 Michelle Zavala-Páez1*
Michelle Zavala-Páez1* Leila do Nascimento Vieira1
Leila do Nascimento Vieira1 Valter Antônio de Baura2
Valter Antônio de Baura2 Eduardo Balsanelli2
Eduardo Balsanelli2 Emanuel Maltempi de Souza2
Emanuel Maltempi de Souza2 Marco Cerna Cevallos3
Marco Cerna Cevallos3 Mark W. Chase4,5
Mark W. Chase4,5 Eric de Camargo Smidt1*†
Eric de Camargo Smidt1*†- 1Departamento de Botânica, Universidade Federal do Paraná, Curitiba, Brazil
- 2Departamento de Bioquímica e Biologia Molecular, Universidade Federal do Paraná, Curitiba, Brazil
- 3Grupo de Investigación Nunkui Wakan, Universidad Politécnica Salesiana, Quito, Ecuador
- 4Royal Botanic Gardens, Kew, Richmond, United Kingdom
- 5Department of Environment and Agriculture, Curtin University, Perth, WA, Australia
Pantropical Bulbophyllum, with ∼2,200 species, is one of the largest genera in Orchidaceae. Although phylogenetics and taxonomy of the ∼60 American species in the genus are generally well understood, some species complexes need more study to clearly delimit their component species and provide information about their evolutionary history. Previous research has suggested that the plastid genome includes phylogenetic markers capable of providing resolution at low taxonomic levels, and thus it could be an effective tool if these divergent regions can be identified. In this study, we sequenced the complete plastid genome of eight Bulbophyllum species, representing five of six Neotropical taxonomic sections. All plastomes conserve the typical quadripartite structure, and, although the general structure of plastid genomes is conserved, differences in ndh-gene composition and total length were detected. Total length was determined by contraction and expansion of the small single-copy region, a result of an independent loss of the seven ndh genes. Selection analyses indicated that protein-coding genes were generally well conserved, but in four genes, we identified 95 putative sites under positive selection. Furthermore, a total of 54 polymorphic simple sequence repeats were identified, for which we developed amplification primers. In addition, we propose 10 regions with potential to improve phylogenetic analyses of Neotropical Bulbophyllum species.
Introduction
Bulbophyllum Thouars is the largest Pantropical genus of Orchidaceae, with ∼2,200 species (WCSP, 2018). However, its distribution is not homogeneous over the entire range. With hundreds of species, the Paleotropics are by far the richest, followed by Africa and the Neotropics, the last with just ∼60 species (Smidt et al., 2011; Pridgeon et al., 2014). In the Neotropics, the genus is represented by six clades, two north of the Equator and four mainly in the Brazilian Atlantic Rainforest and Cerrado (Smidt and Borba, 2007; Smidt et al., 2011).
Bulbophyllum species are recognized worldwide not only for their diversity but also for the potential use of their compounds in the fields of traditional medicine and agricultural pest management. These orchids contain aromatic compounds that have properties of use in human health, such as in treatment of tumors and conception problems and production of antifebriles (Wu et al., 2006; Chen et al., 2008; Lalitharani et al., 2011). Furthermore, the volatile compounds of Bulbophyllum species are used in integrated pest management strategies. Tephritid flies, pests of many crops, can be attracted to traps using floral fragrances of Bulbophyllum (Tan et al., 2006; Tan and Nishida, 2007; Jaleel et al., 2018).
Owing to their species diversity and high levels of endemism, Bulbophyllum species have been widely studied in the Neotropics (Smidt and Borba, 2007; Smidt et al., 2007; Mancinelli and Smidt, 2012). As a result, multiple taxonomic (Borba et al., 1998; Ribeiro et al., 2008; Nunes et al., 2014, 2015, 2017) and phylogenic (Smidt et al., 2011, 2013) studies have been published. In molecular phylogenetic studies of Neotropical Bulbophyllum species, plastid [psbA-trnH and trnS-trnG intergenic spacers (IGSs)] and nuclear [internal transcribed spacer (ITS) spacer] markers have been employed (Smidt et al., 2011, 2013). However, despite these studies, some species complexes need further investigation to aid in species delimitation and provide evidence of natural hybridization/introgression (Borba and Semir, 1998; Azevedo et al., 2006; Mancinelli and Smidt, 2012).
Previous research (Zhitao et al., 2017) has demonstrated that the plastid genome can be an effective tool in population genetics, but ancestral polymorphisms can be problematic in some taxa (Dodsworth et al., 2020). In addition, the study of the plastid genome for phylogeographical analyses has great potential (Mariac et al., 2014; Smidt et al., 2020). In Orchidaceae, with the exception of several mycoheterotrophic species (Feng et al., 2016; Yuan et al., 2018), the plastid genome has the typical angiosperm quadripartite structure: two inverted repeated (IR) regions separated by a small (SSC) and large single-copy (LSC) regions (Chang et al., 2006; Delannoy et al., 2011; Pan et al., 2012; Yang et al., 2013; Luo et al., 2014; Zhitao et al., 2017; Smidt et al., 2020). The size of the plastid genome within orchids is relatively small, ranging from 0.01 to 0.17 Mb (Lin et al., 2015; Schelkunov et al., 2015). Gene content and order are generally conserved, but some variations have been reported in length, composition of the ndh-gene complex, and loss of many photosynthesis-related genes in mycoheterotrophic species (Graham et al., 2017; Zhitao et al., 2017).
In this article, we present complete plastid genomes for eight Bulbophyllum species, representing five of six Neotropical taxonomic sections. First, we compared gene content and order and codon use. Second, codons under positive selection in protein-coding genes are identified. Third, we determined locations of long- and short repeated sequences in these genomes, including those with potential as microsatellite markers. Fourth, sequence variability (SV) is calculated to determine the 10 most-variable regions, for which we provide PCR primer sequences. Finally, performance of the most-variable regions in these Bulbophyllum species is evaluated using a phylogenetic analysis. Our results provide information for species identification and increase phylogenetic resolution among Bulbophyllum species. Also, more effective molecular markers can produce results with implications for species management and conservation.
Materials and Methods
Plastid Genome Sequencing and Assembly
Plastid-enriched DNA (ptDNA) was extracted from fresh leaves of eight Bulbophyllum species (Table 1) using the Sakaguchi et al. (2017) methodology, reducing the amount of material required for plastid isolation. Afterward, ptDNA was extracted following the Doyle and Doyle (1987) protocol. Purification was performed with DNA Clean and Concentrator kit (Zymo Research, Orange, CA, United States).

Table 1. Bulbophyllum species sequenced, taxonomy, and collection data.
DNA was sequenced on an Illumina MiSeq® following instructions of DNA Nextera XT Sample Prep Kit (IlluminaTM). The raw data were trimmed according to default parameters (i.e., quality limit = 0.05; ambiguous limit = 2; minimum read length = 30 bp) of CLC Genomics Workbench 8.01, and a hybrid reference-guided de novo assembly approach was performed to assemble the genomes. First, the trimmed pair-end reads were mapped onto the complete plastid genome sequence of Dendrobium officinale Kimura & Migo (NC_024019.1) as a reference to generate a consensus sequence. The consensus sequence was extracted, excluding regions with low coverage (threshold < 8) and length fraction of 0.5. Afterward, the trimmed pair-end reads were de novo assembled using the following parameters: automatic word size, automatic bubble size, minimum contig length = 300, map reads back to contigs using mismatch cost = 2, insertion cost = 3, deletion cost = 3, length fraction = 0.5, and similarity fraction = 0.8. The multiple contigs created through the de novo assembly strategy were used in the gap-closing process, creating a consensus sequence developed with these high-quality contigs.
At this stage, there were six physical gaps in Bulbophyllum exaltatum Lindl. (3), Bulbophyllum epiphytum Barb.Robr (2), and Bulbophyllum steyermarkii Foldats (1). Three of them were resolved by amplification with primers specific to the flanking regions and Sanger sequencing on an ABI 3500 (Applied Biosystems). The others were located in AT-rich regions of repetitive sequences. These three gaps are small (9, 40, and 63 bp), and their length was estimated by reference mapping to a closely related Bulbophyllum species. Tentative errors sites were identified and manually checked by mapping the Illumina pair-end raw reads onto the assembled plastid genome. Assembled errors were characterized by mismatch readings or an abnormal read-mapping depth.
Dual Organellar GenoMe Annotator software (Wyman et al., 2004) and Geneious R7 (Kearse et al., 2012) were used to annotate the plastid genomes, using default values to predict genes coding for proteins (CDSs), transfer RNAs (tRNAs), and ribosomal DNAs (rDNAs). All genomes were checked manually, and the codon positions were determined by BLASTX against the National Center for Biotechnology Information (NCBI) protein database using the D. officinale plastid genome as a reference. Gene maps were drawn by Organellar Genome DRAW V1.1 (Lohse et al., 2007).
The newly assembled plastomes were deposited in GenBank (Table 2; accession numbers MN604056, MN737573, MN580547, MN604059, MN604054, MN604055, MN604057, and MN604058).

Table 2. General characteristics of the plastid genomes of eight Bulbophyllum species.
Plastid Genome Features
For each plastid genome, length, numbers of CDSs, tRNAs, and rDNAs were plotted. Genes in the inverted repeats and with two or more introns were identified. Junction positions between single-copy and IR regions were compared. Guanine-cytosine (GC) content was calculated using Geneious R7 (Kearse et al., 2012).
Codon Usage
All CDSs for each plastid genome were extracted using Geneious R7 (Kearse et al., 2012) to determine the distribution of codon usage. The relative synonymous codon usage (RSCU) ratio was calculated using the R package SeqinR (Charif and Lobry, 2007). Relative synonymous codon usage values greater than 1 represent codons used more frequently than expected, and values less than 1 signify the opposite. RSCU distributions were illustrated in the form of heatmaps using Heatmapper (Babicki et al., 2016).
Molecular Evolution of Protein-Coding Genes
A Bayesian inference (BI) approach was applied to identify amino acid sites in all CDSs under positive or purifying selection. The evolutionary model M8 was run on The Selecton server website (Stern et al., 2007). When positive selection was detected, a likelihood ratio test was run with the M8a (null) and M8 (alternative) models to test whether positive selection was significant. Furthermore, gene divergence analysis was performed with Selecton using the default parameters. Pairwise distances and branch lengths were computed using the maximum likelihood (ML) criterion under a codon model (Nielsen and Yang, 1998).
Characterization of Repeat Sequences and Simple Sequence Repeats
Direct, reverse, and palindromic repeat sequences were identified by REPuter online program (Kurtz et al., 2001) according to a Hamming distance of 3.90% sequence identity and minimum repeat size of 30 bp. Simple sequence repeats (SSRs) were located using the MISA-web program (Beier et al., 2017) with search parameters set to the following: ≥10 repeat units for mononucleotide SSRs, ≥5 repeat units for dinucleotide SSRs, and ≥3 repeat units for dinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide SSRS. Microsatellites primers were designed using Websat (Martins et al., 2009) for polymorphic SSRs present in at least four species. The following parameters were used: product length of 100–500 bp, primer length of 18–27 bp, and GC content of 40–60% with 1°C as the maximum difference between primer melting temperatures.
Hypervariable Regions
To identify the 10 most-variable regions, all plastid genomes were aligned with the progressive Mauve algorithm (Darling et al., 2004). Then, the CDS, introns, and IGSs with minimum length of 150 bp flanked by the same region were manually extracted. The number of mutations and indel events was calculated by DnaSP v5 (Rozas et al., 2004), and their rate was calculated per 100 bp. Sequence variability was calculated as follows: (number of mutations + number of indel events)/(conserved sites + number of mutations + number of indel events) × 100% (Shaw et al., 2014). Due to indels having higher levels of homoplasy than mutations, indels were considered as events instead of sites in the alignment (Ingvarsson et al., 2003). Also, potentially parsimony informative sites were counted by DnaSP v5 program.
Specific primers were designed for the 10 most-variable regions using PRIMER32 with the following parameters: maximum product length of 1,000 bp, primer length of 18–27 bp, and GC content of 40–60% with 2°C as the maximum difference between primer melting temperatures.
Phylogenetic Analysis
To evaluate the performance of the marker regions identified in this study, phylogenetic analyses were performed for two datasets: (1) complete plastid genome sequences and (2) the 10 most-variable regions. Individual sequence alignments were performed using MAFFT and then combined for each dataset using SequenceMatrix (Vaidya et al., 2011).
Maximum parsimony (MP), BI, and ML analyses were run for each set. Maximum parsimony analyses were performed with PAUP v4b10 (Swofford, 2003), with a member of the sister genus of Bulbophyllum, Dendrobium [Dendrobium aphyllum (Roxb.) C.E.C.Fisch; NC_035322.1], as the outgroup. Heuristic searches were conducted with 1,000 bootstrap replicates (BS), tree bisection–reconnection (TBR) branch swapping, and simple stepwise addition. All characters had equal weight; gaps were treated as missing data.
Maximum likelihood trees with 1,000 BS were produced with IQ-tree 1.6.11 (Trifinopoulos et al., 2016). The best fit models for each dataset were calculated by ModelFinder (Kalyaanamoorthy et al., 2017). The K3Pu + F + R2 model was used for the complete plastid genome matrix. For the second matrix, the model of evolution was calculated for each sequence with IQ-tree 1.6.11 (Supplementary Table 1). The resulting trees were produced using the FigTree software v1.4.13.
BI used MrBayes 3.2.7 (Ronquist and Huelsenbeck, 2003), and the best-fit model for each set was determined with the Bayesian information criterion (BIC) in jModeltest version 2.1.10 (Darriba et al., 2012; Supplementary Table 1). Four independent Markov chain Monte Carlo (MCMC) were run twice, each with 3,000,000 million generations. Trees were sampled every 1,000 generations, and the first 25% of these were discarded. The remaining trees were used to build the Bayesian tree of posterior probabilities.
Results
Plastid Genome Features
The eight Bulbophyllum plastid genomes were 146,401–151,493 bp long; the smallest was that of Bulbophyllum plumosum Barb.Rodr., whereas the largest was that of Bulbophyllum regnellii Rchb.f. The average coverage was between 45.26 and 247.55x for B. epiphytum and B. steyermarkii (Supplementary Table 2). The genomes have the typical angiosperm quadripartite structure (Figure 1 and Supplementary Figure 1): IRs = 25,465 bp (Bulbophyllum granulosum Barb.Rodr) to 26,340 bp (Bulbophyllum mentosum Barb.Robr), LSC = 82,354 bp (B. epiphytum) to 84,868 bp (B. regnellii), and SSC = 11,088 bp (B. plumosum) to 16,048 bp [Bulbophyllum weddellii (Lindl.) Rchb.f; Supplementary Figure 2]. GC content was similar (36.6–36.8%; Table 2).
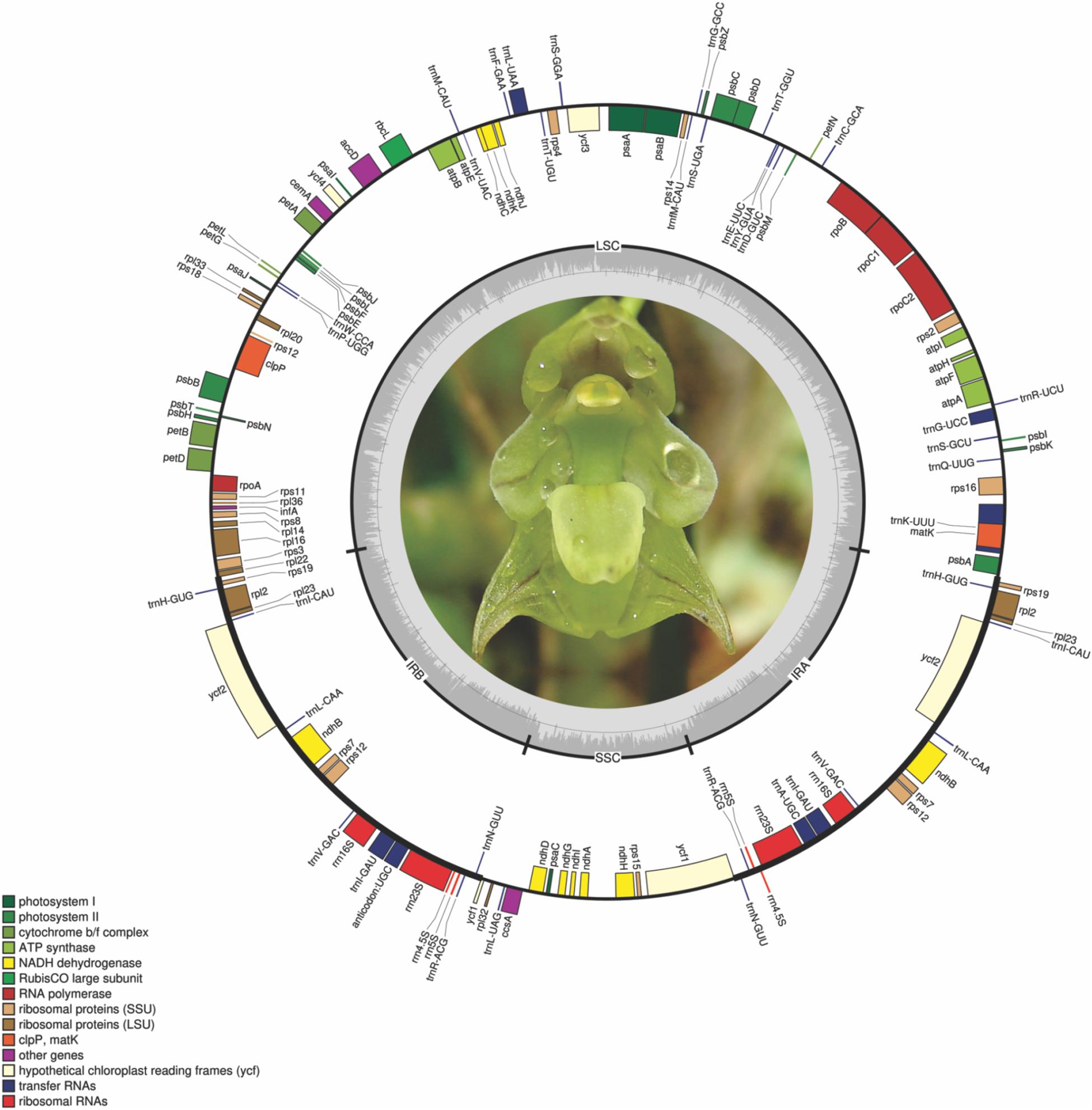
Figure 1. Bulbophyllum regnellii plastid genome. Genes shown on the outside of the circle are transcribed clockwise; genes on the inside are transcribed counterclockwise. The color of the gene boxes indicates the functional group to which the gene belongs. The thick lines indicate the length of the inverted repeats (IRA and IRB), separated by the large single-copy (LSC) and small single-copy (SSC) regions. The dashed darker gray area in the inner circle indicates genome GC content, whereas lighter gray area indicates AT content.
For each plastid genome, 102 genes were predicted: 68 CDSs, 30 tRNAs, and four rDNAs. Eight CDSs, eight tRNAs, and all four rDNAs were present in the IRs (Supplementary Table 3). A small fragment of ycf1 was found in IRb/SSC junction (Figure 2).
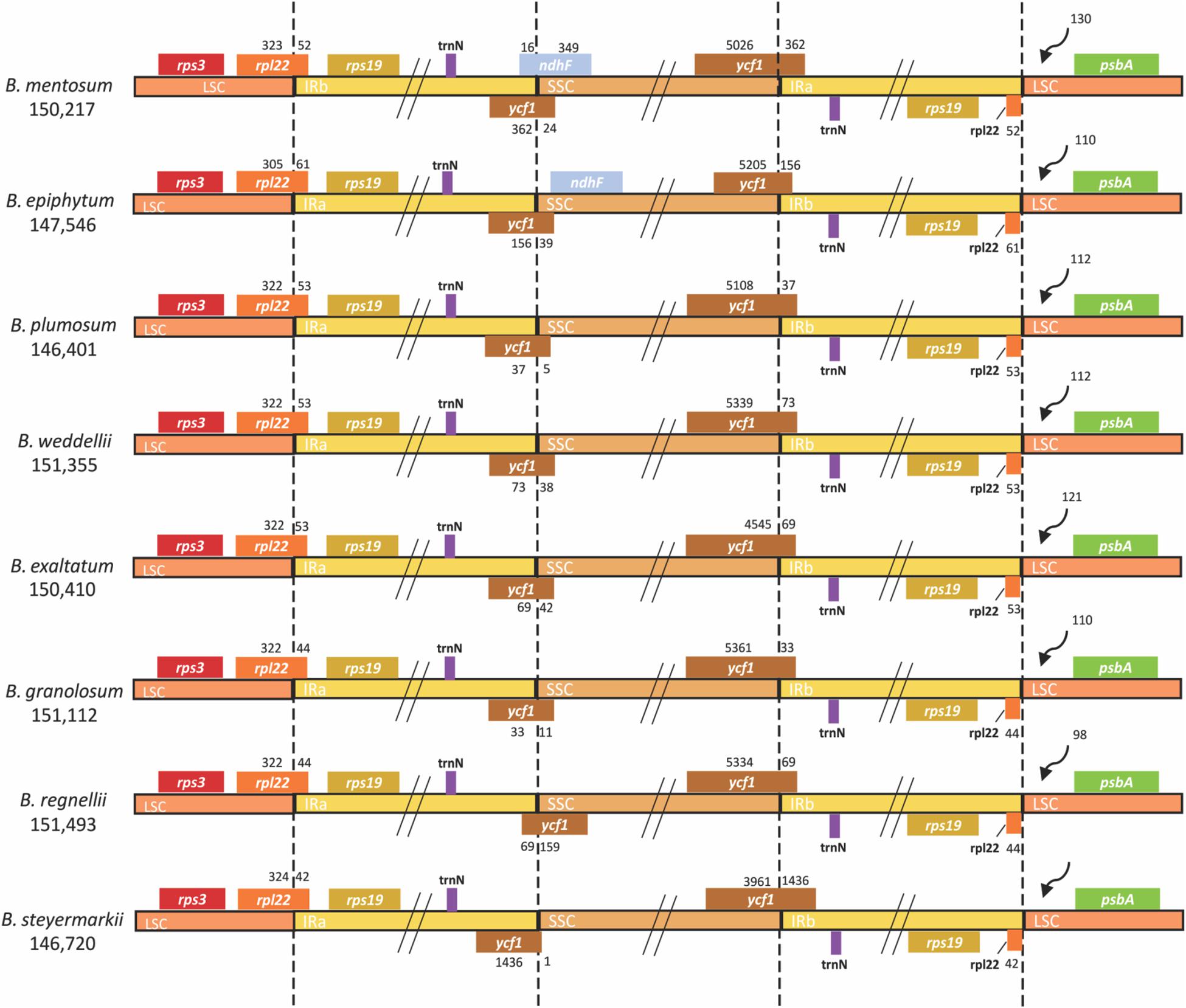
Figure 2. Comparison of LSC, IR, and SSC junctions in eight Bulbophyllum plastid genomes. Genes shown on the top are transcribed in reverse, and those shown underneath are forward. LSC, large single copy; IR, inverted repeated; SSC, small single copy.
Additionally, most ndh genes were truncated or completely lost. However, ndhG retained a full reading frame in B. exaltatum. Seven of the 11 ndh genes were located in the SSC, and their lengths were correlated with SSC expansion and contraction (r = 0.94, p ≤ 0.001). SSC expansion and contraction also determined the total plastid genome size (r2 = 0.89, F1, 6 = 57.27, p ≤ 0.001). SSC length differences between the largest and smallest genomes were the 4,960 bp, in which the value corresponds to 97% of the total length differences among these plastid genomes (Figure 3).
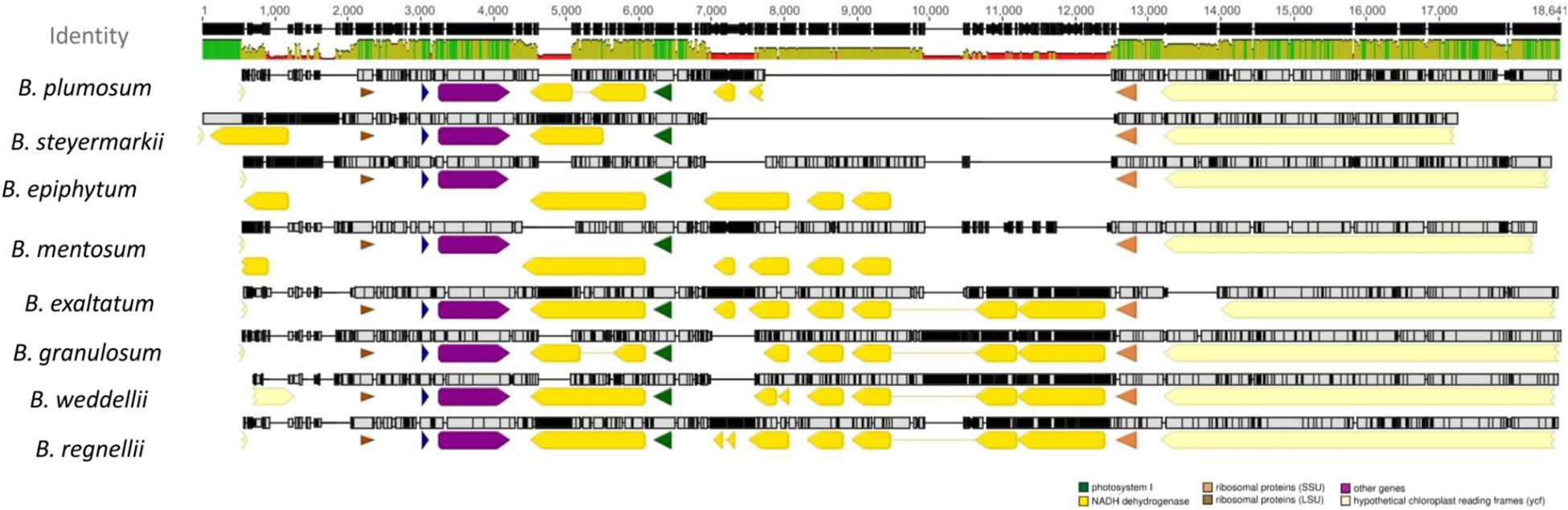
Figure 3. MAFFT alignment of the small single-copy (SSC) region of all plastid genomes. The yellow blocks represent the NDH genes. The green lines on the top represent the high similarity between the sequences, whereas red indicates the opposite. Genomes are organized in descending lengths.
In all plastid genomes, LSC/IRb and IRb/SSC junctions were in rpl22 and ycf1 (pseudogene), respectively, whereas the IRa/SSC and IRa/LSC junctions were in ycf1 and rpl22. At the LSC/IRb border, IRb expanded up to 61 bp toward the rpl22 gene and in the IRb/SSC border up to 1,436 bp toward the ycf1 pseudogene. Furthermore, the IRb/SSC border of only B. mentosum was located in the ndhF pseudogene, with just 16 bp located in the IRb region. The ndhF pseudogene was close to but did not overlap with the IRb/SSC junction in B. epiphytum (Figure 2).
Codon Usage
Codon use frequency and RSCU assessed 68 unique CDSs among the eight plastid genomes. A range of 18,831–19,306 codons were examined, among which the most common codon was AAA (4.27%), whereas CGC (0.36%) was the rarest. ATG was the initiation codon for most protein-coding genes, but in the rps19 (GTG) and rpl2 genes (ACG), alternative initiation codons were found. Leucine was the most abundant amino acid, ranging between 1,258 and 2,687 codons in B. mentosum and B. plumosum, respectively, whereas cysteine was the least abundant, ranging between 209 and 776 codons in B. exaltatum and B. mentosum, respectively (Supplementary Table 4). RSCU analysis indicated that 29 of 32 codons ending with G/C were not frequently used, whereas 28 of 32 codons that ended with A/T had high RSCU (Figure 4 and Supplementary Table 5).
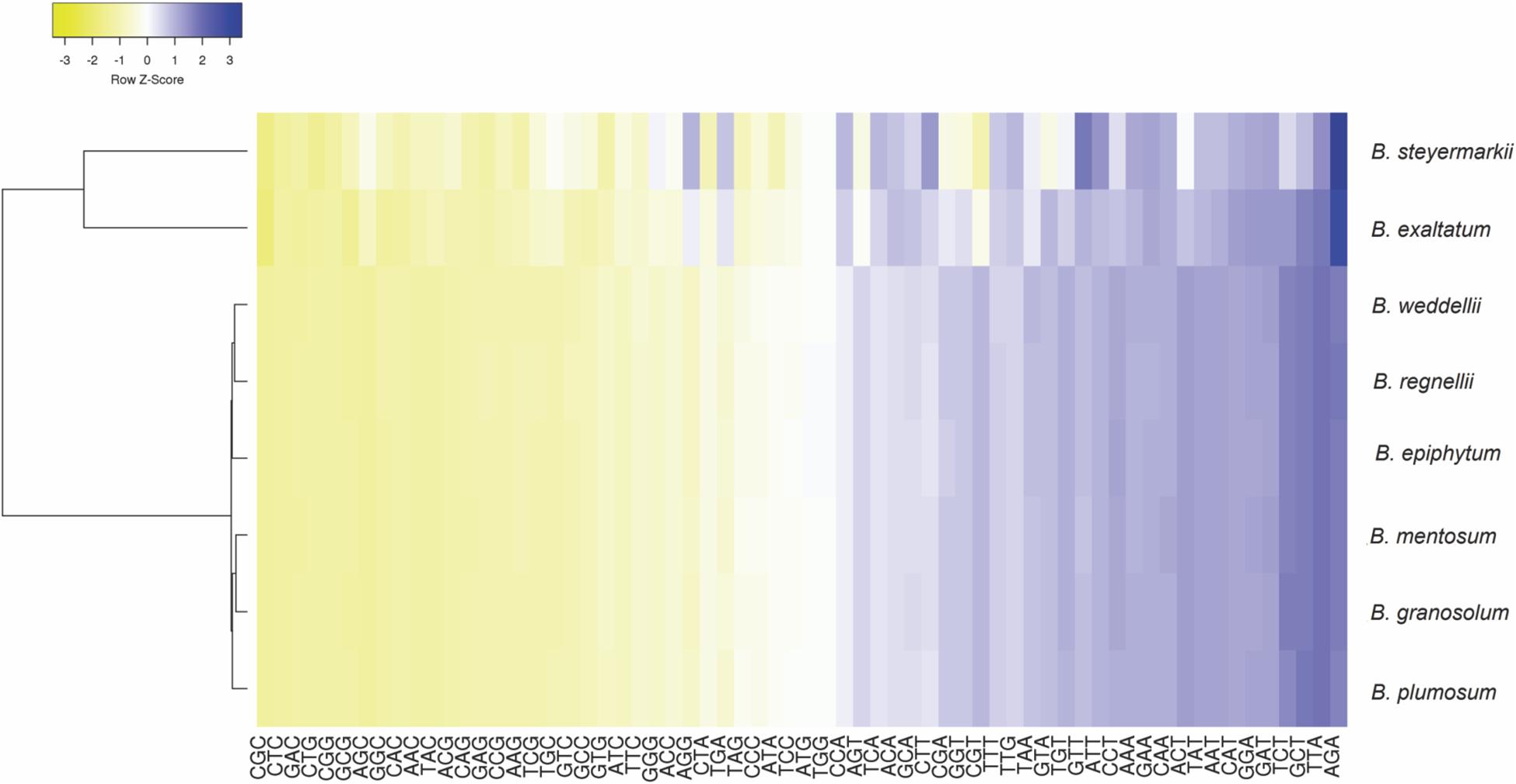
Figure 4. Relative synonymous codon usage ratio (RSCU) of protein-coding genes of the eight Bulbophyllum plastid genomes. Blue indicates a high RSCU ratio, and yellow indicates low.
Molecular Evolution Analysis of Protein-Coding Genes
Analyses to investigate selection indicated that 89 putative sites were under positive selection. These sites were distributed in four out of 68 shared CDSs in these plastid genomes. The putatively most selected sites (adaptative selection) were found in ycf1 (68 sites) followed by ycf2, accD, and rps3 genes with 10, 10, and 1, respectively. Moreover, according to the average branch length of each protein-coding gene tree, the most divergent genes were ycf1, rps15, accD, psbE, rpl36, and ycf2 (Figure 5).
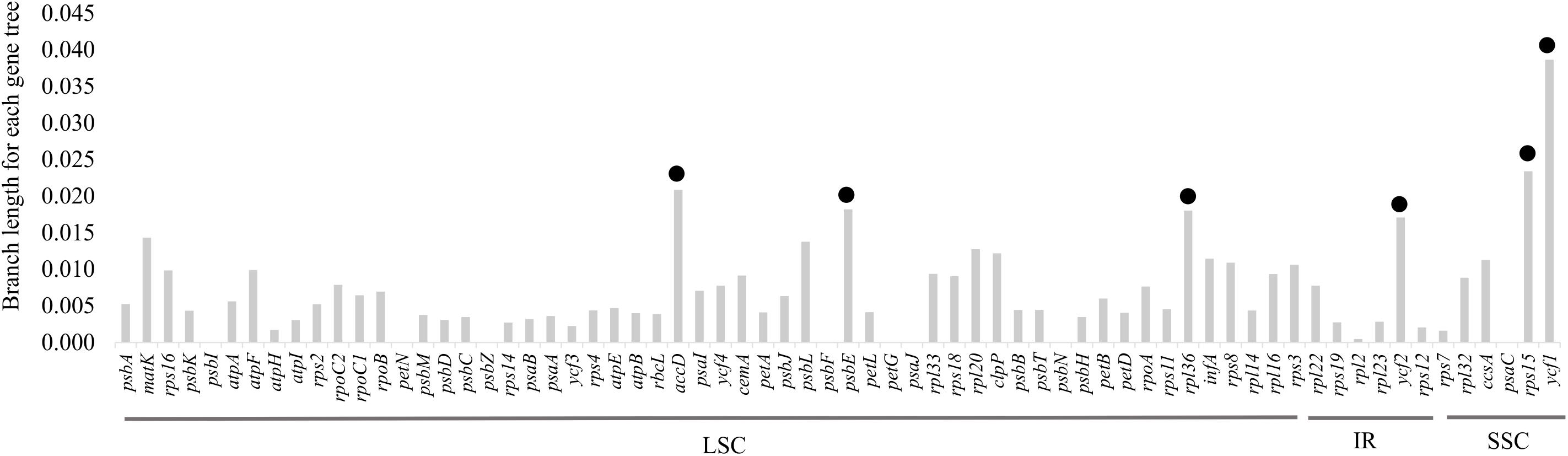
Figure 5. Molecular evolution of 68 protein-coding genes shared among the eight plastid genomes. The black circles indicate the six most divergent genes.
Characterization of Repeat Sequences and Simple Sequence Repeat
Forward, reverse, and palindromic repeats (>30 bp) were compared for the eight plastid genomes. In total, 249 repeats were identified in these plastid genomes, including 78 simple repeats, 153 palindromic repeats, and 18 reverse repeats (Figure 6A). B. steyermarkii exhibited the most (36), whereas B. mentosum had the fewest (25; Supplementary Table 6). The majority of repeat sequences were distributed in IGSs (54.62%), and the most frequent lengths were 30–40 bp (Figure 6B and Supplementary Table 7). Four palindromic repeats were shared by these eight plastid genomes (Table 3).
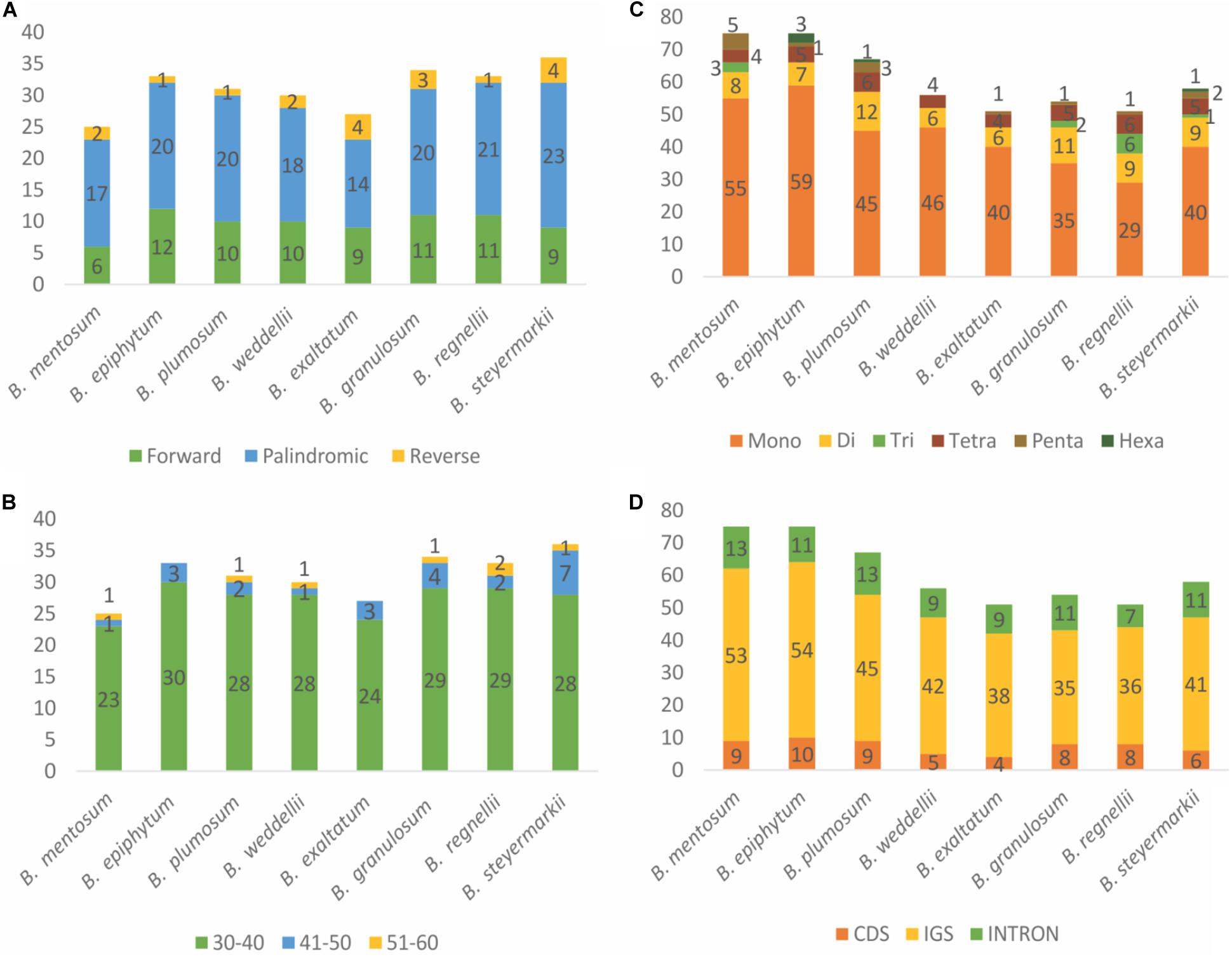
Figure 6. (A) Frequency of forward, reverse, and palindromic repeats. (B) Length frequency of tandem repeats. (C) Distribution and frequency of simple sequence repeat (SSR) types. (D) Distributions of SSRs for each region.

Table 3. Palindromic repeat sequences shared by Bulbophyllum species.
MISA analysis was performed to identify single SSRs in the plastid genomes. We detected 527 SSRs (Supplementary Table 8). The number of SSRs per species ranged from 51 to 75. Among these, there were 31–61 mononucleotide repeats, 8–14 dinucleotide repeats, 1–6 trinucleotide repeats, 5–7 tetranucleotide repeats, 1–5 pentanucleotide repeats, and 1–3 hexanucleotide repeats. Trinucleotide, pentanucleotide, and hexanucleotide repeats were absent from several plastid genomes (Figure 6C). Most SSRs were in the LSC (78%), followed by SSC (14%) and IR (8%). Most 527 SSRs were located in IGSs (347 SSRs), followed by CDS (95 SSRs) and introns (84 SSRs, Figure 6D). We designed 46 primer pairs for these microsatellites, which were present in at least four species (Supplementary Table 9).
Hypervariable Regions
With the use of B. steyermarkii as a reference, the number of mutations and indels events were compared among these Bulbophyllum plastid genomes. There were 148 events (CDSs, IGSs, and introns) flanked by the same exon and longer than 150 bp: 4,064 mutations and 624 indels (Figures 7A,B). Intergenic spacers had the highest mutation rate, 5.21 mutations per 100 bp, following by introns and CDSs with 3.56 and 2.58, respectively (Table 4). Also, when indels were considered, IGSs had the most, 1.11 indels per 100 bp.
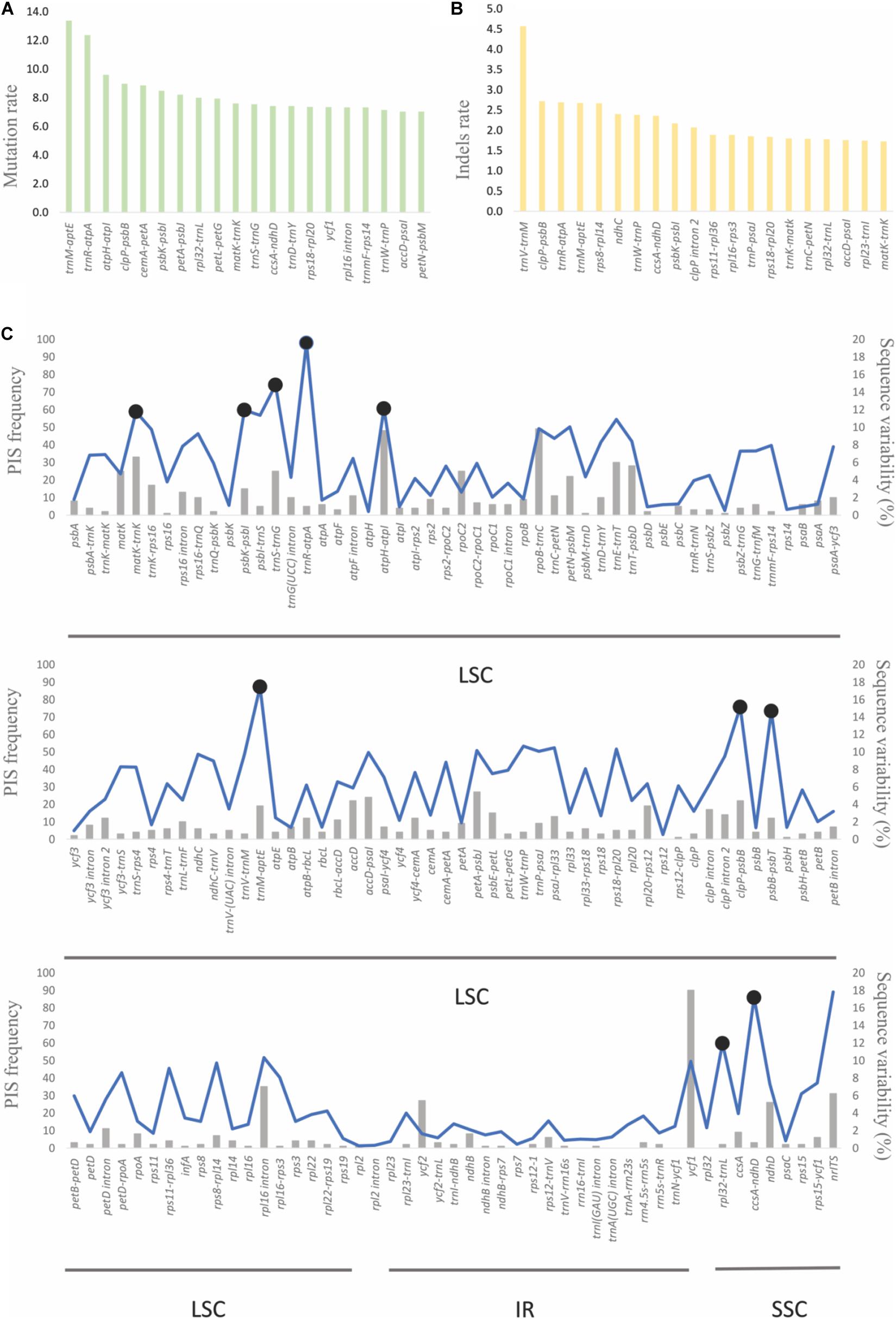
Figure 7. (A) The 20 sequences with the highest substitution and (B) indel rate. (C) Sequence variability and parsimony-informative sites (PIS) of the 148 plastid sequences and internal transcribed spacer (ITS). The blue line represents the sequence variability (SV), and the 10 most-variable regions are marked with black circles. The gray bars indicate the frequency of PIS of each sequence.
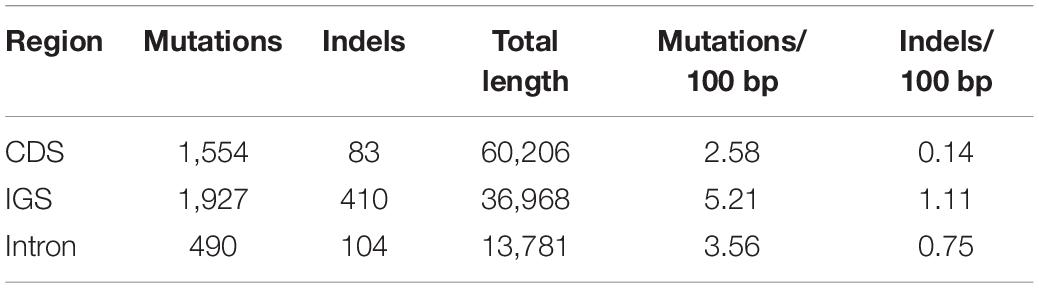
Table 4. Mutations, indel events, and mutation rates for each region type: protein coding genes (CDS), intergenic spacers (IGS), and introns.
Furthermore, genes with the greatest number of potentially parsimony informative sites (PIS; Figure 7C) and greatest SV (Figure 7C, Supplementary Table 10; 15%, 184 of the total) were similar. Correlation analysis showed that SV was positively correlated with AT content (r = 0.81, p < 0.001). Primer pairs were developed for the 10 most-variable markers (Supplementary Table 11).
Phylogenetic Analysis
Maximum parsimony, BI, and ML analyses were performed using (i) the complete plastid genomes of the eight Bulbophyllum species and (ii) the 10 most-variable regions; D. aphyllum was selected as the outgroup. The eight plastid genomes and outgroup were aligned in a single data matrix with a total of 163,916 nucleotide sites, of which 2,429 (1.48%) were identified as potentially PIS. The 10 most-variable regions matrix had 7,087 nucleotide sites with 285 (4%) potentially PIS (Figure 7C).
The tree topologies generated in the MP, BI, and ML analyses were identical for the two datasets. The majority of nodes received high support, but two nodes in the tree produced from the 10 most-variable regions had only moderate support (Figure 8).
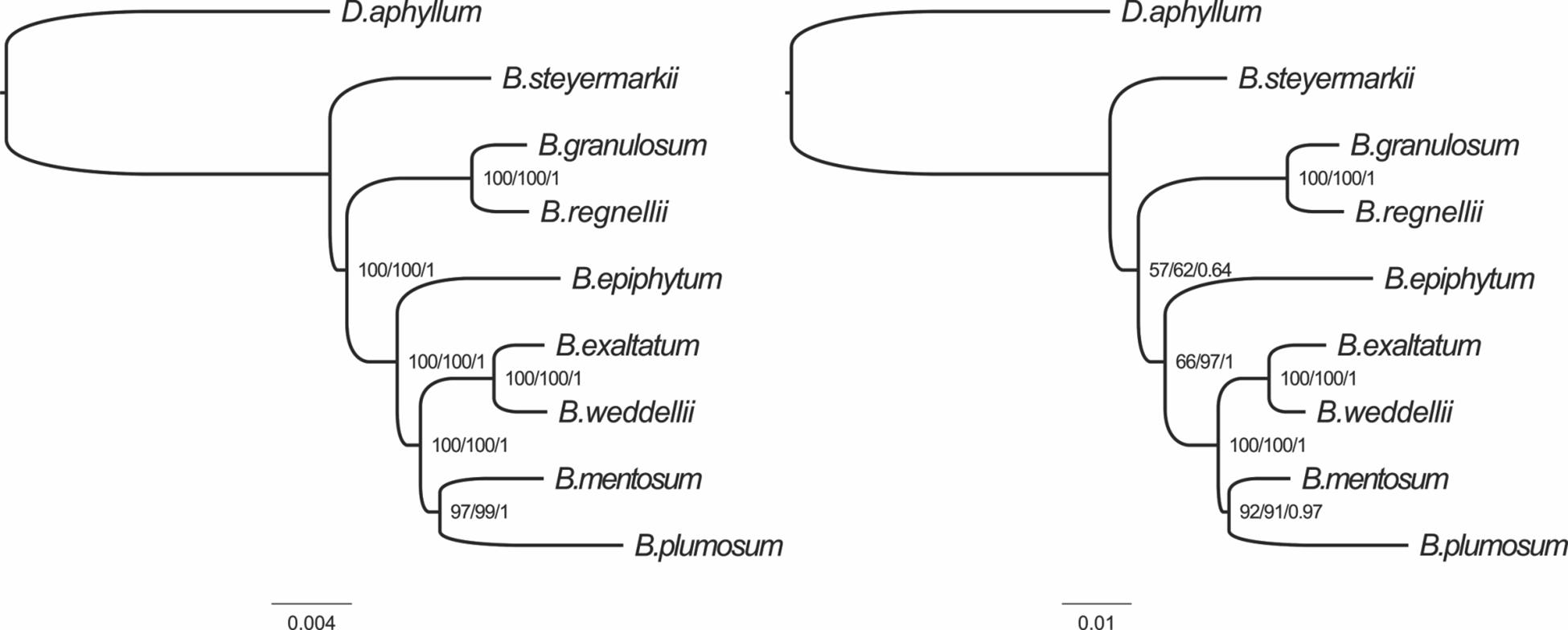
Figure 8. Phylogenetic relationships of the eight Bulbophyllum species constructed from whole genome (left) and the 10 most-variable regions (right) with maximum parsimony (MP), maximum likelihood (ML), and Bayesian inference (BI) methods. ML topology and branch length are represented. Numbers associated with branches are bootstrap percentages of MP/ML and BI posterior probabilities (in this order).
Discussion
Plastid Genome Features
In this study, all eight plastid genomes of Neotropical Bulbophyllum species conserved the typical quadripartite structure previously reported in Orchidaceae (Zhitao et al., 2017), with exception of Aphyllorchis montana Rchb.f and Gastrodia elata Blume, heterotrophic orchids that have lost one IR (Feng et al., 2016; Yuan et al., 2018). Furthermore, gene arrangement and content were similar in the Bulbophyllum and Dendrobium (outgroup) genomes, with 68 CDS, 30 tRNA genes, and four ribosomal rRNA genes (Zhitao et al., 2017).
Although the general structure of Bulbophyllum plastid genomes is conserved, differences in ndh gene composition and total length were detected. The ndh family is composed of 11 genes involved in respiratory electron transport and chlororespiration (Ohyama et al., 1986; Martín and Sabater, 2010; Ueda et al., 2012). Ten of the 11 ndh genes in Bulbophyllum were deleted or truncated by point mutations or deletions, generating stop codons. A full reading frame for ndhG was observed only in B. exaltatum. Loss of ndh genes has been previously reported in orchids (Chang et al., 2006; Yang et al., 2013; Luo et al., 2014; Kim et al., 2015; Zhitao et al., 2017; Kim and Chase, 2017; Mauad et al., 2019; Smidt et al., 2020) and is useful in comparative analyses but not in phylogenetics owing to high levels of homoplasy depicted by the independent loss of some ndh genes in a number of species of Orchidaceae (Kim et al., 2015; Kim and Chase, 2017).
Previous studies concluded that the total length of angiosperm plastid genomes is influenced by contractions and expansions of the IRs (Chumley et al., 2006; Green, 2011; Weng et al., 2017). In contrast, here, we found that the length of Bulbophyllum plastid genomes is determined by the contraction and expansion of the SSC due to the independent loss of the seven ndh genes present in this region. The pronounced effect of the gain/loss of ndh genes in the SSC has been previously reported (Chang et al., 2006; Jheng et al., 2012; Kim et al., 2015; Zhu et al., 2018), but only in Zhitao et al. (2017) was the influence of LSC, IR, and SSC length on the genome size measured. As a result, they found that variation in LSC length plays an important role in plastid genome-size variation.
In general, gene content of the IR borders among Bulbophyllum orchids was similar. However, some differences in the IR/SSC junction were detected. In all Bulbophyllum plastid genomes, we identified the trnH-rps19 gene cluster within the IR, and IR/LSC junctions were located 5’ of rpl22. In agreement with our findings, Luo et al. (2014); Kim et al. (2015), and others detected the same IR/LSC pattern among species of orchids, which is the type III of Wang et al. (2008) based on the trnH-rps19 position among 123 angiosperms and suggests that this IR/LSC junction is one of those conserved among orchids.
However, different IR/SSC patterns have been reported in Orchidaceae (Yang et al., 2013; Luo et al., 2014; Niu et al., 2017a; Dong et al., 2018; Zhu et al., 2018). According to Kim et al. (2015), the stability of the IR/SSC junctions is highly affected by loss/gain of the ndh genes, especially ndhF, the absence of which could create complicated modifications like shifts in the IR border gene, length reduction of ycf1 (less than 1 kb), and changes in ycf1 position within the IR. Related to presence/absence of the ndhF pseudogene, three types of IR/SSC junctions were identified. The first was found in B. epiphytum, in which the ndhF pseudogene is present near IRb, but it does not overlap with the IRb/SSC junction. B. mentosum has the second type with the IRb expanded by 16 and 362 bp toward ndhF and ycf1, respectively. The other six genomes exhibit the third type in which the ndhF gene was lost, and the IR expands toward ycf1 by 33–1,436 bp (Figure 2). Owing to the Bulbophyllum species with different IR border types not being grouped as in the phylogenetic tree, we suggest that IR borders do not have a simple phylogenetic signal.
Codon Usage
Codon usage analysis revealed substitutions in the start codons of rps19 and rpl2 genes (putatively making these pseudogenes) and preferences for codons ending in A/T. These and RNA editing sites in rps12 and ycf15 genes have been previously reported in Orchidaceae (Chang et al., 2006; Yang et al., 2013; Luo et al., 2014; Barthet et al., 2015; Schelkunov et al., 2015), and they are probably involved in adaption in some manner (He et al., 2016). Additionally, RSCU analysis indicated that codons ending in A/T are more frequently used, similar to what has been found in most angiosperms (Meng et al., 2018; Tian et al., 2018; Raman et al., 2019), perhaps a result of compositional AT bias in plastid genomes (Morton, 1993; Zhou et al., 2008; Niu et al., 2017b).
Molecular Evolution in Protein-Coding Genes
Protein-coding genes with sites putatively under positive selection, in general, occurred in the most divergent genes. Eighty-nine sites putatively under positive selection were located in ycf1, ycf2, accD, and rps3, the first being the second-largest plastid gene, essential for photosynthetic protein import (Kikuchi et al., 2013). Also, it is one of the most variable genes among the land plants, and therefore, it has been used in phylogenetic analyses (Dong et al., 2015). Other orchid plastid genomes studied (Niu et al., 2017b; Dong et al., 2018; Li et al., 2019) detected similar patterns.
Additionally, ycf2 and accD had 10 putative sites under adaptative selection. The function of ycf2 is still unknown, but accD regulates fatty acid synthesis, which is essential to maintain plastids (Kode et al., 2005). Additional study is needed to understand why these genes might be under adaptative selection.
Characterization of Repeat Sequences and Simple Sequence Repeat
Repeat sequences may play an important role in genome recombination and rearrangements, size, and structure through illegitimate recombination and slipped-strand mispairing (Gemayel et al., 2010). In this study, 249 repeats were identified, the majority in non-coding regions. This result is similar to that of Yang et al. (2013), in which analyses of eight Cymbidium species found 232 repeat sequences, again mostly in non-coding regions. However, in Cymbidium, reverse repeats were the most abundant, whereas in Bulbophyllum palindromic repeats were the most common.
Owing to their high mutation rates (Gemayel et al., 2010), SSRs have been extensively used in populations genetic and phylogenetic studies (Kartzinel et al., 2013; Minasiewicz et al., 2018). Here, a total of 54 polymorphic SSRs were identified. The most abundant SSR type was the mononucleotide repeat (A/T), similar to the findings of Zhitao et al. (2017). Also, like other plastid genome studies (Zhang et al., 2016; Niu et al., 2017a), the majority of SSRs in Bulbophyllum is composed of polyT and polyA repeats (Supplementary Table 8). Owing to the great potential of plastid SSRs in genetic diversity studies, we provide here a complete set of Bulbophyllum microsatellite–primers pairs.
Hypervariable Regions
As expected, the 148 sequences longer than 150 bp with the highest mutation and indel rates were located in non-coding regions, probably a result of their high AT content (Supplementary Figure 3). High AT content may contribute to replication errors creating mutations or deletions (Niu et al., 2017a). Based on number of mutations, indels, and conserved sites, we advocate use of 10 markers with barcoding and phylogenetic potential in Bulbophyllum (Figure 7). In recent years, numerous orchid plastid genomes have been sequenced. As a result, various plastid markers have been proposed for Orchidaceae (Yang et al., 2013; Niu et al., 2017a, c; Zhitao et al., 2017; Dong et al., 2018; Zhu et al., 2018; Li et al., 2019; Smidt et al., 2020). For instance, Niu et al. (2017c) suggested that trnK-rps16, trnS-trnG, and rps16-trnQ IGSs could be used for genera within Epidendroideae, and clpP-psbB and rps16-trnQ for Cypripedioideae. In addition, Niu et al. (2017a) recommended several for use in Apostasioideae. The identification of different highly variable markers among the orchid subfamilies suggests inconsistent divergence among taxa, a result corroborated here. Only four of the 10 most-variable markers identified for Bulbophyllum are shared with its sister genus, Dendrobium (trnR-atpA, psbB-psbT, rpl32-trnL, and clpP-psbB). Among the other six regions, two (matK-trnK and trnS-trnG) were identified by Niu et al.(2017a; 2017c) and Zhu et al. (2018), whereas four (atpH-atpI, ccsA-ndhD, psbK-psbI, and trnM-aptE) are reported for the first time here as highly variable in Orchidaceae.
Previous markers used in Bulbophyllum analyses proved here not to be highly variable; for example, Smidt et al. (2011) used psbA-trnH and trnS-trnG for the Neotropical sections of Bulbophyllum. Although, trnS-trnG had high statistics in this study, the SV of psbA-trnH was only 6.5%. Another example is Fischer et al. (2007), who studied Madagascan Bulbophyllum using four plastid IGSs (and nrITS). Among these four, one (trnF-ndhJ) was not considered in this study because it is missing from the plastid genomes of these Neotropical Bulbophyllum species. Use of markers with low SV is one of the reasons why in both previous studies some terminal nodes were unresolved and poorly supported.
In general, nrITS has been widely used in angiosperm phylogenetics, in particular in orchids (Fischer et al., 2007; Smidt et al., 2011, 2018), owing to its high variability and PIS. Here, we found one IGS, trnR-atpA, with greater variability than nrITS and two markers (atpH-atpI and matK-trnK) with more PIS than the nrITS but lower overall variability (Supplementary Figure 4). Thus, there are plastid markers with similar variability to nrITS that have been not thus far used in Bulbophyllum studies.
Phylogenetic Analysis
We examined phylogenetic relationships among the eight Bulbophyllum species with the complete plastid genomes and the top 10 most-variable markers. Both datasets produced the same topology with similar support for all three phylogenetic methods. Also, this tree is congruent with that of Neotropical Bulbophyllum in Smidt et al. (2011). Furthermore, in Smidt et al. (2011), the position of B. mentosum was unclear. In the nrITS tree, B. mentosum was a member of Bulbophyllum section Micranthae, but in the plastid tree from that analysis, it should be in Bulbophyllum section Xiphizusa, agreeing with results here for both matrices analyzed. The incongruence between nuclear and plastid data found in Smidt et al. (2011) may be a result of ancient hybridization (Barber et al., 2007).
Conclusion
In summary, we sequenced here eight complete plastomes of Bulbophyllum species, representing five of the six Neotropical taxonomic sections. In general, the plastomes were similar in gene content and structure, except for ndh gene composition. However, despite this general high similarity, we detected several regions with higher variability than the nuclear and plastid molecular markers previously used in this genus. We have thus provided important molecular resources for Bulbophyllum, comprising 10 highly variable regions and 54 microsatellites (and primers to amplify them). These molecular resources for Bulbophyllum will be useful to improve our understanding of phylogenic relationships, population genetics, and phylogeography and aid in species identification.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MN604056, MN737573, MN580547, MN604059, MN604054, MN604055, MN604057, MN604058.
Author Contributions
ECS designed the study. MCC collected specimens. MZ-P, LV, VB, EB and ES performed the laboratory work. MZ-P and LV performed all analyses. All authors contributed to writing of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sistema de Autorização e Informação em Biodiversidade/Instituto Chico Mendes de Conservação da Biodiversidade (SISBIO/ICMBio) for the collecting permits and the Universidad Politécnica Salesiana for the financial support. ECS would like to thank Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for grant Bolsa de Produtividade em Pesquisa CNPq-Nível 2 (proc.311001/2014-9, 308460/2017-0) and Pos-doc grant (proc. 203304/2018-7) and Brazilian Program of National Institutes of Science and Technology-INCT/Brazilian Research Council-CNPq/MCT. Ana Victoria Mauad is thanked for her help in the lab and phylogenetic analyses.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00799/full#supplementary-material
Footnotes
- ^ https://www.qiagenbioinformatics.com/
- ^ http://bioinfo.ut.ee/primer3-0.4.0/
- ^ http://tree.bio.ed.ac.uk
References
Azevedo, C. O., Borba, E. L., and Van Den Berg, C. (2006). Evidence of natural hybridization and introgression in Bulbophyllum involutum Borba, Semir & F. Barros and B. weddellii (Lindl.) Rchb. f. (Orchidaceae) in the Chapada Diamantina, Brazil, by using allozyme markers. Braz. J. Bot. 29, 415–421. doi: 10.1590/s0100-84042006000300008
Babicki, S., Arndt, D., Marcu, A., Liang, Y., Grant, J. R., Maciejewski, A., et al. (2016). Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 44, 147–153. doi: 10.1093/nar/gkw419
Barber, J. C., Finch, C. C., Francisco-Ortega, J., Santos-Guerra, A., and Jansen, R. K. (2007). Hybridization in Macaronesian Sideritis (Lamiaceae): evidence from incongruence of multiple independent nuclear and chloroplast sequence datasets. Taxon 56, 74–88. doi: 10.2307/25065737
Barthet, M. M., Moukarzel, K., Smith, K. N., Patel, J., and Hilu, K. W. (2015). Alternative translation initiation codons for the plastid maturase MatK: unraveling the pseudogene misconception in the Orchidaceae. BMC Evol. Biol. 15:491. doi: 10.1186/s12862-015-0491-491
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Borba, E., and Semir, J. (1998). Bulbophyllum cipoense (Orchidaceae), a new natural hybrid from the Brazilian” campos rupestres”: description and biology. Lindleyana 13, 113–120.
Borba, E. L., Semir, J., and de Barros, F. (1998). Bulbophyllum involutum Borba, Semir & F. Barros (Orchidaceae), a new species from the Brazilian “campos rupestres. Novon 8, 225–229. doi: 10.2307/3392005
Chang, C. C., Lin, H. C., Lin, I. P., Chow, T. Y., Chen, H. H., Chen, W. H., et al. (2006). The chloroplast genome of Phalaenopsis aphrodite (Orchidaceae): comparative analysis of evolutionary rate with that of grasses and its phylogenetic implications. Mol. Biol. Evol. 23, 279–291. doi: 10.1093/molbev/msj029
Charif, D., and Lobry, J. R. (2007). “SeqinR 1. 0-2 : a contributed package to the R Project for statistical computing devoted to biological sequences retrieval and analysis,” in Structural Approaches to Sequence Evolution, eds V. M. Bastolla, U. M. Porto, and H. E. Roman, (New York, NY: Springer Science & Business Media), 1–26. doi: 10.1007/978-3-540-35306-5
Chen, Y., Xu, J., Yu, H., Qing, C., Zhang, Y., Wang, L., et al. (2008). Cytotoxic phenolics from Bulbophyllum odoratissimum. Food Chem. 107, 169–173. doi: 10.1016/j.foodchem.2007.07.077
Chumley, T. W., Palmer, J. D., Mower, J. P., Fourcade, H. M., Calie, P. J., Boore, J. L., et al. (2006). The complete chloroplast genome sequence of Pelargonium × hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. doi: 10.1093/molbev/msl089
Darling, A., Mau, B., Blattner, F., and Perna, N. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Darriba, D., Taboada, G., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Delannoy, E., Fujii, S., Colas Des Francs-Small, C., Brundrett, M., and Small, I. (2011). Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol. Biol. Evol. 28, 2077–2086. doi: 10.1093/molbev/msr028
Dodsworth, S., Christenhusz, M. J., Conran, J. G., Guignard, M. S., Knapp, S., and Struebig et al. (2020). Extensive plastid-nuclear discordance in a recent radiation of Nicotiana section Suaveolentes (Solanaceae). Bot. J. Linn. Soc. boaa024. doi: 10.1093/botlinnean/boaa024
Dong, W., Xu, C., Li, C., Sun, J., Zuo, Y., Shi, S., et al. (2015). ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5:8348. doi: 10.1038/srep08348
Dong, W. L., Wang, R. N., Zhang, N. Y., Fan, W. B., Fang, M. F., and Li, Z. H. (2018). Molecular evolution of chloroplast genomes of orchid species: insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 19:716. doi: 10.3390/ijms19030716
Doyle, J., and Doyle, J. (1987). A rapid DNA isolation procedure for small amounts of leaf tissue. Phytochem. Bull. 19, 810–815.
Feng, Y. L., Wicke, S., Li, J. W., Han, Y., Lin, C. S., Li, D. Z., et al. (2016). Lineage-specific reductions of plastid genomes in an orchid tribe with partially and fully mycoheterotrophic species. Genome Biol. Evol. 8, 2164–2175. doi: 10.1093/gbe/evw144
Fischer, G. A., Gravendeel, B., Sieder, A., Andriantiana, J., Heiselmayer, P., Cribb, P. J., et al. (2007). Evolution of resupination in Malagasy species of Bulbophyllum (Orchidaceae). Mol. Phylogenet. Evol. 45, 358–376. doi: 10.1016/j.ympev.2007.06.023
Gemayel, R., Vinces, M. D., Legendre, M., and Verstrepen, K. J. (2010). Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 44, 445–477. doi: 10.1146/annurev-genet-072610-155046
Graham, S. W., Lam, V. K. Y., and Merckx, V. S. F. T. (2017). Plastomes on the edge: the evolutionary breakdown of mycoheterotroph plastid genomes. New Phytol. 214, 48–55. doi: 10.1111/nph.14398
Green, B. R. (2011). Chloroplast genomes of photosynthetic eukaryotes. Plant J. 66, 34–44. doi: 10.1111/j.1365-313X.2011.04541.x
He, P., Huang, S., Xiao, G., Zhang, Y., and Yu, J. (2016). Abundant RNA editing sites of chloroplast protein-coding genes in Ginkgo biloba and an evolutionary pattern analysis. BMC Plant Biol. 16:257. doi: 10.1186/s12870-016-0944-948
Ingvarsson, P. K., Ribstein, S., and Taylor, D. R. (2003). Molecular evolution of insertions and deletion in the chloroplast genome of Silene. Mol. Biol. Evol. 20, 1737–1740. doi: 10.1093/molbev/msg163
Jaleel, W., Lu, L., and He, Y. (2018). Biology, taxonomy, and IPM strategies of Bactrocera tau Walker and complex species (Diptera; Tephritidae) in Asia: a comprehensive review. Environ. Sci. Pollut. Res. 25, 19346–19361. doi: 10.1007/s11356-018-2306-2306
Jheng, C. F., Chen, T. C., Lin, J. Y., Chen, T. C., Wu, W. L., and Chang, C. C. (2012). The comparative chloroplast genomic analysis of photosynthetic orchids and developing DNA markers to distinguish Phalaenopsis orchids. Plant Sci. 190, 62–73. doi: 10.1016/j.plantsci.2012.04.001
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14:587. doi: 10.1038/nmeth.4285
Kartzinel, T. R., Shefferson, R. P., and Trapnell, D. W. (2013). Relative importance of pollen and seed dispersal across a Neotropical mountain landscape for an epiphytic orchid. Mol. Ecol. 22, 6048–6059. doi: 10.1111/mec.12551
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kikuchi, S., Bédard, J., Hirano, M., Hirabayashi, Y., Oishi, M., Imai, M., et al. (2013). Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 339, 571–574. doi: 10.1126/science.1229262
Kim, H. T., and Chase, M. W. (2017). Independent degradation in genes of the plastid ndh gene family in species of the orchid genus Cymbidium (Orchidaceae; Epidendroideae). PLoS One 12:e187318. doi: 10.1371/journal.pone.0187318
Kim, H. T., Kim, J. S., Moore, M. J., Neubig, K. M., Williams, N. H., Whitten, W. M., et al. (2015). Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across orchids and associated instability of the inverted repeat/small single-copy region boundaries. PLoS One 10:e142215. doi: 10.1371/journal.pone.0142215
Kode, V., Mudd, E. A., Iamtham, S., and Day, A. (2005). The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 44, 237–244. doi: 10.1111/j.1365-313X.2005.02533.x
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lalitharani, S., Mohan, V. R., and Maruthupandian, A. (2011). Pharmacognostic investigations on Bulbophyllum albidum (Wight) Hook. F. Int. J. PharmTech Res. 3, 556–562.
Li, Z. H., Ma, X., Wang, D. Y., Li, Y. X., Wang, C. W., and Jin, X. H. (2019). Evolution of plastid genomes of Holcoglossum (Orchidaceae) with recent radiation. BMC Evol. Biol. 19:63. doi: 10.1186/s12862-019-1384-1385
Lin, C. S., Chen, J. J. W., Huang, Y. T., Chan, M. T., Daniell, H., Chang, W. J., et al. (2015). The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 5, 1–10. doi: 10.1038/srep09040
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Luo, J., Hou, B. W., Niu, Z. T., Liu, W., Xue, Q. Y., and Ding, X. Y. (2014). Comparative chloroplast genomes of photosynthetic orchids: insights into evolution of the Orchidaceae and development of molecular markers for phylogenetic applications. PLoS One 9:e99016. doi: 10.1371/journal.pone.0099016
Mancinelli, W., Smidt, E., and de, C. (2012). O gênero Bulbophyllum (Orchidaceae) na Região Sul do Brasil. Rodriguésia 63, 803–815. doi: 10.1590/s2175-78602012000400006
Mariac, C., Scarcelli, N., Pouzadou, J., Barnaud, A., Billot, C., Faye, A., et al. (2014). Cost-effective enrichment hybridization capture of chloroplast genomes at deep multiplexing levels for population genetics and phylogeography studies. Mol. Ecol. Resour. 14, 1103–1113. doi: 10.1111/1755-0998.12258
Martín, M., and Sabater, B. (2010). Plastid ndh genes in plant evolution. Plant Physiol. Biochem. 48, 636–645. doi: 10.1016/j.plaphy.2010.04.009
Martins, W. S., Lucas, D. C. S., Neves, K. F., de, S., and Bertioli, D. J. (2009). WebSat–a web software for microsatellite marker development. Bioinformation 3, 282–283. doi: 10.6026/97320630003282
Mauad, A. V. S. R., do Nascimento Vieira, L., Bolson, M., de Baura, V. A., Balsanelli, E., Maltempi de Souza, E., et al. (2019). Complete chloroplast genome of Anathallis obovata (Orchidaceae: Pleurothallidinae). Rev. Bras. Bot. 42, 345–352. doi: 10.1007/s40415-019-00524-523
Meng, J., Li, X., Li, H., Yang, J., Wang, H., and He, J. (2018). Comparative analysis of the complete chloroplast genomes of four Aconitum medicinal species. Molecules 23, 2–15. doi: 10.3390/molecules23051015
Minasiewicz, J., Znaniecka, J. M., Górniak, M., and Kawiñski, A. (2018). Spatial genetic structure of an endangered orchid Cypripedium calceolus (Orchidaceae) at a regional scale: limited gene flow in a fragmented landscape. Conserv. Genet. 19, 1449–1460. doi: 10.1007/s10592-018-1113-1114
Morton, B. R. (1993). Chloroplast DNA codon use: evidence for selection at the psbA locus based on tRNA availability. J. Mol. Evol. 37, 273–280. doi: 10.1007/BF00175504
Nielsen, R., and Yang, Z. (1998). Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148, 929–936.
Niu, Z., Pan, J., Zhu, S., Li, L., Xue, Q., Liu, W., et al. (2017a). Comparative analysis of the complete plastomes of Apostasia wallichii and Neuwiedia singapureana (Apostasioideae) reveals different evolutionary dynamics of IR/SSC boundary among photosynthetic orchids. Front. Plant Sci. 8:1713. doi: 10.3389/fpls.2017.01713
Niu, Z., Xue, Q., Wang, H., Xie, X., Zhu, S., Liu, W., et al. (2017b). Mutational biases and GC-biased gene conversion affect GC content in the plastomes of Dendrobium genus. Int. J. Mol. Sci. 18:2307. doi: 10.3390/ijms18112307
Niu, Z., Xue, Q., Zhu, S., Sun, J., Liu, W., and Ding, X. (2017c). The complete plastome sequences of four orchid species: insights into the evolution of the Orchidaceae and the utility of plastomic mutational hotspots. Front. Plant Sci. 8:715. doi: 10.3389/fpls.2017.00715
Nunes, E. L. P., Maldonado, P. E., Smidt, E. C., Stützel, T., and Coan, A. I. (2017). Floral micromorphology and anatomy and its systematic application to Neotropical Bulbophyllum section Micranthae (Orchidaceae). Bot. J. Linn. Soc. 183, 294–315. doi: 10.1093/botlinnean/bow007
Nunes, E. L. P., Smidt, E. C., Stützel, T., and Coan, A. I. (2014). What do floral anatomy and micromorphology tell us about Neotropical Bulbophyllum section Didactyle (Orchidaceae: Bulbophyllinae)? Bot. J. Linn. Soc. 175, 438–452. doi: 10.1111/boj.12176
Nunes, E. L. P., Smidt, E. C., Stützel, T., and Coan, A. I. K. E. (2015). Comparative floral micromorphology and anatomy of species of Bulbophyllum section Napelli (Orchidaceae), a Neotropical section widely distributed in forest habitats. Bot. J. Linn. Soc. 177, 378–394. doi: 10.1111/boj.12253
Ohyama, K., Fukuzawa, H., Kohchi, T., Shirai, H., Sano, T., Sano, S., et al. (1986). Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 322, 572–574. doi: 10.1038/322572a0
Pan, I. C., Liao, D. C., Wu, F. H., Daniell, H., Singh, N. D., Chang, C., et al. (2012). Complete chloroplast genome sequence of an orchid model plant candidate: Erycina pusilla apply in tropical Oncidium breeding. PLoS One 7:e34738. doi: 10.1371/journal.pone.0034738
Pridgeon, A. M., Cribb, P. J., Chase, M. W., and Rasmussen, F. N. (2014). Genera Orchidacearum, Epidendroideae, Vol. 6, Part 3. Oxford: Oxford University Press.
Raman, G., Park, S., Lee, E. M., and Park, S. J. (2019). Evidence of mitochondrial DNA in the chloroplast genome of Convallaria keiskei and its subsequent evolution in the Asparagales. Sci. Rep. 9, 1–11. doi: 10.1038/s41598-019-41377-w
Ribeiro, P. L., Borba, E. L., De Camargo Smidt, E., Lambert, S. M., Schnadelbach, A. S., and van den Berg, C. (2008). Genetic and morphological variation in the Bulbophyllum exaltatum (Orchidaceae) complex occurring in the Brazilian “campos rupestres”: implications for taxonomy and biogeography. Plant Syst. Evol. 270, 109–137. doi: 10.1007/s00606-007-0603-605
Ronquist, F., and Huelsenbeck, J. (2003). MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinfomatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rozas, J., Sánchez-DelBarrio, J. C., Messeguer, X., and Rozas, R. (2004). DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19, 2496–2497. doi: 10.1079/9780851994758.0139
Sakaguchi, S., Ueno, S., Tsumura, Y., Setoguchi, H., Ito, M., Hattori, C., et al. (2017). Application of a simplified method of chloroplast enrichment to small amounts of tissue for chloroplast genome sequencing. Appl. Plant Sci. 5:1700002. doi: 10.3732/apps.1700002
Schelkunov, M. I., Shtratnikova, V. Y., Nuraliev, M. S., Selosse, M. A., Penin, A. A., and Logacheva, M. D. (2015). Exploring the limits for reduction of plastid genomes: a case study of the mycoheterotrophic orchids Epipogium aphyllum and Epipogium roseum. Genome Biol. Evol. 7, 1179–1191. doi: 10.1093/gbe/evv019
Shaw, J., Shafer, H. L., Leonard, O. R., Kovach, M. J., Schorr, M., and Morris, A. B. (2014). Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. Am. J. Bot. 101, 1987–2004. doi: 10.3732/ajb.1400398
Smidt, E. C., and Borba, E. (2007). Bulbophyllums in Brazil: collection history and distribution. Orchids 76, 130–133.
Smidt, E. C., Borba, E. L., Gravendeel, B., Fischer, G. A., and van den Berg, C. (2011). Molecular phylogeny of the Neotropical sections of Bulbophyllum (Orchidaceae) using nuclear and plastid spacers. Taxon 60, 1050–1064. doi: 10.1002/tax.604009
Smidt, E. C., De Brito, A. L. V. T., Martins, A. C., Royer, C. A., Whitten, W. M., and Chase, M. W. (2018). Phylogenetics, biogeography and character evolution in the Ornithocephalus clade (Orchidaceae. Oncidiinae). Bot. J. Linn. Soc. 188, 339–354. doi: 10.1093/botlinnean/boy067
Smidt, E. C., Gallo, L. W., and Scatena, V. L. (2013). Leaf anatomical and molecular studies in Bulbophyllum section Micranthae (Orchidaceae) and their implications for systematics. Rev. Bras. Bot. 36, 75–82. doi: 10.1007/s40415-013-0008-3
Smidt, E. C., Páez, M. Z., Vieira, L. D. N., Viruel, J., De Baura, V. A., Balsanelli, E., et al. (2020). Characterization of sequence variability hotspots in Cranichideae plastomes (Orchidaceae, Orchidoideae). PLoS One 15:e227991. doi: 10.1371/journal.pone.0227991
Smidt, E. C., Silva-Pereira, V., Borba, E. L., and van den Berg, C. (2007). Richness, distribution and important areas to preserve Bulbophyllum in the Neotropics. Lankesteriana 7, 107–113.
Stern, A., Doron-Faigenboim, A., Erez, E., Martz, E., Bacharach, E., and Pupko, T. (2007). Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res. 35, W506–W511. doi: 10.1093/nar/gkm382
Swofford, D. L. (2003). PAUP∗. Phylogenetic Analysis Using Parsimony (and Other Methods). Sunderland, MA: Sinauer.
Tan, K. H., and Nishida, R. (2007). Zingerone in the floral synomone of Bulbophyllum baileyi (Orchidaceae) attracts Bactrocera fruit flies during pollination. Biochem. Syst. Ecol. 35, 334–341. doi: 10.1016/j.bse.2007.01.013
Tan, K. H., Tan, L. T., and Nishida, R. (2006). Floral phenylpropanoid cocktail and architecture of Bulbophyllum vinaceum orchid in attracting fruit flies for pollination. J. Chem. Ecol. 32, 2429–2441. doi: 10.1007/s10886-006-9154-9154
Tian, N., Han, L., Chen, C., and Wang, Z. (2018). The complete chloroplast genome sequence of Epipremnum aureum and its comparative analysis among eight Araceae species. PLoS One 13:e192956. doi: 10.1371/journal.pone.0192956
Trifinopoulos, J., Nguyen, L.-T., von Haeseler, A., and Minh, B. Q. (2016). IQTREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, 232–235. doi: 10.1093/nar/gkw256
Ueda, M., Kuniyoshi, T., Yamamoto, H., Sugimoto, K., Ishizaki, K., Kohchi, T., et al. (2012). Composition and physiological function of the chloroplast NADH dehydrogenase-like complex in Marchantia polymorpha. Plant J. 72, 683–693. doi: 10.1111/j.1365-313X.2012.05115.x
Vaidya, G., Lohman, D. J., and Meier, R. (2011). SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. doi: 10.1111/j.1096-0031.2010.00329.x
Wang, R. J., Cheng, C. L., Chang, C. C., Wu, C. L., Su, T. M., and Chaw, S. M. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8:36. doi: 10.1186/1471-2148-8-36
WCSP (2018). World Checklist of Selected Plant Families. Facilitated by the Royal Botanic Gardens, Kew. Kew: WCSP.
Weng, M. L., Ruhlman, T. A., and Jansen, R. K. (2017). Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 214, 842–851. doi: 10.1111/nph.14375
Wu, B., He, S., and Pan, Y. J. (2006). New dihydrodibenzoxepins from Bulbophyllum kwangtungense. Planta Med. 72, 1244–1247. doi: 10.1055/s-2006-947200
Wyman, S., Jansen, R., and Boore, J. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Yang, J. B., Tang, M., Li, H. T., Zhang, Z. R., and Li, D. Z. (2013). Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 13:84. doi: 10.1186/1471-2148-13-84
Yuan, Y., Jin, X., Liu, J., Zhao, X., Zhou, J., Wang, X., et al. (2018). The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nat. Commun. 9:1615. doi: 10.1038/s41467-018-03423-3425
Zhang, Y., Du, L., Liu, A., Chen, J., Wu, L., Hu, W., et al. (2016). The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 7:306. doi: 10.3389/fpls.2016.00306
Zhitao, N., Shuying, Z., Jiajia, P., Ludan, L., Jing, S., and Xiaoyu, D. (2017). Comparative analysis of Dendrobium plastomes and utility of plastomic mutational hotspots. Sci. Rep. 7, 1–11. doi: 10.1038/s41598-017-02252-2258
Zhou, M., Long, W., and Li, X. (2008). Patterns of synonymous codon usage bias in chloroplast genomes of seed plants. For. Stud. China 10:235. doi: 10.1007/s11632-008-0047-41
Keywords: plastid genome, Neotropical orchids, molecular evolution, next-generation sequencing, molecular markers
Citation: Zavala-Páez M, Vieira LN, Baura VA, Balsanelli E, Souza EM, Cevallos MC, Chase MW and Smidt EC (2020) Comparative Plastid Genomics of Neotropical Bulbophyllum (Orchidaceae; Epidendroideae). Front. Plant Sci. 11:799. doi: 10.3389/fpls.2020.00799
Received: 18 March 2020; Accepted: 19 May 2020;
Published: 03 July 2020.
Edited by:
Nina Rønsted, National Tropical Botanical Garden, United StatesReviewed by:
Cássio Van Den Berg, State University of Feira de Santana, BrazilEduardo Ruiz-Sanchez, Universidad de Guadalajara, Mexico
Copyright © 2020 Zavala-Páez, Vieira, Baura, Balsanelli, Souza, Cevallos, Chase and Smidt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michelle Zavala Páez, bWljaGVsbGV6YXZhbGFwYWV6QG91dGxvb2suZXM=; Eric de Camargo Smidt, ZWNzbWlkdEBnbWFpbC5jb20=
†ORCID: Eric de Camargo Smidt, orcid.org/0000-0002-1177-1682