 Lei Shang1,2
Lei Shang1,2 Lin-Feng Li1*
Lin-Feng Li1* Zhi-Ping Song1
Zhi-Ping Song1 Yi Wang3
Yi Wang3 Ji Yang4
Ji Yang4 Chuan-Chao Wang5Shi-Yun Qiu1
Chuan-Chao Wang5Shi-Yun Qiu1 Jing-Xin Huang1,6Ming Nie1Lorne M. Wolfe1,7
Jing-Xin Huang1,6Ming Nie1Lorne M. Wolfe1,7 Bo Li1*
Bo Li1*- 1Coastal Ecosystems Research Station of Yangtze River Estuary, Ministry of Education Key Laboratory for Biodiversity Science and Ecological Engineering, Institute of Biodiversity Science, Institute of Eco-Chongming (IEC), Fudan University, Shanghai, China
- 2Institute of Forensic Science, Ministry of Public Security, Beijing, China
- 3Ministry of Education Key Laboratory of Contemporary Anthropology, School of Life Sciences and Institutes of Biomedical Sciences, Fudan University, Shanghai, China
- 4Key Laboratory of Biodiversity and Evolution, Fudan University, Shanghai, China
- 5Department of Anthropology and Ethnology, Xiamen University, Xiamen, China
- 6School of Energy and Environment Science, Yunan Normal University, Kunming, China
- 7School of Life Sciences, University of KwaZulu-Natal, Scottsville, South Africa
Biological invasion represents a global issue of concern due to its large negative impacts on native ecosystems and society. Elucidating the evolutionary history and genetic basis underpinning invasiveness is critical to understanding how alien species invade and adapt to novel environments. Smooth cordgrass (Spartina alterniflora, 2n = 6x = 62) is a notorious invasive species that causes heavily negative effects on native ecosystems worldwide. Here we addressed the evolutionary mechanisms underlying the invasion and dispersal history of this species along the China coast in the past decades. We employed nine microsatellites and three chloroplast fragments to investigate phylogeographic structure and genetic diversity of 11 native US and 11 invasive Chinese S. alterniflora populations. Demographic history simulation was also performed for both the native and invasive populations, respectively. Comparative genetic analyses of these natural populations revealed that although all the Chinese populations were introduced only once, high level of genetic diversity with weak geographic structure was observed. In particular, both the genetic features and mathematical simulation illustrated very recent population expansion in the Chinese populations. We found that genetic variants identified in native US populations were mixed in the Chinese populations, suggesting the recombination of these original variants during the invasion process. These genetic attributes indicate that Chinese populations might not have experienced a genetic bottleneck during the invasion process. High genetic diversity and genetic admixture might have contributed to the success of invasion of S. alterniflora in China. Our study provides a framework of how the smooth cordgrass spreads along the China coast as well as its potential genetic mechanisms underlying the invasion.
Introduction
Invasive alien species (IAS) represent one of the greatest threats to global biodiversity due to their negative impacts on the functioning of native ecosystems (IUCN, 1999; Mack et al., 2000; Lee, 2002; Prentis et al., 2008; Vilà et al., 2011). Current strategies for IAS control and management include the use of chemical toxins and pesticides, classical biological control, and habitat removal (Myers et al., 2000; Courchamp et al., 2003; Harvey-Samuel et al., 2017). However, these deployments often cause significant off-target effects so that the IAS are only temporally mitigated rather than being eradicated from the local ecosystems (Simberloff, 2009; Zarnetske et al., 2010). To this end, further investigations focusing on the genetic basis of invasiveness can not only offer vital information for elucidating evolutionary mechanisms underlying the successful establishment of IAS, but also provide an efficient guideline for controlling and management of the IAS in the native ecosystems. Indeed, many attempts have been made to explain how the genetic variability contributes to the colonization of IAS to novel environments (Tsutsui et al., 2000; Kolbe et al., 2004; Hodgins et al., 2013; Ueno et al., 2015). For example, high level of genetic diversity is supposed to contribute high adaptability to IAS to cope with diverse stresses caused by natural selection (Ahlroth et al., 2003; Prentis et al., 2008; Wang et al., 2012; Hufbauer et al., 2013). Likewise, additional work by comparing the genetic differences between the native and invasive populations has also revealed the crucial roles of genetic variability in shaping life history and reproductive and defensive characters (Blair and Wolfe, 2004). These empirical studies support the notion that genetic variability is a critical determinant in the invasion process of IAS.
The genus Spartina (Poaceae) includes 13–15 species that all are perennial herbs and native to the Atlantic and northeastern Pacific coasts (Ainouche et al., 2009). Cytological studies indicate that all extant Spartina species are either tetraploid, hexaploid, or dodecaploid, most of which exhibit highly invading ability (Marchant, 1968; Ayres et al., 2008a; Ainouche et al., 2012). Among these polyploid species, Spartina alterniflora (2n = 6x = 62) is originally distributed in the Atlantic and Gulf coasts of North America but have become one of the most aggressive invaders in global coastal ecosystems (Wang et al., 2006; Strong and Ayres, 2013). In the United Kingdom (UK), for example, the invasive species S. alterniflora was accidentally introduced during the 19th century (Ainouche et al., 2003). Homoploid hybridization between S. alterniflora and native Spartina maritima (2n = 6x = 60) gave rise to a sterile hexaploid hybrid S. x townsendii. Thereafter, chromosome doubling of the F1 hybrid of the two hexaploid species resulted in the formation of a fertile and high aggressive dodecaploid species Spartina anglica (2n = 12x = 100, 122, or 124) (Marchant, 1968). In the western coast of France, reciprocal crosses between the same two parental species had also led to the establishment of a morphologically distinct hexaploid species Spartina x neyrautii (Marchant, 1977). These recently formed polyploid species show relatively high tolerance to biotic and abiotic stresses so that they can occupy a wide range of habitats (An et al., 2007; Ainouche et al., 2009). Similar phenomenon has also been observed in the western coast of United States (US) where interspecific hybridization occurs frequently between the invasive S. alterniflora and native hexaploid hybrid Spartina foliosa (2n = 6x = 62) (Daehler and Strong, 1997). The resulting hybrids are genetically diverse and usually possess highly aggressive traits compared to their parental species in the salt marsh systems (Ayres et al., 1999; Ayres et al., 2003; Sloop et al., 2005; Ayres et al., 2008b).
In China, four Spartina species were introduced for the purpose of wetland restoration, tidal land reclamation, and saline soil mitigation (Zhang et al., 2004; Zuo et al., 2012). Of the four Spartina species, the dodecaploid S. anglica was introduced from England and Denmark in 1963 (Chung, 2006). Thereafter, the other three species, S. alterniflora, Spartina patens (2n = 4x = 40), and Spartina cynosuroides (2n = 4x = 40), were introduced from North American Atlantic coast in 1979 (Zuo et al., 2009; Zuo et al., 2012). To date, the three species, S. anglica, S. patens, and S. cynosuroides, are restricted to narrow ranges or almost disappeared from natural environments (An et al., 2007; Zuo et al., 2012). In contrast, the other species S. alterniflora has rapidly spread along the Chinese coast over the past decades and was officially listed as one of the top 16 invasive species in China (Wang et al., 2006; Zuo et al., 2012). These attributes offer the S. alterniflora an ideal system to address evolutionary mechanisms underlying the successful invasion of alien species. To this end, several previous studies attempted to elucidate how the genetic, morphological, and physiological factors contributed to the invasion of S. alterniflora. For example, historical investigation of the original sample collection records (both seeds and alive plants) revealed that all Chinese S. alterniflora populations were primarily originated from North Carolina, Georgia, and Florida of the US (Xu et al., 1989; Chen and Zhong, 1990; Bernik et al., 2016). While these original seeds of S. alterniflora were primarily transplanted in southeastern China, long-distance colonizing ability with fast growth rate has eventually resulted in a continuous distribution pattern across the coastal regions (Wang et al., 2006). In the US, high genetic diversity is observed in the native populations of S. alterniflora, especially most of the variants are explained by the differences among individuals within populations (Blum et al., 2007). As an invasive species originated through a single introduction, it is surprising that high genetic variability is observed in the Chinese populations (Wang et al., 2012; Bernik et al., 2016). However, these observations relied mainly on few samples and limited molecular markers.
The overarching goal of this study was to explore the evolutionary mechanisms underlying the introduction and subsequent invasion history of S. alterniflora in China. In this study, we collected a total of 1,227 accessions from 11 US native and 11 Chinese invasive populations. Based on this large sample size, we employed nine microsatellites and three chloroplast fragments to investigate the population structure and genetic variability between the US native and Chinese invasive populations. These genomic data allow us to evaluate if both the native and invasive populations show geographic structure and how these S. alterniflora populations spread along the China coast. In addition, we also performed a mathematical simulation to test whether or not the inter-population mixing among the three original populations contributed to the invasion success of S. alterniflora in China. Our study therefore provides an evolutionary perspective on how the invasive species S. alterniflora evolves in China.
Materials And Methods
Plant Materials and DNA Extraction
The species S. alterniflora possesses both sexual (self-compatible) and asexual reproductive strategies (Wang et al., 2006). Its seeds facilitate long-distance colonization through tidal water (Elsey-Quirk et al., 2009), and its rhizomes can promote strong clonal growth (Zhang et al., 2006). To obtain representative accessions from each wild population, two to five young leaves were sampled from each individual that spaced to each other at least 40 m apart. With this sampling strategy, we collected a total of 1,227 accessions from 11 US native and 11 Chinese invasive populations (Supplementary Table 1). In brief, the 11 invasive Chinese populations were collected along the coastline to represent its current distribution range in China. In addition, we also collected samples from 11 naturally distributed populations along the US eastern and southern coastline. Among the 11 native US populations, the 3 populations localized in North Carolina (U-MC), Georgia (U-SI), and Florida (U-TB) are believed to be the source of all Chinese S. alterniflora accessions (Chen and Zhong, 1990). Leaf samples were dried with silica gel and stored at −20°C. Genomic DNA was extracted from each accession using a Plant Genomic DNA Kit (Tiangen, Beijing, China).
Microsatellite Genotyping and Chloroplast Fragment Sequencing
Previous studies have characterized a series of polymorphic microsatellites from S. alterniflora (Blum et al., 2004; Sloop et al., 2005). We tested the transferability of 35 microsatellites using 24 accessions collected from different populations. Nine of these microsatellite loci (SPAR.02, SPAR.05, SPAR.06, SPAR.07, SPAR.09, SPAR.10, SPAR.11, SPAR.20, SPAR.27) that generated clear bands were selected for genotyping in this study (Supplementary Table 2). PCR amplifications were performed in a volume of 10 µl including 0.2 µl of DNA (20–50 ng), 1 unit Taq DNA polymerase (Takara, Dalian, China), 0.8 µl of dNTPs, 0.3 µl of each primer (10 µM), 1 µl of 10× PCR buffer (Mg2+ included), and 7.3 µl ddH2O. The forward primer for each primer pair was fluorescently labeled with FAM, HEX, or ROX (Supplementary Table 2). The PCR reactions were conducted on an ABI 9700 thermocycler: an initial denaturation of 5 min at 94°C, 30 cycles of 30 s at 94°C, annealing temperature (Ta) for 30 s (Ta for each primer pair is listed in Supplementary Table 2), 30 s at 72°C, and a final extension of 72°C for 5 min. The labeled PCR amplicons were then sized against ABI GS500LIZ standard on an ABI 3730 automated sequencer and scored with GeneMapper v.4.0 (Applied Biosystems, California, USA). As a complementary step, we also sequenced three chloroplast fragments (trnT2-rps4, trnT-L, and rbcL-psaI) of 207 representative samples, of which 92 and 115 accessions were selected from the USA and Chinese populations, respectively (Supplementary Table 1). All PCRs were carried out in a thermal volume of 50 µl: 2 µl of genomic DNA, 0.25 µl of Taq polymerase (Takara, Dalian, China), 4 µl of dNTP, 2 µl of each primer, and 5 µl of 10× buffer. PCR amplifications were generated by an ABI thermal cycler following a regime of one cycle at 94°C for 2 min, followed by 35 cycles of 94°C for 1 min, Ta (55°C for the trnT-trnL and 56°C for the trnT2-rps4 and rbcL-psaI) for 1 min, 72°C for 2 min, and a final extension at 72°C for 5 min. All PCR amplicons were then purified and sequenced using an ABI 3730 automated sequencer (Applied Biosystems, USA).
Data Correction, Population Genetic, and Phylogeographic Analyses
The program Microchecker v.2.2.3 (van Oosterhout et al., 2004) was used to correct the null allele in the microsatellite matrix. In addition, the species S. alterniflora can spread rapidly through its rhizomes. We therefore eliminated the data bias caused by the asexual reproduction based on the clonal diversity (the number of genotypes/sample size). A total of 173 accessions with genotype occurring more than once were excluded from the data matrix. To further minimize the effects of sample size on genetic indices, these populations with relatively large sample size (>43 samples) were recollected. To further explore the tendency that genetic diversity of native populations might vary with sample size, we performed a simulation of sampling, of which individuals from US populations were randomly sampled along a gradient of sample sizes: 2, 5, 25, 50, 100, 200, and 400. Ten replicates were conducted for each simulation for a given sample size. We also calculated Shannon’s information index (I) for each sampling and determined the proportion of I (I for the sampled population/I for US populations as a whole). The fitted curve was then added after a simulation of nonlinear model in the software R v.3.0.2 (R Core Team, 2013). Related computer source codes are given in Supplementary Text 1.
The corrected microsatellite data set was subjected to subsequent analyses of genetic diversity and population structure. Hardy–Weinberg equilibrium was tested for each microsatellite using Genepop v.4 (Rousset, 2008). It has been suggested that all Chinese S. alterniflora populations were introduced from the three US populations (U-MC, U-SI, and U-TB). To this end, we defined the 22 Chinese and US populations as three groups: (1) the source group containing the 3 USA populations (U-MC, U-SI, and U-TB) that were supposed to be the ancestors of all Chinese populations; (2) the non-source group including all the other 8 native USA populations; and (3) the introduced group that consisted of all the 11 Chinese populations (Supplementary Table 1). The software GenAlex v.6.5b3 (Peakall and Smouse, 2006; Peakall and Smouse, 2012) was employed to calculate the genetic diversity parameters at both the population and group levels, including the number of effective alleles (Ae), Shannon’s information index (I), individual inbreeding coefficient (Fi), and private allele (Pa). To further assess the genetic variation pattern within and between populations, analysis of molecular variation (AMOVA) and principal coordinate analysis (PCoA) were performed for all 22 populations using Arlequin v.3.5.1.2 (Excoffier et al., 2005). We also estimated genetic assignments of the 22 Chinese and US populations using STRUCTURE v.2.3.4 (Pritchard et al., 2000). The K values were calculated from 1 to 23 with 20 independent iterations. Each run was executed using the admixture model with an initial burn-in period of 104 and followed by 105 Markov Chain Monte Carlo (MCMC) iterations, and the delta K was used to determine the best genetic assignment.
Chloroplast sequences were checked against the original graphic files and aligned using MEGA v.5 (Tamura et al., 2011) with correction if necessary. Sequences of the three chloroplast fragments were combined as a single matrix for subsequent data analyses. In detail, haplotypes were identified on the basis of variable sites (including both of the single nucleotide polymorphism and insertion/deletion). Haplotype network was constructed using the program NETWORK v.4.6 (http://www.fluxus-engineering.com;Bandelt et al. 1999). Phylogeographic structure was tested for both native and introduced groups by comparing the values of Gst and Nst using Haplonst (Raymond and Rousset, 1995) and U-statistics (Pons and Petit, 1996). To further reveal the geographic distribution of these chloroplast haplotypes, we calculated the number of haplotypes within each of the 22 populations. As a complementary of the above microsatellite data analyses, we also estimated the average gene diversity at both the population (HS) and group (HT) levels using Haplonst (Raymond and Rousset, 1995). In addition, haplotype diversity (Hd), nucleotide diversity (π), and AMOVA were also calculated using Arlequin v.3.5.1.2 (Excoffier et al., 2005).
Demographic History Simulation
Theoretically, invasive species are expected to show relatively low genetic diversity due to the bottleneck effect during the invasion process. To experimentally test whether the invasive populations found in China have undergone a genetic bottleneck, we used the microsatellite data set to simulate the effective population size for both the native and introduced populations using Bottleneck v.1.2.02 (Cornuet and Luikart, 1996), with the infinite alleles model (IAM), stepwise mutation model (SMM), and two-phased model (TPM). Both the sign and Wilcoxon sign rank tests were applied to assess the deviation from mutation-drift equilibrium. In addition, we also applied the Bayesian evolutionary analysis by sampling trees (BEAST) to estimate the effective population size changes based on the chloroplast data set (Drummond et al., 2002; Drummond and Rambaut, 2007), with the HKY substitution model without I+G and strict clock (Richardson et al., 2001). The piecewise-linear skyline coalescence model was chosen with the number of groups (bins) setting as 10. Single MCMC chain was run for 20 million steps and sampled for every 1,000 steps. The log file was inspected in tracer for convergence of the chain and the effective sample size (ESS) values, and the Bayesian skyline reconstruction was run. Using the Bayesian skyline plots generated from BEAST, we calculated the rate of population growth per year. Each skyline plot consisted of 100 smoothed data points. The initial population size was set as the minimum population size during the period immediately preceding population growth. We also estimated the population growth rate for the interval in our data where the growth was the fastest. We chose the exponential growth equation for this analysis as suggested in Gignoux et al. (2011): r=ln(Nt/N0)/t, where r represents the population growth rate per year, N0 is the initial population size, and t is the time since growth began. To further estimate the genetic contribution rate from the native populations to the introduced populations, we used a, b, (1 – a − b) to respectively represent the relative genetic contributions of the 3 source populations (U-MC, U-SI, and U-TB) to all the 11 Chinese populations. The probability obtained from observed alleles and their frequencies in Chinese populations was calculated as follows:
where i ranges from the first microsatellite locus SP02 to the ninth locus SP27; j ranges from the first allele to the last allele for each locus; fij,MC, fij,SI, and fij,TB represent the frequencies of allele j at locus i in populations U-MC, U-SI, and U-TB, respectively; and nij,China represents the number of allele j at locus i in Chinese populations. With the feasibility of calculation considered, we applied the logarithmic transformation to equation (1). We then obtained:
A grid search was then conducted to find the optimal solution to equation (2), with a, b, and (1 – a − b) ranging from 0 to 1. To further simulate population genetic mixing, we explored the effect of admixture on amount of genetic variation with the assumption that the three native source populations were mixed evenly. Thus, N was taken as the original sample size for each native source population. We then conducted a simulation of genetic mixing to produce observed alleles frequencies (where expected = admixture from three source populations and observed = Chinese) and then generated the optimal value of N. Based on allele frequency data of native source populations estimated above, we next calculated the expected distribution of alleles at the nine microsatellite loci for Chinese populations based on the formulas a×2N for U-MC, b×2N for U-SI, and (1 – a − b)×2N for U-TB, respectively. All these simulations were performed using the approximate Bayesian computation (ABC), with the times of repetition for each N being set to 1 billion. The computer codes for simulation are given in the Supplementary Text 2, which were run directly on LINUX operating system.
Results
Genetic Diversity and Variation Pattern
All nine microsatellite markers used in this study were successfully amplified across all the 1,227 samples. As the S. alterniflora is an allohexaploid species, we examined whether or not the null alleles existed in the microsatellite matrix. At the population level, all the 22 populations showed different degrees of null alleles in the nine microsatellites, with the Chinese population C-NF only possessing null alleles in the locus SP27 but the source US population U-SI having null alleles across all microsatellite markers (Supplementary Table 3). At the locus level, the nine microsatellites harbored null alleles, ranging from 5 populations at the locus SP20 to 20 populations at the locus SP27 (Supplementary Table 3). While the above statistics identified null alleles in the microsatellite matrix, most of which exhibited low null allele frequency (<0.2) across the data matrix. To eliminate data bias in subsequent data analyses, all these detected null alleles were corrected at each locus. In addition, we also assessed clonal index to examine the effects of asexually reproductive strategy on the estimation of genetic diversity. Our results revealed that while the clonal diversity varied among the 22 populations, both US (average value = 0.87) and Chinese (average value = 0.85) groups showed similar level of clonal diversity (Table 1). To further test whether the sampling size affected the assessment of genetic diversity, we simulated the Shannon’s information index with different numbers of sampling size. The variation tendency of the proportion of the Shannon’s information index reached saturation curve very fast (sampling size < 25) (Supplementary Figure 1). These features suggest that the estimation of genetic diversity was not significantly affected by the difference in sampling size.
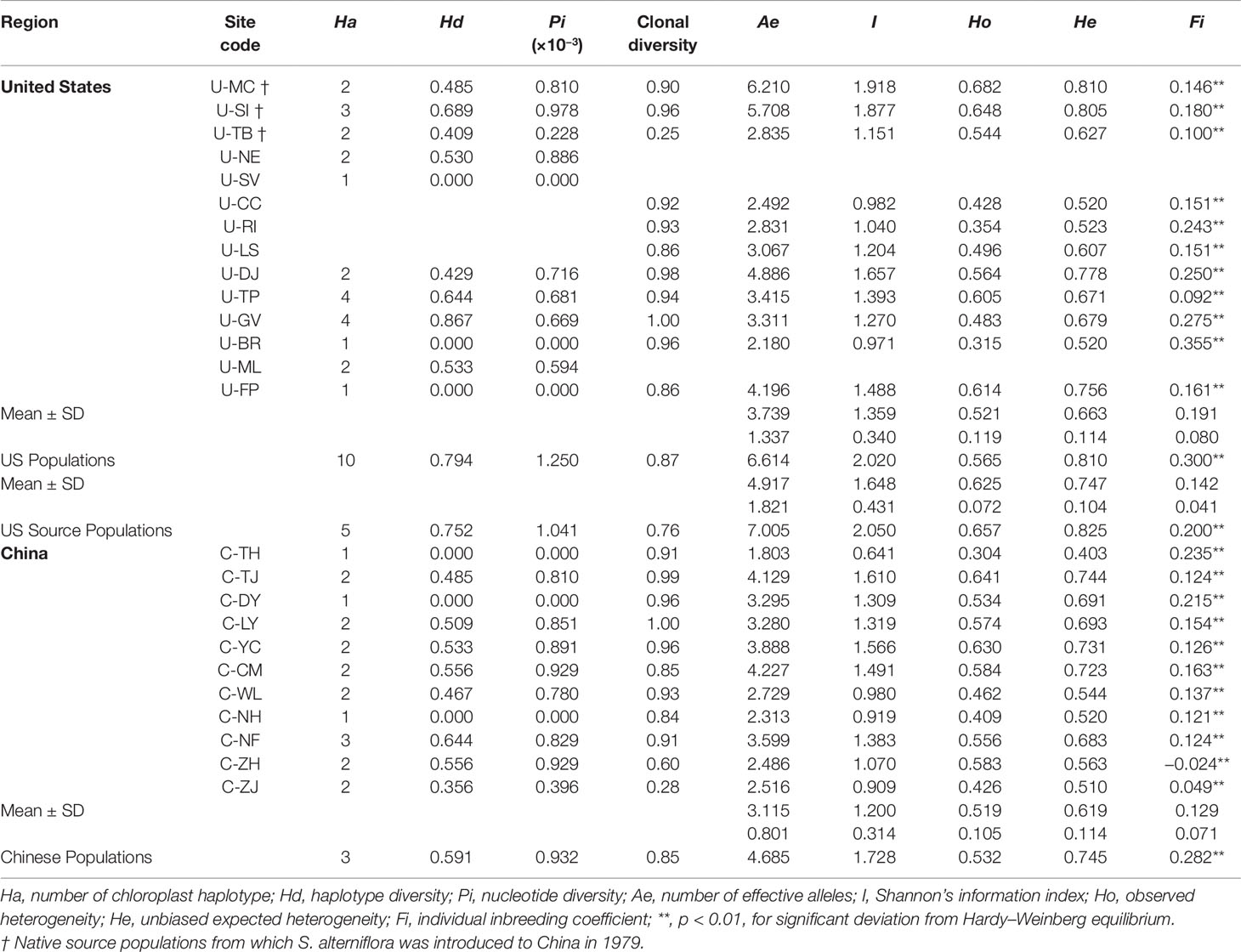
Table 1 Measures of clonal and genetic diversity of native and invasive Spartina alterniflora.
Based on the corrected microsatellite data matrix, we calculated the genetic diversity parameters at both the population and group levels. At the population level, the number of mean Ae varied from 1.803 in the Chinese population C-TJ to 6.210 in the US population U-MC (Table 1). At the group level, compared to the 11 Chinese populations (Ae = 4.685), the 11 US populations (Ae = 6.614) harbored relatively higher genetic diversity, and this trend was more pronounced in the 3 US source populations (Ae = 7.005). Similar phenomena were also observed in the other three genetic diversity parameters (I, Ho, and He) where the population C-TJ and U-MC harbored the lowest and highest genetic diversity among the 22 populations, respectively; and the US group showed relatively high genetic diversity compared to the Chinese group (Table 1). Through analyzing the individual inbreeding coefficient value (Fi), all these samples were found to show significant deviation from Hardy–Weinberg equilibrium at both the population and group levels. For the three chloroplast fragments, a total of nine sequence polymorphisms corresponding to 10 haplotypes were identified across the 207 individuals (Supplementary Table 4). All the 10 haplotypes were identified in the US populations, but the Chinese population only possessed 3 haplotypes (Table 1). The level of genetic diversity (Hd and π) varied among the 22 populations, with 6 populations (U-SV, U-BR, U-FP, C-TH, C-DY, and C-NH) showing no sequence polymorphisms and the populations U-GV (Hd = 0.867) and U-SI (π = 0.978 × 10 −3) harboring higher Hd and π values (Table 1). Being consistent with above microsatellite data set, the overall 11 US populations (Hd = 0.794, π = 1.250 × 10−3) exhibited relatively higher genetic diversity compared to all the 11 Chinese populations (Hd = 0.591, π = 0.932 × 10−3) (Table 1).
Population Genetic and Phylogeographic Structure
Bayesian genetic assignments revealed that the three northeastern US populations possessed a genetic cluster (mostly in green color) that is different from the four Caribbean populations (mostly in red color), and the remaining four populations exhibited a mixed genetic structure (Figure 1A). In contrast, while both genetic clusters (red and green colors) were shared between the US and Chinese populations, 9 of the 10 Chinese populations showed basically identical genetic assignments (mostly in green color) (Figure 1A). To further examine this genetic structure, we performed PCA analysis at both the population and group levels. Being consistent with the results of Bayesian genetic assignments, no obvious genetic divergence was found between the US and Chinese groups (Figure 1B). At the population level, moderate genetic divergence was observed between the four northeastern and three Caribbean populations (Figure 1C). In contrast, no genetic divergence was found between the source US populations and Chinese populations (Figure 1D). We also estimated the distribution pattern of the 22 populations at both the population and group levels. As revealed by the AMOVA, 80.83–92.74% of the variances were explained by the within-population variation for the source US native, non-source US native, Chinese invasive, and overall populations (Table 2). In contrast, 7.26–17.44% of the total variance was accounted by the among-population variation. In addition, we found that only 2.37% of the variance was from the between US and Chinese groups, being consistent with above genetic assignment and PCA results that no clearly genetic divergence is found between the US and Chinese populations.
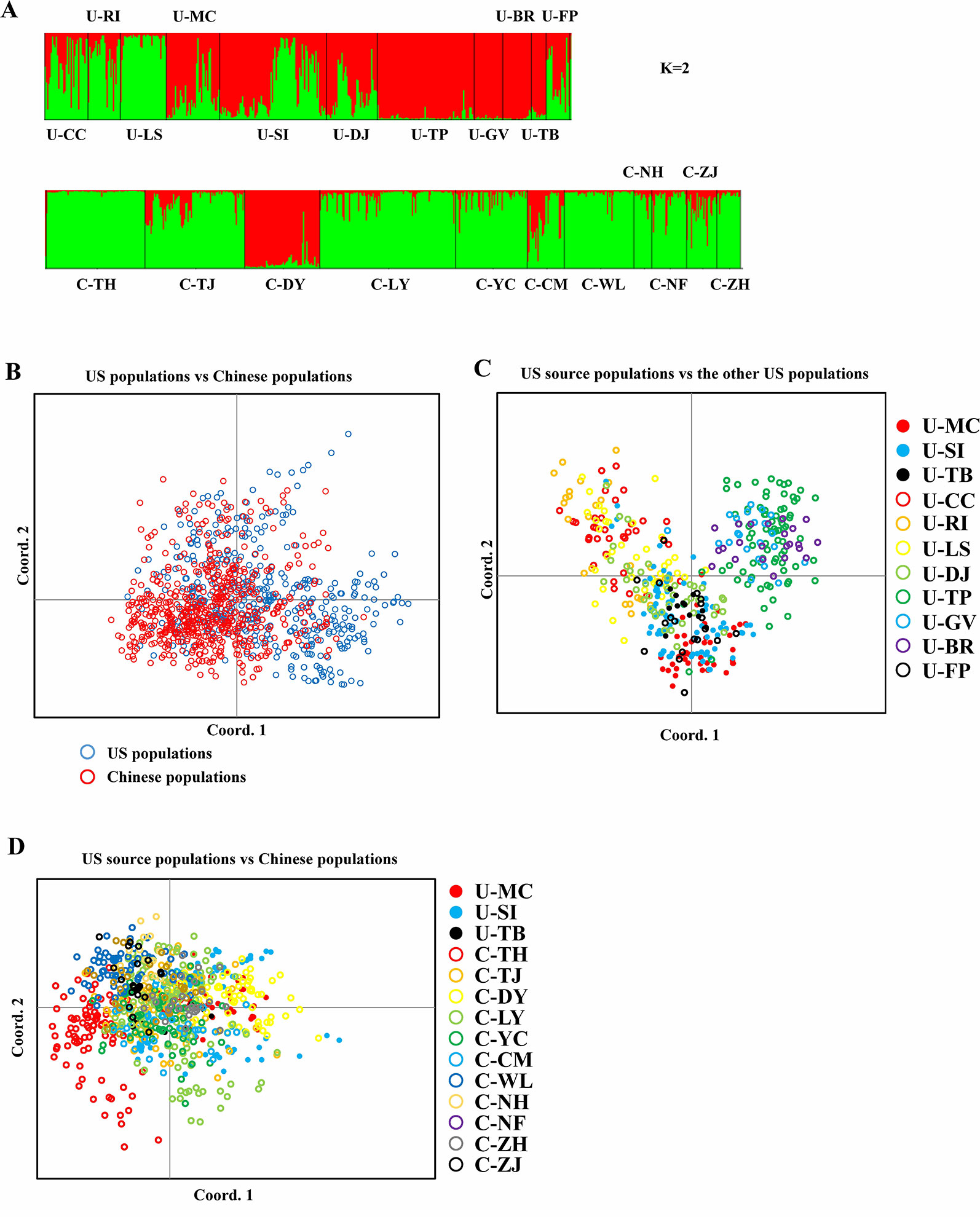
Figure 1 Spartina alterniflora populations. (A) Bar plot estimated by STRUCTURE representing assignments of genotypes of S. alterniflora from all populations to each cluster (K = 2). (B) PCoA of S. alterniflora between US and Chinese populations. (C) PCoA of S. alterniflora between US source populations and the other US populations. (D) PCoA of S. alterniflora between US source populations and Chinese populations.
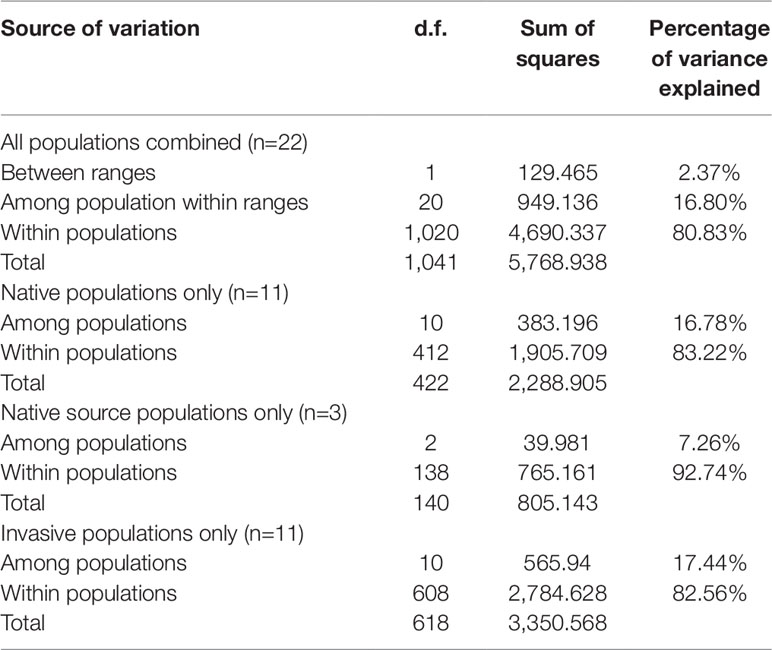
Table 2 Results of AMOVA showing distribution of genetic variation of S. alterniflora in US and China based on microsatellite dataset.
Although the above-population genetic analyses revealed moderate genetic differentiation between northern US and Caribbean populations, no significant phylogeographic structures were found within either of the US and Chinese groups (U-statistics, p > 0.05). However, the haplotype network showed that seven minor haplotypes (H4-H10) were specific to the US populations, whereas the three major haplotypes were shared between the US and Chinese groups (Supplementary Figure 2). We also examined the correlations between the 10 chloroplast haplotypes and their geographic distributions. In the US group, the haplotypes found in the northeastern US populations were highly different from the Caribbean populations (Figure 2A). In the Chinese group, a weak correlation between the haplotypes and geographic distribution was also found, with the northern and southern populations mainly consisting of the H1 and H3 haplotypes respectively, and the H2 haplotype widely distributed across the China coast (Figure 2B). Comparing the 11 Chinese populations with the 3 source US populations revealed that the haplotype H1 was found to exhibit a relatively higher proportion in the northern populations and the haplotype H3 was specific to the southern populations (Figures 2A, B). Unlike the microsatellite data set, percentages of variances explained by the among-population variation (39.58–43.86%) were slightly lower than by within-population variation (56.14–60.42%) (Table 3). In addition, the genetic differentiation between populations was higher in the chloroplast fragments (FST = 0.400–0.439) compared to the microsatellite data set (FST = 0.070 – 0.170) (Tables 2 and 3).
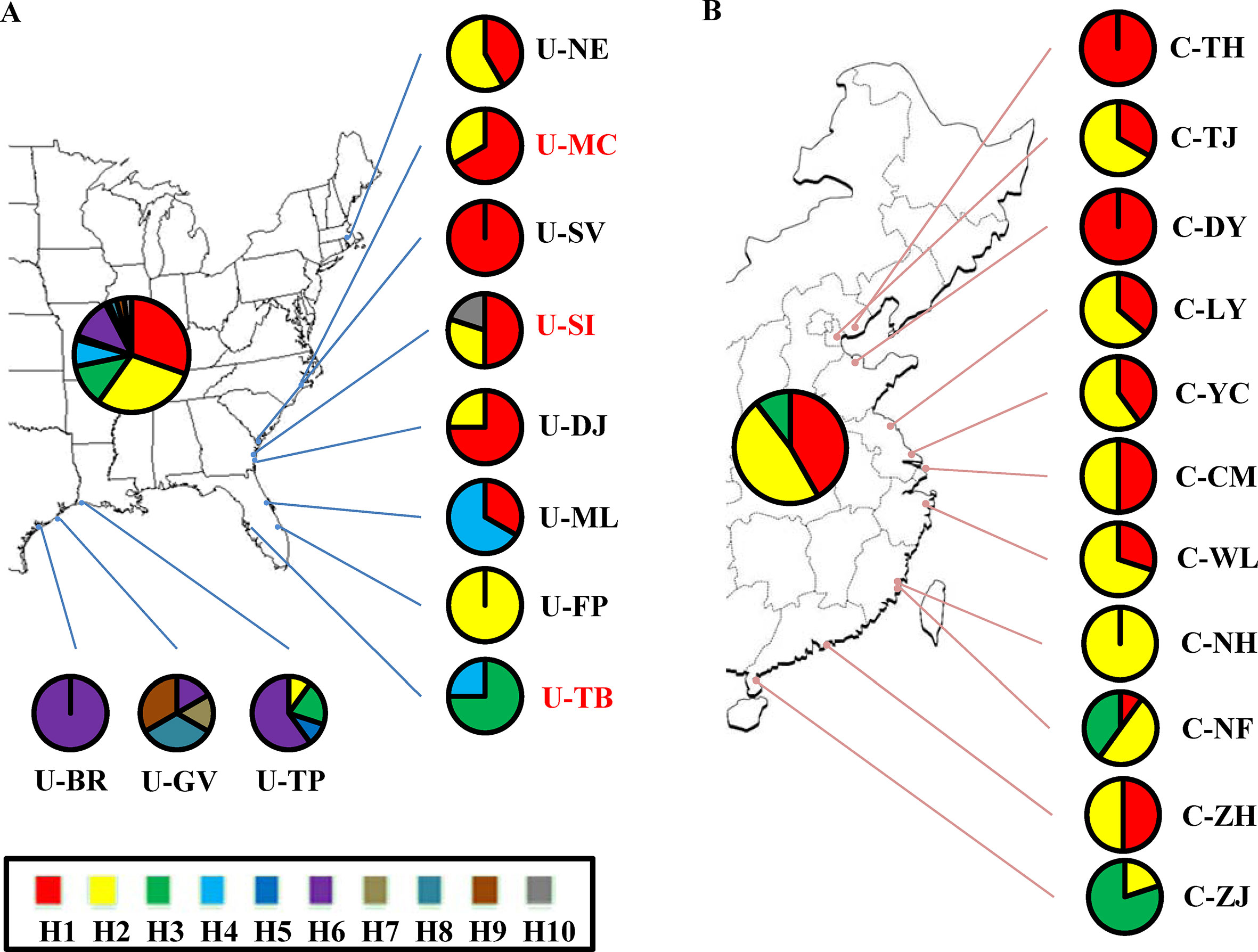
Figure 2 Geographic distribution of 10 chloroplast haplotypes detected in S. alterniflora. Pie charts represent the proportion and distribution of haplotypes across (A) native US and (B) invasive China ranges. Population and haplotype names in the map are the same as those in Table 1 and Supplementary Figure 2.
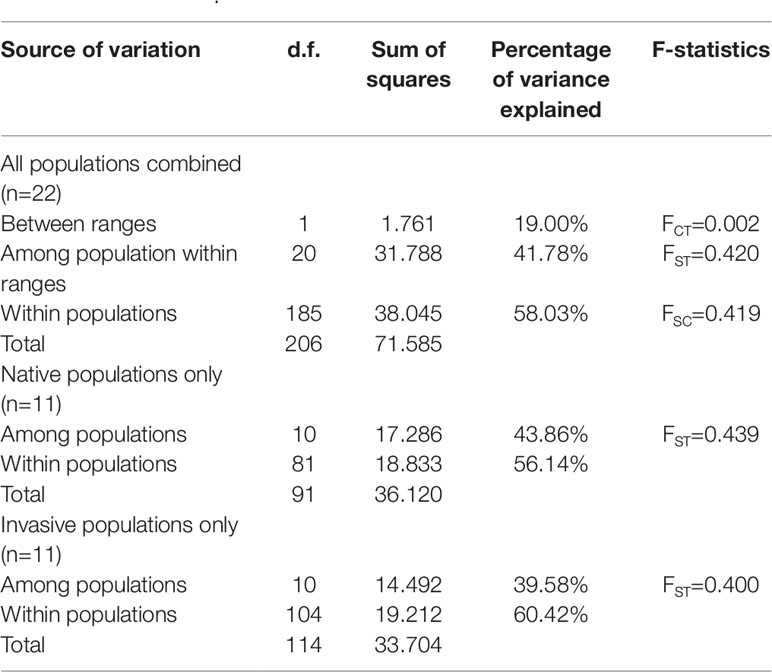
Table 3 Analysis of molecular variance for populations of S. alterniflora based on chloroplast dataset.
Demographic History and Colonization Route
We simulated how the S. alterniflora spreads in China and whether it experienced a genetic bottleneck during the invasion process. The simulation of the bottleneck effect based on microsatellite data set indicated that none of the 22 native and invasive populations has experienced significant bottleneck across three models (Table 4). We also estimated contributions of the 3 source US populations to the 11 Chinese populations. Results from the ABC simulation revealed a bell-shaped tendency for matching frequency at different sample sizes (Supplementary Table 5). The optimal sample size of 61 showed the highest matching frequency for each native source population, indicating that only a small sample size could result in the genetic diversity of current Chinese populations (Supplementary Table 5). Based on the simulation results, we found that the US source population U-SI contributed 76% of genetic constitution to the Chinese populations, which was obviously higher than those of U-MC (22%) and U-TB (2%). To further address the colonization history of these Chinese populations, we identified a total of 20 private alleles that were present in the three source US populations but absent in the other US populations (Supplementary Table 6). Analyzing the distribution pattern of these private alleles in the Chinese populations, we found that five accessions of the population C-YC have novel combinations (genotypes) of these private alleles (Supplementary Table 6), which suggested the possibility of admixture history in the Chinese populations. To assess whether or not the native US and invasive Chinese groups had undergone genetic bottleneck in the long-term evolutionary process, we estimated the effective population size for each of the two groups and at the whole species level. Our Bayesian simulations revealed that the native US group as well as the total species experienced a rapid increase in effective population size during the past 1,500 and 2,500 years (Supplementary Figures 3A, B). In contrast, the curve of the Chinese populations stretched very slowly, suggesting that S. alterniflora did not experience population expansion until 35 years ago (Supplementary Figure 3C). In addition, we also found that the growth rate of Chinese populations (growth rate = 0.02) was much higher than those of the native US group (growth rate = 0.0005) and the whole species (growth rate = 0.0016) (Supplementary Figure 4). It was broadly matched with the rapid invasion history of S. alterniflora in China.
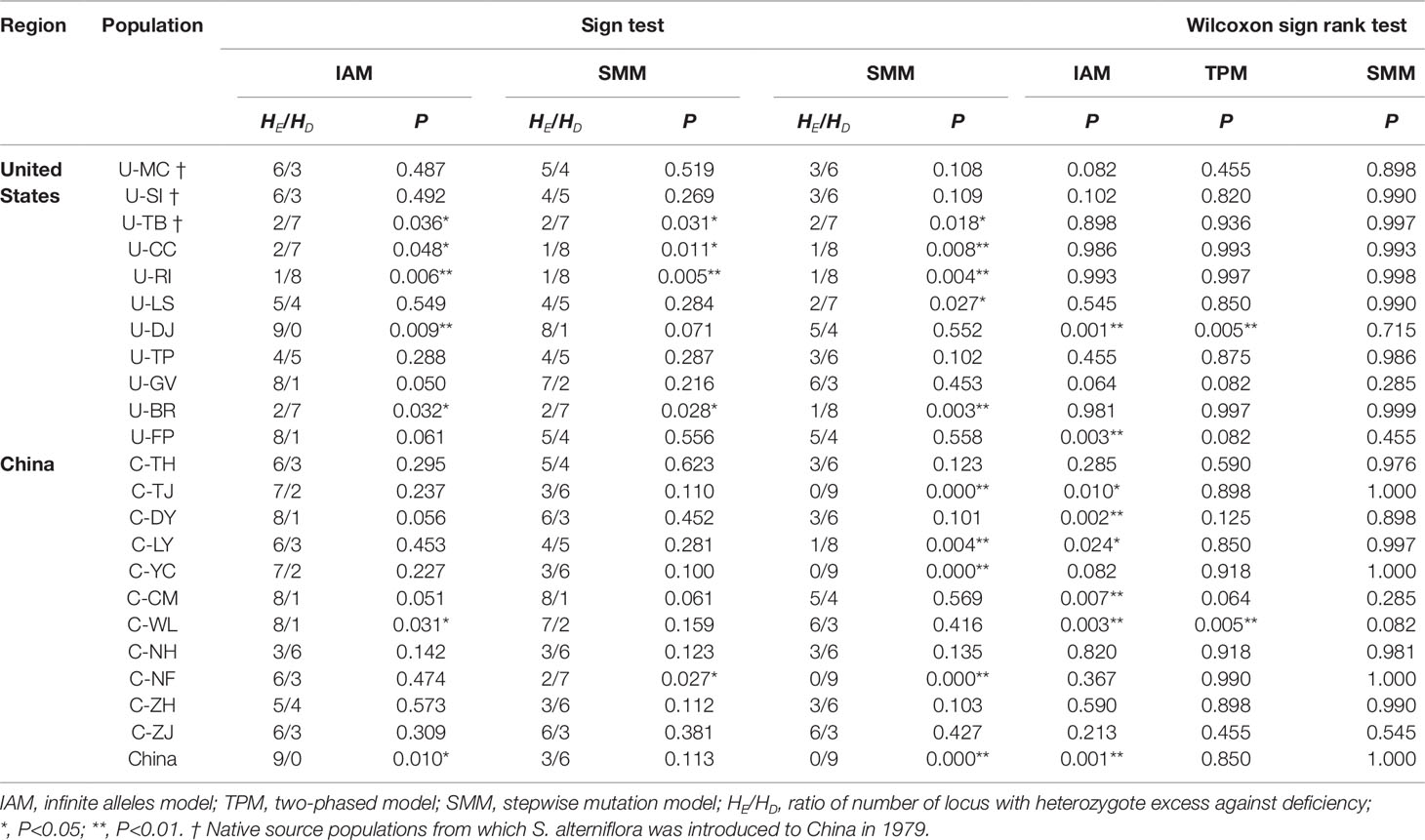
Table 4 Sign test and Wilcoxon sign rank test of bottleneck estimates in S. alterniflora populations.
Discussion
Demographic and Invasion History of Chinese S. alterniflora Populations
S. alterniflora is a worldwide invasive species that possesses several mechanisms to enhance salt tolerance (e.g., ion exclusion at root level and secretion in leaves through salt glands), all of which confer it exceptionally high adaptability to salinity and tidal submersion (Bradley and Morris, 1991; Daehler and Strong, 1997; Bedre et al., 2016). Therefore, it is not only a dominant salt marsh grass in its native range of Atlantic and Gulf coasts of the North America but also a successful colonizer in the other coastal regions as an invasive species (Ainouche et al., 2012). In case of China, S. alterniflora has spread very fast along the coast over the past 40 years, whereas all current invasive Chinese populations are originally planted in the Luoyuan town of Fujian province (Southeastern China) (Xu et al., 1989; Wang et al., 2006). In this study, our results from both the microsatellite and chloroplast data sets revealed weak geographic structure between the northeastern Atlantic and Caribbean S. alterniflora populations, being broadly consistent with a previous study that found the two geographic groups to be genetically diverged from each other (Blum et al., 2007). This phylogeographical structure observed between the two native geographic groups can be explained by either distinct environmental conditions or demographic histories. As expected, our demographic simulations revealed very recent population expansion (∼2,500 years) of S. alterniflora in North America. Given that the southern Atlantic coast of US (e.g., Florida) is possibly an independent refugee during the last glacial maximum, we propose that distinct recolonization routes of the two geographic groups might be partially responsible for the observed genetic divergence. In addition, environmental differences between the local habitats as well as migration barriers are also potential factors that might have contributed to the observed geographic patterns (Blum et al., 2007).
The three source US populations (U-MC, U-SI, and U-TB) showed mixed genetic structure, and most of the genetic variability was identified in the Chinese populations. This observation is further confirmed by the demographic simulation that no significant genetic bottleneck was found in the Chinese populations. Instead, obvious recent population expansion with high growth rate was detected in these invasive populations, which is consistent with the rapid invasion history of S. alterniflora in China. This demographic attribute is different from that observed in S. alterniflora of the US west coast as well as some other invasive species, in which founder effect and genetic bottleneck are common features during the colonization and range expansion process (Meimberg et al., 2006; Blum et al., 2007; Zhang et al., 2010). High genetic diversity of invasive species is possibly due to the multiple introductions or admixture genetic stock of the source populations (Genton et al., 2005; Kelager et al., 2013). In our case, all the Chinese populations were introduced only once and primarily planted together in the Fujian province of southeastern China (Xu et al., 1989). To this end, while the three source US populations showed differentially genetic contributions, it is more likely that high genetic diversity observed in the Chinese invasive populations is possibly because of admixture of the independent genetic stocks of the three US source populations. With regard to the invasive routes of S. alterniflora, the chloroplast network analyses revealed weak geographic structure among these Chinese populations, with the northern Chinese populations possessing chloroplast haplotypes highly similar to the two northern source US populations (U-MC and U-SI) and the southern Chinese populations harboring the unique haplotype identified in the U-TB population. Of significance, the two southern invasive populations, C-NH and C-NF, are geographically very close to the primary transplanting location. These findings suggest independent dispersal routes of the northern and southern Chinese populations during the invasion process.
High Genetic Diversity Followed By Admixture of Independent Genetic Stocks Enhances the Species Invasiveness
The importance of high genetic diversity to the successful establishment of alien species has been well recognized in many different invasive species (Prentis et al., 2008; Wang et al., 2012; Hufbauer et al., 2013). As reported in the European and western US coastal regions, hybridization between the invasive S. alterniflora and native congeneric species not only reunites diverged genomes within the same nucleus but also confers aggressive traits that allow the alien species to adapt to local environments (Strong and Ayres, 2013). A similar model of hybridization with adaptation is also widely observed in the other invasive plants [reviewed in Schierenbeck and Ellstrand (2009); Rius and Darling (2014)]. On the other hand, multiple introductions or single origin from various sources offer another strategy to increase the adaptability of IAS (Genton et al., 2005; Kelager et al., 2013). For example, high allelic diversity detected in the introduced populations of garlic mustard (Alliaria periolata) is thought to result from multiple introductions (Durka et al., 2005). However, our population genetic and demographic history analyses revealed very recent single origin followed by rapid population expansion of these Chinese populations, indicating that high adaptability of the Chinese populations cannot be explained solely by the above evolutionary mechanisms. Given that all plants collected from the three source US populations were mixed and grown within the same marsh area, we propose that intraspecific genetic admixture might be one of the potential factors acting as a stimulus in the invasion process. In fact, evidence of inter-population mixing has been reported in the other invasive species (Rosenthal et al., 2008; Culley and Hardiman, 2009). In our case, genetic evidence that supports this hypothesis is the observed recombination of private microsatellite alleles in the Chinese population. In addition, similar evidence is further observed in the chloroplast data set that haplotypes of the three source US populations co-occur in the same population. While this kind of genetic evidence is not widely present in all the Chinese populations, it at least suggests the possibility of intraspecific genetic admixture of the three source US populations in the process of invasion. Correlations between the interspecific/intraspecific hybridization and successful invasion have also been reported in other recent studies (Fitzpatrick and Shaffer, 2007; Strong and Ayres, 2009; Keller and Taylor, 2010; Rius and Darling, 2014).
While above analyses demonstrate the importance of intraspecific genetic admixture, the contribution of high genetic diversity harbored in the source US populations cannot be ignored. It has been well documented that high level of genetic diversity shows positive effects on the adaptability of species in responses to varying environments and colonization (Forsman, 2014). As demonstrated in previous experimental study (Wang et al., 2012), high genetic diversity of the invasive S. alterniflora populations can not only increase the ability to adapt to local environments, but also reduce the species richness of native species such as Scirpus mariqueter (Cyperaceae). In this study, our results illustrate that all the Chinese populations harbor high genetic diversity and show no significant genetic differentiation from the source US populations. In particular, mathematical simulation indicates that sampling size of the three source US populations is enough to represent the genetic constitution of the S. alterniflora in China. In addition, an opposite example is the congeneric invasive species S. anglica in which low genetic diversity is thought to be a potential factor that has resulted in its rapid decrease in its range in China (An et al., 2004). Taken together, our findings based on multiple microsatellite and chloroplast markers suggest that high genetic diversity of the source US populations followed by genetic admixture of the three independent genetic stocks contributed to the successful establishment of S. alterniflora in China. Further investigations by using genome-wide scanning might be able to provide more comprehensive evidence to the successful establishment of S. alterniflora in China. Specifically, four Spartina species (S. alterniflora, S. anglica, S. patens, and S. cynosuroides) have been introduced in China and co-occur in natural habitats. It remains a worthwhile issue to examine whether interspecific hybridizations have conferred adaptability to the S. alterniflora.
Data Availability Statement
All data sets generated in this study are included in the article/Supplementary Material. All DNA sequences can be accessed in GenBank with the accession numbers MK675746–MK675756.
Author Contributions
LS, L-FL, and BL designed the study. LS, S-YQ, and J-XH performed the experiment. Genetic data analysis was done by LS, with the help of Z-PS and C-CW, and the mathematical simulation was done by YW. The paper was written by LS and L-FL, and revised by Z-PS, LW, JY, C-CW, MN, and BL.
Funding
This project was financially supported by the National Key Research and Development Program of China (grant no. 2017YFC1200100), National Science Foundation of China (grant no. 41630528 and 31670491), and Shanghai Pujiang Scholar Program (grant no. 19PJ1401500 and 16PJ1400900).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mark Bertness, Andrew Altieri, Tyler Coverdale, Wilson Freshwater, Steve Pennings, Hongyu Guo, Junda Lin, Edward Proffitt, and Glenn Coldren for their help with sampling. We are grateful to Donald R. Strong for his numerous suggestions about the data interpretation, and Hui Guo, Xuyan Geng, Shang Jiang, Tingting Yang, Xizhong Cai, Zhuoxian He, Zhe Wang, and Yao Zhao for their technical help in the laboratory or data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01467/full#supplementary-material
References
Ahlroth, P., Alatalo, R. V., Holopainen, A., Kumpulainen, T., Suhonen, J. (2003). Founder population size and number of source populations enhance colonization success in waterstriders. Oecologia 137 (4), 617–620. doi: 10.1007/s00442-003-1344-y
Ainouche, M. L., Baumel, A., Salmon, A., Yannic, G. (2003). Hybridization, polyploidy and speciation in Spartina (Poaceae). New Phytol. 161 (1), 165–172. doi: 10.1046/j.1469-8137.2003.00926.x
Ainouche, M. L., Fortune, P. M., Salmon, A., Parisod, C., Grandbastien, M. A., Fukunaga, K. (2009). Hybridization, polyploidy and invasion: lessons from Spartina (Poaceae). Biol. Invasions 11 (5), 1159. doi: 10.1007/s10530-008-9383-2
Ainouche, M. L., Chelaifa, H., Ferreira, J., Bellot, S., Ainouche, A., Salmon, A. (2012). “Polyploid evolution in Spartina: dealing with highly redundant hybrid genomes,” in Polyploidy and genome evolution. Eds. Soltis, P. S., Soltis, D. E. (Berlin, Heidelberg: Springer), 225–243. doi: 10.1007/978-3-642-31442-1_12
An, S., Xiao, Y., Qing, H., Wang, Z., Zhou, C., Li, B. (2004). “Varying success of Spartina spp. invasions in China: genetic diversity of differentiation?,” in Proceedings of the Third International Conference on Invasive Spartina. Eds. Ayres, D. R., Kerr, D. W., Ericson, S. D., Olofson, P. R. (San Francisco, CA).
An, S. Q., Gu, B. H., Zhou, C. F., Wang, Z. S., Deng, Z. F., Zhi, Y. B. (2007). Spartina invasion in China: implications for invasive species management and future research. Weed Res. 47 (3), 183–191. doi: 10.1111/j.1365-3180.2007.00559.x
Ayres, D. R., Garcia-Rossi, D., Davis, H. G., Strong, D. R. (1999). Extent and degree of hybridization between exotic (Spartina alterniflora) and native (S. foliosa) cordgrass (Poaceae) in California, USA determined by random amplified polymorphic DNA (RAPDs). Mol. Ecol. 8 (7), 1179–1186. doi: 10.1046/j.1365-294x.1999.00679.x
Ayres, D. R., Strong, D. R., Baye, P. (2003). Spartina foliosa-a common species on the road to rarity?. Madrono 50, 209–213.
Ayres, D., Grotkopp, E., Zaremba, K., Sloop, C. M., Blum, M. J., Bailey, J. P. (2008a). Hybridization between invasive Spartina densiflora (Poaceae) and native S. foliosa in San Francisco Bay, California, USA. Am. J. Bot. 95 (6), 713–719. doi: 10.3732/ajb.2007358
Ayres, D. R., Katherine, Z., Sloop, C. M., Strong, D. R. (2008b). Sexual reproduction of cordgrass hybrids (Spartina foliosa× alterniflora) invading tidal marshes in San Francisco Bay. Diversity Distrib. 14 (2), 187–195. doi: 10.1111/j.1472-4642.2007.00414.x
Bandelt, H. J., Forster, P., Röhl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16 (1), 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
Bedre, R., Mangu, V. R., Srivastava, S., Sanchez, L. E., Baisakh, N. (2016). Transcriptome analysis of smooth cordgrass (Spartina alterniflora Loisel), a monocot halophyte, reveals candidate genes involved in its adaptation to salinity. BMC Genomics 17 (1), 657. doi: 10.1186/s12864-016-3017-3
Bernik, B. M., Li, H., Blum, M. J. (2016). Genetic variation of Spartina alterniflora intentionally introduced to China. Biol. Invasions 18, 1485–1498. doi: 10.1007/s10530-016-1096-3
Blair, A. C., Wolfe, L. M. (2004). The evolution of an invasive plant: an experimental study with Silene latifolia. Ecol. 85 (11), 3035–3042. doi: 10.1890/04-0341
Blum, M. J., Sloop, C. M., Ayres, D. R., Strong, D. R. (2004). Characterization of microsatellite loci in Spartina species (Poaceae). Mol. Ecol. Notes 4 (1), 39–42. doi: 10.1046/j.1471-8286.2003.00556.x
Blum, M. J., Jun Bando, K., Katz, M., Strong, D. R. (2007). Geographic structure, genetic diversity and source tracking of Spartina alterniflora. J. Biogeogr. 34 (12), 2055–2069. doi: 10.1111/j.1365-2699.2007.01764.x
Bradley, P. M., Morris, J. T. (1991). Relative importance of ion exclusion, secretion and accumulation in Spartina alterniflora Loisel. J. Exp. Bot. 42 (245), 1525–1532. doi: 10.1093/jxb/42.12.1525
Chen, J. Q., Zhong, C. X. (1990). Genecological studies of Spartina alterniflora Loisel I. the difference in physiological reaction among the three kinds of Spartina alterniflora in saline environments. Acta Phytoecol. Geobot. Sin. 14 (1), 33–39. doi: 10.1007/BF03008874
Chung, C. H. (2006). Forty years of ecological engineering with Spartina plantations in China. Ecol. Eng. 27 (1), 49–57. doi: 10.1016/j.ecoleng.2005.09.012
Cornuet, J. M., Luikart, G. (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genet. 144 (4), 2001–2014. doi: 10.0000/PMID8978083
Courchamp, F., Chapuis, J., Pascal, M. (2003). Mammal invaders on islands: impact, control and control impact. Biol. Rev. 78 (3), 347–383. doi: 10.1017/S1464793102006061
Culley, T. M., Hardiman, N. A. (2009). The role of intraspecific hybridization in the evolution of invasiveness: a case study of the ornamental pear tree Pyrus calleryana. Biol. Invasions 11 (5), 1107–1119. doi: 10.1007/s10530-008-9386-z
Daehler, C., Strong, D. (1997). Hybridization between introduced smooth cordgrass (Spartina alterniflora; Poaceae) and native California cordgrass (S. foliosa) in San Francisco Bay, California, USA. Am. J. Bot. 84 (5), 607–611. doi: 10.2307/2445896
Drummond, A., Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7 (1), 214. doi: 10.1186/1471-2148-7-214
Drummond, A. J., Nicholls, G. K., Rodrigo, A. G., Solomon, W. (2002). Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genet. 161 (3), 1307–1320. doi: 10.0000/PMID12136032
Durka, W., Bossdorf, O., Prati, D., Auge, H. (2005). Molecular evidence for multiple introductions of garlic mustard (Alliaria petiolata, Brassicaceae) to North America. Mol. Ecol. 14 (6), 1697–1706. doi: 10.1111/j.1365-294X.2005.02521.x
Elsey-Quirk, T., Middleton, B. A., Proffitt, C. E. (2009). Seed flotation and germination of salt marsh plants: The effects of stratification, salinity, and/or inundation regime. Aquat. Bot. 91 (1), 40–46. doi: 10.1016/j.aquabot.2009.02.001
Excoffier, L., Laval, G., Schneider, S. (2005). Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinf. Online 1, 47–50. doi: 10.1177/117693430500100003
Fitzpatrick, B. M., Shaffer, H. B. (2007). Hybrid vigor between native and introduced salamanders raises new challenges for conservation. Proc. Natl. Acad. Sci. 104 (40), 15793–15798. doi: 10.1073/pnas.0704791104
Forsman, A. (2014). Effects of genotypic and phenotypic variation on establishment are important for conservation, invasion, and infection biology. Proc. Natl. Acad. Sci. 111 (1), 302–307. doi: 10.1073/pnas.1317745111
Genton, B., Shykoff, J., Giraud, T. (2005). High genetic diversity in French invasive populations of common ragweed, Ambrosia artemisiifolia, as a result of multiple sources of introduction. Mol. Ecol. 14 (14), 4275–4285. doi: 10.1111/j.1365-294X.2005.02750.x
Gignoux, C. R., Henn, B. M., Mountain, J. L. (2011). Rapid, global demographic expansions after the origins of agriculture. Proc. Natl. Acad. Sci. 108 (15), 6044–6049. doi: 10.1073/pnas.0914274108
Harvey-Samuel, T., Ant, T., Alphey, L. (2017). Towards the genetic control of invasive species. Biol. Invasions 19 (6), 1683–1703. doi: 10.1007/s10530-017-1384-6
Hodgins, K. A., Lai, Z., Nurkowski, K., Huang, J., Rieseberg, L. H. (2013). The molecular basis of invasiveness: differences in gene expression of native and introduced common ragweed (Ambrosia artemisiifolia) in stressful and benign environments. Mol. Ecol. 22 (9), 2496–2510. doi: 10.1111/mec.12179
Hufbauer, R. A., Rutschmann, A., Serrate, B., Vermeil de Conchard, H., Facon, B. (2013). Role of propagule pressure in colonization success: disentangling the relative importance of demographic, genetic and habitat effects. J. Evol. Biol. 26 (8), 1691–1699. doi: 10.1111/jeb.12167
IUCN. (1999). IUCN Red List Criteria review provisional report: draft of the proposed changes and recommendations. Species 31–32, 43–57.
Kelager, A., Pedersen, J. S., Bruun, H. H. (2013). Multiple introductions and no loss of genetic diversity: invasion history of Japanese Rose, Rosa rugosa, in Europe. Biol. Invasions 15, 1125–1141. doi: 10.1007/s10530-012-0356-0
Keller, S., Taylor, D. (2010). Genomic admixture increases fitness during a biological invasion. J. Evol. Biol. 23 (8), 1720–1731. doi: 10.1111/j.1420-9101.2010.02037.x
Kolbe, J. J., Glor, R. E., Schettino, L. R., Lara, A. C., Larson, A., Losos, J. B. (2004). Genetic variation incresases during biological invasion by a Cuban lizard. Nat. 431 (9), 177–181. doi: 10.1038/nature02807
Lee, C. E. (2002). Evolutionary genetics of invasive species. Trends In Ecol. Evol. 17 (8), 386–391. doi: 10.1016/S0169-5347(02)02554-5
Mack, R. N., Simberloff, D., Mark Lonsdale, W., Evans, H., Clout, M., Bazzaz, F. A. (2000). Biotic invasions: causes, epidemiology, global consequences, and control. Ecol. Appl. 10 (3), 689–710. doi: 10.1890/1051-0761(2000)010[0689:BICEGC]2.0.CO;2
Marchant, C. J. (1968). Evolution in Spartina (Gramineae): III. Species chromosome numbers and their taxonomic significance. Bot. J. Linn. Soc. 60 (383), 411–417. doi: 10.1111/j.1095-8339.1968.tb00097.x
Marchant, C. J. (1977). Hybrid characteristics in Spartina X neyrautii Fouc., a taxon rediscovered in northern Spain. Bot. J. Linn. Soc. 74 (4), 289–296. doi: 10.1111/j.1095-8339.1977.tb01182.x
Meimberg, H., Hammond, J. I., Jorgensen, C. M., Park, T. W., Gerlach, J. D., Rice, K. J. (2006). Molecular evidence for an extreme genetic bottleneck during introduction of an invading grass to California. Biol. Invasions 8 (6), 1355–1366. doi: 10.1007/s10530-005-2463-7
Myers, N., Mittermeier, R. A., Mittermeier, C. G., da Fonseca, G. A. B., Kent, J. (2000). Biodiversity hotspots for conservation priorities. Nat. 403 (6772), 853–858. doi: 10.1038/35002501
Peakall, R., Smouse, P. E. (2006). GenALEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6 (1), 288–295. doi: 10.1111/j.1471-8286.2005.01155.x
Peakall, R., Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinf. 28 (19), 2537–2539. doi: 10.1093/bioinformatics/bts460
Pons, O., Petit, R. J. (1996). Measuring and testing genetic differentiation with ordered versus unordered alleles. Genet. 144 (3), 1237–1245. doi: 10.1016/S1050-3862(96)00162-3
Prentis, P. J., Wilson, J. R. U., Dormontt, E. E., Richardson, D. M., Lowe, A. J. (2008). Adaptive evolution in invasive species. Trends In Plant Sci. 13 (6), 288–294. doi: 10.1016/j.tplants.2008.03.004
Pritchard, J. K., Stephens, M., Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genet. 155 (2), 945–959.
R Core Team. (2013).R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
Raymond, M., Rousset, F. (1995). An exact test for population differentiation. Evol. 49 (6), 1280–1283. doi: 10.2307/2410454
Richardson, J. E., Pennington, R. T., Pennington, T. D., Hollingsworth, P. M. (2001). Rapid diversification of a species-rich genus of neotropical rain forest trees. Sci. 293 (5538), 2242–2245. doi: 10.1126/science.1061421
Rius, M., Darling, J. A. (2014). How important is intraspecific genetic admixture to the success of colonising populations?. Trends In Ecol. Evol. 29 (4), 233–242. doi: 10.1016/j.tree.2014.02.003
Rosenthal, D. M., Ramakrishnan, A. P., Cruzan, M. B. (2008). Evidence for multiple sources of invasion and intraspecific hybridization in Brachypodium sylvaticum (Hudson) Beauv. in North America. Mol. Ecol. 17 (21), 4657–4669. doi: 10.1111/j.1365-294X.2008.03844.x
Rousset, F. (2008). GENEPOP’007: a complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 8 (1), 103–106. doi: 10.1111/j.1471-8286.2007.01931.x
Schierenbeck, K. A., Ellstrand, N. C. (2009). Hybridization and the evolution of invasiveness in plants and other organisms. Biol. Invasions 11 (5), 1093–1105. doi: 10.1007/s10530-008-9388-x
Simberloff, D. (2009). The role of propagule pressure in biological invasions. Annu. Rev. Ecol. Evol. Syst. 40 (1), 81–102. doi: 10.1146/annurev.ecolsys.110308.120304
Sloop, C. M., McGray, H. G., Blum, M. J., Strong, D. R. (2005). Characterization of 24 additional microsatellite loci in Spartina species (Poaceae). Conserv. Genet. 6 (6), 1049–1052. doi: 10.1007/s10592-005-9084-7
Strong, D. R., Ayres, D. R. (2009). “Spartina introductions and consequences in salt marshes,” in Human impacts on salt marshes: a global perspective. Eds. Bertness, M. D., Grosholz, E. D. (Berkeley, CA, USA: University of California Press), 3–22.
Strong, D. R., Ayres, D. R. (2013). Ecological and evolutionary misadventures of Spartina. Annu. Rev. Ecology Evol. Syst. 44 (1), 389–410. doi: 10.1146/annurev-ecolsys-110512-135803
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28 (10), 2731–2739. doi: 10.1093/molbev/msr121
Tsutsui, N. D., Suarez, A. V., Holway, D. A., Case, T. J. (2000). Reduced genetic variation and the success of an invasive species. Proc. Natl. Acad. Sci. 97 (11), 5948–5953. doi: 10.1073/pnas.100110397
Ueno, S., Rodrigues, J. F., Alves-Pereira, A., Pansarin, E. R., Veasey, E. A. (2015). Genetic variability within and among populations of an inasieve, exotic orchid. AoB Plants 7, plv077. doi: 10.1093/aobpla/plv077
van Oosterhout, C., Hutchinson, W. F., Wills, D. P., Shipley, P. (2004). Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4 (3), 535–538. doi: 10.1111/j.1471-8286.2004.00684.x
Vilà, M., Espinar, J. L., Hejda, M., Hulme, P. E., Jarošík, V., Maron, J. L. (2011). Ecological impacts of invasive alien plants: a meta-analysis of their effects on species, communities and ecosystems. Ecol. Lett. 14 (7), 702–708. doi: 10.1111/j.1461-0248.2011.01628.x
Wang, Q., An, S. Q., Ma, Z. J., Zhao, B., Chen, J. K., Li, B. (2006). Invasive Spartina alterniflora: biology, ecology and management. Acta Phytotaxonomica Sin. 44, 559–588. doi: 10.1360/aps06044
Wang, X. Y., Shen, D. W., Jiao, J., Xu, N. N., Yu, S., Zhou, X. F. (2012). Genotypic diversity enhances invasive ability of Spartina alterniflora. Mol. Ecol. 21 (10), 2542–2551. doi: 10.1111/j.1365-294X.2012.05531.x
Xu, G. W., Zhuo, R. Z., Cao, H., Li, X. G. (1989). Annual changes of biomass of Spartina alterniflora and the relationships between biomass and tidal land habits. Acta Phytoecol. Geobot. Sin. 13 (3), 230–235. doi: 10.1007/BF02009745
Zarnetske, P. L., Seabloom, E. W., Hacker, S. D. (2010). Non-target effects of invasive species management: beachgrass, birds, and bulldozers in coastal dunes. Ecosphere 1 (5), art13. doi: 10.1890/ES10-00101.1
Zhang, R. S., Shen, Y. M., Lu, L. Y., Yan, S. G., Wang, Y. H., Li, J. L. (2004). Formation of Spartina alterniflora salt marshes on the coast of Jiangsu Province, China. Ecol. Eng. 23 (2), 95–105. doi: 10.1016/j.ecoleng.2004.07.007
Zhang, D., Yang, M., Li, J., Chen, X. (2006). Vegetative dispersal ability of Spartina alterniflora in eastern end of Chongming island. J. East China Normal Univ. (Natural Science) (2), 130–135. doi: 10.3969/j.issn.1000-5641.2006.02.020
Zhang, Y. Y., Zhang, D. Y., Barrett, S. C. H. (2010). Genetic uniformity characterizes the invasive spread of water hyacinth (Eichhornia crassipes), a clonal aquatic plant. Mol. Ecol. 19 (9), 1774–1786. doi: 10.1111/j.1365-294X.2010.04609.x
Zuo, P., Liu, C. A., Zhao, S. H., Wang, C. H., Liang, Y. B. (2009). Distribution of Spartina plantations along the China's coast. Acta Oceanologica Sin. 31 (5), 101–111.
Keywords: genetic admixture, mathematical simulation, population variation, plant invasion, rapid evolution, Spartina alterniflora
Citation: Shang L, Li L-F, Song Z-P, Wang Y, Yang J, Wang C-C, Qiu S-Y, Huang J-X, Nie M, Wolfe LM and Li B (2019) High Genetic Diversity With Weak Phylogeographic Structure of the Invasive Spartina alterniflora (Poaceae) in China. Front. Plant Sci. 10:1467. doi: 10.3389/fpls.2019.01467
Received: 28 July 2019; Accepted: 22 October 2019;
Published: 20 November 2019.
Edited by:
Yuannian Jiao, Chinese Academy of Sciences, ChinaReviewed by:
Yelin Huang, Sun Yat-sen University, ChinaItzi Fragoso-Martínez, National Autonomous University of Mexico, Mexico
Jun Rong, Nanchang University, China
Copyright © 2019 Shang, Li, Song, Wang, Yang, Wang, Qiu, Huang, Nie, Wolfe and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin-Feng Li, bGlsaW5mZW5nQGZ1ZGFuLmVkdS5jbg==; Bo Li, Ym9vbEBmdWRhbi5lZHUuY24=