process in plant reproduction and seed dispersal, and hazelnut kernel quality has been characterized previously (Ferreira et al., 2009; Ferreira et al., 2010; Xu and Hanna, 2010; Solar and Stampar, 2011). Furthermore, a set of genes that might be involved in regulating blank-fruit formation has been identified though transcriptome analysis of abortive and developing ovules; these differentially expressed genes were significantly enriched in the following Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways: metabolism, plant hormone signal transduction, and RNA transport (Cheng et al., 2015a; Cheng et al., 2017). These studies provide clues about the mechanisms regulating hazel fruit quality. However, whether or not blank-fruit formation is induced at the transcriptome level remains unclear. Furthermore, the key genes involved in embryo development require further genetic evidence and verification.
Forward genetic screens can help elucidate the biological mechanisms in model species. Their success relies on the feasibility of mutant gene isolation (Nordström et al., 2013). Identification of causal mutations typically begins with genetic mapping, followed by candidate gene sequencing and complementation studies based on genetic transformation. Whole-genome re-sequencing of two Italian tomato landraces revealed sequence variations in genes associated with stress tolerance, fruit quality, and long shelf-life traits, and elucidated the genetic and molecular bases of fruit metabolism and storability in tomato (Tranchida-Lombardo et al., 2017). Using whole-genome re-sequencing and homozygosity mapping, it was found that the SHELL gene is responsible for the tenera phenotype (thin-shelled) in both cultivated and wild palms in sub-Saharan Africa (Singh et al., 2013). Thus, advances in DNA sequencing technologies have transformed genetic mapping and gene identification.
Hazel has unique flower and fruit developmental characteristics. The ovary is absent when the female pistillate inflorescence blooms. After pollination, pollen germination, and pollen tube growth, several layers of early ovary-primordium cells begin to differentiate. Thereafter, the ovary, ovule, and embryo sac take shape successively. Corylus heterophylla blooms in early- or mid-April in northeast China, and requires approximately two months from pollination to complete fertilization (Liu et al., 2014a). Subsequently, two ovules in the ovary begin to grow rapidly, but blank nuts form if embryo abortion occurs at this stage (Liu et al., 2012). In four artificial hazel orchards modified from natural forest, we found four mutant hazelnut trees that have apparently been producing only blank nuts for years. Their unique germplasm with the blank-nut phenotype could be valuable for mining key genes implicated in kernel development. In the present study, we therefore aimed to determine the genes involved in hazel embryo abortion, thus providing genetic and molecular bases for this process. The results should also promote the development of seedless germplasm in other fruit species.
Materials and Methods
Plant Materials
In 2017, we found four hazel mutants that had borne only blank nuts for several years, in Northeast China. These mutant plants were distributed in four artificial orchards modified from hazel nature forest, in four different locations, namely Siping, Tieling, Kaiyuan, and Huludao Cities. In the orchard in Siping City, one plant bearing normal edible nuts, and another that had borne only blank nuts for several years, were selected; they were named Full1 and Empty1, respectively. To make the genetic background as close as possible, the spatial distance between the Empty1 plant and reference Full1 was less than 3.0 m. Similarly, Full2 and Empty2 (mutant) from Tieling City, Full3 and Empty3 (mutant) from Kaiyuan City, and Full4 and Empty4 (mutant) from Huludao City were sampled and analyzed. Blank nuts contain only small and unformed ovules, with no edible content (Figure 1). The pollen of several Corylus heterophylla × C. avellana cultivars (“Dawei,” “Bokehong,” and “Yuzhui”) and of C. heterophylla was collected and equally mixed following the method of Liu et al. (2014b). Primary artificial pollination was carried out, and the nut quality-analysis results confirmed that the blank-nut ratios were 100% for the four mutants, and that blank-nut formation in these mutants could not be relieved by artificial pollination. These chosen plants were subjected to molecular analysis to confirm their identity as C. heterophylla, using a simple sequence repeat (SSR)-based technique at Jilin Normal University. Seven primer pairs were used, according to the method of Cheng et al. (2018a).
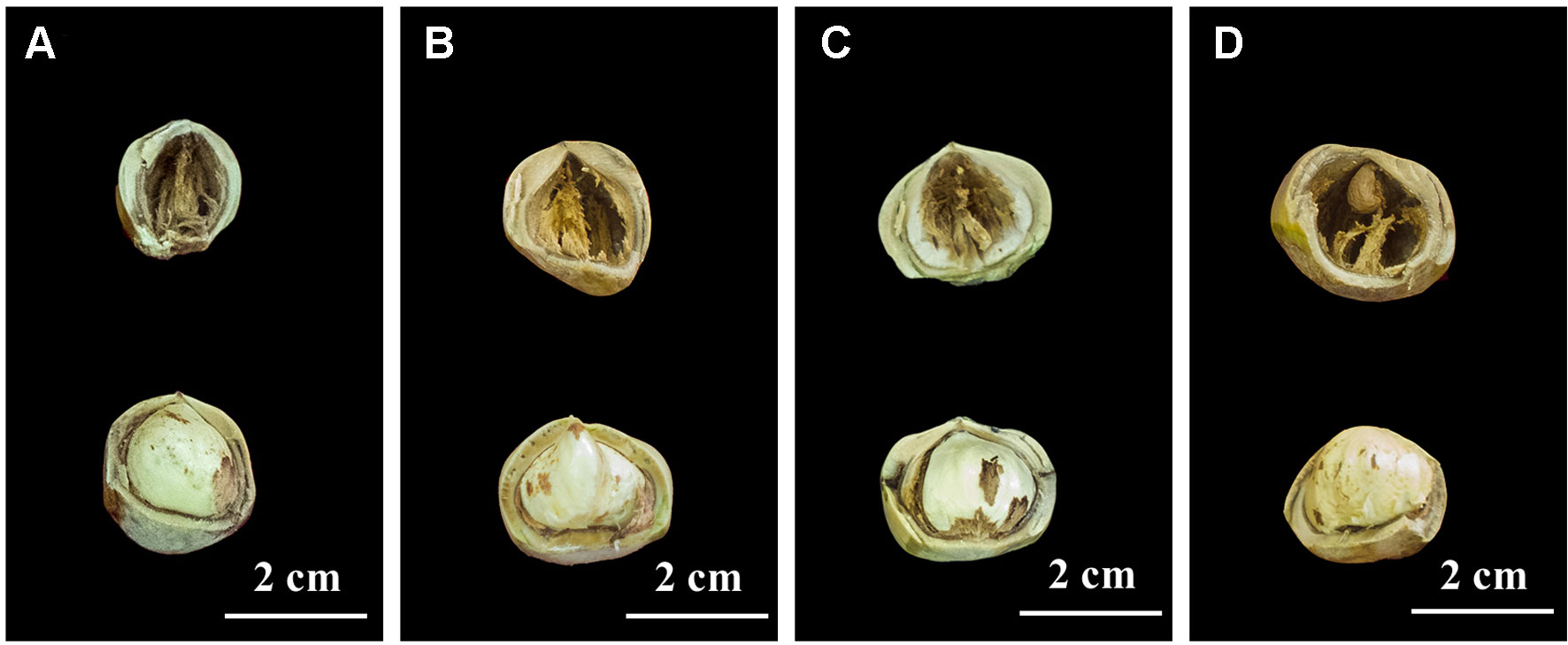
Figure 1 Nut characters of the eight hazel (Corylus heterophylla) trees sampled in northeastern China. (A) Empty1 (the first line) with blank nut trait and Full1 (the second line) with edible seed from Siping; (B) Empty2 (the first line) with blank nut trait and Full2 (the second line) with edible seed from Tieling; (C) Empty3 (the first line) with blank nut trait and Full3 (the second line)with edible seed from Kaiyuan; (D) Empty4 (the first line) with blank-nut trait and Full4 (the second line) with edible seed from Huludao.
In 2018, young leaves of the eight C. heterophylla plants mentioned above were collected and stored in liquid nitrogen for whole-genome re-sequencing. Meanwhile, some nut quality indexes were investigated, including shell weight, kernel weight, nut total weight, kernel ratio (kernel weight × 100/nut total weight), diameter, and blank-nut ratio.
Isolation and Sequencing of DNA
DNA was isolated using the DNeasy Plant Mini Kit (QIAGEN, Valencia, CA, USA) according to manufacturer’s recommendations. DNA quality was evaluated by electrophoresis on a 0.8% agarose gel, and analyzed using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Wilmington, USA) and Qubit 2.0 Fluorometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Genomic DNA was fragmented randomly using ultrasound. Fragments ranging from 200 to 500 bp in length were recycled by electrophoresis, and were connected with adaptors to generate clusters for sequencing. Eight paired-end fragment libraries averaging 300 bp were generated and sequenced on an Illumina HiSeq 2000 platform (Biomarker Technologies, Beijing, China) following Illumina protocols. The raw data were submitted to the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/Traces/study) (Accession number: PRJNA529018).
Filtering and Assembling of Sequencing Data
The sequenced raw reads were filtered to obtain clean reads for further analysis. The following reads were removed: (1) paired reads with adaptor sequences; (2) paired reads with more than 10% unknown bases (N); (3) paired reads with a base quality value less than Q20; (4) paired reads shorter than 25 bp in length; and (5) paired reads with other than A, T, C, or G at the 5′ end. The filtered sequences were aligned to the reference Jefferson hazelnut genome (https://www.cavellanagenomeportal.com/) to detect the genomic mutations in our samples using Burrows–Wheeler Aligner software (Li and Durbin, 2009). Picard tools (https://broadinstitute.github.io/picard/) were used to remove duplicate reads, and the BAM file generated was used to calculate sequencing depth and coverage. Genome Analysis Toolkit (GATK) and SAMtools were used to map the obtained genome sequence data to the reference hazelnut genome and call high-quality SNPs (Li et al., 2009; Van der Auwera et al., 2013). GATK software was used to locally re-align reads near indels and generate BAM files after realignment; this can eliminate false positive SNPs around indels. Then, GATK was used to detect SNP, and filter out the sites with lower sequence depth and quality score. The genotype information of each point was obtained in the final VCF file. Based on the comparison results of GATK, VarScan 2 software (version 2.4.1) was used to obtain information about SNPs (Koboldt et al., 2009) and remove variable sites with relatively low sequencing depth and quality, to obtain a highly reliable data set. Filtering parameters were as follows: lowest coverage depth, 30 reads; minimum mutation frequency, 20%; minimum mutation base number, 15 reads; lowest quality value for mutant sites, Q20; and both positive and reverse reads supported the mutation site, and the difference between the number of positive and reverse reads was less than 10%. ANNOVAR software was used to obtain SNP annotations (Wang et al., 2010). Based on the SNP information for each sample, online software PHYLIP version 3.697 (http://evolution.genetics.washington.edu/phylip.html) was used to construct the phylogenetic tree of the eight hazel samples, using neighbor-joining method (Figure S1).
Identification of Major Genes Regulating Embryo Abortion
To narrow the range of candidate genes, several analyses were performed in succession on detected SNPs and their positional data. First, online Venn Diagram tools (http://bioinformatics.psb.ugent.be/webtools/Venn/) were used to make SNP-paired comparisons of Empty1 vs. Full1, Empty2 vs. Full2, Empty3 vs. Full3, and Empty4 vs. Full4, thereby identifying the SNPs unique to the blank-nut mutants. A common intersection of SNPs was also generated from the comparisons. Candidate genes with common and unique SNPs in their exon regions were selected for Gene Ontology (GO) analysis using WEGO 2.0 (http://wego.genomics.org.cn/). Genes associated with embryo development were considered to be those involved in transporter activity function (Liu et al., 2013), and those with homozygous SNPs in Empty1–4. Finally, cis elements in the starch synthase 4 (SS4) promoter (a 2 000 bp region upstream of the start codon) were analyzed using PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/).
Validation of SNP Assays
Genomic DNA from each sample was PCR-amplified. Three primer pairs designed using the reference sequence of SS4 (g16468) (forward primer: 5′-TGTTGAAAAGACTGTCGCTGAGAAG; reverse primer: 5′-CTTCCATCTCTCTTCCACACCATTT), acetyl-CoA carboxylase 1 (ACC1, g11831) (forward primer: 5′-TTTGGACTCTAATATTGCTGAAG; reverse primer: 5′-TAAGGTAGATCAGCCATGCAGTA), sodium/hydrogen exchanger 2 (NHX2, g12219) (forward primer: 5′-TGGTGGAAGAAGGTATATAAAGAA; reverse primer: 5′-GGTTCTTTGTTCACATCTCTTTAA). Approximately 30 ng of genomic DNA was amplified using a Bio-Rad thermal cycler (Bio-Rad, Hercules, CA). The reaction volume was 20 µl, comprising 1.0 µl genomic DNA template, 0.4 µM primers, and 10.0 µl 2 × Taq plus Master Mix (Tiangen Biotech Co., Ltd., Beijing, China). The thermocycling protocol was as follows: 95°C for 5 min, followed by 35 cycles of 95°C for 30 s, 55°C for 20 s, and 72°C for 60°s. Amplicons were subsequently cloned into the pMD19-T vector (Takara), transformed into Escherichia coli DH5a cells following manufacturer protocol, and sequenced by Sangon Biotech Co., Ltd (Shanghai, China).
Cytological Analysis of Starch Granules in Cotyledons
The cotyledons were isolated from the seeds under a stereoscope, and cut into cubes of edge length 1–2 mm. These cubes were fixed in 4% polyformaldehyde solution for 24 h and stored in 70% ethanol at −20°C until further use. The samples were then dehydrated in gradient ethanol solutions. Dehydration, embedding, and aggregation were performed using the LR White Resin Embedding Kit (Electron Microscopy Sciences, USA), following the protocol of the manufacturer; 0.5% periodic acid and Schiff reagent were used to stain starch granules in the cotyledons. Cytogenetic observation of starch granules in the sections was performed and photographed using optical light microscopy.
Results
Overview of Sequencing Data
Whole-genome re-sequencing of our eight samples generated 42.01 Gb of clean data; statistical analysis results are shown in Table 1. These data were aligned to the 289.80-Mb reference genome. The sequencing depth ranged from 16.38× to 25.03×, covering 67%–73% of the reference genome bases. The Illumina sequence data were assembled using software SPAdes 3.0 (Bankevich et al., 2012), and 97.9% of the assembled sequences, on average, could be mapped to the reference genome. This indicates that the C. heterophylla samples have a close genetic relationship with C. avellana.The base error rate was lower than 0.02%. The percentage of bases with Phred value >20 was more than 94%. The GC count per read followed a normal distribution. These results indicate that library construction and quality by Illumina sequencing were reliable.
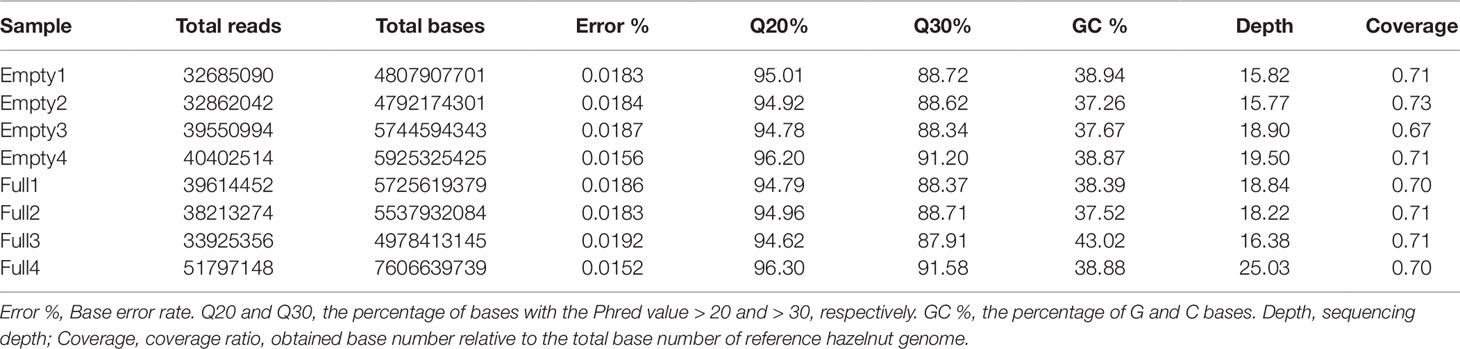
Table 1 Statistical analysis results of clean sequencing data, for eight hazel (Corylus heterophylla) trees sampled in Northeast China.
Single-Nucleotide Polymorphism Detection
Mapping of our genome sequencing data to the reference hazelnut genome using GATK was performed to call high-quality SNPs. In the samples of Empty1, Empty2, Empty3, and Empty4, 1 494 330, 1 539 833, 2 065 212, and 214 6447 SNPs were identified, respectively (Table 2); in the samples of Full1, Full2, Full3, and Full4, 1 982 873, 1 979 751, 965 214, and 2 775 577 SNPs were identified, respectively (Table 2). In total, 3 391 427 SNPs were identified using our eight samples. The distribution of SNPs showed an notable preference, and the ratio of SNPs located in the intergenic region was the highest (43%–47%), followed by that in the exonic (14%–24%), intronic (16%–19%), upstream (7%–9%), and downstream regions (6%–8%) (Table 2). The SNP mutation sites in the exon regions might have potential effects on protein translation. Approximately 52% of the SNP mutations in the exon regions can lead to nonsynonymous mutation at the translation level (Table 3).
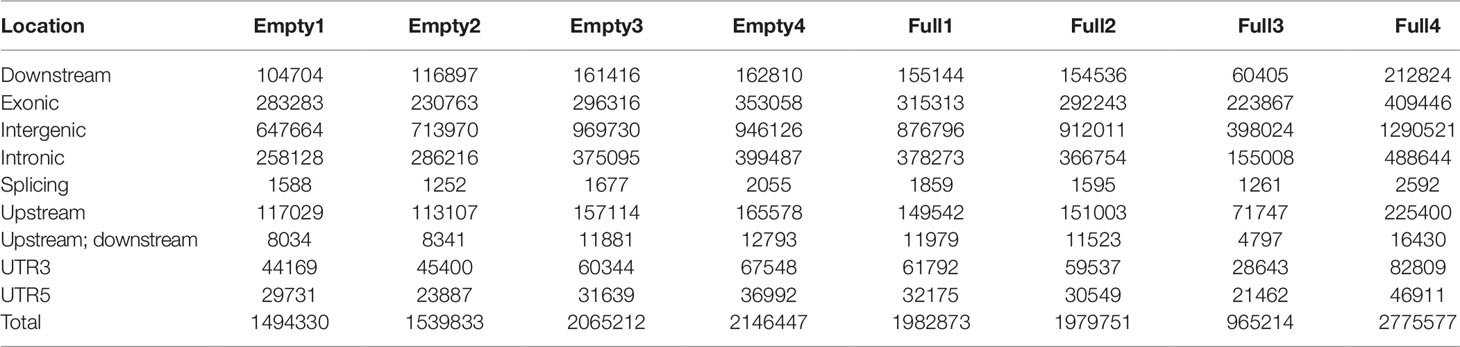
Table 2 Statistical analysis results of SNP annotation, for eight hazel (Corylus heterophylla) trees sampled in Northeast China.
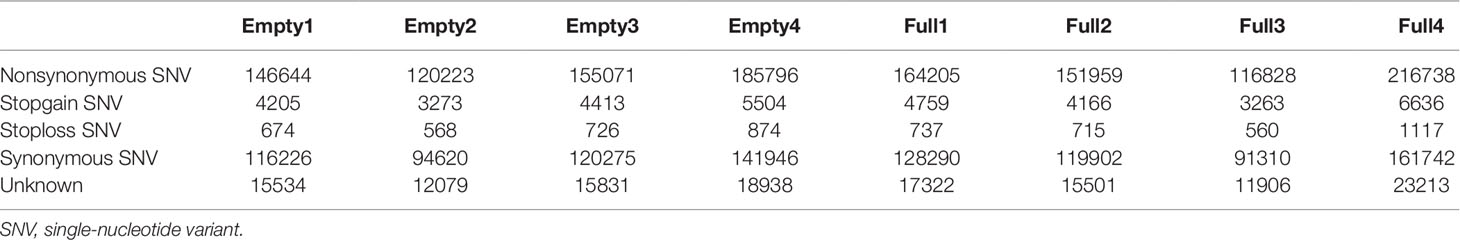
Table 3 Statistical analysis results of the effect of SNP mutation sites in the exon regions on protein translation, for eight hazel (Corylus heterophylla) trees sampled in Northeast China.
Unique and Common SNPs in the Blank-Nut Mutants
Paired comparisons of SNPs between two samples from the same orchard were carried out to identify unique SNPs in the blank-nut mutants using Venn Diagrams tools. Consequently, 820 705, 797 299, 1 578 029, and 880 405 were identified by paired comparison of Empty1 vs. Full1, Empty2 vs. Full2, Empty3 vs. Full3, and Empty4 vs. Full4, respectively (Figures 2A–D). Based on these findings, common and unique SNPs in the blank-nut mutants were identified, using the four groups of SNPs mentioned above. In total, 3 081 common and unique SNPs were detected using the Venn Diagrams tools (Figure 2E). For SNP mutations to occur in the exon regions, the nonsynonymous single-nucleotide variants (SNVs) should induce changes in amino acids at the protein translation level. Among the 3,081 common and unique SNPs (Table S1), 215 SNPs belonged to nonsynonymous SNVs in the exon region, and they were distributed among 178 candidate genes (Table S2).
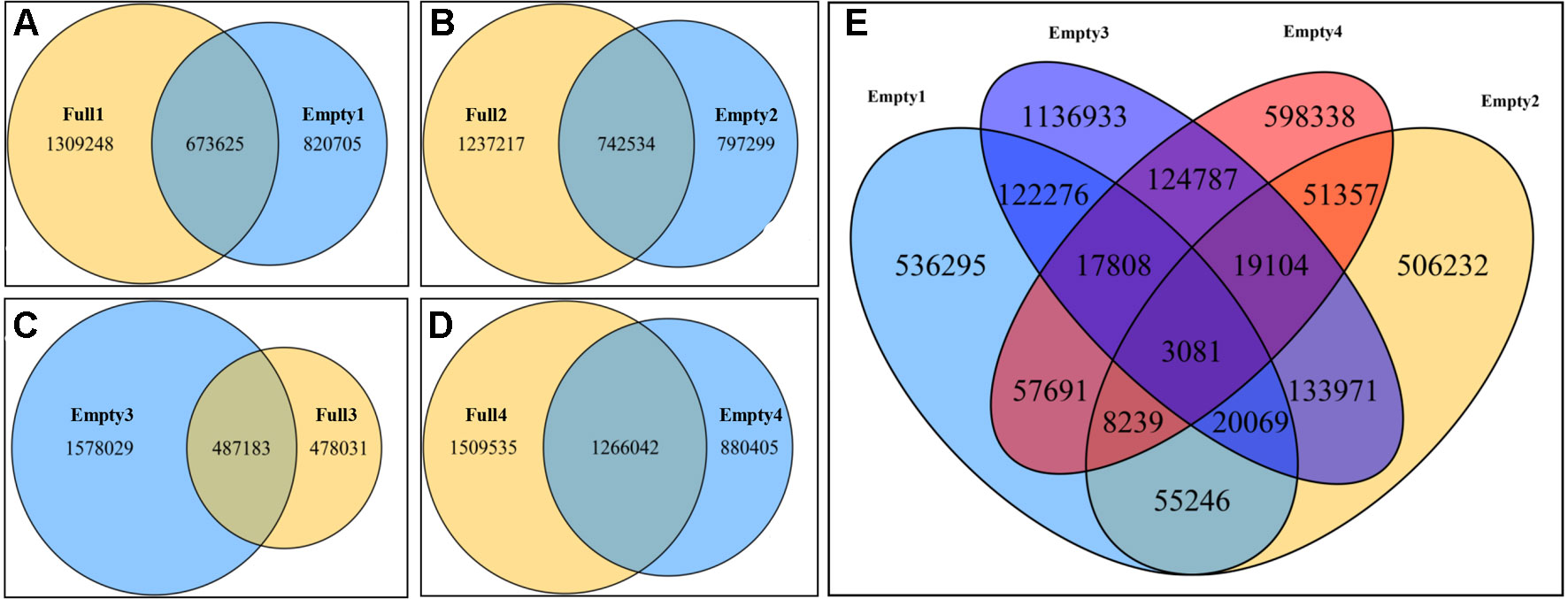
Figure 2 Common and unique single-nucleotide polymorphisms (SNPs) in blank-nut mutants of hazel (Corylus heterophylla) sampled in Northeast China. (A) Paired comparison of Full1 vs. Empty1; 82 075 unique SNPs were found in the blank-nut mutant Empty1; (B) paired comparison of Full2 vs. Empty2; 797 299 unique SNPs were found in the blank-nut mutant Empty2; (C) paired comparison of Full3 vs. Empty3; 478 031 unique SNPs were found in the blank-nut mutant Empty3; (D) paired comparison of Full3 vs. Empty3; 880 405 unique SNPs were found in the blank-nut mutant Empty4; (E) unique SNPs in the blank-nut mutants revealed using Venn Diagrams.
Analysis of SNP Heterozygosity and Validation of SNP Assays
We identified 14 908 159 SNPs in all eight samples. Among these samples, homozygous (1/1) SNP ratios ranged from 0.377 to 0.445, while heterozygous (0/1) SNP ratios ranged from 0.552 to 0.629. The 1/2 heterozygous genotype was scarce, with a ratio of <0.004 in all samples. Average homozygous and heterozygous SNP ratios were 0.409 and 0.591, respectively (Figure 3). Among the 178 candidate genes (Table S3), 18 had the homozygous SNP genotype in Empty1, Empty2, Empty3, and Empty4 (Table S4). Three genes of interest were selected for PCR validation: starch synthase 4 (SS4, g16468), Acetyl-CoA carboxylase 1 (ACC1, g11831), and sodium/hydrogen exchanger 2 (NHX2, g12219). The results were consistent with those from whole-genome re-sequencing.
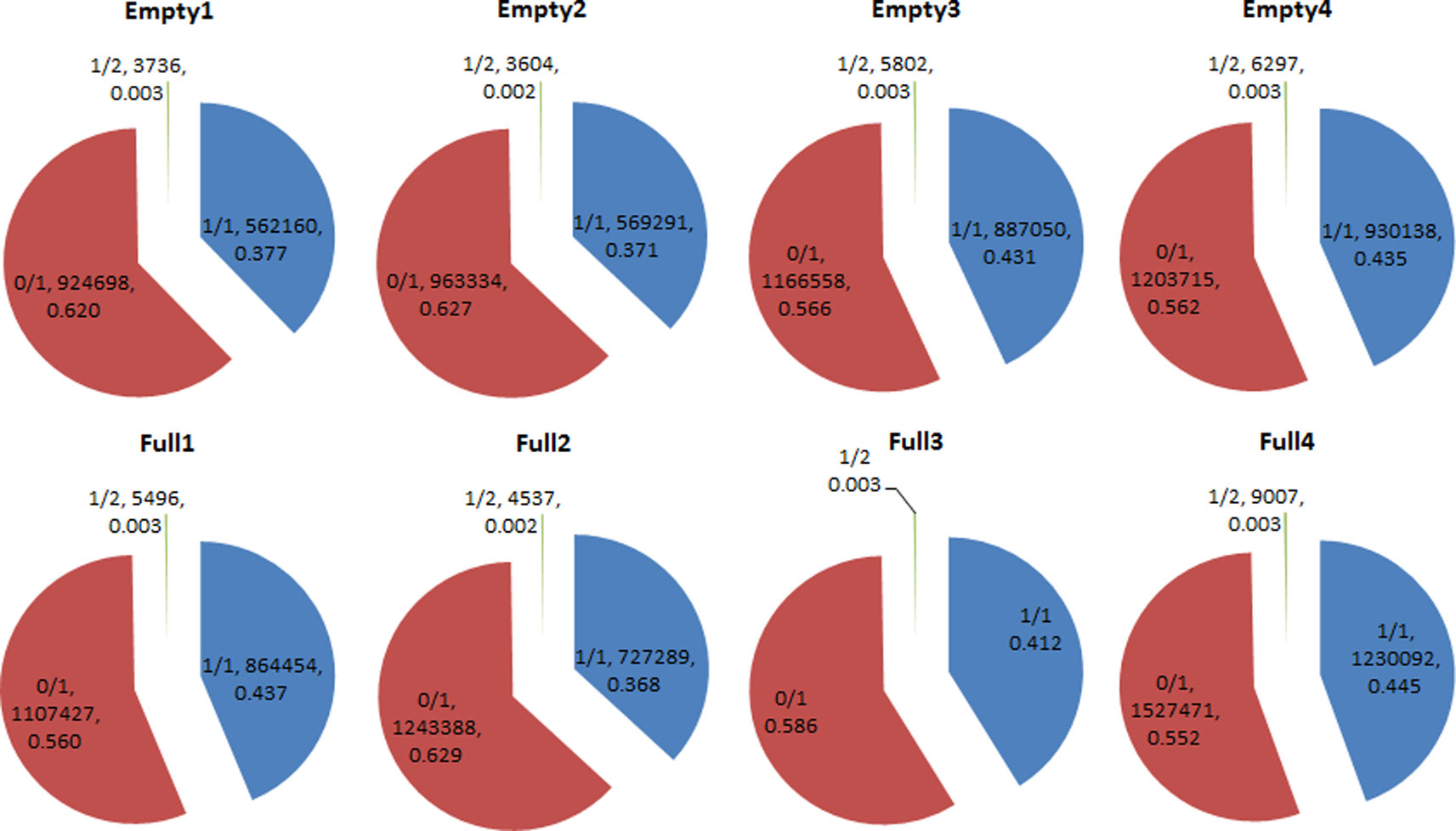
Figure 3 Heterozygosity statistical results of single-nucleotide polymorphisms (SNPs) in hazel (Corylus heterophylla) trees sampled in Northeast China. 1/1: homozygous genotype; 0/1 and 1/2: heterozygous genotypes. The pie charts each show three parts representing three genotypes, and three parameters that represent the genotype of SNP, the number of SNPs, and the corresponding percentage of SNPs, respectively.
Gene Ontology Analysis of Candidate Genes
We assigned 178 candidate genes to the following three GO categories: biological processes, cellular components, and molecular functions (Figure 4). The majority of these genes were involved in binding, catalysis, molecular transducers, transporters, and molecular function regulators. They were also enriched in the categories cellular and metabolic processes, response to stimulus, developmental process, regulation of biological process, multicellular organismal process, cellular component organization or biogenesis, multi-organism process, reproductive process, localization, signaling, and positive regulation of biological process. Most corresponding proteins were located in organelles and the cell membrane.
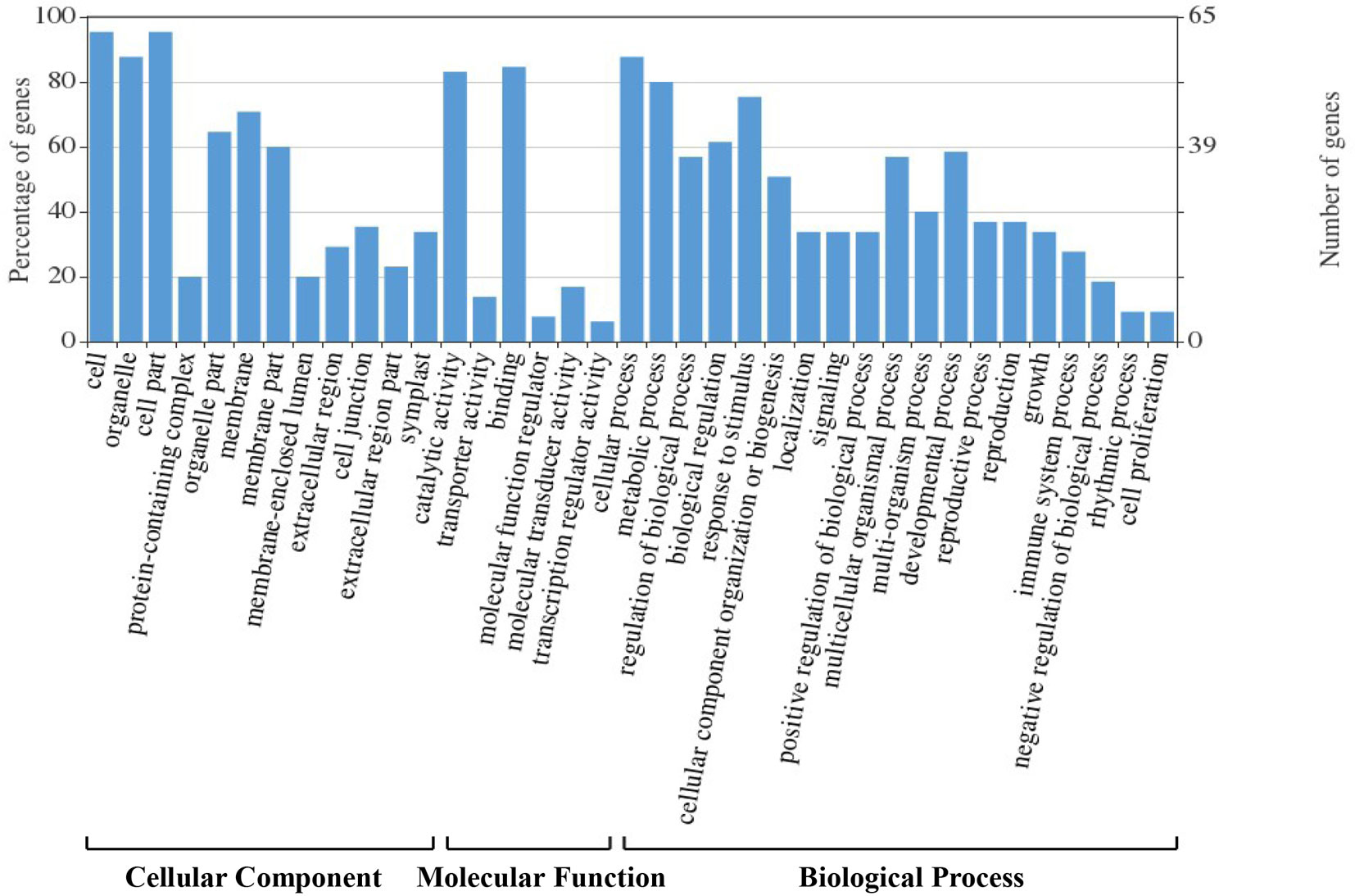
Figure 4 Gene Ontology (GO) classification analysis of the 178 candidate genes identified in hazel (Corylus heterophylla) sampled in Northeast China.
Cis Elements in the Gene Promoter
The nine genes involved in transporter molecular functions according to GO analysis were chosen for further cis element analysis. These genes were thought to be involved in regulating embryo abortion and are expected to be expressed in seeds. Only SS4 (g16468) had the RY-element indicative of seed-specific regulation, however (Table 4). In the SS4 promoter region, we found other cis elements including ABRE, Box 4, G-Box, GARE-motif, and P-box, suggesting that abscisic acid, light, and gibberellin regulate SS4 expression.
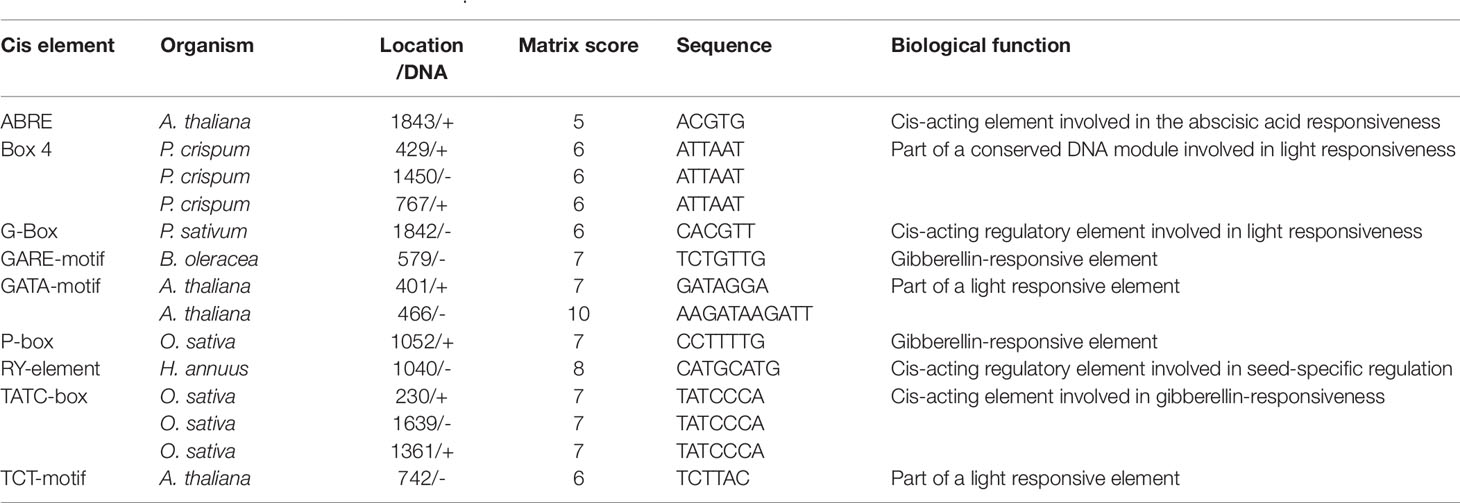
Table 4 Cis element in the promoter region of SS4.
Single-Nucleotide Variant in SS4
BLASTP searches of the eight SS4 genes were performed against a protein database containing the model plant Arabidopsis thaliana and related species, such as Corylus avellana, Quercus suber, and Juglans regia. The results identified four coiled-coil domains at the N terminal of SS4. Additionally, the SNV of the blank-nut mutants occured in the CC1 region (Figure 5). The sequence from the 40th to 49th amino acid in the CC1 region was highly conserved in all tested species. In the four blank-nut mutants, the 45th amino acid residue mutated from a hydrophilic glutamine (Q) to an alkaline arginine (R). At the same position, Full1–4 maintained a high degree of similarity with related sequences in C. avellana, Q. suber, and J. regia. The results of SS4 SNP heterozygosity analysis show that Empty1, 2, and 4 were heterozygous (0/1), while Empty3 was homozygous (1/1) at this SNV loci (Table S3).
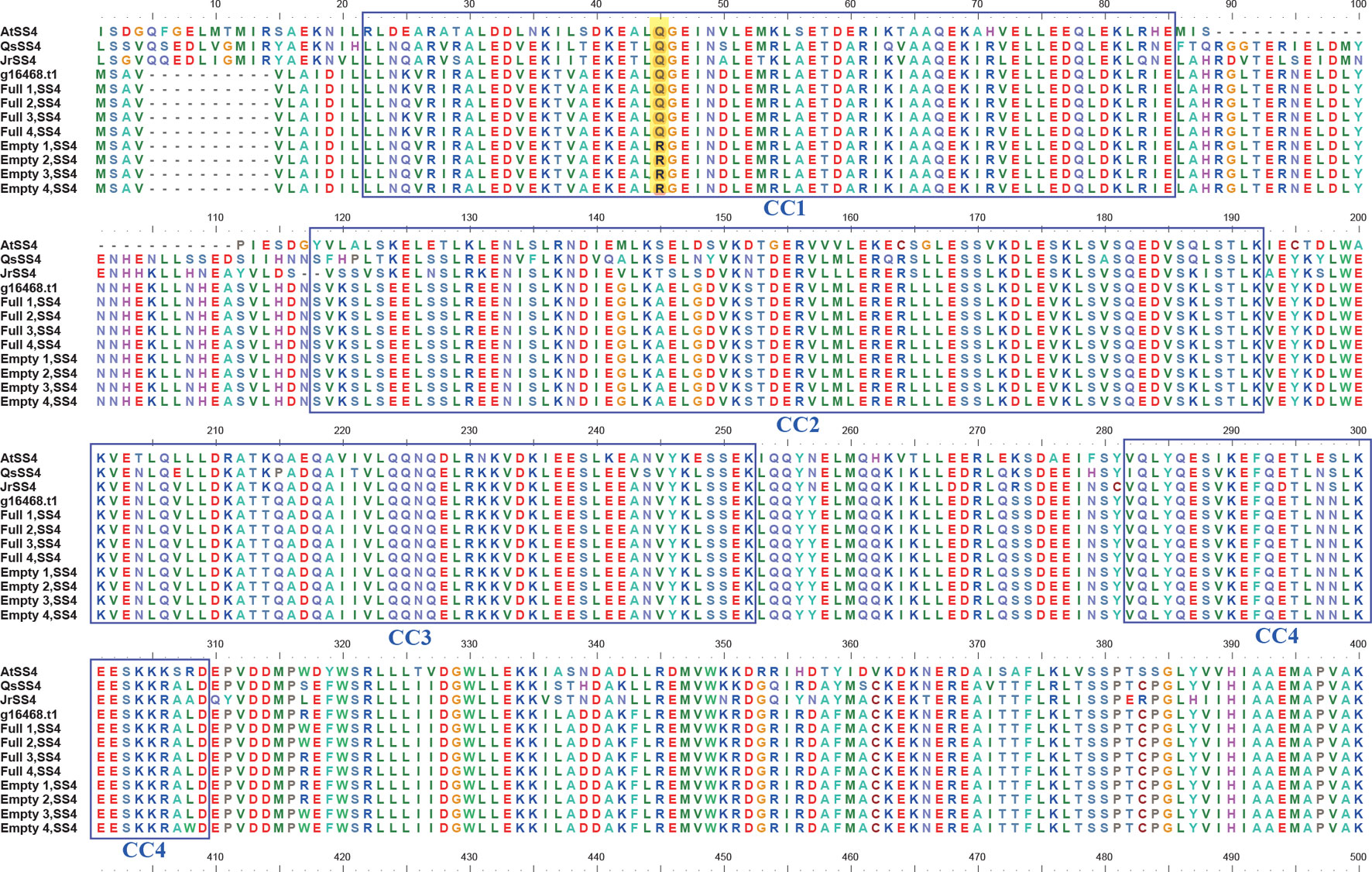
Figure 5 Comparison of the amino acid sequence of starch synthase 4 (SS4) in hazel (Corylus heterophylla) sampled in northeastern China with SS4 from other plants. AtSS4, Arabidopsis thaliana SS4; QsSS4, Quercus suber SS4; JrSS4, Juglans regia SS4; g16468.t1, Corylus avellana SS4; Empty, blank-nut mutant; Full, edible seed.
Observation of Starch Granules in Cotyledons
To demonstrate that the SS4 mutation causes embryo abortion, we used resin sections stained with periodic acid and Schiff reagent to analyze starch granule distribution in cotyledons. We observed multiple starch granules in all cotyledon cells of Full1–4 (Figures 6A–D). However, Empty1–4 cotyledons were nearly devoid of starch granules (Figures 6E–H). These results strongly suggest that an SNV of SS4 impaired starch biosynthesis in Empty1–4 embryos.
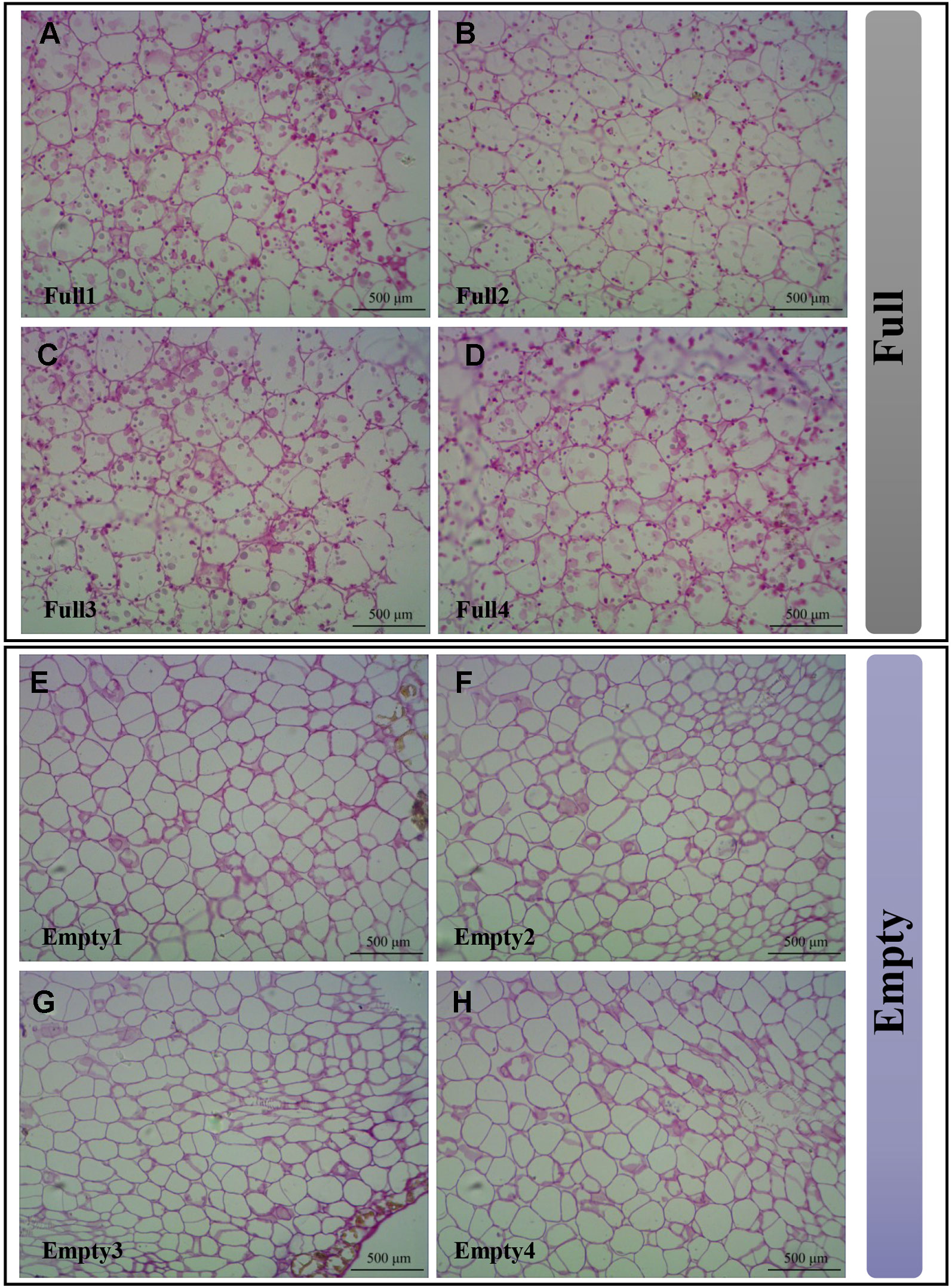
Figure 6 Starch granules observed in cotyledons of hazel (Corylus heterophylla) sampled in Northeast China. (A) Full1; (B) Full2; (C) Full3; (D) Full4; (E) Empty1; (F) Empty2; (G) Empty3; (H) Empty4. Empty, blank-nut mutant; Full, edible seed.
Statistical Analysis of Nut Characteristics
We found clear differences in blank versus wild-type nut characteristics from plants of the same orchard. Mutants had significantly higher nutshell weight, along with significantly lighter kernel and total nut weight. Consequently, mutant nuts exhibited a greater shell ratio and smaller kernel ratio (Table 5). These results suggested that photosynthate transport to nuts occurred preferentially in the shell rather than the kernel, leading to heavier shell weight among blank nuts. However, blank and wild-type nuts did not differ in diameter (Table 5). Occasionally, Full1 and Full 2 produced blank nuts, while the four mutants produced 100% blanks without exception (Table 5).
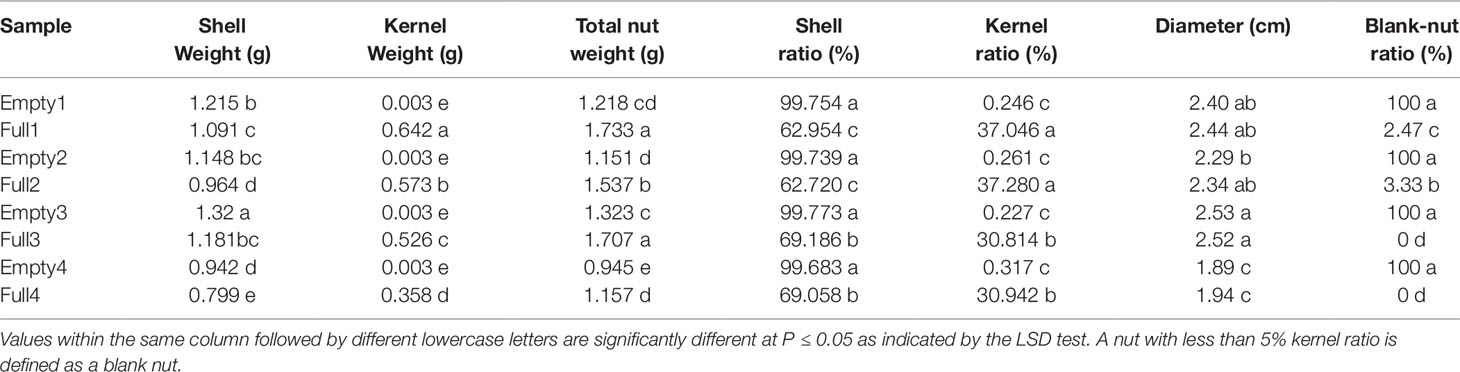
Table 5 Characteristics of nuts used in the present study, from eight hazel (Corylus heterophylla) trees sampled in northeastern China.
Discussion
Corylus heterophylla (family Betulaceae) is unique to China and the major source of hazelnut products in China (Liu et al., 2014b). We currently have little genetic information about this important agricultural product. Here, our assembled genomic sequences, mapped to the reference Jefferson hazelnut (C. avellana) genome, revealed that the two plants are closely related. Thus, the C. avellana genome information can also be used for C. heterophylla gene identification and mining. In Northeast China, many artificial hazel orchards have been created by modifying natural C. heterophylla forests, for the purposes of protecting forests. Compared with the nuts of C. heterophylla × C. avellana cultivars, those of C. heterophylla possess thicker shells, lower kernel ratio, higher blank-nut ratio, and poorer nut quality. However, C. heterophylla has exceptional cold resistance and thus the potential to be cultivated in more regions. Therefore, this was an unprecedented opportunity to identify important genes regulating kernel development, using our four blank-nut mutant germplasms. Evolutionary tree analysis based on obtained SNP information showed that Full2, Empty2, Full3, and Empty3 are on four different genetic branches (Figure S1), indicating that sexual propagation may play an important role in C. heterophylla population formation, despite the common use of tiller propagation.
Corylus heterophylla is an anemophilous species with pollen self-incompatibility and a heterozygous genome (2n = 22). Consistent with this, we found that most SNPs were heterozygous. The fact that 37.7–44.5% of SNPs were homozygous implies that selfing likely plays an important role in hazelnut population formation and evolution.
Important Candidate Genes That May Regulate Embryo Abortion
In the present study, paired comparisons based on whole-genome re-sequencing and SNP screening revealed 3,081 common SNPs in Empty1, Empty2, Empty3, and Empty4. Among these, 215 nonsynonymous SNPs, distributed across 178 candidate genes, appear to induce amino acid changes at the translation level, while 18 genes had the 1/1 homozygous SNP genotype. Homozygous SNP mutation in C. heterophylla indicates simultaneous base mutation in both alleles, which appears more likely than heterozygous SNP mutation to induce observable phenotypes. After filtering unannotated genes, integrated biological functions, and embryo-abortion characteristics, we identified a set of gene products implicated in regulating blank-nut formation, including ACC1, NHX2, UDP-glycosyltransferase 74E2 (UGT74E2), DEFECTIVE IN MERISTEM SILENCING 3 (DMS3), and DETOXIFICATION 43 (FRD3) (Table 6). We discuss each of these proteins briefly below.
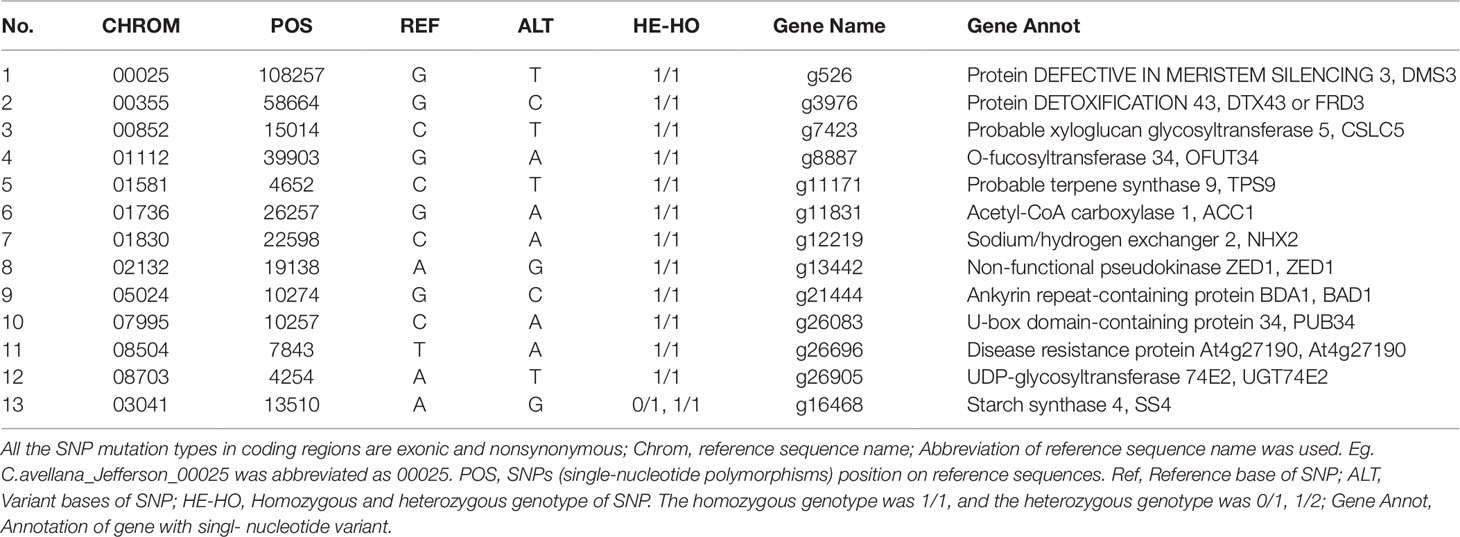
Table 6 Interest common, unique, and nonsynonymous SNPs in coding regions of Empty1, Empty2, Empty3, and Empty4.
Mature hazelnut is rich in unsaturated fatty acids. The homozygous SNP mutation of ACC1 may impair citrate conversion to long-chain fatty acids and trigger physiological disorders in ovules. Furthermore, ACC1 loss-of-function led to abnormal embryo morphogenesis and embryo lethality in transgenic Arabidopsis seeds (Baud et al., 2003).
NHX2 has vital effects on cellular pH and Na+/K+ homeostasis. The double-knockout mutants of nhx1/nhx2 caused significantly reduced growth, smaller cells, and shorter hypocotyls in etiolated seedlings, as well as abnormal stamens in mature flowers (Bassil et al., 2011).
UDP-glycosyltransferase 74E2 (UGT74E2) is an auxin glycosyltransferase. In Arabidopsis, UGT74E2 overexpression disrupted indole-3-butyric acid and auxin homeostasis, thus altering plant architecture while improving stress tolerance (Tognetti et al., 2010). Immunohistochemical analysis showed that auxin distribution was in enriched at the growth center of ovaries during early ovule formation, implying that the enzyme is important in regulating ovule development (Cheng et al., 2018b).
DNA methylation is an epigenetic alteration related to gene silencing. In the RNA-directed DNA methylation (RdDM) pathway, DMS3 is required in producing Pol V-dependent transcripts, suggesting a regulatory role in DNA methylation and gene silencing (Law et al., 2010). The total methylation ratio of hazel ranges from 44.61 to 48.68% before pollination, but then decreases by ∼4% after pollination, suggesting that epigenetic changes are an important mechanism for initiating ovary and ovule development (Cheng et al., 2015b).
FRD3 encodes a membrane protein belonging to the multidrug and toxin efflux family, which is involved in transporting small organic molecules. In Arabidopsis, frd3 mutant plants were defective in either iron-deficiency signaling or iron distribution, indicating that FRD3 is an important component of iron homeostasis (Rogers and Guerinot, 2002).
Taken together, homozygous SNP mutations in ACC1, NHX2, UGT74E2, DMS3, and FRD3 impair numerous important biological processes that would induce embryo abortion in ovules.
SS4 May Play an Important Role in Regulating Hazelnut Embryo Abortion
Starch is important in the metabolism of photosynthetic organisms, accumulating in chloroplasts during the day and degrading at night to supply energy for growth (Stitt and Zeeman, 2012). Over the long term, starch can also be stored in seed endosperm, tubers, and other storage organs, providing energy for plant growth, seed germination, and other biological processes (Toyosawa et al., 2016). Starch synthases are transglycosylases that elongate the α-1,4-glycoside bond through glycosyl transfer, using ADP as a glucose donor (Miura et al., 2018). SS4 specifically controls starch-grain abundance in the chloroplast. Deletion mutations of SS4 result in severely restrict starch grains per chloroplast. Moreover, the core of starch grains in the mutants also differed significantly from those of the wild type, indicating that SS4 participates in the initiation of starch-grain biosynthesis (Crumpton-Taylor et al., 2013; Raynaud et al., 2016). The N-terminal part of Arabidopsis SS4 (At4g18240) contains 543 amino acids, and the sequence is well conserved across the SS4 proteins of species sequenced to date (Raynaud et al., 2016). The N-terminal part of At4g18240 is divided into five segments: CC1 (coiled-coil domain 1), CC2, CC3, CC4, and CR (conserved region). The SS4 fragment containing CC1 and CC2 interacts with the FBN1b protein located on the plastoglobule surface, potentially facilitating SS4’s association with the thylakoid membrane (Gámez-Arjona et al., 2015). Deletion of a long coiled-coil region at the N terminus of SS4 (Roldán et al., 2010) prevents the enzyme from binding to fibrillin 1 and alters its localization in the chloroplast. Thus, SS4 localization and its interaction with fibrillins are mediated by the N-terminal segment (Gámez-Arjona et al., 2015). A rice (Oryza sativa L.) double-mutant SSIIIa and SSIVb (ss3a ss4b) generated spherical starch granules in seeds, whereas single mutants produced polyhedral starch granules similar to wild-type (Toyosawa et al., 2016). Overexpressing SSIV increased starch content in Arabidopsis leaves by 30% to 40%. For long-term storage of starch in potato tubers, SSIV overexpression increases tuber starch content and yield (Gámez-Arjona et al., 2011). Thus, SS4 is responsible for the initiation of starch-grain biosynthesis in both chloroplasts and storage organs, and its N-terminal determines localization in cells.
Our previous study indicated that photosynthates were not transported to abortive ovules in blank nuts (Liu et al., 2013). Our current findings imply that genes involved in transport could also regulate embryo abortion in hazelnut, and those involved in photosynthate transport might induce ovule developmental failure. Only the SS4 promoter region contained the RY-element, implying its seed-specific expression pattern. An SNV of SS4 showed that the 45th amino acid residue in the highly conserved CC region of N-terminal mutates from a hydrophilic glutamine (Q) to an alkaline arginine (R) in Empty1, Empty2, Empty3, and Empty4, whereas this region in Full1, Full2, Full3, and Full4 was similar to those in C. avellana, Q. suber, and J. regia. An SNV of SS4 at the N terminal might impair appropriate protein localization and induce the failure of starch-grain biosynthesis. This hypothesis is consistent with previous data showing ovule abortion soon after fertilization (Liu et al., 2012). We also confirmed that starch granules were absent in cotyledon cells of Empty1–4 and present in cotyledon cells of Full1–4. Therefore, the starch content in abortive ovaries was significantly lower than in developing ovule.
Of the four blank-nut mutants, three were heterozygous and one was homozygous at the 45th amino acid residue of the SS4 N-terminal. As it is diploid, mutation in one of paired chromosomes of C. heterophylla may be enough to induce the blank-nut phenotype, due to gene dosage effects. Thus, even in commercial hazel cultivars, a certain proportion of blank-nut formation is common (Beyhan and Marangoz, 2007). Consistent with a cis element in the SS4 promoter, comparative transcriptome analysis of developing and abortive hazelnut ovules showed that inhibiting gibberellin and activating abscisic acid biosynthesis may contribute to ovule abortion (Cheng et al., 2017). Finally, blank nuts possess heavier shells, implying that photosynthates were preferentially transported to the shell instead of the kernel. In summary, our results suggest that improper cellular localization of SS4 plays a vital role in regulating embryo abortion in hazelnut. These results further verify the biological function of SS4 in seed development through gain or loss mutation. Given that seedlessness is considered a valuable breeding trait in some species, our study provides a new idea for seedless cultivar development.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
JL and YC contributed to study conception and design, collection and/or assembly of data, data analysis and interpretation, and manuscript writing. SJ, XZ, and HH prepared samples and observed starch granules in cotyledons.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 31670681; 31770723) and the Science and Technology Research Project of The Education Department of Jilin Province (No. JJKH20191012KJ; JJKH20190996KJ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all donors who supported this research.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01465/full#supplementary-material
References
Amaral, J. S., Susana, C., Seabra, R. M., Oliveira, B. P. P. (2006). Effects of roasting on hazelnut lipids. J. Agric. Food Chem. 54, 1315–1321. doi: 10.1021/jf052287v
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bassil, E., Tajima, H., Liang, Y. C., Ohto, M. A., Ushijima, K., Nakano, R., et al. (2011). The Arabidopsis Na+/H+ antiporters NHX1 and NHX2 control vacuolar pH and K+ homeostasis to regulate growth, flower development, and reproduction. Plant Cell 23 (9), 3482–3497. doi: 10.1105/tpc.111.089581
Baud, S., Guyon, V., Kronenberger, J., Wuillème, S., Miquel, M., Caboche, M., et al. (2003). Multifunctional acetyl-CoA carboxylase 1 is essential for very long chain fatty acid elongation and embryo development in Arabidopsis. Plant J. 33 (1), 75–86. doi: 10.1046/j.1365-313X.2003.016010.x
Beyhan, N., Marangoz, D. (2007). An investigation of the relationship between reproductive growth and yield loss in hazelnut. Sci. Hortic. 113, 208–215. doi: 10.1016/j.scienta.2007.02.007
Cheng, Y., Liu, J., Zhang, H., Wang, J., Zhao, Y., Geng, W. (2015a). Transcriptome analysis and gene expression profiling of abortive and developing ovules during fruit development in hazelnut. PloS One 10 (4), e0122072. doi: 10.1371/journal.pone.0122072
Cheng, Y., Wang, J., Liu, J., Zhao, Y., Geng, W., Zhang, H. (2015b). Analysis of ovary DNA methylation during delayed fertilization in hazel using the methylation-sensitive amplification polymorphism technique. Acta Physiol. Plant 37 (11), 231. doi: 10.1007/s11738-015-1984-7
Cheng, Y., Zhao, Y., Liu, J., Yang, B., Ming, Y. (2017). Comparison of phytohormone biosynthesis and signal transduction pathways in developing and abortive hazelnut ovules. Plant Growth Regul. 81 (1), 147–157. doi: 10.1007/s10725-016-0196-5
Cheng, Y., Zhang, L., Zhao, Y., Liu, J. (2018a). Analysis of SSR markers information and primer selection from transcriptome sequence of Hybrid Hazelnut Corylus heterophylla × C. avellana. Acta Hortic. Sin. 45 (1), 139–148. doi: 10.16420/j.issn.0513-353x.2017-0281
Cheng, Y., Zhang, Y., Liu, C., Ai, P., Liu, J. (2018b). Identification of genes regulating ovary differentiation after pollination in hazel by comparative transcriptome analysis. BMC Plant Biol. 18 (1), 84. doi: 10.1186/s12870-018-1296-3
Crumpton-Taylor, M., Pike, M., Lu, K. J., Hylton, C. M., Feil, R., Eicke, S., et al. (2013). Starch synthase 4 is essential for coordination of starch granule formation with chloroplast division during Arabidopsis leaf expansion. New Phytol. 200, 1064–1075. doi: 10.1111/nph.12455
Ferreira, J. J., Garciagonzález, C., Tous, J., Rovira, M. (2010). Genetic diversity revealed by morphological traits and ISSR markers in hazelnut germplasm from northern Spain. Plant Breed. 129, 435–441. doi: 10.1111/j.1439-0523.2009.01702.x
Ferreira, J. J., Garcia, C., Tous, J., Rovira, M. (2009). Structure and genetic diversity of local hazelnut collected in Asturias (Northern Spain) revealed by ISSR markers. Acta Hortic. 845, 163–168. doi: 10.17660/ActaHortic.2009.845.20
Gámez-Arjona, F. M., Sandy, R., Paula, R., Angel, M. (2015). Starch Synthase 4 is located in the thylakoid membrane and interacts with plastoglobule-associated proteins in Arabidopsis. Plant J. 80, 305–316. doi: 10.1111/tpj.12633
Gámez-Arjona, F. M., Li, J., Raynaud, S., Baroja-Fernández, E., Muñoz, F. J., Ovecka, M., et al. (2011). Enhancing the expression of starch synthase class IV results in increased levels of both transitory and long-term storage starch. Plant Biotechnol. J. 9, 1049–1060. doi: 10.1111/j.1467-7652.2011.00626.x
Koboldt, D. C., Chen, K., Wylie, T., Larson, D. E., McLellan, M. D., Mardis, E. R., et al. (2009). VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285. doi: 10.1093/bioinformatics/btp373
Law, J. A., Ausin, I., Johnson, L. M., Vashisht, A. A., Zhu, J. K., Wohlschlegel, J. A., et al. (2010). A protein complex required for polymerase V transcripts and RNA-directed DNA methylation in Arabidopsis. Curr. Biol. 20 (10), 951–956. doi: 10.1016/j.cub.2010.03.062
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 1653–1654. doi: 10.1046/j.1440-1665.1999.0178e.x
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Liu, J., Cheng, Y., Yan, K., Qiang, L., Wang, Z. (2012). The relationship between reproductive growth and blank fruit formation in Corylus heterophylla Fisch. Sci. Hortic Amsterdam 136, 128–134. doi: 10.1016/j.scienta.2012.01.008
Liu, J., Cheng, Y., Liu, C., Zhang, C., Wang, Z. (2013). Temporal changes of disodium fluorescein transport in hazelnut during fruit development stage. Sci. Hortic Amsterdam 150, 348–353. doi: 10.1016/j.scienta.2012.12.001
Liu, J., Zhang, H., Cheng, Y., Kafkas, S., Güney, M. (2014a). Pistillate flower development and pollen-tube growth mode during the delayed fertilization stage in Corylus heterophylla Fisch. Plant Reprod. 27 (3), 145–152. doi: 10.1007/s00497-014-0248-9
Liu, J., Zhang, H., Cheng, Y., Wang, J., Zhao, Y., Geng, W. (2014b). Comparison of ultrastructure, pollen tube growth pattern and starch content in developing and abortive ovaries during the progamic phase in hazel. Front. Plant Sci. 5, 528. doi: 10.3389/fpls.2014.00528
Miura, S., Crofts, N., Saito, Y., Hosaka, Y., Oitome, N. F., Watanabe, T., et al. (2018). Starch synthase IIa-deficient mutant rice line produces endosperm starch with lower gelatinization temperature than japonica rice cultivars. Front. Plant Sci. 9, 645. doi: 10.3389/fpls.2018.00645
Nordström, K. J., Albani, M. C., James, G. V., Gutjahr, C., Hartwig, B., Turck, F., et al. (2013). Mutation identification by direct comparison of whole-genome sequencing data from mutant and wild-type individuals using k-mers. Nat. Biotechnol. 31, 325. doi: 10.1038/nbt.2515
Raynaud, S., Ragel, P., Rojas, T., Mérida, Á.(2016). The N-terminal part of Arabidopsis thaliana Starch Synthase 4 determines the localization and activity of the enzyme. J. Biol. Chem. 291, 10759. doi: 10.1074/jbc.M115.698332
Rogers, E. E., Guerinot, M. L. (2002). FRD3, a member of the multidrug and toxin efflux family, controls iron deficiency responses in Arabidopsis. Plant Cell 14 (8), 1787–1799. doi: 10.1105/tpc.001495
Roldán, I., Wattebled, F., Mercedes Lucas, M., Delvallé, D., Planchot, V., Jiménez, S., et al. (2010). The phenotype of soluble starch synthase IV defective mutants of Arabidopsis thaliana suggests a novel function of elongation enzymes in the control of starch granule formation. Plant J. 49, 492–504. doi: 10.1111/j.1365-313X.2006.02968.x
Silva, A. P., Ribeiro, R. M., Santos, A., Rosa, E. (1996). Blank fruits in hazelnut (Corylus avellana L.) cv. ‘Butler’: characterization and influence of climate. J. Hortic. Sci. 71, 709–720. doi: 10.1080/14620316.1996.11515451
Singh, R., Low, E. T., Ooi, L. C., Ong-Abdullah, M., Ting, N. C., Nagappan, J., et al. (2013). The oil palm SHELL gene controls oil yield and encodes a homologue of SEEDSTICK. Nature 500, 340–344. doi: 10.1038/nature12356
Solar, A., Stampar, F. (2011). Characterisation of selected hazelnut cultivars: phenology, growing and yielding capacity, market quality and nutraceutical value. J. Sci. Food. Agric. 91, 1205–1212. doi: 10.1002/jsfa.4300
Stitt, M., Zeeman, S. C. (2012). Starch turnover: pathways, regulation and role in growth. Curr. Opin. Plant Biol. 15, 282–292. doi: 10.1016/j.pbi.2012.03.016
Tognetti, V. B., Van Aken, O., Morreel, K., Vandenbroucke, K., de Cotte, B., De Clercq, I., et al. (2010). Perturbation of indole-3-butyric acid homeostasis by the UDP-glucosyltransferase UGT74E2 modulates Arabidopsis architecture and water stress tolerance. Plant Cell 22 (8), 2660–2679. doi: 10.1105/tpc.109.071316
Toyosawa, Y., Kawagoe, Y., Matsushima, R., Crofts, N., Ogawa, M., Fukuda, M., et al. (2016). Deficiency of starch synthase IIIa and IVb alters starch granule morphology from polyhedral to spherical in rice endosperm. Plant Physiol. 170, 1255. doi: 10.1104/pp.15.01232
Tranchida-Lombardo, V., Aiese Cigliano, R., Anzar, I., Landi, S., Palombieri, S., Colantuono, C., et al. (2017). Whole-genome re-sequencing of two Italian tomato landraces reveals sequence variations in genes associated with stress tolerance, fruit quality and long shelf-life traits. DNA Res. 25 (2), 149–160. doi: 10.1093/dnares/dsx045
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinf. 11 (1110), 11–33. doi: 10.1002/0471250953.bi1110s43
Wang, K., Li, M., Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, 16. e164. doi: 10.1093/nar/gkq603
Keywords: whole-genome re-sequencing, Corylus heterophylla, blank-nut mutants, embryo abortion, single-nucleotide polymorphism
Citation: Cheng Y, Jiang S, Zhang X, He H and Liu J (2019) Whole-Genome Re-Sequencing of Corylus heterophylla Blank-Nut Mutants Reveals Sequence Variations in Genes Associated With Embryo Abortion. Front. Plant Sci. 10:1465. doi: 10.3389/fpls.2019.01465
Received: 09 September 2019; Accepted: 22 October 2019;
Published: 13 November 2019.
Edited by:
Luigi Cattivelli, Council for Agricultural and Economics Research, ItalyReviewed by:
Paolo Boccacci, Italian National Research Council (IPSP-CNR), ItalyAna Paula Silva, University of Trás-os-Montes and Alto Douro, Portugal
Copyright © 2019 Cheng, Jiang, Zhang, He and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianfeng Liu, amlhbmZlbmdsaXUxOTc2QDE2My5jb20=
†ORCID: Jianfeng Liu, orcid.org/0000-0003-3220-8941