 Matias Köhler
Matias Köhler Marcelo Reginato
Marcelo Reginato Tatiana Teixeira Souza-Chies
Tatiana Teixeira Souza-Chies Lucas C. Majure
Lucas C. Majure- 1Programa de Pós-Graduação em Botânica, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 2Florida Museum of Natural History, University of Florida Herbarium (FLAS), Gainesville, FL, United States
- 3Department of Research, Conservation and Collections, Desert Botanical Garden, Phoenix, AZ, United States
Chloroplast genomes (plastomes) are frequently treated as highly conserved among land plants. However, many lineages of vascular plants have experienced extensive structural rearrangements, including inversions and modifications to the size and content of genes. Cacti are one of these lineages, containing the smallest plastome known for an obligately photosynthetic angiosperm, including the loss of one copy of the inverted repeat (∼25 kb) and the ndh gene suite, but only a few cacti from the subfamily Cactoideae have been sufficiently characterized. Here, we investigated the variation of plastome sequences across the second-major lineage of the Cactaceae, the subfamily Opuntioideae, to address (1) how variable is the content and arrangement of chloroplast genome sequences across the subfamily, and (2) how phylogenetically informative are the plastome sequences for resolving major relationships among the clades of Opuntioideae. Our de novo assembly of the Opuntia quimilo plastome recovered an organelle of 150,347 bp in length with both copies of the inverted repeat and the presence of all the ndh gene suite. An expansion of the large single copy unit and a reduction of the small single copy unit was observed, including translocations and inversion of genes, as well as the putative pseudogenization of some loci. Comparative analyses among all clades within Opuntioideae suggested that plastome structure and content vary across taxa of this subfamily, with putative independent losses of the ndh gene suite and pseudogenization of genes across disparate lineages, further demonstrating the dynamic nature of plastomes in Cactaceae. Our plastome dataset was robust in resolving three tribes with high support within Opuntioideae: Cylindropuntieae, Tephrocacteae and Opuntieae. However, conflicting topologies were recovered among major clades when exploring different assemblies of markers. A plastome-wide survey for highly informative phylogenetic markers revealed previously unused regions for future use in Sanger-based studies, presenting a valuable dataset with primers designed for continued evolutionary studies across Cactaceae. These results bring new insights into the evolution of plastomes in cacti, suggesting that further analyses should be carried out to address how ecological drivers, physiological constraints and morphological traits of cacti may be related with the common rearrangements in plastomes that have been reported across the family.
Introduction
Cacti comprise one of the most charismatic plant clades of the world, exhibiting an enormous variety of growth forms, morphology and intriguing niche occupancy across the Americas (Britton and Rose, 1919; Anderson, 2001; Hunt et al., 2006; Hernández-Hernández et al., 2011). This diversity is reflected in a high number of species and heterogeneous diversification rates across the clade (Arakaki et al., 2011; Hernández-Hernández et al., 2014). Some uncommon features in most angiosperms, such as succulent tissues, Crassulacean acid metabolism (CAM), betalain pigments and the reduction of or absence of leaves are typical characters of cacti that have long captured the attention of plant biologists and have been suggested as adaptations to allow survival in harsh environments (Mooney et al., 1977; Mauseth, 1999; Landrum, 2002; Nobel, 2002). Indeed, members of the family are conspicuous elements of the arid and semiarid succulent biome of the New World, but they are also found in subtropical and tropical forests, especially as epiphytes (Taylor and Zappi, 2004; Hunt et al., 2006). Besides major morphological and physiological adaptations, genetic and genomic-level changes are also expected. For example, whole genome duplication events have long been suggested to be associated with adaptations to extreme environments (e.g., Stebbins, 1971; Soltis and Soltis, 2000; Brochmann et al., 2004), and significant gene family expansion in genes related to stress adaptation, as well as more restricted events of gene duplications were reported in lineages of Caryophyllales adapted to severe environments including in cacti (Wang et al., 2019).
Although gene content, structural organization and size of the chloroplast genome (plastomes) of land plants is often considered highly conserved (Raubeson and Jansen, 2005; Chumley et al., 2006; Wicke et al., 2011), deviations have been increasingly reported in some clades and have challenged the generality of this phenomenon (Daniell et al., 2016; Mower and Vickrey, 2018; Ruhlman and Jansen, 2018). Astonishing variety of size have been observed across land plants, from 19 kb in a non-photosynthetic Epipogium roseum (D. Don) Lindl. (Orchidaceae) to giant plastomes with 217 kb, as in Pelargonium × hortorum L. H. Bailey (Geraniaceae) (Chumley et al., 2006; Schelkunov et al., 2015), reflected by expansions or contraction of the inverted repeat (IR), large single copy (LSC) or even small single copy (SSC) units. Also, the independent losses of one copy of the inverted repeat region (∼25 kb in size) have been identified across disparate clades, such as Fabaceae, Geraniaceae, Orobanchaceae, and Cactaceae (Cai et al., 2008; Ruhlman and Jansen, 2014; Sanderson et al., 2015), and a variety of taxa have lost particular genes (e.g., ndh genes in parasites, carnivorous plants and xerophytes) (Braukmann et al., 2009; Wicke et al., 2011; Iles et al., 2013; Peredo et al., 2013; Ruhlman et al., 2015; Sanderson et al., 2015).
Members of Cactaceae also have experienced different alterations in their chloroplast genome. A conserved inversion of ∼6 kb on the large single copy unit comprising the trnM-rbcL genes have long been suggested (Wallace, 1995) and more recently confirmed (Sanderson et al., 2015; Majure et al., 2019; Solórzano et al., 2019). Besides that, the first cactus plastome assembled from the saguaro cactus [Carnegiea gigantea (Engelm.) Britton & Rose] exhibited an exceptional reduction in size (113 kb) and gene content, including the loss of one of the two inverted repeat regions and nine of the 11 ndh genes (Sanderson et al., 2015). More recently, newly assembled plastomes of seven species of the short-globose cacti of Mammillaria Haw. revealed three different plastome structures across the genus, all with two copies of a divergent inverted repeat, including (i) an extreme reduction in size of IRs [<1 kb, typically ranging from 15 to 30 kb in land plants (Zhu et al., 2016)]; (ii) an intermediate reduction of IR (∼7 kb) with translocation of some typical LSC genes to the IR; and (iii) a structure with a divergent IR structure and a surprisingly reduced plastome (∼107 kb), being now the putative smallest plastome known for an obligately photosynthetic angiosperm (Solórzano et al., 2019). However, considering these dissimilar patterns between the few described plastomes of cacti, a broad sampling including other lineages may shed new insights into chloroplast genome evolution across the family.
The classification of Cactaceae has been long proposed based on morphological characters (Schumann, 1899; Britton and Rose, 1919; Backeberg, 1958; Hunt et al., 2006), and further tested with the aid of molecular phylogenies (Nyffeler, 2002; Bárcenas et al., 2011; Hernández-Hernández et al., 2011). Three major well-supported clades are currently circumscribed as subfamilies: Opuntioideae, Maihuenioideae and Cactoideae, while the traditional “Pereskioideae” has been revealed as a basal grade including the two leafy lineages of the cacti, which are subsequent sisters to the rest, i.e., Leuenbergeria Lodé and Pereskia Mill. (Edwards et al., 2005, reviewed in Guerrero et al., 2019). Opuntioideae (∼350 spp.) is the most widespread subfamily with members occurring from southern South America (Argentina) to northern North America (Canada) (Britton and Rose, 1919; Anderson, 2001; Hunt et al., 2006; Ritz et al., 2012; Majure and Puente, 2014; Majure et al., 2019). The group shows interesting morphological synapomorphies, such as the small brushlike, barbed spines (i.e., glochids) and a bony aril surrounding a campylotropous ovule (Stuppy, 2002; Taylor et al., 2002). However, the delimitation of taxa within Opuntioideae is still not settled, and the controversy is observed across different taxonomic levels, from species to tribes (Schumann, 1899; Britton and Rose, 1919; Hunt, 2002; Stuppy, 2002; Taylor et al., 2002). Traditional classifications based on general morphology – such as growth form, stem and leaf morphology, as well as floral, fruit, pollen, and seed characters – were used to divide the subfamily from few to up to 20 smaller genera (Britton and Rose, 1919; Stuppy, 2002; Hunt et al., 2006; Griffith and Porter, 2009). Nonetheless, molecular phylogenetic studies, mainly based on chloroplast (rpl16 intron and trnL-trnF region) and nuclear ribosomal ITS sequences, revealed that the most comprehensive genus, Opuntia s.l. (L.) Mill., was paraphyletic, which reinforced the recognition of numerous smaller genera corresponding to well-supported clades (Stuppy, 2002; Taylor et al., 2002; Wallace and Dickie, 2002; Griffith and Porter, 2009; Majure et al., 2012; Ritz et al., 2012; Majure and Puente, 2014). Likewise, the tribal classification of Opuntioideae has been controversial based on different approaches, with up to six tribes proposed (Hunt, 2011). While Doweld (1999) and Wallace and Dickie (2002) proposed five tribes, with different circumscriptions from each other — four were recognized as monophyletic in the last comprehensive molecular study of Opuntioideae (Griffith and Porter, 2009). Despite great improvement in our phylogenetic understanding in Opuntioideae (Griffith and Porter, 2009; Ritz et al., 2012; Majure et al., 2019), support for the relationships among those clades, as well as a better taxon sampling with more molecular markers, still needs to be strengthened.
Apart from the external and internal transcribed spacer (ETS and ITS) of the nuclear ribosomal repeats (NRR) and ppc marker, most molecular phylogenies of cacti have been historically based on a few plastid markers (trnL-trnF, rpl16, trnK, and matK) (Nyffeler, 2002; Edwards et al., 2005; Korotkova et al., 2010; Arakaki et al., 2011; Bárcenas et al., 2011; Demaio et al., 2011; Hernández-Hernández et al., 2011, 2014; Ritz et al., 2012; Bárcenas, 2016; Vargas-Luna et al., 2018). While these markers have shown to be potentially able to resolve some clades, some relationships are still lacking support (Nyffeler, 2002; Griffith and Porter, 2009; Bárcenas et al., 2011; Hernández-Hernández et al., 2011). In this case, next-generation sequencing (NGS) could be a useful tool, since it has transformed the study of non-model plant taxa in phylogenetic inferences with high throughput data allowing deep resolution across major plant clades (Xi et al., 2012; Ma et al., 2014; Gardner et al., 2016; Zong et al., 2019). NGS data are also showing to be extremely useful for discovering informative regions across genomes, for marker development (Wu et al., 2010; Dong et al., 2012; Ripma et al., 2014; Reginato et al., 2016; Abdullah et al., 2019), as well as to investigate chloroplast genome evolution (Dong et al., 2013; Mower and Vickrey, 2018; Yao et al., 2019). Nevertheless, this approach is still in its infancy across Cactaceae (Majure et al., 2019) and remains a path to be explored.
Here, we investigate the use of next-generation sequencing across Opuntioideae to address two major questions: (1) how homogenous is the content and arrangement of chloroplast genomes across the subfamily? and (2) how phylogenetically informative are chloroplast genome sequences for resolving major relationships among the clades of Opuntioideae? We used a combination of de novo and reference-guided assemblies to process genome skimming data: (i) assembling and characterizing the first chloroplast genome of an Opuntia species, O. quimilo K. Schum., (ii) investigating overall patterns of reference-guided assemblies and comparative chloroplast genome sequence analyses across the subfamily, (iii) inferring phylogenetic relationships with assembled sequences and (iv) surveying plastomes for highly informative phylogenetic markers for Sanger-based studies for future use.
Materials and Methods
Taxon Sampling, DNA Extraction, and Sequencing
All currently recognized genera in Opuntioideae (sensu Hunt et al., 2006, plus Majure et al., 2019 for Grusonia s.l.), with the exception of Punotia (see Ritz et al., 2012), were sampled with one accession per genus, resulting in a dataset of 17 taxa, which were sequenced via genome-skimming (Straub et al., 2012; Majure et al., 2019). All seven genera of tribe Opuntieae were included; five of the six genera in Tephrocacteae were sampled, and all five genera in Cylindropuntieae were included in our analyses. Three additional samples were selected as outgroup taxa [Cactoideae: Parodia magnifica (F. Ritter) F. H. Brandt and Coryphantha macromeris (Engelm.) Lem.; and Pereskia: Pereskia sacharosa Griseb.] based on previous studies (Arakaki et al., 2011; Hernández-Hernández et al., 2014). Plant materials were from wild collections or from the Desert Botanical Garden’s living collection (see Supplementary Table S1 for details). DNA was extracted from silica-dried or fresh epidermal tissues using a standard CTAB incubation (Doyle and Doyle, 1987) followed by chloroform/isoamyl alcohol precipitation and silica column-based purification steps, as described in Neubig et al. (2014) and Majure et al. (2019). Whole genomic DNAs were quantified using the Qubit dsDNA BR Assay Kit and Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, United States); high-molecular-weight DNA (>15 kb) samples showing no degradation were considered suitable and sent to Rapid Genomics LLC1 (Gainesville, FL, United States) for library preparation and high-throughput sequencing using the Illumina HiSeq X platform with 150 bp paired-end reads. A total of sixty samples were included per lane for sequencing.
De novo Assembly and Data Processing for Chloroplast Genome Sequences
Raw reads were imported into Geneious 11.1.5 (Biomatters, Auckland, New Zealand), and paired reads were set with an expected insert size of 300 bp calculated with BBMap using default setting (Bushnell, 2016). Low quality bases (Q < 20) were trimmed, and all reads shorter than 20 bp were discarded using BBDuk for quality control (Bushnell, 2016). Different methods were employed to assemble the chloroplast genome of the diploid Opuntia quimilo. First, a de novo assembly was performed with 40% of the reads using the Geneious de novo assembler (low/fast sensitivity option, plus default settings). A consensus sequence (with a majority threshold for sequence matching – fewest ambiguities) of each contig greater than 1,000 bp in length was saved. Considering that the Cactaceae plastomes already published have unusual rearrangements, we looked for plastid contigs searching those saved contigs against the Portulaca oleracea L. plastome (Portulacaceae, one of the closest relatives of Cactaceae; see Walker et al., 2018) (GenBank accession KY490694, Liu et al., 2017) using MegaBLAST (following parameters proposed from Ripma et al., 2014). Additional chloroplast genome de novo assemblies of O. quimilo were performed using a set of different pipelines, such as GetOrganelle (Jin et al., 2019) and NOVOPlasty (Dierckxsens et al., 2017) to cross-validate and compare among the assemblies. After checking convergence of the assemblies from the different pipelines and the plastid contig recovered from the Geneious de novo assembly, we used the NOVOPlasty circular contig for downstream analyses. Annotations were performed with GeSeq (Tillich et al., 2017), using default parameters to predict protein-coding genes by HMMER profile search and ARAGORN v1.2.38 (Laslett and Canback, 2004); tRNA genes were annotated with tRNAscan-SE v2.0 (Lowe and Chan, 2016), and BLAST searches were used to annotate ribosomal RNA (rRNA), tRNA, and DNA genes conserved in embryophyte plastomes (Wommack et al., 2008). All annotations were cross checked with the “Annotate from” feature in Geneious, transferring annotations with a 50% or greater similarity from the P. oleracea plastome, and eventual start/stop codons were manually adjusted with the “Open Read Frame (ORF)” feature from Geneious. The genes that had their structures affected by the insertion of internal stop codons and/or a small ORF, thus did not form their respective full coding sequence (CDS), were annotated as putative pseudogenes. The graphical representation of the O. quimilo circular annotated plastome was created in OGDRAW (Lohse et al., 2013; Greiner et al., 2019). To visualize changes in gene order and content, we compared the O. quimilo assembly with the canonical gene order of the P. oleracea plastome via multiple whole genome alignments using MAUVE (default options, assuming colinearity; Darling et al., 2004). Boundaries between the IRa, IRb, LSC, SSC and putative inversions were visually checked in Geneious using an in silico approach adapted from Oliver et al. (2010).
Comparative Chloroplast Genome Sequence Analyses Across Opuntioideae
The newly annotated plastome of Opuntia quimilo, with one of the inverted repeats (IRa) manually stripped to avoid data duplication, was then used for a reference guided assembly on the trimmed reads from all other taxa using Geneious mapper with a medium-low sensitivity iterating up to five times (adapted from Ripma et al., 2014). Each of the assemblies mapped had a majority threshold consensus sequence generated and annotations transferred from the O. quimilo reference, and manually adjusted. To identify highly variable regions across the subfamily, the 17 assembled Opuntioideae chloroplast genome sequences were compared using mVista (Frazer et al., 2004) in Shuffle-LAGAN alignment mode (Brudno et al., 2003) using the annotated plastome of O. quimilo as a reference. We also used the full chloroplast genome sequence alignment (see below) to calculate nucleotide diversity values (π) to detect highly variable sites among Opuntioideae chloroplast genome sequences. DNA polymorphism analysis was performed on DnaSP v.6.10 (Rozas et al., 2017) using the sliding window analysis with a step size of 200 bp and window length of 800 bp. Assembly maps of raw read coverages from Geneious mapper of each taxon to the O. quimilo plastome were also used to visualize and compare the gene content of the chloroplast genome sequences across the subfamily.
Phylogenetic Analyses and Informative Regions
The assembled chloroplast genome sequences, obtained as described in the previous section, were aligned using MAFFT v. 7 with an automatic strategy search for algorithm selection (Katoh and Standley, 2013), using 200PAM scoring matrix and an open gap penalty of 1.53 (offset value 0.123). The alignment was manually examined for misaligned areas following a similarity criterion (Simmons, 2004). Sequence portions that contained gaps and/or ambiguities across more than 80% of the taxa were stripped using the “Mask Alignments” feature in Geneious. Phylogenetic inference was performed using Maximum Likelihood implemented in RAxML 8.2.4 (Stamatakis, 2014) in the CIPRES Portal (Miller et al., 2010). As RAxML is mainly designed to implement generalized time-reversible molecular models (GTR), we employed the GTR + G model for the entire sequence, which have been suggested for topological reconstruction skipping model selection (Abadi et al., 2019), and GTR + I + G is not recommended by Stamatakis (see RAxML v8.2 manual) given the potential interaction between the I and G parameters. Support values were estimated implementing 1,000 bootstrap pseudoreplicates.
To identify and rank highly phylogenetically informative regions in the Opuntioideae plastomes, we split the full plastome alignment into protein coding sequences (cpCDS – pseudogenes were included here), non-coding sequences (cpNCDS) and intergenic spacers (cpIGS) using the annotated O. quimilo plastome. Each individual marker (cpCDS, cpNCDS, cpIGS) was extracted from the above-mentioned alignment, and a Maximum Likelihood tree was inferred with RAxML using GTR + G model (see reasons above) and 100 bootstrap replicates. For each marker, we report the number of variable sites, number of parsimony informative sites (PIS), mean sequence distance (under K80 model), alignment length, mean sequence length, mean bootstrap support and distance to the full chloroplast genome sequence tree (RF distance; Robinson and Foulds, 1981). The metrics were retrieved using functions of the R packages ape and phangorn (Paradis et al., 2004; Schliep, 2011). Markers were ranked by phylogenetic information using a weighted mean of relative values of the following metrics: number of variable sites (weight = 1), mean bootstrap (weight = 2) and distance to the full plastid tree (weight = 3). We designed primer pairs for the top five markers identified in the previous step with suitable size for PCR amplification (< ∼900 bp). Primers flanking the target regions were designed with Primer3, using the default settings (Rozen and Skaletsky, 2000). All metrics reported, as well as primer design, were considered only for the ingroup (the 17 Opuntioideae chloroplast genome sequences). Further phylogenetic inferences (RAxML, GTR + G, 1000 bootstrap), were performed for a dataset concatenating: (1) the top five markers, (2) the top 10 markers, and (3) the five markers which have primers designed.
Results
DNA Sequencing
Runs on Illumina HiSeq X resulted in 227,003,814 reads from 20 samples (17 Opuntioideae and three outgroups), between 5,624,110 and 20,219,350 reads per sample, for a mean read number of 11,350,190 sequences. Reads per sample following quality control were between 5,360,990 and 19,863,298 with a mean post-quality control read pool number of 11,084,834. The GC content of the raw reads ranged from 37.4 to 40.6% with a mean of 38.45% and following quality control were between 36.9 and 40% with a mean of 38%. Detailed results with the number of raw reads, post-quality control and %GC content per taxa are presented in Supplementary Table S1.
Opuntia quimilo Plastome
The complete chloroplast genome of Opuntia quimilo was sequenced, assembled, annotated and deposited in GenBank (accession number MN114084). The length of the Opuntia quimilo plastome is 150,374 bp, including a 101,475 bp LSC region, a 4,115 bp SSC region and 22,392 bp of two IR (IRa and IRb) regions (Figure 1 and Table 1). A total of 701,318 reads were assembled, with an average organelle depth of 844x. The GC content varies from 33% in the SSC, to 35.5% in LSC and 39.6% in the IR regions, while 38% in coding regions (CDS) and 35.6% in non-coding regions (Table 1).
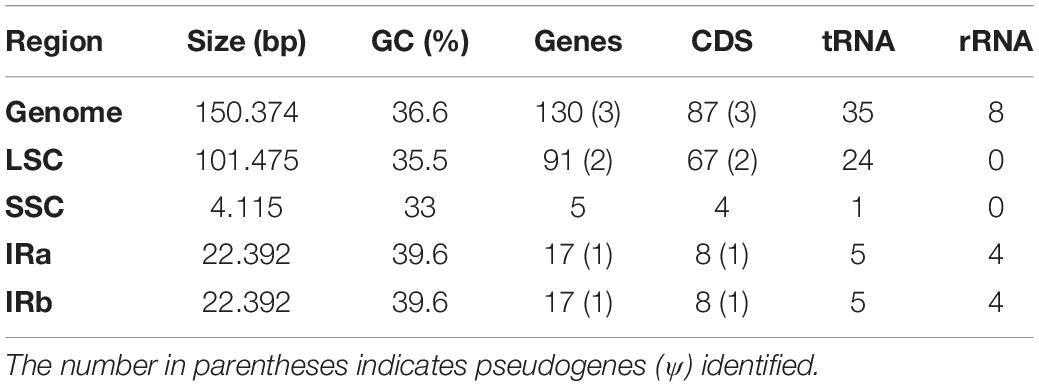
Table 1. Chloroplast genome composition of Opuntia quimilo.
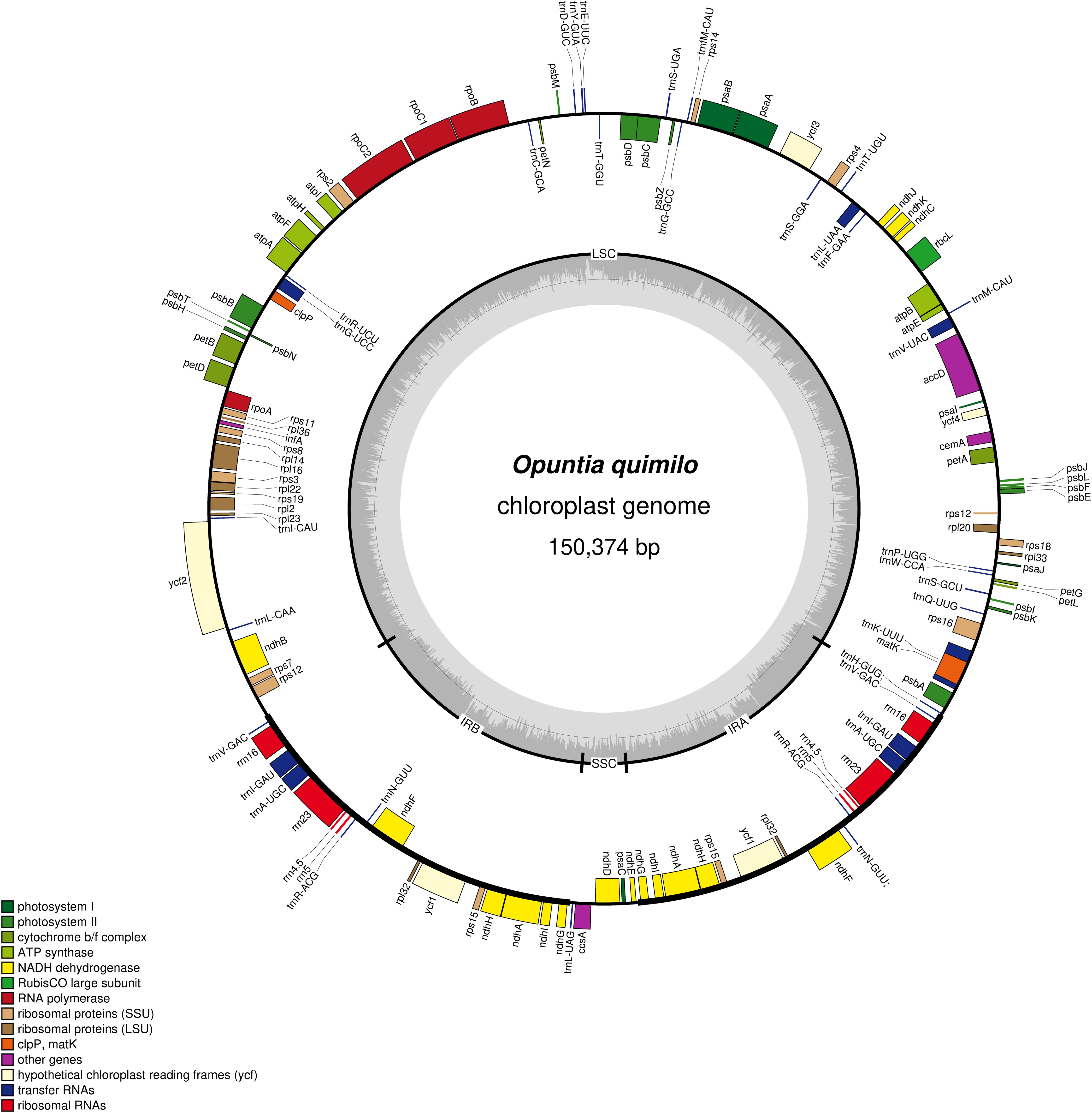
Figure 1. Circular map of chloroplast genome of Opuntia quimilo with annotated genes. The genes transcribed clockwise are shown inside of the circle, whereas genes transcribed counter clockwise are shown outside of the circle. The borders of chloroplast genome are defined with LSC, SSC, IRa, and IRb. The dashed gray color of inner circle shows the GC content.
The de novo assembly of the Geneious assembler produced 1,000 contigs; of these, 988 were higher than 1,000 bp in length from a minimum length of 1,026 bp to a maximum of 283,150 bp. MegaBLAST search found one consensus plastid contig of 128,909 bp that included the full chloroplast sequence with two putative inverted repeats assembled as a single IR unit (∼22 kb). The GetOrganele and NOVOPlasty pipelines both yielded one plastid contig of 150,374 bp with the same gene content, order, and structure as the plastid contig of the Geneious assembler, except for the two inverted repeats that were interleaved by the LSC and SSC on the first ones, while in Geneious these were merged as one IR.
The Opuntia quimilo plastome encodes 87 protein-coding genes (CDS), 35 transfer RNA genes (tRNA) and eight ribosomal RNA (rRNA) genes, totaling 130 genes (Tables 1, 2). Three canonical CDS from angiosperm chloroplast genomes were annotated as putative pseudogenes (Ψ) based on their structure: accD, ycf1, and ycf2. Two of them (accD and ycf2) are in the LSC, and ycf1 in the IRs. Duplicated CDS in the IRs included ndhA, ndhF, ndhG, ndhH, ndhI, rpl32, ycf1(Ψ), and rps15; and all four rRNA genes and five of the 35 tRNAs were duplicated in the IR regions. The O. quimilo plastome includes 16 intron-containing genes, of which 15 contain one intron (atpF, ndhA, ndhB, petB, petD, rpl16, rpoC1, rps12, rps16, trnAUGC, trnGUCC, trnIGAU, trnKUUU, trnLUAA, trnVUAC), while one gene contains two introns (ycf3), and the clpP gene has lost its two introns, reduced to an exon of 609 bp.

Table 2. Structural and functional gene composition of Opuntia quimilo chloroplast genome.
The LSC of the Opuntia quimilo plastome appears to have experienced an expansion, with surprisingly 101 kb, while the SSC was shown to have exceptional reduction (4 kb). The LSC contains 24 tRNA genes and 67 CDS, and the SSC contains a unique tRNA gene (trnLUAG), and four CDS: ccsA, ndhE, ndhD and psaC (Figures 1, 2 and Tables 1, 2). A total of eight genes (ndhB, rpl2, rpl23, rps7, rps19, trnICAU, trnLCAA, and ycf2) that are usually reported occurring in the IR regions of canonical angiosperm plastomes are apparently present as unique genes – not repeated – in the LSC region of the O. quimilo plastome (Figure 2, region V). On the contrary, seven genes (ndhA, ndhF, ndhG, ndhH, ndhI, rpl32, and rps15), usually from the SSC, are duplicated into the IR regions of the O. quimilo plastome (Figure 2, orange genes).
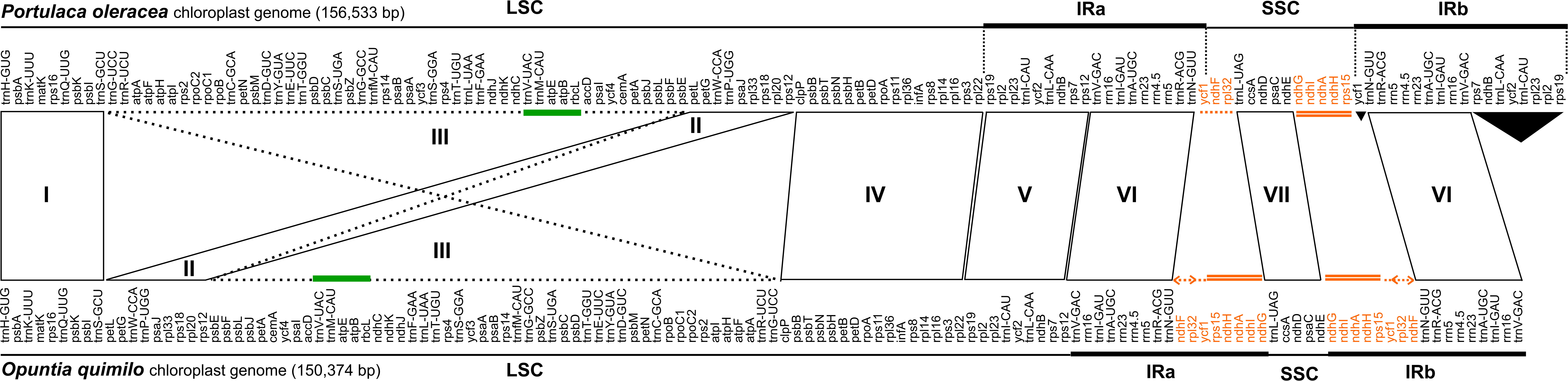
Figure 2. Plastid genome structure and gene order in Opuntia quimilo compared with purslane (Portulaca oleracea). Purslane has the canonical order typical of most angiosperms. For simplicity, the circular map has been linearized. Green line highlights the trnMCAU-rbcL synapomorphic inversion of Cactaceae, which in O. quimilo also includes the trnVUAC gene. Regions I, IV, V, VI, and VII are colinear in both plastomes. Region II is colinear but is translocated in the O. quimilo plastome, while region III is inverted and translocated. Region V comprise the genes that are typically in the IR region but are translocated to the large single copy in O. quimilo. Genes highlighted in orange are those typically found in the SSC but transferred to the IR region in O. quimilo. Orange dashed line indicate the double inversion on the ycf1-rpl32 genes, placing ycf1 gene adjacent to rpl32. Black triangles represent duplicated genes present in purslane but absent in O. quimilo; LSC, large single-copy region; SSC, small single-copy region; IR, Inverted repeat.
When compared to the canonical angiosperm chloroplast genome of Portulaca oleracea, two block translocations in the LSC are present in the O. quimilo plastome: the first (Figure 2, region II) is a simple colinear translocation of nine genes (Figure 2, region II); while the second one is a big block inversion and translocation comprising 50 genes within the trnGUCC-psbE region (Figure 2, region III). Inside that block (region III), the putative synapomorphic inversion of cacti encompassing the trnM-rbcL genes is confirmed for Cactaceae, but in the O. quimilo plastome this inversion also encompassed the trnVUAC gene (Figure 2, green bars). Further gene order is mainly colinear (Figure 2, regions I, IV, V, VI, VII), except for the rearrangement comprising the SSC genes that were transferred to the IR regions, including a double inversion on the ycf1-rpl32 region, placing ycf1 gene adjacent to rpl32 (Figure 2, orange genes).
Reference-Guided Assemblies and Comparative Chloroplast Sequence Analyses
The reference-guided assembles of the remaining Opuntioideae and outgroup taxa to the Opuntia quimilo plastome (one inverted repeat stripped) mapped an average of 616,615 reads with a mean genome depth of 721x (Supplementary Table S2). The consensus sequence length varied between 126,925 bp [Pereskiopsis diguetii (F.A.C. Weber) Britton & Rose] to 129,181 bp [Tacinga palmadora (Britton & Rose) N.P. Taylor & Stuppy] and the GC content between 35.8% (Pterocactus gonjianii R. Kiesling) to 36.3% [Austrocylindropuntia cylindrica (Lam.) Backeb. and Cylindropuntia bigelovii] (Supplementary Table S2).
Pairwise comparison of divergent regions within the Opuntioideae chloroplast genome sequences using mVISTA with O. quimilo as a reference revealed both strikingly conserved and divergent regions across the chloroplast genome sequences (Figure 3). Overall, the alignment uncovered sequence divergence across assemblies, suggesting that chloroplast genome sequences are not conserved. Divergences were observed both in non-coding regions and coding regions. Among coding regions (CDS), non-conserved regions were frequent on genes of the ndh gene suite (i.e., ndhA, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ) as well clpP, ycf3 and particularly highlighted on ycf1, ycf2, and accD genes (Figure 3). Ten non-coding regions show substantial divergence, being all intergenic spacers: ndhE-psaC, rpl32-ndhF, trnVGAC-rps12, psbB-clpP, rpoB-trnCGCA, psbM-trnDGUC, trnTGGU-psbD, psbE-rpl20, ndhC-rbcL (Figure 3).
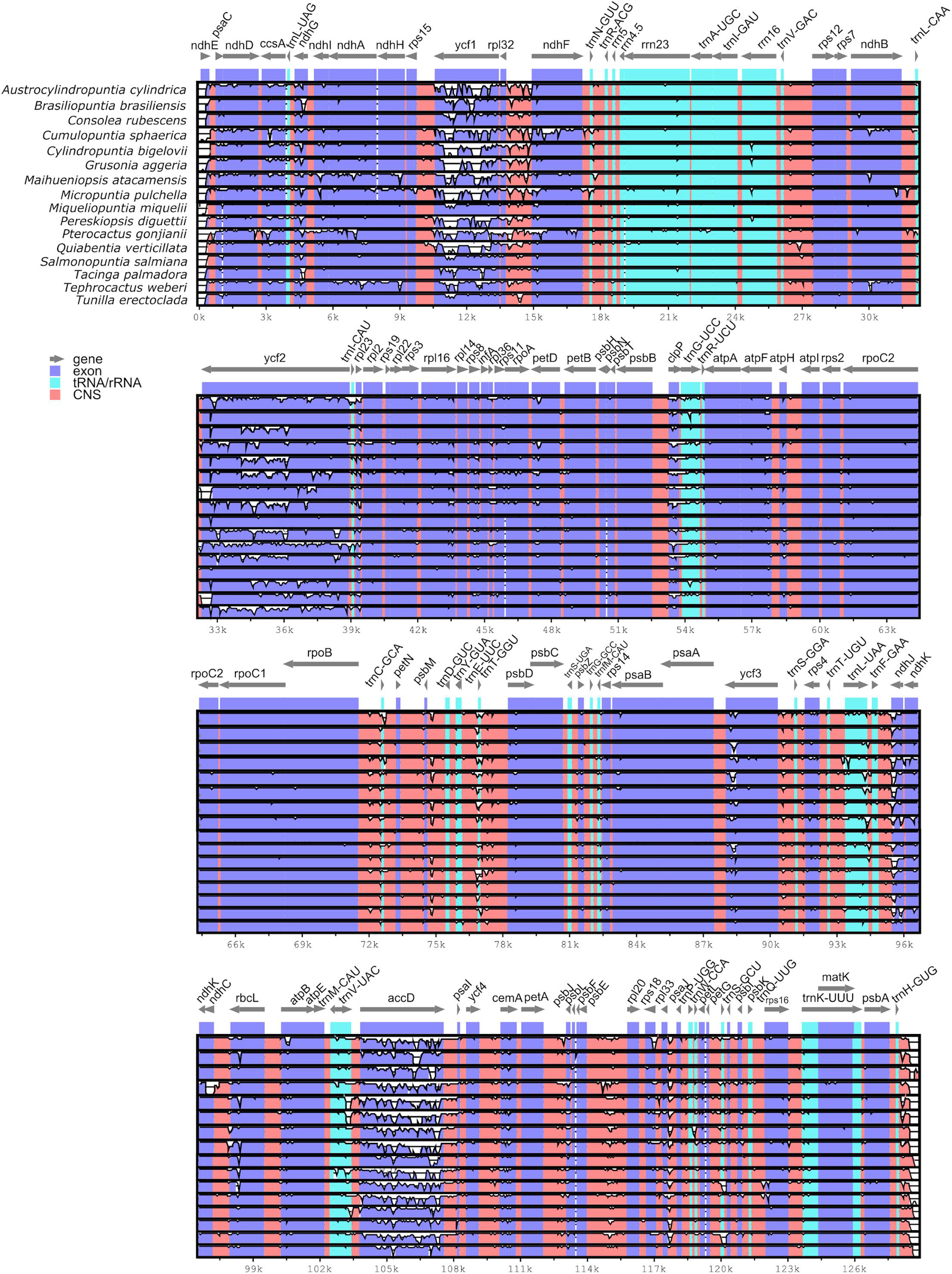
Figure 3. Visualized alignment of the Opuntioideae chloroplast genome sequences (one IR stripped) with annotations using mVISTA. Each horizontal lane shows the graph for the sequence pairwise identity with Opuntia quimilo as reference. The x-axis represents the base sequence of the alignment and the y-axis represents the pairwise percent identity within 50–100%. Gray arrows represent the genes and their orientations. Dark-blue boxes represent exon regions; light-blue boxes represent tRNA and rRNA regions; red boxes represent non-coding sequence (CNS) regions.
The nucleotide diversity values (π) within the 17 Opuntioideae chloroplast genome sequences ranged from 0.00191 to 0.18551, with a mean value of 0.02201, indicating the sequences as highly variable. Three major regions were identified as hypervariable (π > 0.1), which comprises ycf1 and accD genes and an intergenic spacer rpl32-ndhF (Figure 4); while six regions were observed as moderately-variable (π > 0.05), those being four genes: ycf2, ccsA, clpP and trnLUAA; and two intergenic spacers rps18-rpl33 and trnFGAA-ndhJ (Figure 4).
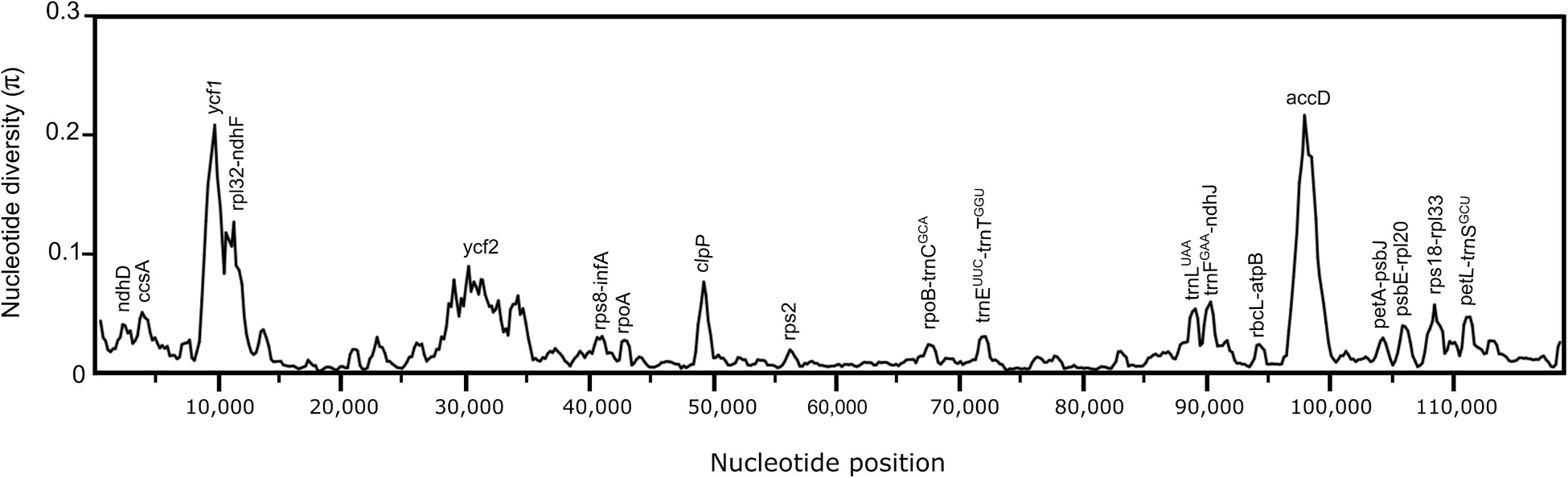
Figure 4. Nucleotide diversity graphs of the 17 Opuntioideae chloroplast genome sequences from the sliding windows analysis performed in DnaSP (windows length: 800 bp, step size: 200 bp). The x-axis represents the base sequence of the alignment, and the y-axis represents the nucleotide diversity (π value). Each variation hotspot for the chloroplast genome sequences of the Opuntioideae alignment is annotated on the graph.
Reference-guided assembled maps of Opuntioideae and outgroups to the Opuntia quimilo chloroplast genome as a reference revealed regions with extremely low coverage or even gaps across different taxa (Figure 5). The regions highlighted with this feature are related with genes of the ndh suite, ycf1, ycf2 and accD, suggesting gene loss, transfer to nuclear genomes and/or pseudogenization (Figure 5). Several members of Opuntioideae appear to have missing ndh genes in their chloroplast genome (Micropuntia, Maihueniopsis, Pterocactus, Tephrocactus), especially in the Tephrocacteae clade, but without a clear pattern across lineages.
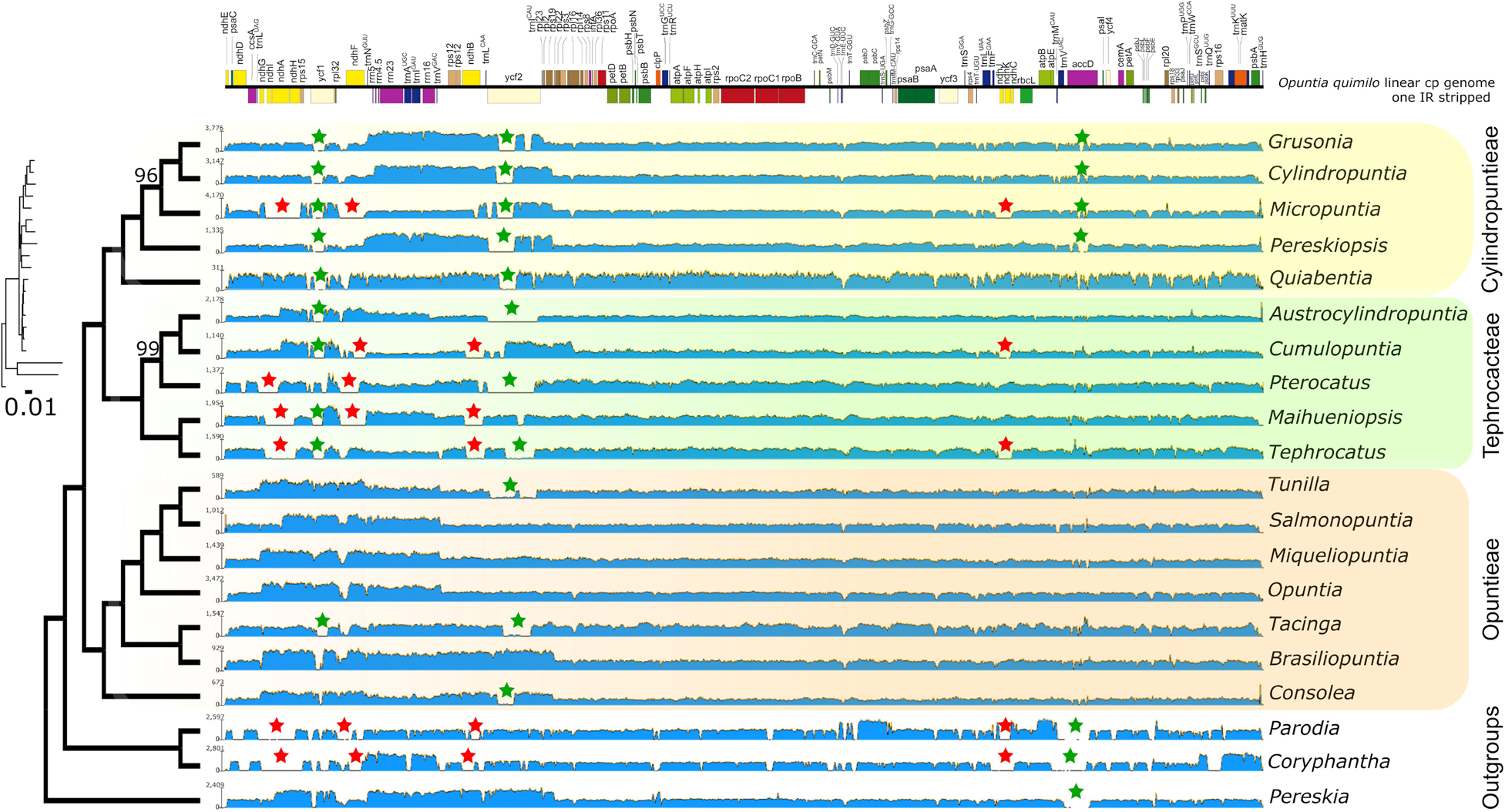
Figure 5. Maximum likelihood phylogenetic tree from RAxML analysis transformed in cladogram with the phylogram represented in small size with substitution rate scaled. All nodes have total bootstrap values (bs = 100) with exception for those that are shown above the branch. Each tip is represented with the assembly map of raw read coverages from Geneious mapper to the Opuntia quimilo chloroplast genome (one IR stripped, represented on the top with annotated genes). Red stars represent low coverage mapping and putative losses associated with the ndh gene suite; green stars represent partial low coverages associated with putative pseudogenization of ycf1, ycf2, and accD genes. Tribe Opuntieae is highlighted in orange, Tephrocacteae in green and Cylindropuntieae in yellow.
Phylogenetic Analyses and Informative Regions
The full chloroplast genome sequences resulted in an alignment of 118,930 bp with 86,484 identical sites (72.7%), a pairwise identity of 94.5% and 8,694 distinct alignment patterns. There were 8,922 parsimony informative sites (PIS) and 11,509 sites with gaps. Maximum Likelihood analyses resolved a well-supported Opuntioideae (bs = 100), with three major subclades (those currently circumscribed as tribes), Opuntieae, Cylindropuntieae and Tephrocacteae (Figure 6A). Opuntieae, consisting of the seven genera Consolea, Brasiliopuntia, Tacinga, Opuntia, Miqueliopuntia, Salmonopuntia and Tunilla, was resolved as sister to a Tephrocacteae (Tephrocactus, Maihueniopsis, Pterocactus, Cumulopuntia, and Austrocylindropuntia) + Cylindropuntieae (Quiabentia, Pereskiopsis, Micropuntia, Grusonia, and Cylindropuntia) clade. All nodes had full bootstrap support values (bs = 100), except at two nodes, which were still higher than 90% (Figure 6A).
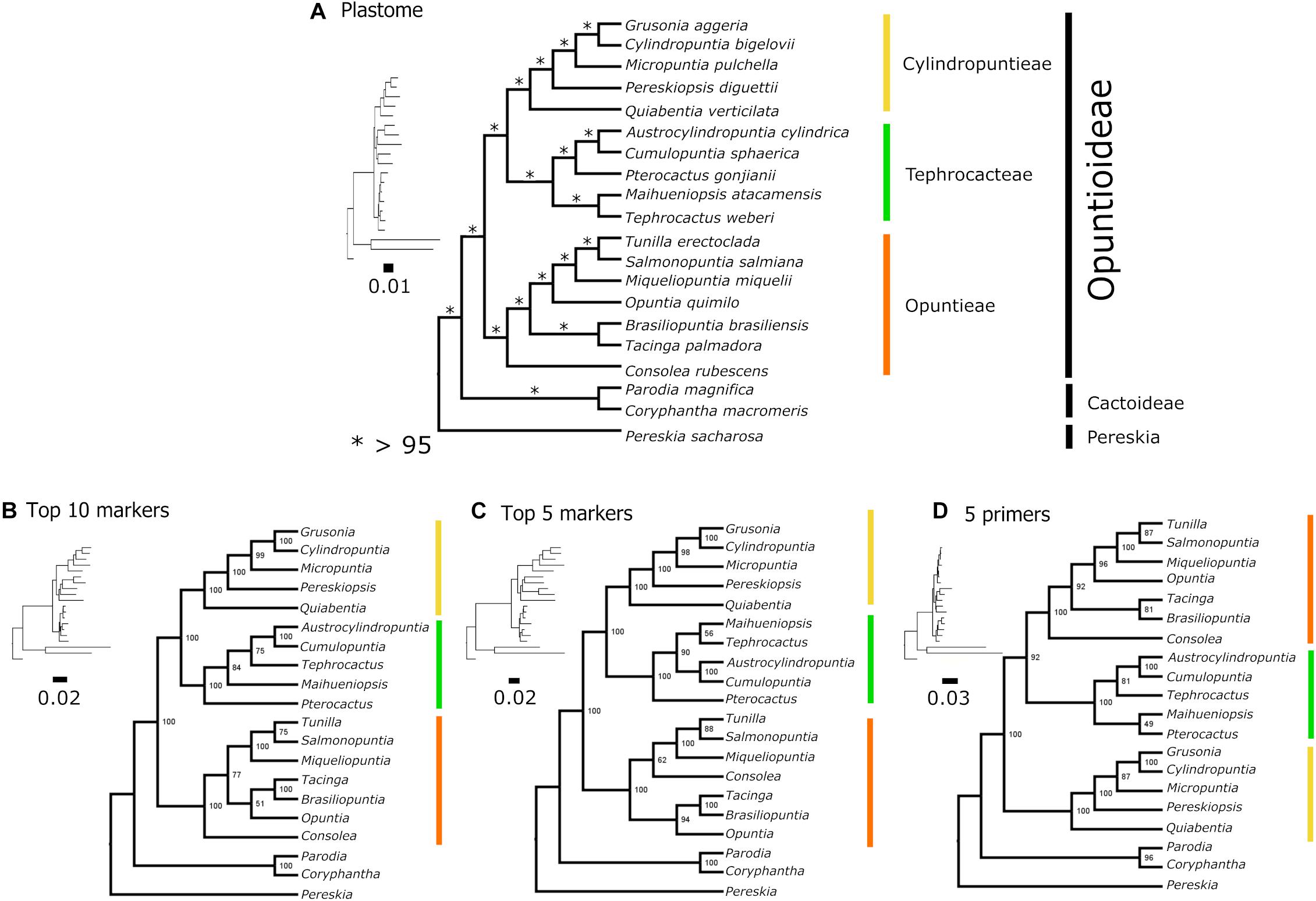
Figure 6. Topological comparisons of different datasets based on ML analyses. (A) Plastome dataset topology, (B) top 10 marker dataset topology, (C) top five marker dataset topology, and (D) five marker dataset topology for which primers were designed. The Cylindropuntieae sister to Tephrocacteae + Opuntieae topology was recovered only in the five-marker primer dataset (D). Generic relationships are highly variable in Tephrocacteae among the datasets used. ∗bootstrap support.
The summary statistics for all markers (cpCDS, cpNCDS, cpIGS) are presented in Supplementary Table S3. A list of the top 10 markers ranked by phylogenetic information considering topological distance to the plastome tree, mean bootstrap support and number of parsimony informative sites is given in Table 3. All single marker phylogenies presented some disagreement to the plastome tree (RF tree distance ranging from 6 to 28), with bootstrap support ranging from 0 to 89 (mean = 37), and number of PIS from 0 to 619 (mean = 25), revealing many markers as not useful for phylogenetic inference (Supplementary Table S3). Phylogenetic trees of the top 10 individual markers are shown in Supplementary Figures S1, S2, and all trees are available as Supplementary Material. Primer pair sequences for PCR amplification are provided for the top five markers with suitable Sanger sequencing size (maximum ∼900 bp) in Table 4.
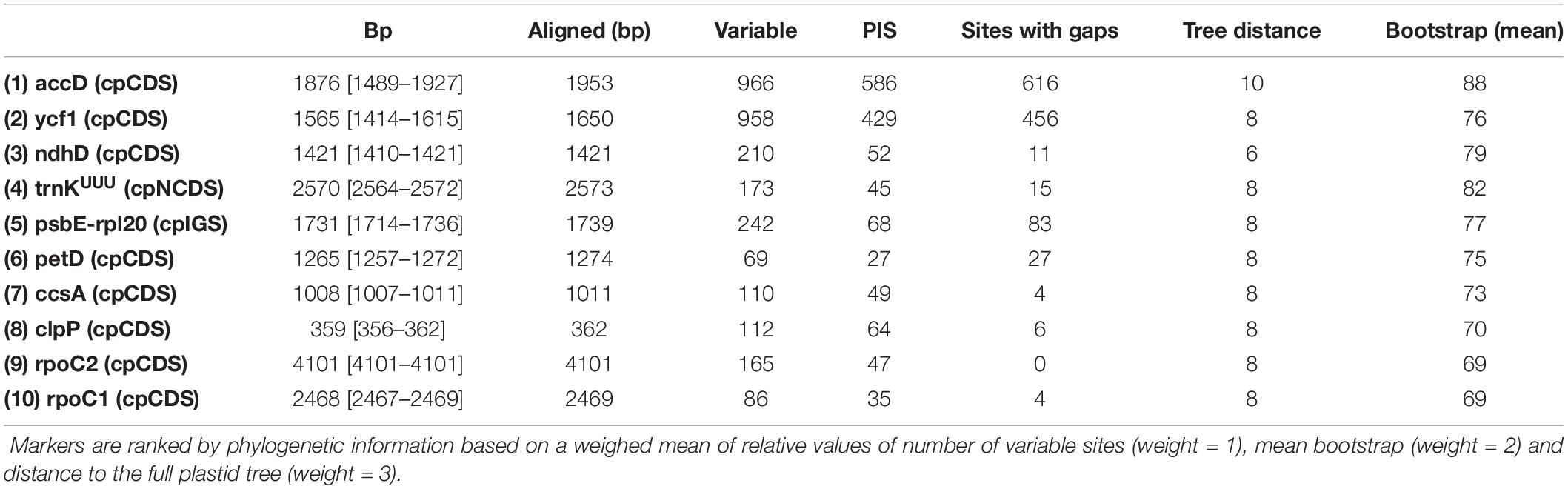
Table 3. Summary statistics for the top 10 markers.
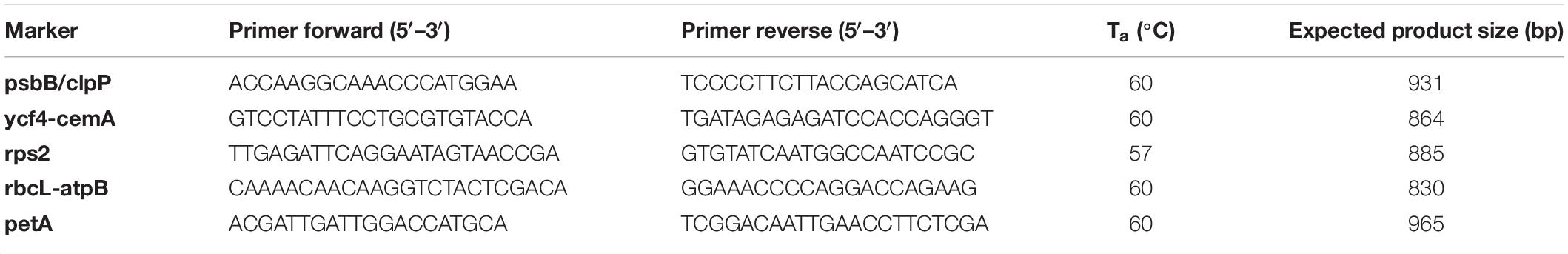
Table 4. Primer pair sequences for the identified top 5 highly informative markers across the 17 chloroplast genome sequences of Opuntioideae.
Phylogenetic inferences from the top 5 and top 10 markers concatenated yield similar topologies compared with the plastome tree, supporting three tribes (bs = 100) and Opuntieae as sister to (Tephrocacteae + Cylindropuntieae), although, there were minor incongruences within Tephrocacteae and Opuntieae (Figures 6B,C). Contrarily, phylogenetic inference from the five markers, which had primers designed (< ∼900 bp) revealed a conflicting topology, with Cylindropuntieae as sister to (Tephrocacteae + Opuntieae) with high support (bs = 92), and all three tribes with full support (bs = 100) (Figure 6D).
Discussion
Insights From Chloroplast Genome Assemblies in Opuntioideae and Cactaceae
The first chloroplast genome of a member of subfamily Opuntioideae and a species of Opuntia is here reported. Although the bulk of its gene content is not far from canonical angiosperm plastomes, it deviates in some cases from the typical chloroplast genome structure, showing: (i) an expansion of the LSC incorporating genes that are typically in the IRs; (ii) a reduction of the SSC translocating some common genes of the SSC into the IR region; and (iii) at least one massive translocation with an inversion of a block of genes in the LSC (Figure 2). Part of the content of the IRs in the O. quimilo plastome remained remarkably constant, including all four rRNA and five tRNA genes that are nearly universally reported in IRs of land plants (Mower and Vickrey, 2018). The GC content observed in the O. quimilo plastome is regular as expected based on other chloroplast genomes, being an AT rich organelle, with differences observed between coding/non-coding regions, where selection may be acting to preserve GC content for amino acid coding (Raubeson and Jansen, 2005; Downie and Jansen, 2015; Daniell et al., 2016).
Successive expansion–contraction events or even multiple contractions have been recurrently reported as one of the main ways of developing structural changes across angiosperm plastomes (Downie and Jansen, 2015; Daniell et al., 2016; Fonseca and Lohmann, 2017; Weng et al., 2017; Mower and Vickrey, 2018) and may also be one way in which genes are translocated to different regions of the genome, as suggested in adzuki bean (Perry and Wolfe, 2002). The atypical reduction of the SSC (∼4 kb), reported here for the O. quimilo plastome, has also been noticed in Viviana marifolia (Francoaceae, Geraniales), and a slightly similar reduced size for the SSC (∼6 kb) have been inferred for the ancestral chloroplast genome of Geraniaceae (Weng et al., 2014). A partial deletion of the SSC region has also been reported in two hemiparasitic Taxillus (Loranthaceae) species resulting in a ∼6 kb region with only two genes (Li et al., 2017), and the smallest SSC hitherto reported is for the hemiparasitic Pedicularis ishidoyana (Orobanchaceae), with only 27 bp (Cho et al., 2018). A model to explain the major rearrangements observed in the Lamprocapnos spectabilis (Papaveraceae) plastome, involving at least six IR boundary shifts and five inversions resulting in a SSC of just 1,645 bp with a partial ndhF gene, was recently provided by Park et al. (2018). The SSC contains most ndh genes, and previous studies have shown that boundary shifts of the IR and SSC regions are correlated with transformations of ndhF and ycf1 genes (Logacheva et al., 2014; Kim et al., 2015; Li et al., 2017).
The Opuntia quimilo plastome reinforces some different putative structural synapomorphies that have been reported in Caryophyllales. The loss of the rpl2 intron, previously suggested to be absent throughout the Centrospermae (Palmer et al., 1988), is supported in our study and other newly assembled plastomes in Caryophyllales (Yao et al., 2019). The trnM-rbcL inversion is again recovered in the O. quimilo plastome, although also involving the trnVUAC gene, as in Cylindropuntia bigelovii (Majure et al., 2019), providing further support for this inversion as a synapomorphy in the family. Additionally, Sanderson et al. (2015) and Solórzano et al. (2019), inspecting plastomes of Cactoideae, reported a gene orientation of ycf2-trnLCAA-ycf1 in the SSC as a synapomorphy of Cactoideae. Our results corroborate this observation, since this feature is not present in the O. quimilo plastome, strengthening this gene order as a putative synapomorphy for Cactoideae. On the other hand, the ycf1-rpl32-ndhF orientation, reported in the Cylindropuntia bigelovii chloroplast sequence (Majure et al., 2019), is recovered in the O. quimilo plastome and is here suggested as a putative synapomorphy for Opuntioideae.
Reference-guided assemblies and comparative analyses revealed insights for plastome rearrangements across disparate Opuntioideae. The differences of depth and coverage among specific chloroplast genes suggest that gene presence or structure may vary over species in Opuntioideae, as have been observed in other Cactaceae, specifically Cactoideae (Sanderson et al., 2015; Solórzano et al., 2019). The putative independent losses of several ndh genes in all Cactoideae plastomes assembled hitherto, such as the saguaro cactus and several Mammillaria species, can be also inferred for our Cactoideae outgroups sampled (Parodia magnifica and Coryphantha macromeris; Figure 5, red stars). Likewise, some members of Cylindropuntieae and Tephrocacteae (Micropuntia, Cumulopuntia, Pterocactus, Maihueniopsis, and Tephrocactus) also likely experienced independent losses of several genes of the ndh suite in their chloroplast genomes, although this was not so for tribe Opuntieae, where those genes were found to be intact (Figure 5, red stars), indicating putative homoplasious events. The putative loss of one of the inverted repeat regions in Quiabentia must be further investigated through rigorous de novo assemblies (Figure 5).
Loss of ndh genes or the ndh gene suite has been reported in both gymnosperms (Wakasugi et al., 1994; McCoy et al., 2008; Wu et al., 2009) and angiosperms, as well as some other photosynthetic organisms. The loss of such genes is well-known and is often associated with hemi- or holoparasitism where genes necessary for photosynthesis are often unessential (e.g., Epifagus, Orobanchaceae, De Pamphilis and Palmer, 1990; Santalales, Shin and Lee, 2018). However, a number of autotrophic plants have also shown a similar trend with losses or pseudogenization of ndh genes. For example, Lin et al. (2017) showed the repeated loss of ndh genes across several different autotrophic orchid species and suggested that those losses could have been a step toward heterotrophy. Ruhlman et al. (2015) suggested that the evolution and retention of the NDH (NADH dehydrogenase-like) complex was associated with the transition of plants to environmentally stressful environments, and that ndh gene loss may be associated with a relaxed reliance on the complex based on decreased environmental stressors (e.g., through reliance on host species for resources in parasites).
Contrastingly, there are numerous reports of ndh loss or pseudogenization in angiosperms associated with the presence of CAM photosynthesis, which has evolved as a response to water limited habitats (i.e., water stress), such as in desert or other edaphically arid areas where cacti occur or also associated with an epiphytic habit, for instance in orchids (Luo et al., 2014; Givnish et al., 2015; Sanderson et al., 2015). Whether or not the absence of those ndh genes in the chloroplast corresponds to their integration into the nuclear genome often remains to be determined, but there are some studies showing that those genes likewise, have not been incorporated into the nucleus (Lin et al., 2017) and thus are totally lost. Certain species of Opuntioideae have been shown to be facultatively CAM species (Winter et al., 2011), whereas other species appear to be obligate CAM. Perhaps the putative loss or pseudogenization of ndh genes across members of Opuntioideae coincides with the transition to more water limited habitats and thus a stronger association with obligate CAM photosynthesis. Although assumed that most derived cacti (Cactoideae, Opuntioideae) are obligate CAM, there are actually very little data to show photosynthetic pathways across Cactaceae, and the retention of large leaves in Opuntioideae bring into question this assumption (Majure et al., 2019). Likewise, our knowledge of CAM photosynthesis is in a state of flux, and it is clear that there are taxa that do not clearly fit into basic photosynthetic pathways as traditionally defined (Edwards, 2019). The putative connection with ndh gene loss and CAM photosynthesis needs to be rigorously tested.
The major plastid regions marked by pseudogenization in the Opuntia quimilo plastome (ycf1, ycf2, and accD) are visually highlighted as non-conserved regions in reference-guided maps (Figure 5, green stars), as in the mVista alignment across Opuntioideae (Figure 3). These regions are also emphasized as with hyper or moderate variability regarding nucleotide diversity values (Figure 4). All genes here reported as pseudogenes in the O. quimilo plastome (accD, ycf1, and ycf2) have also been reported as pseudogenes in the Mammillaria plastomes (Solórzano et al., 2019), while the accD was described as a pseudogene in Carnegiea gigantea (Sanderson et al., 2015). Pseudogenization of these genes has been repeatedly reported across different angiosperm lineages, such as Malpighiales, Campanulales, Ericales, Poales, Solanales, Geraniales, Santalales, and Myrtales (Haberle et al., 2008; Fajardo et al., 2013; Harris et al., 2013; Weng et al., 2014; Li et al., 2017; Machado et al., 2017; Bedoya et al., 2019; Cui et al., 2019). Even though these genes have been identified with essential functions beyond photosynthesis and retained in the plastome of most embryophytes (Drescher et al., 2000; Kuroda and Maliga, 2003; Kode et al., 2005; Kikuchi et al., 2013; Parker et al., 2014; Dong et al., 2015), there are several other plants where these genes are missing from the chloroplast genome (Magee et al., 2010; Kim et al., 2014; Liu et al., 2016; Graham et al., 2017). The pseudogenization or loss of the accD, rpl22 and several genes of the ndh suite from the plastids has been reported to be a consequence of them being transferred to the nuclear genome (Jansen et al., 2011; Jansen and Ruhlman, 2012; Sanderson et al., 2015; Liu et al., 2016; Cauz-Santos et al., 2017). Plastid gene transfer to the nucleus remains to be examined in O. quimilo and related Opuntioideae.
Several regions highlighted as hyper or moderately variable regarding the nucleotide diversity values across Opuntioideae chloroplast sequences (i.e., accD, ycf1, clpP, petD, rpl32, and ccsA) have been reported to be putatively under positive selection in some lineages, such as Brassicaceae, Bignoniaceae, Rutaceae, Orchidaceae, Geraniaceae, and Poaceae (Carbonell-Caballero et al., 2015; Hu et al., 2015; Weng et al., 2016; Park et al., 2017; Dong et al., 2018; Piot et al., 2018; Ruhlman and Jansen, 2018; Thode and Lohmann, 2019). Positive selection may come into play in response to environmental changes (Piot et al., 2018). For example, the accD gene, which encodes the β-carboxyl transferase subunit of acetyl-CoA carboxylase, is an essential and required component for plant leaf development (Kode et al., 2005), and it is suggested to have played a pivotal role in the adaptive evolution of orchids (Dong et al., 2018). The signatures of positive selection in the accD gene observed in Brassicaceae and Campanulaceae have also indicated that this gene may have been repeatedly involved in the adaption to specific ecological niches during the radiation of eudicotyledonous plants (Rousseau-Gueutin et al., 2013; Hu et al., 2015). Considering the harsh environment that cacti occupy, their fitness already expressed in its peculiar morphology and physiology, further studies should be carried out to investigate the putative relation of chloroplast rearrangement – such as pseudogenization, loss of genes, translocation and inversion – with ecological features.
Phylogenetic Relationship of Opuntioideae Tribes
The plastome phylogeny of Opuntioideae strongly resolves three major and well-supported clades, the tribes Opuntieae (O), Tephrocacteae (T) and Cylindropuntieae (C) (Figure 6A). Three previously described tribes (Austrocylindropuntieae, Pterocacteae, and Pereskiopsideae) (Doweld, 1999; Wallace and Dickie, 2002), mainly representing lineages of a single genus, are nested within these more broadly circumscribed tribes, and thus have no real practical taxonomic use (Hunt, 2011).
In our phylogenomic analyses, Opuntieae was sister to a Tephrocacteae/Cylindropuntieae clade, as in Majure et al. (2019), who also used plastome data but with a reduced taxon sampling in Opuntieae and Tephrocacteae. This same topology [O + (T + C)] was further uncovered with high support using our top 10 and 5 phylogenetic informative markers concatenated (Figures 6B,C). On the other hand, Walker et al. (2018) and Wang et al. (2019), using transcriptome data, revealed Cylindropuntieae as sister to an Opuntieae/Tephrocacteae clade [C + (O + T)] yet with very limited taxon sampling. Likewise, this alternate topology [C + (O + T)] was recovered in our study when exploring our five markers concatenated, which have primer-pairs designed (Figure 6D), i.e., those best ranked markers with suitable size for PCR amplification (< ∼900 bp) (see further discussion in the next section). Hernández-Hernández et al. (2011, 2014), as well as Bárcenas et al. (2011), although recovering the same three tribes sampling few genera, did not resolve the relationships among them, while Nyffeler (2002) did not have sufficient taxon sampling to infer relationships within Opuntioideae. Thus, this recalcitrant relationship between the three tribes must be further investigated using more genealogies, such as nuclear, plastome and mitochondrial datasets.
Griffith and Porter (2009) previously tackled relationships within Opuntioideae using DNA sequence data with a comprehensive sampling, yet based only on nrITS and trnL-trnF data. Our results partially recovered their topology, with a “flat-stemmed” and a “terete-stemmed” clade, moderately equivalent to our Opuntieae and Cylindropuntieae tribes, respectively. However, many members of Tephrocacteae recovered here were nested within their “terete-stemmed” clade, such as Austrocylindropuntia, Cumulopuntia and Tephrocactus. Likewise, the Griffith and Porter (2009) topology revealed a grade of two clades (Pterocactus and Maihueniopsis), which were sister to the rest of Opuntioideae but that was not recovered in our study. However, as our study is still based on one sample per genus, future studies including a wider sampling should be carried out across the subfamily to further test the relationships here recovered and are currently underway (Majure et al., in preparation).
Tribe Opuntieae is the most diverse and widespread clade among Opuntioideae, consisting of seven accepted genera and around 230 species (Majure and Puente, 2014; Guerrero et al., 2019). Consolea Lem., an endemic tree-like cactus of the Caribbean Islands and neighboring areas (Majure et al., submitted), is sister to the rest of Opuntieae, which consists of two subclades: (i) one comprising Brasiliopuntia (K. Schum.) A. Berger + Tacinga Britton & Rose; and the other comprising (ii) Opuntia (L.) Mill. + [Miqueliopuntia Friè ex F. Ritter + (Salmonopuntia P.V. Heath + Tunilla D.R. Hunt & Iliff.)]. Previous analyses did not resolve this position for Consolea, mostly based on the lack of data/data type (Griffith and Porter, 2009) and/or outgroup taxon sampling (Majure et al., 2012; Majure and Puente, 2014). Likewise, the sister relationship of Opuntia with the (Salmonopuntia + Tunilla) + Miqueliopuntia clade had not been recovered in previous analyses (Majure et al., 2012).
Based on our plastome analysis, Cylindropuntieae and Tephrocacteae are sister tribes, comprised of five and six genera, respectively (Figure 6A). Cylindropuntieae are primarily represented by genera that occur in the western North American desert regions [Cylindropuntia (Engelm.) F.M. Knuth, Grusonia F. Rchb. & K. Schum. and Micropuntia Daston], which formed a well-supported subclade, but they also contain two genera that are found in tropical dry forest of Mexico/Northern Central America (Pereskiopsis Britton & Rose) and Tropical Dry Forest and Chaco of South America (Quiabentia Britton & Rose). Tribe Pereskiopsideae (Doweld, 1999), previous described to only accommodate the leafy Pereskiopsis, is nested within Cylindropuntieae and is redundant, and thus unnecessary. Deeper relationships within Cylindropuntieae were recently untangled using a phylogenomic approach and dense sampling, revealing biogeographic patterns as well as characters evolution (Majure et al., 2019). Our plastome phylogeny here revealed an identical phylogenetic pattern among genera (Figure 6A) of Majure et al. (2019), and equivalent to Bárcenas (2016).
Tephrocacteae is a South American clade adapted to diverse climatic conditions over a wide area of the southern Andes and adjacent lowlands (Ritz et al., 2012; Guerrero et al., 2019; Las Peñas et al., 2019). The tribe includes morphologically diverse species from geophytes and cushion-plants to dwarf shrubs, shrubs or small trees (Anderson, 2001); and probably geophytes and cushion-forming species evolved several times from shrubby-like precursors (Ritz et al., 2012). Tribes Austrocylindropuntieae and Pterocacteae (Wallace and Dickie, 2002) were described to circumscribe Austrocylindropuntia + Cumulopuntia and Pterocactus, respectively, and both are nested within the Tephrocacteae as amplified by Hunt (2011). So, as shown here, their use is mostly redundant. Although our plastome data recovered Maihueniopsis and Tephrocactus as sister to Pterocactus + (Austrocylindropuntia + Cumulopuntia), the phylogenetic topology among genera of the tribe are highly variable when using different datasets (Figures 6A–D). It is probable that increased taxon sampling may ameliorate this topological variability, as we still lack whole plastome data for the monospecific genus Punotia. Greater taxon and data sampling will be necessary to fully test these relationships.
Phylogenetically Informative Regions
Our plastome survey for phylogenetically informative markers revealed a list of potentially highly informative plastid markers for Sanger-based phylogenetic studies in Opuntioideae (Supplementary Table S3). The top 10 markers in our cpCDS dataset are: accD, ycf1, ndhD, petD, ccsA, clpP, rpoC1, rpoC2, including just one intron (the trnK intron comprising the matK gene – trnK/matK) and one intergenic spacer (psbE-rpl20) (Table 3). However, two of the better ranked markers (accD and ycf1) are putative pseudogenes and must be treated apart from traditional protein coding genes. The impact and utility of pseudogenes as markers for phylogenetic inferences must be further investigated (see below).
From the top 10 markers ranked in our list, just one (trnK/matK) has been used in more than one phylogenetic study in cacti (Nyffeler, 2002; Edwards et al., 2005; Korotkova et al., 2010; Arakaki et al., 2011; Bárcenas et al., 2011; Demaio et al., 2011; Hernández-Hernández et al., 2011, 2014; Ritz et al., 2012; Bárcenas, 2016; Vargas-Luna et al., 2018); while Majure et al. (2012) and Franck et al. (2013) used partial sequences of the ycf1 gene. The other top 10 markers have been previously used under phylogenomic approaches in cacti (Arakaki et al., 2011; Majure et al., 2019).
Although the majority of the top 10 markers here reported have not been used in phylogenetic studies of cacti, the relationship of several other groups has been inferred with some of these markers. For example, the accD gene, combined with other plastid regions including rpoC1, was employed for phylogenetic inference of Crocus (Iridaceae), Coptis (Ranunculaceae) and Orchidaceae genera (Petersen et al., 2008; Guo et al., 2012; He et al., 2014). However, accD intergenic spacers, such as rbcL-accD and accD-psaI, have been much more widely used across disparate groups (Barfuss et al., 2005; Miikeda et al., 2006; Reginato et al., 2010; Sun et al., 2012; Michelangeli et al., 2013). The ycf1 gene appears to be moderately used (Gernandt et al., 2009; Guo et al., 2012; Majure et al., 2012; Shi et al., 2013; Whitten et al., 2013; Dastpak et al., 2018), and increasingly reported to be a useful marker in phylogenetic inferences (Neubig et al., 2009; Neubig and Abbott, 2010; Dong et al., 2012; Thomson et al., 2018), and the most promising plastid DNA barcode of land plants (Dong et al., 2015). The petD intron has been used (Löhne et al., 2007; Worberg et al., 2007; Borsch et al., 2009; Scataglini et al., 2014), but in our analysis the entire gene was used (exon + intron) showing phylogenetic utility. The ccsA gene seems to be underexplored as a phylogenetic marker (Marx et al., 2010; Peterson et al., 2012) but was already suggested as convenient for phylogenetic inferences (Logacheva et al., 2007). The rpoC1 and rpoC2 genes have been occasionally used together as markers (Liston and Wheeler, 1994; Kulshreshtha et al., 2004) or combined with other markers (Downie et al., 2000; GPWS, 2001; Zhang et al., 2011; Guo et al., 2012) yielding satisfactory results. The rpoC2 gene was recently found as the best performing marker to recover, with high levels of concordance, the “accepted tree” of the angiosperm phylogeny (Walker et al., 2019). The ndhD gene seems to be scarcely used for phylogenetic inference (Panero and Funk, 2002), while the intergenic spacer of psbE-rpl20 genes has never been used individually to our knowledge.
Eight of the top 10 markers are more than 900 bp, indicating that longer genes are superior for phylogenetic reconstruction, as previous suggested by Walker et al. (2019), although they may require internal primer designing for complete Sanger sequencing. A list of the top 10 markers with less than 900 bp is reported (Supplementary Table S4), and primer pair design for the top five is provided in Table 4. Our phylogenetic inference from the top five markers concatenated, which had primers designed (Figure 6D) recovered a conflicting topology compared with the plastome tree (Figure 6A). The topology with Cylindropuntieae as sister to Tephrocacteae + Opuntieae has also been recovered based on transcriptome data (Walker et al., 2018; Wang et al., 2019). Curiously, we obtained this same topology, although not strongly supported, using the top 10 marker dataset concatenated, when stripping the two pseudogenes accD and ycf1 (Supplementary Figure S3), suggesting that functional constraints of these pseudogenes may influence the underlying topology.
Our top five markers contained intergenic spacers, which influence our alignment, wherein the incorporation of gaps is necessary. Duvall et al. (2020) found that as gaps increased in their alignment of plastomes across Poaceae, differing topologies were increasingly supported. This may also play a role in the incongruent topologies recovered in our analyses. Perhaps a higher level of homoplasy across datasets including gaps may reduce their suitability for resolving deep phylogenetic relationships, however, those same regions (i.e., intergenic spacers) are likely more appropriate for resolving species relationships among closely-related species (Shaw et al., 2005). Likewise, selective pressures on the genes in both our reduced 10 marker datasets, as well as in previously published transcriptome data (Walker et al., 2018), may likewise influence topology. Homoplasy in these reduced datasets may also be a factor leading to conflicting topologies. More research should be focused on the level of utility of specific gene regions (e.g., coding genes, intergenic spacers) across clades.
Chloroplast markers have been used for testing evolutionary relationships among plants for the past 30 years (Gitzendanner et al., 2018). While the assumption that these markers are evolving as a single unit without recombination, routine analyses have used concatenated data producing highly supported phylogenies that have been underlying the current classification of angiosperms (APG, 2016). However, as here reported, no marker as a single unit (gene tree) recovered the same topology of the plastome inference (concatenated tree), and even within the top 10 markers listed, some showed high values of discordance (Table 3, Supplementary Table S4, and Supplementary Figures S1, S2). Such results discourage and call attention to phylogenetic approaches based on one or few markers. While the full chloroplast sequences showed to be the most robust dataset to resolve relationships within Opuntioideae, phylogenies from the top 10 and 5 markers concatenated resolved many relationships with high bootstrap values and few nodes with low support (Figures 6B,C). Although we cannot test how effective these datasets would work in determining closely related species relationships, based on our limited taxon sampling here, it is significant that these smaller datasets resolve relationships among these clades and genera that have not been resolved previously using a similar number of loci (e.g., Hernández-Hernández et al., 2011). Thus, we would expect that using these more highly variable loci, although few, should greatly increase resolution across many subclades in Cactaceae. We also encourage their use across subclades within Cactoideae to test their broader utility.
Recent studies have explored gene tree conflict in plastome-inferred phylogenies and incongruence between gene trees and species trees in plastid genes (Gonçalves et al., 2019; Walker et al., 2019). Gonçalves et al. (2019) emphasized the importance of considering variation in phylogenetic signal across plastid genes and the exploration of plastome data to increase accuracy of estimating relationships; they also revealed that phylogenies inferred with multispecies coalescent (MSC) methods are accurate with plastome matrices and should be considered in future phylogenomic investigations. Walker et al. (2019) highlighted that most plastid genes are largely uninformative and are unlikely to misguide plant systematics. However, the concatenating of plastid genes without some level of scrutiny can mislead branch length estimation (Walker et al., 2019). The causes of discordant topologies across gene trees from chloroplast genome still needs to be better investigated. The main explanations include systematic errors (e.g., poor modeling, stochastic events) or more biologically meaningful processes, such as heteroplasmic recombination that have been invoked to explain discordance in disparate plant clades (Huang et al., 2001; Marshall et al., 2001; Bouillé et al., 2011; Walker et al., 2019).
Conclusion
Chloroplast genomes have long been considered conserved among land plants, but recent generation of 1000s of plastomes through NGS has illuminated that this is not always the case. Cactaceae are no exception to variation that has been observed in other clades. Previous plastomes of cacti have shown to have lost one copy of the inverted repeat regions and several genes of the ndh gene suite, as well as to possess divergent inverted repeat regions and the smallest chloroplast genome known for an obligately photosynthetic angiosperm. We showed that the Opuntia quimilo plastome also presents deviations of canonical angiosperm plastomes with an expansion of the LSC incorporating genes that are typically in the IRs, a reduction of the SSC translocating some common genes of the SSC into the IR region, and one massive translocation with an inversion of a block of genes in the LSC. Strikingly different from other cacti, two copies of the inverted repeat region were recovered in the Opuntia quimilo plastome. Our reference-guided assemblies across Opuntioideae allowed us to infer putative independent losses of some ndh genes across disparate taxa of the subfamily. We did not find synapomorphic plastome features within Opuntioideae clades, thus, we hypothesize that putative rearrangements across the subfamily are from homoplasious events. Further analyses should be carried out to address how ecological drivers and morphological traits of cacti may be related with positive selection of genes and the common rearrangements in chloroplast genomes that have been reported in the family. Phylogenetic analyses of chloroplast genome sequences strongly support Opuntioideae and its three tribes: Opuntieae, Cylindropuntieae, and Tephrocacteae. As computational and budget limitations are still a bottleneck to deal with high throughput data, especially in developing countries, a list of highly informative plastid markers is presented for future use, and several top ranked markers have not been used in phylogenetic studies of cacti. However, conflicting topologies were recovered among major clades when exploring different assemblies of markers, revealing that gene tree discordance among markers must be carefully considered while inferring phylogenies in this remarkable group of plants, especially considering the occurrence of putative pseudogenes. Even so, topological incongruences may actually signal deeper phylogenetic patterns underlying biologically relevant evolutionary history and should be further explored using both nuclear and plastome datasets.
Data Availability Statement
The datasets generated for this study can be found in the GenBank: MN114084 and MT369928–MT369946.
Author Contributions
MK: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing – original draft, writing – review and editing, visualization, project administration, and funding acquisition. MR: methodology, software, validation, formal analysis, and writing – review and editing. TS-C: writing – review and editing. LM: methodology, validation, investigation, resources, data curation, and writing – review and editing, visualization, supervision, project administration, and funding acquisition.
Funding
Partial funding for this project was provided by the Desert Botanical Garden (AZ, United States), start-up funds from the University of Florida to LM, the National Science Foundation (DEB 1735604), and the “Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil” (CAPES) – Finance Code 001.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Grant T. Godden (FLMNH) for providing valuable comments about chloroplast assemblers, Urs Eggli (Sukkulenten-Sammlung Zürich and Alexandra Elbakyan) for provide important bibliography, and Ricardo Reis and Josimar Külkamp (Jardim Botânico do Rio de Janeiro, Brazil) for providing sample. MK is grateful to the American Society of Plant Taxonomists (ASPT), Cactus and Succulent Society of America (CSSA), International Association for Plant Taxonomy (IAPT) and IDEA WILD for supporting part of the research reported here, the Brazilian National Council for Scientific and Technological Development (Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq, process number 141523/2017-4) for his Ph.D. scholarship, and the PDSE/CAPES for supporting his period as Visiting Researcher at the Florida Museum of Natural History (FLMNH, UF, United States, Process 88881.186882/2018-01). We also thank two reviewers for providing valuable comments that improved this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00729/full#supplementary-material
Footnotes
References
Abadi, S., Azouri, D., Pupko, T., and Mayrose, I. (2019). Model selection may not be a mandatory step for phylogeny reconstruction. Nat. Commun. 10, 8822. doi: 10.1038/s41467-019-08822-w
Abdullah, M. F., Shahzadi, I., Waseem, S., Mirza, B., Ahmed, I., and Waheed, M. T. (2019). Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): comparative analyses and identification of mutational hotspots. Genomics 112:10. doi: 10.1016/j.ygeno.2019.04.010
APG (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181:12385. doi: 10.1111/boj.12385
Arakaki, M., Christin, P.-A., Nyffeler, R., Lendel, A., Eggli, U., Ogburn, R. M., et al. (2011). Contemporaneous and recent radiations of the world’s major succulent plant lineages. Proc. Natl. Acad. Sci. U.S.A. 108, 8379–8384. doi: 10.1073/pnas.1100628108
Bárcenas, R. T. (2016). A molecular phylogenetic approach to the systematics of Cylindropuntieae (Opuntioideae, Cactaceae). Cladistics 32:359. doi: 10.1111/cla.12135
Bárcenas, R. T., Yesson, C., and Hawkins, J. A. (2011). Molecular systematics of the Cactaceae. Cladistics 27, 470–489. doi: 10.1111/j.1096-0031.2011.00350.x
Barfuss, M. H. J., Samuel, R., Till, W., and Stuessy, T. F. (2005). Phylogenetic relationships in subfamily Tillandsioideae (Bromeliaceae) based on DNA sequence data from seven plastid regions. Am. J. Bot. 92, 337–351. doi: 10.3732/ajb.92.2.337
Bedoya, A. M., Ruhfel, B. R., Philbrick, C. T., Madriñán, S., Bove, C. P., Mesterházy, A., et al. (2019). Plastid genomes of five species of riverweeds (Podostemaceae): structural organization and comparative analysis in malpighiales. Front. Plant Sci. 10:35. doi: 10.3389/fpls.2019.01035
Borsch, T., Korotkova, N., Raus, T., Lobin, W., and Löhne, C. (2009). The petD group II intron as a species level marker: utility for tree inference and species identification in the diverse genus Campanula (Campanulaceae). Willdenowia 39, 7–33. doi: 10.3372/wi.39.39101
Bouillé, M., Senneville, S., and Bousquet, J. (2011). Discordant mtDNA and cpDNA phylogenies indicate geographic speciation and reticulation as driving factors for the diversification of the genus Picea. Tree Genet. Genomes 7, 469–484. doi: 10.1007/s11295-010-0349-z
Braukmann, T. W. A., Kuzmina, M., and Stefanoviæ, S. (2009). Loss of all plastid ndh genes in Gnetales and conifers: extent and evolutionary significance for the seed plant phylogeny. Curr. Genet. 55, 323–337. doi: 10.1007/s00294-009-0249-7
Brochmann, C., Brysting, A. K., Alsos, I. G., Borgen, L., Grundt, H. H., Scheen, A.-C., et al. (2004). Polyploidy in arctic plants. Biol. J. Linn. Soc. 82, 521–536. doi: 10.1111/j.1095-8312.2004.00337.x
Brudno, M., Malde, S., Poliakov, A., Do, C. B., Couronne, O., Dubchak, I., et al. (2003). Glocal alignment: finding rearrangements during alignment. Bioinformatics 19, i54–i62. doi: 10.1093/bioinformatics/btg1005
Bushnell, B. (2016). BBMap Short Read Aligner, and Other Bioinformatic Tools. Available online at: https://sourceforge.net/projects/bbmap/ (accessed June 4, 2019).
Cai, Z., Guisinger, M., Kim, H.-G., Ruck, E., Blazier, J. C., McMurtry, V., et al. (2008). Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. J. Mol. Evol. 67, 696–704. doi: 10.1007/s00239-008-9180-7
Carbonell-Caballero, J., Alonso, R., Ibañez, V., Terol, J., Talon, M., and Dopazo, J. (2015). A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the Genus Citrus. Mol. Biol. Evol. 32, 2015–2035. doi: 10.1093/molbev/msv082
Cauz-Santos, L. A., Munhoz, C. F., Rodde, N., Cauet, S., Santos, A. A., Penha, H. A., et al. (2017). The chloroplast genome of Passiflora edulis (Passifloraceae) assembled from long sequence reads: structural organization and phylogenomic studies in malpighiales. Front. Plant Sci. 8:334. doi: 10.3389/fpls.2017.00334
Cho, W.-B., Choi, B.-H., Kim, J.-H., Lee, D.-H., and Lee, J.-H. (2018). Complete plastome sequencing reveals an extremely diminished SSC region in hemiparasitic Pedicularis ishidoyana (Orobanchaceae). Ann. Bot. Fenn. 55, 171–183. doi: 10.5735/085.055.0122
Chumley, T. W., Palmer, J. D., Mower, J. P., Fourcade, H. M., Calie, P. J., Boore, J. L., et al. (2006). The complete chloroplast genome sequence of Pelargonium x hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. doi: 10.1093/molbev/msl089
Cui, Y., Zhou, J., Chen, X., Xu, Z., Wang, Y., Sun, W., et al. (2019). Complete chloroplast genome and comparative analysis of three Lycium (Solanaceae) species with medicinal and edible properties. Gene Rep. 17:464. doi: 10.1016/j.genrep.2019.100464
Daniell, H., Lin, C.-S., Yu, M., and Chang, W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Dastpak, A., Osaloo, S. K., Maassoumi, A. A., and Safar, K. N. (2018). Molecular phylogeny of astragalus sect. Ammodendron (Fabaceae) inferred from chloroplast ycf1 gene. Ann. Bot. Fenn. 55, 75–82. doi: 10.5735/085.055.0108
De Pamphilis, C. W., and Palmer, J. D. (1990). Loss of photosynthetic and chlororespiratory genes from the plastid genome of a parasitic flowering plant. Nature 348, 337–339. doi: 10.1038/348337a0
Demaio, P. H., Barfuss, M. H. J., Kiesling, R., Till, W., and Chiapella, J. O. (2011). Molecular phylogeny of Gymnocalycium (Cactaceae): assessment of alternative infrageneric systems, a new subgenus, and trends in the evolution of the genus. Am. J. Bot. 98, 1841–1854. doi: 10.3732/ajb.1100054
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:E18. doi: 10.1093/nar/gkw955
Dong, W., Liu, J., Yu, J., Wang, L., and Zhou, S. (2012). Highly Variable Chloroplast Markers for Evaluating Plant Phylogeny at Low Taxonomic Levels and for DNA Barcoding. PLoS One 7:e35071. doi: 10.1371/journal.pone.0035071
Dong, W., Xu, C., Cheng, T., and Zhou, S. (2013). Complete Chloroplast Genome of Sedum sarmentosum and chloroplast genome evolution in saxifragales. PLoS One 8:e77965. doi: 10.1371/journal.pone.0077965
Dong, W., Xu, C., Li, C., Sun, J., Zuo, Y., Shi, S., et al. (2015). Ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5:8348. doi: 10.1038/srep08348
Dong, W.-L., Wang, R.-N., Zhang, N.-Y., Fan, W.-B., Fang, M.-F., and Li, Z.-H. (2018). Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution. Int. J. Mol. Sci. 19:716. doi: 10.3390/ijms19030716
Doweld, A. B. (1999). Tribal taxonomy of Pereskioideae and Opuntioideae (Cactaceae). Succulents 1, 25–26.
Downie, S. R., and Jansen, R. K. (2015). A Comparative Analysis of Whole Plastid Genomes from the Apiales: Expansion and Contraction of the Inverted Repeat, Mitochondrial to Plastid Transfer of DNA, and Identification of Highly Divergent Noncoding Regions. Syst. Bot. 40, 336–351. doi: 10.1600/036364415X686620
Downie, S. R., Katz–Downie, D. S., and Watson, M. F. (2000). A phylogeny of the flowering plant family Apiaceae based on chloroplast DNA rpl16 and rpoC1 intron sequences: towards a suprageneric classification of subfamily Apioideae. Am. J. Bot. 87, 273–292. doi: 10.2307/2656915
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Drescher, A., Ruf, S., Calsa, T., Carrer, H., and Bock, R. (2000). The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 22, 97–104. doi: 10.1046/j.1365-313x.2000.00722.x
Duvall, M. R., Burke, S. V., and Clark, D. C. (2020). Plastome phylogenomics of Poaceae: alternate topologies depend on alignment gaps. Bot. J. Linn. Soc. 192, 9–20. doi: 10.1093/botlinnean/boz060
Edwards, E. J. (2019). Evolutionary trajectories, accessibility and other metaphors: the case of C4 and CAM photosynthesis. New Phytol. 223, 1742–1755. doi: 10.1111/nph.15851
Edwards, E. J., Nyffeler, R., and Donoghue, M. J. (2005). Basal cactus phylogeny: implications of Pereskia (Cactaceae) paraphyly for the transition to the cactus life form. Am. J. Bot. 92, 1177–1188. doi: 10.3732/ajb.92.7.1177
Fajardo, D., Senalik, D., Ames, M., Zhu, H., Steffan, S. A., Harbut, R., et al. (2013). Complete plastid genome sequence of Vaccinium macrocarpon: structure, gene content, and rearrangements revealed by next generation sequencing. Tree Gen. Genomes 9, 489–498. doi: 10.1007/s11295-012-0573-9
Fonseca, L. H. M., and Lohmann, L. G. (2017). Plastome Rearrangements in the “Adenocalymma-Neojobertia” Clade (Bignonieae, Bignoniaceae) and Its Phylogenetic Implications. Front. Plant Sci. 8:1875. doi: 10.3389/fpls.2017.01875
Franck, A. R., Cochrane, B. J., and Garey, J. R. (2013). Phylogeny, Biogeography, and Infrageneric Classification of Harrisia (Cactaceae). Syst. Bot. 38, 210–223. doi: 10.1600/036364413X662105
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Gardner, A. G., Sessa, E. B., Michener, P., Johnson, E., Shepherd, K. A., Howarth, D. G., et al. (2016). Utilizing next-generation sequencing to resolve the backbone of the Core Goodeniaceae and inform future taxonomic and floral form studies. Mol. Phylogenet. Evol. 94, 605–617. doi: 10.1016/j.ympev.2015.10.003
Gernandt, D. S., Hernández-León, S., Salgado-Hernández, E., and Rosa, J. A. P. (2009). Phylogenetic Relationships of Pinus Subsection Ponderosae Inferred from Rapidly Evolving cpDNA Regions. Syst. Bot. 34, 481–491. doi: 10.1600/036364409789271290
Gitzendanner, M. A., Soltis, P. S., Yi, T.-S., Li, D.-Z., and Soltis, D. E. (2018). “Plastome Phylogenetics: 30 Years of Inferences Into Plant Evolution,” in Advances in Botanical Research Plastid Genome Evolution, eds S.-M. Chaw and R. K. Jansen (Cambridge, MA: Academic Press), 293–313. doi: 10.1016/bs.abr.2017.11.016
Givnish, T. J., Spalink, D., Ames, M., Lyon, S. P., Hunter, S. J., Zuluaga, A., et al. (2015). Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proc. R. Soc. Lond. B Biol. Sci. 282, 20151553. doi: 10.1098/rspb.2015.1553
Gonçalves, D. J. P., Simpson, B. B., Ortiz, E. M., Shimizu, G. H., and Jansen, R. K. (2019). Incongruence between gene trees and species trees and phylogenetic signal variation in plastid genes. Mol. Phylogenet. Evol. 138, 219–232. doi: 10.1016/j.ympev.2019.05.022
GPWS (2001). Phylogeny and Subfamilial Classification of the Grasses (Poaceae). Ann. Mo. Bot. Gard. 88, 373–457. doi: 10.2307/3298585
Graham, S. W., Lam, V. K. Y., and Merckx, V. S. F. T. (2017). Plastomes on the edge: the evolutionary breakdown of mycoheterotroph plastid genomes. New Phytol. 214, 48–55. doi: 10.1111/nph.14398
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Griffith, M. P., and Porter, J. M. (2009). Phylogeny of Opuntioideae (Cactaceae). Int. J. Plant Sci. 170, 107–116. doi: 10.1086/593048
Guerrero, P. C., Majure, L. C., Cornejo-Romero, A., and Hernández-Hernández, T. (2019). Phylogenetic Relationships and Evolutionary Trends in the Cactus Family. J. Hered 110, 4–21. doi: 10.1093/jhered/esy064
Guo, Y.-Y., Luo, Y.-B., Liu, Z.-J., and Wang, X.-Q. (2012). Evolution and biogeography of the slipper orchids: eocene vicariance of the conduplicate genera in the old and new world tropics. PLoS One 7:e38788. doi: 10.1371/journal.pone.0038788
Haberle, R. C., Fourcade, H. M., Boore, J. L., and Jansen, R. K. (2008). Extensive Rearrangements in the Chloroplast Genome of Trachelium caeruleum Are Associated with Repeats and tRNA Genes. J. Mol. Evol. 66, 350–361. doi: 10.1007/s00239-008-9086-4
Harris, M. E., Meyer, G., Vandergon, T., and Vandergon, V. O. (2013). Loss of the Acetyl-CoA Carboxylase (accD) Gene in Poales. Plant Mol. Biol. Rep. 31:21. doi: 10.1007/s11105-012-0461-3
He, Y., Hou, P., Fan, G., Arain, S., and Peng, C. (2014). Comprehensive analyses of molecular phylogeny and main alkaloids for Coptis (Ranunculaceae) species identification. Biochem. Syst. Ecol 56, 88–94. doi: 10.1016/j.bse.2014.05.002
Hernández-Hernández, T., Hernández, H. M., De-Nova, J. A., Puente, R., Eguiarte, L. E., and Magallón, S. (2011). Phylogenetic relationships and evolution of growth form in Cactaceae (Caryophyllales, Eudicotyledoneae). Am. J. Bot. 98, 44–61. doi: 10.3732/ajb.1000129
Hernández-Hernández, T., Brown, J. W., Schlumpberger, B. O., Eguiarte, L. E., and Magallón, S. (2014). Beyond aridification: multiple explanations for the elevated diversification of cacti in the New World Succulent Biome. New Phytol. 202, 1382–1397. doi: 10.1111/nph.12752
Hu, S., Sablok, G., Wang, B., Qu, D., Barbaro, E., Viola, R., et al. (2015). Plastome organization and evolution of chloroplast genes in Cardamine species adapted to contrasting habitats. BMC Genomics 16:306. doi: 10.1186/s12864-015-1498-0
Huang, S., Chiang, Y. C., Schaal, B. A., Chou, C. H., and Chiang, T. Y. (2001). Organelle DNA phylogeography of Cycas taitungensis, a relict species in Taiwan. Mol. Ecol. 10, 2669–2681. doi: 10.1046/j.0962-1083.2001.01395.x
Hunt, D. (2011). Classification of the ‘cylindroid’ opuntias of South America. Cactaceae Systematics Initiatives 25, 5–29.
Iles, W. J. D., Smith, S. Y., and Graham, S. W. (2013). “A well-supported phylogenetic framework for the monocot order Alismatales reveals multiple losses of the plastid NADH dehydrogenase complex and a strong long-branch effect,” in Early Events in Monocot Evolution, eds W. Paul and S. J. Mayo (Cambridge: Cambridge University Press), doi: 10.1017/CBO9781139002950.002
Jansen, R. K., and Ruhlman, T. A. (2012). “Plastid Genomes of Seed Plants,” in Genomics of Chloroplasts and Mitochondria Advances in Photosynthesis and Respiration, eds R. Bock and V. Knoop (Dordrecht: Springer Netherlands), 103–126. doi: 10.1007/978-94-007-2920-9_5
Jansen, R. K., Saski, C., Lee, S.-B., Hansen, A. K., and Daniell, H. (2011). Complete plastid genome sequences of three Rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 28, 835–847. doi: 10.1093/molbev/msq261
Jin, D.-P., Choi, I.-S., and Choi, B.-H. (2019). Plastid genome evolution in tribe Desmodieae (Fabaceae: Papilionoideae). PLoS One 14:e0218743. doi: 10.1371/journal.pone.0218743
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kikuchi, S., Bédard, J., Hirano, M., Hirabayashi, Y., Oishi, M., Imai, M., et al. (2013). Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 339, 571–574. doi: 10.1126/science.1229262
Kim, H. T., Chung, M. G., and Kim, K.-J. (2014). Chloroplast genome evolution in early diverged leptosporangiate ferns. Mol. Cells 37, 372–382. doi: 10.14348/molcells.2014.2296
Kim, H. T., Kim, J. S., Moore, M. J., Neubig, K. M., Williams, N. H., Whitten, W. M., et al. (2015). Seven New Complete Plastome Sequences Reveal Rampant Independent Loss of the ndh Gene Family across Orchids and Associated Instability of the Inverted Repeat/Small Single-Copy Region Boundaries. PLoS One 10:e0142215. doi: 10.1371/journal.pone.0142215
Kode, V., Mudd, E. A., Iamtham, S., and Day, A. (2005). The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 44, 237–244. doi: 10.1111/j.1365-313X.2005.02533.x
Korotkova, N., Zabel, L., Quandt, D., and Barthlott, W. (2010). A phylogenetic analysis of Pfeiffera and the reinstatement of Lymanbensonia as an independently evolved lineage of epiphytic Cactaceae within a new tribe Lymanbensonieae. Willdenowia 40, 151–172. doi: 10.3372/wi.40.40201
Kulshreshtha, S., Creamer, R., and Sterling, T. M. (2004). Phylogenetic relationships among New Mexico Astragalus mollissimus varieties and Oxytropis species by restriction fragment analysis. Weed Sci. 52, 984–988. doi: 10.1614/WS-03-143R1
Kuroda, H., and Maliga, P. (2003). The plastid clpP1 protease gene is essential for plant development. Nature 425, 86–89. doi: 10.1038/nature01909
Landrum, J. V. (2002). Four Succulent Families and 40 Million Years of Evolution and Adaptation to Xeric Environments: What Can Stem and Leaf Anatomical Characters Tell Us about Their Phylogeny? Taxon 51, 463–473. doi: 10.2307/1554859
Las Peñas, M. L., Kiesling, R., and Bernardello, G. (2019). Phylogenetic reconstruction of the genus Tephrocactus (Cactaceae) based on molecular, morphological, and cytogenetical data. Taxon 68, 714–730. doi: 10.1002/tax.12092
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Li, Y., Zhou, J., Chen, X., Cui, Y., Xu, Z., Li, Y., et al. (2017). Gene losses and partial deletion of small single-copy regions of the chloroplast genomes of two hemiparasitic Taxillus species. Sci. Rep. 7:12834. doi: 10.1038/s41598-017-13401-4
Lin, C.-S., Chen, J. J. W., Chiu, C.-C., Hsiao, H. C. W., Yang, C.-J., Jin, X.-H., et al. (2017). Concomitant loss of NDH complex-related genes within chloroplast and nuclear genomes in some orchids. Plant J. 90, 13525. doi: 10.1111/tpj.13525
Liston, A., and Wheeler, J. A. (1994). The phylogenetic position of the genus Astragalus (Fabaceae): Evidence from the chloroplast genes rpoC1 and rpoC2. Biochem. Syst. Ecol. 22, 377–388. doi: 10.1016/0305-1978(94)90028-0
Liu, T.-J., Zhang, C.-Y., Yan, H.-F., Zhang, L., Ge, X.-J., and Hao, G. (2016). Complete plastid genome sequence of Primula sinensis (Primulaceae): structure comparison, sequence variation and evidence for accD transfer to nucleus. PeerJ 4:e2101. doi: 10.7717/peerj.2101
Liu, X., Yang, H., Zhao, J., Zhou, B., Li, T., and Xiang, B. (2017). The complete chloroplast genome sequence of the folk medicinal and vegetable plant purslane (Portulaca oleracea L.). J. Hortic. Sci. Biotechnol. 93:1389308. doi: 10.1080/14620316.2017.1389308
Logacheva, M. D., Penin, A. A., Samigullin, T. H., Vallejo-Roman, C. M., and Antonov, A. S. (2007). Phylogeny of flowering plants by the chloroplast genome sequences: in search of a “lucky gene.” Biochemistry Moscow 72, 1324–1330. doi: 10.1134/S0006297907120061
Logacheva, M. D., Schelkunov, M. I., Nuraliev, M. S., Samigullin, T. H., and Penin, A. A. (2014). The plastid genome of mycoheterotrophic monocot Petrosavia stellaris exhibits both gene losses and multiple rearrangements. Genome Biol. Evol. 6, 238–246. doi: 10.1093/gbe/evu001
Löhne, C., Borsch, T., and Wiersema, J. H. (2007). Phylogenetic analysis of Nymphaeales using fast-evolving and noncoding chloroplast markers. Bot. J. Linn. Soc. 154, 141–163. doi: 10.1111/j.1095-8339.2007.00659.x
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Luo, J., Hou, B.-W., Niu, Z.-T., Liu, W., Xue, Q.-Y., and Ding, X.-Y. (2014). Comparative chloroplast genomes of photosynthetic orchids: insights into evolution of the Orchidaceae and development of molecular markers for phylogenetic applications. PLoS One 9:e99016. doi: 10.1371/journal.pone.0099016
Ma, P.-F., Zhang, Y.-X., Zeng, C.-X., Guo, Z.-H., and Li, D.-Z. (2014). Chloroplast Phylogenomic Analyses Resolve Deep-Level Relationships of an Intractable Bamboo Tribe Arundinarieae (Poaceae). Syst. Biol. 63, 933–950. doi: 10.1093/sysbio/syu054
Machado, L., de, O., Vieira, L., Do, N., Stefenon, V. M., de Oliveira, P. F., et al. (2017). Phylogenomic relationship of feijoa (Acca sellowiana (O.Berg) Burret) with other Myrtaceae based on complete chloroplast genome sequences. Genetica 145, 1–12. doi: 10.1007/s10709-017-9954-1
Magee, A. M., Aspinall, S., Rice, D. W., Cusack, B. P., Sémon, M., Perry, A. S., et al. (2010). Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 20, 1700–1710. doi: 10.1101/gr.111955.110
Majure, L. C., Baker, M. A., Cloud-Hughes, M., Salywon, A., and Neubig, K. M. (2019). Phylogenomics in Cactaceae: A case study using the chollas sensu lato (Cylindropuntieae, Opuntioideae) reveals a common pattern out of the Chihuahuan and Sonoran deserts. Am. J. Bot. 106, 1327–1345. doi: 10.1002/ajb2.1364
Majure, L. C., and Puente, R. (2014). Phylogenetic relationships and morphological evolution in Opuntia s. str. and closely related members of tribe Opuntieae. Succul. Plant Res. 8, 9–30.
Majure, L. C., Puente, R., Griffith, M. P., Judd, W. S., Soltis, P. S., and Soltis, D. E. (2012). Phylogeny of Opuntia s.s. (Cactaceae): clade delineation, geographic origins, and reticulate evolution. Am. J. Bot. 99, 847–864. doi: 10.3732/ajb.1100375
Marshall, H. D., Newton, C., and Ritland, K. (2001). Sequence-repeat polymorphisms exhibit the signature of recombination in lodgepole pine chloroplast DNA. Mol. Biol. Evol. 18, 2136–2138. doi: 10.1093/oxfordjournals.molbev.a003757
Marx, H. E., O’Leary, N., Yuan, Y.-W., Lu–Irving, P., Tank, D. C., Múlgura, M. E., et al. (2010). A molecular phylogeny and classification of Verbenaceae. Am. J. Bot. 97, 1647–1663. doi: 10.3732/ajb.1000144
Mauseth, J. D. (1999). Anatomical adaptations to xeric conditions in Maihuenia (Cactaceae), a relictual, leaf-bearing cactus. J. Plant. Res. 112, 307–315. doi: 10.1007/PL00013886
McCoy, S. R., Kuehl, J. V., Boore, J. L., and Raubeson, L. A. (2008). The complete plastid genome sequence of Welwitschia mirabilis: an unusually compact plastome with accelerated divergence rates. BMC Evol. Biol. 8:130. doi: 10.1186/1471-2148-8-130
Michelangeli, F. A., Guimaraes, P. J. F., Penneys, D. S., Almeda, F., and Kriebel, R. (2013). Phylogenetic relationships and distribution of New World Melastomeae (Melastomataceae). Bot. J. Linn. Soc. 171, 38–60. doi: 10.1111/j.1095-8339.2012.01295.x
Miikeda, O., Kita, K., Handa, T., and Yukawa, T. (2006). Phylogenetic relationships of Clematis (Ranunculaceae) based on chloroplast and nuclear DNA sequences. Bot. J. Linn. Soc. 152, 153–168. doi: 10.1111/j.1095-8339.2006.00551.x
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. New Orleans, LA: Gateway Computing Environments Workshop (GCE), 1–8. doi: 10.1109/GCE.2010.5676129
Mooney, H. A., Gulmon, S. L., and Weisser, P. J. (1977). Environmental adaptations of the atacaman desert cactus Copiapoa haseltoniana. Flora 166, 117–124. doi: 10.1016/S0367-2530(17)32124-2
Mower, J. P., and Vickrey, T. L. (2018). “Structural diversity among plastid genomes of land plants,” in Advances in Botanical Research Plastid Genome Evolution, eds S.-M. Chaw and R. K. Jansen (Cambridge, MA: Academic Press), 263–292. doi: 10.1016/bs.abr.2017.11.013
Neubig, K., Whitten, M., Abbott, R., Elliot, S., Soltis, D., Soltis, P., et al. (2014). Variables affecting DNA Preservation in Archival Plant Specimens. DNA Banking for the 21st Century. Available online at: https://www.idigbio.org/sites/default/files/sites/default/files/Neubig_et_al_2014_.pdf (accessed June 4, 2019).
Neubig, K. M., and Abbott, J. R. (2010). Primer development for the plastid region ycf1 in Annonaceae and other magnoliids. Am. J. Bot. 97, e52–e55. doi: 10.3732/ajb.1000128
Neubig, K. M., Whitten, W. M., Carlsward, B. S., Blanco, M. A., Endara, L., Williams, N. H., et al. (2009). Phylogenetic utility of ycf1 in orchids: a plastid gene more variable than matK. Plant Syst. Evol. 277, 75–84. doi: 10.1007/s00606-008-0105-0
Nyffeler, R. (2002). Phylogenetic relationships in the cactus family (Cactaceae) based on evidence from trnK/matK and trnL-trnF sequences. Am. J. Bot. 89, 312–326. doi: 10.3732/ajb.89.2.312
Oliver, M. J., Murdock, A. G., Mishler, B. D., Kuehl, J. V., Boore, J. L., Mandoli, D. F., et al. (2010). Chloroplast genome sequence of the moss Tortula ruralis: gene content, polymorphism, and structural arrangement relative to other green plant chloroplast genomes. BMC Genomics 11:143. doi: 10.1186/1471-2164-11-143
Palmer, J. D., Jansen, R. K., Michaels, H. J., Chase, M. W., and Manhart, J. R. (1988). Chloroplast DNA Variation and Plant Phylogeny. Ann. Mo. Bot. Gard. 75, 1180–1206. doi: 10.2307/2399279
Panero, J. L., and Funk, V. A. (2002). Toward a phylogenetic subfamilial classification for the Compositae (Asteraceae). Proc. Biol. Soc. Wash. 115, 909–922.
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Park, S., An, B., and Park, S. (2018). Reconfiguration of the plastid genome in Lamprocapnos spectabilis: IR boundary shifting, inversion, and intraspecific variation. Sci. Rep. 8:13568. doi: 10.1038/s41598-018-31938-w
Park, S., Ruhlman, T. A., Weng, M.-L., Hajrah, N. H., Sabir, J. S. M., and Jansen, R. K. (2017). Contrasting patterns of nucleotide substitution rates provide insight into dynamic evolution of plastid and mitochondrial genomes of Geranium. Genome Biol. Evol. 9, 1766–1780. doi: 10.1093/gbe/evx124
Parker, N., Wang, Y., and Meinke, D. (2014). Natural variation in sensitivity to a loss of chloroplast translation in Arabidopsis. Plant Physiol. 166, 2013–2027. doi: 10.1104/pp.114.249052
Peredo, E. L., King, U. M., and Les, D. H. (2013). The plastid genome of Najas flexilis: adaptation to submersed environments is accompanied by the complete loss of the NDH complex in an aquatic angiosperm. PLoS One 8:e68591. doi: 10.1371/journal.pone.0068591
Perry, A. S., and Wolfe, K. H. (2002). Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 55, 501–508. doi: 10.1007/s00239-002-2333-y
Petersen, G., Seberg, O., Thorsøe, S., Jørgensen, T., and Mathew, B. (2008). A phylogeny of the genus Crocus (Iridaceae) based on sequence data from five plastid regions. Taxon 57, 487–499. doi: 10.2307/25066017
Peterson, P. M., Romaschenko, K., Snow, N., and Johnson, G. (2012). A molecular phylogeny and classification of Leptochloa (Poaceae: Chloridoideae: Chlorideae) sensu lato and related genera. Ann. Bot. 109, 1317–1329. doi: 10.1093/aob/mcs077
Piot, A., Hackel, J., Christin, P.-A., and Besnard, G. (2018). One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 247, 2781. doi: 10.1007/s00425-017-2781-x
Raubeson, L. A., and Jansen, R. K. (2005). “Chloroplast genomes of plants,” in Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants, ed. R. J. Henry (Wallingford: CABI), 45–68. doi: 10.1079/9780851999043.0045
Reginato, M., Michelangeli, F. A., and Goldenberg, R. (2010). Phylogeny of Pleiochiton (Melastomataceae, Miconieae): total evidence. Bot. J. Lin. Soc. 162, 423–434. doi: 10.1111/j.1095-8339.2009.01022.x
Reginato, M., Neubig, K. M., Majure, L. C., and Michelangeli, F. A. (2016). The first complete plastid genomes of Melastomataceae are highly structurally conserved. PeerJ 4, e2715. doi: 10.7717/peerj.2715
Ripma, L. A., Simpson, M. G., and Hasenstab-Lehman, K. (2014). Geneious! Simplified genome skimming methods for phylogenetic systematic studies: a case study in Oreocarya (Boraginaceae). Appl. Plant. Sci. 2:1400062. doi: 10.3732/apps.1400062
Ritz, C. M., Reiker, J., Charles, G., Hoxey, P., Hunt, D., Lowry, M., et al. (2012). Molecular phylogeny and character evolution in terete-stemmed Andean opuntias (Cactaceae-Opuntioideae). Mol. Phylogenet. Evol. 65, 668–681. doi: 10.1016/j.ympev.2012.07.027
Robinson, D. F., and Foulds, L. R. (1981). Comparison of phylogenetic trees. Math. Biosci. 53, 131–147. doi: 10.1016/0025-5564(81)90043-2
Rousseau-Gueutin, M., Huang, X., Higginson, E., Ayliffe, M., Day, A., and Timmis, J. N. (2013). Potential functional replacement of the plastidic acetyl-CoA carboxylase subunit (accD) gene by recent transfers to the nucleus in some angiosperm lineages. Plant. Physiol. 161, 1918–1929. doi: 10.1104/pp.113.214528
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Rozen, S., and Skaletsky, H. (2000). Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132, 365. doi: 10.1385/1-59259-192-2
Ruhlman, T. A., Chang, W.-J., Chen, J. J., Huang, Y.-T., Chan, M.-T., Zhang, J., et al. (2015). NDH expression marks major transitions in plant evolution and reveals coordinate intracellular gene loss. BMC Plant Biol. 15:100. doi: 10.1186/s12870-015-0484-7
Ruhlman, T. A., and Jansen, R. K. (2014). “The plastid genomes of flowering plants,” in Chloroplast Biotechnology: Methods and Protocols Methods in Molecular Biology, ed. P. Maliga (Totowa, NJ: Humana Press), 3–38. doi: 10.1007/978-1-62703-995-6_1
Ruhlman, T. A., and Jansen, R. K. (2018). “Aberration or Analogy? The Atypical Plastomes of Geraniaceae,” in Advances in Botanical Research Plastid Genome Evolution, eds S.-M. Chaw and R. K. Jansen (Cambridge, MA: Academic Press), 223–262. doi: 10.1016/bs.abr.2017.11.017
Sanderson, M. J., Copetti, D., Búrquez, A., Bustamante, E., Charboneau, J. L. M., Eguiarte, L. E., et al. (2015). Exceptional reduction of the plastid genome of saguaro cactus (Carnegiea gigantea): Loss of the ndh gene suite and inverted repeat. Am. J. Bot. 102, 1115–1127. doi: 10.3732/ajb.1500184
Scataglini, M. A., Zuloaga, F. O., Giussani, L. M., Denham, S. S., and Morrone, O. (2014). Phylogeny of New World Paspalum (Poaceae, Panicoideae, Paspaleae) based on plastid and nuclear markers. Plant Syst. Evol. 300, 1051–1070. doi: 10.1007/s00606-013-0944-1
Schelkunov, I., Shtratnikova, V. Y., Nuraliev, M. S., Selosse, M.-A., Penin, A. A., and Logacheva, M. D. (2015). Exploring the limits for reduction of plastid genomes: a case study of the mycoheterotrophic orchids Epipogium aphyllum and Epipogium roseum. Genome Biol. Evol. 7, 1179–1191. doi: 10.1093/gbe/evv019
Schliep, K. P. (2011). phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593. doi: 10.1093/bioinformatics/btq706
Schumann, K. (1899). Gesamtbeschreibung der Kakteen (Monographia cactacearum). Dȩbno, Poland: Neudamm, Verlag J. Neumann. doi: 10.5962/bhl.title.10394
Shaw, J., Lickey, E. B., Beck, J. T., Farmer, S. B., Liu, W., Miller, J., et al. (2005). The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 92, 142–166. doi: 10.3732/ajb.92.1.142
Shi, S., Li, J., Sun, J., Yu, J., and Zhou, S. (2013). Phylogeny and Classification of Prunus sensu lato (Rosaceae). J. Integr. Plant Biol. 55, 1069–1079. doi: 10.1111/jipb.12095
Shin, H. W., and Lee, N. S. (2018). Understanding plastome evolution in Hemiparasitic Santalales: Complete chloroplast genomes of three species, Dendrotrophe varians, Helixanthera parasitica, and Macrosolen cochinchinensis. PLoS One 13:e0200293. doi: 10.1371/journal.pone.0200293
Simmons, M. P. (2004). Independence of alignment and tree search. Mol. Phylogenet. Evol. 31, 874–879. doi: 10.1016/j.ympev.2003.10.008
Solórzano, S., Chincoya, D. A., Sanchez-Flores, A., Estrada, K., Díaz-Velásquez, C. E., González-Rodríguez, A., et al. (2019). De Novo Assembly Discovered Novel Structures in Genome of Plastids and Revealed Divergent Inverted Repeats in Mammillaria (Cactaceae, Caryophyllales). Plants (Basel) 8:392. doi: 10.3390/plants8100392
Soltis, P. S., and Soltis, D. E. (2000). The role of genetic and genomic attributes in the success of polyploids. Proc. Natl. Acad. Sci. U.S.A. 97, 7051–7057. doi: 10.1073/pnas.97.13.7051
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Straub, S. C. K., Parks, M., Weitemier, K., Fishbein, M., Cronn, R. C., and Liston, A. (2012). Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 99:335. doi: 10.3732/ajb.1100335
Stuppy, W. (2002). Seed characters and the generic classification of the Opuntioideae (Cactaceae). Succul. Plant Res. 6, 25–58.
Sun, Y., Wang, A., Wan, D., Wang, Q., and Liu, J. (2012). Rapid radiation of Rheum (Polygonaceae) and parallel evolution of morphological traits. Mol. Phylogenet. Evol. 63, 150–158. doi: 10.1016/j.ympev.2012.01.002
Taylor, N. P., Stuppy, W., and Barthlott, W. (2002). Realignment and revision of the opuntioideae of Eastern Brazil. Succul. Plant Res. 6, 99–132.
Taylor, N. P., and Zappi, D. C. (2004). Cacti of Eastern Brazil. Richmond: Royal Botanic Gardens, KEW.
Thode, V. A., and Lohmann, L. G. (2019). Comparative chloroplast genomics at low taxonomic levels: a case study using Amphilophium (Bignonieae, Bignoniaceae). Front. Plant Sci. 10:796. doi: 10.3389/fpls.2019.00796
Thomson, A. M., Vargas, O. M., and Dick, C. W. (2018). Complete plastome sequences from Bertholletia excelsa and 23 related species yield informative markers for Lecythidaceae. Appl. Plant Sci. 6:e01151. doi: 10.1002/aps3.1151
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Vargas-Luna, M. D., Hernández-Ledesma, P., Majure, L. C., Puente-Martínez, R., Macías, H. M. H., and Bárcenas, R. T. (2018). Splitting Echinocactus: morphological and molecular evidence support the recognition of Homalocephala as a distinct genus in the Cacteae. PhytoKeys 13, 31–59. doi: 10.3897/phytokeys.111.26856
Wakasugi, T., Tsudzuki, J., Ito, S., Nakashima, K., Tsudzuki, T., and Sugiura, M. (1994). Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. Proc. Natl. Acad. Sci. U.S.A. 91, 9794–9798.
Walker, J. F., Walker-Hale, N., Vargas, O. M., Larson, D. A., and Stull, G. W. (2019). Characterizing gene tree conflict in plastome-inferred phylogenies. PeerJ 7:e7747. doi: 10.7717/peerj.7747
Walker, J. F., Yang, Y., Feng, T., Timoneda, A., Mikenas, J., Hutchison, V., et al. (2018). From cacti to carnivores: Improved phylotranscriptomic sampling and hierarchical homology inference provide further insight into the evolution of Caryophyllales. Am. J. Bot. 105, 446–462. doi: 10.1002/ajb2.1069
Wallace, R. S. (1995). Molecular systematic study of the Cactaceae: using chloroplast DNA variation to elucidate cactus phylogeny. Bradleya 13, 1–12. doi: 10.25223/brad.n13.1995.a1
Wallace, R. S., and Dickie, S. L. (2002). Systematic implication of chloroplast DNA sequence variation in subfamily Opuntioideae (Cactaceae). Succul. Plant Res. 6, 9–24.
Wang, N., Yang, Y., Moore, M. J., Brockington, S. F., Walker, J. F., Brown, J. W., et al. (2019). Evolution of portulacineae marked by gene tree conflict and gene family expansion associated with adaptation to harsh environments. Mol. Biol. Evol. 36, 112–126. doi: 10.1093/molbev/msy200
Weng, M.-L., Blazier, J. C., Govindu, M., and Jansen, R. K. (2014). Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 31, 645–659. doi: 10.1093/molbev/mst257
Weng, M.-L., Ruhlman, T. A., and Jansen, R. K. (2016). Plastid-nuclear interaction and accelerated coevolution in plastid ribosomal genes in geraniaceae. Genome Biol. Evol. 8, 1824–1838. doi: 10.1093/gbe/evw115
Weng, M.-L., Ruhlman, T. A., and Jansen, R. K. (2017). Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 214, 842–851. doi: 10.1111/nph.14375
Whitten, W. M., Neubig, K. M., and Williams, N. H. (2013). Generic and Subtribal relationships in neotropical Cymbidieae (Orchidaceae) based on matK/ycf1 plastid data. Lankesteriana 13, 375–392. doi: 10.15517/lank.v13i3.14425
Wicke, S., Schneeweiss, G. M., De Pamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Winter, K., Garcia, M., and Holtum, J. A. M. (2011). Drought-stress-induced up-regulation of CAM in seedlings of a tropical cactus, Opuntia elatior, operating predominantly in the C3 mode. J. Exp. Bot. 62, 4037–4042. doi: 10.1093/jxb/err106
Wommack, K. E., Bhavsar, J., and Ravel, J. (2008). Metagenomics: Read Length Matters. Appl. Environ. Microbiol. 74, 1453–1463. doi: 10.1128/AEM.02181-07
Worberg, A., Quandt, D., Barniske, A.-M., Löhne, C., Hilu, K. W., and Borsch, T. (2007). Phylogeny of basal eudicots: Insights from non-coding and rapidly evolving DNA. Org. Divers. Evol. 7, 55–77. doi: 10.1016/j.ode.2006.08.001
Wu, C.-S., Lai, Y.-T., Lin, C.-P., Wang, Y.-N., and Chaw, S.-M. (2009). Evolution of reduced and compact chloroplast genomes (cpDNAs) in gnetophytes: selection toward a lower-cost strategy. Mol. Phylogenet. Evol. 52, 115–124. doi: 10.1016/j.ympev.2008.12.026
Wu, F.-H., Chan, M.-T., Liao, D.-C., Hsu, C.-T., Lee, Y.-W., Daniell, H., et al. (2010). Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC Plant Biol. 10:68. doi: 10.1186/1471-2229-10-68
Xi, Z., Ruhfel, B. R., Schaefer, H., Amorim, A. M., Sugumaran, M., Wurdack, K. J., et al. (2012). Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc. Natl. Acad. Sci. U.S.A. 109, 17519–17524. doi: 10.1073/pnas.1205818109
Yao, G., Jin, J.-J., Li, H.-T., Yang, J.-B., Mandala, V. S., Croley, M., et al. (2019). Plastid phylogenomic insights into the evolution of Caryophyllales. Mol. Phylogenet. Evol. 134, 74–86. doi: 10.1016/j.ympev.2018.12.023
Zhang, S., Soltis, D. E., Yang, Y., Li, D., and Yi, T. (2011). Multi-gene analysis provides a well-supported phylogeny of Rosales. Mol. Phylogenet. Evol. 60, 21–28. doi: 10.1016/j.ympev.2011.04.008
Zhu, A., Guo, W., Gupta, S., Fan, W., and Mower, J. P. (2016). Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 209, 1747–1756. doi: 10.1111/nph.13743
Keywords: cacti, de novo assembly, Opuntia, plastid structural rearrangements, plastome, pseudogenization, reference-guided assembly
Citation: Köhler M, Reginato M, Souza-Chies TT and Majure LC (2020) Insights Into Chloroplast Genome Evolution Across Opuntioideae (Cactaceae) Reveals Robust Yet Sometimes Conflicting Phylogenetic Topologies. Front. Plant Sci. 11:729. doi: 10.3389/fpls.2020.00729
Received: 27 February 2020; Accepted: 06 May 2020;
Published: 19 June 2020.
Edited by:
Tingshuang Yi, Kunming Institute of Botany, Chinese Academy of Sciences, ChinaReviewed by:
Hengchang Wang, Wuhan Botanical Garden, Chinese Academy of Sciences, ChinaChun-Lei Xiang, Kunming Institute of Botany, Chinese Academy of Sciences, China
Copyright © 2020 Köhler, Reginato, Souza-Chies and Majure. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matias Köhler, bWF0aWFzLmtAdWZyZ3MuYnI=; Lucas C. Majure, bG1hanVyZUBmbG9yaWRhbXVzZXVtLnVmbC5lZHU=