Abstract
In phylogenetic studies across angiosperms, at various taxonomic levels, polytomies have persisted despite efforts to resolve them by increasing sampling of taxa and loci. The large amount of genomic data now available and statistical tools to analyze them provide unprecedented power for phylogenetic inference. Targeted sequencing has emerged as a strong tool for estimating species trees in the face of rapid radiations, lineage sorting, and introgression. Evolutionary relationships in Cyperaceae have been studied mostly using Sanger sequencing until recently. Despite ample taxon sampling, relationships in many genera remain poorly understood, hampered by diversification rates that outpace mutation rates in the loci used. The C4 Cyperus clade of the genus Cyperus has been particularly difficult to resolve. Previous studies based on a limited set of markers resolved relationships among Cyperus species using the C3 photosynthetic pathway, but not among C4 Cyperus clade taxa. We test the ability of two targeted sequencing kits to resolve relationships in the C4 Cyperus clade, the universal Angiosperms-353 kit and a Cyperaceae-specific kit. Sequences of the targeted loci were recovered from data generated with both kits and used to investigate overlap in data between kits and relative efficiency of the general and custom approaches. The power to resolve shallow-level relationships was tested using a summary species tree method and a concatenated maximum likelihood approach. High resolution and support are obtained using both approaches, but high levels of missing data disproportionately impact the latter. Targeted sequencing provides new insights into the evolution of morphology in the C4 Cyperus clade, demonstrating for example that the former segregate genus Alinula is polyphyletic despite its seeming morphological integrity. An unexpected result is that the Cyperus margaritaceus-Cyperus niveus complex comprises a clade separate from and sister to the core C4 Cyperus clade. Our results demonstrate that data generated with a family-specific kit do not necessarily have more power than those obtained with a universal kit, but that data generated with different targeted sequencing kits can often be merged for downstream analyses. Moreover, our study contributes to the growing consensus that targeted sequencing data are a powerful tool in resolving rapid radiations.
Introduction
Since the late 1980s, molecular phylogenetics has yielded major new insights into the evolution of land plants, especially for flowering plants (e.g., Chase et al., 1993; Ruhfel et al., 2014; Wickett et al., 2014; APG IV, 2016). However, uncertainty in topologies has persisted, particularly for deep nodes (Wickett et al., 2014) and for ancient and recent rapid radiations, which are often inferred as polytomies (Fishbein et al., 2001; Whitfield and Lockhart, 2007; Snak et al., 2016; Spalink et al., 2016). Researchers have attempted to resolve these issues by increasing taxon sampling, the number of DNA loci sampled, or both (e.g., Philippe et al., 2011; Nabhan and Sarkar, 2012; Nicholls et al., 2015).
Targeted sequencing of genomic libraries can yield hundreds to thousands of DNA loci across multiple individuals and species, depending on the targeted sequencing kit used (e.g., Faircloth et al., 2018; Johnson et al., 2018; Couvreur et al., 2019), providing sequencing data suitable to addressing challenging and outstanding problems in plant systematics. It is an extremely versatile technique that can be used to solve ancient and recent species radiations (Nicholls et al., 2015; Stevens et al., 2015; Mitchell et al., 2017; Kadlec et al., 2017), as well as to bridge micro- and macroevolutionary levels (Kates et al., 2018; Villaverde et al., 2018). Additionally, it works well with degraded DNA template, e.g., herbarium material (Hart et al., 2016; McKain et al., 2018; Brewer et al., 2019). Targeted sequencing is rapidly becoming a standard phylogenomic method for flowering plants (McKain et al., 2018).
Evolutionary relationships in the sedge family (Cyperaceae) have mainly been studied using Sanger sequencing (e.g., Simpson et al., 2007; Muasya et al., 2009; Jung and Choi, 2012; Escudero and Hipp, 2013; Spalink et al., 2016; Semmouri et al., 2019; Figure 1A). To date, high-throughput sequencing approaches in sedges include a targeted sequencing study using anchored hybrid enrichment or anchored phylogenomics (Lemmon et al., 2012; Buddenhagen et al., 2016), focusing on the Scirpo-Caricoid clade (Léveillé-Bourret et al., 2018), and two other studies using reduced-representation phylogenomic methods, restriction-site associated DNA sequencing (RAD-Seq; Baird et al., 2008), and genotyping-by-sequencing (GBS; Elshire et al., 2011; Deschamps et al., 2012), to resolve fine-scale relationships in the megadiverse genus Carex (Escudero et al., 2014; Maguilla et al., 2017). Hyb-Seq, i.e., targeted sequencing combined with genome skimming (Weitemier et al., 2014; Dodsworth et al., 2019), was recently used to investigate the broad-scale relationships in Carex (Villaverde et al., in review). Additionally, relationships at tribal and generic levels in Cyperaceae are being investigated using targeted sequencing (I. Larridon et al., unpubl. data). Relationships among Cyperaceae taxa that use the C4 photosynthetic pathway remain ill-understood, hampered by an apparent faster rate of diversification leading to limited topological resolution (e.g., Larridon et al., 2013; Bauters et al., 2014; Roalson et al., 2019). In particular, the relationships between C4 Cyperus L. species are still unresolved.
Figure 1
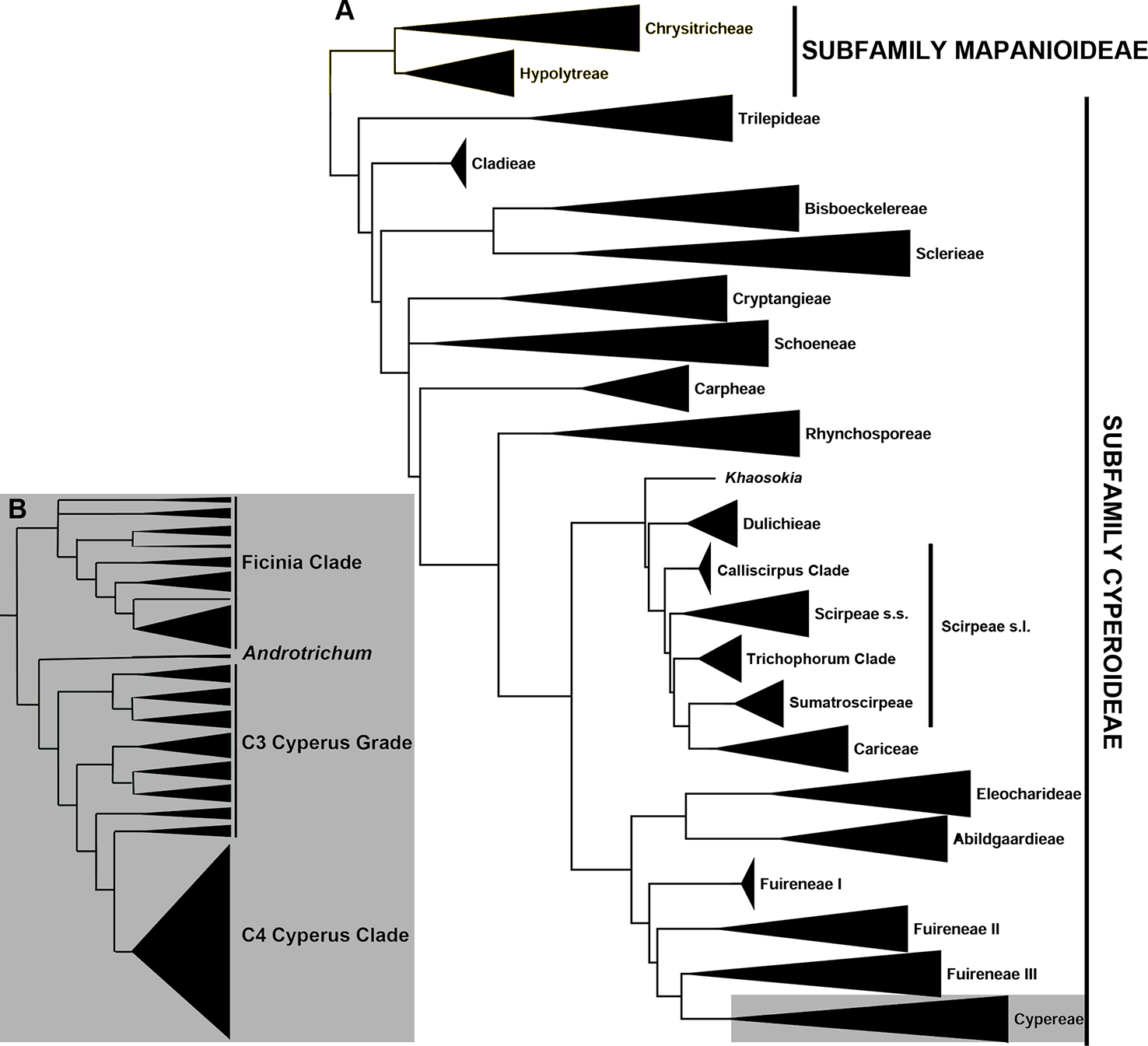
Phylogenetic relationships in Cyperaceae (A) Summary phylogeny of the family Cyperaceae based on Semmouri et al. (2019). (B) Summary phylogeny of the tribe Cypereae based on Larridon et al. (2011a, 2013).
Within tribe Cypereae, the Cyperus clade includes two genera, i.e., the giant genus Cyperus (950 species) and the small genus Androtrichum (Brongn.) Brongn. (two species). Thirteen segregate genera recognized by Goetghebeur (1998) have since been subsumed into Cyperus (Larridon et al., 2011a; Larridon et al., 2011b; Larridon et al., 2013; Larridon et al., 2014; Bauters et al., 2014), comprising three genera that use C3 photosynthesis—Courtoisina Soják, Kyllingiella R.W. Haines & Lye, and Oxycardium Nees—and 10 genera that use C4 photosynthesis—Alinula J. Raynal, Ascolepis Nees ex Steud., Ascopholis C.E.C. Fisch., Kyllinga Rottb., Lipocarpha R. Br., Pycreus P. Beauv., Queenslandiella Domin, Remirea Aubl., Sphaerocyperus Lye, and Volkiella Merxm. & Czech. The small genus Androtrichum (C3 photosynthesis) has not yet been subsumed into Cyperus because only rbcL sequences are available to date, and the phylogenetic placement of the genus is consequently unresolved (Muasya et al., 2009; Jung and Choi, 2012; Hinchliff and Roalson, 2013; Semmouri et al., 2019). Given the ecological importance of Cyperus, which achieves high diversity and biomass in ecoregions across the tropics (Larridon et al., 2013; Kipkemboi and van Dam, 2018), and its ethnobotanical significance (Simpson and Inglis., 2001), understanding the diversity of the clade is of high societal importance.
Previous studies based on a limited set of plastid and nuclear ribosomal DNA (nrDNA) markers resolved relationships among species of Cyperus using the C3 photosynthetic pathway (Larridon et al., 2011a; Larridon et al., 2011b), but not among sections and species using C4 photosynthesis (Larridon et al., 2013; Larridon et al., 2014; Bauters et al., 2014). In these studies, the species-poor lineages of genus Cyperus, which diverge from deeper nodes, form a grade of generally well-circumscribed Cyperus sections that all use C3 photosynthesis (hereafter C3 Cyperus grade, c. 190 species); while the more highly derived clade (hereafter C4 Cyperus clade) represents a radiation of c. 760 Cyperus species that use C4 photosynthesis (Figure 1B). The C4 Cyperus clade is a particularly challenging lineage taxonomically (Huygh et al., 2010; Larridon et al., 2011c; Reynders et al., 2011; Bauters et al., 2014) and previous attempts to resolve relationships within it have resulted in a polytomy (Muasya et al., 2002; Larridon et al., 2011a; Larridon et al., 2013; Bauters et al., 2014). Spalink et al. (2016) showed an increased diversification rate for the C4 Cyperus clade commencing c. 20 Ma (million years ago).
It is still unclear under what conditions universal targeted sequencing kits, which target low-copy nuclear markers conserved across a wide phylogenetic range (e.g., angiosperms; Buddenhagen et al., 2016), can be used to infer relationships in fast evolving lineages. If they can, then there may be little benefit to developing custom probes for studies of small numbers of taxa, and there are obvious downstream benefits in using universal probes, data from which can be readily combined across labs. A recent study (Kadlec et al., 2017) on the heather genus Erica (Ericaceae, with well over 800 species) concluded that data from markers that are custom-designed using existing pipelines (e.g., MarkerMiner; Chamala et al., 2015) may deliver better results than those obtained using a more universal approach. As Liu et al. (2019) have shown, capture success drops significantly when probe sequences used in a targeted sequencing kit diverge >30% from their intended targets (Supplementary Figures 5 and 6 in Liu et al., 2019). Recently, an angiosperm-wide targeted sequencing kit, i.e., Angiosperms-353 (Johnson et al., 2018), has been designed using a k-medoids clustering algorithm (Bauckhage, 2015) from a much larger dataset, including several published genomes (available from https://phytozome.jgi.doe.gov) plus 655 angiosperm transcriptomes generated by the one thousand plant transcriptomes (1KP) initiative (Matasci et al., 2014), in the context of the Plant and Fungal Trees of Life (PAFTOL) research program at Royal Botanic Gardens, Kew (https://www.kew.org/science/who-we-are-and-what-we-do/strategic-outputs-2020/plant-and-fungal-trees-life). The Angiosperms-353 kit (Johnson et al., 2018) targets 353 putatively single-copy protein-coding genes (spanning 260,802 bp in total) and was designed using transcriptome data from representatives of all major clades in angiosperms (three accessions belong in Cyperaceae—Cyperus, Lepidosperma, and Mapania—out of 128 monocots in total), to keep expected divergence between all potential taxa and the probes below the 30% divergence threshold beyond which capture is no longer efficient, as Liu et al. (2019) experimentally determined. This kit includes multiple probes for each locus (3x tiling) to optimize its performance with low-quality template (e.g., historical herbarium collections; Brewer et al., 2019). The aforementioned reasons may result in the Angiosperms-353 kit being more successful than other universal targeted sequencing kits for flowering plants.
Here, we present novel data from the C4 Cyperus clade obtained using both the Angiosperms-353 targeted sequencing kit and a Cyperaceae-specific kit designed by Villaverde et al. (in review) using the MarkerMiner pipeline (Chamala et al., 2015), with transcriptome data for Cyperus papyrus L. (1KP) and Carex siderosticta Hance (S. Kim et al., unpubl. data). The Cyperaceae-specific kit targets 554 low-copy nuclear orthologous loci, spanning c. 1 Mbp. We use these data to: 1) test the effectiveness of the Angiosperms-353 kit to resolve hitherto intractable relationships, 2) compare the relative effectiveness of these universal probes to the Cyperaceae-specific probes, and 3) establish well-supported relationships among this ecologically important group of sedges.
Materials and Methods
Taxon Sampling
Sampling for enrichment with the Angiosperms-353 kit consisted of 38 Cyperus accessions (one C3 Cyperus species, i.e., Cyperus kyllingiella Larridon, and 37 species from the C4 Cyperus clade) (Supplementary Table 1A). Sampling for enrichment with the Cyperaceae-specific kit consisted of eight species of the C4 Cyperus clade and Schoenoplectus pungens (Vahl) Palla (tribe Fuireneae) used as outgroup (Supplementary Table 1B). Cyperus esculentus L., Cyperus mindorensis (Steud.) Huygh, and Cyperus richardii Steud. were enriched with both kits.
Deoxyribonucleic Acid Extraction, Library Preparation, Hybridization, and Sequencing
The voucher information and treatment of each accession is provided (Supplementary Tables 1 and 2). Both the Angiosperms-353 (Johnson et al., 2018) and the Cyperaceae-specific (Villaverde et al. in review) kits are available from Arbor Biosciences (Ann Arbor, MI, USA).
Molecular work for accessions enriched with the Angiosperms-353 probes was carried out at the Sackler Phylogenomic Laboratory, within the Jodrell Laboratory at Royal Botanic Gardens, Kew (Richmond, Surrey, UK). Genomic DNA was extracted from leaf tissue obtained from herbarium specimens or silica collected samples, using either a modified cetyl trimethylammonium bromide (CTAB) approach (Doyle and Doyle, 1987) or a CTAB protocol, based on Beck et al. (2012), modified for optimal simultaneous extraction of 96 to 192 samples (i.e., one or two plates) from suboptimal (i.e., herbarium) tissue (Fairlie & Pokorny protocol provided in Supplementary Data Sheet 1). Lastly, two accessions were sourced from the Kew DNA Bank (http://dnabank.science.kew.org/) (Supplementary Table 1A). The samples extracted using a CTAB approach were purified using Agencourt AMPure XP Bead Clean-up (Beckman Coulter, Indianapolis, IN, USA). All DNA extracts were quantified using a Quantus™ Fluorometer (Promega Corporation, Madison, WI, USA) and then run on a 1% agarose gel to assess the average fragment size. Samples with very low concentration (not visible on a 1% agarose gel), were assessed on an Agilent Technologies 4200 TapeStation System using Genomic DNA ScreenTape (Santa Clara, CA, USA). DNA extracts with average fragment sizes above 350 bp were sonicated using a Covaris M220 Focused-ultrasonicator™ (Covaris, Woburn, MA, USA) following the manufacturer’s protocol to obtain an average fragment size of 350 bp. Dual-indexed libraries for Illumina® sequencing were prepared using the DNA NEBNext® Ultra™ II Library Prep Kit and the NEBNext® Multiplex Oligos for Illumina® (Dual Index Primers Set 1 and 2) from New England BioLabs® (Ipswich, MA, USA) following the manufacturer’s instructions but at half the recommended volumes. The quality of the libraries was evaluated on the TapeStation using High Sensitivity D1000 ScreenTape and the libraries were quantified using a Quantus Fluorometer. The final average library size including the adapters was c. 500 bp. Afterwards, the samples were pooled (8–24 samples/reaction) and enriched with the Angiosperms-353 probes (Johnson et al., 2018) following the manufacturer’s instructions (myProbes® Manual v4.01, Arbor Biosciences, Ann Arbor, MI, USA) setting the hybridization temperature to 65°C for 24 h. Final products were again run on the TapeStation to assess quality (i.e., average fragment size) so they could be pooled equimolarly for sequencing (48–96 samples/pool). After multiplexing library pools, sequencing was performed on an Illumina® MiSeq instrument (San Diego, CA, USA)—with v2 (300-cycles at 2 × 150 bp) or v3 (600-cycles at 2 × 300 bp) chemistry at Royal Botanic Gardens, Kew (Richmond, Surrey, UK)—or on an Illumina® HiSeq (San Diego, CA, USA)—at either Macrogen (Seoul, South Korea) or GENEWIZ® (Leipzig, Germany), producing 2 × 150 bp long reads.
Molecular work for the accessions enriched with the Cyperaceae-specific probes was carried out at The Morton Arboretum (Lisle, IL, USA) and the Pritzker Laboratory of the Field Museum of Natural History (Chicago, IL, USA). Genomic DNA was extracted from leaf tissue obtained from silica preserved samples (Supplementary Table 1B) using the QIAGEN DNeasy Plant Mini Kit following the manufacturer’s protocols (QIAGEN, Valencia, CA, USA) or a modified CTAB protocol (Doyle and Doyle, 1987). Samples were sonicated to a target fragment size of 550 bp using a Covaris E220 Focused-ultrasonicator™ (Wohurn, MA, USA). Sequencing libraries were prepared using the Illumina® TruSeq Nano HT DNA kit (San Diego, CA, USA). DNA libraries were checked for quality using an Agilent Technologies 2100 Bioanalyzer (Santa Clara, CA, USA) and their concentration quantified using a Qubit 2.0 Fluorometer (Life Technologies, Grand Island, NY, USA). Indexed samples were pooled in approximately equal quantities and the pool was enriched using the custom Cyperaceae-specific probes (Villaverde et al. in review) following the manufacturer’s protocols for myBaits® kit (v3), i.e., we hybridized at 65°C for 16 h. The paired-end libraries were sequenced in one run (including a total of 88 accessions; Villaverde et al. in review) on an Illumina MiSeq (2 × 300 bp; 600 cycle v3) at the Pritzker Laboratory.
Bioinformatics, Contig Assembly, and Multi-Sequence Alignment
Raw reads were trimmed to remove adapter sequences and to remove portions of low quality with Trimmomatic v.0.36 (Bolger et al., 2014) using the setting LEADING:20 TRAILING:20 SLIDINGWINDOW:4:20 MINLEN:50. HybPiper v1.3.1 (Johnson et al., 2016) was used with default settings to process the quality-checked, trimmed reads. Paired reads of samples enriched with two different kits independently (Angiosperms-353 and Cyperaceae-specific) were mapped to targets using BWA v0.7.17 (Li and Durbin, 2009) and their respective nucleotide (DNA) target file (Supplementary Data Sheet 2); additionally, we also used BLASTx (Altschul et al., 1990) when using the Angiosperms-353 target loci with amino acid (AA) sequences (Supplementary Data Sheet 3). Summary statistics such as the percentage of reads mapping were generated using SAMtools flagstat v1.8 (Li et al., 2009). Mapped reads were then assembled into contigs with SPAdes v3.13.1 (Bankevich et al., 2012). Subsequently, exonerate v2.2 (Slater and Birney, 2005) was used to align the assembled contigs to their associated target sequence and remove intronic regions. Only exon data were analyzed in the current study in order to directly compare the information content provided by the targeted loci. HybPiper flags potential paralogs when multiple contigs are discovered mapping well to a single reference sequence. The program uses coverage and identity to a reference to choose a “main” sequence and denotes the others as potential paralogs. All loci flagged as potential paralogs were removed from downstream analyses. A list of the potential paralogs is provided (Supplementary Table 3).
The paralog-filtered consensus sequences for each locus were used to generate eight different datasets (Table 1). This allowed us to investigate the phylogenetic informativeness of the data generated by the two kits separately and allowed us to test the mergeability of the data generated by both kits. Four unmerged datasets were created: (dataset 1) loci targeted with the Angiosperms-353 kit for the 37 C4 Cyperus clade accessions enriched with this kit plus Cyperus kyllingiella (C3 Cyperus grade) as outgroup; (dataset 2) loci targeted with the Cyperaceae-specific baits for the eight C4 Cyperus clade accessions enriched with this kit plus S. pungens as outgroup; (dataset 3) loci targeted with the Angiosperms-353 kit for a subset of eight C4 Cyperus clade accessions enriched with this kit; (dataset 4) loci targeted with the Cyperaceae-specific baits for the eight C4 Cyperus clade accessions enriched with this kit. Four merged datasets were assembled: (dataset 5) loci targeted with the Angiosperms-353 baits for all 46 Cyperus accessions; (dataset 6) loci targeted with the Cyperaceae-specific kit for all 46 Cyperus accessions; (dataset 7) all targeted loci for all 46 Cyperus accessions; and (dataset 8) the 57 overlapping loci targeted by both bait kits for all 46 Cyperus accessions (retrieved from the Angiosperms-353 data). The overlapping loci are listed in Supplementary Table 4. Contigs were aligned using MAFFT v7 (Katoh and Standley, 2013) with the “–auto” option. The number of parsimony informative sites were calculated for each contig alignment using AMAS (Borowiec, 2016).
Table 1
| Loci targeted by | Accessions enriched with | Accessions included | |
|---|---|---|---|
| Unmerged datasets | Angiosperms-353 | Angiosperms-353 | 37 C4 Cyperus clade + Cyperus kyllingiella as outgroup |
| Cyperaceae-specific | Cyperaceae-specific | 8 C4 Cyperus clade + Schoenoplectus pungens as outgroup | |
| Angiosperms-353 | Angiosperms-353 | 8 C4 Cyperus clade | |
| Cyperaceae-specific | Cyperaceae-specific | 8 C4 Cyperus clade | |
| Merged datasets | Angiosperms-353 | All accessions | 46 Cyperus accessions |
| Cyperaceae-specific | All accessions | 46 Cyperus accessions | |
| All loci | All accessions | 46 Cyperus accessions | |
| 57 overlapping loci | All accessions | 46 Cyperus accessions |
The eight datasets analyzed in this study.
Dataset 3 and dataset 4 were analyzed to account for the difference in sampling size when comparing the number of PIS retrieved across locus alignments for the two targeted sequencing kits. In these datasets, the eight C4 Cyperus clade accessions from dataset 2 (representing loci targeted with the Cyperaceae-specific baits for the accessions enriched with this kit) were compared with a taxonomically equivalent subset of eight accessions from dataset 1 (representing loci targeted with the Angiosperms-353 kit for the accessions enriched with this kit). The accessions selected for this subsampling are indicated by an asterisk in Supplementary Tables 1 and 2, and are represented respectively in dataset 3 and dataset 4 by 1) four species of the former segregate genus Kyllinga plus a closely related species to match five species of former segregate genus Kyllinga (two of which are represented by the same species in both datasets); 2) one species of the former segregate genus Ascolepis to match one species of the former segregate genus Ascolepis; 3) C. esculentus (represented by the same accession in both datasets); and 4) C. papyrus L. to match Cyperus rotundus L. which are closely related species.
Phylogenetic Inference
Trees were inferred using either a summary method that is statistically consistent under the Multiple Species Coalescent (MSC) (i.e., ASTRAL-III) or a total evidence approach, in which maximum likelihood (ML) inference was conducted on a concatenated matrix of all loci. Both approaches were used to analyze the eight different datasets described above (Table 1). For the summary approach under the MSC, individual gene trees were constructed using RAxML v8 (Stamatakis, 2014) applying GTRCAT and 200 bootstrap replicates followed by slow ML optimization with the “-f a” option. We then ran ASTRAL-III v5.5.11 (Zhang et al., 2018) to infer a species tree using “-t 2” to output quartet support values visualizing gene tree conflict. For the total evidence approach, phylogenetic inference of the targeted sequencing data was executed in IQ-TREE v1.6.10 (Nguyen et al., 2015) with 1,000 ultrafast bootstraps using the “- bb” and “-m TEST” options. To investigate gene tree versus species tree concordance, we calculated quartet distance between each individual gene tree and the concatenated total evidence tree obtained using the R package Quartet v1.0.2 (Sand et al., 2014; Smith, 2019), which yields a measure of the similarity of each gene tree versus the species tree based on shared four-taxon subtrees. We also calculated two measures of genealogical concordance in our dataset, the gene concordance factor (gCF) and the site concordance factor (sCF), using the options “-gcf” and “-scf” in IQ-TREE v1.7beta (Nguyen et al., 2015; Minh et al., 2018). This approach provides a description of possible disagreement among loci and across sites.
Results
Targeted Sequencing Kits and Data Quality
Summary statistics are available in Supplementary Table 5. When comparing the summary statistics between the two equally sized datasets 3 and 4 (Supplementary Table 5C), on average 324,655 (42,386–832,980) paired reads were produced for the accessions enriched with Angiosperms-353 probes vs. 199,961 (52,978–420,228) for the accessions enriched with the Cyperaceae-specific probes. Raw reads for all accessions are available from GenBank Sequence Read Archive (SRA) under BioProject numbers PRJNA553989 (Cyperus BioProject) and PRJNA553631 (S. pungens—Carex BioProject) (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA553989; http://www.ncbi.nlm.nih.gov/bioproject/PRJNA553631).
Universal Versus Family-Specific Probes
Recovery success and sequence length of the targeted loci, with both targeted sequencing kits, are provided in Supplementary Table 2 and visualized in Figure 2 and Supplementary Images 1–3. Percentage of recovery, i.e., the percentage of summed captured length of all target loci per individual divided by the summed mean length of all loci, was highest when running HybPiper for accessions enriched with the Cyperaceae-specific kit with its corresponding nucleotide target file (42.1%). We recovered on average 396 loci (324–476; Supplementary Table 2C) with the Cyperaceae-specific kit. For accessions enriched with the Angiosperms-353 kit, capture success was higher with the AA target file (33.23%) than with the DNA target file (21.7%). We recovered on average 215 loci (39–309; Supplementary Table 2B) with the AA target file and 162 loci (35–235) with the DNA target file. For data generated with the Angiosperms-353 kit, post-alignment length of contigs ranged from 87 to 3,103 bp long, with 751 bp mean length (Table 2; Supplementary Table 6A). For data generated with the Cyperaceae-specific kit, post-alignment length of contigs ranged from 93 to 7,527 bp long, with 1,608 bp mean length (Table 2; Supplementary Table 6B). In both cases, longer contigs had more Parsimony Informative Sites (PIS) (Figure 3).
Figure 2
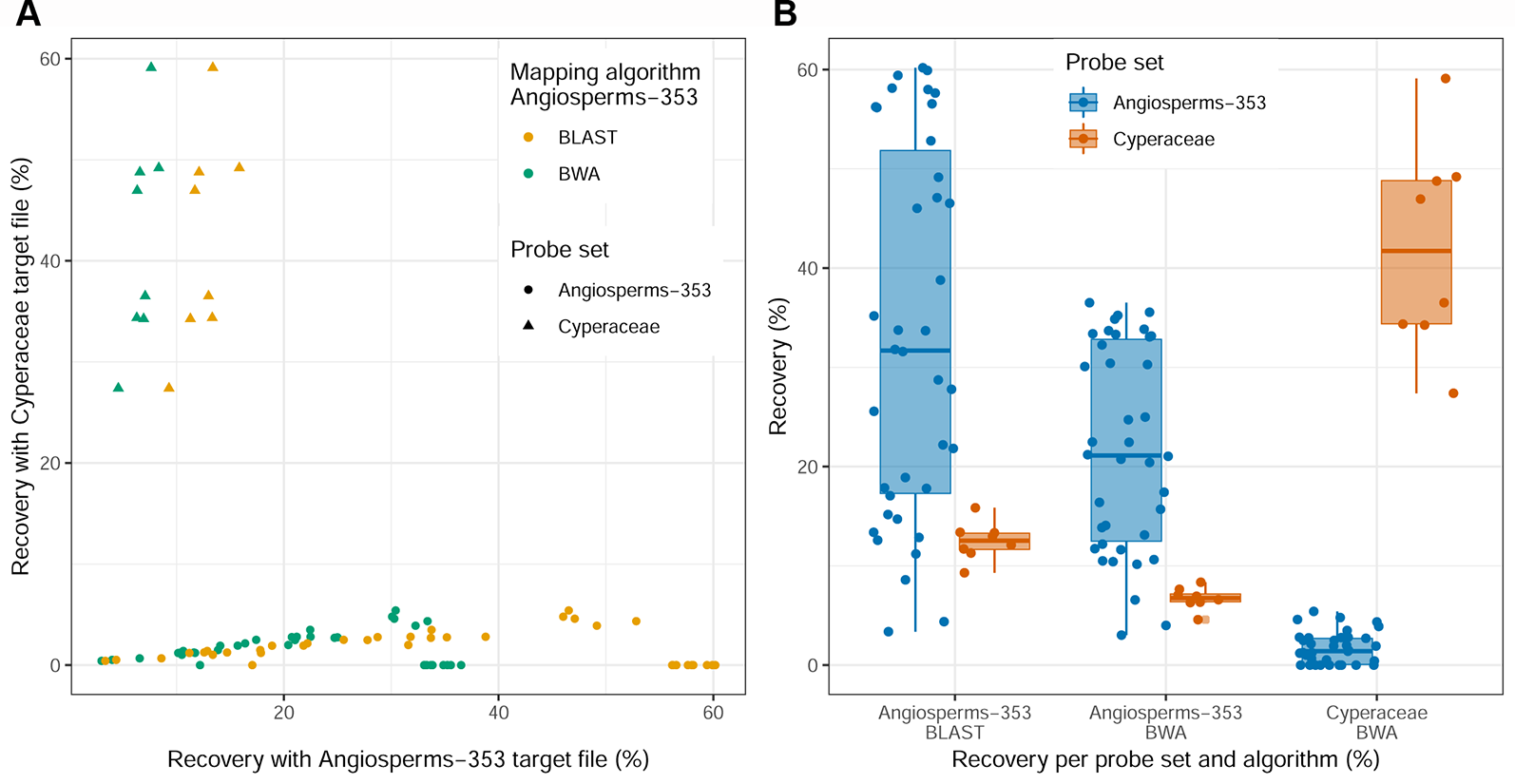
Recovery success for the Angiosperms-353 kit vs. the Cyperaceae-specific kit. (A) Percentage recovery with Angiosperms-353 (AA) target file from the accessions enriched with the Angiosperms-353 kit vs. percentage recovery of the Cyperaceae-specific targets from the accessions enriched with the Cyperaceae-specific probes. (B) Summary of recovery success per kit and across kits.
Table 2
| Contig | PIS | ||
|---|---|---|---|
| Angiosperms-353 | Mean | 751 | 75 |
| (38 accessions) | SD | 438 | 65 |
| Dataset 1 | Min | 87 | 0 |
| Max | 3,103 | 439 | |
| Total | 233,429 | 23,217 | |
| Cyperaceae-specific | Mean | 1,608 | 63 |
| (9 accessions) | SD | 830 | 59 |
| Dataset 2 | Min | 93 | 0 |
| Max | 7,527 | 479 | |
| Total | 683,427 | 26,630 | |
| Angiosperms-353 | Mean | 717 | 25 |
| (subset of 8 | SD | 411 | 28 |
| accessions) | Min | 150 | 0 |
| Dataset 3 | Max | 2,826 | 147 |
| Total | 221,564 | 7,613 | |
| Cyperaceae-specific | Mean | 1,471 | 50 |
| (8 accessions) | SD | 818 | 51 |
| Dataset 4 | Min | 162 | 0 |
| Max | 7,524 | 400 | |
| Total | 667,945 | 22,767 |
Length of the aligned contigs and number of parsimony informative sites (PIS) for data obtained after enrichment with the Angiosperms-353 and Cyperaceae-specific probes.
Figure 3
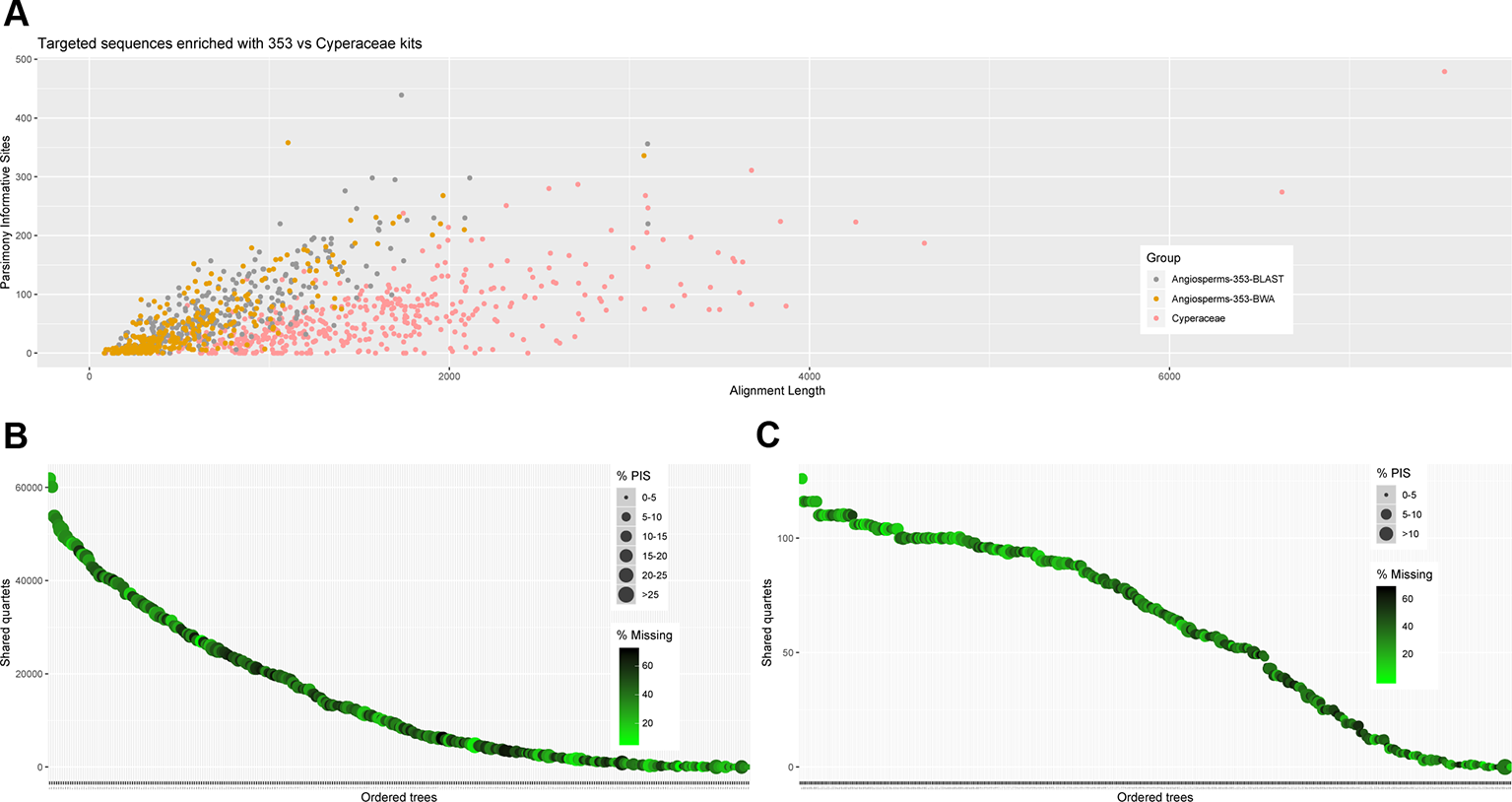
(A) Scatter plot of aligned contig length versus number of parsimony informative sites. (B) Number of gene trees that support the inferred species tree for data generated with the Angiosperms-353 probes, and (C) for data generated with the Cyperaceae-specific kit.
We investigated three measures of resolution power when dealing with shallow-level phylogenetic relationships for the Angiosperms-353 kit versus the Cyperaceae-specific kit: 1) the proportion of gene trees that support the inferred species tree (under the MSC) (Figure 3; Figures 4A, C), 2) the disagreement among loci and across sites in the total evidence tree (Figures 4B, D), and 3) the number of PIS retrieved across locus alignments (Figure 3; Supplementary Images 4 and 5). Addressing the first measure, many nodes are well supported in the ASTRAL tree generated for dataset 1 (loci targeted with the Angiosperms-353 kit for the accessions enriched with this kit) (Figure 4A) having local posterior probability (LPP) values greater than 0.9. Addressing the second measure, many nodes are similarly well supported in the IQ-TREE tree generated with the same data (Figure 4B) with most nodes having bootstrap (BS) values greater than 90%. However, some of the branches that received low LPP or BS value support have quartet scores indicating gene tree conflict and/or have low gCF scores, which indicates that few gene trees support the grouping. These branches occur among some species of the Cyperus margaritaceus-Cyperus niveus complex (clade A) and in the main backbone of clade B, which represents species of C4 Cyperus s.s. and the 10 C4 segregate genera accepted by Goetghebeur (1998). In the phylogenetic hypotheses obtained for dataset 2 (loci targeted with the Cyperaceae-specific probes for the accessions enriched with this kit) (Figures 4C, D), most nodes are equally well supported having LPP values greater than 0.9 or BS values greater than 90%. Addressing the third measure, when comparing the probe sets in terms of PIS by comparing dataset 4 (loci targeted by the Cyperaceae-specific probes for the eight C4 Cyperus clade accessions enriched with this kit; Supplementary Table 6D) with the equally sized and taxonomically equivalent dataset 3 (loci targeted by the Angiosperms-353 probes for a subset of eight C4 Cyperus clade accessions enriched with this kit; Supplementary Table 6C), the former has an average contig length of 1,471 (162–7,524), while the latter has shorter average length of 717 (150–2,826). However, the relative number of PIS is the same at 0.03 PIS/bp. This is also shown in Supplementary Image 4. A comparison of the support provided by the gene trees for the species tree between the two datasets of eight accessions is provided in Supplementary Image 5.
Figure 4
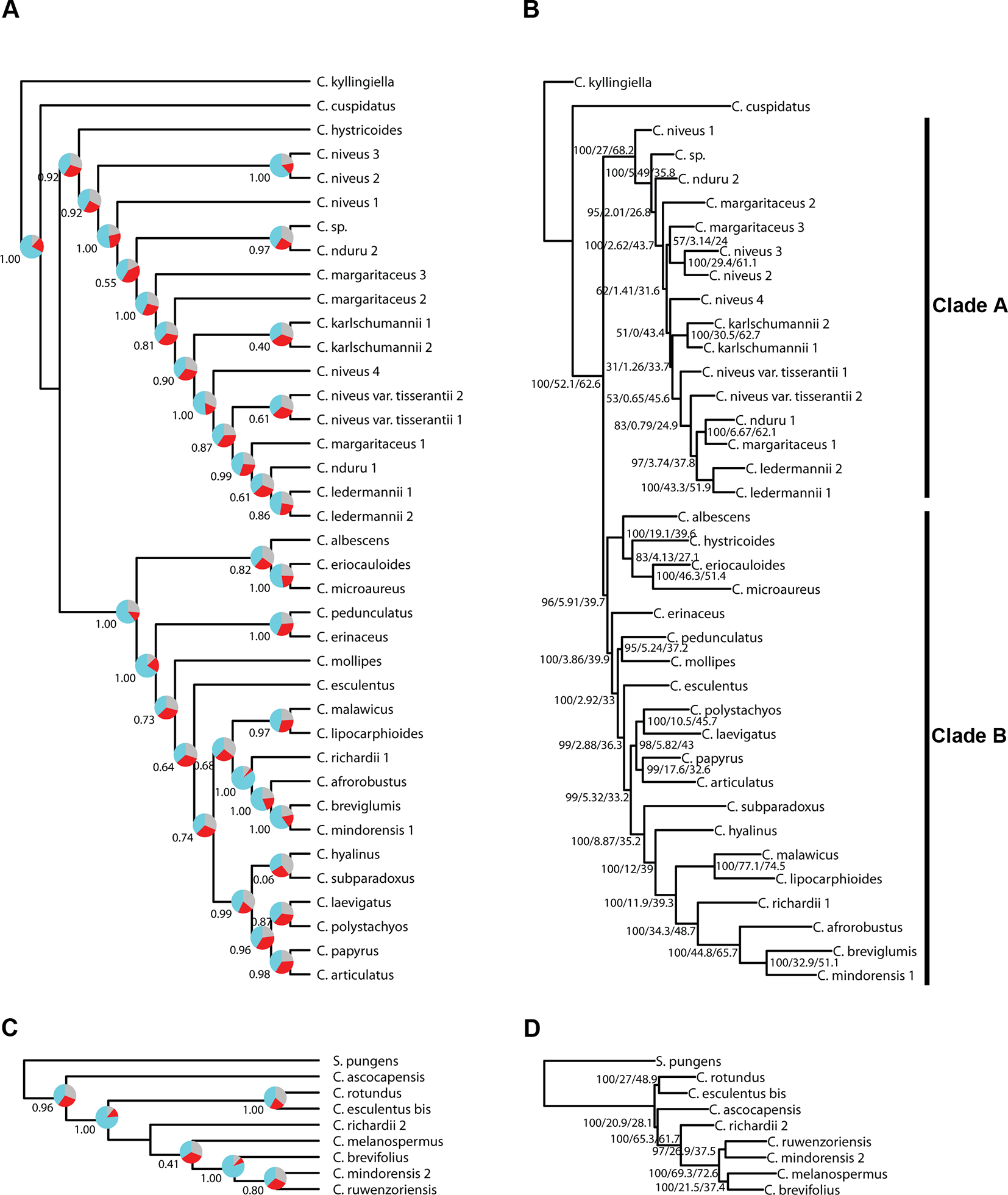
Phylogenetic reconstructions using ASTRAL (A, C) and using IQ-TREE (B, D) of the relationships in the C4 Cyperus clade inferred for (A, B) dataset 1, i.e., 38 samples enriched with the Angiosperms-353 probes, and (C, D) dataset 2, i.e., 9 samples enriched with the Cyperaceae-specific probes. The ASTRAL trees show local posterior probability values and pie charts visualizing quartet support values at the nodes (blue = agreeing loci; red = disagreeing loci; gray = uninformative), while the IQ-TREE trees show BS/gCF/sCF values at the nodes.
Mergeability of Data Obtained With Different Targeted Sequencing Kits
Different percentages of recovery were obtained using the AA or the DNA target files of the Angiosperms-353 kit in the accessions enriched with the Cyperaceae-specific kit. With the AA target file, we were able to recover 12.5% of the total target size from accessions enriched with the Cyperaceae-specific kit (Supplementary Table 2B). This percentage decreases to 6.73% when using the DNA target file (Supplementary Table 2A). On average 44 loci (35–53) were retrieved from accessions enriched with the Cyperaceae-specific kit using the Angiosperms-353 AA target file, and 32 (21–41) with the DNA target file. Capture success was very low when targeting Cyperaceae-specific loci from accessions enriched with the Angiosperms-353 probes (1.7%) using the DNA target file; however, sequence data was still retrieved from an average of 37 loci (0–106; Supplementary Table 2C). This information is summarized in Figure 2.
We tested the mergeability of the data generated for all Cyperus samples produced after enrichment with the two targeted sequencing kits, by inferring trees using ASTRAL and IQ-TREE for four merged datasets. Table 3 provides the length of the aligned contigs and number of PIS for the four merged datasets. The number of PIS is positively correlated with the size of the dataset (Table 3; Supplementary Tables 6E–H).
Table 3
| Contig | PIS | ||
|---|---|---|---|
| Angiosperms-353 | Mean | 739 | 85 |
| Dataset 5 | SD | 492 | 75 |
| Min | 87 | 0 | |
| Max | 4,211 | 539 | |
| Total | 259,321 | 27,656 | |
| Cyperaceae-specific | Mean | 1,458 | 76 |
| Dataset 6 | SD | 805 | 81 |
| Min | 153 | 0 | |
| Max | 7,520 | 554 | |
| Total | 720,324 | 37,645 | |
| All loci | Mean | 1,192 | 74 |
| Dataset 7 | SD | 775 | 74 |
| Min | 87 | 0 | |
| Max | 7,520 | 554 | |
| Total | 910,926 | 56,615 | |
| 57 overlapping loci | Mean | 1,206 | 152 |
| Dataset 8 | SD | 687 | 100 |
| Min | 312 | 6 | |
| Max | 4,211 | 539 | |
| Total | 68,719 | 8,686 |
Length of the aligned contigs and number of parsimony informative sites (PIS) for the four merged datasets: 1) aligned contigs of the loci targeted with the Angiosperm-353 probes, 2) aligned contigs of the loci targeted with the Cyperaceae-specific probes, 3) aligned contigs of all loci (without duplicating the overlapping genes), and 4) aligned contigs of the 57 overlapping loci targeted by both kits.
The amount of data retrieved targeting the Cyperaceae-specific loci from the data generated for all samples is larger when comparing the length of the alignments (Table 3; Figure 5) and the total number of PIS is also higher (c. 26.5% more PIS). The 57 overlapping loci are on average longer than those of most loci targeted by the Angiosperms-353 probes (1,206 versus 793) but shorter than the average for loci targeted with the Cyperaceae-specific kit (1,206 versus 1,458). These overlapping loci have a higher proportion of PIS per alignment in comparison with the other merged datasets (152 versus 74–85) (Supplementary Image 6; Supplementary Tables 6E–H). Although it includes much less data than the other analyzed datasets, the dataset of the 57 overlapping loci still includes a high number of PIS (8,686 PIS out of 68,719 bp or 0.12 PIS/bp; Table 3; Supplementary Table 6H).
Figure 5
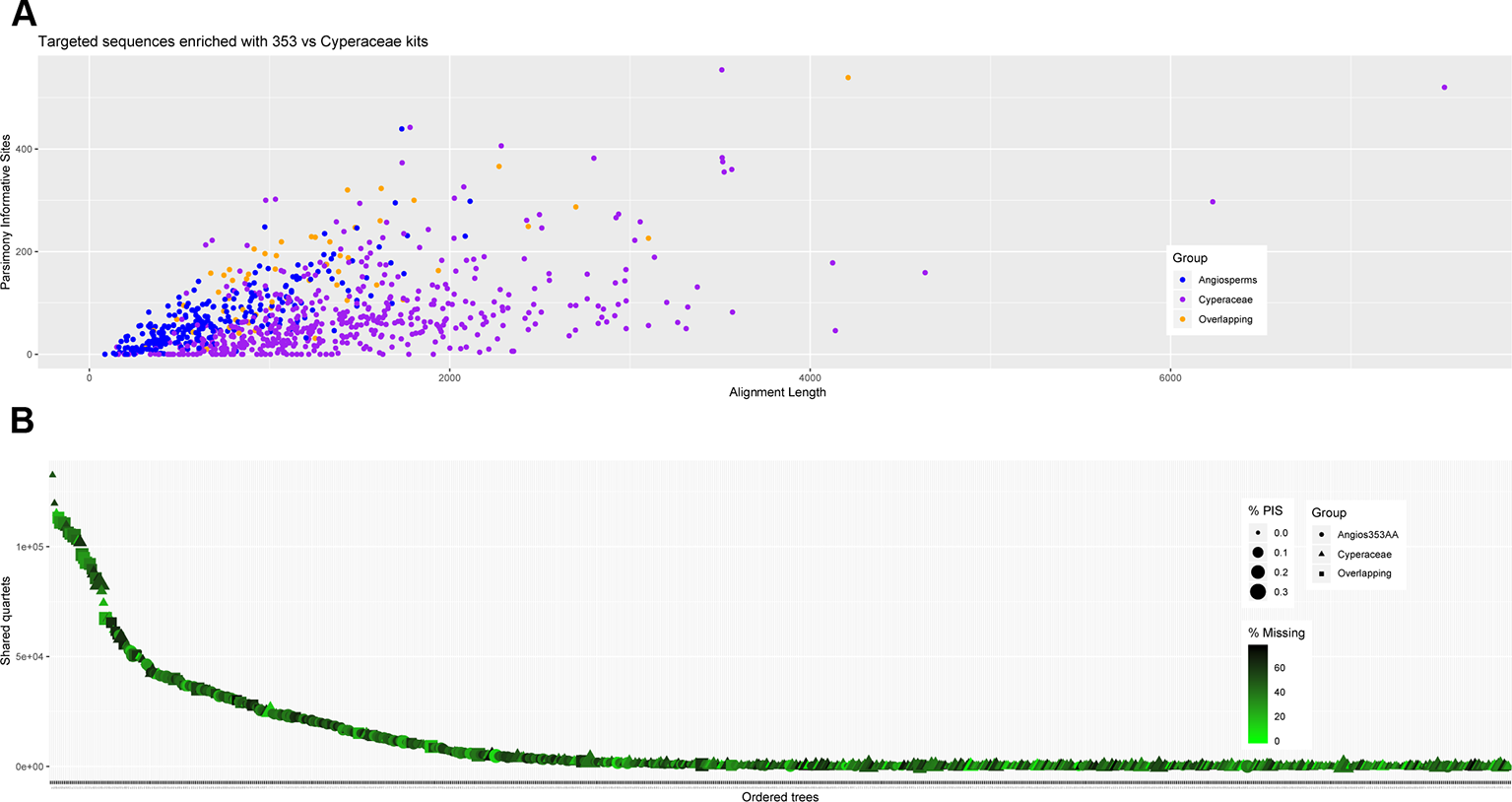
Number of gene trees that support the inferred species tree (A); and (B) scatter plot of aligned contig length vs. parsimony informative sites (PIS) for the data generated for all samples recovered targeting the Angiosperms-353 genes, the Cyperaceae-specific genes, and indicating the overlapping genes.
Supplementary Table 4 lists the names of the overlapping loci for both targeted sequencing kits and a comparison of statistics between the recovery of these loci is provided in Supplementary Table 6I. The average contig length of the overlapping genes retrieved with the Angiosperms-353 AA target file is shorter than when they are retrieved with the Cyperaceae-specific DNA target file (1,206 vs. 1,482). However, the average number of PIS retrieved with the Angiosperms-353 AA target file is higher than when the data are retrieved with the Cyperaceae-specific DNA target file (152 vs. 126).
Resolving Relationships in the C4 Cyperus Clade
Topologies produced with both approaches for dataset 1 (loci targeted with the Angiosperms-353 kit for the accessions enriched with this kit; Figures 4A, B) are very similar, except for the position of Cyperus hystricoides (B. Nord.) Bauters, which is retrieved as sister to clade A in the ASTRAL analysis (Figure 4A) and as part of clade B in the IQ-TREE analysis (Figure 4B). Likewise, the trees generated with both approaches, using dataset 2 (loci targeted with the Cyperaceae-specific probes for the accessions enriched with this kit; Figures 4C, D), result in similar topologies, except for the placement of Cyperus ascocapensis Bauters.
The topologies in the ASTRAL trees resulting from the four merged datasets are very similar (Figure 6), with generally high levels of node support. As in Figure 4, the placement of C. hystricoides was unstable, being reconstructed as sister to clade A in the tree based on the loci targeted by the Angiosperms-353 kit (Figure 6A), and among the first branching lineages of clade B in the other analyses (Figures 6B, D). Other differences in topology were found among taxa of the C. margaritaceus-C. niveus complex (clade A) and in the backbone of clade B where node support is lower and quartet scores indicate higher gene tree discordance (Figure 6). The proportion of gene trees supporting the retrieved topology is similar in Figures 6A–C, but the ASTRAL analysis of dataset 8 (overlapping loci for all accessions), yields a tree with higher locus concordance at most nodes.
Figure 6
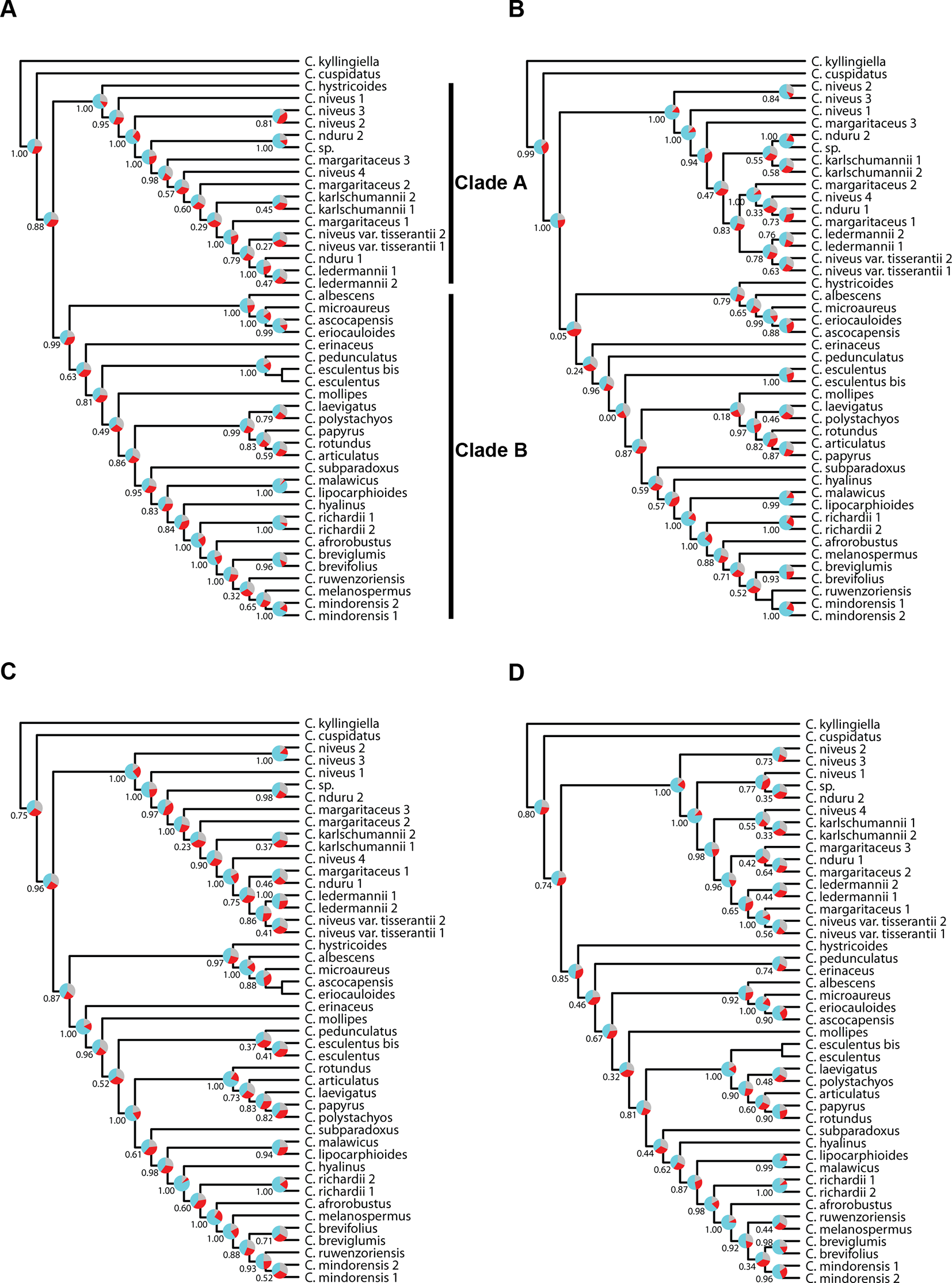
Phylogenetic reconstructions using ASTRAL of the relationships in the C4 Cyperus clade inferred for all accessions from aligned contigs of (A) dataset 5, i.e., the loci targeted with the Angiosperms-353 probes, (B) dataset 6, i.e., the loci targeted with the Cyperaceae-specific kit, (C) dataset 7, i.e., all targeted loci, and (D) dataset 8, i.e., the overlapping loci targeted by both kits. The trees show local posterior probability values and pie charts visualizing quartet support values at the nodes (blue = agreeing loci; red = disagreeing loci; gray = uninformative).
The analyses performed with IQ-TREE yielded topologies similar (Figure 7) to those retrieved with ASTRAL for the respective datasets. For clade A, a morphologically homogeneous species complex in the C4 Cyperus clade, resolution, and support are comparable between the ASTRAL and IQ-TREE results (Figures 6 and 7). However, for clade B, a morphologically heterogeneous subset of the C4 Cyperus clade, the IQ-TREE analyses provided higher support for some nodes, although the IQ-TREE topology is often less well resolved (Figures 6B, Dvs.Figures 7B, D). For dataset 6 (loci targeted with the Cyperaceae-specific probes for all accessions) and dataset 7 (all loci for all accessions), ASTRAL, a summary species-tree method, seems the handle high levels of missing data better in that the obtained results retrieve conspecific accessions or closely related species as sister taxa (Figures 6B, C). On the other hand, the total evidence trees inferred from concatenated data matrices in IQ-TREE under ML does not accurately place several samples in clade B (Figures 7B, C), i.e., C. esculentus (same accession sequenced with both targeted sequencing kits) is not reconstructed as monophyletic, and neither is C. mindorensis (different accessions but the same species). Similarly, the IQ-TREE analysis of dataset 8 (overlapping loci for all accessions; Figure 7D), did not reconstruct Cyperus ledermannii (Kük.) S.S. Hooper, C. niveus var. tisserantii (Cherm.) Lye, C. mindorensis, and C. richardii as monophyletic. This issue is not found for taxa of clade B in the IQ-TREE ML analysis of dataset 5 (loci targeted with the Angiosperms-353 kit for all accessions), although placement of Cyperus karlschumannii C.B. Clarke and C. niveus var. tisserantii were not reconstructed as expected in clade A (Figure 7A).
Figure 7
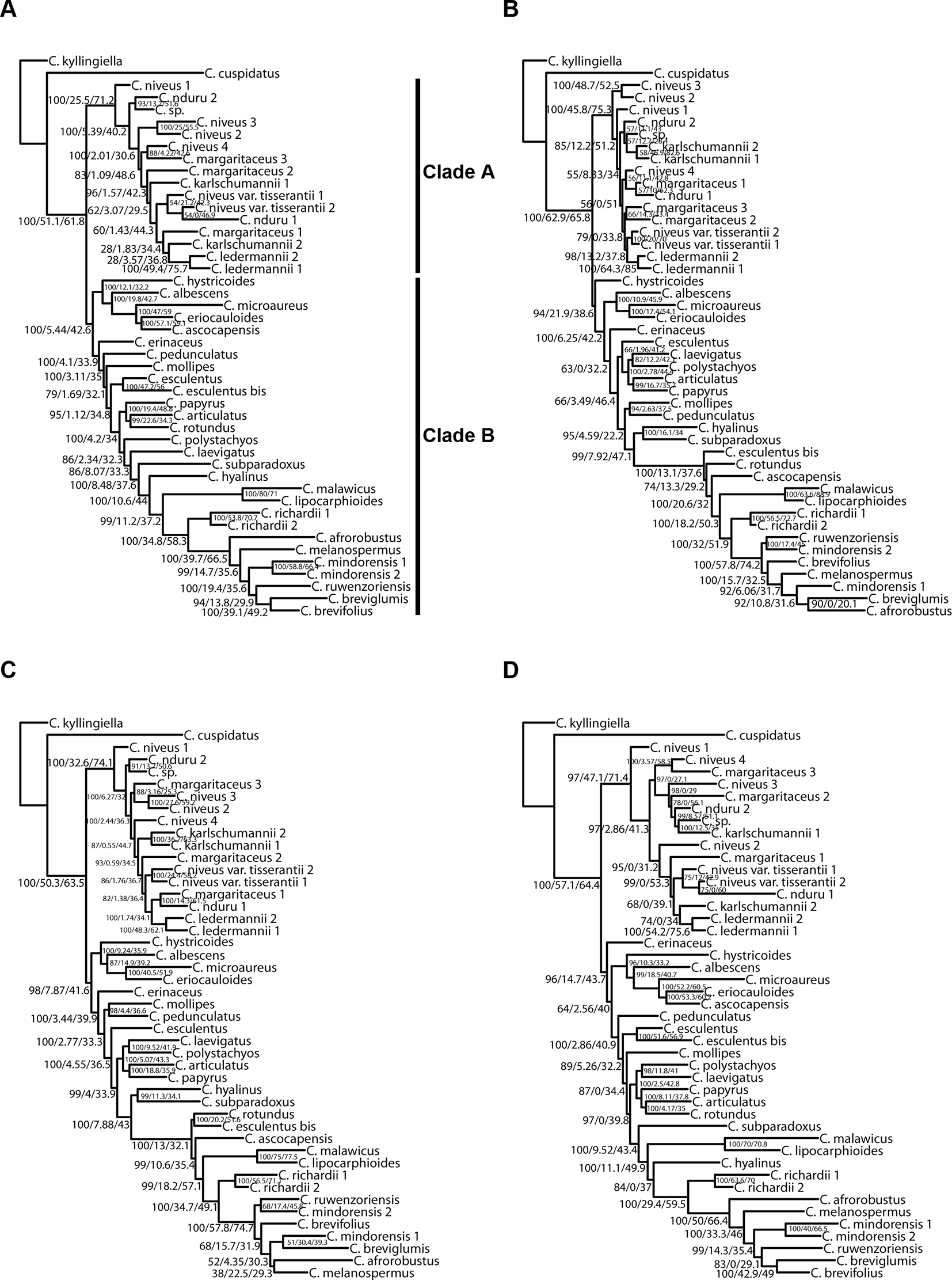
Phylogenetic reconstructions using IQ-TREE of the relationships in the C4 Cyperus clade inferred for all accessions from aligned contigs of (A) dataset 5, i.e., the loci targeted with the Angiosperms-353 probes, (B) dataset 6, i.e., the loci targeted with the Cyperaceae-specific kit, (C) dataset 7, i.e., all targeted loci, and (D) dataset 8, i.e., the overlapping loci targeted by both kits. The trees show bootstrap/gene concordance factor/site concordance factor values at the nodes.
When considering gene tree concordance for the analyses of dataset 5 (loci targeted with the Angiosperms-353 kit for all accessions), the monophyly of conspecific accessions or accessions of closely related species [e.g., Cyperus lipocarphioides (Kük.) Lye and Cyperus malawicus (J. Raynal) Lye] in clade B is supported by a high number of gene trees (Figure 7A). In clade A, only a few taxa are resolved as monophyletic (Figure 7A). In contrast, in the IQ-TREE results of dataset 6 (loci targeted with the Cyperaceae-specific probes for all accessions), and in the analysis of dataset 7 (all loci for all accessions), conspecific accessions in clade A tend to be retrieved as monophyletic and supported by a significant number of gene trees, while phylogenetic relationships between the taxa of clade B are not well resolved (Figures 7B, C). In the analyses based on dataset 8 (overlapping loci for all accessions), most nodes in clade B are supported by a proportion of gene trees, while many nodes in clade A have a gCF value of 0 (Figure 7D). This result contrasts with the higher locus concordance observed at most nodes in the ASTRAL analysis of this dataset (Figure 6D).
Discussion
Data Quality: Herbarium Versus Tissue Preserved for Deoxyribonucleic Acid Extraction
Most tissue samples in this study (30 out of 47) were obtained from herbarium specimens. The remainder were from silica dried samples (17 samples). The high quality of the sequence capture across types illustrates the potential of targeted sequencing to generate genomic level data from fragmented DNA (Hart et al., 2016; McKain et al., 2018; Villaverde et al., 2018; Brewer et al., 2019). Differences in extraction and sequencing methods did not appear to influence capture success or recovered target length with respect to total length targeted (Supplementary Table 2).
Universal Versus Family-Specific Probes
Recovery of loci targeted with the Cyperaceae-specific probes from accessions enriched with this kit was higher than for loci targeted with the universal Angiosperms-353 kit (Supplementary Table 2). Although more data was retrieved with the Cyperaceae-specific kit (dataset 2) than with the Angiosperms-353 kit (dataset 1) (total length aligned contigs was 683,427 bp and 233,429 bp, respectively), the number of PIS was comparable (26,630 vs. 23,217, respectively), yielding a lower ratio of PIS per total length aligned for the Cyperaceae-specific kit (0.04 PIS/bp) than for the Angiosperms-353 kit (0.1 PIS/bp) (Table 2). However, when comparing the values between the equally sized and taxonomically equivalent datasets 3 and 4, a comparable ratio of PIS per total aligned contig length is obtained (both with 0.03 PIS/bp). This indicates that the power of both targeted sequencing kits (off-target reads notwithstanding) to resolve shallow-level relationships for the C4 Cyperus clade is quite similar (Figure 3; Table 2).
Our finding belies our expectation going into this study that our family-specific kit would recover more variable loci. A recent study (Kadlec et al., 2017) on the species-rich heather genus Erica concluded that data from markers, custom-designed using the MarkerMiner pipeline, deliver better results than those obtained using a more universal approach. In another recent study, Chau et al. (2018) developed a custom-designed targeted sequencing kit using data from two Buddleja species (Scrophulariaceae) from 1KP using a modified version of the Sondovac marker development pipeline (Schmickl et al., 2016). They compared these taxon-specific loci with three universal loci sets (a conserved ortholog set [COSII] by Wu et al., 2006, shared single-copy nuclear [APVO SSC] genes by Duarte et al., 2010; and pentatricopeptide repeat [PPR] genes by Yuan et al., 2009). Chau et al. (2018) conclude that targeted sequencing is an effective method for increasing resolution and support in phylogenetics compared to Sanger sequencing, and that universal target loci can be as effective as taxon-specific loci in terms of capture success and phylogenetic informativeness. Our results support the conclusions of Chau et al. (2018). The advantage of a universal off-the-shelf targeted sequencing kit like the Angiosperms-353 kit is that it opens the opportunity to use targeted sequencing in plant groups with few genomic resources (Chau et al., 2018). Furthermore, universal kits are attractive in terms of reduced cost and effort, because they generate data suitable for wider analyses across angiosperms and may be applied as a DNA barcoding tool (Blattner, 2016; Kadlec et al., 2017), and predesigned kits can often be purchased at a discount from the producers (https://arborbiosci.com/products/mybaits-plant-angiosperms/).
Mergeability of Data Obtained with Different Targeted Sequencing Kits
Villaverde et al. (in review) combined accessions enriched with the anchored phylogenomics probes (Buddenhagen et al., 2016; Léveillé-Bourret et al., 2018) in their analyses of Carex using the Cyperaceae-specific kit. Here, we go one step further: we combine accessions enriched with two different kits, and we merge the data in both directions. Our study may be the first to do so, at least in angiosperms. As expected, sequence data recovery is higher when analyses are performed with the target file that matches the loci for which the samples were enriched (Figure 2). Nonetheless, even with limited overlap (57 genes) between targeted sequencing kits and a high level of missing data, data analyses under the MSC recover the same overall topology irrespective of dataset, with strong support. However, combined data analyses using a concatenated ML approach appear less robust to dataset differences and result in conspecific accessions not always being reconstructed as monophyletic when analyzing the merged datasets.
Resolving Relationships in the C4 Cyperus Clade
Most nodes are well supported in all analyses we conducted in the C4 Cyperus clade (Figures 4, 6, and 7), except for the branches near the backbone of clade B, as has been observed in earlier studies (e.g., Larridon et al., 2013; Bauters et al., 2014; Semmouri et al., 2019). These nodes show a higher gene tree discordance (based on ASTRAL quartet score and IQ-TREE gCF and sCF values), which likely resulted from an increased diversification rate (Spalink et al., 2016). Still the resolution and support retrieved in the backbone of the C4 Cyperus clade from targeted sequencing data is an important improvement over the polytomy obtained with Sanger sequencing results (e.g., Larridon et al., 2013).
The relationships retrieved in the C4 Cyperus clade, here investigated for the first time using phylogenomic data, largely match those obtained in previous studies (e.g., Larridon et al., 2011b; Larridon et al., 2013), with Cyperus cuspidatus Kunth sister to all other taxa in the C4 Cyperus clade. This species has an inflorescence of digitately clustered spikelets, which is characteristic of species in the C3 Cyperus grade and C4 Cyperus section Amabilis C.B. Clarke. Previously, sections in the C3 Cyperus grade + the C4 Cyperus section Amabilis were placed together in Cyperus subgenus Pycnostachys C.B. Clarke based on this shared inflorescence type versus the remaining sections in the C4 Cyperus clade, which are characterized by having spikes of spikelets. A notable difference with earlier studies is that remaining species in the C4 Cyperus clade form two well-supported clades (indicated as clade A and B; Figures 4, 6, and 7). One of the two groups of closely related species included in this study—i.e., the white-glumed Cyperus species or the C. margaritaceus-C. niveus complex (clade A)—is here reconstructed as sister to a clade (B) comprising the rest of the C4 Cyperus clade. Species of clade A had not been included in previous molecular studies. More research is needed to confirm that this is not a sampling artifact, however, although sampling in this study is limited, it adequately covers the range of morphological diversity observed in the C4 Cyperus clade, as it encompasses both species of C4 Cyperus s.s. (e.g., type species C. esculentus L.) and all 10 of the C4 segregate genera recognized by Goetghebeur (1998).
The position of C. hystricoides is unstable, being inferred either as sister to clade A or as part of the species-poor lineages in clade B (Figures 4, 6, and 7). This species was placed in Rikliella J. Raynal (Raynal, 1973) and later merged into Lipocarpha by Goetghebeur and Van den Borre (1989), who interpreted the inflorescence as a head of several spikes of spirally arranged single-flowered spikelets lacking a spikelet prophyll and glumes. However, an ontogenetic study (Bauters et al., 2014) showed that the inflorescence should be interpreted as a head of several spikelets with multiple spirally arranged flowers that have both a spikelet prophyll and glumes. With the new interpretation, the inflorescence type in C. hystricoides is similar to that of species previously placed in the C3 segregate genus Kyllingiella (now part of Cyperus sect. Leucocephali, incl. Cyperus kyllingiella; Larridon et al., 2011a), which is sister to the C4 Cyperus clade. This could provide morphological arguments for the placement of C. hystricoides among the species-poor lineages, away from the crown, of the C4 Cyperus clade.
Besides the C. margaritaceus-C. niveus complex, the other group of closely related species included in this study are seven species of Cyperus section Kyllinga (Rottb.) J. Kern (e.g., its type species C. mindorensis). Nodes within Cyperus sect. Kyllinga are well supported (Figures 4, 6, and 7), demonstrating the utility of the data obtained with both targeted sequencing kits to resolve low-level relationships in the C4 Cyperus clade. However, in the C. margaritaceus-C. niveus complex relationships between taxa are poorly supported although most morphologically defined taxa are retrieved as monophyletic, at least in the ASTRAL analyses (Figures 4, 6, and 7).
The results confirm the close relationship between Cyperus laevigatus (placed in the former segregate genus Juncellus C.B. Clarke) and Cyperus polystachyos (type species of the former segregate genus Pycreus) found in previous studies (e.g., Larridon et al., 2013; Semmouri et al., 2019). As in C3 Cyperus and C4 Cyperus s.s., the species previously placed in Juncellus and Pycreus have spikelets with multiple distichously arranged glumes each bearing a flower. However, in contrast to Cyperus s.s. with trigonous nutlets, Juncellus was recognized by dorsiventrally flattened nutlets, while in Pycreus nutlets are laterally compressed. In Cyperoideae, the development of the gynoecium from an annular primordium facilitates the shift in localization of stigma primordia (Vrijdaghs et al., 2011). Together with the decoupled development of the ovary and ovule (Reynders et al., 2012), this enables shifts between trigonous and dorsiventrally and laterally flattened nutlets in related species.
Our study is also the first to include all four species of the former segregate genus Alinula (C. lipocarphioides, C. malawicus, Cyperus microaureus Lye, Cyperus subparadoxus Kük.; Figures 4, 6, and 7). Earlier efforts to include all species in a Sanger sequencing study had failed due to degraded DNA extracted from herbarium specimens. This illustrates the advantage of targeted sequencing over Sanger sequencing for degraded DNA. Alinula sensu Goetghebeur (1998) is clearly polyphyletic (three groups). The first species to be published in Alinula was C. lipocarphioides by Raynal (1977) when he described the new genus. Later, another species, C. malawicus, was suggested to be its closest relative (Haines and Lye, 1983; Goetghebeur, 1986; Goetghebeur and Vorster, 1988). Our results confirm this close relationship. The species Cyperus microaureus was originally described in the segregate genus Ascolepis, but Goetghebeur (1977) relegated it to its own monotypic genus Marisculus Goetgh. because some of its inflorescence and spikelet characteristics are peculiar. Later, it was placed in Alinula (Goetghebeur and Vorster, 1988). In our results, the species appears sister to Ascolepis [represented by C. ascocapensis and Cyperus eriocauloides (Steud.) Bauters]. In his doctoral thesis, Vorster (1978) placed the fourth species, Cyperus subparadoxus, in a monotypic genus Pseudolipocarpha (not validly published) before moving it to Alinula (Goetghebeur and Vorster, 1988. It is here retrieved as a lineage separate from the other species formerly placed in Alinula.
Conclusion
We show the utility of two targeted sequencing kits, the universal Angiosperms-353 kit and a Cyperaceae-specific kit, in resolving relationships in a fast-evolving and taxonomically complex plant lineage, i.e., the C4 Cyperus clade. The probes from both kits work well with the often-degraded DNA-template obtained from herbarium material and allow the resolution of long-standing questions in Cyperaceae systematics (e.g., concerning the former segregate genus Alinula), where Sanger sequencing was previously either unsuccessful or provided no resolution. Generally, high support is retrieved using data of either or both kits, but some issues remain for the shortest branches where either significant conflict in gene trees or lack of signal occurs as shown by quartet scores, and gene and sCF. Potentially, adding off-target flanking regions and retrieving off-target high-copy sequence data such as the plastid genome, may provide added resolution. Our results demonstrate that data generated with a family-specific kit do not necessarily have more power than those obtained with a universal kit, at least in the C4 Cyperus clade, but that data generated with different targeted sequencing kits can often be merged for downstream analyses. Moreover, our study contributes to the growing consensus that targeted sequencing data are a powerful tool in resolving rapid radiations. We encourage ongoing studies to use targeted sequencing in lieu of Sanger sequencing to investigate the evolutionary history of Cyperaceae. The short-term costs in the lab will surely be mediated by long-term savings, as data can be repurposed for population genetics and phylogenetics with no return to the lab to sequence just one more locus.
Funding
IL is supported by the B.A. Krukoff Fund for the Study of African Botany and a pilot study grant from the Royal Botanic Gardens, Kew. Phylogenomic work at Kew was funded by grants from the Calleva Foundation, the Sackler Trust and the Garfield Weston Foundation. The research of TV is supported by the Spanish Ministry of Economy and Competitiveness (project CGL2016-77401-P). The research of EM was supported by the Spanish Government, through Juan de la Cierva-Formación contract (FJCI-2017-32314). Carex NSF grant (NSF-DEB award #1255901) supported the lab work and sequencing at the Morton Arboretum. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Statements
Data availability statement
The data generated for this study can be found in Genbank SRA under Bioproject numbers PRJNA553989 (Cyperus Bioproject), PRJEB35281 (Cyperus Baits Bioproject) and PRJNA553631 (Schoenoplectus pungens – Carex Bioproject).
Author contributions
IL and TV contributed equally as first authors. IL, TV, and AZ conceived the project design. IL and MX performed the sampling. IL, TV, LP, GB, NE, IF, MH, EM, and OM were responsible for generating the sequence data. IL, TV, AZ, and LP conducted the bioinformatic and molecular evolutionary analyses and wrote the manuscript. WB, FF, and AH supervised the research. All authors read and commented on the manuscript.
Acknowledgments
We would like to thank Sidonie Bellot for her help in visualizing the ASTRAL output and for always being happy to respond to lab and analysis related questions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01655/full#supplementary-material
References
1
Altschul S. F. Gish W. Miller W. Myers E. W. Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol.215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
2
Angiosperm Phylogeny Group (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc181, 1–20. doi: 10.1111/boj.12385
3
Baird N. A. Etter P. D. Atwood T. S. Currey M. C. Shiver A. L. Lewis Z. A. et al . (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PloS One3, e3376. doi: 10.1371/journal.pone.0003376
4
Bankevich A. Nurk S. Antipov D. Gurevich A. A. Dvorkin M. Kulikov A. S. et al . (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol.19, 455–477. doi: 10.1089/cmb.20120021
5
Bauckhage C. (2015). Numpy/scipy recipes for data science: k-medoids clustering (Published on ResearchGate). doi: 10.13140/RG.2.1.46020564
6
Bauters K. Larridon I. Reynders M. Huygh W. Asselman P. Vrijdaghs A. et al . (2014). A new classification for Lipocarpha and Volkiella as infrageneric taxa of Cyperus s.l. (Cypereae, Cyperoideae, Cyperaceae): insights from species tree reconstruction supplemented with morphological and floral developmental data. Phytotaxa166, 1–32. doi: 10.11646/phytotaxa.166.1.1
7
Beck J. B. Alexander P. J. Applhin L. Al-Shehbaz I. A. Rushworth C. Bailey C. D. et al . (2012). Does hybridization drive the transition to asexuality in diploid Boechera? Evol.66, 985–995. doi: 10.1111/j.1558-5646.2011.01507.x
8
Blattner F. R. . (2016). TOPO6: a nuclear single-copy gene for plant phylogenetic inference. Plant Syst. Evol.302, 239–244. doi: 10.1007/s00606-015-1259-1
9
Bolger A. M. Lohse M. Usadel B. (2014). Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics30, 2114–2120. doi: 10.1093/bioinformatics/btu170
10
Borowiec M. L. (2016). AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ4, e1660. doi: 10.7717/peerj1660
11
Brewer G. E. Clarkson J. J. Maurin O. Zuntini A. R. Barber V. Bellot S. et al . (2019). Factors affecting targeted sequencing of 353 nuclear genes from herbarium specimens spanning the diversity of angiosperms. Front. Plant Sci.10, 1102. doi: 10.3389/fpls.2019.01102
12
Buddenhagen C. Lemmon A. R. Lemmon E. M. Bruhl J. J. Cappa J. Clement W. L. et al . (2016). Anchored phylogenomics of angiosperms I: assessing the robustness of phylogenetic estimates. BioRxiv [Preprint], 1–61. doi: 10.1101/086298
13
Chamala S. García N. Godden G. T. Krishnakumar V. Jorden-Thaden I. E. De Smet R. et al . (2015). MarkerMiner 1.0: A new application for phylogenetic marker development using angiosperm transcriptomes. Appl. Plant Sci.3, 1400115. doi: 10.3732/apps.1400115
14
Chase M. W. Soltis D. E. Olmstead R. G. Morgan D. Les D. H. Mishler B. D. et al . (1993). Phylogenetics of Seed Plants: An Analysis of Nucleotide Sequences from the Plastid Gene rbcL. Ann. Missouri Bot. Garden80, 528–548 & 550–580. doi: 10.2307/2399846
15
Chau J. H. Rahfeldt W. A. Olmstead R. G. (2018). Comparison of taxon-specific versus general locus sets for targeted sequence capture in plant phylogenomics. Appl. Plant Sci.6, e1032. doi: 10.1002/aps3.1032
16
Couvreur T. L. P. Helmstetter A. J. Koenen E. J. M. Bethune K. Brandão R. D. Little S. A. et al . (2019). Phylogenomics of the major tropical plant family Annonaceae using targeted enrichment of nuclear genes. Front. Plant Sci.9, 1941. doi: 10.3389/fpls.2018.01941
17
Deschamps S. Llaca V. May G. D. (2012). Genotyping-by-Sequencing in Plants. Biol.1, 460–483. doi: 10.3390/biology1030460
18
Dodsworth S. Pokorny S. Johnson M. G. Kim J. T. Maurin O. Wickett N. J. et al . (2019). Hyb-Seq for Flowering Plant Systematics. Trends Plant Sci.24, 887–891. doi: 10.1016/j.tplants.2019.07.011
19
Doyle J. J. Doyle J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull.19, 11–15.
20
Duarte J. M. Wall P. K. Edger P. P. Landherr L. L. Ma H. Pires J. C. et al . (2010). Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol.10, 61. doi: 10.1186/1471-2148-10-61
21
Elshire R. J. Glaubitz J. C. Sun Q. Poland J. Kawamoto K. Buckler E. S. et al . (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PloS One6, e19379. doi: 10.1371/journal.pone.0019379
22
Escudero M. Hipp. A. L. (2013). Shifts in diversification rates and clade ages explain species richness in higher-level sedge taxa (Cyperaceae). Am. J. Bot.100, 2403–2411. doi: 10.3732/ajb.1300162
23
Escudero M. Eaton D. A. R. Hahn M. Hipp A. L. (2014). Genotyping-by-sequencing as a tool to infer phylogeny and ancestral hybridization: A case study in Carex (Cyperaceae). Mol. Phyl. Evol.79, 359–367. doi: 10.1016/j.ympev.2014.06.026
24
Faircloth B. C. Alda F. Hoekzema K. Burns M. D. Oliveira C. Albert J. S. et al . (2018). A target enrichment bait set for studying relationships among ostariophysan fishes. BioRxiv [Preprint], 1–53. doi: 10.1101/432583
25
Fishbein M. Hibsch-Jetter C. Soltis D. E. Hufford L. (2001). Phylogeny of saxifragales (Angiosperms, Eudicots): analysis of a rapid, ancient radiation. Syst. Biol.50, 817–847. doi: 10.1080/106351501753462821
26
Goetghebeur P. Van den Borre A. (1989). Studies in Cyperaceae 8. A revision of Lipocarpha, including Hemicarpha and Rikliella. Wageningen Agr. Univ. Pap.89, 1–87. http://edepot.wur.nl/283696.
27
Goetghebeur P. (1977). Studies in Cyperaceae 1. Taxonomic notes on Ascolepis and Marisculus, a new genus of the tribe Cypereae. Bull. Nat. Plantentuin Belg.47, 435–447. doi: 10.2307/3667910
28
Goetghebeur P. (1986). “Genera Cyperacearum,” in Een bijdrage tot de kennis van morfologie, systematiek en fylogenese van de Cyperaceae. PhD thesis (Gent: Ghent University), 1164.
29
Goetghebeur P. Vorster P . (1988). Studies in Cyperaceae 7. The genus Alinula J. Raynal: A reappraisal. Bull. Jard. Bot. Natl. Belg.58, 457–465. doi: 10.2307/3668298
30
Goetghebeur P. (1998). “Cyperaceae,” in The families and genera of vascular plants 4. Ed. KubitzkiK. (Berlin: Springer-Verlag), 141–190.
31
Haines R. Lye K. A. (1983). The sedges and rushes of East Africa. (Nairobi: East African Natural History Society), p. 404.
32
Hart M. L. Forrest L. L. Nicholls J. A. Kidner C. A. (2016). Retrieval of hundreds of nuclear loci from herbarium specimens. Taxon65, 1081–1092. doi: 10.12705/655.9
33
Hinchliff C. E. Roalson E. H. (2013). Using supermatrices for phylogenetic inquiry: an example using the sedges. Syst. Biol.62, 205–219. doi: 10.1093/sysbio/sys088
34
Huygh W. Larridon I. Reynders M. Muasya A. M. Govaerts R. Simpson D. A. et al . (2010). Nomenclature and typification of names of genera and subdivisions of genera in Cypereae (Cyperaceae): 1. Names of genera in the Cyperus clade. Taxon59, 1883–1890. doi: 10.1002/tax.596021
35
Johnson M. G. Gardner E. M. Liu Y. Medina R. Goffinet B. Shaw A. W. et al . (2016). HybPiper: Extracting coding sequence and introns for phylogenetics from high-throughput sequencing reads using target enrichment. Appl. Plant Sci.4, 1600016. doi: 10.3732/apps.1600016
36
Johnson M. G. Pokorny L. Dodsworth S. Botigué L. R. Cowan R. S. Devault A. et al . (2018). A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering. Syst. Biol.68, 594–606. doi: 10.1093/sysbio/syy086
37
Jung J. Choi H.-K. (2012). Recognition of two major clades and early diverged groups within the subfamily Cyperoideae (Cyperaceae) including Korean sedges. J. Plant Res.126, 335–349. doi: 10.1007/s10265-012-0534-2
38
Kadlec M. Bellstedt D. U. Le Maitre N. C. Pirie M. D. (2017). Targeted NGS for species level phylogenomics: “made to measure” or “one size fits all”? PeerJ5, e3569. doi: 10.7717/peerj3569
39
Kates H. R. Johnson M. G. Gardner E. M. Zerega N. J. C. Wickett N. J. (2018). Allele phasing has minimal impact on phylogenetic reconstruction from targeted nuclear gene sequences in a case study of Artocarpus. Am. J. Bot.105, 404–416. doi: 10.1002/ajb21068
40
Katoh K. Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol.30, 772–780. doi: 10.1093/molbev/mst010
41
Kipkemboi J. van Dam A. A. (2018). “Papyrus Wetlands,” in The Wetland Book: II: Distribution, Description, and Conservation. Eds. FinlaysonC. M.MiltonG. R.PrenticeR. C.DavidsonN. C. (Dordrecht: Springer Netherlands), 183–197. doi: 10.1007/978-94-007-4001-3_218
42
Larridon I. Reynders M. Huygh W. Bauters K. Vrijdaghs A. Leroux O. et al . (2011a). Taxonomic changes in C3 Cyperus (Cyperaceae) supported by molecular phylogenetic data, morphology, embryology, ontogeny and anatomy. Plant Ecol. Evol.144, 327–356. doi: 10.5091/plecevo.2011.653
43
Larridon I. Reynders M. Huygh W. Bauters K. Van de Putte K. Muasya A. M. et al . (2011b). Affinities in C3 Cyperus lineages (Cyperaceae) revealed using molecular phylogenetic data and carbon isotope analysis. Bot. J. Linn. Soc167, 19–46. doi: 10.1111/j.1095-8339.2011.01160.x
44
Larridon I. Reynders M. Huygh W. Muasya A. M. Govaerts R. Simpson D. A. et al . (2011c). Nomenclature and typification of names of genera and subdivisions of genera in Cypereae (Cyperaceae): 2. Names of subdivisions of Cyperus. Taxon60, 868–884. doi: 10.1002/tax.603021
45
Larridon I. Bauters K. Reynders M. Huygh W. Muasya A. M. Simpson D. A. et al . (2013). Towards a new classification of the giant paraphyletic genus Cyperus (Cyperaceae): phylogenetic relationships and generic delimitation in C4 Cyperus. Bot. J. Linn. Soc172, 106–126. doi: 10.1111/boj.12020
46
Larridon I. Bauters K. Huygh W. Reynders M. Goetghebeur P. (2014). Taxonomic changes in C4 Cyperus (Cypereae, Cyperoideae, Cyperaceae): combining the sedge genera Ascolepis, Kyllinga and Pycreus into Cyperus s.l. Phytotaxa166, 33–48. doi: 10.11646/phytotaxa.166.1.2
47
Lemmon A. R. Emme S. A. Lemmon E. M. (2012). Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst. Biol.61, 727–744. doi: 10.1093/sysbio/sys049
48
Léveillé-Bourret É. Starr J. R. Ford B. A. Lemmon E. M. Lemmon A. R. (2018). Resolving rapid radiations within angiosperm families using anchored phylogenomics. Syst. Biol.67, 94–112. doi: 10.1093/sysbio/syx050
49
Li H. Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics25, 1754–1760. doi: 10.1093/bioinformatics/btp324
50
Li H. Handsaker B. Wysoker A. Fennell T. Ruan J. Homer N. et al . (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics25, 2078–2079. doi: 10.1093/bioinformatics/btp352
51
Liu Y. Johnson M. G. Cox C. J. Medina R. Devos N. Vanderpoorten A. et al . (2019). Resolution of the ordinal phylogeny of mosses using targeted exons from organellar and nuclear genomes. Nat. Commun.10, 1485. doi: 10.1038/s41467-019-09454-w
52
Maguilla E. Escudero M. Hipp A. L. Luceño M. (2017). Allopatric speciation despite historical gene flow: Divergence and hybridization in Carex furva and C. lucennoiberica (Cyperaceae) inferred from plastid and nuclear RAD-seq data. Mol. Ecol.26, 5646–5662. doi: 10.1111/mec.14253
53
Matasci N. Hung L.-H. Yan Z. Carpenter E. J. Wickett N. J. Mirarab S. et al . (2014). Data access for the 1,000 Plants (1KP) project. GigaScience3, 10–17. doi: 10.1186/2047-217X-3-17
54
McKain M. R. Johnson M. G. Uribe-Convers S. Eaton D. Yang Y. (2018). Practical considerations for plant phylogenomics. Appl. Plant Sci.6, e1038. doi: 10.1002/aps31038
55
Minh B. Q. Hahn M. Lanfear R. (2018). New methods to calculate concordance factors for phylogenomic datasets. BioRXiv [Preprint], 1–7. doi: 10.1101/487801
56
Mitchell N. Lewis P. O. Lemmon E. M. Lemmon A. R. Holsinger K. E. (2017). Anchored phylogenomics improves the resolution of evolutionary relationships in the rapid radiation of Protea L. Am. J. Bot.104, 102–115. doi: 10.3732/ajb.1600227
57
Muasya A. M. Simpson D. A. Chase M. W. (2002). Phylogenetic relationships in Cyperus L. s.l. (Cyperaceae) inferred from plastid DNA sequence data. Bot. J. Linn. Soc138, 145–153. doi: 10.1046/j.1095-8339.2002.138002145.x
58
Muasya A. M. Simpson D. A. Verboom G. A. Goetghebeur P. Naczi R. F. C. Chase M. W. et al . (2009). Phylogeny of Cyperaceae based on DNA sequence data: current progress and future prospects. Bot. Rev.75, 2–21. doi: 10.1007/s12229-008-9019-3
59
Nabhan A. R. Sarkar I. N. (2012). The impact of taxon sampling on phylogenetic inference: a review of two decades of controversy. Brief Bioinform.13, 122–134. doi: 10.1093/bib/bbr014
60
Nguyen L.-T. Schmidt H. A. van Haeseler A. Minh B. Q. (2015). IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol.32, 268–274. doi: 10.1093/molbev/msu300
61
Nicholls J. A. Pennington R. T. Koenen E. J. M. Hughes C. E. Hearn J. Bunnefeld L. et al . (2015). Using targeted enrichment of nuclear genes to increase phylogenetic resolution in the neotropical rain forest genus Inga (Leguminosae: Mimosoideae). Front. Plant Sci.6, 710. doi: 10.3389/fpls.2015.00710
62
Philippe H. Brinkmann H. Lavrov D. V. Littlewood T. J. Manuel M. Wörheide Gert et al . (2011). Resolving Difficult Phylogenetic Questions: Why More Sequences Are Not Enough. PloS Biol.9, e1000602. doi: 10.1371/journal.pbio.1000602
63
Raynal J. . (1973). Cyperological notes 19. Contribution to the classification of the Cyperoideae subfamily. Adansonia Ser.2, 13 (2), 145–171.
64
Reynders M. Huygh W. Larridon I. Muasya A. M. Govaerts R. Simpson D. A. et al . (2011). Nomenclature and typification of names of genera and subdivisions of genera in the Cypereae (Cyperaceae): 3. Names in segregate genera of Cyperus. Taxon60, 885–895. doi: 10.1002/tax.603022
65
Reynders M. Vrijdaghs A. Larridon I. Huygh W. Leroux O. Muasya A. M. et al . (2012). Gynoecial anatomy and development in Cyperoideae (Cyperaceae, Poales): congenital fusion of carpels facilitates evolutionary modifications in pistil structure. Plant Ecol. Evol.145, 96–125. doi: 10.5091/plecevo.2012.675
66
Roalson E. H. Prata A. P. Mesterházy A. Chase M. W. Simpson D. A. Thomas W. W. et al . (2019). A broader circumscription of Bulbostylis, to include Nemum (Abildgaardieae: Cyperaceae). Phytotaxa395, 199–208. doi: 10.11646/phytotaxa.395.3.4
67
Ruhfel B. R. Gitzendanner M. A. Soltis P. S. Soltis D. E. Burleigh J. G. (2014). From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol. Biol.14, 23. doi: 10.1186/1471-2148-14-23
68
Sand A. Holt M. K. Johansen J. Brodal G. S. Mailund T. Pedersen C. N. S. (2014). tqDist: a library for computing the quartet and triplet distances between binary or general trees. Bioinformatics30, 2079–2080. doi: 10.1093/bioinformatics/btu157
69
Schmickl R. Liston A. Zeisek V. Oberlander K. Weitemier K. Straub S. C. K. et al . (2016). Phylogenetic marker development for target enrichment from transcriptomic and genome skim data: The pipeline and its application in southern African Oxalis (Oxalidaceae). Mol. Ecol. Resour.16, 1124–1135. doi: 10.1111/1755-0998.12487
70
Semmouri I. Bauters K. Léveillé-Bourret É. Starr J. R. Goetghebeur P. Larridon I. (2019). The phylogeny and systematics of Cyperaceae, the evolution and importance of embryo morphology. Bot. Rev.85, 1–39. doi: 10.1007/s12229-018-9202-0
71
Simpson D. A. Inglis C. A. (2001). Cyperaceae of economic, ethnobotanical and horticultural importance: a checklist. Kew Bull.56, 257–360. doi: 10.2307/4110962
72
Simpson D. A. Muasya A. M. Alves M. Bruhl J. J. Dhooge S. Chase M. W. et al . (2007). Phylogeny of Cyperaceae based on DNA sequence data—a new rbcL analysis. Aliso23, 72–83. doi: 10.5642/aliso.20072301.09
73
Slater G. S. Birney E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinform.6, 31. doi: 10.1186/1471-2105-6-31
74
Smith M. R. (2019). Quartet: comparison of phylogenetic trees using quartet and bipartition measures. doi: 10.5281/zenodo.2536318
75
Snak C. Vatanparast M. Christian S. Lewis G. P. Lavin M. Kajita T. et al . (2016). A dated phylogeny of the papilionoid legume genus Canavalia reveals recent diversification by a pantropical liana lineage. Mol. Phyl. Evol.98, 133–146. doi: 10.1016/j.ympev.2016.02.001
76
Spalink D. Drew B. T. Pace M. C. Zaborsky J. G. Starr J. R. Cameron K. M. et al . (2016). Biogeography of the cosmopolitan sedges (Cyperaceae) and the area-richness correlation in plants. J. Biogeogr.43, 1893–1904. doi: 10.1111/jbi.12802
77
Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics30, 1312–1313. doi: 10.1093/bioinformatics/btu033
78
Stevens J. D. Rogers W. L. Heyduk K. Cruse-Sanders J. M. Determann R. O. Glenn T. C. et al . (2015). Resolving phylogenetic relationships of the recently radiated carnivorous plant genus Sarracenia using target enrichment. Mol. Phyl. Evol.85, 76–87. doi: 10.1016/j.ympev.2015.01.015
79
Villaverde T. Pokorny L. Olsson S. Rincón-Barrado M. Johnson M. G. Gardner E. M. et al . (2018). Bridging the micro- and macroevolutionary levels in phylogenomics: Hyb-Seq solves relationships from populations to species and above. New Phytol.220, 636–650. doi: 10.1111/nph.15312
80
Villaverde T. Jiménez-Mejías P. Luceño M. Roalson E. H. Hipp A. L. the Global Carex Group (in review). A new classification of Carex subgenera supported by a Hyb-Seq backbone phylogeny. Bot. J. Linn. Soc.
81
Vorster P. J. (1978). Revision of the taxonomy of Mariscus Vahl and related genera in southern Africa. PhD thesis (Pretoria: University of Pretoria), 384.
82
Vrijdaghs A. Reynders M. Muasya A. M. Larridon I. Goetghebeur P. Smets E. (2011). Morphology and development of spikelets and flowers in Cyperus and Pycreus (Cyperaceae). Plant Ecol. Evol.144, 44–63. doi: 10.5091/plecevo.2011.436
83
Weitemier K. Straub S. C. K. Cronn R. C. Fishbein M. Schmickl R. McDonnell A. et al . (2014). Hyb-Seq: Combining target enrichment and genome skimming for plant phylogenomics. Appl. Plant Sci.2, 1400042. doi: 10.3732/apps.1400042
84
Whitfield J. B. Lockhart P. J. (2007). Deciphering ancient rapid radiations. Trends Ecol. Evol.22, 258–265. doi: 10.1016/j.tree.2007.01.012
85
Wickett N. J. Mirarab S. Nguyen N. Warnow T. Carpenter E. Matasci N. et al . (2014). Phylotranscriptomic analysis of the origin and early diversification of land plants. PNAS111, E4859–E4868. doi: 10.1073/pnas.1323926111
86
Wu F. Mueller L. A. Crouzillat D. Pétiard V. Tanksley S. D. (2006). Combining bioinformatics and phylogenetics to identify large sets of single-copy orthologous genes (COSII) for comparative evolutionary and systematic studies: A test case in the euasterid plant clade. Genetics174, 1407–1420. doi: 10.1534/genetics.106.062455
87
Yuan Y.-W. Liu C. Marx H. E. Olmstead R. G. (2009). The pentatricopeptide repeat (PPR) gene family, a tremendous resource for plant phylogenetic studies. New Phytol.182, 272–283. doi: 10.1111/j.1469-8137.2008.02739.x
88
Zhang C. Rabiee M. Sayyari E. Mirarab S. (2018). ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinf.19, 153. doi: 10.1186/s12859-018-2129-y
Summary
Keywords
C4 Cyperus clade, Cyperaceae, Plant and Fungal Trees of Life, phylogenomics, polytomy, targeted sequencing
Citation
Larridon I, Villaverde T, Zuntini AR, Pokorny L, Brewer GE, Epitawalage N, Fairlie I, Hahn M, Kim J, Maguilla E, Maurin O, Xanthos M, Hipp AL, Forest F and Baker WJ (2020) Tackling Rapid Radiations With Targeted Sequencing. Front. Plant Sci. 10:1655. doi: 10.3389/fpls.2019.01655
Received
04 July 2019
Accepted
22 November 2019
Published
09 January 2020
Volume
10 - 2019
Edited by
Michael R. McKain, University of Alabama, United States
Reviewed by
Angela Jean McDonnell, Chicago Botanic Garden, United States; Ryan Folk, Mississippi State University, United States
Updates
Copyright
© 2020 Larridon, Villaverde, Zuntini, Pokorny, Brewer, Epitawalage, Fairlie, Hahn, Kim, Maguilla, Maurin, Xanthos, Hipp, Forest and Baker.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabel Larridon, i.larridon@kew.org; Tamara Villaverde, t.villaverde@rjb.csic.es
This article was submitted to Plant Systematics and Evolution, a section of the journal Frontiers in Plant Science
†These authors share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.