 Sean R. Asselin
Sean R. Asselin Anita L. Brûlé-Babel
Anita L. Brûlé-Babel David L. Van Tassel
David L. Van Tassel Douglas J. Cattani
Douglas J. Cattani- 1Department of Plant Science, University of Manitoba, Winnipeg, MB, Canada
- 2Agriculture and Agri-Food Canada, Swift Current Research and Development Centre, Swift Current, SK, Canada
- 3The Land Institute, Salina, KS, United States
Parallels exist between the domestication of new species and the improvement of various crops through selection on traits which favor the sowing, harvest and retention of yield potential and the directed efforts to improve their agronomics, disease resistance and quality characteristics. Common selection pressures may result in the parallel selection of orthologs underlying these traits and homologies between crop species can be exploited by plant breeders to improve germplasm. Perennial grains and oilseeds are a class of proposed crops for improving the diversity and sustainability of agricultural systems. Maximilian sunflower (Helianthus maximiliani Schrad.) is a perennial crop wild relative of sunflower (Helianthus annuus L.) and a candidate perennial oilseed species. Understanding parallels between cultivated H. annuus and H. maximiliani may provide new tools for the development of Maximilian sunflower and other wild relatives of sunflower as crops to enhance functional diversity in cropping systems. F2 populations of Maximilian sunflower segregating for traits associated with the domestication ideotype of cultivated sunflower including branching architecture, capitulum morphology and flowering time were developed to investigate parallels between H. maximiliani and H. annuus. Genotype-by-sequencing (GBS) was employed to genotype novel Maximilian sunflower populations and perform quantitative-trait-loci (QTL) analysis. A total of 11 QTL in five regions were identified across 21 linkage groups using 4142 GBS derived single nucleotide polymorphism markers called using the sunflower reference genome as a guide. A major QTL on linkage group 17b, associated with aspects of floral development and apical dominance, was discovered and corresponds with a known domestication QTL hotspot in H. annuus and candidate genes were identified. This suggests the potential to exploit orthologs for neo-domestication of H. maximiliani for traits such as branching architecture, timing of anthesis, and capitulum size and morphology for the development of a perennial oilseed crop from wild relatives of cultivated sunflower.
Introduction
Annual crops comprise an estimated 60–80% of global cropland and approximately 75% of calories consumed by humans come from four annual grain crops: maize, wheat, rice, and soybean (Lobell et al., 2011). Targeting perennial species and integrating them into agroecosystems dominated by annual crops has been suggested as a method of enhancing functional diversity (i.e., the number of functionally disparate species), ecosystem function (Isbell et al., 2011), and ultimately, productivity through the introduction of new ecosystem services (Asbjornsen et al., 2014). A number of candidate perennial grain and oilseeds crops have been proposed through both the hybridization with annual crops and the identification of wild species with favorable characteristics for neo-domestication (Wagoner and Schaeffer, 1990; DeHaan et al., 2016).
Maximilian sunflower (Helianthus maximiliani Schrad.) is a perennial crop wild relative of cultivated sunflower (Helianthus annuus) and is native to much of the interior plains of North America, with a range extending from southern Canada to northern Mexico (Kawakami et al., 2011, 2014; Tetreault et al., 2016). Maximilian sunflower has been used in range seeding mixtures for high quality livestock forage, as a perennial filter strip to reduce agricultural run-off, and as a source of wildlife food and habitat (Dietz et al., 1992; USDA-NRCS, 2017). Maximilian sunflower produces an oil rich seed safe for human consumption primarily composed of linoleic and oleic acids and surveys of wild populations have shown an oil content of 31.1% (Dorrell and Whelan, 1978; Seiler, 1994; Seiler and Brothers, 1999). The natural range of Maximilian sunflower, along with its phenotypic divergence (Kawakami et al., 2011; Asselin et al., 2018), high seed production potential, and documented resistance to known common pathogens of annual sunflower (H. annuus L.) such as Sclerotinia rot [Sclerotinia sclerotiorum (Lib.) de Bary] (Taski-Ajdukovic et al., 2006; Liu et al., 2011), has attracted attention to the species for the development of a perennial oilseed. Maximilian sunflower is a candidate species for the development of dual-use perennial crop for edible oil, forage and bioenergy applications and is targeted for domestication and improvement (Cox et al., 2002; Van Tassel et al., 2014; Asselin et al., 2018).
Efforts to improve of Maximilian sunflower has, until recently, been focused on its use for conservation and rangeland applications or as a trait donor for H. annuus. Two open pollinated commercialized cultivars, “Aztec” and “Prairie Gold” were released by the United States Department of Agriculture Natural Resources Conservation Service (USDA-NRCS) in 1978 (Texas Agricultural Experiment Station, 1979; Dietz et al., 1992; USDA-NRCS, 2017). Aztec was developed for wildlife feed, livestock forage cover, use as a natural hedge, as filter-strips, and as an ornamental landscape plant. Prairie Gold was released for landscape reclamation and wildlife food plantings. Both cultivars were selected for vigor and stand establishment in Oklahoma and Texas (Aztec), or Kansas and further north (Prairie Gold). Agronomic research on Maximilian sunflower as a perennial grain candidate began at The Land Institute in the 1980s (Jackson, 1990). The first breeding program focused on developing Maximilian sunflower as a perennial grain was launched in 2002 at The Land Institute (Cox et al., 2002) with the intention of domesticating the species as a perennial oilseed. Selection for seed size and apical dominance has been effective and following three rounds of recurrent selection, the average seed size was increased by 240% and plants exhibiting a highly restricted branching habit were developed by 2012 (Van Tassel et al., 2014).
Crop domestication shows many parallels between species. Vavilov in the development of the Law of Homologous Series first recognized that heritable variation in common traits will occur in different species based on parallel selection pressures (Vavilov, 1922). The Law of Homologous Series states that closely related species, genera and families exhibit parallel variation in shared traits. In grains and oilseeds such as sunflower, parallel selection for a more determinate growth habit, larger inflorescence size, and increased grain size has occurred through domestication (Zohary, 2004; Purugganan and Fuller, 2009; Lin et al., 2012). A number of domestication syndrome and quality traits in annual grains are controlled by alleles with large effects and relatively simple genetic control. Domestication has led to the selection of common orthologs such as sh1 genes which contribute to shattering tolerance in sorghum, rice and maize (Lin et al., 2012); Q genes in wheat (Triticum aestivum L.) and barley (Hordeum vulgare L.) which confer the brittle rachis/free-threshing trait (Simons, 2005); and variations in fatty acid desaturase genes in oilseeds such as canola (Brassica napus L.) and sunflower (Peng et al., 2010; Chapman and Burke, 2012). Strong apical dominance and restricted branching were key traits identified for the domestication of H. annuus (Burke et al., 2002; Wills and Burke, 2007). Wild type Maximilian sunflower has many similarities to wild type H. annuus, which exhibits profuse branching, small achenes, indeterminate flowering and lacks shattering resistance (Burke et al., 2002; Wills and Burke, 2007). Maximilian sunflower individuals with restricted branching, resembling the unbranched ideotype of cultivated sunflower, have been identified in germplasm developed at The Land Institute (Cox et al., 2010; Van Tassel et al., 2014) and offer a unique opportunity to examine parallels in the domestication process in Helianthus. H. annuus and H. maximiliani are both diploid species (2n = 17) capable of hybridization and cytogenetic analysis has shown a high proportion of normal bivalents in hybrids, suggestive of high homology between parental chromosomes (Jan, 1997; Binsfeld et al., 2001). As an extension of Vavilov’s Law of Homologous Series, knowledge of the domestication and genomic resources available in annual sunflower could be applied as a model for the neo-domestication and improvement of Maximilian sunflower through marker-assisted-selection (MAS) for domestication orthologs.
Genotype-by-sequencing (GBS) has successfully been utilized for species in which heterologous reference genomes are available for single nucleotide polymorphism (SNP) discovery, such as for wild crop relatives of soybean (Chang et al., 2014), wheat (Edae et al., 2016), and sunflower (Baute et al., 2016; Asselin et al., 2018). The goals of this research were to examine parallels between the domestication of cultivated sunflower and the development of Maximilian sunflower as a perennial oilseed crop. This work involved applying GBS to Maximilian sunflower using annual cultivated sunflower as a reference genome and performing quantitative-trait-loci (QTL) analysis in a novel Maximilian sunflower population segregating for apical dominance.
Materials and Methods
Development of Maximilian Sunflower With Restricted Branching at the Land Institute
Seed from 96 wild Kansas populations were collected in the autumns of 1999 and 2000. Most of these were roadside populations in areas dominated by native rangeland. Ten plants from each population were transplanted to the field at The Land Institute in 2001 and evaluated for two growing seasons. One hundred and fifty individuals with higher than average seed production were selected and clonally replicated by rhizome division in 2003 for further assessment. Seed yield and seed mass data from the 2004–2005 growing seasons were used to select 20 genotypes for polycrossing. Polycrosses were performed by pollen bulking and subsequent manual pollination in 2006. Polycross seed from each of the 20 genotypes was sprouted in the greenhouse and 112 vigorous seedlings from each maternal family were transplanted to a new field in 2007. Although the purpose of this cycle was originally to estimate trait heritability using the 20 half-sib families, a single unbranched individual was found in 2007. Assuming that the unbranched trait was recessive, we planted 100 open-pollinated (half-sibling) seeds from this genotype in 2008, none of which exhibited restricted branching. Half-sibling plants were allowed to intermate and progeny were evaluated in the 2009 growing season for branching characteristics. Reduced branching individuals reappeared in the progeny, however, many of these plants exhibited reduced seed fertility. Plants with the least amount of branching and normal capitula morphology were polycrossed in 2009 and repeated cycles of visual selection for restricted branching and intermating occurred in 2010 and 2011 to develop seeds stock for populations with restricted branching. In 2012 crosses were performed between plants exhibiting restricted branching to generate full sibling families exhibiting limited to no branching.
Development of F2 Mapping Populations
The mapping populations described in this study were produced through crossing highly-branched wild-type H. maximiliani plants from Manitoba to advanced, restricted branching populations developed by The Land Institute (TLI), Salina, Kansas. Manitoban plants were selected as a parent to introduce genetic diversity to alleviate potential inbreeding depression observed in TLI germplasm and to initialize the introduction of the restricted branching trait into an earlier flowering genetic background better suited for northern growing conditions. Maximilian sunflower exhibits clinal variation in life history traits, most notably timing of flowering which occurs in July–August in Manitoban genotypes and September–October in Kansas genotypes (Heiser et al., 1969; Kawakami et al., 2011; Cattani and Van Tassel, personal communication). Full-sib families were developed for mapping traits associated with branching, capitulum size and flowering time. Five restricted branching populations from TLI (“o/i,” “I/P,” “I/H,” “h/L,” and “h/e”) were initially screened under controlled environments. Plants exhibiting a complete restriction of branches, late flowering genetic background and a single large capitulum (30–40 mm in diameter) were selected as a parents for the development of the mapping populations. The Manitoba parents consisted of wild plants collected from roadside sites across southern Manitoba. Manitoban populations, previously described under growth chamber conditions in Asselin et al. (2018), exhibit extensive branching (∼11–15 branches), an early flowering genetic background and a large number (∼12–20) of small capitula which are ∼7–21 mm in diameter. A series of crosses were produced and F1 seed derived from a cross between an individual from the TLI population “o/i” and a wild Manitoban accession collected near Brunkild, Manitoba (49.49°N, 97.57W) were grown for sib-mating. The F1 plants were clonally propagated from rhizome cuttings to produce materials for sib-crossing. Three F1 individuals (herein denoted as F1A, F1B, and F1C) were intercrossed through reciprocal sib-mating to generate two F2 populations which segregated for branching, herein denoted as crosses AB (F1A/F1B) and BC (F1B/F1C).
Phenotypic Evaluation
Plant Propagation
Seeds of the F2 were surface sterilized using a 70% ethanol solution for 10 min, allowed to air dry, and seed coats were then manually removed to break seed dormancy. Seeds were placed in 9 cm petri dishes on filter paper moistened with distilled water containing a 0.1% solution of plant preservative mixture (PPMTM, Plant Cell Technology, Washington, DC, United States). Petri plates were placed in the dark at room temperature for 48 h to germinate. Germinated seeds were transplanted once radicles had emerged. All seedlings were started in 6-well 122.9 mL volume seeding trays filled with Sunshine #4TM soilless potting mix (SunGro Horticulture Ltd., Agawam, MA, United States) and transferred to 1 L pots containing a 2:2:1 ratio of soil:sand:peat by volume once they had reached the three to four true-leaf stage. Plants were grown in a growth chamber under a 23°C 16-h day/18°C 8-h night cycle. Plant positions were assigned randomly and rotated on a weekly basis to account for potential differences in airflow, humidity, and light intensity across benches within the growth chamber. Phenotypic traits measured can be broadly classified as capitulum size traits, traits relating to anthesis, traits associated with branching architecture, traits related to total plant size. Seed yield and size traits were not examined in this study directly due to the constraints of the testing location and ability to cross-pollinate all F2 progeny examined.
Trait Evaluation
Sixteen traits were phenotyped on 163 and 177 individuals from the AB and BC crosses, respectively, for a total 340 F2 plants (Table 1). As the parental and F1 individuals underwent photoperiod induction (resulting in skewed phenotypes), complete phenotypic analysis was conducted solely on the F2 generation. Phenotyping of the appearance of reproductive buds and date of the first capitulum to reach anthesis through fifth capitulum to reach anthesis were conducted three times a week. As plants segregated for flowering time, the date of fifth anthesis was employed as a common physiological milestone at which all other traits were assessed. All nodes were counted starting at the cotyledonary node moving upwards along the main stem. Branches were measured as the total number of branches on the main stem greater than 2 cm in length. The highest branching node, a measurement of apical branching, was measured as the total number of unbranched nodes which were found above the highest branch on the apical portion of the stem. The lowest branching node, a measurement of basal branching, was measured as the number of unbranched nodes on the basal region of the stem between the cotyledonary node and lowest branch. Capitulum morphology was characterized to infer potential traits associated with shattering resistance. Shattering in wild H. annuus is in part due to the continued growth of the capitulum resulting in a convex shape (increased depth: width ratio). Domesticated H. annuus exhibits a low depth: width ratio, resulting in a flatter capitulum, less prone to shattering (Burke et al., 2002). As plants were not evaluated for seed traits with the depth: width ratio employed as a proxy for this trait.

Table 1. Descriptions of 16 phenotypic traits measured under growth chamber conditions for 340 H. maximiliani F2 plants following a 23°C 16-h day/18°C 8-h night cycle.
Data were analyzed for normality using the SAS UNIVARIATE procedure to assure data meet assumptions of normality for downstream analysis. Principal component analysis was conducted to explore trait relationships on centered and scaled data using the R function PRCOMP. The number of principal components to retain was determined by examining a breakdown of eigenvectors following Cattell’s rule (Cattell, 1966) and Horn’s parallel analysis (Horn, 1965) was run using 1000 permutations in the R package paran (Dinno, 2012) and adjusted eigenvectors >1 were retained for analysis.
DNA Extraction
Leaf tissues from all plants grown under growth chamber conditions were sampled for DNA extraction. Samples from each plant were labeled with an identity number and frozen in liquid N within 1 h of collection prior to lyophilization and storage at room temperature. Genomic DNA was extracted from the parental, F1 and a subset consisting of 190 F2 individuals, consisting of 78 individuals from the AB cross and 112 individuals from the BC cross. A modified single-tube cetyltrimethylammonium bromide (CTAB) extraction protocol (Doyle, 1987; Li et al., 2007) was employed with 1 μL of 10 mg mL–1 solution of proteinase K (Promega) added to the initial CTAB incubation step. DNA quantity was determined using a dsDNA broad Range Fluorescence Assay kit and a Qubit 2.0 Fluorometer (Life Technologies) following the manufacturers’ instructions for 1 μL sample sizes. DNA quality was assessed using 260/280 and 260/230 nm wavelength absorbance ratios measured using a Nanodrop 1000 spectrophotometer (Thermo Fischer Scientific). Samples that fell below a minimum DNA concentration of 50 μg/μL or absorbance ratios below 1.7 for either 260/230 or 260/280 nm were discarded and extractions were repeated to meet quality requirements.
Genotype-by-Sequencing and SNP Calling
One-hundred and ninety-five DNA samples consisting of the Manitoba and Kansas parentals, F1 parents and 190 F2 plants were submitted to Data2bio LLC (Ames, IA, United States) for GBS and SNPs calling. A tunable genotype-by-sequencing (tGBS) protocol was employed, which differs from conventional GBS through the use of a greater selective genome reduction procedure (Ott et al., 2017). Fewer sites are sequenced in tGBS relative to the conventional GBS, resulting in greater read depth. The advantage of this approach is that greater depth of coverage allows for more effective calling of heterozygous genotypes and reduces false homozygote calls, leading to a lower level of missing data per site.
Sequencing via tGBS was carried out using a three selective base (TGT) protocol as described in Ott et al. (2017) for genome reduction. Fragments were sequenced via eight runs on an Ion ProtonTM sequencer (Thermo Fisher Scientific, Waltham, MA, United States). The conservative three selective base approach was chosen as H. maximiliani is an obligate outcrossing species and exhibit a high degree of heterozygosity (Asselin et al., 2018). Trimmed reads were aligned to the cultivated H. annuus reference genome, HA412.v.1.1.bronze.20142015,1 using GSNAP (Wu and Nacu, 2010) with polymorphisms within the first and last 3 bp for the read being ignored (Ott et al., 2017). Polymorphisms with base calls with a PHRED score below 20 were removed from the analyses. The SNPs were called as homozygous if the most common allele was supported by a minimum of 80% aligned reads, while heterozygous SNPs were called if the two most common alleles were supported by a minimum of 30% of all aligned reads and the sum of the two most common alleles accounted for a minimum of 80% of the aligned reads. Single nucleotide polymorphisms that could be genotyped in a minimum of half of the samples, that contained an allele number of 2, a minor allele frequency >1% and a heterozygous rate of <70% were maintained for downstream analysis. Imputation was conducted separately on the AB and BC crosses using Beagle4.1 (Browning and Browning, 2016) using default parameters.
Linkage Map Construction
The AB and BC crosses were examined jointly as a single population employing markers segregating in the same fashion between the two crosses for analysis in Joinmap 5.0 (Van Ooijen, 2018). Common segregation patterns between the crosses were determined using the F1 genotypes and observed segregation patterns in the F2 generation to allow joint analysis. Markers were coded as three possible segregation types (<lmxll>, <nnxnp>, and <hkxhk>) corresponding to backcross-like or F2-like segregation patterns. Homozygous allele states accounting for less than 10% of the observed total calls were considered likely genotyping errors and set to missing data prior to analysis. Markers classes in which either the parental genotype could not be accurately determined or were non-informative were removed. The remaining markers were tested for distorted segregation using a chi-square goodness-of-fit test through the test segregation function of Joinmap at p < 0.05 for downstream analysis. Due to the presence of a high degree of segregation distortion, markers exhibiting distortion were retained within the dataset and initially grouped based upon their assigned linkage groups in the cultivated H. annuus HA412.v.1.1.bronze.20142015 reference genome for ordering. Segregation distortion has been reported as having little effect on marker order (Zhang et al., 2010), but erroneously called genotypes, which may result in the appearance of highly-distorted markers, may cause pseudo-linkages between biologically unlinked sequences (Ronin et al., 2010).
Linkage map construction was conducted using the cross-pollinated (CP) analysis option in Joinmap 5.0. Identical markers were removed and reference genome positions were utilized in initial maps using the “start order” option to position markers and linkage groups were numbered based upon their reference genome analog. Markers were ordered using the regression mapping algorithm with map distances in cM being calculated using Kosambi’s genetic distance. Markers which produced negative map distances were excluded from the map and maps were recalculated. In instances were insufficient linkage was present within a linkage group markers were split into separate groups (i.e., LG 3a and 3b) for analysis to produce the final map.
QTL Analysis
Restricted multiple-qtl model (rMQM) analysis was conducted using MapQTL 6.0. As a subset of the genotyped F2 plants exhibited an abnormal “die-back” of the central capitulum, 15 plants were removed from the dataset, leaving 175 individuals for QTL analysis. An initial analysis was conducted to identify putative QTL and possible cofactors for rMQM using the interval mapping procedure at 1 cM intervals. A non-parametric one-way analysis of variance Kruskal–Wallis (K–W) rank-sum test was also conducted. The K–W test method does not require the incorporation of a genetic map, providing marker-trait associations independently of neighboring markers and was used to confirm putative co-factors and QTLs. To account for multiple testing, a stringent alpha of 0.005 was used to reduce potential spurious associations as suggested by Van Ooijen (2018). Possible rMQM cofactors were limited to no more than two per linkage group per trait and an automatic cofactor selection procedure in MapQTL 6.0 was performed using default settings. Subsequent rounds of rMQM mapping were performed for each trait until co-factors stabilized. To account for multiple testing and control of family-wise-error rate for type I errors, whole-genome LOD thresholds were calculated for each trait independently (Churchill and Doerge, 1994). Appropriate genome-wide LOD threshold values were estimated using 1000 permutations and an alpha threshold of p < 0.05. A genome-wide LOD threshold score of p < 0.05 was employed to declare significant QTL. Confidence intervals were calculated using the two-LOD drop-off method which approximates roughly a 95% confidence interval of QTL position (Lander and Botstein, 1989). Strongly overlapping/pleiotropic QTL between two or more traits were given a common name to designate the QTL region.
To infer candidate genes and provide functional annotation of the candidate SNPs generated from the analyses, H. maximiliani SNPs falling within the two-LOD QTL confidence interval were compared with the cultivated H. annuus reference genome HA412.v.1.1.bronze.20142015 assembly using Jbrowse (Skinner et al., 2009), available via the HeliaGene bioinformatics portal (Carrere et al., 2008). Significant SNPs found within a 1kb distance were considered a common entry for annotation. SNPs were compared to their positions within the H. annuus reference genome and a search was conducted for possible candidate genes within 100 kb of each entry. To annotate the presence of putative orthologs, a literature search of sunflower domestication studies was conducted to determine if QTL controlling similar traits have previously been reported on the same linkage groups in H. annuus.
Results
Phenotypic Analysis
The initial TLI populations screened for restricted branching exhibited either a single capitulum and a complete lack of branching, or a complete lack of mid-plant and basal branches and a small number (2–3) short apical branches bearing capitula. It has been suggested that branching restriction in Maximilian sunflower exhibits a leaky phenotype, and this observation is consistent with previous observations from restricted branching populations (Van Tassel, personal communication). The F1 plants were uniform in appearance, and exhibited a branched phenotype consistent with that of wild Manitoban populations. The range of phenotypes observed amongst both crosses was extensive in the F2 generation and the phenotypic distributions are suggestive of quantitative genetic control (Table 2). The unbranched phenotype observed in the TLI parent was not recovered in the F1 or F2 generations although a number of plants exhibiting highly restricted branching were observed in the F2 generation. Branch number ranged from one to 21 branches and the percentage of branch bearing nodes ranged from 2.38 to 48.97%. Timing of first anthesis ranged from 61 to 198 days. Plant height ranged from 90to 192 cm and diameter of the central capitulum ranged from 6.2to 25.6 mm. The total number of capitulum exhibited the highest coefficient of variation relative to other traits examined, ranging from 4 to 126 capitula per plant likely due to the presence of secondary and tertiary branches.
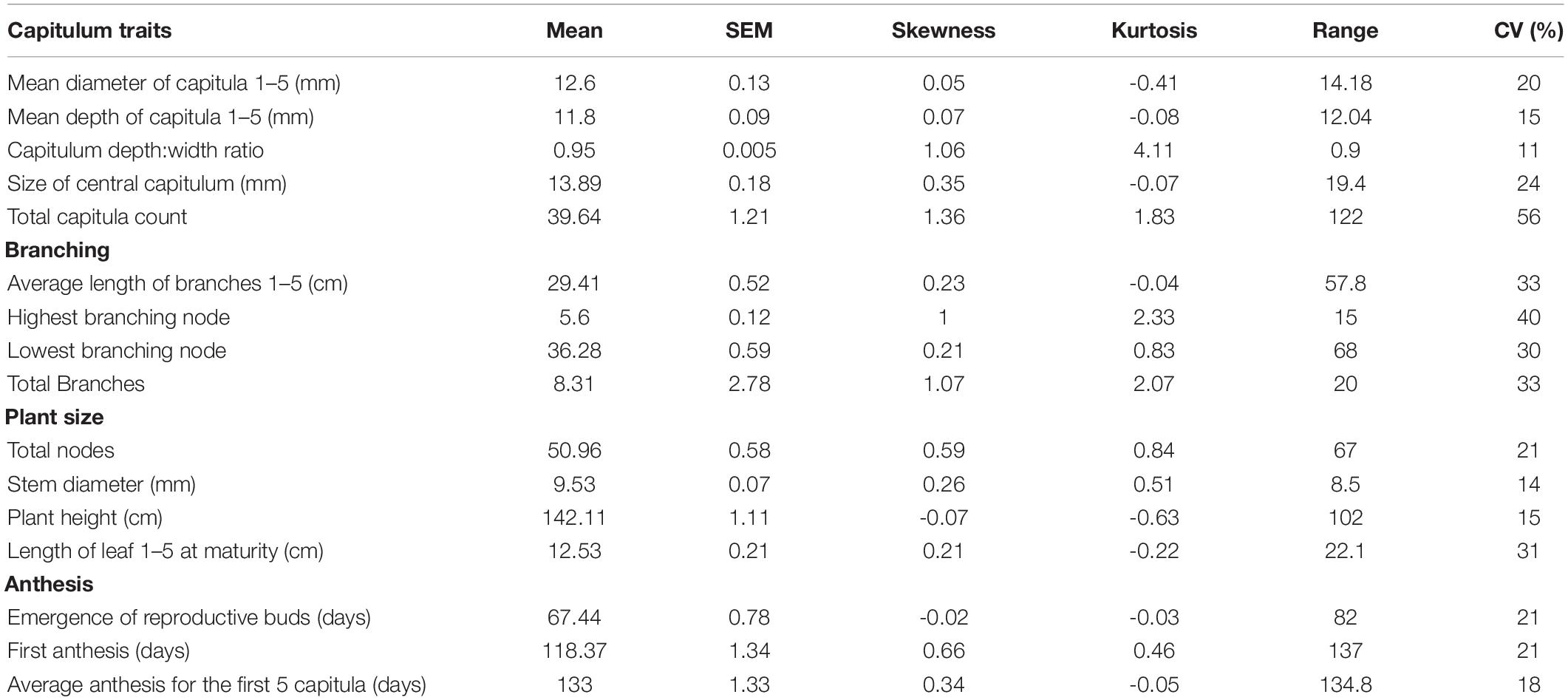
Table 2. Phenotypic mean, standard error of the mean (SEM) and coefficient of variation (CV) and other summary statistics of 340 F2 H. maximiliani plants phenotyped for 21 traits under growth chamber conditions following a 23°C 16-h day/18°C 8-h night cycle.
Principal component analysis revealed four significant components and patterns suggestive of a resource allocation trade-off relating to the degree of apical dominance (Figure 1 and Supplementary Table S1). Along the first two principal components axes a negative relationship between capitulum size traits (greater apical dominance) and the total number of branches and capitula (reduced apical dominance) is apparent. The first component (PC1) which explained 30.9% of the phenotypic variance appears to be dominated by trait differences in the parental materials, with positive loadings for later anthesis and increased capitulum size and negative loadings for capitulum and branch number. The second component (PC2), explaining 21.64% of the phenotypic variance revealed patterns between timing and anthesis and capitulum size traits which contradict the first component, suggesting these traits are not strongly associated and may represent recombination between parental types. The third and fourth components (Supplementary Figure S1), explaining 9.84 and 8.36% of the phenotypic variance respectively were dominated by branching characteristics (PC3) and other aspects of apical dominance (highest branching node, average branch length and capitulum count) as well as stem diameter (PC4).
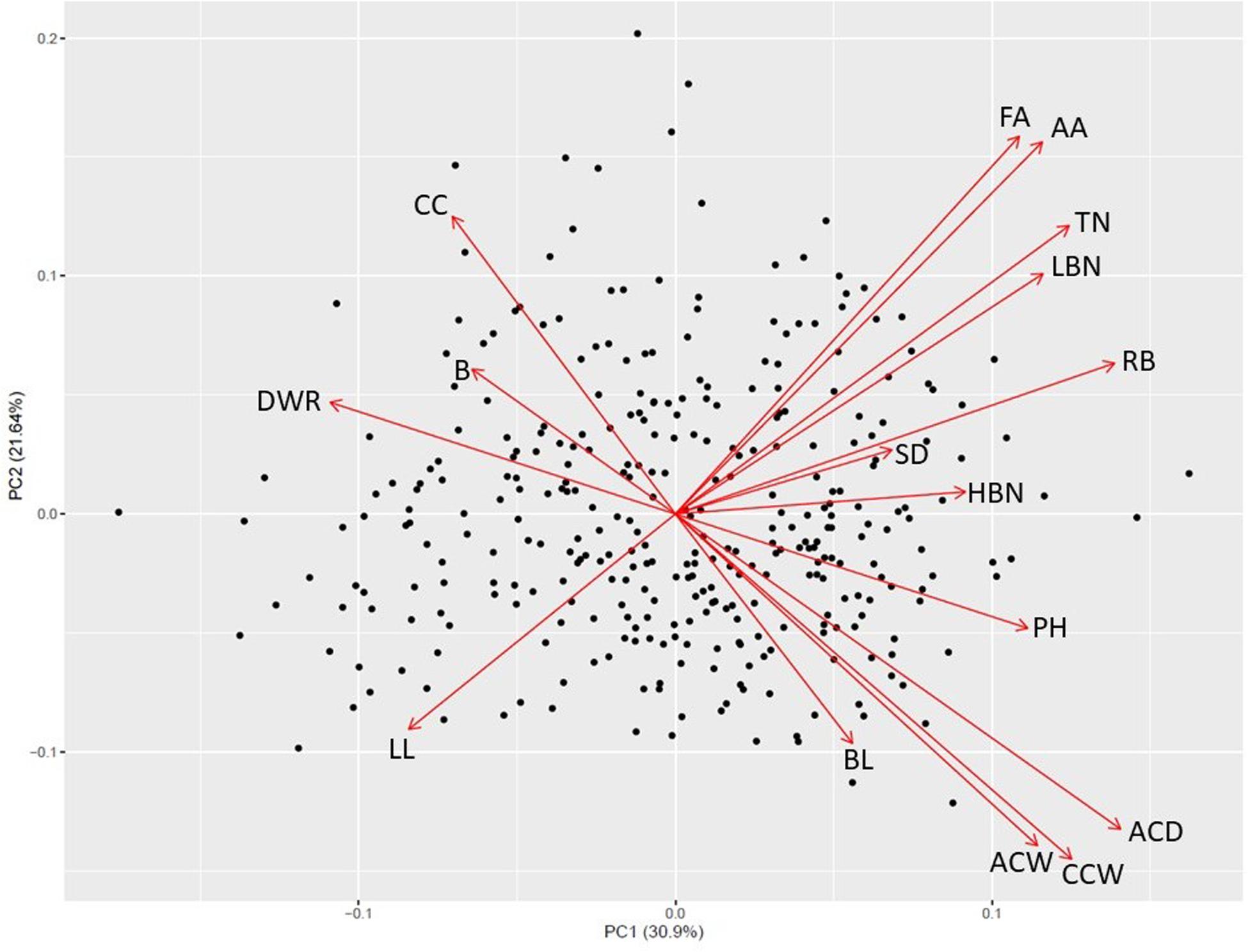
Figure 1. First and second principal component axes for 340 Maximilian sunflower individuals segregating for apical dominance. FA, First anthesis; AA, average date of anthesis; TN, total nodes; LBN, lowest branching node; RB, reproductive budding; SD, stem diameter; HBN, highest branching node; PH, plant height; ACD, average capitula depth; CCW, central capitulum width; ACW, average capitula width; BL, branch length; LL, leaf length; DWR, capitulum depth:width ratio; B, branches; CC, total capitula count.
SNP Calling and Linkage Map Development Using the Homeologous H. annuus Genome as a Reference Guide
A total of 6094 polymorphic SNPs were aligned to the homeologous reference genome of H. annuus for QTL identification spanning all linkage groups. Following filtering 5323 SNPs were considered for joint linkage map development and QTL analysis. Due to insufficient linkage more than the expected 17 linkage groups of H. maximiliani were recovered, linkage groups 3, 5, 13, and 17 were split into separate groups resulting in 21 linkage groups (Supplementary Figure S2). To date, this is the first reported linkage map of Maximilian sunflower and spans a total of 3159.29 cM of the Maximilian sunflower genome, with an average marker interval of 1.31 cM. A total of 4142 markers were incorporated into the map, with 42–300 markers per linkage group (Supplementary Table S2).
QTL Analysis and Identification of Putative Candidate Genes Associated With Domestication
The Genome-wide QTL scans detected 11 significant QTL that were supported by both the rMQM and K-W analysis which corresponded to five genomic regions. A major co-localized QTL (herein referred to as ultra1) was detected on linkage group 17b explaining 12.1–21.5% of the variation in traits associated with timing of anthesis (timing of reproductive budding, first anthesis and average date of anthesis) as aspects of apical dominance (number of branches, average leaf length, average capitulum size, and average capitulum depth) (Table 3). Further QTL for timing of reproductive budding were found on linkage groups 6 (rb1) and 15 (rb2), and total capitulum count on linkage groups 2 (cc1) and 3b (cc2).
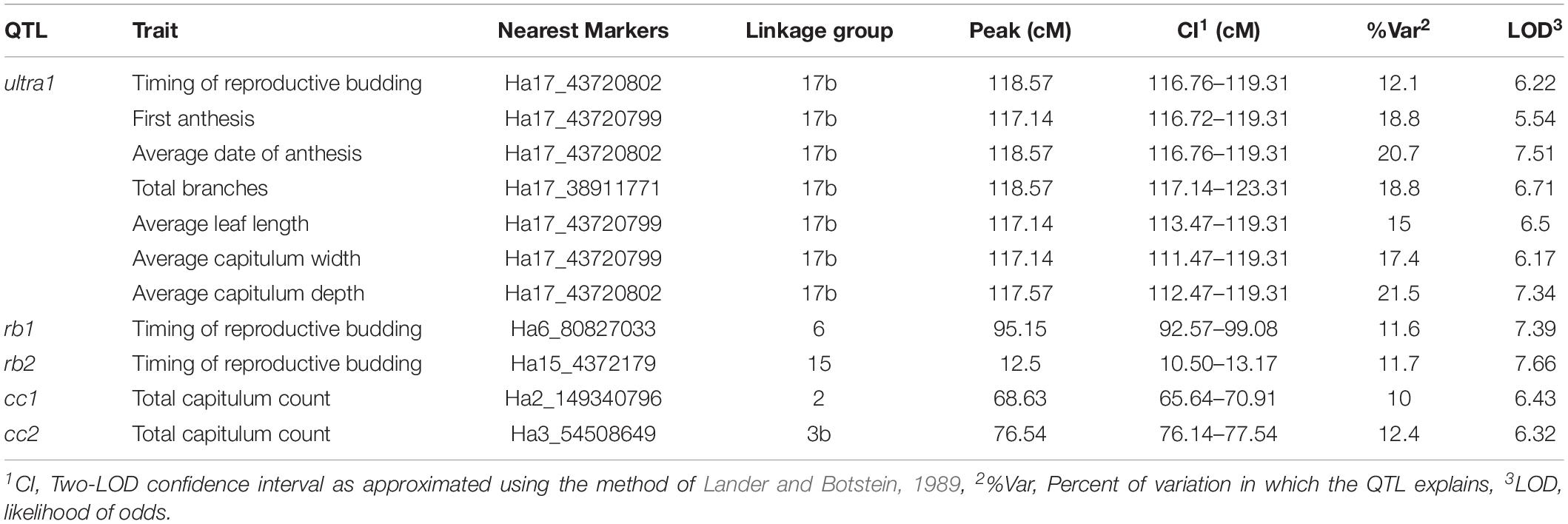
Table 3. Significant genome-wide QTL in the tested Maximilian sunflower populations (p < 0.05).
To identify putative genes associated with the domestication phenotype of H. annuus 100 kb flanking regions surrounding SNPs within the two-LOD QTL support intervals were manually examined using the sunflower genome JBrowse (INRA Sunflower Bioinformatics Resources, 2014). None of the QTL associated SNPs were found within gene sequences but several were within close proximity to annotated genes (Supplementary Table S3).
The SNPs associated with the QTL ultra1 on linkage group 17b of H. maximiliani correspond to a gene-rich region of the H. annuus reference genome. A variety of putative functions were found within this region relating to hormone response and regulation and meristem development. Nearby genes in the region of ultra1 include plant GH3 auxin-responsive promotors (IPR004993) and a ULTRAPETALA developmental regulator (IPR020533) (Supplementary Table S3) which is known as a key negative regulator of cell accumulation in shoot and floral meristems in Arabidopsis thaliana (Fletcher, 2001; Carles et al., 2004). Genes encoding Cytochrome P450 (IPR001128) and F-box domain proteins (IPR001810), both of which are candidate genes for branching in H. annuus (Mandel et al., 2013; Nambeesan et al., 2015), were also identified.
Interestingly, the SNPs associated with ultra1 are approximately 200 kb downstream to a region containing a number of potentially influential genes associated with AUX/IAA proteins (IPR003311), Auxin response factors (IPR010525) and self-incompatibility S1 proteins (IPR010264) suggesting potential genetic linkage between genes associated with the domestication phenotype of H. annuus and self-compatibility as noted in previous studies (Burke et al., 2002; Gandhi et al., 2005; Tang et al., 2006).
The SNPs associated with cc1 were found to be in proximity to genes which code proteins related to cell proliferation, differentiation and development (ARID/BRIGHT DNA-binding domain, IPR001606) and plant hormone production and regulation (AUX/IAA protein, IPR003311; Auxin response factor, IPR015300; Terpenoid synthase, IPR0089494 and Cytokinin riboside 5′-monophospahte phophoribohydrolase LOG, IPR005269). The annotations for cc2 included a fibroblast growth factor receptor (IPR016248). While SNPs associated with rb1 were not found to be within 100 kb of putative candidate genes, these SNPs flank a genomic region containing a phytochrome domain (IPR001294) which is associated with plant light response, the control of photoperiodism and flowering. The presence of a flowering-time QTL on this linkage group is also in line with previous studies which have identified a cluster of paralogous HaFT genes on LG 6 which are major contributors to photoperiod sensitivity in H. annuus (Blackman et al., 2011). Annotations for rb2 which corresponds to LG 15 of H. annuus did not yield obvious functional candidates within close proximity to the associated SNPs though a QTL for flowering is known on this linkage group in wild X landrace crosses of H. annuus (Wills and Burke, 2007; Blackman et al., 2011).
Discussion
Genetic Parallels With H. annuus and Putative S-Locus Linkage
The release of the sunflower reference genome has enhanced the ability to generate SNP libraries for analyzing both natural and segregating populations of cultivated sunflower as well as their wild crop relatives. The use of the sunflower reference genome greatly assisted in the development of a genetic map of Maximilian sunflower and in the identification of candidate genes.
Several promising candidate genes within 100 kb of ultra1 were identified in this study, one of which is documented in Arabidopsis to influence the phenotypic traits associated with ultra1. ULTRAPETALA (Ult1) has been shown to regulate a number of development processes including the timing of vegetative to floral transition, maintenance of the shoot apical meristem and termination of floral meristem development in A. thaliana (Fletcher, 2001; Carles et al., 2004). This is in line with the associations between ultra1 and timing of anthesis, overall capitulum size and branching in the present study, which presents a strong case for a causative candidate gene. Plants that carry a Utl1 loss-of-function mutation show restricted branching and an increase in floral meristem size similar to the Kansas populations utilized in this study.
Another promising candidate gene in proximity to ultra1 is a GH3 auxin-responsive promotor. The role of auxins, cytokinins, and strigolactones in the control of shoot branching and maintenance of apical dominance is well-documented (Bennett et al., 2006; Shimizu-Sato et al., 2009). Several candidate genes have been identified in sunflower domestication studies relating to the production, regulation, and transport of these hormones (Nambeesan et al., 2015).
Additional candidates underlying ultra1 include cytochrome P450 and F-box proteins. The cytochrome P450 and F-box gene families include several well-known genes associated with the control of axillary meristem initiation and growth such as the more axillary growth (MAX) genes of A. thaliana (Stirnberg et al., 2002). One of these genes, MAX2 encodes an F-box protein which has been shown to co-localize with a branching QTL on LG 17 of H. annuus (Mandel et al., 2013).
An interesting parallel between H. maximiliani and H. annuus was observed in the QTL analysis. The QTL ultra1 on LG 17b corresponds to a region in the H. annuus reference genome which harbors annotations for a number of S-locus associated proteins, suggesting potential linkage between ultra1 and self-compatibility in H. maximiliani. Previous studies looking at wild X crop crosses in H. annuus have observed a preponderance for crop alleles on this linkage group and a clustering of upwards of 20 domestication and post-domestication QTL in tight linkage with the S-locus on LG 17 (Burke et al., 2002; Gandhi et al., 2005; Tang et al., 2006).
The role of ULTRAPETALA (Ult1) genes in sunflower has yet to be explored but several studies have identified domestication QTL in tight linkage with the S-locus on LG 17 of H. annuus. Burke et al. (2002) observed a clustering of QTL for flowering time, stem diameter, capitulum number, number of capitula/branch, plant height, number of leaves, peduncle length, achene weight, shattering and total number of selfed seeds on LG 17. Gandhi et al. (2005) subsequently mapped the S-locus to the same region on LG 17. Tang et al. (2006) observed several overlapping QTL for seed size and seed oil concertation in close proximity to the S-locus on LG 17. Wills and Burke (2007) also observed overlapping QTL for capitulum diameter, branch number, ray number, total capitula number and number of selfed seeds again in the same region on LG 17.
The presence of an S-locus on LG 17b in Maximilian sunflower may impact breeding efforts and is one explanation that helps explain the reduced fertility observed in the initial Kansas populations selected for apical dominance. This observation warrants further investigation as other forms of inbreeding depression reducing fertility cannot be ruled out at this stage. Tight linkage between domestication QTL such as ultra1 and an S-locus could impose restrictions on domestication if linkage cannot be broken. Stringent selection for a given variant of a ultra1 allele may result in a genetic bottleneck and reduced S-locus allelic diversity. Limited genetic diversity in breeding pools of Maximilian sunflower may limit seed yield through the lack of compatibility in pollen sources, reducing to ability to generate novel germplasm through directed crosses. Fine-scale genetic mapping of ultra1 and the S-locus and development of further SNP markers in Maximilian sunflower would provide an efficient way to break the putative genetic linkage between these loci through the identification of recombinant progeny carrying different S-locus alleles.
Phenotypic Trade-Offs in the Context of Neo-Domestication
A trade-off between capitulum size and capitulum number was observed along both the first and second principal component axis in the F2 populations and is suggestive of a putative resource allocation trade-off between capitulum size and capitulum number. In H. annuus, unbranched biotypes have larger capitula and seeds than their branched counterparts.
The presence of a major QTL such as ultra1 influencing flowering time, branching and capitulum morphology would explain the first principal component of the PCA which is dominated by type differences observed in the parental materials. The presence of ultra1 helps explain the phenotypic correlations between traits such as later flowering and increased capitulum width and depth. Interestingly, the second principal component shows the opposite relationship between these traits suggesting this trait correlation is not absolute.
While selection for a single large capitulum is a defining domestication syndrome characteristic of cultivated annual sunflower, the path perennial oilseeds will take through domestication will ultimately depends on the how standing genetic variation will contribute to yield. In sunflower, branching not only interacts with capitulum size and total capitula number but also seed weight and oil content amongst other traits (Fick et al., 1974; Dedio, 1980; Tang et al., 2006; Bachlava et al., 2010).
While there are inherent benefits to restricting branching, such as the facilitation of mechanical harvest and uniform maturity, restricting branching may also limit yield potential if selected for too stringently. Branched Maximilian sunflower plants are capable of producing a large number of capitula per stem–1 (>100) while completely unbranched plants exhibited a single, central capitulum. The relationship between capitulum size and capitula number in Maximilian sunflower is not a 1:1 ratio. Plants with restricted branching have the tendency to exhibit larger capitula, though this increase in capitulum size does not appear to fully compensate for the loss of capitulum number and it has been suggested that this may limit seed yield (Van Tassel et al., 2014; DeHaan et al., 2016). Selection for a single central capitulum, akin to the domestication ideotype of cultivated sunflower, could decrease seed production in H. maximiliani if the loss of capitula through restricted branching is not compensated for by a reciprocal increase in capitulum size. Conversely, the single capitulum ideotype may be better suited for polyculture use as opposed to a monoculture, as the loss of yield associated with an unbranched stem may be compensated for by diversification within the field (Picasso et al., 2008; Picasso et al., 2011).
Helianthus annuus was initially domesticated for its edible seed, pigments and medicinal compounds (Heiser, 1951) with selection for oil content and composition occurring more recently (Burke et al., 2005; Chapman and Burke, 2012). Therefore, the defining characteristics of its domestication may not necessarily apply to the neo-domestication of Maximilian sunflower as an oilseed crop. Selection for a Maximilian sunflower ideotype as an oilseed may parallel other small-seeded Compositae oilseeds such as safflower (Carthamus tinctorius) and noug (Guizotia abyssinica) both of which exhibit contrasting domestication syndromes to annual sunflower (Pearl et al., 2014; Dempewolf et al., 2015), having retained a branched architecture and selection for greater seed production. Similarly, successful oilseed crops in Western Canada such as canola (B. napus L.) retain a branched architecture, small seed size, and are still prone to considerable shattering losses under certain conditions (Gulden et al., 2003; Cavalieri et al., 2016). Despite these characteristics, these crops are capable of sustaining economic yields supporting their use as crops. Increased branching and seed number may constrain seed size, in H. annuus and smaller seeds tend to bear a higher concentration of oil. Estimates of 6.7–8.5% greater oil concentration in the seed of branched individuals relative to unbranched individuals in segregating populations have been observed, presumably due to a thinner pericarp (Tang et al., 2006; Bachlava et al., 2010). Therefore, increasing branching and seed number may prove beneficial in increasing oil yield per unit area.
Though selection for restricted branching may limit the total production of capitula, and by extension yield potential on a single plant basis, branching restriction may enhance yield on a per unit area. It has been observed that in species such as safflower lower order branches are often infertile and that in general, plants that produced many branches produced smaller capitula, fewer florets per branch, and smaller seeds in smaller quantities (Bidgoli et al., 2006). Branching restriction may enhance harvestable yield indirectly through greater synchronicity of flowering, maturity of capitula and through changes in capitulum morphology, both of which may alleviate shattering losses. In the present study, the capitulum depth:width ratio appears to decrease with branching restriction and the increase in capitulum size. Plants with a wider, flatter capitulum, more akin to those of domesticated sunflower may exhibit reduced shattering losses if these patterns are common between species. Furthermore, it may be possible to compensate for the loss of secondary capitula through increasing stem density on a per unit area, either through selection for a greater number of stems per plant or sowing higher density plantings coupled with tolerance to crowding. Ultimately, increasing the harvest index of Maximilian sunflower through conventional as well as marker-assisted breeding approaches appears to be possible through multiple pathways and further investigation into these factors under realistic field conditions is required to define the optimal ideotype for production.
In summary, we were successful in identifying QTL for important traits associated with domestication in Maximilian sunflower and identified candidate genes using the existing genomic resources of cultivated sunflower. Results presented in this study have implications for the defining an appropriate ideotype for perennial oilseed crops in the Compositae and the development of genomic resources in crop wild relatives. Further work is needed to compare contrasting Maximilian sunflower ideotypes and characterization is required to verify the presence of putative S-locus linkage and the development of markers for this and other domestication QTL. Future work will focus on the field based evaluation of yield components in proposed Maximilian sunflower ideotypes and the development of a de novo genetic map for Maximilian sunflower and fine mapping of genomic regions surrounding ultra1. Understanding both the genetic and agronomic parallels of domestication will play an important role in the neo-domestication of Maximilian sunflower and related species.
Data Availability Statement
The SNP dataset generated in this study is available in Dryad digital repository (doi: 10.5061/dryad.tdz08kpwt). Any other datasets and germplasm used and/or analyzed during the current study will be made available through contact with the corresponding author on reasonable request.
Author Contributions
SA generated the mapping populations, conducted the greenhouse and laboratory work, analyzed the data, and wrote the manuscript as a graduate student. DC and AB-B provided laboratory resources and supervision of experimental design and aided in the editing of the manuscript. DV developed the initial restricted branching populations and provided germplasm and feedback on the manuscript. All authors have reviewed and agreed on the contents of the manuscript.
Funding
This work was supported by the Western Grains Research Foundation graduate scholarship, by grants from Manitoba Agriculture [Advanced Rural Development Initiative (ARDI) Project No. 12-1136], and The Land Institute, Salina, KS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge Ardelle Slama, Robert Visser, Kathy Bay, and Ian Brown for providing greenhouse support throughout the length of the experiment and Dr. Maria Stoimenova and Dr. Curt McCartney for advice and technical support in the areas of molecular biology and genetic mapping respectively. Dr. Robert Gulden, Dr. Barbara Sharanowski, and Dr. Khalid Rashid are acknowledged for serving on SA’s graduate student committee and providing feedback throughout the length of the experiment.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00834/full#supplementary-material
Footnotes
References
Asbjornsen, H., Hernandez-Santan, V., Liebman, M., Bayala, J., Chen, J., Helmers, M., et al. (2014). Targeting perennial vegetation in agricultural landscapes for enhancing ecosystem services. Renew. Agric. Food Syst. 29, 101–125. doi: 10.1017/S1742170512000385
Asselin, S. R., Brûlé-Babel, A. L., and Cattani, D. J. (2018). Genetic characterization of maximilian sunflower for the development of a locally adapted perennial grain oilseed. Crop Sci. 58, 2241–2260. doi: 10.2135/cropsci2017.10.0611
Bachlava, E., Tang, S., Pizarro, G., Schuppert, G. F., Brunick, R. K., Draeger, D., et al. (2010). Pleiotropy of the branching locus (B) masks linked and unlinked quantitative trait loci affecting seed traits in sunflower. Theor. Appl. Genet. 120, 829–842. doi: 10.1007/s00122-009-1212-1
Baute, G. J., Owens, G. L., Bock, D. G., and Rieseberg, L. H. (2016). Genome-wide genotyping-by-sequencing data provide a high-resolution view of wild Helianthus diversity, genetic structure, and interspecies gene flow. Am. J. Bot. 103, 2170–2177. doi: 10.3732/ajb.1600295
Bennett, T., Siberer, T., Willett, B., Book, J., Luschnig, C., and Leyser, O. (2006). The Arabidopsis MAX pathway controls shoot branching by regulating auxin transport. Curr. Biol. 16, 553–563. doi: 10.1016/j.cub.2006.01.058
Bidgoli, A. M., Akbari, G. A., Mirhadi, M. J., Zand, E., and Soufizadeh, S. (2006). Path analysis of the relationships between seed yield and some morphological and phenological traits in safflower (Carthamus tinctorius L.). Euphytica 148, 261–268. doi: 10.1007/s10681-005-9019-x
Binsfeld, P. C., Wingender, R., and Schnabl, H. (2001). Cytogenetic analysis of interspecific sunflower hybrids and molecular evaluation of their progeny. Theor. Appl. Genet. 102, 1280–1285. doi: 10.1007/s001220000517
Blackman, B. K., Rasmussen, D. A., Strasburg, J. L., Raduski, A. R., Burke, J. M., Knapp, S. J., et al. (2011). Contributions of flowering time genes to sunflower domestication and improvement. Genetics 187, 271–287. doi: 10.1534/genetics.110.121327
Browning, B. L., and Browning, S. R. (2016). Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 98, 116–126. doi: 10.1016/j.ajhg.2015.11.020
Burke, J. M., Knapp, S. J., and Rieseberg, L. H. (2005). Genetic consequences of selection during the evolution of cultivated sunflower. Genetics 171, 1933–1940. doi: 10.1534/genetics.104.039057
Burke, J. M., Tang, S., Knapp, S. J., and Rieseberg, L. H. (2002). Genetic analysis of sunflower domestication. Genetics 161, 1257–1267.
Carles, C. C., Lertpiriyapong, K., Reville, K., and Fletcher, J. C. (2004). The ULTRAPETALA1 gene functions in early Arabidopsis development to restrict shoot apical meristem activity and acts through WUSCHEL to regulate floral meristem determinacy. Genetics 167, 1893–1903. doi: 10.1534/genetics.104.028787
Carrere, S., Gouzy, J., Langlade, N., Gamas, P., and Vincourt, P. (2008). “HELIAGENE, a bioinformatics portal for Helianthus sp genomics,” in Proceeding of the 17th International Sunflower Conference, Cordoba, Spain. 8–12 June 2008 (Paris: International Sunflower Association), 611–615.
Cattell, R. B. (1966). The scree test for the number of factors. Multivariate Behav. Res. 1, 245–276. doi: 10.1207/s15327906mbr0102_10
Cavalieri, A., Harker, K. N., Hall, L. M., Willenborg, C. J., Haile, T. A., Shirtliffe, S. J., et al. (2016). Evaluation of the causes of on-farm harvest losses in canola in the northern great plains. Crop Sci. 56, 2005–2015. doi: 10.2135/cropsci2016.01.0014
Chang, S., Thurber, C. S., Brown, P. J., Hartman, G. L., Lambert, K. N., and Domier, L. L. (2014). Comparative mapping of the wild perennial Glycine latifolia and Soybean (G. max) reveals extensive chromosome rearrangements in the genus Glycine. PLoS One 9:e99427. doi: 10.1371/journal.pone.0099427
Chapman, M. A., and Burke, J. M. (2012). Evidence of selection on fatty acid biosynthetic genes during the evolution of cultivated sunflower. Theor. Appl. Genet. 125, 897–907. doi: 10.1007/s00122-012-1881-z
Churchill, G. A., and Doerge, R. W. (1994). Empirical threshold value for quantitative trait mapping. Genetics 138, 963–971. doi: 10.1534/genetics.107.080101
Cox, T. S., Bender, M., Picone, C., Van Tassel, D. L., Holland, J. B., Brummer, E. C., et al. (2002). Breeding perennial grain crops. Crit. Rev. Plant Sci. 21, 59–91.
Cox, T. S., Van Tassel, D. L., Cox, C. M., and Dehaan, L. R. (2010). Progress in breeding perennial grains. Crop Pasture Sci. 61, 513–521. doi: 10.1071/CP09201
Dedio, W. (1980). Comparison of achene characteristics and combining ability of branching and non-branching near-isogenic sunflower restorer lines. Crop Sci. 20, 189–190. doi: 10.2135/cropsci1980.0011183X002000020010x
DeHaan, L. R., Van Tassel, D. L., Anderson, J. A., Asselin, S. R., Barnes, R., Baute, G. J., et al. (2016). A pipeline strategy for grain crop domestication. Crop Sci. 56, 917–930. doi: 10.2135/cropsci2015.06.0356
Dempewolf, H., Tesfaye, M., Teshome, A., Bjorkman, A. D., Andrew, R. L., Scascitelli, M., et al. (2015). Patterns of domestication in the Ethiopian oil-seed crop noug (Guizotia abyssinica). Evol. Appl. 8, 464–475. doi: 10.1111/eva.12256
Dietz, D. R., Wasser, C. H., Dittberner, P. L., and Martin, C. O. (1992). Defense Natural Resources Program: Maximilian Sunflower (Helianthus maximiliani), Section 7.4.3. Washington, D.C: US Army Corps of Engineers Wildlife Resources Management.
Dorrell, D. G., and Whelan, E. D. P. (1978). Chemical and Morphological Characteristics of Seeds of Some Sunflower Species 1. Crop Sci. 18, 969–971. doi: 10.2135/cropsci1978.0011183X001800060015x
Doyle, J. J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Edae, E. A., Olivera, P. D., Jin, Y., Poland, J. A., and Rouse, M. N. (2016). Genotype-by-sequencing facilitates genetic mapping of a stem rust resistance locus in Aegilops umbellulata, a wild relative of cultivated wheat. BMC Genomics 17:1039. doi: 10.1186/s12864-016-3370-2
Fick, G. N., Zimmer, D. E., and Zimmerman, D. C. (1974). Correlation of seed oil content in sunflowers with other plant and seed characteristics. Crop Sci. 14, 755–757. doi: 10.2135/cropsci1974.0011183x001400050042x
Fletcher, J. C. (2001). The ULTRAPETALA gene controls shoot and floral meristem size in Arabidopsis. Development 128, 1323–1333.
Gandhi, S. D., Heesacker, A. F., Freeman, C. A., Argyris, J., Bradford, K., and Knapp, S. J. (2005). The self-incompatibility locus (S) and quantitative trait loci for self-pollination and seed dormancy in sunflower. Theor. Appl. Genet. 111, 619–629. doi: 10.1007/s00122-005-1934-7
Gulden, R. H., Shirtliffe, S. J., and Thomas, A. G. (2003). Harvest losses of canola (Brassica napus) cause large seedbank inputs. Weed Sci. 51, 83–86. doi: 10.1614/0043-1745(2003)051[0083:hlocbn]2.0.co;2
Heiser, C. B. (1951). The sunflower among the North American Indians. Proc. Am. Philos. Soc. 95, 432–448.
Heiser, C. B., Smith, D. M., Clevenger, S. B., and Martin, W. C. (1969). “The North American sunflowers (Helianthus),” in Memoirs of the Torrey Botanical Club, Vol. 22 (Durham, NC: Club by the Seeman Printery), 1–218.
Horn, J. L. (1965). A rationale and test for the number of factors in factor analysis. Psychometrika 30, 179–185. doi: 10.1007/BF02289447
INRA Sunflower Bioinformatics Resources (2014). HA412-HO Genome Portal (Bronze version). Available online at: https://www.heliagene.org/HA412.v1.1.bronze.20141015/
Isbell, F., Calcagno, V., Hector, A., Connolly, J., Harpole, W. S., Reich, P. B., et al. (2011). High plant diversity is needed to maintain ecosystem services. Nature 477, 199–202. doi: 10.1038/nature10282
Jackson, W. (1990). “Agriculture with nature as analogy,” in Sustainable Agriculture in Temperate Zones, eds C. A. Francis, C. Butler, and L. D. King (New York, NY: John Wiley & Sons, Inc), 381–422.
Jan, C. C. (1997). Cytology and interspecific hybridization. Sunflow. Technol. Prod. 35, 497–558. doi: 10.2134/agronmonogr35.c10
Kawakami, T., Darby, B. J., and Ungerer, M. C. (2014). Transcriptome resources for the perennial sunflower Helianthus maximiliani obtained from ecologically divergent populations. Mol. Ecol. Resour. 14, 812–819. doi: 10.1111/1755-0998.12227
Kawakami, T., Morgan, T. J., Nippert, J. B., Ocheltree, T. W., Keith, R., Dhakal, P., et al. (2011). Natural selection drives clinal life history patterns in the perennial sunflower species, Helianthus maximiliani. Mol. Ecol. 20, 2318–2328. doi: 10.1111/j.1365-294X.2011.05105.x
Lander, E. S., and Botstein, D. (1989). Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 136:705.
Li, J. T., Yang, J., Chen, D. C., Zhang, X. L., and Tang, Z. S. (2007). An optimized mini-preparation method to obtain high-quality genomic DNA from mature leaves of sunflower. Genet. Mol. Res. 6, 1064–1071.
Lin, Z., Li, X., Shannon, L. M., Yeh, C.-T., Wang, M. L., Bai, G., et al. (2012). Parallel domestication of the Shattering1 genes in cereals. Nat. Genet. 44, 720–724. doi: 10.1038/ng.2281
Liu, Z., Cai, X., Seiler, G. J., Gulya, T. J., Rashid, K. Y., and Jan, C. C. (2011). “Transferring sclerotinia stalk rot resistance genes from wild Helianthus species into cultivated sunflower,” in Proceedings of the 33rd Sunflower Research Forum, Fargo, ND, 12–13.
Lobell, D. B., Schlenker, W., and Costa-Roberts, J. (2011). Climate trends and global crop production since 1980. Science 333, 616–620. doi: 10.1126/science.1204531
Mandel, J. R., Nambeesan, S., Bowers, J. E., Marek, L. F., Ebert, D., Rieseberg, L. H., et al. (2013). Association mapping and the genomic consequences of selection in sunflower. PLoS Genet. 9:e1003378. doi: 10.1371/journal.pgen.1003378
Nambeesan, S. U., Mandel, J. R., Bowers, J. E., Marek, L. F., Ebert, D., Corbi, J., et al. (2015). Association mapping in sunflower (Helianthus annuus L.) reveals independent control of apical vs. basal branching. BMC Plant Biol. 15:84. doi: 10.1186/s12870-015-0458-9
Ott, A., Liu, S., Schnable, J. C., Yeh, C. T. E., Wang, K. S., and Schnable, P. S. (2017). tGBS® genotyping-by-sequencing enables reliable genotyping of heterozygous loci. Nucleic Acids Res. 45:e178. doi: 10.1093/nar/gkx853
Pearl, S. A., Bowers, J. E., Reyes-Chin-Wo, S., Michelmore, R. W., and Burke, J. M. (2014). Genetic analysis of safflower domestication. BMC Plant Biol. 14:43. doi: 10.1186/1471-2229-14-43
Peng, Q., Hu, Y., Wei, R., Zhang, Y., Guan, C., Ruan, Y., et al. (2010). Simultaneous silencing of FAD2 and FAE1 genes affects both oleic acid and erucic acid contents in Brassica napus seeds. Plant Cell Rep. 29, 317–325. doi: 10.1007/s00299-010-0823-y
Picasso, V. D., Brummer, E. C., Liebman, M., Dixon, P. M., and Wilsey, B. J. (2008). Crop species diversity affects productivity and weed suppression in perennial polycultures under two management strategies. Crop Sci. 48, 331–342. doi: 10.2135/cropsci2007.04.0225
Picasso, V. D., Brummer, E. C., Liebman, M., Dixon, P. M., and Wilsey, B. J. (2011). Diverse perennial crop mixtures sustain higher productivity over time based on ecological complementarity. Renew. Agric. Food Syst. 26, 317–327. doi: 10.1017/s1742170511000135
Purugganan, M. D., and Fuller, D. Q. (2009). The nature of selection during plant domestication. Nature 457, 843–848. doi: 10.1038/nature07895
Ronin, Y., Mester, D., Minkov, D., and Korol, A. (2010). Building reliable genetic maps: different mapping strategies may result in different maps. Nat. Sci. 02, 576–589. doi: 10.4236/ns.2010.26073
Seiler, G. J. (1994). Oil concentration and fatty acid composition of achenes of North American Helianthus (Asteraceae) species. Econ. Bot. 48, 271–279. doi: 10.1007/bf02862328
Seiler, G. J., and Brothers, M. E. (1999). Oil concentration and fatty acid composition of achenes of Helianthus species (Asteraceae) from Canada. Econ. Bot. 53, 273–280. doi: 10.1007/bf02866637
Shimizu-Sato, S., Tanaka, M., and Mori, H. (2009). Auxin-cytokinin interactions in the control of shoot branching. Plant Mol. Biol. 69, 429–435. doi: 10.1007/s11103-008-9416-3
Simons, K. J. (2005). Molecular characterization of the major wheat domestication gene Q. Genetics 172, 547–555. doi: 10.1534/genetics.105.044727
Skinner, M. E., Uzilov, A. V., Stein, L. D., Mungall, C. J., and Holmes, I. H. (2009). JBrowse: a next-generation genome browser. Genome Res. 19, 1630–1638. doi: 10.1101/gr.094607.109
Stirnberg, P., van De Sande, K., and Leyser, H. M. O. (2002). MAX1 and MAX2 control shoot lateral branching in Arabidopsis. Development 129, 1131–1141.
Tang, S. X., Leon, A., Bridges, W. C., and Knapp, S. J. (2006). Quantitative trait loci for genetically correlated seed traits are tightly linked to branching and pericarp pigment loci in sunflower. Crop Sci. 46, 721–734. doi: 10.2135/cropsci2005.0006-7
Taski-Ajdukovic, K., Vasic, D., and Nagl, N. (2006). Regeneration of interspecific somatic hybrids between Helianthus annuus L. and Helianthus maximiliani (Schrader) via protoplast electrofusion. Plant Cell Rep. 25, 698–704. doi: 10.1007/s00299-006-0134-5
Tetreault, H. M., Kawakami, T., Ungerer, M. C., and Levy, C. (2016). Low temperature tolerance in the perennial sunflower Helianthus maximiliani. Am. Midl. Nat. 175, 91–102. doi: 10.1674/amid-175-01-91-102.1
Van Ooijen, J. W. (2018). JoinMap ® 5, Software for the Calculation of Genetic Linkage Maps in Experimental Populations of Diploid Species. Wageningen: Kyazma B.V.
Van Tassel, D. L., Asselin, S. R., Cox, S. A., Sideli, G., and Cattani, D. J. (2014). “Evaluating perennial candidates for domestication: lessons from wild sunflower relatives,” in Proceedings of the FAO Expert Workshop: Perennial Crops for Food Security. 28–30 Aug. 2013, eds C. Batello, L. Wade, S. Cox, N. Pagna, A. Bozzini, and J. Choptiany (Rome: FAO), 112–140.
Vavilov, N. I. (1922). The law of homologous series in variation. J. Genet. 12, 47–89. doi: 10.1007/bf02983073
Wagoner, P., and Schaeffer, J. R. (1990). Perennial grain development: past efforts and potential for the future. Crit. Rev. Plant Sci. 9, 381–408. doi: 10.1080/07352689009382298
Wills, D. M., and Burke, J. M. (2007). Quantitative trait locus analysis of the early domestication of sunflower. Genetics 176, 2589–2599. doi: 10.1534/genetics.107.075333
Wu, T. D., and Nacu, S. (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881. doi: 10.1093/bioinformatics/btq057
Zhang, L., Wang, S., Li, H., Deng, Q., Zheng, A., Li, S., et al. (2010). Effects of missing marker and segregation distortion on QTL mapping in F2 populations. Theor. Appl. Genet. 121, 1071–1082. doi: 10.1007/s00122-010-1372-z
Keywords: domestication, oilseeds, perennial grains, comparative genomics, ecosystem services, genotype-by-sequencing, functional diversity, sunflower
Citation: Asselin SR, Brûlé-Babel AL, Van Tassel DL and Cattani DJ (2020) Genetic Analysis of Domestication Parallels in Annual and Perennial Sunflowers (Helianthus spp.): Routes to Crop Development. Front. Plant Sci. 11:834. doi: 10.3389/fpls.2020.00834
Received: 23 March 2020; Accepted: 25 May 2020;
Published: 12 June 2020.
Edited by:
Petr Smýkal, Palacký University Olomouc, CzechiaReviewed by:
Aleksandra Dimitrijevic, Institute of Field and Vegetable Crops, SerbiaAlejandro Presotto, Universidad Nacional del Sur, Argentina
Copyright © 2020 Asselin, Brûlé-Babel, Van Tassel and Cattani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sean R. Asselin, U2Vhbi5Bc3NlbGluQENhbmFkYS5jYQ==; c2Vhbi5hc3NlbGluQGdtYWlsLmNvbQ==