 Weijie Xu
Weijie Xu Yiyang Zhao
Yiyang Zhao Sisi Chen1,2,3
Sisi Chen1,2,3 Deqiang Zhang
Deqiang Zhang- 1Beijing Advanced Innovation Center for Tree Breeding by Molecular Design, College of Biological Sciences and Technology, Beijing Forestry University, Beijing, China
- 2National Engineering Laboratory for Tree Breeding, College of Biological Sciences and Technology, Beijing Forestry University, Beijing, China
- 3Key Laboratory of Genetics and Breeding in Forest Trees and Ornamental Plants, College of Biological Sciences and Technology, Beijing Forestry University, Ministry of Education, Beijing, China
New kinase has emerged throughout evolution, but how new kinase evolve while maintaining their functions and acquiring new functions remains unclear. Fructokinase (FRK), the gateway kinase to fructose metabolism, plays essential roles in plant development, and stress tolerance. Here, we explored the evolution of FRK gene family in 20 plant species (from green algae to angiosperms) and their functional roles in Populus. We identified 125 putative FRK genes in the 20 plant species with an average of 6 members per species. Phylogenetic analysis separated these 125 genes into 8 clades including 3 conserved clades and 5 specific clades, the 5 of which only exist in green algae or angiosperms. Evolutionary analysis revealed that FRK genes in ancient land plants have the largest number of functional domains with the longest amino acid sequences, and the length of FRK genes became shorter during the transition to vascular plants. This was accompanied by loss, acquisition, and diversification of functional domains. In Populus, segmental duplication appears to be the main mechanism for the expansion of FRK genes. Specially, most FRK genes duplicated in salicoids are regulated by Populus-specific microRNAs. Furthermore, compared with common FRKs, Populus-specific FRKs have showed higher expression specificity and are associated with fewer growth and wood property traits, which suggests that these FRKs may have undergone functional divergence. Our study explores the specific roles of FRKs in the Populus genome and provides new insights for functional investigation of this gene family.
Introduction
Evolution is accompanied by various changes including morphological, physiological and genome, which allow organisms to deal with challenging conditions, including increased CO2 concentration, desiccation, changes in light intensity, high temperature, marked seasonal changes, and limited nutrient availability (Delaux et al., 2012; Romani et al., 2018). At the cellular level, changes of these physiological are regulated by kinase, which are major drivers of evolution (Michelson et al., 1985; Mehlgarten et al., 2018). New kinase classes evolved to orchestrate the complex development and environmental responses of higher organisms. Meanwhile, the birth, loss, and the expansion of kinase families has been demonstrated in each fully sequenced eukaryote genome (Mehlgarten et al., 2018).
As an important kinase to catalyze the key metabolic step of fructose phosphorylation, fructokinase (FRK) helps phosphorylates fructose to form fructose 6-phosphate (F6P) and functions in all stages of plant development (Plaxton, 1996). In addition, the FRK gene family is characterized by the presence of a phosphofructokinase-B (PfkB) domain and exists in all living organism. The PfkB domain possesses two notable motifs: a di-GLY (GG) motif in the N-terminal region and a GXGD or AXGD motif in the C-terminal region (Mcmahan et al., 1997; Kuettel et al., 2011). The function of each motif has been identified by structural and mutational analyses. For example, the aspartate in the GXGD or AXGD motif acts as a factor during catalysis, and it activates the C6 fructose hydroxyl group for nucleophilic attack on the γ-phosphate in ATP; the GG motif which connects the lid supplies flexibility in the hinge region (Riggs et al., 2017).
The evolution of FRK gene families across the plant kingdom has not been extensively studied. A phylogenetic analysis clustered FRK genes from dicotyledons and monocotyledons into six clades (Chen et al., 2017) and FRK genes from angiosperms into three clades (Riggs et al., 2017). In the moss Physcomitrella patens, FRK genes are clustered into two clades (Stein and Granot, 2018). These observations may be biased because of a limited number of organisms used in the studies. Therefore, the evolutionary history and origin of FRKs remains to be fully investigated. Furthermore, the availability of complete annotations of several plant genomes helped us to embark on a comprehensive evolutionary study of FRKs.
By changing in the native expression levels of FRK genes in transgenic plants through knock-down, knock-out and ectopic expression, FRK genes of different clades may have different or divergent physiological charactercharacters in angiosperm species, which can induce different phenotypes (Kanayama et al., 1997). For example, the growth of different tomato (Solanum lycopersicum) FRK knock-down lines was affected to different degrees, as knock-down FRK2 affected more plant size than knock-down FRK1 (Saori et al., 2002; Stein et al., 2017). FRK2 is involved in xylem and phloem development, and RNA interference RNAi-mediated knock-down of tomato FRK3 resulted in reduced water conductance (Saori et al., 2002). Simultaneous knock-down of both FRK2 and FRK3 led to huge and severe defects in plant growth and development, and it was more acute than those seen in FRK2 knock-down plants alone (Stein and Granot, 2018). The diverse roles of FRK members suggest that the functions of these members in FRK family may be diverged.
Populus is a genus of woody perennial plants, currently it represents the most important and available tree model system for plant genomics (Tuskan et al., 2006). Throughout evolution, Populus has undergone three whole-genome duplication events (Tuskan et al., 2006), leading to the increase of genes which may significantly associate with lignocellulosic wall biosynthesis, disease resistance and metabolite transport (Tuskan et al., 2006). Trees have a long generation time but are short of characterized mutants, which impedes potential functional analysis of genes or gene families (Shanfa et al., 2013). However, single nucleotide polymorphism (SNP) based on the association mapping analysis has emerged as an effective and reliable way to explore the effect of genes on variation of phenotypic in perennial woody plants (Thumma et al., 2005). For example, in the previous studies of candidate gene-based association, several pivotal SNPs within candidate genes which associated with tree wood property and growth traits in Populus and E. nitens have been identified (Thumma et al., 2005).
In this work, we mainly focused on the evolution and functional divergence of the FRK gene family and the generation of functional novelty of new FRK gene. We strictly performed an analysis of phylogenetic of the FRK genes in diverse plant species and analyzed the predicted gene structures, motif composition and auxiliary domains of the FRKs, aiming to explore the functional divergence of this gene family in evolution. What’s more, these auxiliary domains will help us to track and analyze the evolutionary process of the FRK gene family, including gene loss and gene duplication events; and help us to associate these domains with the functional role of each class of FRK genes. Finally, we profiled the expression of FRKs in P. tomentosa and examined their genetic effects by association analysis. Our results help people to have a better understanding of evolution of lineage-specific FRK gene and provide a framework for the study of the functional divergence of gene families.
Materials and Methods
Identification of the FRK Genes in Viridiplantae
High-confidence FRK genes of Arabidopsis thaliana, FRK1–FRK7 (Riggs et al., 2017) were used as reference sequences to identify FRK genes from Phytozome v11.01 using a BLASTp search (E-value < 0.01) (Zaykin et al., 2002). Amino acid sequences, in which we identified a PfkB domain (PF00294) by using the PFAM2 database, were considered as putative members of FRK gene. Subsequently, we calculated the protein length, molecular mass, and theoretical pIs of FRKs in 20 plant species using ExPASy3 (Gasteiger et al., 2005).
Amino Acid Sequence Alignment of FRKs and Phylogenetic Analysis
We performed phylogenetic analyses of the FRK gene family from 20 plant species: six chlorophytes: Chlamydomonas reinhardtii, Dunaliella salina, Volvox carteri, Coccomyxa subellipsoidea, Micromonas pusill, and Micromonas sp. RCC299; two embryophytes: Marchantia polymorpha and P. patens; one lycophyte: Selaginella moellendorffii; one gymnosperms: Picea abies; one basal angiosperm: Amborella trichopoda; eight Eudicot: Brachypodium distachyon, Brassica napus, A. thaliana, S. lycopersicum, Eutrema salsugineum, Vitis vinifera, Eucalyptus grandis and Populus trichocarpa; and two monocots: Zea mays and Oryza sativa. We used MAFFT software with default settings to perform multiple sequence alignments (Szerypo et al., 2003), after which alignments were manually adjusted in BioEdit software (Tippmann, 2004). Phylogenetic trees were constructed using the maximum likelihood (ML) method implemented in RAxML (Silvestro and Michalak, 2012) software; branch support was estimated with 100 bootstrap replicates.
Inference of the Gain and Loss of FRK Clades
Under the Dollo parsimony rule, the gain and loss of different clades of FRK families, as well as the ancestral state, were assessed by using the COUNT software (Csűös, 2010) based on the presence or absent of FRK clades.
Gene Structure and Domain Analyses
To better display the characteristics of FRK gene structure, genomic sequences were downloaded from Phytozome.11 database, and the conserved motifs and domains of the full-length amino acid sequences of FRKs were identified using the Multiple Expectation Maximization for Motif Elicitation (MEME)4 database and Pfam database. Relative parameters used in the analysis were as follow: the maximum number of motifs was 10 and the optimum width of motifs was set from 15 to 50. Furthermore, all identified motifs were annotated according to InterProScan database5 (Zdobnov and Apweiler, 2001). The structures of FRK genes were examined by the GSDS6 based on the genomic sequence and coding sequence of each FRK gene (Hu et al., 2014).
Identification of FRK Genes Which Originated From Different Duplication Mechanisms
By using the methods of a previous study (Yun et al., 2011), FRK genes originated from tandem duplication or inverted duplication in Populus tomentosa were identified. FRK genes originated from segmental duplication were detected by using McscanX software (Yupeng et al., 2012).
Identification of Potential miRNAs Regulating FRK Genes in Populus tomentosa
Regulatory mechanisms and the potential target site of miRNAs were predicted by using PsRNATarget7 based on 401 published miRNA sequences in P. trichocarpa (Puzey et al., 2012). Parameters were set as follows: maximum expectation score, 3; penalty for other mismatches, 1.0 (Dai et al., 2018). Populus-specific miRNAs were identified in a previous study (Xie et al., 2017). To further understand the potential regulation site of the FRK genes in Populus, we used MEME v.4.10.24 in zoop mode (zero or one occurrence per sequence) to predict all conserved sequences in the transcripts. The parameters were set as follows: maximum number of motifs, 5; maximum and minimum motif width, 50 and 6, respectively; and default value for other parameters (Bailey et al., 2006).
Reverse Transcription Quantitative PCR Analysis
All RNA was isolated from the mature leaf, immature leaf, elongated leaf, petiole, immature xylem, mature xylem, cambium, phloem, apex, and root of a 4-year-old Populus tomentosa clone “LM50” which planted in Guan Xian Country, by using the Plant RNA Kit (Magen, China). For biological replicates, this study used three individuals from one genotype of P. tomentosa. RNA was collected from third to eighth leaves of 1-year-old P. tomentosa clone “LM50,” and it was treated with the following stress treatments (three biological replicates per treatment): Drought stress was simulated by osmotic stress from treatment with 30% polyethylene glycol (PEG) solution. For the high temperature treatment, the seedlings were maintained at 42°C. The abscisic acid (ABA) treatment was applied by growing the seedlings on media supplemented with 100 mM/L ABA. The heavy metal treatment used lead (Pb) and was applied by growing the seedlings on a Pb (lead salt) 200 mg/L solution. The plants were treated with all stresses for 0, 1, 3, 6, and 12 h, after which leaf tissues samples were collected, frozen immediately in liquid nitrogen, and stored at 980°C for RNA isolation.
Primers were designed according to the corresponding FRK genes’ sequences from P. trichocarpa genome. RNA samples were treated with DNase I (TaKaRa, Beijing, China), and then the PrimeScript RT reagent Kit (Perfect Real Time) with gDNA Eraser (Takara, Beijing, China) was used for cDNA synthesis with oligo (dT) as the primer. The 7500 Fast Real-Time PCR System (Applied BIOsystems, Foster City, CA, United States) and the SYBR Green Premix Ex TaqTM (TaKaRa, Beijing, China) were used for all the reverse transcription quantitative PCR (RT-qPCR) assays. All the PCRs were performed using the two-step RT-qPCR procedure, and three replications per sample were performed to reduce the experimental error. We plotted a heat map of the transcription level of FRK genes across different tissues by using the pheatmap package in R. The genes and corresponding primers are listed in Supplementary Table S1.
Measurement of Tissue Specificity
To analyze the tissue specificity of FRK gene expression,the tissue specificity score (Liao and Zhang, 2006) was computed as follows: With aij indicating the average expression level of gene i in tissue j, and the tissue specificity of gene i is defined by:
With n indicating the number of tissues we used. Therefore, when the expression of a gene is the same in all tissues we used, the score is 0; when a gene is expressed in just one tissue, the score is 1.
Population and Phenotypic Data
Correlative association population used in this study was composed of 435 unrelated Populus tomentosa individuals from a collection in Guan Xian town, Shandong province (China, 36°23′N, 115°47′E), covering nearly all the natural distribution individuals of Populus tomentosa species, including the northwestern, southern, and northeastern regions of China (30–40°N, 10–125°E) (Du et al., 2012). These 435 individuals were used to discover single nucleotide polymorphism (SNP)s within candidate FRK genes.
We used three growth traits and seven wood property traits for the association analysis. The seven wood properties were as follow: holocellulose content (HC, %), lignin content (LC, %), α-cellulose content (CC, %), hemicellulose content (HEC, %), fiber length (FL, mm), fiber width (FW, μm), and microfibril angle (MFA, °). The three growth traits were as follow: diameter at breast height (DBH, cm), tree height (H, m), and stem volume (V, m3). Relevant measurement methods of the phenotypic data for the 435 genotypes were described in previous study (Du et al., 2014).
Discovery and Genotyping of SNP
The 435 unrelated individuals were resequenced to raw data (>15× coverage) by using the sequencing platform Illumina GA II. Then filtered reads were mapped to the genome of P. trichocarpa (a reference genome) by using SOAPaligner.2.20 with default value (Gu et al., 2013). The depth of effective mapping was ∼11× for most individuals and the rate of mapping in different accessions varied from 81 to 92%.
In order to acquire high quality SNPs, we uniquely used mapped paired end reads to execute SNP calling process. We calculated the genotype likelihood parameter of the genomic site for each individual by using SOAPsnp software of default parameters (Gu et al., 2013). To test the accuracy of our results of SNP calling, we compared it with our previous study of SNP data within 10 candidate genes of 120 individuals from genome re-sequencing, and that data was calculated by using PCR-Sanger sequencing (Du et al., 2014) randomly. The relative accuracy rate of SNP calling was more than 99%, indicating the high quality of platform of the SNP calling. According to the annotation of the P. trichocarpa genome (version.3.0) which were downloaded from Phytozome database, the gene-derived double-alleles SNPs in the full-length genes, including 1 kb downstream and 2 kb upstream of this genes, were extracted by using VCFtools (Petr et al., 2011)8 (Goodstein et al., 2012).
Data Analysis
Analysis of Single SNP-Based Association
We used TASSEL.v.5.0 (Bradbury et al., 2007) with mixed linear model (MLM) for exploring single nucleotide polymorphism (SNP)-based association in the association population of Populus tomentosa. The formula of MLM was as follow: y = μ + e + Qv + Zu, with μ indicating the intercepts vector, e indicating random experimental error, e indicating error of random experimental, Q matrixes indicating the structure of population, v indicating a vector for population effects, and Z being the matrixes relating y to u.
Furthermore, the pairwise kinship (K) and estimated membership probability (Q) were used to assess the population structure and relatedness among marker trait associations of individuals. We assessed the K matrix with previous methods (Grynko and Shkuratov, 2003; Evanno et al., 2010) and the Q matrix was assessed depending on subpopulations (k = 3) based on the previous related analysis (Evanno et al., 2010) by STRUCTURE V.2.3.4 (Evanno et al., 2010; Nick et al., 2013). Finally, we used positive false discovery rate (FDR) to make a correcting multiple test by employing the Q value package in R (Reiss et al., 2012).
Analysis of Haplotype-Based Association
We conducted haplotype trend regression analysis to assess the haplotype frequencies from the genotype data, and estimate the potential haplotype-based associations (Zaykin et al., 2002; Gad and Ron, 2005). Based on 104 permutation tests in our study, the significance of singleton alleles and the frequencies of haplotype-based associations which lower than 0.05 were abandoned in this analysis.
Results
Identification of FRK Genes in 20 Plant Species
To perform a comprehensive analysis of FRK gene family in plant species, we firstly performed an analysis of phylogeny on the FRK genes of 20 plant species (Phytozome.v11.0, see text footnote 9), including six chlorophytes: C. reinhardtii, D. salina, V. carteri, C. subellipsoidea, M. pusill and Micromonas sp. RCC299; two embryophytes: M. polymorpha and P. patens; one lycophyte: Selaginella moellendorffii; one gymnosperms: Picea abies; one basal angiosperm: A. trichopoda; eight rosids: B. distachyon, B. napus, A. thaliana, S. lycopersicum, E. salsugineum, V. vinifera, E. grandis and P. trichocarpa; and two monocots: Zea mays and Oryza sativa. FRK genes of A. thaliana (Riggs et al., 2017; Supplementary Dataset S1) were used as reference sequences to identify FRK genes from the Phytozome database using a BLASTp search, generating 143 putative FRK genes in total. Then the Pfam database was used to verify the presence of the PfkB domain and 18 genes were eliminated in this process. Thus, we identified 125 FRK genes across 20 diverse plant species (Supplementary Dataset S1), with the largest number of FRK genes in P. abies (a gymnosperm; 12) and the smallest number in C. subellipsoidea (green algae; 1). Notably, we found that 15 FRK genes were already present in green algae (Supplementary Dataset S1), suggesting that FRK genes appeared early in evolution.
We also calculated and analyzed the percentage (%) of all protein-coding genes represented by FRK genes and found that FRK genes account for 0.023% of all genes in P. trichocarpa, 0.018% in S. moellendorffii (a basal angiosperm), and 0.007% in V. carteri (a green algae). Interestingly, the percentages of FRK genes among all P. trichocarpa genes are 1.3–2.8 times more than the percentages of that in ancient plants. This may be the result of a Populus lineage-specific paleotetraploidization (Tuskan et al., 2006).
We analyzed the features of FRK genes and our results showed a high variability of genetic features across the 20 plant species (Supplementary Dataset S1). The FRK genes of the 20 species were characterized as having 2–19 exons, with the fewest exons detected in green algae. As shown in our dataset, the FRK genes in the 20 plant species encode FRK proteins ranging from 71 to 1,207 amino acids in length with the longest in P. patens and the shortest in P. abies. The relative molecular mass varied from 16.12 kDa (green algae) to 68.98 kDa (A. thaliana) and the pIs ranged from 4.22 (P. abies) to 7.13 (V. vinifera) (Supplementary Dataset S1). Together, our phylogenetic analysis of the FRK genes indicates that an event of domain loss took place early in the evolution of algae, and before the evolution of land plant lineages.
Extensive FRK Gene Structure Reorganization During the Transition to Land Plants
To examine the phylogenetic relationships among the land plants, we constructed a phylogenetic tree by using a maximum likelihood (ML) method (Figure 1). In addition, other methods, such as, neighbor-joining (NJ) method, were also used to reconstruct the phylogenetic tree of FRK family. As a result, the topology of NJ and ML trees were very similar. We also noticedthe topology of NJ and ML trees were very simila that the intron-exon structures supported the classification of FRK clades. All FRK genes were classified into eight clades (Figure 1). Each clade contained 5–20 members. Interestingly, five FRK clades (I, II, VI, VII, and VIII) were already present in green algae with high supporting values. Remarkably, clade VIII is specific to green algae with a high specific intron-exon structure and specific motif composition (Supplementary Figure S2).
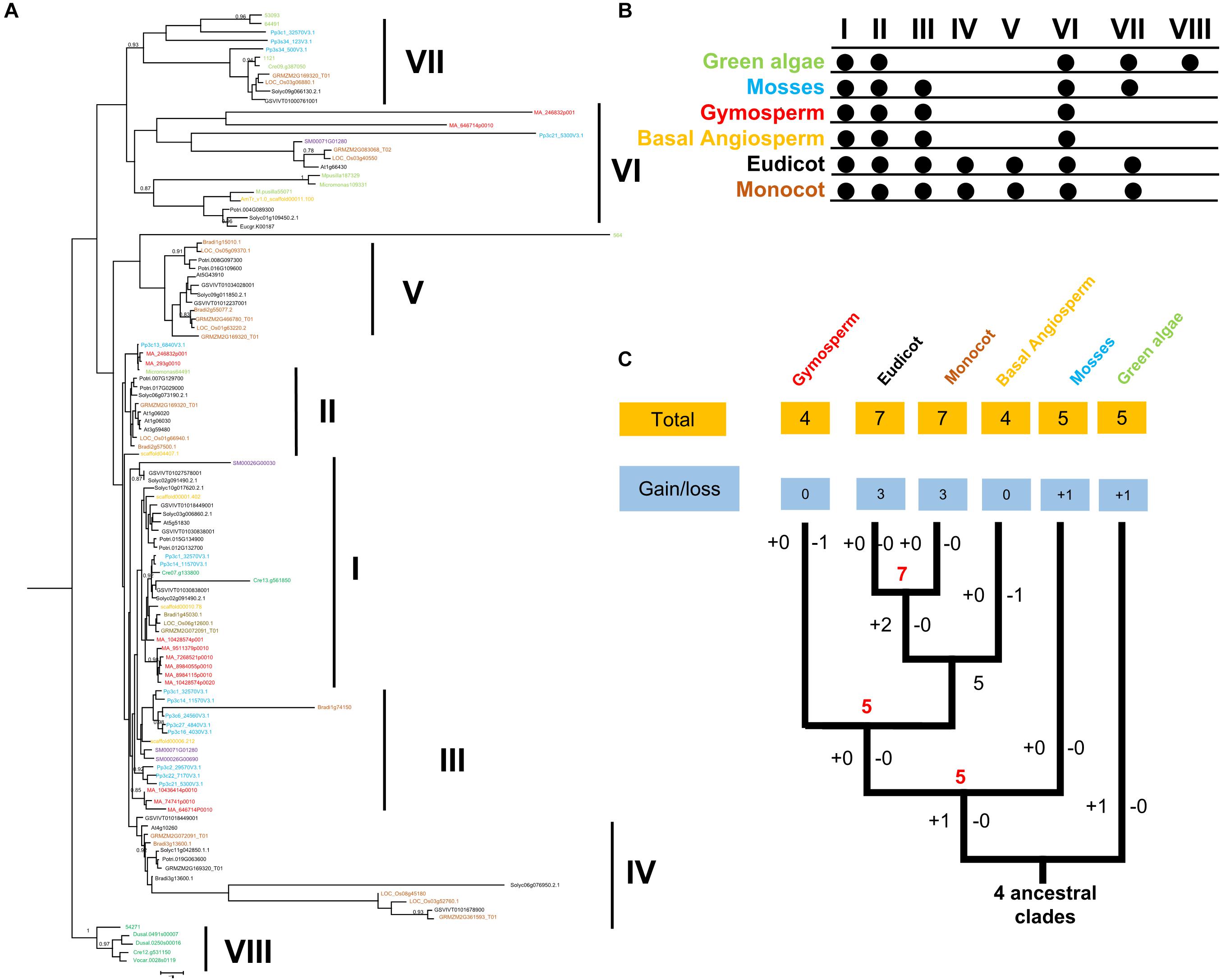
Figure 1. (A) Phylogeny of FRK proteins. Bootstrap values are shown; values < 0.80 were omitted. The scale bar indicates the number of changes per site. The tree was constructed using amino acid alignment. The eight clades of FRKs are indicated at the right of the tree. The tree is accompanied by a table to summarize the presence (∙) or absence (empty) of the clades in the plant family. (B) Birth and death clades of FRK families. The (+) on the branch represents the increase of clade and (–) represents the decrease of clade, and the number on the node represents the number of clade of ancestor species. (C) Plant divisions are color coded depending on their taxonomy classification: green algae (green), embryophytes (dark blue), basal angiosperms (dark yellow), gymnosperms (red), eudicot (black), and monocot (gray).
In an embryophyte, an ancient land plant, five FRK clades (I, II, III, VI, and VII) were detected, suggesting that clade VIII was lost and clade III was gained before the divergence between green algae and embryophytes. In gymnosperms, basal angiosperms, and lycophytes, only four clades (I, II, III, and VI) were observed, suggesting that clade VII was lost during the transition (Figure 1). Notably, in the angiosperms including eudicot and monocot, we observed an extensive diversity of FRK genes with seven clades, including two angiosperm-specific clades (IV and V) (Figure 1). This may suggest that FRK genes of eudicot and monocot may have the same origin.
In general, the same motif composition was observed in the same clade, implying functional similarities in the same clades (Figure 1 and Supplementary Figure S2). However, we observed some clade-specific motifs across all the clades, which is highly consistent with topology of phylogenetic tree. For example, as shown in Figure 1, the LVXC motif, a sequence of 20 amino acid residues, was identified in the N-terminus of amino acid of FRKs, and it almost appeared in all the FRKs of different clades. In contrast, the LVXC motif-less lineage was only conserved in clade IV of angiosperm species (Supplementary Figure S2), indicating that clade IV may appear later in evolution. The ILK motif was conserved in all FRK of clade VIII, VII, and VI (Supplementary Figure S2), which indicated that the three clades may origin from the same ancestors. In this regard, there were most diverse motifs found in clade VIII (Supplementary Figure S2). Specially, PAFK motif appeared in clade VIII and was not found in any other clades (Supplementary Figure S2).
The auxiliary domains in FRKs can be useful to track and analyze the potential evolutionary process and functional divergence of the FRK gene family, including gene loss and gene duplication events. We then explored the conservation and the evolution of auxiliary domains between the different protein clades. The AP_endonuc_2 domain (Xylose isomerase-like TIM barrel; PF01261) and ABC_tran domain (ATP-binding domain of ABC transporters; PF00005) were detected in FRK genes in the ancient clades VIII and VII (Figure 2 and Table 1). Following the parsimony rule, we inferred that the incorporation of these domains occurred in the early stage of plant species evolution, and before the green algae-land plant split (550 Mya). Likewise, the bact-PGI_C (Glucose-6-phosphate isomerase; PF10432), ROK (PF00480), FucU transport (PF05025), and ABC_tran domain (PF00005) were found in an ancient land plant, P. patens (Figure 2 and Table 1). Importantly, the ABC_tran domain was found to be conserved in the green algae and P. patens, indicating that the ABC (ATP-binding cassette) transporter domain may play essential roles in communication across cell membranes (Mccormick and Johnstone, 1990). This suggests that the AP_endonuc_2 domain was lost and two domains (the FucU transport domain and the ROK domain) were gained after the split of green algae and P. patens (Figure 2 and Table 1).
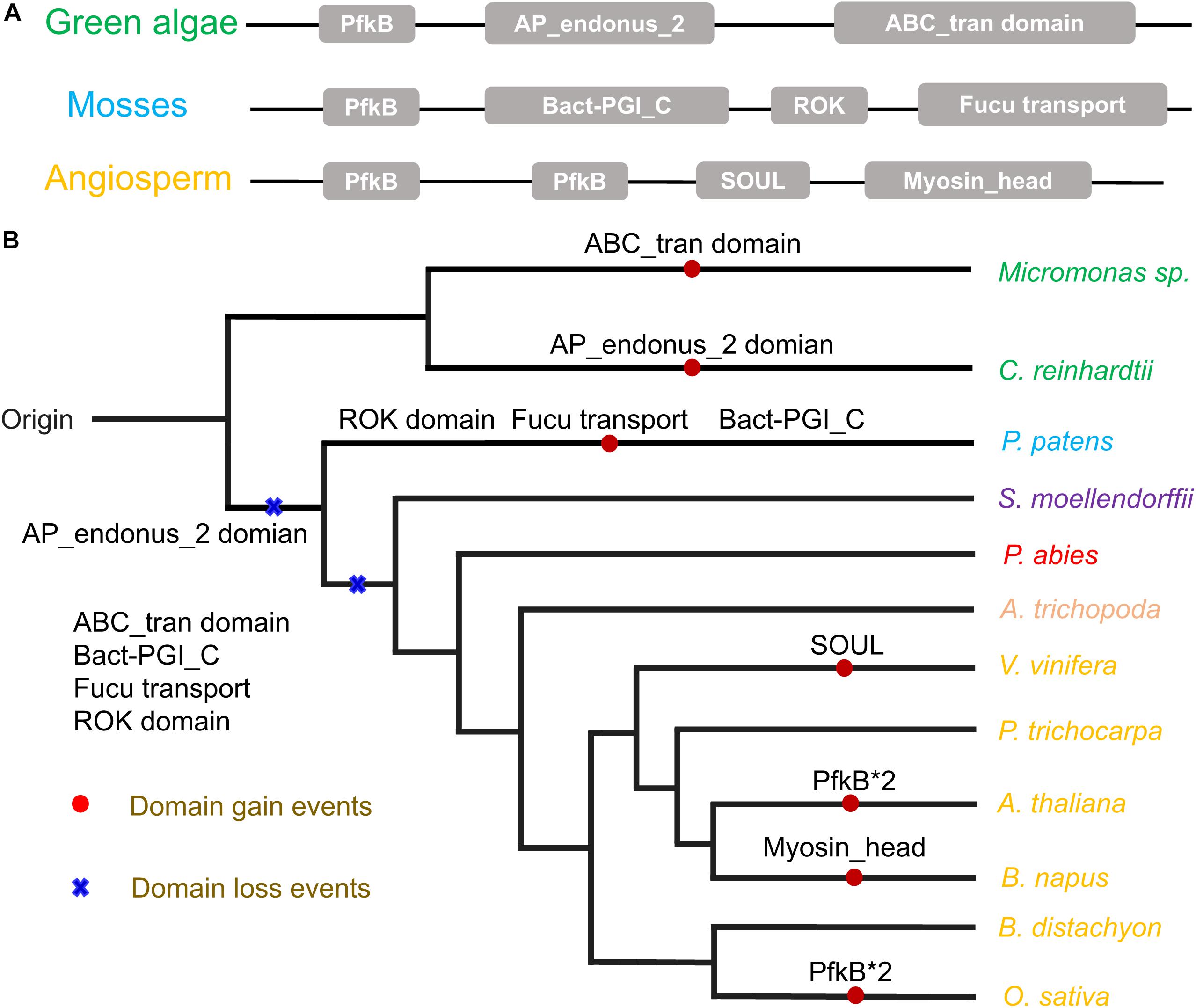
Figure 2. The auxiliary domain structure and the hypothetical evolutionary model in the 125 FRKs. (A) Domain structure of the FRK proteins found in green algae, mosses, and angiosperms. (B) Hypothetical evolutionary model for domain structure in the FRK proteins. Red circles represent domain gain events and blue crosses represent domain loss events inferred from the Pfam database. Plant divisions are color coded depending on their taxonomy classification: green algae (green), mosses (dark blue), lycophytes (violet), gymnosperms (red), basal angiosperms (flesh), and angiosperms (yellow).
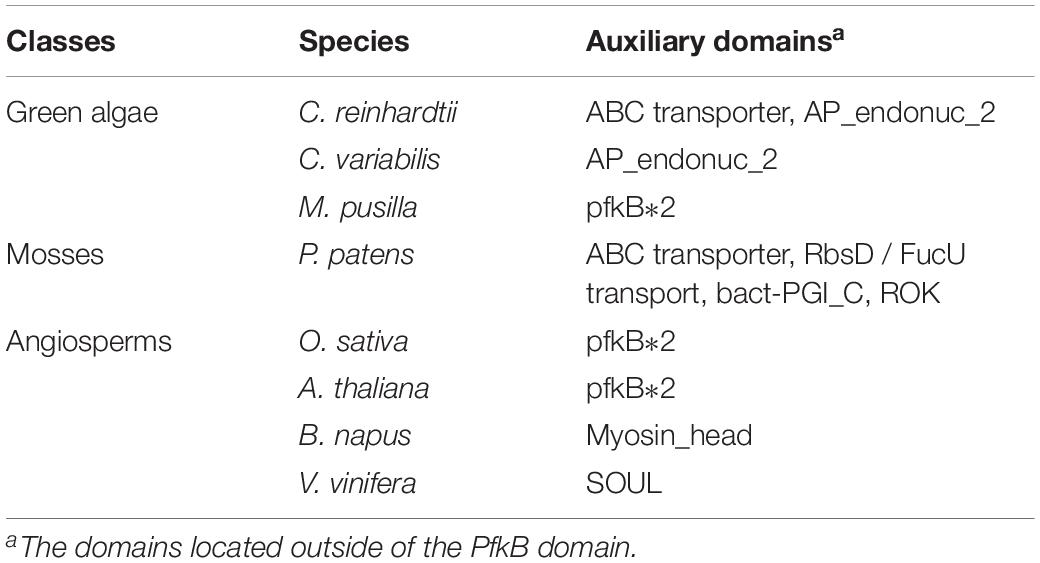
Table 1. Auxiliary domains in FRK proteins from different plants.
However, we did not identify an auxiliary domain which located outside of these conserved domains in S. moellendorffii, P. abies, and A. trichopoda. Interestingly, some of the auxiliary domains were found in angiosperm plants (Figure 2B). For example, the SOUL heme-binding domain (PF04832) was found in V. vinifera, and the Myosin_head (motor domain; PF00063) domain was found in B. napus. Also, a duplication of the PfkB domain (PfkB∗2 domain) event took place in O. sativa and A. thaliana (Figure 2B and Table 1). These results further support the idea that the diversification of special gene families occurred during the origin and rapid diversification of the angiosperms (Hileman et al., 2006). Most importantly, combined with the phylogenetic tree analysis, we found that auxiliary domains tend to appear in specific lineage clades (Figure 1). The result suggests that the ancient FRK genes may perform multiple physiological functions and have experienced a stepwise loss of domains during the transition to land plants, and a rapid diversification before the divergence of angiosperms.
Functional Divergence of Paralogous FRK Pairs Derived From Segmental Duplication
Based on previous studies, duplication of genome has always been seen as a contributor to gene family expansion (Freeling, 2009). There are five types of duplication events: singleton, dispersed, proximal, tandem, and segmental duplication (Xu et al., 2018). In order to explore the type of FRK gene duplication that occurred in Populus, we used the P. trichocarpa genome as a reference to assess which type of duplications was the way to the expand FRK genes in Populus. Then we compared the number of PtoFRK genes originated from different mechanisms in P. tomentosa. This analysis suggested that 88.89% (8 of 9) of the PtoFRK genes originated via duplication events, including 6 from segmental duplication, and 2 from dispersed duplication (Table 2). To verify this result, we detected the chromosome distribution of PtoFRKs and found that the chromosome sites of 6 PtoFRK genes were consistent with the regions where the whole genome duplication took place (Tuskan et al., 2006). Furthermore, according to the Populus dilatancy parameter (λ = 9.1 × 10–9), it is estimated that 6 FRK genes that originated from segmental duplication may have been produced from 1 Mya, which suggests that they appeared recently in evolution. Furthermore, low Ka/Ks values (<0.3) were detected among the six PtoFRK genes, suggesting a strong purified selective pressure (Table 2). Our findings showed that the segmental duplication is the main and key driving mechanism for the increased members of the FRK gene family, and the newly originated PtoFRK genes were found to have originated over a short period of time.
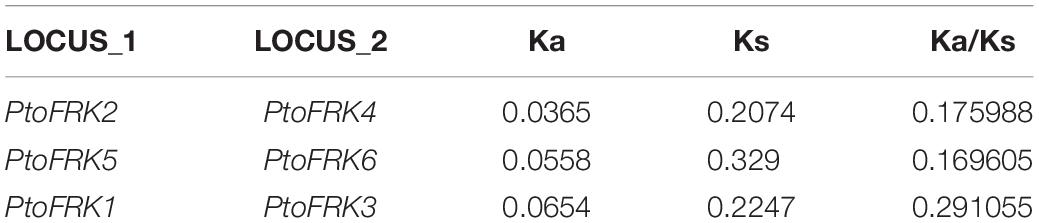
Table 2. Paralogous FRK genes.
Consistent with gene balance hypothesis, previous studies showed that gene families of duplication-resistant can correspond to regulators or transcriptional factors that are overrepresented after events of whole genome duplication (WGD) (Freeling, 2009). We thus expected that salicoid duplicates, which were produced during whole-genome duplications in the lineage that produced the Salix (willow) and Populus genera, would be targeted by more Populus-specific miRNAs (Xie et al., 2017). As expected, all the Populus FRK genes were predicted to be regulated by Populus-specific miRNAs (miR7822, miR6421, miR6423, miR6448, miR6439, miR6452, and miR6462) and only two FRK genes are regulated by Populus-conserved miRNAs (miR171 and miR172) (Table 3). The result supported the idea that 90% of Populus-specific miRNAs target salicoid duplicates (Xie et al., 2017). Analysis of mVISTA alignment of FRK targets of miRNA revealed that cleavage sites of Populus-specific miRNAs are widely distributed in the range of 189 to 1,026 bp of transcript sequence and 25% of cleavage sites are located in non-conserved regions, while all of the cleavage sites of Populus-conserved miRNAs are located in highly conserved regions (Figure 3A and Table 3). Interestingly PtoFRK1, PtoFRK3, PtoFRK2, and PtoFRK4 which derived from a segmental duplication are regulated by the same miRNA. However, PtoFRK5 and PtoFRK6 which derived from a segmental duplication are regulated by the different miRNA and the cleavage sites of miRNAs are different (Tables 2, 3). Consistent with this, they also have different transcription level in selected tissues (P < 0.01) (Figure 3). It infers that forced by segmental duplication in evolution, the regulating mechanism of gene-pairs may diverge.
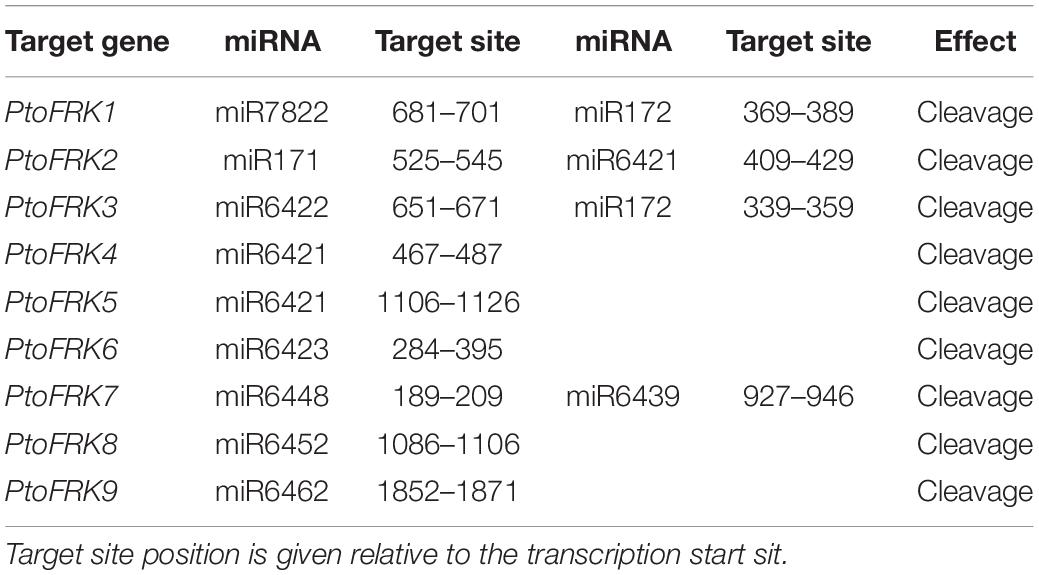
Table 3. Potential regulation of PtoFRKs by miRNAs.
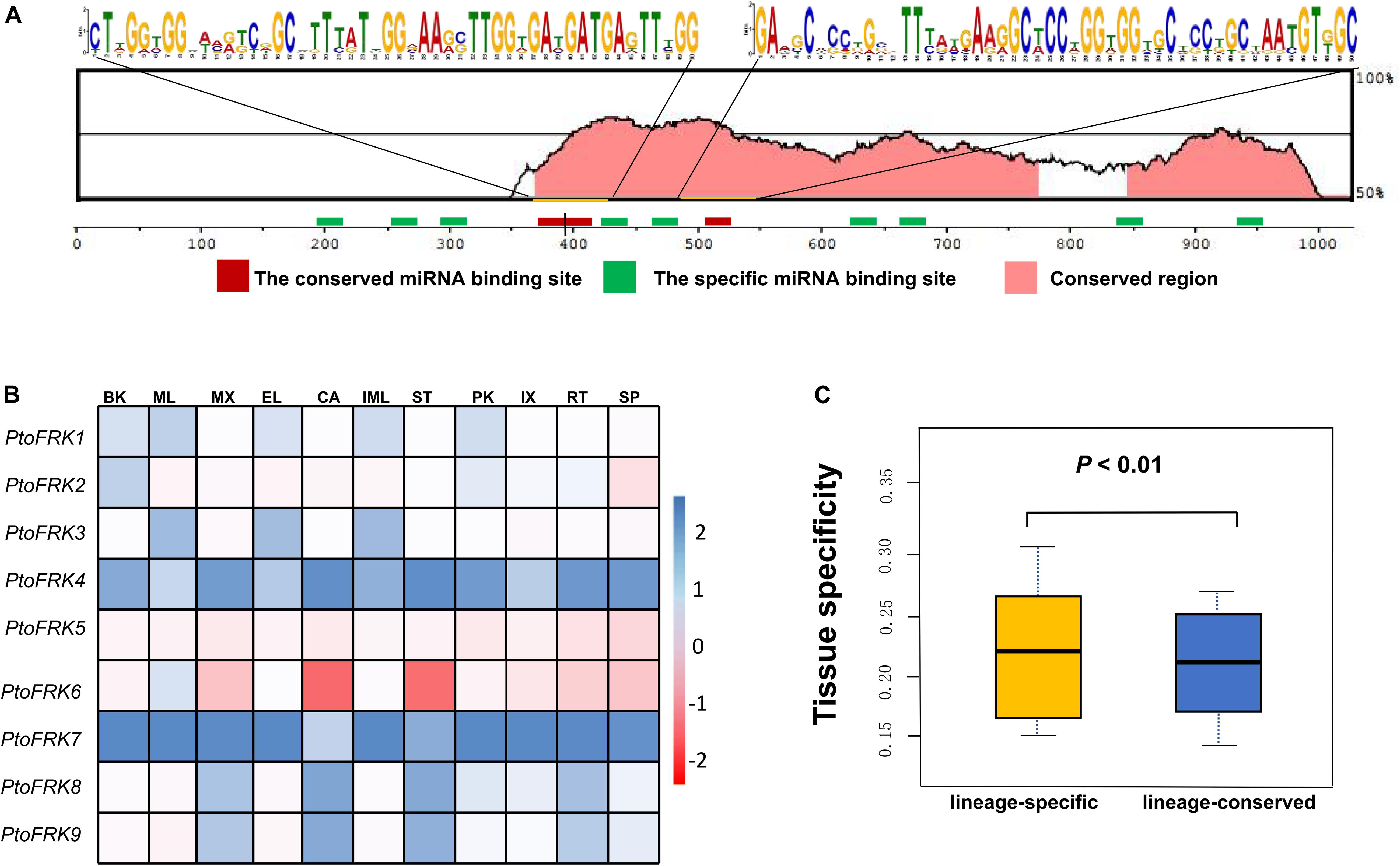
Figure 3. The miRNA target sites of PtoFRK genes. (A) mVISTA alignment of transcript sequences of PtoFRKs. The vertical bar indicates the sequence conservation level, the green rectangles indicate the binding site of specific miRNAs, and the red rectangles indicate the binding site of conserved miRNAs, and the conserved motifs were predicted by MEME. (B) RT-qPCR analysis of 9 PtoFRKs in 11 tissues in P. tomentosa including BK, bark; ML, mature leaf; MX, mature xylem; EL, elongated leaf; CA, cambium; IML, immature leaf; PK, leaf peak; IX, immature xylem; ST, phloem; RT, root; SP, stipe; the color scale represents the expression values. Each data point is the average of three biological replicates. (C) Comparison of tissue specificity of lineage-conserved PtoFRKs with lineage-specific PtoFRKs.
Overall, the regulation patterns of FRK genes support the gene dosage balance hypothesis.
Redundant and Divergent Patterns of FRK Expression During Populus Development and in Response to Stress
Previous reports showed that lineage-specific genes rapidly evolve and uniquely exist in plant lineages or individual species, and often perform unique biological functions (Yu et al., 2004). To further compare the functions between lineage-conserved and lineage-specific clades, we first analyzed the expression patterns of 9 P. tomentosa (Pto) FRK genes across various tissues, including mature leaf, immature leaf, elongated leaf, petiole, mature xylem, immature xylem, cambium, phloem, apex, and root. As a result, we found that most of the PtoFRKs showed a relative higher level in leaves tissues (Figure 3B), and that’s also consistent with a previous study which found that the FRK genes express highly in source tissue (Granot, 2007). Remarkably, most of the PtoFRK genes also showed a high expression level in xylem tissues, suggesting that FRKs may mainly function in wood formation. Then we contrasted the expression specificity of Populus-conserved PtoFRKs (PtoFRK1, PtoFRK2, PtoFRK3, PtoFRK5, and PtoFRK6) and Populus-specific PtoFRKs (PtoFRK4, PtoFRK7, PtoFRK8, and PtoFRK9). As a result, Populus-specific PtoFRK genes have lower expression levels and appear to be preferentially expressed in specific tissues (Figures 3B,C; P < 0.01), indicating a striking difference between Populus-conserved and Populus-specific PtoFRKs. In the same clades, PtoFRK genes showed a similar expression level and patterns, suggesting redundancy of function in the same clades. Overall, these observations suggest that newly originated PtoFRKs have high expression specificity, but generally lower expression levels.
To further examine the specificity of expression level of PtoFRK genes, we tested the expression of these genes under different conditions, such as treatment with the hormone ABA, or different stresses (drought, heavy metal, and high temperature). Indeed, some of these genes do show different expressions in response to distinct stress conditions (Supplementary Figure S1). Notably, PtoFRK1 and PtoFRK2 were differentially expressed in at least one of the four time points measured (1, 3, 6, or 12 h) under all abiotic stresses, suggesting that their functions may be biased toward more central roles in networks. However, under these stresses, no differences in expression level were detected in PtoFRK6, 7, 8, and 9 from lineage-specific clades.
Lineage-Specific PtoFRK Genes Have Higher Potential for Functional Divergence
We used association analysis to further elucidate the potential functions of lineage-specific PtoFRKs in growth and wood formation. To this end, we have identified SNPs in the PtoFRK genes within the regions 1 kb downstream and 2 kb upstream of each gene. Totally, 4,446 common SNPs with frequency ≥ 0.05 within 7 full length of PtoFRK genes were detected, which approximate 19 SNPs per kb. The MLM module in TASSEL 5.0 was used to find significant associations between different phenotypes, including seven wood property traits and three growth (HC, holocellulose content; HEC, hemicellulose content; CC, cellulose content; LC, lignin content; FW, fiber width; FL, fiber length; and MFA, microfibril angle; H, tree height; V, stem volume; DBH, diameter at breast height), and genotypes for the SNPs, which were verified the accuracy with multiple testing by using FDR. We have identified 45 significant single marker associations (P < 0.01; Q ≤ 0.1) which represents 34 SNPs found in 7 candidate genes, and these associations would associate with two wood property and two growth traits. The individual SNPs explained between 10% (PtoFRK3-SNP24) and 22% (PtoFRK6-SNP87) (Supplementary Table S2) of the phenotypic variation, which was the highest contribution to phenotype, suggesting that the PtoFRK genes may play important roles in wood property traits and tree growth.
We contrasted the association of PtoFRKs of different tissues, which revealed a striking difference between Populus-conserved and Populus-specific PtoFRK genes. As expected, the marker-trait association analyses revealed that Populus-conserved FRK genes were more associated with growth and wood property traits. For Populus-conserved FRKs, 31 significant single-marker associations (P < 0.01, Q < 0.1), which represent 25 unique SNPs in PtoFRK genes, were detected, and they associated with four traits including two growth and two wood property traits. For example, 18 SNPs were significantly associated (P < 0.01, Q < 0.1) with D, FW, and V with 10.0% (PtoFRK3-SNP24) to 21.0% (PtoFRK2-SNP82) of the phenotypic variance (R2) (Figure 4A and Table 4). Eight SNPs (PtoFRK5-SNP14, PtoFRK5-SNP32, PtoFRK5-SNP59, PtoFRK6-SNP23, PtoFRK6-SNP44, PtoFRK6-SNP56, PtoFRK6-SNP87, and PtoFRK6-SNP92) associated with four traits (D, V, FW, and LC) with 20.0% (PtoFRK5-SNP14) to 22.0% (PtoFRK6-SNP87) of the phenotypic variance (Figure 4A and Table 4).
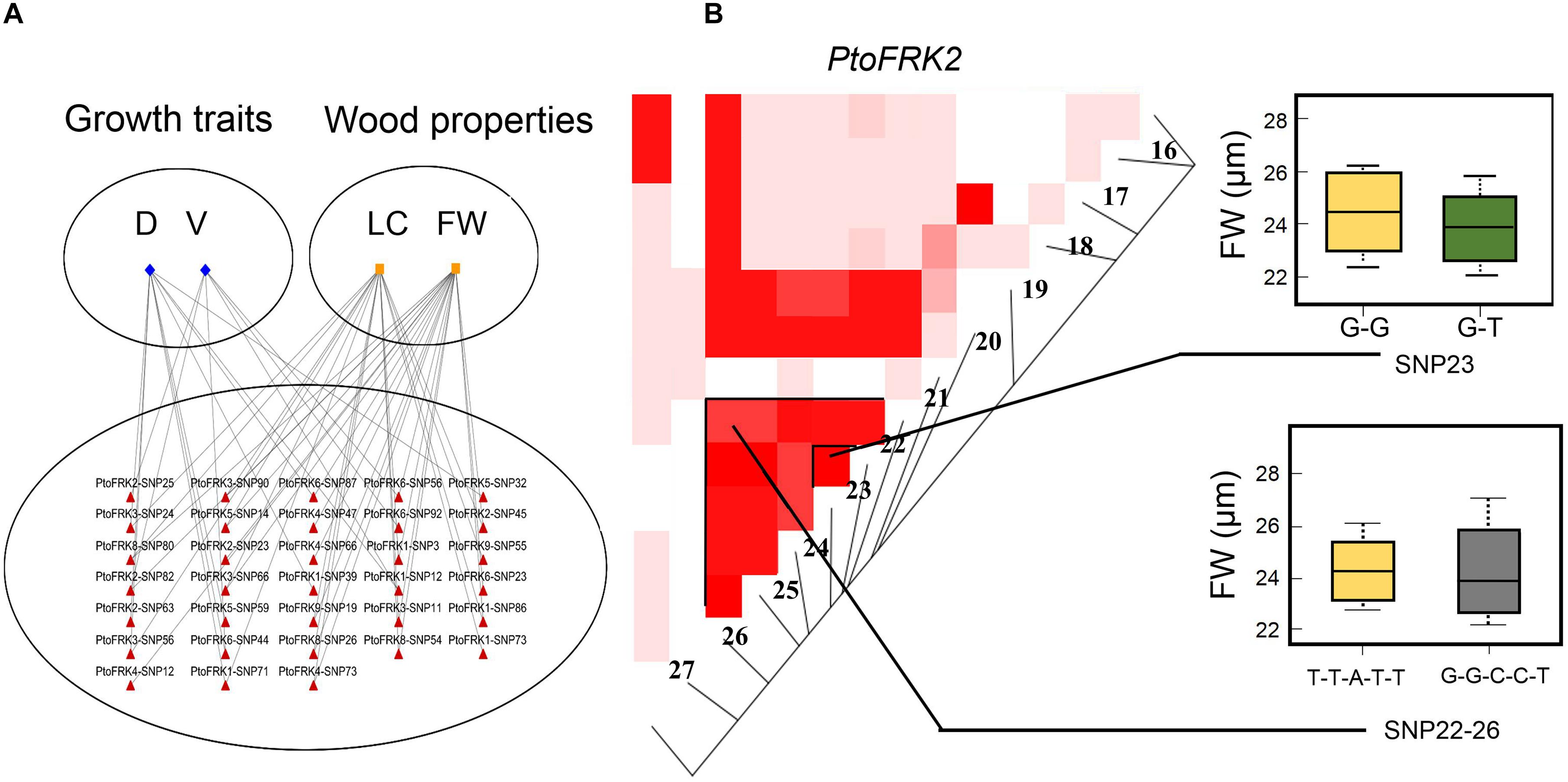
Figure 4. The single SNP-based associations and the haplotype-based associations in the association population of P. tomentosa. (A) The single SNP based associations between significant SNPs from PtoFRKs and their associated traits. The lines represent the significant associations. (B) The genotypic effect for the significant haplotypes of PtoFRK2-SNP22-26 and single SNP-based association of PtoFRK2-SNP23.
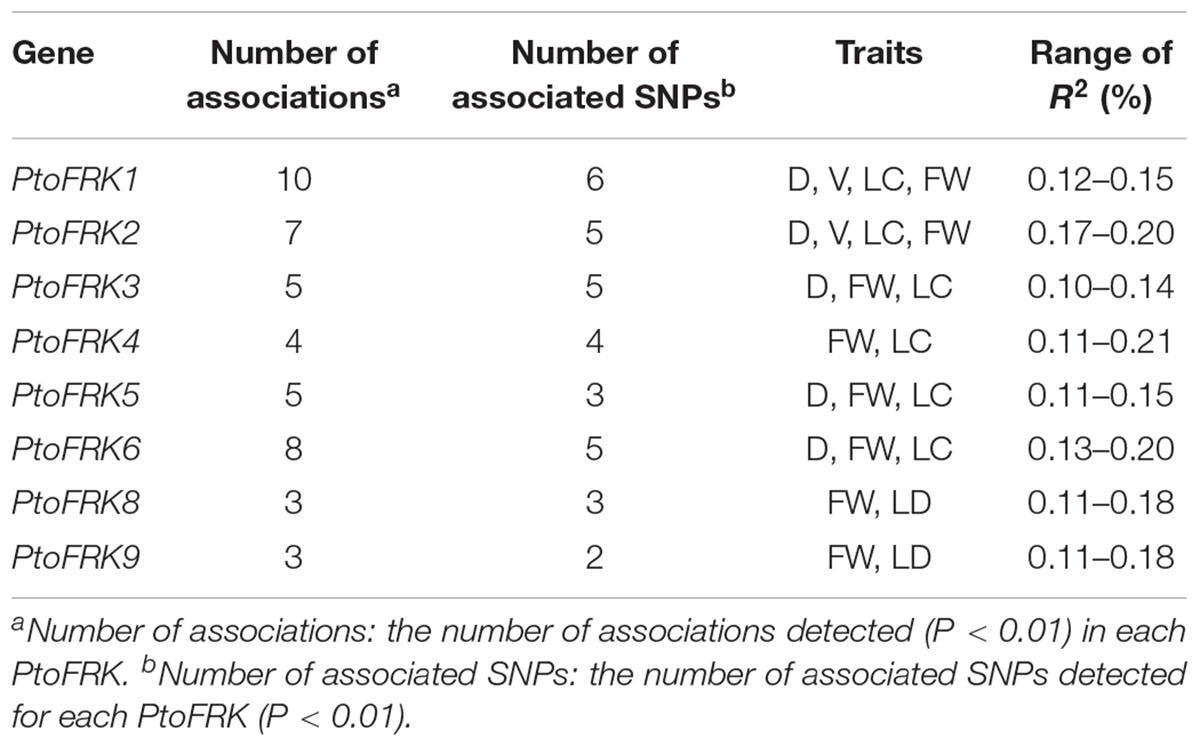
Table 4. Summary of significant SNPs within PtoFRKs associated with growth and wood properties in the association population of P. tomentosa.
Moreover, for lineage-specific PtoFRK genes, only 14 significant associations (P < 0.01, Q < 0.1) representing 9 unique SNPs in 4 PtoFRK genes were detected to be associated with two wood property traits (FW and LC). For example, four SNPs in PtoFRK4 (PtoFRK4-SNP12, PtoFRK4-SNP47, PtoFRK4-SNP66, and PtoFRK4-SNP73) associated with FW and LC with 11.0% (PtoFRK4-SNP12) to 16.0% (PtoFRK4-SNP73) of the phenotypic variance (R2). In clade V, six SNPs in PtoFRKs (PtoFRK8-SNP26, PtoFRK8-SNP54, PtoFRK8-SNP80, PtoFRK9-SNP19, PtoFRK9-SNP32, and PtoFRK9-SNP55) associated with FW and LC with 12.0% (PtoFRK8-SNP80) to 13.0% (PtoFRK8-SNP54) of the phenotypic variance (Figure 4A and Table 4).
Furthermore, haplotype-based associations, performed by haplotype trend regression, were used to explore the genetic effect of PtoFRKs. Specially, one haplotype (T-T-A-T-T) from PtoFRK2 associated with FW, which was supported by significant single SNP-based associations of PtoFRK genes with the same wood property traits (Figure 4B). This further supported the idea that this FRK can play an important role in wood formation (Figure 4B). Together, the significant marker-trait association analyses indicated that FRK, a core kinase in glycolysis, plays an important role in wood formation and growth in P. tomentosa.
Discussion
Domain Loss and Gain Events of the FRK Gene Family
To what we know today, radiation, and colonization of land plant are major or main keystones in the evolutionary process of living organisms (Graham et al., 2012), which can shape the diversity of plant species, landscape and atmosphere on Earth (Kenrick and Crane, 1997). This was accompanied by the changes including genome, physiological, and morphological to deal with the challenging conditions (Graham et al., 2012; Harholt et al., 2015). Therefore, the transition of plants from the oceans to land required several functional adaptations, and it must have been accompanied by multiple protein gain and loss events of motif or domain, for functional domains were the key elements of functional divergence, and they are the main components of proteins and are essential for protein function and protein interactions (Bagowski et al., 2010).
During wood formation, fructose is always phosphorylated to fructose-6-phosphate (F6P) by a fructokinase (FRK) in the beginning, which is necessary for sucrose-derived fructose metabolism because it facilitates carbon allocation to wood cell walls (Melissa et al., 2012). Here, by using publicly available genome databases, we first analyzed the phylogenetic relationships of FRK genes in 6 green algae and 14 land plants. The AP_endonuc_2 domain (PF01261) and ABC_tran domain (ATP-binding domain of ABC transporters; PF00005) were found to be present in green algae. Likewise, four auxiliary domains bact-PGI_C (PF10432), ROK (PF00480), FucU transport (PF05025), and ABC_tran domain (PF00005) were found in the moss P. patens. Several auxiliary domains were also detected in angiosperms. We thus speculate that auxiliary domain gain and loss during plant transition to land always played a key and indispensable role in both the functions of divergence of plant colonization and lineages. Moreover, the motif composition of different clades indicates that gain and loss of domain in evolution is a gradual process.
The origin of a new gene family is mainly conferred by the fusion or fission of protein domains in early streptophytes (Margulis, 1992; Simionato et al., 2007). The domain structure of proteins is crucial for their function; thus, the alteration of auxiliary domains is presumed to be related to the alteration of the functions of FRKs in biological processes and environmental adaptation of plants (Romani et al., 2018). Interestingly, some domains of FRK proteins were only found in green algae and moss, for example, the ABC transporter domain (PF00005), a water-soluble domain of transmembrane ABC transporters, which are often involved in the transmembrane transport of various molecules (the PDR-type ABC transporter strongly mediates cellular uptake of the phytohormone ABA; the ABC transporter (AtABCG25) is always involved in ABA responses and transport) (Danuta et al., 2003). In angiosperms, the Myosin_head domain (PF00063), which has been found in B. napus, mainly associates with converting chemical energy, in the form of ATP, to mechanical energy (Kawakubo et al., 2007). To verify our speculative history of evolution (Figure 2), FRK genes of some other plant which were not often used to evolutionary research have been provided in Supplementary Dataset S3, and we did not find auxiliary domians of FRK protein in other plant species (Figure 2).
Thus, it is likely that these auxiliary domains have the potential to contribute to the evolution of phenotypes and harbor many potential innovations essential for terrestrial life.
Functional and Regulating Redundancy of FRK Genes in Populus
In contrast to green algae, functional redundancy of gene families is a ubiquitous phenomenon in land plants (Pérez-Pérez et al., 2013). Consistent with a previous study, there are only two members of the FRK gene family on average in green algae, which suggested that almost no duplication events occurred. By contrast, three whole-genome duplication (WGD) events were found during Populus evolution, which resulted in overrepresentation of genes associated with disease resistance, metabolite transport, and lignocellulosic wall biosynthesis (Tuskan et al., 2006). In our study, we have identified nine PtoFRK genes in the genome of P. tomentosa, whereas there are 12 FRK genes in A. thaliana. The most common domains occur in P. tomentosa compared with A. thaliana in a ratio ranging from 1.4:1 to 1.8:1 (Tuskan et al., 2006). The low ratio of FRK genes in P. tomentosa compared with A. thaliana might point to a stepwise reduction in FRK genes.
Based on the previous studies, segmental duplication and tandem duplication always played a key role in the expansion of FRKs in A. thaliana. Here, detailed analyses revealed that segmental duplication was the main mechanism of PtoFRK gene expansion in P. tomentosa, as we did not find any tandem duplication of PtoFRK gene clusters in the P. tomentosa genome. Phylogenetic analyses revealed that three lineage-conserved clades and two lineage-specific clades are present in P. tomentosa. Among them, 6 PtoFRK genes are apparently paralogous, suggesting functional redundancy of FRK genes in P. tomentosa. Supporting this, mostly PtoFRKs from the same clade had similar expression patterns, suggesting potential functional redundancy of FRK genes within the same clade. Also, most of PtoFRK paralogous pairs in P. tomentosa are regulated by the same PtomiRNAs, and the target site of regulating miRNA alignment also exhibited an obvious conservation structure of the canonical –G–(6X)–G– repeats, suggesting a possible functional redundancy between the recently duplicated PtoFRK gene pairs. Interestingly, a gene-pair (PtoFRK5 and PtoFRK6) is regulated by the different miRNA and the cleavage sites of miRNAs are different (Tables 2, 3). It infers that regulating mechanism of gene-pairs may also be forced by segmental duplication in evolution.
Functional Novelty of Lineage-Specific Gene Clades
After duplication, one gene copy often accumulates mutations, which can probably lead to functional divergence (Liu et al., 2018). One copy within a paralogous pair which originated from segmental duplication may often evolve in different mechanisms, such as neofunctionalization, subfunctionalization (Lynch and Force, 2000), or non-functionalization (Ohno, 1971). In our study, FRK genes of clades VIII, VII, and IV have been lost in the embryophyte, gymnosperm, and lycophyte genomes, respectively, and two lineage-specific clades (IV and V) emerged in angiosperms. It remains unknown today what the functional contribution of these different clades is to the evolutionary history and process. It is often assumed that mutations in evolutionarily conserved genes are mostly to be deleterious, therefore genes of lineage-conserved clades are biased toward more central roles in biological networks (Konstantin et al., 2007; Gershoni and Pietrokovski, 2014). In our study, we detected significant associations in PtoFRKs of Populus-conserved clades with four wood property and growth traits in trees. Moreover, the low tissue expression specificity and high expression level indicate that FRK genes of lineage-conserved clades often underwent strong selection pressure during evolution (Gramer and Huber, 1991; Plange et al., 2014).
Furthermore, highly conserved functional domains in lineage-conserved genes are often the targets of conserved miRNAs, which also play central roles in biological networks. By contrast, lineage-specific genes are often targeted by many newly originated miRNAs in a random way, and they often appear and choose to be expressed in a specific tissue in a preferential and special way, and are associated with less lineage-specific phenotypes. Therefore, lineage-specific genes have a greater chance to contribute to phenotypic variations because their roles are not absolutely essential. Furthermore, auxiliary domains and specific motifs are preferentially found in lineage-specific genes, suggesting functional differentiation and novelty of lineage-specific gene clades. Thus, the expansion of FRK genes seems to be associated with phenotypic changes and lineage-specific processes that shape the development of plants. Our study extends the exploration of more specific roles of members of large gene families throughout the plant genome, and highlights the important and key selective pressures which can influence regulatory networks.
Conclusion
In summary, we explored hypothetical evolutionary pathways of FRK gene family in 20 plant species and the regulating mechanism and functional divergence in Populus. Totally, 125 putative FRK genes can be organized into 8 clades, including 3 conserved clades and 5 specific clades. Evolutionary analysis revealed that FRK genes transient to vascular plants, accompanied by loss, acquisition and diversification of functional domains. In Populus, segmental duplication often appears to be the major mechanisms of the expansion of FRK genes, and most FRK genes that were duplicated in salicoids can be regulated by Populus-specific miRNAs. Furthermore, we detected the FRK gene family expanded accompanying lineage-specific traits and they often experienced multiple duplication events which help to promote fundamental neofunctionalizations and subfunctionalizations in evolution. All in all, our results contribute to explore more specific roles of members in gene families throughout the genome and provide a new framework for further functional investigation of gene family.
Accession Numbers
The raw data of genome resequencing have been deposited in the Genome Sequence Archive in BIG Data Center (BIG, CAS, China) under accession number of CRA000903.
Data Availability Statement
The variation data of this manuscript have been deposited in Big Data Center, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under the accession number GVM000056 that are publicly accessible at https://bigd.big.ac.cn/gvm/getProjectFile?t=56b6b35f.
Author Contributions
DZ and JX designed the experiment and conception, and obtained the funding and were responsible for this manuscript. WX tested the experiments and wrote the manuscript. YZ helped to analyze and assess the data. JX and SC provided the valuable suggestions on the manuscript. JX, SC, YZ, and DZ helped to revise the manuscript. All authors read and approved the manuscript.
Funding
This study was funded by the Fundamental Research Funds for the Central Universities (No. BLYJ201604), the Project of the National Natural Science Foundation of China (31600537 and 31972954), and the Young Elite Scientists Sponsorship Program by CAST (2018QNRC001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00484/full#supplementary-material
Footnotes
- ^ http://www.phytozome.net/
- ^ http://pfam.xfam.org/
- ^ https://web.expasy.org/protparam/
- ^ http://meme-suite.org/tools/meme
- ^ http://www.ebi.ac.uk/Tools/pfa/iprscan/
- ^ http://gsds.cbi.pku.edu.cn/
- ^ http://plantgrn.noble.org/psRNATarget/
- ^ https://phytozome.jgi.doe.gov/pz/portal.html
References
Bagowski, C. P., Bruins, W., and te Velthuis, A. J. W. (2010). The nature of protein domain evolution: shaping the interaction network. Curr. Genomics 11, 368–376. doi: 10.2174/138920210791616725
Bailey, T. L., Nadya, W., Chris, M., and Li, W. W. (2006). MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, 369–373. doi: 10.1093/nar/gkl198
Bradbury, P., Zhang, Z., Kroon, D., Casstevens, T., Ramdoss, Y., and Buckler, E. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Chen, Y., Zhang, Q., Hu, W., Zhang, X., Wang, L., Hua, X., et al. (2017). Evolution and expression of the fructokinase gene family in Saccharum. BMC Genomics 18:197. doi: 10.1186/s12864-017-3535-7
Csűös, M. (2010). Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 26, 1910–1912. doi: 10.1093/bioinformatics/btq315
Dai, X., Zhuang, Z., and Zhao, P. X. (2018). psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res. 46:W49. doi: 10.1093/nar/gky316
Danuta, K., Kaminski, W. E., Gerhard, L., Armin, P., Wenzel, J. J., Christoph, M. H., et al. (2003). Adenosine triphosphate binding cassette (ABC) transporters are expressed and regulated during terminal keratinocyte differentiation: a potential role for ABCA7 in epidermal lipid reorganization. J. Invest. Dermatol. 121, 465–474. doi: 10.1046/j.1523-1747.2003.12404.x
Delaux, P. M., Nanda, A. K., Mathé, C., Sejalon-Delmas, N., and Dunand, C. (2012). Molecular and biochemical aspects of plant terrestrialization. Perspect. Plant Ecol. Evol. Syst. 14, 49–59. doi: 10.1016/j.ppees.2011.09.001
Du, Q., Wang, B., Wei, Z., Zhang, D., and Li, B. (2012). Genetic diversity and population structure of Chinese White poplar (Populus tomentosa) revealed by SSR markers. J. Heredity 103, 853–862. doi: 10.1093/jhered/ess061
Du, Q., Xu, B., Gong, C., Yang, X., Wei, P., Tian, J., et al. (2014). Variation in growth, leaf, and wood property traits of Chinese white poplar (Populus tomentosa), a major industrial tree species in Northern China. Can. J. Forest. Res. 44, 326–339. doi: 10.1139/cjfr-2013-0416
Evanno, G., Regnaut, S., and Goudet, J. (2010). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Freeling, M. (2009). Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453. doi: 10.1146/annurev.arplant.043008.092122
Gad, K., and Ron, S. (2005). A block-free hidden markov model for genotypes and its application to disease association. J. Comput. Biol. 12, 1243–1260. doi: 10.1089/cmb.2005.12.1243
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S. E., Wilkins, M. R., Appel, R. D., et al. (2005). “Protein identification and analysis tools on the ExPASy server,” in The Proteomics Protocols Handbook, ed. J. M. Walker (Totowa, NJ: Humana Press).
Gershoni, M., and Pietrokovski, S. (2014). Reduced selection and accumulation of deleterious mutations in genes exclusively expressed in men. Nat. Commun. 5:4438. doi: 10.1038/ncomms5438
Goodstein, D. M., Shengqiang, S., Russell, H., Rochak, N., Hayes, R. D., Joni, F., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Graham, L. E., Patricia, A. A., Taylor, W. A., Strother, P. K., and Cook, M. E. (2012). Aeroterrestrial coleochaete (Streptophyta, Coleochaetales) models early plant adaptation to land. Am. J. Bot. 99, 130–144. doi: 10.3732/ajb.1100245
Gramer, M., and Huber, H. (1991). Specifity and stability of cardiovascular responses to stress. Int. J. Psychophysiol. 11:35. doi: 10.1016/0167-8760(91)90151-m
Granot, D. (2007). Role of tomato hexose kinases. Funct. Plant Biol. 34, 564–570. doi: 10.1071/FP06207
Grynko, Y., and Shkuratov, Y. (2003). Scattering matrix calculated in geometric optics approximation for semitransparent particles faceted with various shapes. J. Quant. Spectrosc. Ra 78, 319–340. doi: 10.1016/s0022-4073(02)00223-6
Gu, S., Fang, L., and Xu, X. (2013). Using soapaligner for short reads alignment. Curr. Protoc. Bioinformatics 11, 1–17. doi: 10.1002/0471250953.bi1111s44
Harholt, J., and Moestrup, Ø, and Ulvskov, P. (2015). Why plants were terrestrial from the beginning. Trends Plant Sci. 21, 96–101. doi: 10.1016/j.tplants.2015.11.010
Hileman, L. C., Sundstrom, J. F., Litt, A., Chen, M., Shumba, T., and Irish, V. F. (2006). Molecular and phylogenetic analyses of the mads-box gene family in tomato. Mol. Biol. Evol. 23, 2245–2258. doi: 10.1093/molbev/msl095
Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J., and Gao, G. (2014). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Kanayama, Y., Dai, N., Granot, D., Petreikov, M., Schaffer, A., and Bennett, A. B. (1997). Divergent fructokinase genes are differentially expressed in tomato. Plant Physiol. 113, 1379–1384. doi: 10.1104/pp.113.4.1379
Kawakubo, T., Okada, O., and Minami, T. (2007). “Dynamic structure change due to ATP hydrolysis in the motor domain of myosin: molecular dynamics simulations,” in AIP Conference Proceedings 922, Tokyo, 651.
Kenrick, P., and Crane, P. R. (1997). The Origin and Early Diversification of Land Plants: A Cladistic Study. (Washington DC, Smithsonian Institution Press).
Konstantin, P., Polishchuk, L. V., Leila, M., Dmitry, K., and Konstantin, G. (2007). Accumulation of slightly deleterious mutations in mitochondrial protein-coding genes of large versus small mammals. Proc. Natl. Acad. Sci. U.S.A. 104, 13390–13395. doi: 10.1073/pnas.0701256104
Kuettel, S., Greenwald, J., Kostrewa, D., Ahmed, S., Scapozza, L., and Perozzo, R. (2011). Crystal structures of T. b. rhodesiense adenosine kinase complexed with inhibitor and activator: implications for catalysis and hyperactivation. PLoS Negl. Trop. Dis. 5:e1164. doi: 10.1371/journal.pntd.0001164
Liao, B.-Y., and Zhang, J. (2006). Evolutionary conservation of expression profiles between human and mouse orthologous genes. Mol. Biol. Evol. 23, 530–540. doi: 10.1093/molbev/msj054
Liu, P. L., Huang, Y., Shi, P. H., Yu, M., Xie, J. B., and Xie, L. (2018). Duplication and diversification of lectin receptor-like kinases (LecRLK) genes in soybean. Sci. Rep. U. K. 8:5861. doi: 10.1038/s41598-018-24266-6
Lynch, M., and Force, A. (2000). The probability of duplicate gene preservation by subfunctionalization. Genetics 154, 459–473. doi: 10.1023/A:1004191625603
Margulis, L. (1992). Biodiversity: molecular biological domains, symbiosis and kingdom origins. Biosystems 27, 39–51. doi: 10.1016/0303-2647(92)90045-z
Mccormick, J., and Johnstone, R. M. (1990). Evidence for an essential sulfhydryl group at the substrate binding site of the A-system transporter of Ehrlich cell plasma membranes. Biochem. Cell Biol. 68, 512–519. doi: 10.1139/o90-073
Mcmahan, C. J., Difilippantonio, M. J., Rao, N., and Spanopoulou, E. (1997). A basic motif in the N-terminal region of RAG1 enhances V(D)J recombination activity. Mol. Cell. Biol. 17:4544. doi: 10.1128/mcb.17.8.4544
Mehlgarten, C., Eggeling, R., Gohr, A., Bönn, M., Lemnian, I., Nettling, M., et al. (2018). “Evolution of the AMP-activated protein kinase controlled gene regulatory network,” in Information- and Communication Theory in Molecular Biology, ed. M. Bossert (Cham: Springer), 211–238. doi: 10.1007/978-3-319-54729-9_9
Melissa, R., Lorenz, G., David, S., András, G., Mattias, H. M., Manoj, K., et al. (2012). Fructokinase is required for carbon partitioning to cellulose in aspen wood. Plant J. 70, 967–977. doi: 10.1111/j.1365-313x.2012.04929.x
Michelson, A. M., Blake, C. C., Evans, S. T., and Orkin, S. H. (1985). Structure of the human phosphoglycerate kinase gene and the intron-mediated evolution and dispersal of the nucleotide-binding domain. Proc. Natl. Acad. Sci. U.S.A. 82, 6965–6969. doi: 10.1073/pnas.82.20.6965
Nick, P., Price, A. L., and David, R. (2013). Population structure and eigenanalysis. PLoS Genet. 2:e190. doi: 10.1371/journal.pgen.0020190
Pérez-Pérez, J. M., Esteve-Bruna, D., González-Bayón, R., Kangasjärvi, S., Caldana, C., and Hannah, M. A., et al. (2013). Functional redundancy and divergence within the arabidopsis reticulata-related gene family. Plant Physiol. 162, 589–603.
Petr, D., Adam, A., Goncalo, A., Albers, C. A., Eric, B., Depristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Plange, N., Hirsch, T., Bienert, M., and Remky, A. (2014). Specifity of optic disc evaluation in healthy subjects with large optic discs and physiologic cupping using confocal scanning laser ophthalmoscopy. Klin. Monbl. Augenheilkd 231, 164–169. doi: 10.1055/s-0032-1328094
Plaxton, W. C. (1996). The organization and regulation of plant glycolysis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47, 185–214. doi: 10.1146/annurev.arplant.47.1.185
Puzey, J. R., Karger, A., Axtell, M., and Kramer, E. M. (2012). Deep annotation of Populus trichocarpa microRNAs from diverse tissue sets. PLoS One 7:e33034. doi: 10.1371/journal.pone.0033034
Reiss, P. T., Armin, S., Feihan, L., Lei, H., and Erika, P. (2012). Paradoxical results of adaptive false discovery rate procedures in neuroimaging studies. Neuroimage 63, 1833–1840. doi: 10.1016/j.neuroimage.2012.07.040
Riggs, J. W., Cavales, P. C., Chapiro, S. M., and Callis, J. (2017). Identification and biochemical characterization of the fructokinase gene family in Arabidopsis thaliana. BMC Plant Biol. 17:83. doi: 10.1186/s12870-017-1031-5
Romani, F., Reinheimer, R., Florent, S. N., Bowman, J. L., and Moreno, J. E. (2018). Evolutionary history of HOMEODOMAIN LEUCINE ZIPPER transcription factors during plant transition to land. New Phytol. 219, 408–421. doi: 10.1111/nph.15133
Saori, O., Bennett, A. B., and Yoshinori, K. (2002). Distinct physiological roles of fructokinase isozymes revealed by gene-specific suppression of Frk1 and Frk2 expression in tomato. Plant Physiol. 129, 1119–1126. doi: 10.2307/4280538
Shanfa, L., Quanzi, L., Hairong, W., Mao-Ju, C., Sermsawat, T. A., Hoon, K., et al. (2013). Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc. Natl. Acad. Sci. U.S.A. 110, 10848–10853. doi: 10.1073/pnas.1308936110
Silvestro, D., and Michalak, I. (2012). raxmlGUI: a graphical front-end for RAxML. Org. Divers. Evol. 12, 335–337. doi: 10.1007/s13127-011-0056-0
Simionato, E., Ledent, V., Richards, G., Thomas-Chollier, M., Kerner, P., Coornaert, D., et al. (2007). Origin and diversification of the basic helix-loop-helix gene family in metazoans: insights from comparative genomics. BMC Evol. Biol. 7:33. doi: 10.1186/1471-2148-7-33
Stein, O., and Granot, D. (2018). Plant fructokinases: evolutionary, developmental, and metabolic aspects in sink tissues. Front. Plant Sci. 9:339. doi: 10.3389/fpls.2018.00339
Stein, O., Secchi, F., German, M. A., Damari-Weissler, H., Aloni, R., Holbrook, N. M., et al. (2017). The tomato cytosolic fructokinase FRK1 is important for phloem fiber development. Biol. Plantarum 62, 353–361. doi: 10.1007/s10535-017-0762-3
Szerypo, J., Habs, D., Heinz, S., Neumayr, J., Thirolf, P., Wilfart, A., et al. (2003). MAFFTRAP: ion trap system for MAFF. Nucl. Instrum. Meth. A 204, 512–516. doi: 10.1016/S0168-583X(02)02123-7
Thumma, B. R., Nolan, M. F., Robert, E., and Moran, G. F. (2005). Polymorphisms in cinnamoyl CoA reductase (CCR) are associated with variation in microfibril angle in Eucalyptus spp. Genetics 171, 1257–1265. doi: 10.1534/genetics.105.042028
Tippmann, H. F. (2004). Analysis for free: comparing programs for sequence analysis. Brief. Bioinform. 5, 82–87. doi: 10.1093/bib/5.1.82
Tuskan, G. A., Difazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604. doi: 10.1126/science.1128691
Xie, J., Yang, X., Song, Y., Du, Q., Li, Y., Chen, J., et al. (2017). Adaptive evolution and functional innovation of Populus-specific recently evolved microRNAs. New Phytol. 213, 206–219. doi: 10.1111/nph.14046
Xu, C., Nadon, B. D., Kim, K. D., and Jackson, S. A. (2018). Genetic and epigenetic divergence of duplicate genes in two legume species: duplicate gene evolution in two legume species. Plant Cell Environ. 41, 2033–2044. doi: 10.1111/pce.13127
Yu, Z., Roberts, R. J., and Simon, K. (2004). Segmentally variable genes: a new perspective on adaptation. PLoS Biol. 2:e81. doi: 10.1371/journal.pbio.0020081
Yun, Z., Wen-Kai, J., and Li-Zhi, G. (2011). Evolution of microRNA genes in Oryza sativa and Arabidopsis thaliana: an update of the inverted duplication model. PLoS One 6:e28073. doi: 10.1371/journal.pone.0028073
Yupeng, W., Haibao, T., Debarry, J. D., Xu, T., Jingping, L., Xiyin, W., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Zaykin, D. V., Westfall, P. H., Young, S. S., Karnoub, M. A., Wagner, M. J., and Ehm, M. G. (2002). Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum. Hered 53, 79–91. doi: 10.1159/000057986
Keywords: FRK gene family, lineage-specific gene, adaptation, functional novelty, Populus tomentosa
Citation: Xu W, Zhao Y, Chen S, Xie J and Zhang D (2020) Evolution and Functional Divergence of the Fructokinase Gene Family in Populus. Front. Plant Sci. 11:484. doi: 10.3389/fpls.2020.00484
Received: 27 October 2019; Accepted: 31 March 2020;
Published: 14 May 2020.
Edited by:
Yuannian Jiao, Chinese Academy of Sciences, ChinaReviewed by:
Xiyin Wang, North China University of Science and Technology, ChinaLiangsheng Zhang, Fujian Agriculture and Forestry University, China
Copyright © 2020 Xu, Zhao, Chen, Xie and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianbo Xie, amJ4aWVAYmpmdS5lZHUuY24=; Deqiang Zhang, RGVxaWFuZ1poYW5nQGJqZnUuZWR1LmNu