 Eva YuHua Kuo1,2†
Eva YuHua Kuo1,2† Tse-Min Lee
Tse-Min Lee- 1Department of Marine Biotechnology and Resources, National Sun Yat-sen University, Kaohsiung, Taiwan
- 2Doctoral Degree Program in Marine Biotechnology, National Sun Yat-sen University, Kaohsiung, Taiwan
Autophagy plays a role in regulating important cellular functions in response to stress conditions. The role of nitric oxide (NO) in the regulation of autophagy in Chlamydomonas reinhardtii has been not studied. Illumination of C. reinhardtii cells under a high light (HL, 1,600 μmol m–2 s–1) condition induced a NO burst through NO synthase- and nitrate reductase-independent routes, and cell death. The abundance of CrATG8 protein, an autophagy marker of C. reinhardtii, increased after HL illumination along with a linear increase in the transcript abundance of autophagy-associated genes (CrVPS34, CrATG1, CrATG3, CrATG4, CrATG6, CrATG7, CrATG8, and CrATG12), which were suppressed in the presence of an NO scavenger, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (cPTIO). The cells were treated with NO donors, S-nitroso-N-acetyl-penicillamine, and S-nitrosoglutathione, under a normal light (50 μmol m–2 s–1) condition to elucidate the role of NO in autophagy activation and cell death. Treatment with 0.05 mM or 0.1 mM NO donors increased the abundance of ATG8 protein and CrATG transcripts, which were suppressed in the presence of cPTIO. Moreover, treatment with 0.05 mM NO donors did not affect cell viability, while 0.1 mM NO donors elicited a transient decrease in cell growth and death that recovered after 12 h. The transient effect could be prevented by the presence of cPTIO. However, treatment with 1 mM H2O2 and 0.1 mM NO donors enhanced autophagy induction and resulted in cell death after 24 h. The interaction of H2O2 and NO can be prevented by cPTIO treatment. This implies that NO is critical for the interaction of H2O2 and NO that induces cell death and autophagy. Furthermore, exposure to 0.1 mM NO donors under a non-lethal HL condition (750 μmol m–2 s–1) evoked autophagy and cell death. In conclusion, the present findings demonstrated that the NO-mediated autophagy pathway is activated in C. reinhardtii under lethal high intensity illumination and may interact with H2O2 for HL-induced cell death. The relationships between autophagy and cell death are discussed.
Introduction
Nitric oxide (NO), a short-lived, gaseous molecule that can be either enzymatically or non-enzymatically synthesized in plants (Besson-Bard et al., 2008; Palavan-Unsal and Arisan, 2009), has been recognized as a novel biological messenger in the regulation of various biochemical and physiological activities and stress responses (Anbar, 1995; Neill et al., 2008; Hasanuzzaman et al., 2010; Hayat et al., 2010; Siddiqui et al., 2011; Bajguz, 2014). Recently, increasing importance has been attached to the role of NO in the regulation of high intensity light responses. However, whether NO plays a protective or a harmful role in the response of plants to excessive light energy depends on the plant species. Illumination of the leaves of tall fescue (Festuca arundinacea Schreb.) at an intensity of 500 μmol m–2 s–1 triggers NO production against oxidative stress by increasing the activity of antioxidant enzymes and the content of antioxidants (Xu et al., 2010). Foresia et al. (2010) has reported for a unicellular marine alga Ostreococcus tauri Gen et Sp-NOV that illumination at 400 μmol m–2 s–1 induces an NO burst, which is proposed to be a signal triggering a photoprotection mechanism against high light (HL)-induced oxidative damage. We have recently found a contrasting result in Chlamydomonas reinhardtii P.A. Dangeard that NO generated under very high intensity light (VHL; 3,000 μmol m–2 s–1) conditions is associated with VHL-induced cell death (Chang et al., 2013).
There is accumulating evidence that the generation of NO is crucial for the regulation of developmentally regulated and environmentally induced programmed cell death (PCD) in plants, either its promotion or its inhibition (Delledonne et al., 2001; Wang et al., 2013). NO delays the onset of cell death in gibberellin (GA)-induced PCD in barley aleurone layers (Beligni et al., 2002), while NO at high concentrations induces DNA fragmentation, membrane breakdown, and cell death (Pedroso et al., 2000; Yamasaki, 2000; Romero-Puertas et al., 2004). Moreover, NO is involved in the regulation of hypersensitive cell death (Clarke et al., 2000; de Pinto et al., 2002) and stress-induced cell death (Ahlfors et al., 2009; de Michele et al., 2009). NO also triggers cell death in algae; for example, the aldehyde-induced cell death in diatoms (Vardi et al., 2006), the heat-induced cell death of symbiotic alga Symbiodinium microadriaticum Freudenthal (Bouchard and Yamasaki, 2008), and the mastoparan (MP)-induced cell death of C. reinhardtii (Yordanova et al., 2010).
Reactive oxygen species (ROS) and oxidative stress modulate the autophagy process in plants (Pérez-Pérez et al., 2010, 2012b; Liu and Bassham, 2012; Bassham and Crespo, 2014). Stresses, including methyl viologen (MV)- or hydrogen peroxide (H2O2)-induced oxidative stress, nitrogen deficiency, carbon starvation by dark incubation, endoplasmic reticulum stress, and disordered chloroplast protein homeostasis due to a depletion of ClpP1 protease, are known to trigger autophagy in C. reinhardtii cells (Pérez-Pérez et al., 2010, 2012a,b, 2014; Ramundo et al., 2014). Moreover, a transfer of C. reinhardtii cells from dim light (5–10 μmol m–2 s–1) to high intensity light (1,200 μmol m–2 s–1) caused a transient increase of autophagy-related protein 8 (ATG8) abundance with a peak at 6 h, followed by a gradual decline to the control level when the high intensity illumination was prolonged to 24 h (Pérez-Pérez et al., 2012a). In comparison with wild type, the induction of autophagy by high intensity light illumination, MV, or H2O2, is more pronounced in C. reinhardtii lts1-204 and npq1 lor1 mutants, which exhibit a higher sensitivity to oxidative stress due to low carotenoid levels (Pérez-Pérez et al., 2012a).
Reactive nitrogen species (RNS) are also known to modulate autophagy. In animal system, NO activates autophagy in HeLa cells (Yang et al., 2008) and neurons (Barsoum et al., 2006) but suppresses autophagy in neurodegenerative diseases (Sarkar et al., 2011). In contrast, NO does not affect autophagy in cardiac myocytes (Rabkin and Klassen, 2007). This suggests that the differential regulation of autophagy by NO depends on the type of animal tissue. Apart from ROS and oxidative stress, the role of RNS in the control of autophagy has not previously been reported in C. reinhardtii cells, as far as we know. Therefore, the present study has examined whether NO modulates autophagy in C. reinhardtii cells under very high intensity illumination (HL, 1,600 μmol m–2 s–1), which can induce cell death. First, the time-course changes in NO production detected by 4-amino-5-methylamino-2′,7′-difluororescein (DAF-FM), the level of ATG8 detected using western blots, and the transcript abundance of autophagy-associated genes were determined. Furthermore, the role of NO was confirmed by experiments in the presence or absence of an NO scavenger, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (cPTIO). Then, the NO donors including S-nitrosoglutathione (GSNO) and S-nitroso-N-acetyl-penicillamine (SNAP) were treated under normal light (NL, 50 μmol m–2 s–1) illumination with or without cPTIO to confirm the role of NO in autophagy activation and cell death. Next, the NO donors were treated under non-lethal HL conditions (i.e., a moderate high light condition, ML, 750 μmol m–2 s–1) to examine whether NO can confer sensitivity of C. reinhardtii cells to the induction of autophagy and cell death under moderate high light illumination. In addition, the interaction of NO with H2O2 accumulated under HL illumination in the modulation of autophagy and cell death was investigated by the application of H2O2 together with SNAP or GSNO under NL conditions.
Materials and Methods
Algal Culture and Treatments
Chlamydomonas reinhardtii P.A. Dangeard strain CC125 (mt+) and CC124 (mt−) obtained from the Chlamydomonas Resource Center1 (University of Minnesota, St. Paul, MN, United States), were photoheterotrophically cultured in 50 mL of Tris-acetate phosphate (TAP) medium (Harris, 1989) containing a trace element solution in 125 mL flasks. The cultures were agitated on an orbital shaking incubator (model OS701, TKS company, Taipei, Taiwan) (150 rpm) under continuous illumination with a fluorescent white light at the NL intensity of 50 μmol m–2 s–1 at 28°C. After 18–24 h of incubation, algal cells that grew to the density of approximately 3 × 106 cells mL–1 were centrifuged at 4,000 × g for 5 min at 28°C. The pellet was resuspended in fresh TAP medium as 3 × 106 cells mL–1. Ten mL of the resuspended culture was transferred to a 100 mL beaker and incubated at 28°C under an NL condition for 1.5 h in an orbital shaker (model OS701, TKS Company, Taipei, Taiwan) at a speed of 150 rpm. Then, the algal cells were exposed to HL, 1,600 μmol m–2 s–1, or subjected to chemical treatments at 28°C. Each treatment had three independent biological replicates (n = 3) with each beaker as a biological replicate. The treatment has been repeated three times. Samples taken before (0 h) and after chemical treatment were imaged, then centrifuged at 4,000 × g for 5 min, and the pellet was fixed in liquid nitrogen and stored at −80°C until analysis.
To explore whether NO modulates the HL-induced autophagy and cell death, cPTIO was applied to scavenge NO at a final concentration of 400 μM. Because we have found that cPTIO lost its ability to scavenge NO after 2–3 h, 400 μM cPTIO was added again to the algal culture at 2.5 h. To mimic the effect of NO bursts under the HL condition, the NO donors, SNAP and GSNO (Sigma-Aldrich, St. Louis, MO, United States), were applied to the cells illuminated with 50 μmol m–2 s–1 intensity in the presence or absence of 400 μM cTPIO. In addition, to elucidate whether NO can promote the susceptibility of C. reinhardtii cells to high intensity illumination, SNAP and GSNO were applied separately at concentrations of 0.05 or 0.1 mM under a moderate high light condition (ML, 750 μmol m–2 s–1). In addition, the interaction of NO and H2O2 in the induction of autophagy and cell death was also investigated by treatment with 1 mM H2O2 with and without 0.1 mM NO donors under an NL condition for 3 h. In this experiment, the role of NO was confirmed by the treatment of cPTIO with an NO donor and H2O2.
In the attempts to elucidate the involvement of NO synthase (NOS) or nitrate reductase (NR) in the HL-induced NO production, the inhibitors of NOS and NR, Nω-nitro-L-arginine methyl ester (L-NAME), Nω-monomethyl-L-arginine (L-NMMA), and sodium tungstate, respectively, were added to the TAP medium 1.5 h before the HL treatment.
Determination of Cell Density and Viability
For cell density estimation, 10 μL of algal culture was mixed with 30 μL of Lugol’s solution (Sigma-Aldrich, St. Louis, MO, United States) and the cell number was counted in duplicate using a light microscope (BX43, Olympus, Tokyo, Japan) and a hemocytometer (Improved Neubauer, Boeco, Germany). The cell density was calculated according to the manufacturer’s manual and expressed as units of 106 mL–1.
After treatments, the viability of the algal cells was estimated by loading 2 μL of cell suspension on TAP agar plates, and incubating the plates for 72 h at 28°C under illumination at 50 μmol m–2 s–1 intensity. The colonies were imaged with a digital Nikon camera, and the final composite images were constructed using Adobe Photoshop (Adobe Systems, San Jose, CA, United States). Together with a cell density curve, the cell viability was assessed by growth ability (the color and size of the colony) and was used to evaluate the effects of HL illumination or chemical challenges.
Detection of Dead Cells Using Sytox Green Fluorescence
Cell death was assessed using the SYTOX-Green fluorescent probe (Molecular Probes Inc., Eugene, OR, United States). The SYTOX-Green stock solution of 5 mM in 100% DMSO was added to 1 mL of algal culture at a final concentration of 5 μM, and the mixture was incubated for 5 min at room temperature in the dark. The fluorescence level was detected by a fluorescence spectrophotometer at 525 nm (excitation: 488 nm) (Sato et al., 2004). Based on the blank (TAP medium without algal cells), the relative SYTOX-Green fluorescence level was estimated and expressed as relative fluorescence⋅(106 cells)–1.
Then, the cells were observed with a fluorescence microscope (Eclipse Ni, Nikon, Tokyo, Japan) with excitation at 488 nm using FITC (excitation wavelength: 465–495 nm, emission wavelength: 515–555 nm) and B-2A (excitation wavelength: 450–490 nm, emission wavelength: >520 nm) filters (Nikon, Tokyo, Japan). Fluorescent images were acquired using a CCD camera (Nikon’s Digital Sight DSU3, Tokyo, Japan) and imported into Adobe Photoshop.
Detection of NO Production
An NO-sensitive fluorescent dye, DAF-FM diacetate (Kojima et al., 1998), purchased from Invitrogen Life Technologies (Carlsbad, CA, United States) was employed to measure NO production according to the procedure described in our previous study (Chang et al., 2013). DAF-FM diacetate was preloaded into cell cultures prior to HL and chemical treatments. Algal cells (1 mL) were preincubated in TAP medium containing 5 μM DAF-FM diacetate for 60 min at 25°C under NL to allow the penetration of DAF-FM acetate into the cells (Chang et al., 2013). Then, the algal cells were washed twice with fresh TAP medium and exposed to HL or treated with a chemical. The fluorescence in the cells was determined with a fluorescence spectrophotometer (excitation/emission: 492 nm/525 nm; F-2500, Hitachi, Tokyo, Japan) and observed with a fluorescence microscope (Eclipse Ni, Nikon, Tokyo, Japan) using a FITC filter (Nikon, Tokyo, Japan). Based on the fluorescence of the blank (TAP medium without algal cells), the relative DAF-FM fluorescence level was estimated and expressed as relative fluorescence⋅(106 cells)–1.
Protein Extraction and Western Blots
Soluble protein was extracted according to Pérez-Pérez et al. (2012a). After treatment for 3 h under NL or HL conditions, algal cells (n = 3, three independent biological replicates) were collected by centrifugation (4,000 × g, 5 min), washed once with lysis buffer (50 mM Tris-HCl, pH 7.5) and resuspended in 50 μL lysis buffer. Cells were lysed by two cycles of slow freezing to −80°C, followed by fast thawing to room temperature. The soluble cell extract was then separated from the insoluble fraction by centrifugation (15,000 × g, 15 min) at 4°C and immediately used for western-blot analysis and protein quantitation. Protein concentrations were quantified by the Coomassie Blue dye binding method (Bradford, 1976) using the concentrated dye purchased from BioRad (500-0006, Hercules, CA, United States). Samples were heated to 100°C for 5 min and then used for western blot analysis. For each sample, 30 μg of protein was loaded into each lane, resolved by 15% SDS-PAGE, and transferred to a polyvinylidene fluoride membrane for antibody binding. After being blocked with 5% BSA (Sigma, St. Louis, MO, United States), the membranes were incubated with a rabbit polyclonal anti-ATG8 antibody (ab77003; Abcam, Cambridge, United Kingdom) and a mouse monoclonal antibody against α-tubulin (ab11304; Abcam, Cambridge, United Kingdom) for 90 min at room temperature. After being washed, the membranes were incubated for 90 min with horseradish peroxidase-conjugated secondary antibodies (MD20878; KPL, Gaithersburg, MD, United States), and then the immunoblots were visualized using the ECL Plus chemiluminescence system (Thermo Scientific Inc., Chicago, IL, United States). A LAS-3000 Mini Lumino-image Analyzer (LAS-3000, Fujifilm, Tokyo, Japan) was used for imaging the chemiluminescent western blots. The relative abundance of CrATG8 protein was estimated on the basis of α-tubulin intensity.
RNA Isolation, cDNA Synthesis, and mRNA Quantification by Real-Time Quantitative PCR
For RNA isolation, the algal cells in 5 mL of culture sampled from each of the independent biological replicates (n = 3) were harvested by centrifugation as described above. Total RNA was extracted with TriPure Isolation Reagent (Roche Applied Science, Mannheim, Germany) according to the manufacturer’s instructions. The integrity of the RNA was checked by visual inspection of the two ribosomal RNAs, 18S and 28S, on an ethidium bromide stained 1% agarose (MDBio Inc., Taipei, Taiwan) gel. The RNA sample concentration was adjusted to 2.95 μg total RNA⋅μL–1. After treatment with DNase (TURBO DNA-freeTM Kit, Ambion Inc., The RNA Company, United States) to remove residual DNA, 1.5 μg of total RNA was used for the preparation of cDNA following the protocol previously described (Chang et al., 2013). The cDNA was amplified from the poly-(A+) end using oligo (dT) 12–18 from a VersoTM cDNA Kit (Thermo Fisher Scientific Inc., Waltham, MA, United States). The volume was adjusted to provide a concentration of 30 ng mL–1 of the original RNA for each sample. The primers for the CrTOR, CrVSP34, and CrATG genes (Supplementary Table S1) were designed according to the Phytozome genome using LightCycler Probe Design2 (Roche Applied Science, Mannheim, Germany). The real-time quantitative PCR was performed using the LightCycler 480 system (Roche Applied Science, Mannheim, Germany). A master mix for PCR was prepared with a LightCycler® 480 SYBR Green I Master Kit (Roche Applied Science, Mannheim, Germany). To optimize the primer concentration, real-time PCR analyses using different primer concentrations and a constant template cDNA concentration of 30 or 50 ng were performed. To optimize the cDNA template concentration, each pair of primers was tested across a log dilution series of a positive control DNA sample. After optimization of the real-time PCR conditions, a primer concentration of 3 μM and a cDNA template concentration of 30 ng μL–1 were used for the detection of CrATG transcript abundance, and a primer concentration of 6 μM and a cDNA template concentration of 30 ng μL–1 were used for the detection of the CrVSP34 transcript abundance. Each reaction was performed in a total volume of 10 μL, which contained LightCycler® 480 SYBR Green I Master Mix, the selected concentration of each primer, and cDNA corresponding to 30 or 50 ng μL–1 RNA in the reverse transcriptase reaction. The amplification program was started by first denaturation at 95°C for 5 min, followed by 50 amplification cycles of annealing at 60°C for 10 s, and elongation at 72°C for 5 s for real time fluorescence measurements. The dissociation curves were performed after the PCR, and the fluorescence was analyzed using the LightCycler 480 system. Software with auto CT (cycle threshold) was used to determine the threshold of each gene, and the 2–Δ Δ CT method was used to calculate CT values, in which the relative change in transcript abundance was normalized to reference genes, ubiquitin-conjugating enzyme E2 isoform (CrUBC, NCBI: AY062935) and elongation factor 1 alpha (CrEF-1α, NCBI: XM_001696516.1) (see Supplementary Table S1 for the primers). The fold increase was calculated relative to the RNA sample from the control without light or chemical treatment at 0 h. Because the results based on the CrEF-1α internal control were similar to those based on CrUBCX, the relative changes in the transcript abundance on the basis of CrUBCX are shown in the present manuscript. All of the results presented are the averages of three independent biological replicates.
Statistics
Three independent biological replicates (n = 3) were used for each experiment and all of the experiments were repeated at least three times. Because they showed a similar trend, only the results from one of them are shown in this paper. Statistical analyses were performed using SPSS (SPSS 15.0 for Windows Evaluation Version, Chicago, IL, United States). Significant differences between sample means were analyzed using Student’s t-test or Duncan’s new multiple range test and a subsequent significant analysis of variance for the controls and treatments at P < 0.05.
Results
HL Illumination Induces NO Burst and Cell Death
An NO-sensitive fluorescent dye, DAF-FM diacetate, was loaded into the TAP medium before CC125, the nit1 nit2 mt− genotype that cannot use nitrate and nitrite, was transferred to a HL condition. The production of NO increased linearly during 1–3 h after HL illumination, followed by a significant increase at 5 h and a small increment during 5–8 h (Figure 1A). Most of the algal cells observed under the microscope bleached 5 h after HL illumination and concomitantly emitted intense DAF-FM fluorescence (NO) (Figure 1B). Whether the DAF-FM fluorescence was attributable to NO was confirmed by the treatment with an NO scavenger, cPTIO (400 μM). cPTIO was added twice, at 0 and 2.5 h, to remove the NO generated during HL illumination. Although cPTIO can cause an increase in the DAF-FM fluorescence under certain conditions due to oxidization of NO to the NO2 radical with the subsequent formation of N2O3 (NO2 + NO → N2O3), which can react with the DAF-FM dye to form the fluorescent DAF-2T (Mur et al., 2011; D’Alessandro et al., 2013), our current results showed that the presence of cPTIO effectively reduced the DAF-FM fluorescence in the HL-treated cells (Figures 1A,B). This provides evidence supporting the hypothesis that DAF-FM fluorescence emittance under the HL condition is a result of NO.
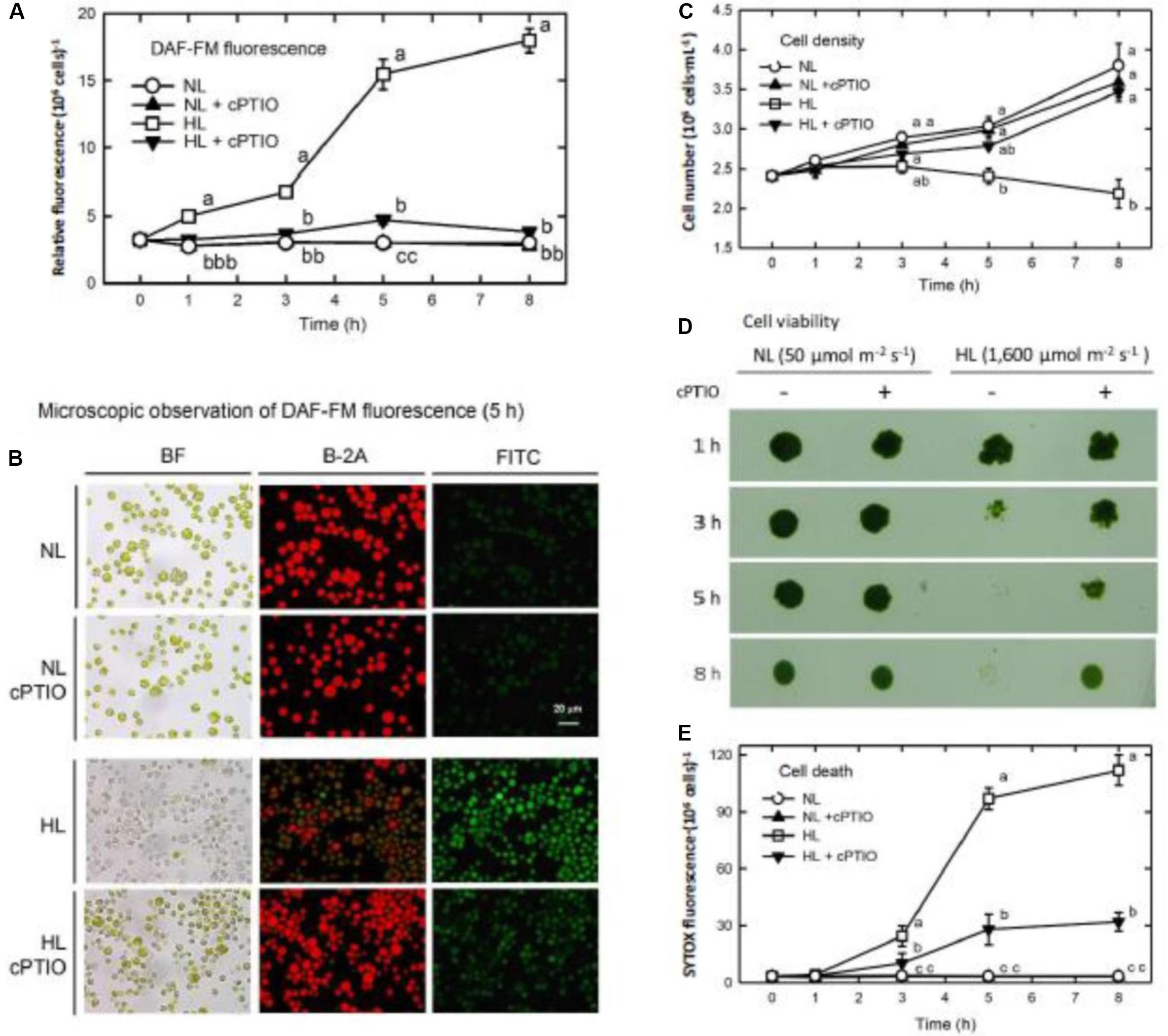
Figure 1. DAF-FM acetate detection of NO production, cell growth, and death in Chlamydomonas reinhardtii cells under NL (50 μmol m–2 s–1) and HL (1,600 μmol m–2 s–1). (A) Quantitation of NO production. (B) Microscopic observation of DAF-FM fluorescence after 5 h of treatment. (C) Cell density. (D) Cell viability. (E) Cell death (SYTOX Green fluorescence). The data in (A), (C), and (E) are expressed as the mean ± SD (n = 3) from three independent biological replicates, and different letters indicate the statistical significance set at P < 0.05 according to ANOVA. In (B), BF represents bright field, B-2A represents the autofluorescence of cells, and FITC represents DAF-FM fluorescence.
The level of DAF-FM fluorescence determined in this study represented the cumulative NO production because the DAF-FM dye was loaded prior to the treatment. Thus, the NO production rate can be estimated from the difference in relative fluorescent units (RFU) between the two time points. As calculated from the data shown in Figure 1A, the NO production rate of NL-grown cells was 0 RFU h–1 over the incubation time, whereas that of the HL-treated cells was 0, 1.43, 0.98, 2.85, and 0.52 RFU h–1 from 0–0.5, 0.5–1, 1–3, 3–5, and 5–8 h, respectively. It is obvious that a transfer of C. reinhardtii cells to a HL condition triggers transient NO production with a profound burst when the time of exposure extends from 0.5 to 5 h with the maximum released during 3–5 h, followed by a decrease.
The cell growth expressed as cell density was inhibited upon exposure to HL illumination (Figure 1C), in which the cell viability decreased after 3 h and then the cells completely died after 5 h (Figure 1D). Similarly, using SYTOX-Green dye staining, the extent of dead cells increased 3 h after HL illumination and reached the maximum after 5 h (Figure 1E). The presence of cPTIO can prevent the HL-induced cell bleaching [see bright field (BF) in Figure 1B], growth inhibition (Figures 1C,D), and cell death (Figure 1E). The effect of cPTIO in the mitigation of HL-induced cell death demonstrates that the large quantity of NO released during prolonged HL illumination is associated with cell death and, in turn, results in a decline in cell growth.
We found that the illumination with moderate light intensity of 750 μmol m–2 s–1 did not induce NO generation (DAF-FM fluorescence) during a 24 h period (Supplementary Figure S1A) and the algal cells can acclimate to this moderate light intensity illumination, reflected by normal growth ability the same as algal cells grown under the NL condition (Supplementary Figure S1B). The extent of cell death evaluated by SYTOX-Green staining was slightly under 750 μmol m–2 s–1 illumination compared to significant cell death in the 1,600 μmol m–2 s–1 condition (Supplementary Figure S1C and Figure 1E). These correlative results reveal that the burst of NO is dependent on the light intensity and the NO production appears to have a negative relationship with cell molarity.
Nitric oxide can be synthesized in plants by an enzymatic process involving NOS or NR, or by non-enzymatic reactions (Besson-Bard et al., 2008; Palavan-Unsal and Arisan, 2009). Whether NO was synthesized via NOS- or NR-dependent routes in C. reinhardtii cells under HL illumination was studied by treating them with the NOS inhibitors, L-NAME (100 or 500 μM) and L-NMMA (100 or 300 μM), and the NR inhibitor, tungstate (300 μM). These inhibitors did not affect the DAF-FM fluorescence of NL- or HL-treated cells (Figure 2). We have also found that the C. reinhardtii strain CC124 (nit1 nit2 mt+) that lacks NR and cannot grow in a medium containing nitrate or nitrite exhibited a similar NO production pattern under HL illumination (Supplementary Figure S2). Because C. reinhardtii strains CC125 and CC124 are mutants lacking NR and cannot use nitrate and nitrite, this implies that the HL-induced NO burst is independent of NR. Therefore, our present findings indicate that neither NOS nor NR is involved in the synthesis of NO in the HL-treated C. reinhardtii cells. This agrees with the results of our previous study that NO generated in C. reinhardtii cells exposed to an extremely high-intensity illumination (3,000 μmol m–2 s–1) is derived from sources other than the NOS- or NR-mediated pathways (Chang et al., 2013).
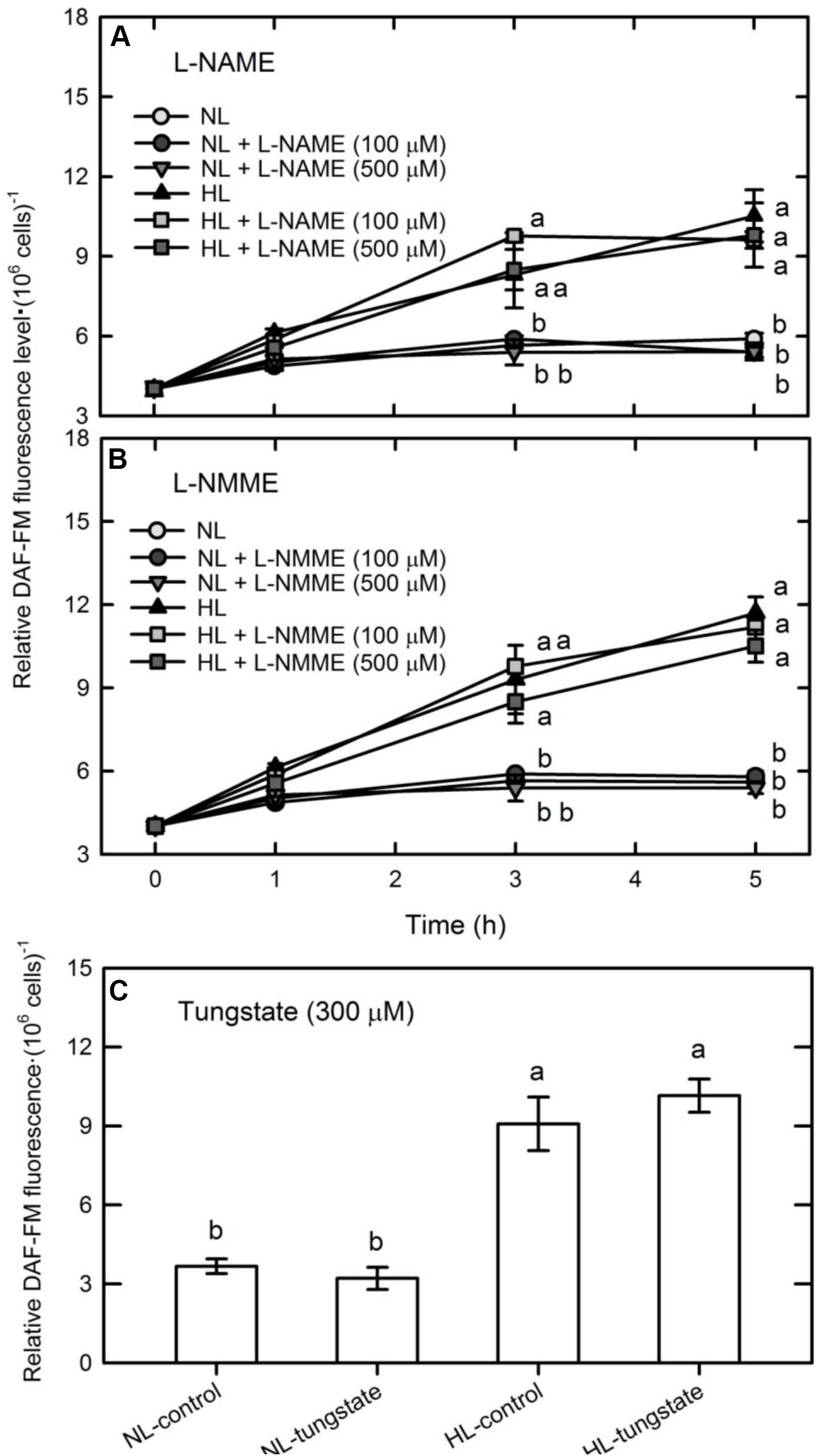
Figure 2. The effect of the NOS inhibitor, L-NAME (A) or L-NMME (B), and the NR inhibitor, tungstate (300 μM) (C), on NO production in Chlamydomonas reinhardtii cells under NL (50 μmol m–2 s–1) or HL (1,600 μmol m–2 s–1) illumination. The data are expressed as the mean ± SD (n = 3) from three independent biological replicates and different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
NO Involvement of Autophagy Induction Under HL Illumination
Because the CrATG8 protein was recognized as an autophagy marker of C. reinhardtii (Pérez-Pérez and Crespo, 2010), a commercial ATG8 antibody raised using recombinant Arabidopsis thaliana APG8A (Abcam, Cambridge, United Kingdom) was used in western blot assays to estimate the abundance of the CrATG8 protein. On a 15% gel, two protein bands with apparent molecular masses of 15 and 14.6 kD were detected in the soluble extract [the solid square symbol represents ATG and the solid circle symbol represents ATG-PE (phosphatidylethanolamine)] (Figure 3). Based on the 0 h control, the abundance of both CrATG8 and CrATG8-PE detected on western blot showed an apparent increase by 3 h of HL treatment (Figure 3A). Based on the normalization by α-tubulin intensity, we found that the relative abundances of CrATG8 and CrATG8-PE remained unchanged for the cells grown under the NL condition over a 0–3 h period while it increased seven-fold 3 h after HL illumination (Figure 3B). The HL-induced increase in the level of CrATG8 protein can be suppressed by the presence of cPTIO (Figure 3). ATG8 plays an essential role in the development of autophagosome formation, target recognition, and vacuole tethering (Mizushima et al., 2011) and is anchored to the autophagosome membrane through covalent binding to the membrane lipid PE to execute its function in autophagosome formation. The present result indicates that the expression of functional ATG8 is activated by accumulated NO under an HL condition.
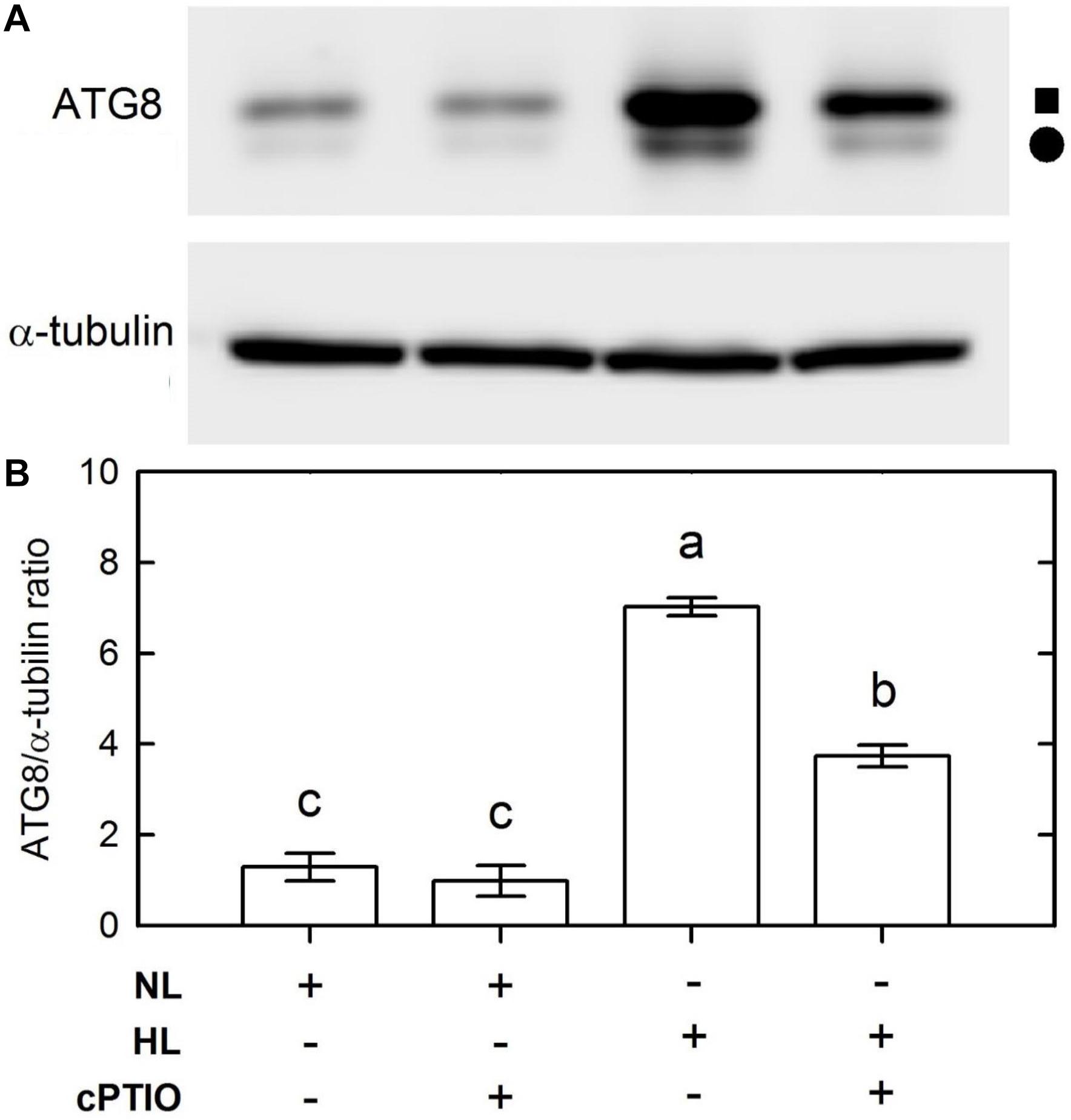
Figure 3. Immunodetection of the CrATG8 protein of Chlamydomonas reinhardtii cells 3 h after NL (50 μmol m–2 s–1) and HL (1,600 μmol m–2 s–1) illumination in the presence or the absence of 400 M cPTIO. (A) Immunodetection using an antibody directed against recombinant Arabidopsis ATG8 (upper part) and α-tubulin (lower part). Triplicate samples were analyzed, and one of them is shown. (B) Relative change of CrATG8 protein expression estimated on the basis of the α-tubulin intensity. A solid square (■) represents ATG8 and a solid circle (∙) represents ATG8-PE. The data in (B) are expressed as the mean ± SD (n = 3) from three independent biological replicates, and different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
High light illumination immediately induced a significant increase in the transcript abundance of CrVPS34 (Figure 4A), CrATG1 (Figure 4B), CrATG3 (Figure 4C), CrATG4 (Figure 4D), CrATG6 (Figure 4E), CrATG7 (Figure 4F), and CrATG8 (Figure 4G), with a peak at 3 h. For CrATG12, although its transcript abundance was statistically increased by HL illumination, it only showed a 1.3-fold increase compared to the NL control (Figure 4H). The presence of cPTIO, which scavenged NO generated under the HL condition (Figure 1A), could suppress the HL-induced increase of CrVPS34 and CrATG transcripts abundance (Figure 4). Accordingly, the HL-induced CrVPS34 and CrATG gene expression is mediated by NO generated under HL illumination.
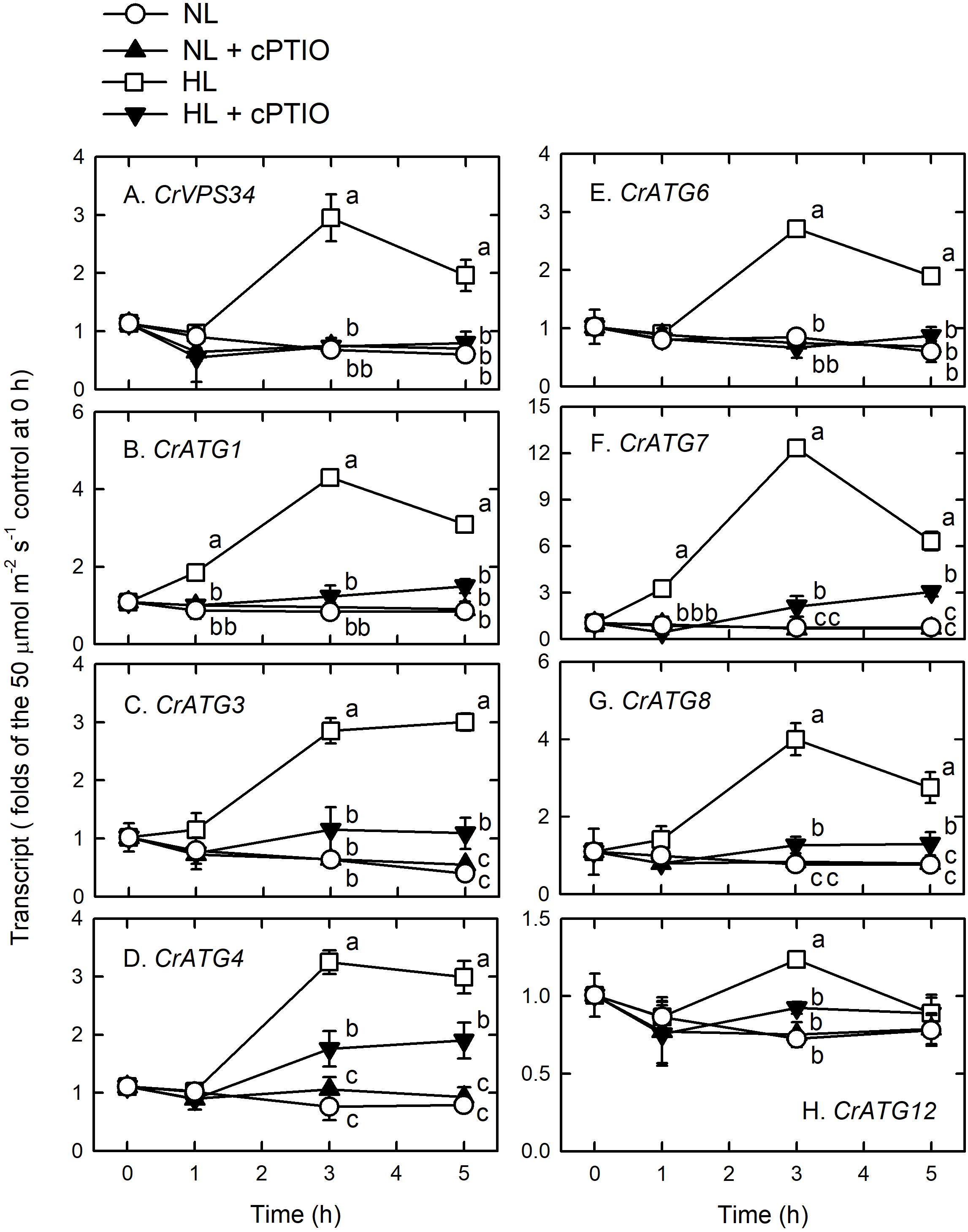
Figure 4. CrVPS34 and CrATG transcripts abundance in Chlamydomonas reinhardtii cells under NL (50 μmol m–2 s–1) and HL (1,600 μmol m–2 s–1) illumination in the presence or the absence of 400 μM cPTIO. (A) CrVPS34. (B) CrATG1. (C) CrATG3. (D) CrATG4. (E) CrATG6. (F) CrATG7. (G) CrATG8. (H) CrATG12. The data are expressed as the mean ± SD (n = 3) from three independent biological replicates. Different letters indicate the statistical significance set at P < 0.05 according to ANOVA. Open circle, NL; solid triangle, NL + cPTIO; open square, HL; inverted triangle, HL + cPTIO.
Treatment With NO Donors Induces Autophagy Under NL Conditions
The role of NO in the modulation of autophagy was further confirmed by the treatment with NO donors, GSNO (0.05 or 0.1 mM) and SNAP (0.05 or 0.1 mM), under NL conditions. The time-course survey of the level of NO released showed that NO was rapidly emitted within 0.5 h after SNAP (Supplementary Figure S3A) or GSNO (Supplementary Figure S3B) treatment and reached a plateau after 1 h. Compared to the treatment with 0.05 mM, the release of NO was approximately two-fold higher for the treatment with 0.1 mM NO donors. The NO released from GSNO or SNAP at either the 0.05 or 0.1 mM concentration can be scavenged in the presence of 400 μM cPTIO (Supplementary Figure S3). The concentrations and the time-course changes of NO released from 0.1 mM SNAP to 0.1 mM GSNO were similar to those occurring under HL conditions. Therefore, to identify the effect of NO on autophagy and cell death without the interference of HL illumination, in the NL condition, NO donor, SNAP, or GSNO was applied at concentrations of either 0.05 or 0.1 mM.
Using western blot assays, the abundance of CrATG8 protein was markedly increased by the treatment with 0.05 mM SNAP (Figure 5A) or 0.05 mM GSNO (Figure 5B) under the NL condition for 3 h. When treated with a higher concentration (0.1 mM), the CrATG8 protein exhibited a higher increment than with 0.05 mM treatment (Figure 6). We also found that the expression of genes related to autophagy were induced by exposure to 0.05 mM (Figure 7) and 0.1 mM (Figure 8) NO donors. In response to 0.05 mM NO donors, the transcript abundances of CrVPS34 (Figure 7A), CrATG1 (Figure 7B), CrATG3 (Figure 7C), CrATG4 (Figure 7C), CrATG6 (Figure 7E), CrATG7 (Figure 7F), and CrATG8 (Figure 7G) increased approximately 3 h after treatment, but these treatments did not affect the CrATG12 transcript abundance (Figure 7H). When the concentration of NO donors was increased to 0.1 mM, the transcript abundances of CrVPS34 and all CrATG genes showed a higher induction compared to 0.05 mM treatment (Figure 8). The transcript abundance of CrATG12 can be slightly increased by 0.1 mM NO donors (Figure 8H) but not by a lower NO donor concentration (Figure 7H). The presence of cPTIO can suppress the SNAP- or GSNO-induced increase in CrATG8 protein abundance (Figures 5, 6) and the transcripts abundance of the CrVPS34 and CrATG genes (Figures 7, 8). The present findings demonstrated that NO released from NO donors can elicit autophagy in C. reinhardtii cells grown under NL conditions.
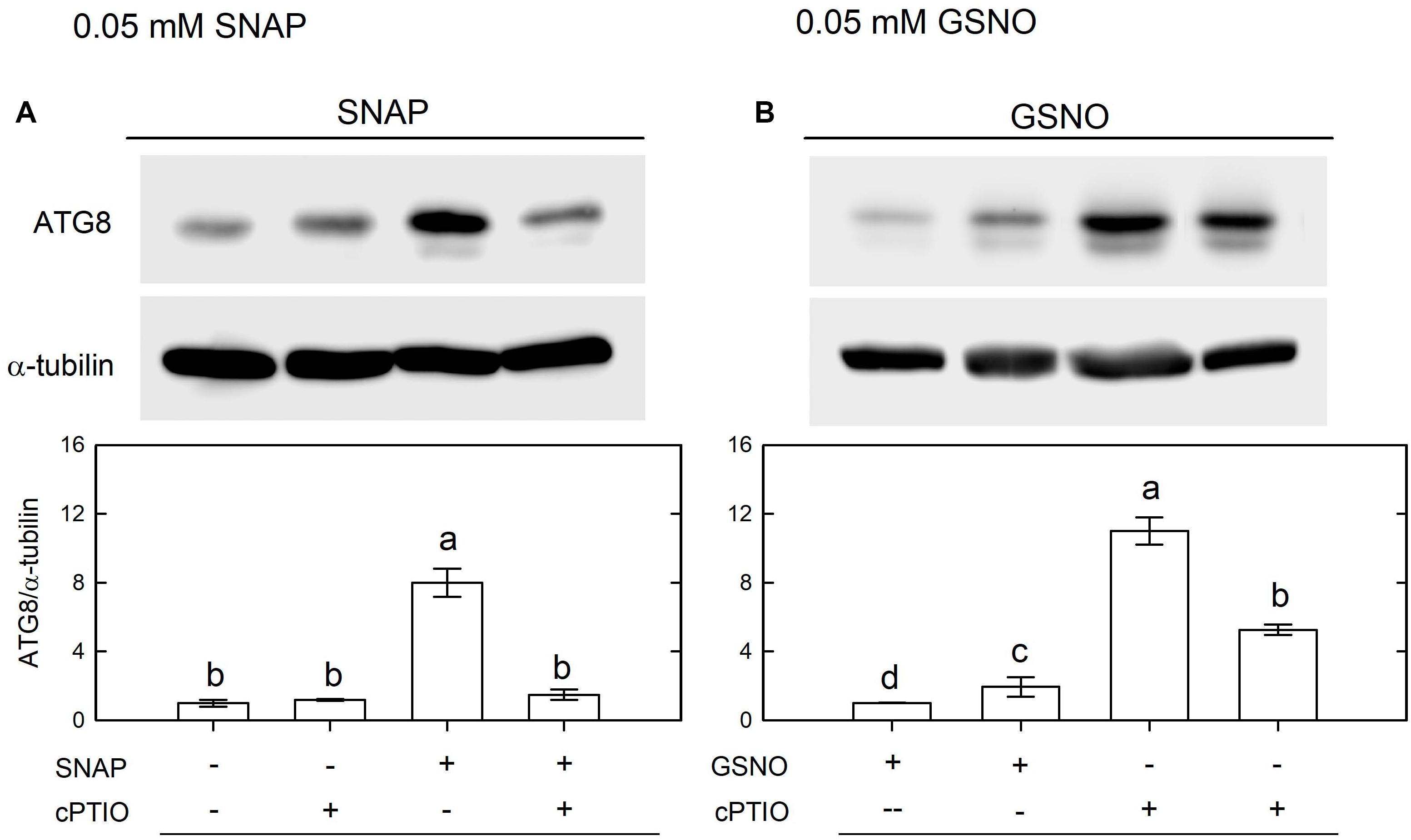
Figure 5. Immunodetection of the CrATG8 protein of Chlamydomonas reinhardtii cells after 0.05 mM SNAP (A) or 0.05 mM GSNO (B) treatment for 3 h in the presence or absence of 400 μM cPTIO under an NL (50 μmol m–2 s–1) condition. Triplicate samples were analyzed, and one of them is shown. The quantitation data are expressed as the mean ± SD (n = 3) from three independent biological replicates and different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
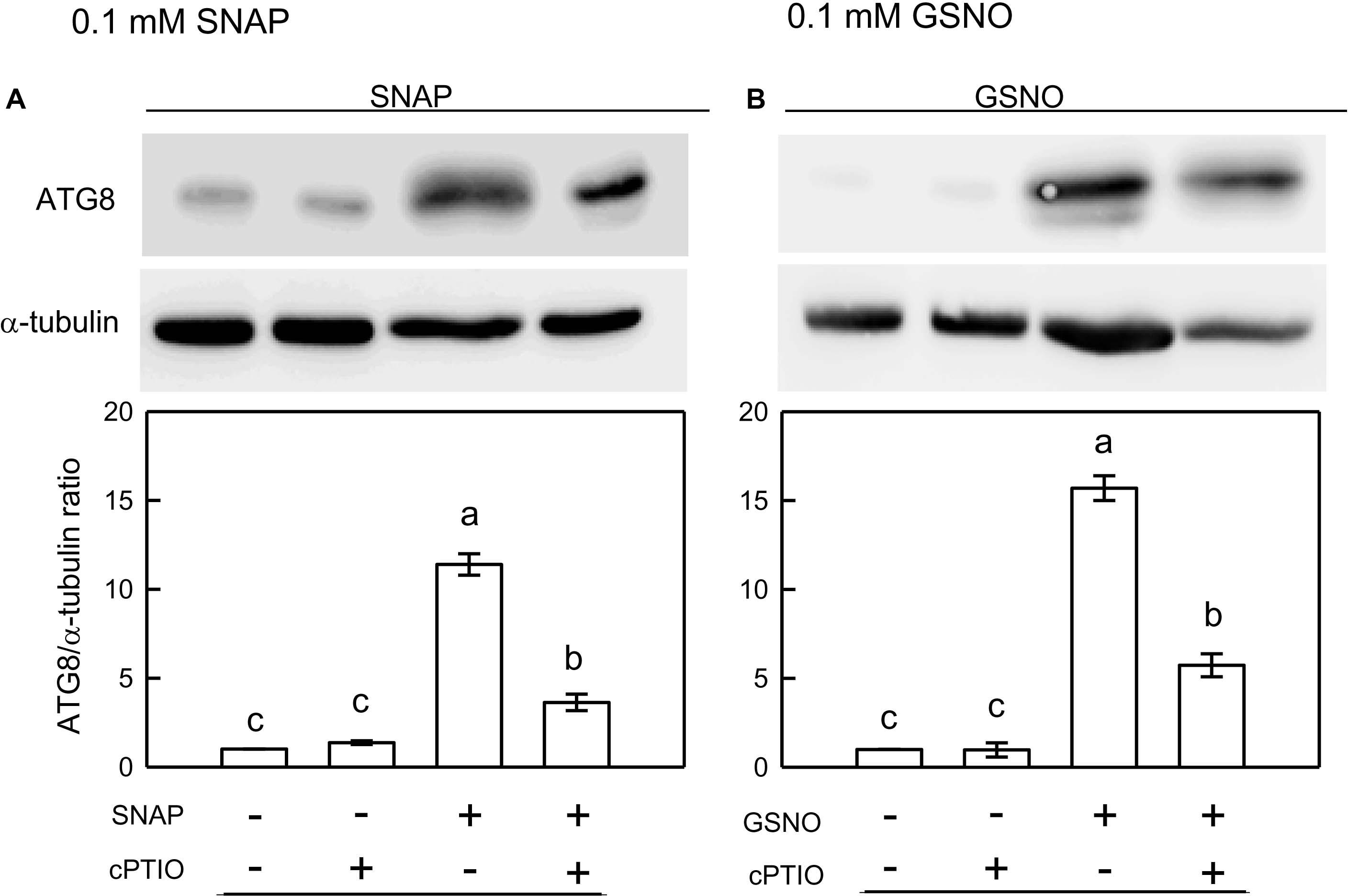
Figure 6. Immunodetection of the CrATG8 protein of Chlamydomonas reinhardtii cells after 0.1 mM SNAP (A) or 0.1 mM GSNO (B) treatment for 3 h in the presence or absence of 400 μM cPTIO under an NL (50 μmol m–2 s–1) condition. Triplicate samples were analyzed, and one of them is shown. The quantitation data are expressed as the mean ± SD (n = 3) from three independent biological replicates and different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
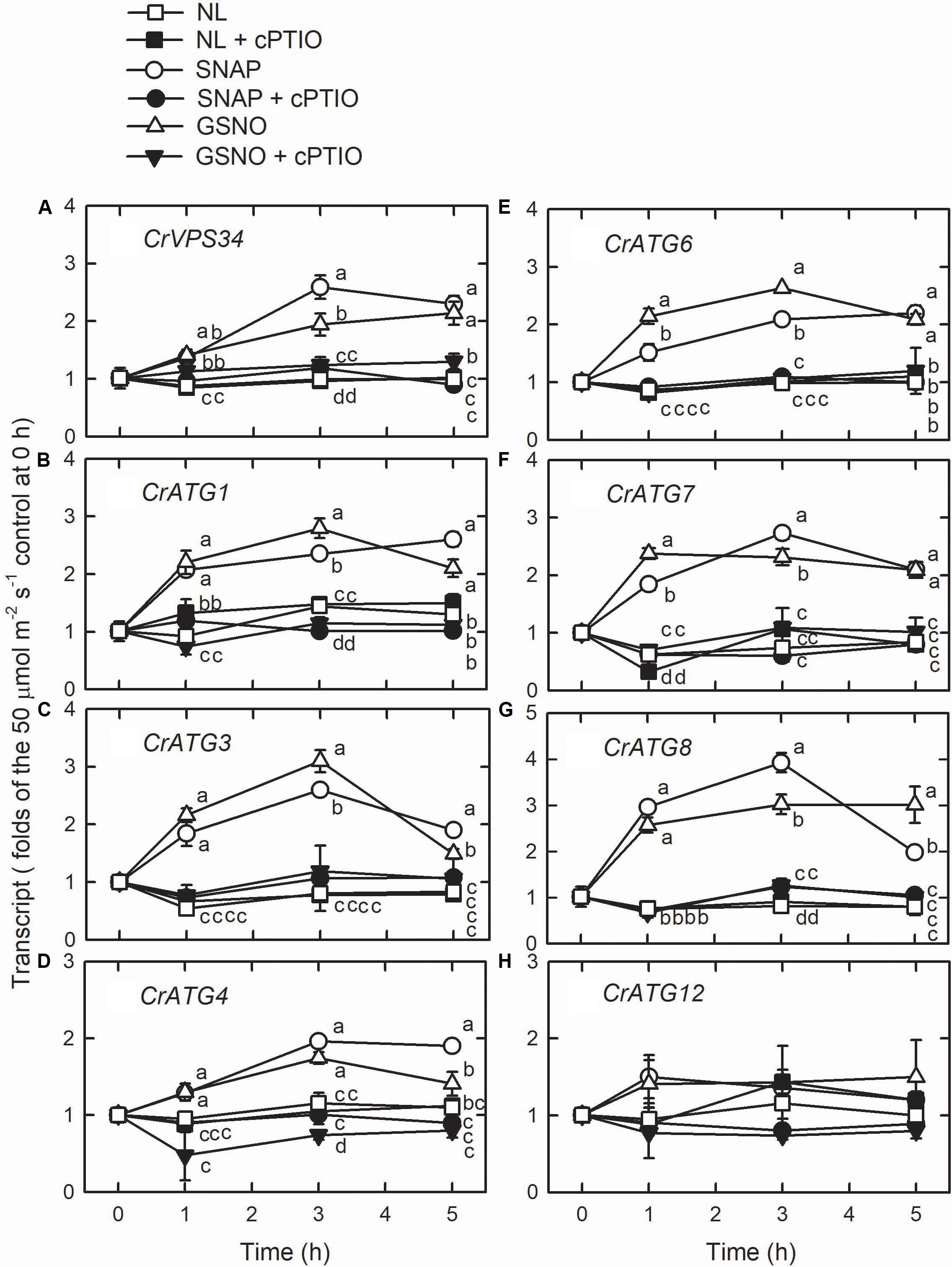
Figure 7. The time-course changes in CrVPS34 and CrATG transcripts abundance in Chlamydomonas reinhardtii cells in response to 0.05 mM SNAP or 0.05 mM GSNO under an NL (50 μmol m–2 s–1) condition. (A) CrVPS34. (B) CrATG1. (C) CrATG3. (D) CrATG4. (E) CrATG6. (F) CrATG7. (G) CrATG8. (H) CrATG12. The data are expressed as the mean ± SD (n = 3) from three independent biological replicates and different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
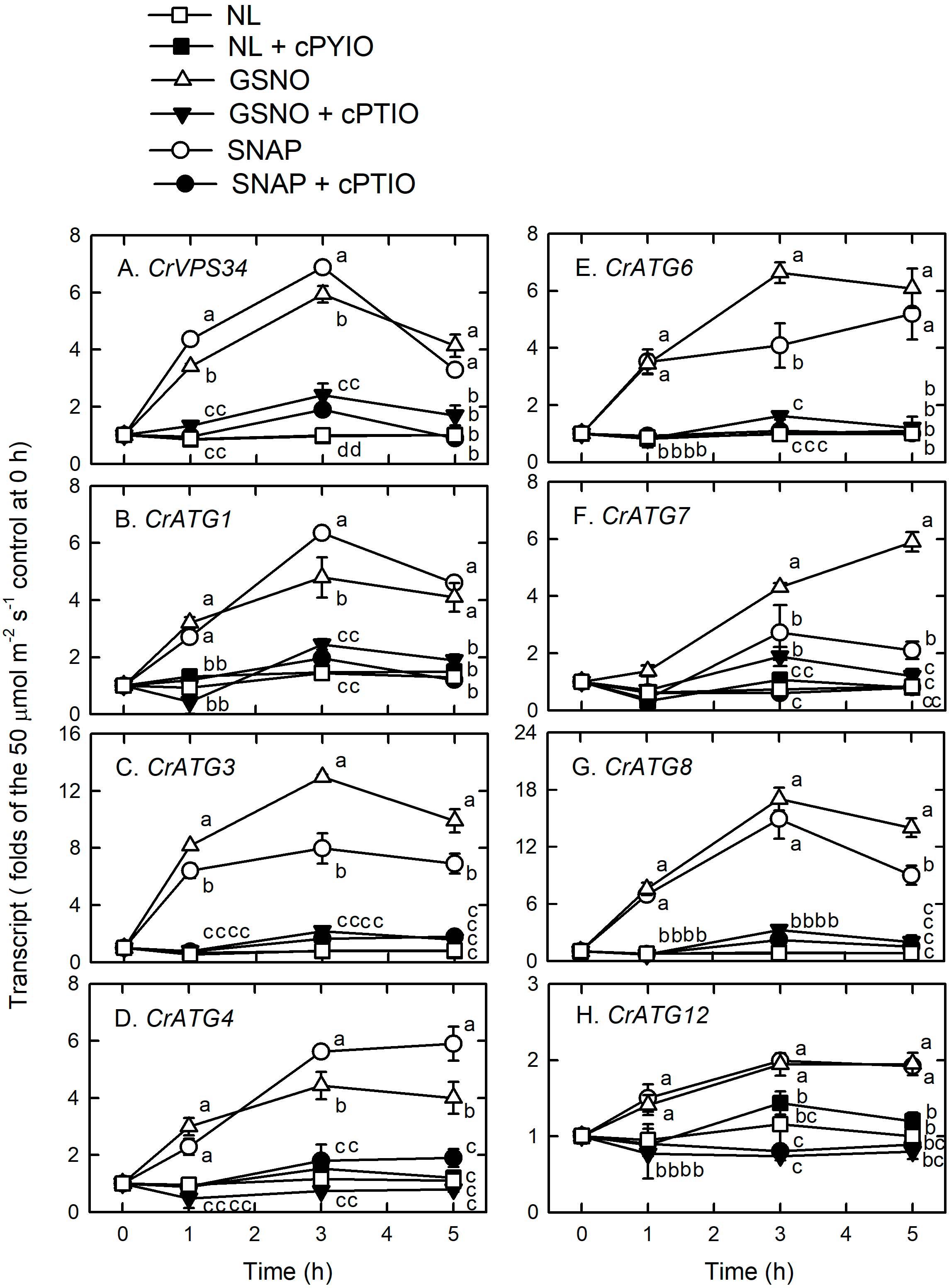
Figure 8. The time-course changes in CrVPS34 and CrATG transcripts abundance in Chlamydomonas reinhardtii cells in response to 0.1 mM SNAP or 0.1 mM GSNO under an NL (50 μmol m–2 s–1) condition. (A) CrVPS34. (B) CrATG1. (C) CrATG3. (D) CrATG4. (E) CrATG6. (F) CrATG7. (G) CrATG8. (H) CrATG12. The data are expressed as the mean ± SD (n = 3) from three independent biological replicates and different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
Treatment With NO Donors at Higher Concentrations Under the NL Condition Causes Cell Death
Whether NO can lead to cell death under the NL condition was determined. The treatment with 0.05 mM SNAP or 0.05 mM GSNO did not affect cell growth (Supplementary Figure S7) or cell viability (Figure 9 for SNAP and Figure 10 for GSNO). However, when the NO donor concentration was increased to 0.1 mM, the cell growth was temporarily inhibited during the 3–5 h after treatment but then it could be recovered to the control level after a prolonged growth period (12–24 h) (Supplementary Figure S7). Furthermore, we also observed that both 0.1 mM SNAP (Figure 9) and 0.1 mM GSNO (Figure 10) treatment for 3 h triggered a minor manifestation of cell death symptoms as reflected by the appearance of SYTOX Green fluorescence in some algal cells, and then the fluorescence was no longer observed after 24 h. In addition, the temporary decrease of cell viability and the induction of cell death by 0.1 mM NO donors at 3 h could be prevented by the presence of cPTIO (Figures 9, 10, the viability in the Figures was for 0.1 mM SNAP or GSNO treatment). The cell viability at 0.05 mM was not affected (data not shown). This demonstrates that the transient viability decline and cell death were caused by higher NO release from 0.1 mM SNAP or 0.1 mM GSNO, but it did not occur under a lower NO donor concentration of 0.05 mM.
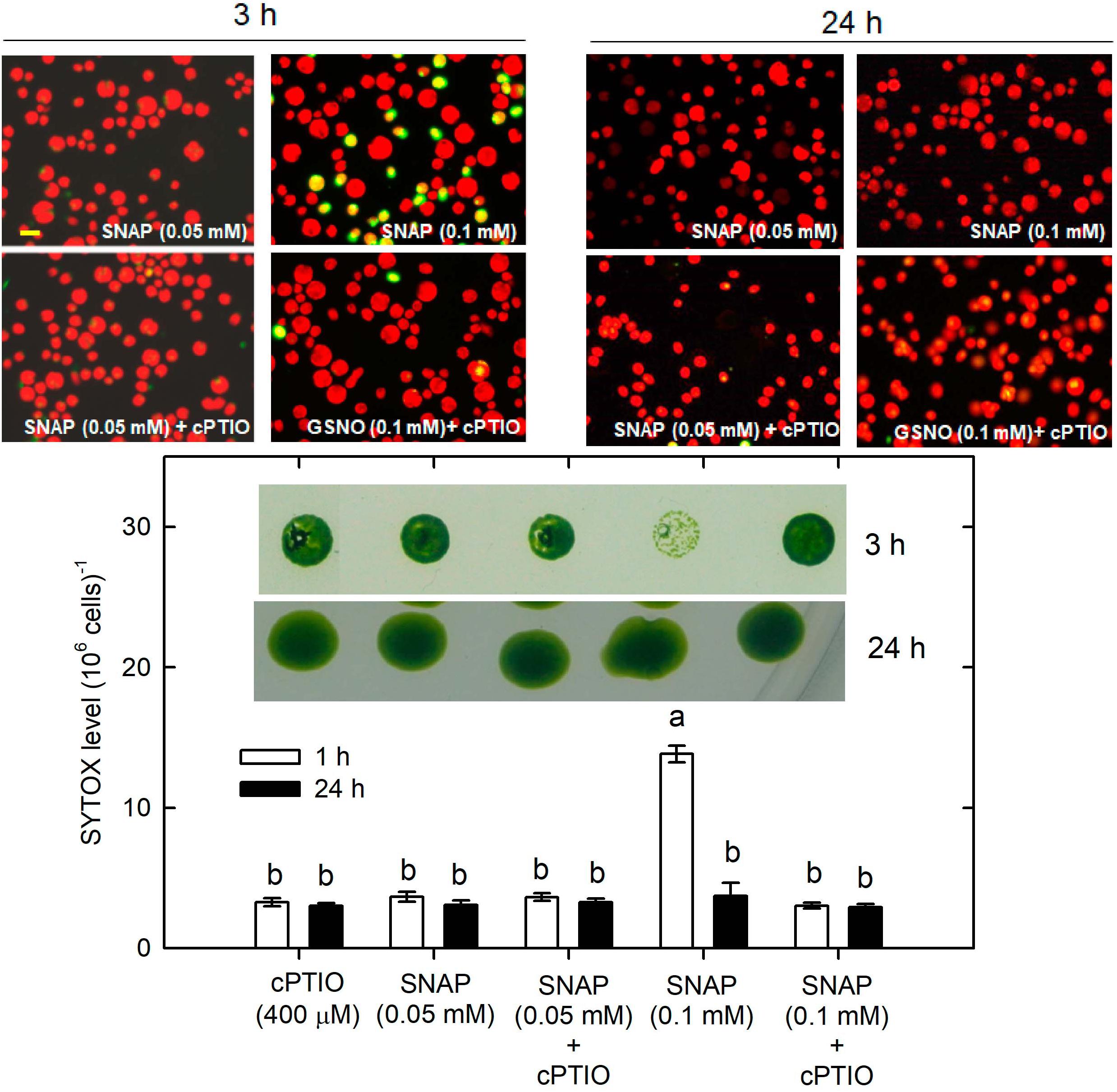
Figure 9. The effect of 0.05 (A) or 0.1 (B) mM SNAP treatment on cell viability and cell death detected by SYTOX Green fluorescence dye in C. reinhardtii cells under an NL (50 μmol m–2 s–1) condition in the presence or the absence of 400 μM cPTIO. The results for 3 and 24 h treatment are shown. The cell viability shown is after the treatment with 0.1 mM NO donors. The data are expressed as the mean ± SD (n = 3) from three independent biological replicates and the vertical bar on the symbols represents SD. Different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
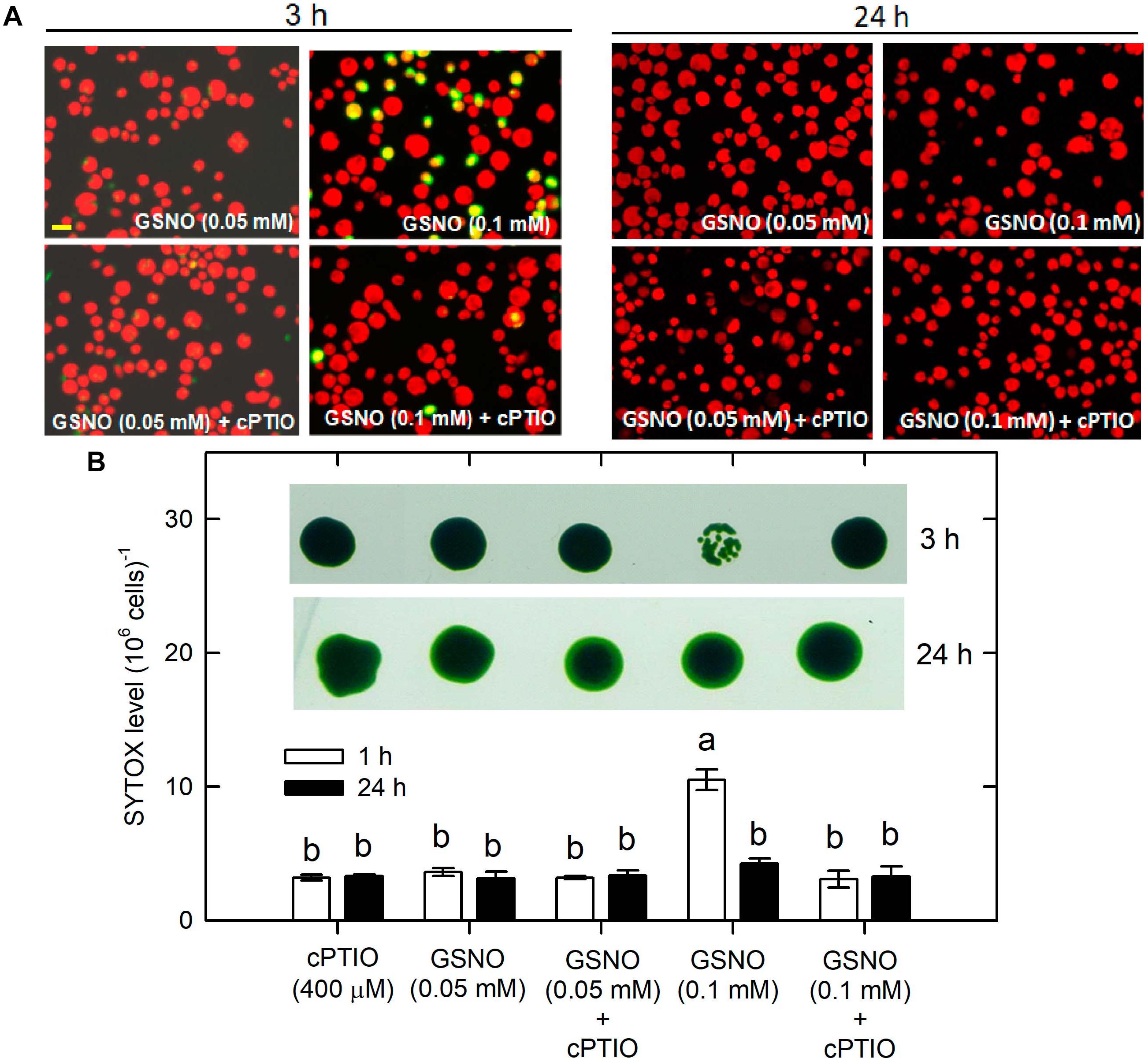
Figure 10. The effect of 0.05 (A) or 0.1 (B) mM GSNO treatment on cell viability and cell death detected by SYTOX Green fluorescence dye in C. reinhardtii cells under an NL (50 μmol m–2 s–1) condition in the presence or absence of 400 μM cPTIO. The results for 3 and 24 h treatment are shown. The cell viability shown is after the treatment with 0.1 mM NO donors. The data are expressed as the mean ± SD (n = 3) from three independent biological replicates and the vertical bar on the symbols represents SD. Different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
Treatment With NO Donors Under a Moderate HL Condition Causes Cell Death and Stimulates Autophagy Activation
Subsequently, in the attempt to elucidate whether the excessive burst of NO confers the susceptibility of C. reinhardtii cells to non-lethal HL illumination in terms of autophagy induction and cell death, the NO donors were applied under a ML condition (750 μmol m–2 s–1). An exposure to ML illumination did not induce NO production (Supplementary Figure S1A), but it triggered a linear increase in H2O2 production as the treatment time advanced (Supplementary Figure S6). However, the level of ML-induced H2O2 production was lower than that under the 1,600 μmol m–2 s–1 condition (Supplementary Figure S6). Furthermore, we found that although H2O2 was produced under ML illumination, the algal cells exhibited a similar growth ability to those grown under an NL condition (Supplementary Figure S1B). This indicates that Chlamydomonas cells can tolerate accumulated H2O2 and acclimate to moderate light stress.
Then, NO donors were administered under the ML condition to clarify whether NO could increase the susceptibility of algal cells under the ML condition. The results depended on the concentration of NO donors and the treatment time. Both cell density (Figure 11A) and viability (Figure 11B) were not affected by the presence of 0.05 mM GSNO or 0.05 mM SNAP but showed a decrease in response to 0.1 mM NO donor treatment. The cell death detected using SYTOX-Green fluorescence did not appear under treatment with 0.05 mM NO donors whereas it exhibited a marked increase under treatment with 0.1 mM NO donors (Figure 11C). The presence of cPTIO can suppress the negative effect of 0.1 mM NO donor treatment on cell activity (Figure 11C). The present findings demonstrate that supplementation with NO confers susceptibility of C. reinhardtii cells to moderate HL illumination.
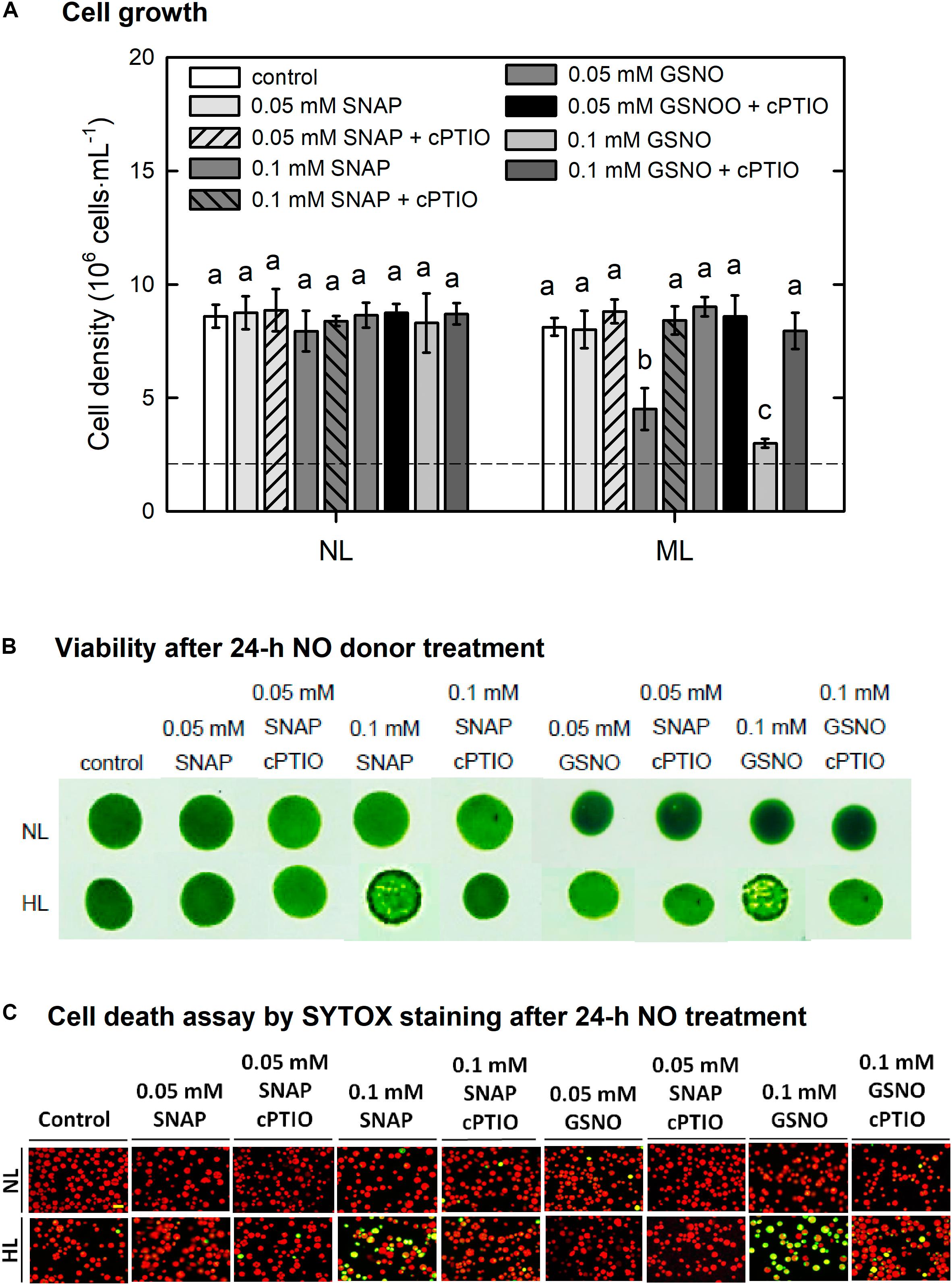
Figure 11. The effect of 0.05 or 0.1 mM SNAP and GSNO treatment on cell growth (A), viability (B), and cell death (C) detected by SYTOX Green fluorescence dye in C. reinhardtii cells under an ML (750 μmol m–2 s–1) condition in the presence or absence of 400 μM cPTIO for 24 h. The data are expressed as the mean ± SD (n = 3) from three independent biological replicates and the vertical bar on the symbols represents SD. Different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
The transcripts abundance of CrVPS34 (Figure 12A), CrATG1 (Figure 12B), CrATG3 (Figure 12C), CrATG4 (Figure 12C), CrATG6 (Figure 12E), CrATG7 (Figure 12F), and CrATG8 (Figure 12G) slightly increased 3 h after ML treatment and their increase can be enhanced by the supplementation with 0.1 mM NO donors, and this enhancement can be suppressed in the presence of cPTIO. Among the CrATG genes, the transcript abundance of CrATG12 was not increased by exposure to ML treatment, while the application of 0.1 mM NO donors induced an increase of its expression and this increase was also suppressed in the presence of cPTIO (Figure 12H).
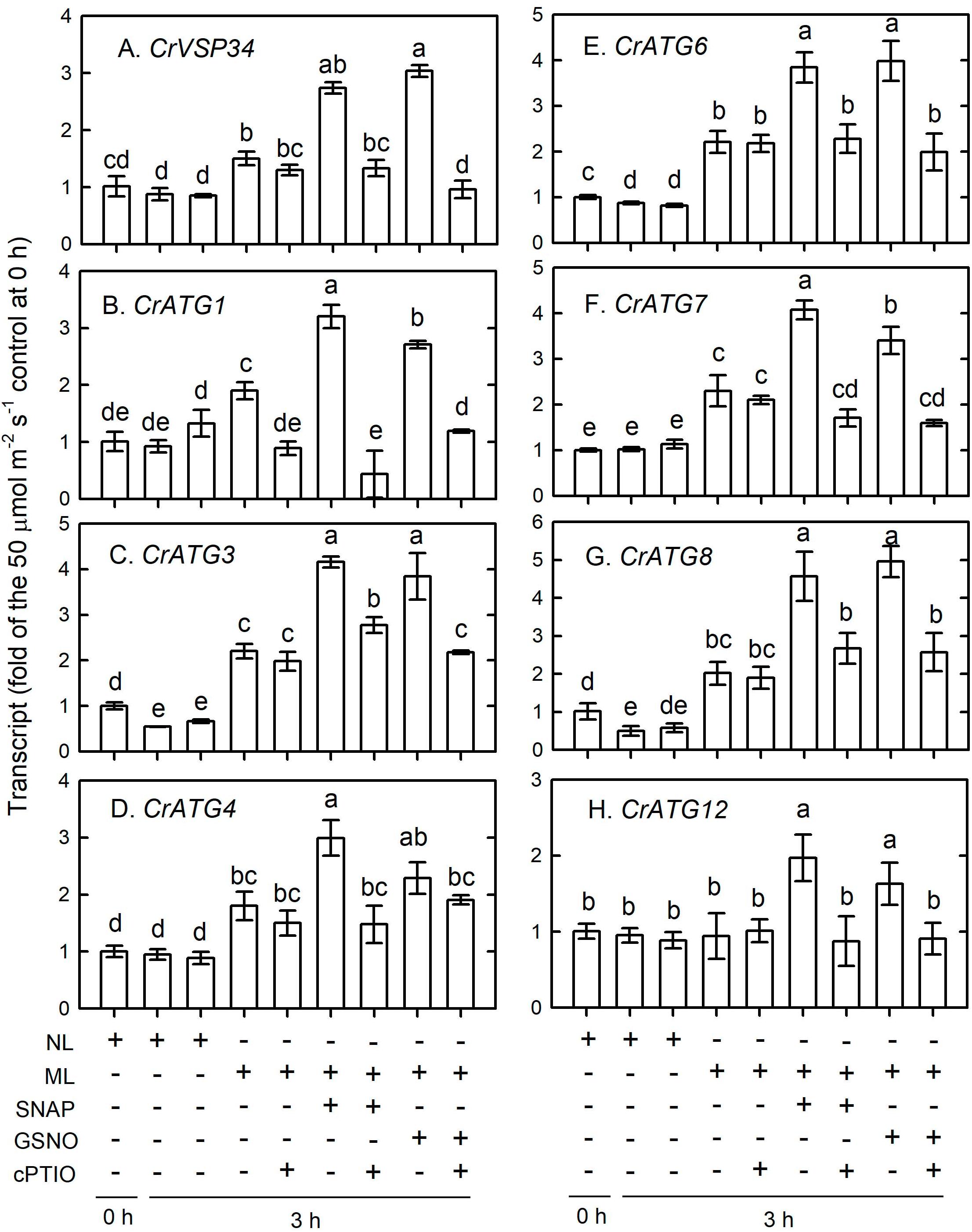
Figure 12. The time-course changes in CrVPS34 and CrATG transcripts abundance in Chlamydomonas reinhardtii cells in response to 0.1 mM SNAP or 0.1 mM GSNO treatment under an ML (750 μmol m–2 s–1) condition for 3 h. (A) CrVPS34. (B) CrATG1. (C) CrATG3. (D) CrATG4. (E) CrATG6. (F) CrATG7. (G) CrATG8. (H) CrATG12. The data are expressed as the mean ± SD (n = 3) from three independent biological replicates and different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
Interaction of NO and H2O2 for Autophagy Induction and Cell Death
The above results indicate that NO may interact with H2O2 for the induction of autophagy and cell death in C. reinhardtii cells. Therefore, NO donors at a concentration of 0.1 mM were applied together with 1 mM H2O2 under an NL condition for 3 h. The results shown in Figure 13A indicate that 1 mM H2O2 slightly increased the transcript abundance of CrATG8, while the combination of H2O2 and NO donors induced a marked increase in CrATG8 transcript abundance, which was inhibited in the presence of cPTIO. The ATG8 protein abundance in response to H2O2 treatment for 3 h slightly increased but the combination of H2O2 and NO donors induced a significant increase (Figure 13B).
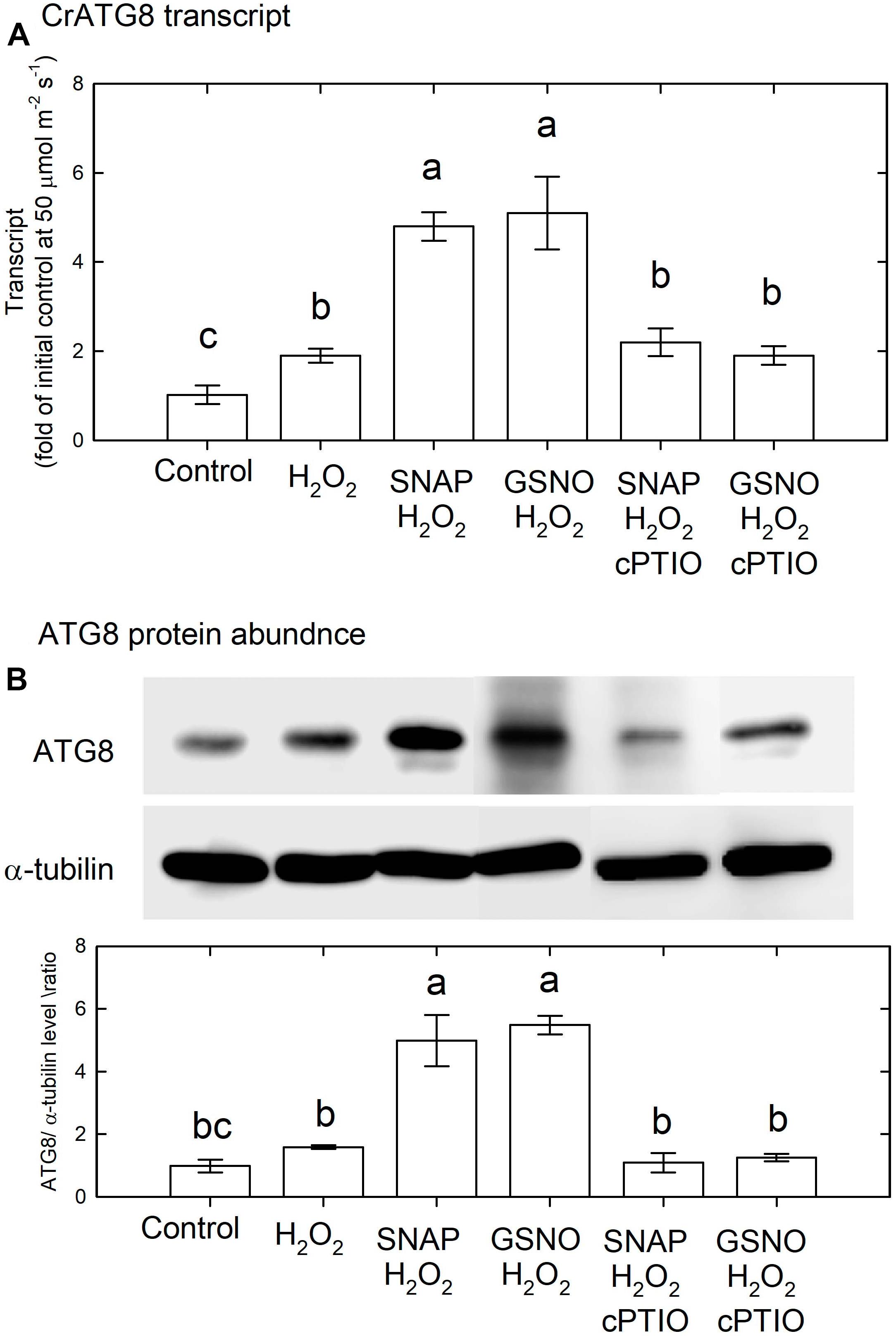
Figure 13. The effect of SNAP (0.1 mM), GSNO (0.1 mM), and 1 mM H2O2 treatment on the CrATG8 transcript abundance (A) and ATG8 protein abundance (B) of Chlamydomonas reinhardtii cells under an NL condition (50 μmol m–2 s–1) for 3 h. The cPTIO (0.4 mM) was added to the SNAP + H2O2 and GSNO + H2O2 treatments. The data are expressed as the mean ± SD (n = 3) from three independent biological replicates and different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
The cell viability and cell death were also determined in response to H2O2 and/or NO donors in the presence or the absence of cPTIO under an NL condition for 24 h. We found that 1 mM H2O2 treatment did not affect cell viability and cell death, while 0.05 mM NO donors also did not affect cell viability and cell death (Figure 14). However, the combination of H2O2 and NO donor treatment induced a significant decrease in cell viability and resulted in significant cell death, which could be inhibited in the presence of 0.4 mM cPTIO (Figure 14). This demonstrated that NO interacts with H2O2 in the induction of cell damage under an NL condition.
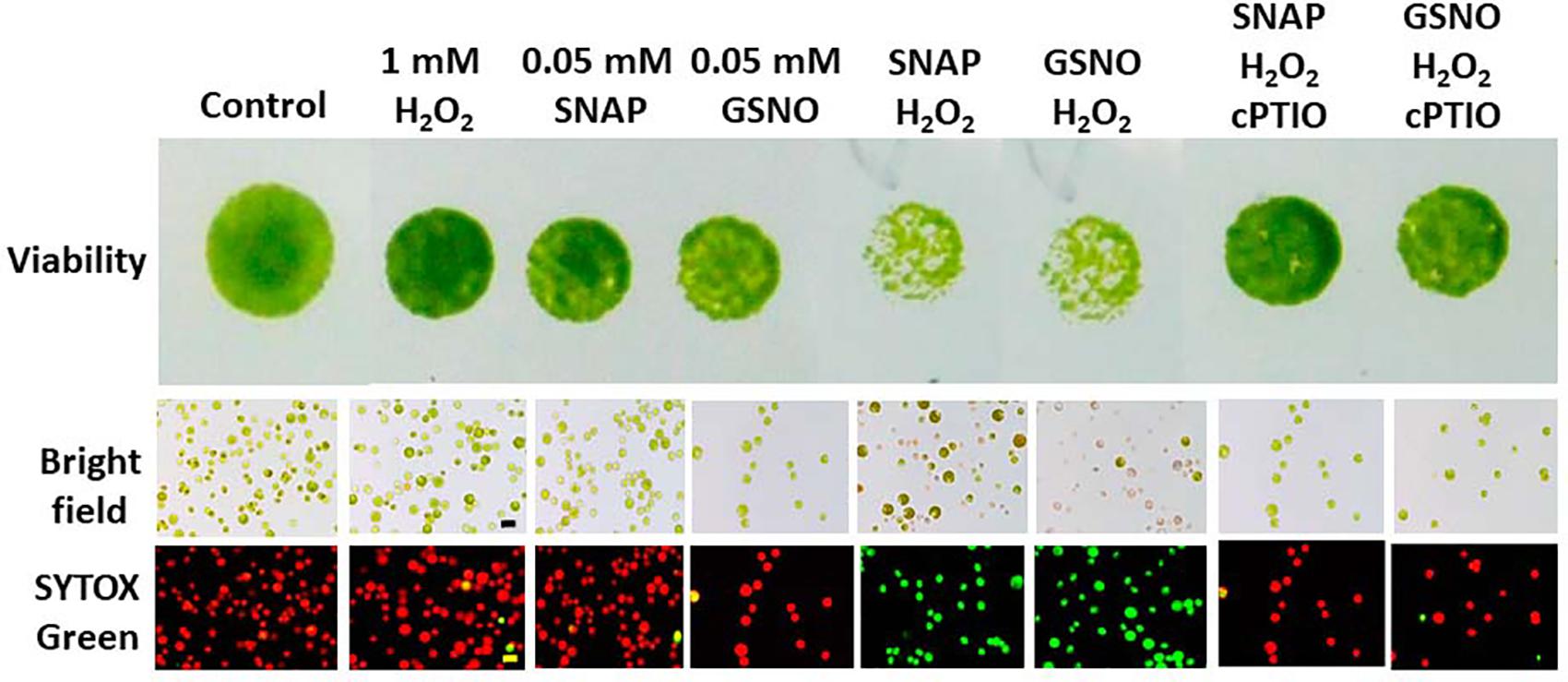
Figure 14. The effect of SNAP (0.1 mM), GSNO (0.1 mM), and 1 mM H2O2 treatment on the growth (cell density) (A) and viability (B) of Chlamydomonas reinhardtii cells under an NL condition (50 μmol m–2 s–1) for 24 h. The cPTIO (400 μM) was applied to the SNAP + H2O2 and GSNO + H2O2 treatments. The data are expressed as the mean ± SD (n = 3) from three independent biological replicates and different letters indicate the statistical significance set at P < 0.05 according to ANOVA.
Discussion
Treatment of C. reinhardtii cells with high intensity illumination of 1,600 μmol m–2 s–1 triggered an NO burst during 3–5 h. An exposure to extremely high-light illumination at 3,000 μmol m–2 s–1 can also induce a significant NO production in C. reinhardtii cells (Chang et al., 2013). However, moderate HL illumination at 750 μmol m–2 s–1 intensity did not trigger NO production. This demonstrates that the generation of NO occurs only when the light intensity is higher than a certain threshold.
Recently, it has been shown that C. reinhardtii can metabolize NO to N2O and this metabolism is dependent on photosynthetic electron transport. These findings are supported by the data obtained using treatment with 3,4-dichlorophenyl-1,1-dimethylurea (DCMU), a photosystem II (PSII) inhibitor, or 2,5-dibromo-3-methyl-6-isopopyl-p-benzoquinone (DBMIB), an inhibitor of electron flow between PSII and PSI (Burlacot et al., 2020). This raises the possibility that the conversion of NO to N2O may lead to the disappearance of NO under the 750 μmol m–2 s–1 condition. However, we found that the application of 10 M DCMO or 2 M DBMIB did not increase NO production under the 750 μmol m–2 s–1 condition (data not shown). Thus, undetectable NO under the 750 μmol m–2 s–1 condition is not due to the degradation of NO to N2O.
The present results that the presence of L-NAME or tungstate cannot suppress the induction of NO production in C. reinhardtii cells by HL illumination suggests that routes other than the NOS and NR pathways are responsible for the HL-induced NO synthesis in C. reinhardtii cells. So far, whether NOS exists in plants is under debate, and the presence of NOS has not been verified in C. reinhardtii, as we know. Furthermore, the strain CC125 used in this study is the 137C mt− carrying the nit1 and nit2 mutations (mt− nit1 nit2) that lacks NR and does not express the proteins required for nitrate assimilation (NIA1, NRT2.1, NRT2.2, NII1, NAR2, and NAR1.1) (Galván and Fernández, 2001). The strain CC124, with genotype mt+ nit1 nit2, also exhibited a similar NO production pattern under 1,600 μmol m–2 s–1 illumination (Supplementary Figure S2). This suggests that the HL-induced NO production in C. reinhardtii cells is independent of NR. In fact, several studies have shown NO production in NR-defective C. reinhardtii mutants under hypoxia conditions (Hemschemeier et al., 2013) or in C. reinhardtii wild-type strain 137C (+) treated with MP (Yordanova et al., 2010). However, Wei et al. (2014) reported that the NR-defective C. reinahrdtii mutants produce less NO in response to nitrogen starvation, while a significant NO release is found in strains of the genotype NIT1 NIT2 with normal NR activity. It is therefore suggested that the NR-dependent route is responsible for the production of NO from C. reinahrdtii cells under nitrogen starvation (Wei et al., 2014). The above results indicate that the routes used for the synthesis of NO in C. reinhardtii cells are different not only between the strains with various genetic backgrounds but also between different treatment conditions. Our present findings demonstrate that C. reinahrdtii lacking NR can generate marked NO production via NR- and NOS-independent routes under illumination at a very high intensity of 1,600 μmol m–2 s–1.
The evidence suggests that NO contributes to the activation of autophagy in C. reinhardtii cells by HL illumination at 1,600 μmol m–2 s–1 intensity. When the marked NO burst occurs during 3–5 h following HL illumination, C. reinhardtii cells concomitantly exhibited marked autophagy induction, supported by an increase in the abundance of CrVPS34 and CrATG transcripts and CrATG8 protein, a Chlamydomonas autophagy marker (Pérez-Pérez et al., 2010). The results of Pérez-Pérez et al. (2012a) also showed that there is a transient activation of autophagy with a peak at 6 h in C. reinhardtii cells subjected to 1,200 μmol m–2 s–1 illumination, reflected by increased CrATG8 protein abundance detected using western blot and immunofluorescence analyses. More importantly, the present finding that the induction of autophagy by illumination at a higher light intensity up to 1,600 μmol m–2 s–1 can be suppressed in the presence of a NO scavenger, cPTIO, which demonstrates that the activation of autophagy under the 1,600 μmol m–2 s–1 condition can be attributable to NO. Similarly, the VHL (3,000 μmol m–2 s–1)-induced increase in NO production (Chang et al., 2013) and the abundance of CrATG8 protein (Supplementary Figure S4) and the CrVPS34, CrATG, and CrTOR transcripts (Supplementary Figure S5) in C. reinhardtii can be suppressed by cPTIO treatment.
Furthermore, treatment with GSNO or SNAP at either 0.05 or 0.1 mM under the 50 μmol m–2 s–1 condition is able to trigger autophagy, reflected by the increase of CrVPS34 and CrATG transcripts abundance and CrATG8 protein abundance, which is suppressed in the presence of cPTIO. Obviously, NO can trigger autophagy without other factors derived from HL illumination. As far as we know, this is the first time that NO has been shown to be involved in the modulation of HL-induced autophagy in C. reinhardtii cells through upregulation of CrVPS34 and CrATG gene expression. The effect of NO on the induction of autophagy is well-known in animal systems (Barsoum et al., 2006; Yang et al., 2008; Sarkar et al., 2011; Duana et al., 2013; Tripathi et al., 2013).
The upregulation of CrVPS34 and CrATG expression by NO and the suppression of their expression by cPTIO indicated that the signaling components are involved in the NO-mediated activation of autophagy under HL illumination. Among the core autophagy machineries, autophagy induction is driven by the association of ATG1 kinase with dephosphorylated ATG13 and other regulatory ATG proteins for the construction of a preautophagosomal structure (Kamada et al., 2000). Subsequently, vesicle nucleation is initiated by the activation of vacuolar-protein-sorting (VPS) 34 PI3K complex containing ATG6 and other VPS proteins (Thompson and Vierstra, 2005). VPS34 is the Class III PI3K, which is conserved from yeasts and plants to mammals (Engelman et al., 2006) for the phosphorylation of phosphatidylinositol to generate phosphatidylinositol 3-phosphate that is a phospholipid central to membrane trafficking processes, is required for the formation of the autophagosome (Itakura et al., 2008; Jaber et al., 2012). Here, the increase of CrVPS34, CrATG1, and CrATG6 transcripts abundance 3 h after HL illumination may indicate that the vesicle nucleation of the phagophore assembly site to form the autophagosome is executed in C. reinhardtii cells under HL stress. Subsequently, an increase in CrATG8 protein abundance and transcripts abundance of CrATG3, CrATG4, CrATG7, and CrATG8 function in the expansion of the autophagosomal membrane and its fusion with the lysosome/vacuolar membrane through the ubiquitin-like protein conjugation system (Ohsumi, 2001; Thompson and Vierstra, 2005; Hanada et al., 2007; Nakatogawa et al., 2007; Xie et al., 2008), which implies the implementation of a mature autophagosome and autolysosome formation in C. reinhardtii cells under the control of NO generated under HL illumination.
Our findings indicate that the expression of CrATG8 is regulated by NO and other factors under very HL conditions of 1,600 μmol m–2 s–1. An increase in CrATG8 transcript abundance by prolonged HL illumination (5 h) is partially inhibited in the presence of cPTIO. Even when cPTIO was administered at a concentration up to 800 M, the CrATG8 transcript abundance still remained at a level approximately 24% of the HL treatment without cPTIO application (data not shown). This may reflect that other factors work with NO for the induction of autophagy in C. reinhardtii under the 1,600 μmol m–2 s–1 condition. Pérez-Pérez et al. (2012a) have shown increased levels of ROS in the chloroplast and a marked increase in autophagic activity due to the absence of photoprotection in C. reinhardtii cells. This may suggest that ROS are also the factors that induce CrATG8 expression (protein abundance) in C. reinhardtii cells exposed to the 1,600 μmol m–2 s–1 condition. In addition, their study showed that the treatment with 1 mM H2O2 under a 20–30 μmol m–2 s–1 condition or a dark condition, or 1 M MV under a 20–30 μmol m–2 s–1 condition for 8 h, can increase the CrATG8 protein abundance (Pérez-Pérez et al., 2012a). In addition, in a study of the responses of a starchless (sta6) mutant to oxidative stress and the induction of autophagy in C. reinhardtii cells, Tran et al. (2019) showed that there is a dose-dependent expression pattern of ATG8 protein detected using western blot analysis with a linear increase in ATG8 protein level in response to a gradual increase in H2O2 concentration from 0, 0.25, 0.50, to 1 mM. Here, we found that the H2O2 concentration was significantly increased by exposure to 1,600 μmol m–2 s–1 illumination (Supplementary Figure S6). Moreover, we also observed that the exposure of C. reinhardtii cells to 1 mM H2O2 for 3 h under an NL condition increased the ATG8 protein abundance and CrATG8 transcript abundance without any adverse effect on cell viability. The application of NO together with H2O2 induces significant increases in ATG8 protein and CrATG8 transcript abundances, which can be suppressed by cPTIO treatment. In addition, although autophagy is not occurring under the ML condition, the administration of 0.1 mM NO donors can induce autophagy under the ML condition. This provides evidence that, in addition to NO, H2O2 is also involved as a signal for the activation of autophagy by 1,600 μmol m–2 s–1 illumination of C. reinhardtii cells.
In addition to the induction of autophagy, 1,600 μmol m–2 s–1 illumination resulted in cell death (bleaching) after 5 h. The prevention of HL-induced cell death by cPTIO treatment supports the hypothesis that NO is involved in the HL-induced cell death. In addition, our evidence shows that other factors, such as ROS, may interact with NO for the induction of cell death in C. reinhardtii cells under lethal HL stress (1,600 μmol m–2 s–1). H2O2 has been shown to be involved in plant cell death (Houot et al., 2001). However, the effect of H2O2 on the induction of cell death works in a concentration-dependent manner. Murik et al. (2014) provided evidence that the growth rate of C. reinhardtii cells is not affected by H2O2 at concentrations below 2 mM whereas it was markedly retarded on exposure to higher H2O2 concentrations in a range between 5 and 10 mM. Here, illumination of C. reinhardtii cells at an intensity of 1,600 μmol m–2 s–1 can induce significant H2O2 accumulation (Supplementary Figure S6). The evidence shows that NO bursts along with H2O2 accumulation occur as the 1,600 μmol m–2 s–1 illumination advances to 5 h (Figure 1A), and treatment with 0.1 mM NO donors can induce a transient growth depression and cell death under an NL condition. More importantly, the presence of the NO scavenger, cPTIO, can effectively prevent growth retardation and cell death caused by the 0.1 mM NO donors. Furthermore, under the ML condition (750 μmol m–2 s–1), the application of 0.1 mM NO donors can cause cell death and growth retardation, which is prevented by cPTIO treatment. Because H2O2 also accumulates under the ML condition (750 μmol m–2 s–1), this again supports that the NO released from the NO donors interacts with H2O2 for the induction of cell death.
To further prove this proposal, we treated C. reinhardtii cells with 1 mM H2O2 and 0.05 mM NO donors together under an NL condition to check whether the application of both chemicals can cause cell death without HL interference. Our results confirmed that exogenously applied NO acts synchronously with accumulated H2O2 to enhance the sensitivity of C. reinhardtii cells to moderate HL illumination, leading to a decrease in cell viability and partial cell death, whereas the treatment with 1 mM H2O2 alone could not affect cell growth and the treatment with 0.05 mM NO donors also did not impact cell activity. Overall, these lines of evidence demonstrate that excessive NO can interact with H2O2 for the induction of cell death in C. reinhardtii cells under lethal HL illumination. In higher plants, NO bursts after a pathogen attack initiate gene-to-gene defense responses and they also interact with H2O2 and other signals in triggering PCD (Delledonne et al., 2001; Hong et al., 2008; Floryszak-Wieczorek and Arasimowicz-Jelonek, 2010). In the investigation of the MP-induced cell death in C. reinhardtii cells, Yordanova et al. (2010) have identified that NO cooperates with ethylene for the induction of cell death caused by MP. Obviously, NO interacts with other factors for the induction of cell death in C. reinhardtii cells, depending on the different stress conditions.
It has long been known that NO exhibits a dual function, beneficial or harmful, in stress conditions depending on its concentration, the type of tissue, age or physiological status, its ability to interact with other signaling molecules, and the type of stress (Delledonne et al., 2001; Arasimowicza and Floryszak-Wieczorek, 2007; Das et al., 2012). For instance, NO at a high concentration causes membrane breakdown, DNA fragmentation, and cell death (Pedroso et al., 2000; Yamasaki, 2000; Romero-Puertas et al., 2004). NO is also involved in the regulation of hypersensitive cell death (Clarke et al., 2000; de Pinto et al., 2002) and stress-induced cell death (Ahlfors et al., 2009; de Michele et al., 2009) in higher plants. In the diatoms, treatment with NO donors mimics aldehyde-induced cell death (Vardi et al., 2006). A negative role of NO on algal growth has been shown in the unicellular green alga Micrasterias denticulate (Brébisson) by exogenous application of a NO donor. The NO suppresses algal cell growth via the inhibition of the activity of enzymes involved in the secretary pathway (Lehner et al., 2009). NO also triggers cell death in algae; for example, aldehyde-induced cell death in diatoms (Vardi et al., 2006) and the heat-induced cell death of symbiotic alga Symbiodinium microadriaticum Freudenthal (Bouchard and Yamasaki, 2008). Previously, we have reported that mass generation of NO at a higher concentration under a 3,000 μmol m–2 s–1 condition contributes to the induction of cell death of C. reinhardtii cells (Chang et al., 2013). Therefore, NO overproduced in C. reinhardtii under extreme HL illumination opens the way to cell death. In contrast, our current study shows that, at lower concentrations, NO does not affect cell growth or cause permanent cell damage to C. reinhardtii cells. Treatment with 0.05 mM NO donors does not affect cell activity while 0.1 mM NO donors can trigger a transient effect on cell activity, followed by a rapid recovery after prolonged culture.
In addition to the concentration effect, the promotion or prevention of cell death is also dependent on the plant systems and conditions. In some plant tissues, NO counteracts PCD. For example, NO delays the onset of GA-induced PCD in barley (Hordeum vulgare cv Himalaya) aleurone layers (Beligni et al., 2002). In a study of a carotenoid deficient mutant, lts1-204, in the regulation of autophagy induction in C. reinhardtii cells by oxidative stress, Pérez-Pérez et al. (2010) demonstrated that 1,200 μmol m–2 s–1 illumination transiently activated autophagy in wild-type Chlamydomonas cells as part of an adaptation response to oxidative stress. In contrast, our current investigation implies that excessive NO generated under lethal HL illumination at the intensity of 1,600 μmol m–2 s–1 cooperates with highly accumulated H2O2 for the modulation of the signaling pathways, causing activation of autophagy, which leads to the death of C. reinhardtii cells.
First, we showed that autophagy induction is positively correlated with cell death in response to a lethal HL condition. This is in contrast to other conditions, such as nutrient starvation, where autophagy is considered to act as a protective process for C. reinhardtii against stress conditions. The review article of Pérez-Pérez et al. (2012b) has concluded from many studies that autophagy is conserved in plants and algae and plays a role in the adaptation to different stress conditions for the recycling of ROS-mediated oxidatively damaged macromolecules and organelles. Similarly, in hypersensitive plants, autophagy functions in the prevention of PCD (Greenberg, 2005). In contrast, in a study of the pathogen causing rice blast disease (Magnaporthe grisea), the use of an Mgatg8 knockout line confirmed that autophagy is responsible for death of the spores (Veneault-Fourrey et al., 2006). Furthermore, in animal cells, certain forms of cell death can be inhibited by treatment with autophagic inhibitors or by reduced ATG gene expression (Shimizu et al., 2004; Yu et al., 2004). It has also been shown that autophagy-induced cell death is linked to unbalanced ROS production in animal cells. Using the mammalian L929 cell line, a widely studied model for TNF-α-induced cell death, caspase inhibitor, benzyloxycarbonyl-valyl-alanyl-aspartic-acid (O-methyl)-fluoromethylketone (zVAD) treatment results in selective autophagic degradation of catalase, a key enzymatic ROS scavenger, and in turn, this leads to marked ROS accumulation and ultimately causes non-apoptotic death (Yu et al., 2006). Thus, these findings imply that autophagy initiates and participates directly in the death process in animal systems. In the green alga Chlamydomonas, our current evidence shows that NO and H2O2 interact in the induction of autophagic cell death when exposed to lethal HL conditions. In the near future, a genetic investigation using ATG mutants is necessary to identify the network linking autophagy and cell death in Chlamydomonas under lethal HL stress.
In conclusion, the present study is the first to clearly demonstrate the interaction between NO and H2O2 in the activation of autophagic cell death in C. reinhardtii under lethal HL stress. This finding provides a background for further examination of the role of NO as a signaling molecule in the control of autophagy induction and the role of ROS interactions with NO during the induction of cell death in C. reinhardtii under lethal HL stress.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
H-LC and EK performed the physiological analysis, RNA extraction, cDNA preparation, and qPCR. S-TL contributed to the preparation of reagents and the determination of biochemical and physiological parameters. T-ML conceived and designed the experiments, interpreted the data, and wrote the manuscript. All authors discussed and reviewed this manuscript.
Funding
This work was supported by grants (NSC99-2311-B-110-001-MY3 and NSC103-2311-B-110-001-MY3) from the Ministry of Science Technology, Executive Yuan, Taiwan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Su-Chiung Fang, Academia Sinica Biotechnology Center in Southern Taiwan, Tainan, Taiwan, for valuable comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00772/full#supplementary-material
Footnotes
References
Ahlfors, R., Brosché, M., Kollist, H., and Kangasjärvi, J. (2009). Nitric oxide modulates ozone-induced cell death, hormone biosynthesis and gene expression in Arabidopsis thaliana. Plant J. 58, 1–12. doi: 10.1111/j.1365-313X.2008.03756.x
Anbar, M. (1995). Nitric oxide: a synchronizing chemical messenger. Experientia 51, 545–550. doi: 10.1007/bf02128740
Arasimowicza, M., and Floryszak-Wieczorek, J. (2007). Nitric oxide as a bioactive signalling molecule in plant stress responses. Plant Sci. 172, 876–887. doi: 10.1016/j.plantsci.2007.02.005
Bajguz, A. (2014). “Nitric oxide: role in plants under abiotic stress,” in Physiological Mechanisms and Adaptation Strategies in Plants Under Changing Environments, eds R. Ahmad and M. R. Wani (New York, NY: Springer), 137–160.
Barsoum, M. J., Yuan, H., Gerencser, A. A., Liot, G., Kushnareva, Y., Graber, S., et al. (2006). Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 25, 3900–3911. doi: 10.1038/sj.emboj.7601253
Bassham, D. C., and Crespo, J. L. (2014). Autophagy in plants and algae. Front. Plant Sci. 5:679. doi: 10.3389/fpls.2014.00679
Beligni, M. V., Fathc, A., Bethke, P. C., Lamattina, L., and Jones, R. L. (2002). Nitric oxide acts as an antioxidant and delays programmed cell death in barley aleurone layers. Plant Physiol. 129, 1642–1650. doi: 10.1104/pp.002337
Besson-Bard, A., Pugin, A., and Wendehenne, D. (2008). New insights into nitric oxide signaling in plants. Annu. Rev. Plant Biol. 59, 21–39. doi: 10.1146/annurev.arplant.59.032607.092830
Bouchard, J. N., and Yamasaki, H. (2008). Heat stress stimulates nitric oxide production in Symbiodinium microadriaticum: a possible linkage between nitric oxide and the coral bleaching phenomenon. Plant Cell Physiol. 49, 641–652. doi: 10.1093/pcp/pcn037
Bradford, M. M. (1976). A rapid method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1016/0003-2697(76)90527-3
Burlacot, A., Richaud, P., Gosset, A., Li-Beisson, Y., and Peltier, G. (2020). Algal photosynthesis converts nitric oxide into nitrous oxide. Proc. Natl. Acad. Sci. U.S.A. 117, 2704–2709. doi: 10.1073/pnas.1915276117
Chang, H. L., Hsu, Y. T., Kang, C. Y., and Lee, T. M. (2013). Nitric oxide down-regulation of carotenoid synthesis and PSII activity in relation to very high light-induced singlet oxygen production and oxidative stress in Chlamydomonas reinhardtii. Plant Cell Physiol. 54, 1296–1315. doi: 10.1093/pcp/pct078
Clarke, A., Desikan, R., Hurst, R. D., Hancock, J. T., and Neill, S. J. (2000). NO way back: nitric oxide and programmed cell death in Arabidopsis thaliana suspension cultures. Plant J. 24, 667–677. doi: 10.1046/j.1365-313x.2000.00911.x
D’Alessandro, S., Posocco, B., Costa, A., Zahariou, G., Schiavo, F. L., Carbonera, D., et al. (2013). Limits in the use of cPTIO as nitric oxide scavenger and EPR probe in plant cells and seedlings. Front. Plant Sci. 4:340. doi: 10.3389/fpls.2013.00340
Das, G., Shravage, B. V., and Baehreck, E. H. (2012). Regulation and function of autophage during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 4:a008813. doi: 10.1101/cshperspect.a008813
de Michele, R., Vurro, E., Rigo, C., Costa, A., Elviri, L., Di Valentin, M., et al. (2009). Nitric oxide is involved in cadmium-induced programmed cell death in Arabidopsis suspension cultures. Plant Physiol. 150, 217–228. doi: 10.1104/pp.108.133397
de Pinto, M. C., Tommasi, F., and De Gara, L. (2002). Changes in the antioxidant systems as part of the signalling pathway responsible for the programmed cell death activated by nitric oxide and reactive oxygen species in tobacco BY-2 cells. Plant Physiol. 130, 698–708. doi: 10.1104/pp.005629
Delledonne, M., Zeier, J., Marocco, A., and Lamb, C. (2001). Signal interactions between nitric oxide and reactive oxygen intermediates in the plant hypersensitive disease resistance response. Proc. Natl. Acad. Sci. U.S.A. 98, 13454–13459. doi: 10.1073/pnas.231178298
Duana, L., Danzerb, B., Levensonb, V. V., and Makia, C. G. (2013). Critical roles for nitric oxide and ERK in the completion of prosurvival autophagy in 4OHTAM-treated estrogen receptor-positive breast cancer cells. Cancer Lett 353, 290–300. doi: 10.1016/j.canlet.2014.07.031
Engelman, J. A., Luo, J., and Cantley, L. C. (2006). The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619.
Floryszak-Wieczorek, J., and Arasimowicz-Jelonek, M. (2010). “Interplay between nitric oxide and other signals involved in plant resistance to pathogens,” in Nitric Oxide in Plant Physiology, eds S. Hayat, M. Mori, J. Pichtel, and A. Ahmad (Hoboken, NJ: Wiley- Blackwell), 139–159. doi: 10.1002/9783527629138.ch10
Foresia, N., Correa-Aragundea, N., Parisib, G., Calóc, G., Salernoc, G., and Lamattina, L. (2010). Characterization of a nitric oxide synthase from the plant kingdom: NO generation from the green alga Ostreococcus tauri is light irradiance and growth phase dependent. Plant Cell 22, 3816–3830. doi: 10.1105/tpc.109.073510
Galván, A., and Fernández, E. (2001). Eukaryotic nitrate and nitrite transporters. Cell. Mol. Life Sci. 58, 225–233. doi: 10.1007/PL00000850
Greenberg, J. T. (2005). Degrade or die: a dual function for autophagy in the plant immune response. Dev. Cell 8, 799–801. doi: 10.1016/j.devcel.2005.05.005
Hanada, T., Noda, N. N., Satomi, Y., Ichimura, Y., Fujioka, Y., Takao, T., et al. (2007). The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 282, 37298–37302. doi: 10.1074/jbc.C700195200
Hasanuzzaman, M., Hossain, M. A., and Fujita, M. (2010). Physiological and biochemical mechanisms of nitric oxide induced abiotic stress tolerance in plants. Am. J. Plant Physiol. 5, 295–324. doi: 10.3923/ajpp.2010.295.324
Hayat, S., Mor, M., Pichtel, J., and Ahmad, A. (2010). Nitric Oxide in Plant Physiology. Weinheim: Wiley-VCH, Verlag GmbH & Co.
Hemschemeier, A., Düner, M., Casero, D., Merchant, S. S., Winkler, M., and Happe, T. (2013). Hypoxic survival requires a 2-on-2 hemoglobin in a process involving nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 110, 10854–10859. doi: 10.1073/pnas.1302592110
Hong, J. K., Yun, B. W., Kang, J. G., Raja, M. U., Kwon, E., Sorhagen, K., et al. (2008). Nitric oxide function and signalling in plant disease resistance. J. Exp. Bot. 59, 147–154. doi: 10.1093/jxb/erm244
Houot, V., Etienne, P., Petitot, A. B., Barbier, S., Blein, J. P., and Suty, L. (2001). Hydrogen peroxide induces programmed cell death features in cultured tobacco BY-2 cells, in a dose-dependent manner. J. Exp. Bot. 52, 1721–1730. doi: 10.1093/jxb/52.361.1721
Itakura, E., Kishi, C., Inoue, K., and Mizushima, N. (2008). Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 19, 5360–5372. doi: 10.1091/mbc.E08-01-0080
Jaber, N., Dou, Z., Chen, J. S., Catanzaro, J., Jiang, Y. P., Ballou, L. M., et al. (2012). Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. U.S.A. 109, 2003–2008. doi: 10.1073/pnas.1112848109
Kamada, Y., Funakoshi, T., Shintani, T., Nagano, K., Ohsumi, M., and Ohsumi, Y. (2000). TOR-mediated induction of autophagy via an Apg1 protein kinas omplex. J. Cell Biol. 150, 1507–1513. doi: 10.1083/jcb.150.6.1507
Kojima, H., Nakatsubo, N., Kikuchi, K., Kawahara, S., Kirino, Y., and Nagoshi, H. (1998). Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Anal. Chem. 70, 2446–2453. doi: 10.1021/ac9801723
Lehner, C., Kerschbaum, H. H., and Luez-Meindl, U. (2009). Nitric oxide suppresses growth and development in the unicellular green alga Micrasterias denticulate. J. Plant Physiol. 166, 117–127. doi: 10.1016/j.jplph.2008.02.012
Liu, Y., and Bassham, D. C. (2012). Autophagy: pathways for self-eating in plant cells. Ann. Rev. Plant Biol. 63, 215–237. doi: 10.1146/annurev-arplant-042811-105441
Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2011). The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132. doi: 10.1146/annurev-cellbio-092910-154005
Mur, L. A. J., Mandon, J., Cristescu, S. M., Harren, F. J. M., and Prats, E. (2011). Methods of nitric oxide detection in plants: a commentary. Plant Sci. 181, 509–519. doi: 10.1016/j.plantsci.2011.04.003
Murik, O., Elboher, A., and Kaplan, A. (2014). Dehydroascorbate: a possible surveillance molecule of oxidative stress and programmed cell death in the green alga Chlamydomonas reinhardtii. New Phytol. 202, 471–484. doi: 10.1111/nph.12649
Nakatogawa, H., Ichimura, Y., and Ohsumi, Y. (2007). Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130, 165–178. doi: 10.1016/j.cell.2007.05.021
Neill, S., Barros, R., Bright, J., Desikan, R., Hancock, J., and Harrison, J. (2008). Nitric oxide, stomatal closure, and abiotic stress. J. Exp. Bot. 59, 165–176. doi: 10.1093/jxb/erm293
Ohsumi, Y. (2001). Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2, 211–216. doi: 10.1038/35056522
Palavan-Unsal, N., and Arisan, D. (2009). Nitric oxide signalling in plants. Bot. Rev. 75, 203–229. doi: 10.1007/si2229-009-9031-2
Pedroso, M. C., Magalhaes, J. R., and Durzan, D. (2000). Nitric oxide induces cell death in Taxus cells. Plant Sci. 157, 173–180. doi: 10.1016/s0168-9452(00)00278-8
Pérez-Pérez, M. E., Couso, I., and Crespo, J. L. (2012a). Carotenoid deficiency triggers autophagy in the model green alga Chlamydomonas reinhardtii. Autophagy 8, 376–388. doi: 10.4161/auto.18864
Pérez-Pérez, M. E., and Crespo, J. L. (2010). Autophagy in the model alga Chlamydomonas reinhardtii. Autophagy 6, 562–563. doi: 10.4161/auto.6.4.11822
Pérez-Pérez, M. E., Florencio, F. J., and Crespo, J. L. (2010). Inhibition of target of rapamycin signaling and stress activate autophagy in Chlamydomonas reinhardtii. Plant Physiol. 152, 1874–1888. doi: 10.1104/pp.109.152520
Pérez-Pérez, M. E., Lemaire, S. D., and Crespo, J. L. (2012b). Reactive oxygen species and autophagy in plants and algae. Plant Physiol. 160, 156–164. doi: 10.1104/pp.112.199992
Pérez-Pérez, M. E., Zaffagnini, M., Marchand, C. H., Crespo, J. L., and Lemaire, S. D. (2014). The yeast autophagy protease Atg4 is regulated by thioredoxin. Autophagy 10, 1953–1964. doi: 10.4161/auto.34396
Rabkin, S. W., and Klassen, S. S. (2007). Nitric oxide differentially regulates the gene expression of caspase genes but not some autophagic genes. Nitric Oxide 16, 339–347. doi: 10.1016/j.niox.2006.10.007
Ramundo, S., Casero, D., Mühlhaus, T., Hemme, D., Sommer, F., Crèvecoeur, M., et al. (2014). Conditional depletion of the Chlamydomonas chloroplast ClpP protease activates nuclear genes involved in autophagy and plastid protein quality control. Plant Cell 26, 2201–2222. doi: 10.1105/tpc.114.124842
Romero-Puertas, M. C., Perazzolli, M., Zago, E. D., and Delledonne, M. (2004). Nitric oxide signalling functions in plant-pathogen interactions. Cell. Microbiol. 6, 795–803. doi: 10.1111/j.1462-5822.2004.00428.x
Sarkar, S., Korolchuk, V., Renna, M., Imarisio, S., Fleming, A., Williams, A., et al. (2011). Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell 43, 19–32. doi: 10.1016/j.molcel.2011.04.029
Sato, M., Murata, Y., Mizusawa, M., Iwahashi, H., and Oka, S. (2004). A simple and rapid dual-fluorescence viability assay for microalgae. Microbiol. Cult. Coll. 20, 53–59.
Shimizu, S., Kanaseki, T., Mizushima, N., Mizuta, T., Arakawa-Kobayashi, S., Thompson, C. B., et al. (2004). Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 6, 1221–1228. doi: 10.1038/ncb1192
Siddiqui, M. H., Al-Whaibi, M. H., and Basalah, M. O. (2011). Role of nitric oxide in tolerance of plants to abiotic stress. Protoplasma 248, 447–455. doi: 10.1007/s00709-010-0206-9
Thompson, A. R., and Vierstra, R. D. (2005). Autophagic recycling: lessons from yeast help define the process in plants. Curr. Opin. Plant Biol. 8, 165–173. doi: 10.1016/j.pbi.2005.01.013
Tran, Q. G., Cho, K., Park, S. B., Kim, U., Lee, Y. J., and Kim, H. H. S. (2019). Impairment of starch biosynthesis results in elevated oxidative stress and autophagy activity in Chlamydomonas reinhardtii. Sci. Rep. 9:9856. doi: 10.1038/s41598-019-46313-6
Tripathi, D. N., Chowdhury, R., Trudel, L. J., Tee, A. R., Slack, R. S., Walker, C. L., et al. (2013). Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. U.S.A. 110, E2950–E2957. doi: 10.1073/pnas.1307736110
Vardi, A., Formiggini, F., Casotti, R., De Martino, A., Ribalet, F., Miralto, A., et al. (2006). A stress surveillance system based on calcium and nitric oxide in marine diatoms. PLoS Biol. 4:e60. doi: 10.1371/journal.pbio.0040060
Veneault-Fourrey, C., Barooah, M., Egan, M., Wakley, G., and Talbot, N. J. (2006). Autophagic fungal cell death in necessary for infection by the rice blast fungus. Science 312, 580–583. doi: 10.1126/science.1124550
Wang, Y., Loake, G. J., and Chu, C. (2013). Cross-talk of nitric oxide and reactive oxygen species in plant programed cell death. Front. Plant Sci. 4:314. doi: 10.3389/fpls.2013.00314
Wei, L., Derrien, B., Gautier, A., Houille-Vernes, L., Boulouis, A., Saint-Marcoux, D., et al. (2014). Nitric oxide–triggered remodeling of chloroplast bioenergetics and thylakoid proteins upon nitrogen starvation in Chlamydomonas reinhardtii. Plant Cell 26, 353–372. doi: 10.1105/tpc.113.120121
Xie, Z., Nair, U., and Klionsky, D. J. (2008). Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 19, 3290–3298. doi: 10.1091/mbc.E07-12-1292
Xu, Y., Sun, X., Jin, J., and Zhou, H. (2010). Protective effect of nitric oxide on light-induced oxidative damage in leaves of tall fescue. J. Plant Physiol. 167, 512–518. doi: 10.1016/j.jplph.2009.10.010
Yamasaki, H. (2000). Nitrite-dependent nitric oxide production pathway: implications for involvement of active nitrogen species in photoinhibition in vivo. Phil. Trans. R. Soc. Lond. Biol. Sci. 355, 1477–1488. doi: 10.1098/rstb.2000.0708
Yang, J., Wu, L. J., Tashino, S. I., Onodera, S., and Ikejima, T. (2008). Reactive oxygen species and nitric oxide regulate mitochondria-dependent apoptosis and autophagy in evodiamine-treated human cervix carcinoma HeLa cells. Free Radic. Res. 42, 492–504. doi: 10.1080/10715760802112791
Yordanova, Z. P., Iakimova, E. T., Cristescu, S. M., Harren, F. J., Kapchina-Toteva, V. M., and Woltering, E. J. (2010). Involvement of ethylene and nitric oxide in cell death in mastoparan-treated unicellular alga Chlamydomonas reinhardtii. Cell Biol. Int. 34, 301–308. doi: 10.1042/CBI20090138
Yu, L., Alva, A., Su, H., Dutt, P., Freundt, E., Welsh, S., et al. (2004). Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304, 1500–1502. doi: 10.1126/science.1096645
Keywords: autophagy, autophagy-related protein, cell death, Chlamydomonas, high light, nitric oxide
Citation: Kuo EY, Chang H-L, Lin S-T and Lee T-M (2020) High Light-Induced Nitric Oxide Production Induces Autophagy and Cell Death in Chlamydomonas reinhardtii. Front. Plant Sci. 11:772. doi: 10.3389/fpls.2020.00772
Received: 27 February 2020; Accepted: 15 May 2020;
Published: 10 June 2020.
Edited by:
Dimitris Petroutsos, UMR 5168 Laboratoire de Physiologie Cellulaire Vegetale (LPCV), FranceReviewed by:
Jose L. Crespo, Consejo Superior de Investigaciones Científicas (CSIC), SpainThomas Roach, University of Innsbruck, Austria
Copyright © 2020 Kuo, Chang, Lin and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tse-Min Lee, dG1sZWVAbWFpbC5uc3lzdS5lZHUudHc=
†These authors have contributed equally to this work