 Edi Sudianto
Edi Sudianto Chung-Shien Wu
Chung-Shien Wu Shu-Miaw Chaw
Shu-Miaw Chaw- Biodiversity Research Center, Academia Sinica, Taipei, Taiwan
Plastome downsizing is rare in photosynthetic seed plants. However, a large-scale study of five cupressophyte families (conifers II) indicated that the plastomes of some Cupressaceous genera are notably reduced and compact. Here, we enriched taxon sampling in Cupressaceae by adding plastomes of ten previously unreported genera to determine the origin, evolution, and consequences of plastome reduction in this family. We discovered that plastome downsizing is specific to Callitroideae (a Southern Hemispheric subfamily). Their plastomes are the smallest, encode the fewest plastid genes, and contain the fewest GC-end codons among Cupressaceae. We show that repeated tRNA losses and shrinkage of intergenic spacers together contributed to the plastome downsizing in Callitroideae. Moreover, our absolute nucleotide substitution rate analyses suggest relaxed functional constraints in translation-related plastid genes (clpP, infA, rpl, and rps), but not in photosynthesis- or transcription-related ones, of Callitris (the most diverse genus in Callitroideae). We hypothesize that the small and low-GC plastomes of Callitroideae emerged ca. 112–75 million years ago as an adaptation to increased competition with angiosperms on the Gondwana supercontinent. Our findings highlight Callitroideae as another case of plastome downsizing in photosynthetic seed plant lineages.
Introduction
Cupressaceae (cypress family) is the most widely distributed family in cupressophytes (conifers II), comprising 30–32 genera and ca. 133 species. Many members of the family are economically and ecologically important. For example, bald cypress (Taxodium), China fir (Cunninghamia), cordilleran cypress (Austrocedrus), and cypress pine (Callitris) are important sources of timber, furniture, and ornamentals (Farjon, 2005). They are distributed across all continents except Antarctica (Farjon, 2005; Brodribb et al., 2012). Cypress genera have been classified into seven (or six) subfamilies (Mao et al., 2012; Yang et al., 2012). Two of the seven subfamilies, Athrotaxidoideae and Callitroideae, are mainly restricted to the Southern Hemisphere, while the other five (Cupressoideae, Cunninghamioideae, Sequoioideae, Taiwanioideae, and Taxodioideae) are mostly distributed in the Northern Hemisphere (Gadek et al., 2000; Farjon and Filer, 2013).
Farjon (2005) maintained that there are only six Cupressaceous subfamilies. He merged Callitroideae into Cupressoideae and ranked the Southern Hemispheric clade as the Callitrideae tribe. However, most molecular phylogenetic studies consider the subfamily Callitroideae as separate from Cupressoideae (Leslie et al., 2012; Mao et al., 2012; Yang et al., 2012). Callitroideae is the least abundant conifer group (Leslie et al., 2012); most (ca. 60%) of its genera are monotypic, except for Callitris, which has more than 10 recognized species (Farjon, 2005). In addition, previous studies on the phylogenetic positions of early-diverging Callitroideae genera (e.g., Austrocedrus, Pilgerodendron, and Libocedrus) have been inconsistent and require further scrutiny (Mao et al., 2012; Yang et al., 2012; Crisp et al., 2019).
Plastid genomes (plastomes) of seed plants are generally conserved in both gene order and content. The average size of photosynthetic seed plant plastomes is ∼145 kb (Jansen and Ruhlman, 2012), but exceptions have been reported in gnetophytes (109–119 kb; Wu et al., 2009), pines (107–122 kb; Lin et al., 2010; Sudianto et al., 2016), saguaro cactus (∼113 kb; Sanderson et al., 2015), cupressophytes (121–136 kb; Wu and Chaw, 2016), and legumes (∼121 kb; Choi et al., 2019). Most of these size reductions were caused by the loss of one copy of inverted repeats (IRs), non-coding DNA, or non-essential plastid genes. Some commonly lost plastid genes are the NADH dehydrogenase-like complex (ndh), various small and large ribosomal proteins (rps and rpl), and several transfer RNA (tRNA) genes (Wu et al., 2009; Sanderson et al., 2015; Chaw et al., 2018).
Photosynthetic seed plant plastomes typically encode 29–32 tRNA genes (Jansen and Ruhlman, 2012), far fewer than the 61 standard sense codons. To account for this disparity, Crick (1966) proposed the “wobble hypothesis,” which posits that a minimum of 32 tRNA species is needed to read all codons. This hypothesis suggests that the third codon position can have a non-Watson-Crick base-pair interaction with the first position of the tRNA anticodon (Crick, 1966). More recently, however, studies on tobacco plastomes further narrowed the minimum set of essential tRNA genes down to 25 tRNA species, calling it the “superwobble hypothesis” (Alkatib et al., 2012a, b).
Yet, Callitris has been documented to have lost some of the essential tRNA genes, including trnG-UCC, trnT-UGU, and trnV-UAC (Wu and Chaw, 2016). The Callitris plastome – the smallest, most compact, and most rearranged plastome in the Cupressaceae (Wu and Chaw, 2016; Chaw et al., 2018) – also shows several unique characteristics that highlight major evolutionary events occurring after Callitris diverged from its sister clade. However, it remains unknown whether these features are specific to Callitris or common to all Southern Hemispheric Cupressaceous genera. Thus, extending taxon sampling to include more Callitroideae genera will offer critical information on the origin, evolution, and implications of the above-mentioned features.
Here, we increase sampling of Cupressaceous plastomes by adding ten previously unreported genera, including six from Callitroideae (8–10 recognized genera), two from Cupressoideae (11–13 recognized genera), and one each from Athrotaxidoideae (one recognized genus) and Sequoioideae (three recognized genera). These new sequences fill in the gaps in our understanding of plastomic variation in Cupressaceae, especially among the Southern Hemispheric lineages. Specifically, we address the following questions: (1) Do all Southern Hemispheric Cupressaceous genera commonly have reduced plastomes and fewer plastid tRNAs? and (2) Do losses of many tRNAs influence the codon compositions or nucleotide substitution rates in the plastomes of Callitroideae?
Materials and Methods
Sample Collection and DNA Extraction
Studied samples were collected from trees growing in the University of California Botanical Garden (Berkeley, United States) and Botanischer Garten der Heinrich-Heine-Universität (Düsseldorf, Germany). The specimens were deposited into the Herbarium of Academia Sinica, Taipei, Taiwan (HAST). Collection information, voucher numbers, and GenBank accession numbers are listed in Supplementary Table S1. We extracted total genomic DNA from each sample following a modified CTAB method (Stewart and Via, 1993).
Sequencing, Assembly, and Annotation
Total DNA was sequenced on an Illumina NextSeq500 platform at Genomics BioSci & Tech (New Taipei City) or Tri-I Biotech (New Taipei City), generating ∼2–4 Gb of paired-end (2 × 150 bp) reads for each species. Trimmomatic 0.36 (Bolger et al., 2014) and FastQC 0.11.5 (Andrew, 2011) were used to trim and check the quality of the reads, respectively. Plastomes were de novo assembled using Ray 2.3.1 (Boisvert et al., 2010) and plastid contigs/scaffolds were BLAST-searched using the Taiwania flousiana plastome (NC_021441) as the reference with an E-value of <10–10. We obtained complete plastomes for most of the sampled taxa using this method. However, plastomic assemblies were fragmented in a few taxa. We therefore used these fragmented assemblies and NOVOPlasty 2.7.2 (Dierckxsens et al., 2017) to generate complete plastomes. Plastome annotation was conducted using the “Transfer Annotation” function in Geneious 11.0.5 (Kearse et al., 2012) with the T. flousiana plastome as the reference, followed by manual corrections. Transfer RNA (tRNA) genes were further confirmed using tRNAscan-SE 2.0 (Lowe and Chan, 2016). Repetitive sequences were identified using REPuter (Kurtz et al., 2001) with a minimum size of 8 bp.
Sequence Alignment and Phylogenetic Tree Reconstruction
We extracted 79 common plastid protein-coding genes from 29 Cupressaceous genera (including 10 newly sequenced in this study) and five closely related genera (one Sciadopityaceae and four Taxaceae). Each gene was aligned using MUSCLE (Edgar, 2004) implemented in MEGA 7.0 (Kumar et al., 2016) with the “Align Codon” option. SequenceMatrix (Vaidya et al., 2011) was used to concatenate the alignment. A matrix with 64,185 nucleotide sites was obtained and used to reconstruct the maximum likelihood (ML) and Bayesian inference (BI) trees using RAxML 8.2.10 (Stamatakis, 2014) and MrBayes 3.2.6 (Huelsenbeck and Ronquist, 2001), respectively, using the GTR + G + I model, as suggested by jModelTest 2.1.10 (Darriba et al., 2012) and MrModelTest 2.4 (Nylander, 2004). The ML tree was assessed from 1,000 bootstrap replicates, and we ran two independent analyses for the BI tree, using 6,000,000 generations for each run and sampling every 300 generations. We discarded the first 25% of the samples as burn-in, assessed the level of convergence (convergence level increases as the Potential Scale Reduction Factor score approaches 1), and ensured that the average standard deviation of split frequencies was below 0.01. To infer the tRNA gene gain/loss history, we performed Dollo’s parsimony using Count (Csurös, 2010) and topology inferred from phylogenetic analyses.
Ancestral Plastome Reconstruction
We identified the locally collinear blocks (LCBs) between the 34 sampled genera and Cycas taitungensis (AP009339) plastomes using progressiveMauve (Darling et al., 2010). The IRA region of the Cycas plastome was removed prior to the analysis because previous studies indicated that cupressophyte plastomes lost their IRA (Wu et al., 2011). The ancestral plastomes were reconstructed based on the LCB order matrices and ML tree topology in MLGO (Hu et al., 2014). Subsequently, the reconstructed ancestral plastomes were compared with their close descendants to infer the plastomic inversion history across the phylogeny in GRIMM (Tesler, 2002).
Codon Usage Bias Calculation
We calculated relative synonymous codon usage (RSCU) scores from 61 non-stop codons in the 79 common plastid protein-coding genes of 29 Cupressaceous, one Sciadopityaceous, and four Taxaceous genera using DAMBE 7.0.58 (Xia, 2018). Next, we performed correspondence analyses on the RSCU values using the FactoMineR package (Lê et al., 2008) in R 3.6.1 (R Core Team, 2019). The 34 genera were further classified into five groups based on the hierarchical clustering of the 61 RSCU values.
Estimation of Nucleotide Substitution Rates and Molecular Dating
Non-synonymous (dN) and synonymous substitution rates (dS) of the plastid genes were estimated using the CODEML program in PAML 4.9i (Yang, 2007) with the following parameters: runmode = 0, seqtype = 1, CodonFreq = 2, estFreq = 0, model = 1, and cleandata = 1. The constrained tree topology was obtained from the ML analysis (Figure 1). Divergence times were estimated using the approximate likelihood method in MCMCTREE (dos Reis and Yang, 2011) in PAML 4.9i. We used six estimated points from TimeTree (Hedges et al., 2015) as the calibrated ages of six specific nodes (Supplementary Figure S4). MCMC sampling was run for 2,500,000 generations and samples were drawn every 500 generations, with a burn-in of 200,000. We performed two independent runs to evaluate convergence. Tracer 1.7 (Rambaut et al., 2018) was used to confirm that the effective sample sizes of all parameters were above 200. Absolute substitution rates were calculated by dividing the dN and dS branch lengths over their respective divergence times. Only terminal branch lengths were considered in this study. The branch-site model test was performed using EasyCodeML (Gao et al., 2019).
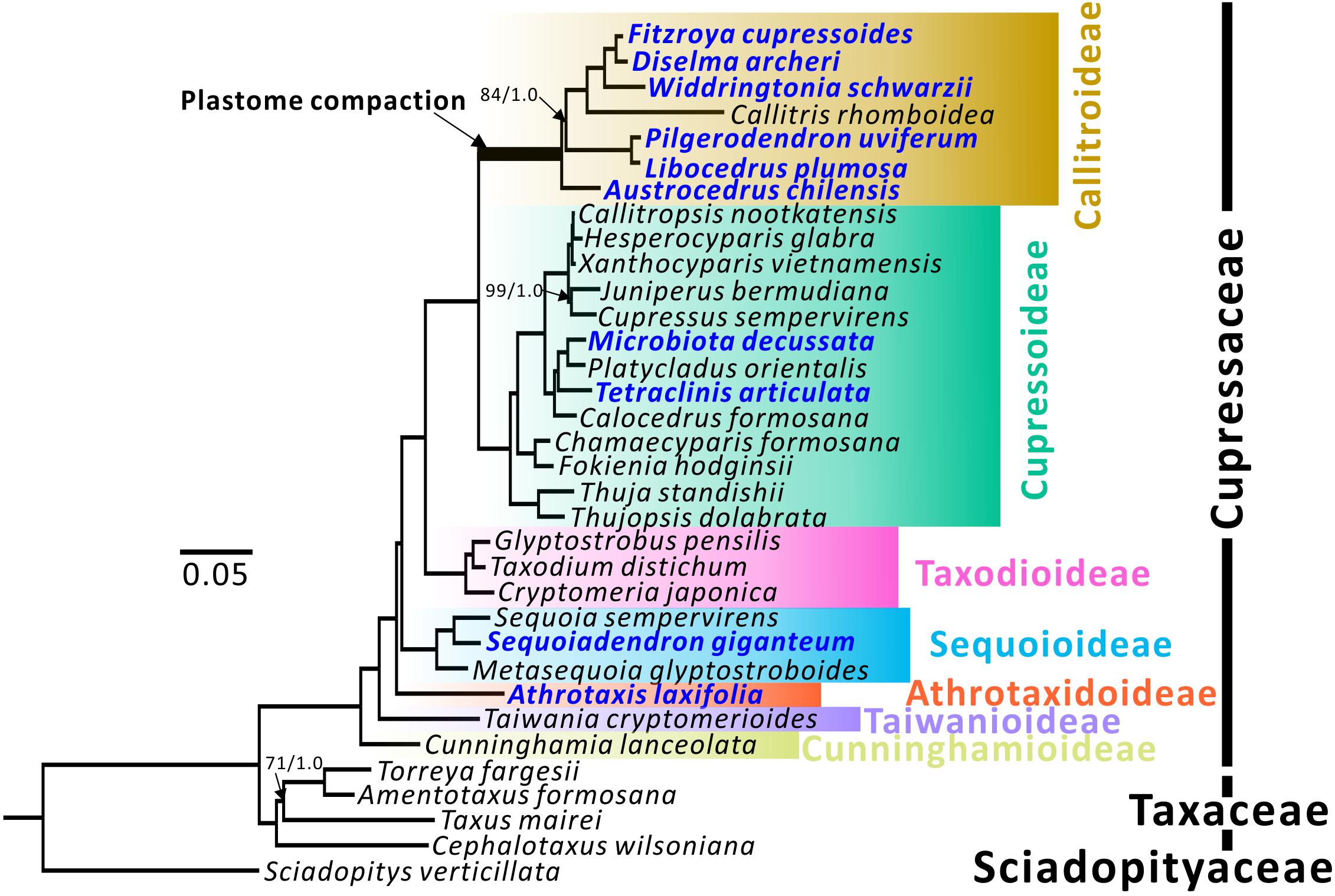
Figure 1. Plastid phylogenomic trees of seven Cupressaceous subfamilies. Sciadopitys verticillata was designated as the outgroup. The tree framework is based on the ML tree. All nodes received full 100% bootstrap support (BS) or 1.0 posterior probability (PP) from ML/BI analyses, except for the three with BS/PP values. Newly sequenced species are in blue.
Results
Support for Seven Distinct Cupressaceous Subfamilies
Topologies of maximum likelihood (ML) and Bayesian inference (BI) trees inferred from 79 concatenated protein-coding genes have 100% bootstrap support (BS) and 1.0 posterior probability (PP) at almost all nodes (Figure 1). Among the seven subfamilies, Cunninghamioideae is the earliest diverging clade, followed by Taiwanioideae, Athrotaxidoideae, Sequoioideae, and Taxodioideae. Taxodioideae is sister to a clade encompassing Cupressoideae and Callitroideae. In Callitroideae, the genus Austrocedrus diverged first and the Pilgerodendron–Libocedrus clade is sister to the clade containing four genera: Fitzroya–Diselma, Widdringtonia, and Callitris. Notably, the branch leading to Callitroideae is remarkably long and contains a signature of positive selection (branch-site model: P < 0.001), suggesting that positive selection resulted in accelerated rates of plastid nucleotide substitutions before the diversification of Callitroideae. Hence, the Southern Hemispheric clade should be treated as a separate subfamily from Cupressoideae.
Callitroideae Has the Smallest and Most Rearranged Plastomes in Cupressaceae
Table 1 compares the newly sequenced plastomes from ten Cupressaceous genera: one Athrotaxidoideae, six Callitroideae, two Cupressoideae, and one Sequoioideae. Their GC contents were similar (34.0–35.5%), but coding gene numbers varied, particularly with tRNA. For example, we detected 28–31 plastid tRNAs in Callitroideae but 33–34 in other Cupressaceous subfamilies (Table 1). In addition, the monotypic species Austrocedrus chilensis (Callitroideae) lacked psaM. This species thus has fewer plastid protein-coding genes than any taxa studied so far. Overall, plastomes of Callitroideae were relatively gene-poor compared to other subfamilies (114–116 genes, compared to 119–120 elsewhere).
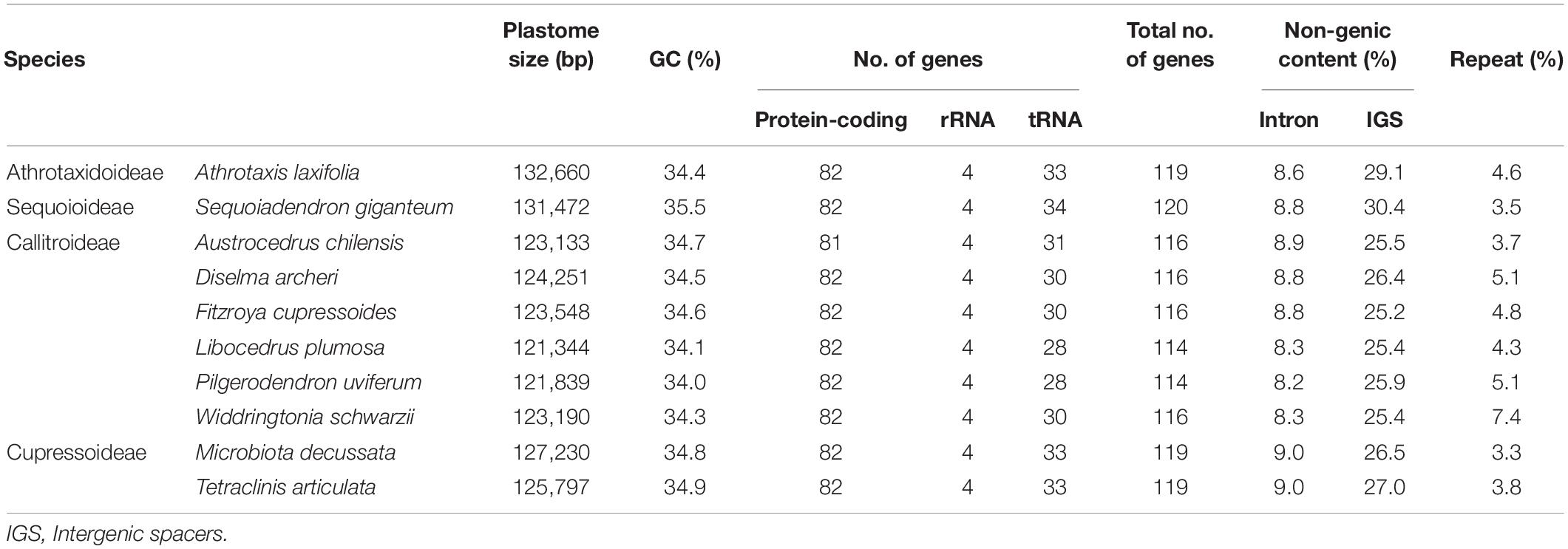
Table 1. Plastomic features of the ten newly sequenced Cupressaceous species.
Plastomes are generally smaller in Callitroideae (121,344–124,251 bp) than other subfamilies (125,797–132,660 bp). An analysis of their introns, intergenic spacers (IGS), and repeats indicates that Callitroideae has lower IGS content (25.2–26.4%) than other subfamilies (26.5–30.4%) but similar intron and repeat content (Table 1). Thus, the shrinkage of IGS resulted in the reduction of plastid non-coding regions in Callitroideae. To create a better picture of the evolution of plastome size in Cupressaceae, available taxa from all living subfamilies were included and compared. As shown in Figure 2, Callitroideae plastomes are significantly smaller and more compact (gene-dense) than those of other subfamilies (Mann-Whitney test; all P < 0.01). Moreover, plastomic rearrangements were the most extensive in Callitroideae (Supplementary Figure S1). We detected three rearrangement hotspots: seven in Callitris, five in the common ancestor of Libocedrus and Pilgerodendron, and five rearrangements in the common ancestor of all Callitroideae genera (Supplementary Figure S1). Together, these results show that Callitroideae plastomes are not only the smallest but also the most rearranged among Cupressaceae.
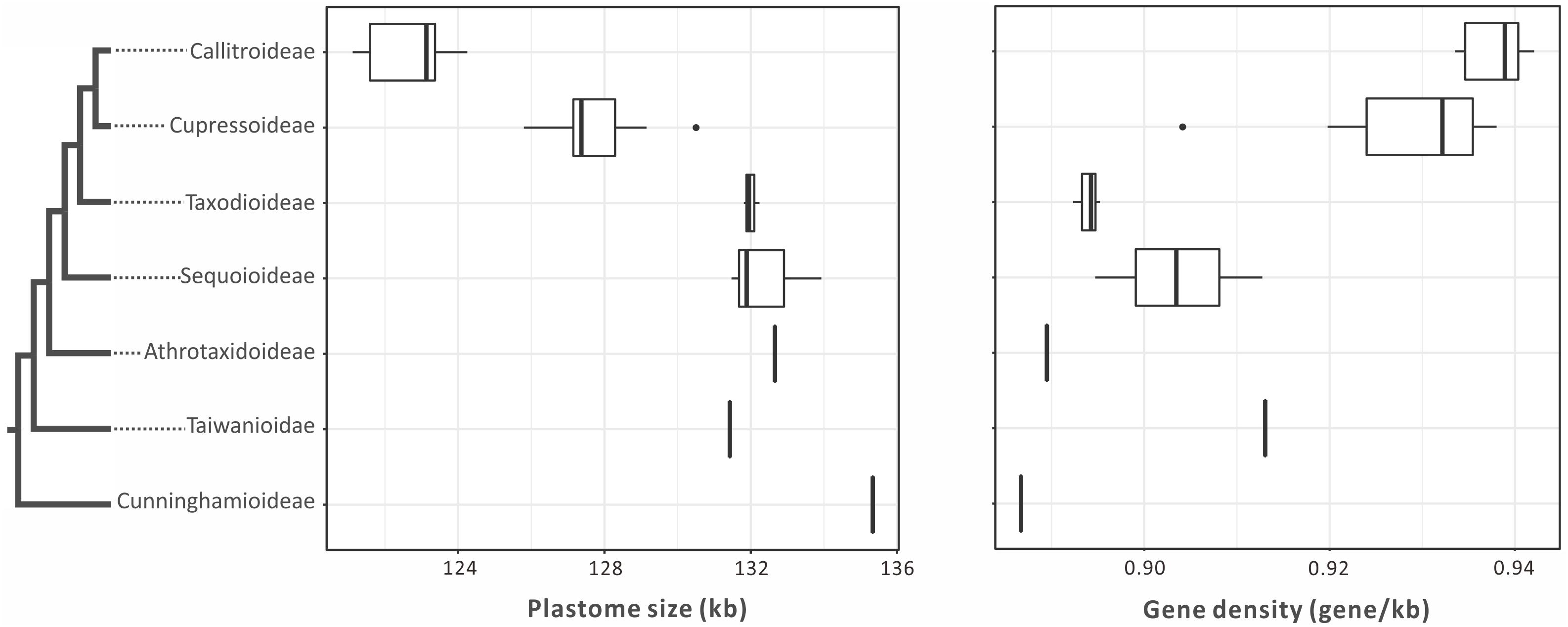
Figure 2. Boxplots for comparing plastome size and gene density among seven Cupressaceous subfamilies. The plastomes of Callitroideae are the smallest and most compact. The black dots represent outliers. The phylogenetic backbone is based on Figure 1.
Callitroideae Plastomes Repeatedly Lost tRNAs and Have Distinct Codon Compositions
To better understand how the plastid tRNA gene repertoire changes across the Cupressaceae, we compared 29 available genera in the seven Cupressaceous subfamilies and found that all genera had 30–31 tRNA species – except for the seven Callitroideae genera, which had only 26–29 tRNA species (Figure 3). Callitroideae genera lost various plastid tRNAs, including trnG-UCC, trnM-CAU, trnP-GGG, trnS-GGA, trnT-GGU, trnT-UGU, and trnV-UAC (Figure 3). Our analysis further supports the assertion that 16 plastid tRNA loss events occurred across Cupressaceae (Supplementary Figure S2). Several tRNAs were repeatedly lost from different lineages. For example, during the evolution of Cupressaceae, there were multiple independent losses of trnG-UCC (2 times), trnM-CAU (2 times), trnP-GGG (3 times), trnT-GGU (4 times), and trnV-UAC (2 times). The majority (10) of these tRNA losses occurred within Callitroideae, suggesting that some driving force differentiates Callitroideae from other subfamilies in the retention of plastid tRNAs.
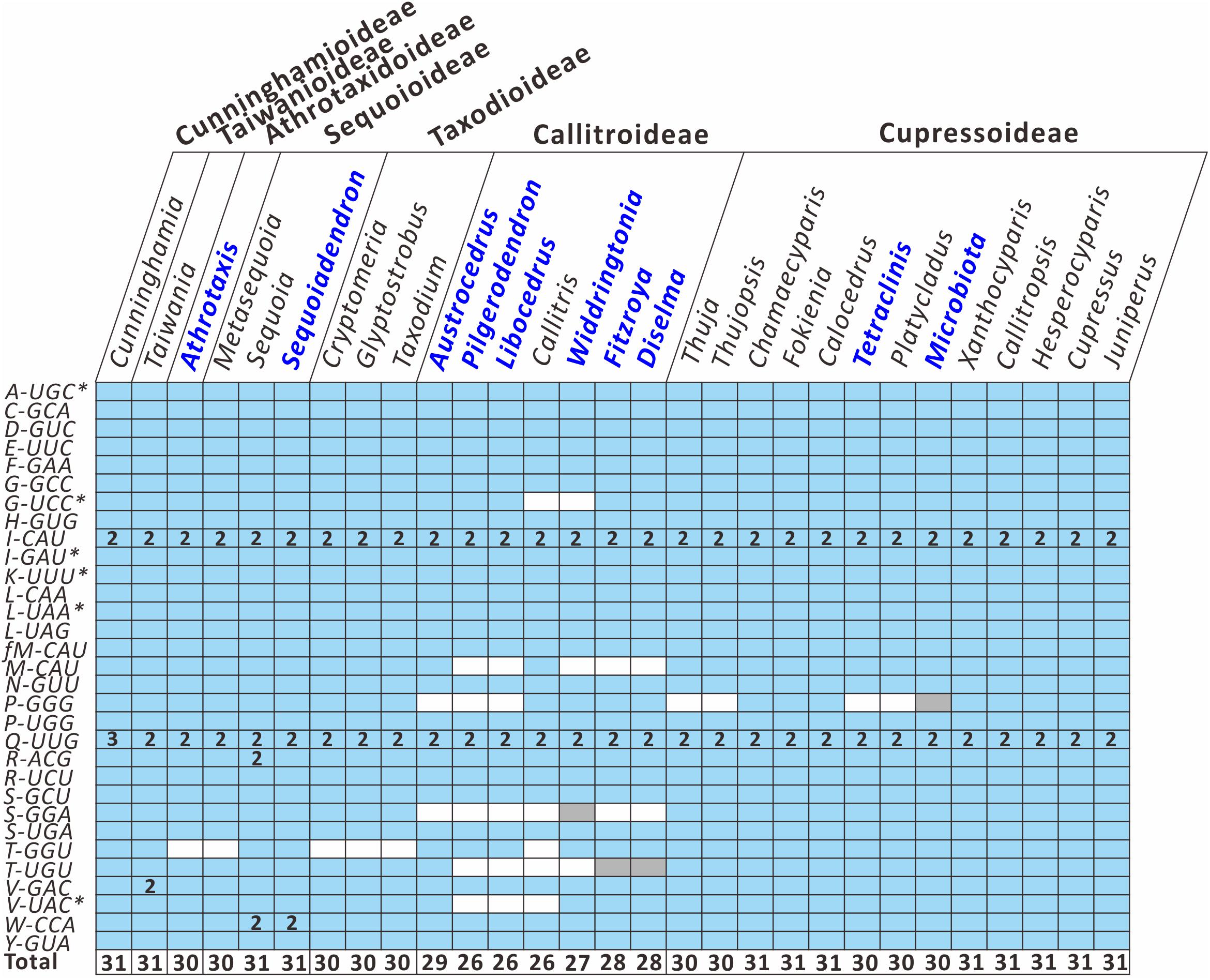
Figure 3. Comparison of plastid tRNA gene repertoires among seven Cupressaceous subfamilies. The functional copy, pseudogene copy, and gene loss are denoted in blue, gray, and white, respectively. Arabic numerals within the boxes indicate the number of gene copies. The total number of distinct tRNA species for each genus is listed in the bottom row. Newly sequenced plastomes are in dark blue font. Intron-containing genes are marked with an asterisk (*).
The loss of many plastid tRNA genes from the Callitroideae prompted us to investigate whether this subfamily has a distinct codon composition. We performed a correspondence analysis (CA) using 61 non-stop relative synonymous codon usage (RSCU) scores from 29 Cupressaceous genera and five genera from two closely related families [Sciadopityaceae (1) and Taxaceae (4)] as the outgroup for comparison. The first two CA dimensions predicted about 59.2% of the RSCU variation. Hierarchical clustering resolved five isolated groups in the 34 sampled genera (Figure 4). The first dimension (38.9%) clearly distinguished Cupressaceae from Sciadopityaceae and Taxaceae, with CGC, UGC, UUC, and AGG codons constituting the largest contributions. The second dimension explained about 20.3% of the RSCU variation and separated Callitroideae from its sister subfamily, Cupressoideae. Our analyses further indicate that this separation is mainly caused by nucleotides at the 3rd codon position – i.e., Callitroideae plastomes have more A/U-ending codons, while Cupressoideae plastomes contain more C/G-ending codons (Figure 4). Moreover, Callitroideae and Athrotaxidoideae plastomes contain the lowest number of GC nucleotides, especially at the 3rd codon position (Supplementary Figure S3). No evidence of plastid codon reassignments was detected in Callitroideae (Supplementary Table S3).
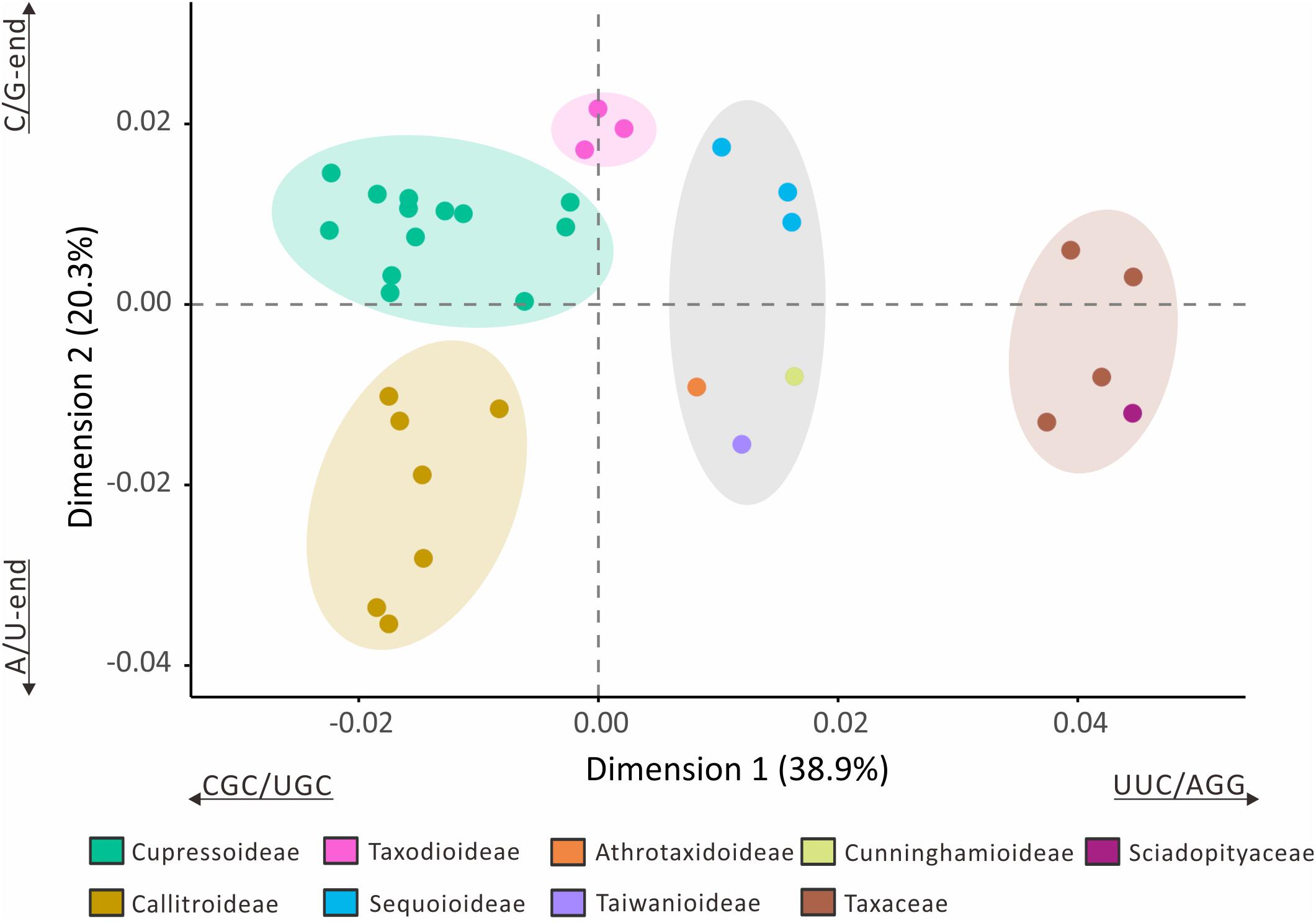
Figure 4. Correspondence analysis of plastid relative synonymous codon usage (RSCU). Thirty-four genera were included in the analysis (29 Cupressaceae and five closely related genera: one Sciadopityaceae and four Taxaceae). Hierarchical clustering clearly reveals five separate groups. The first two dimensions accounted for 59.2% of the RSCU variation among the 34 sampled genera.
Relaxed Functional Constraints on Plastid Translation-Related Genes in Callitris
Figure 5A compares the absolute synonymous (RS) and non-synonymous (RN) substitution rates estimated from the 29 sampled Cupressaceous genera. RS and RN are strongly and positively correlated (R2 = 0.76; P < 0.001). Callitris exhibits the highest values of both RS and RN, noticeably deviating from the expected values based on the regression line. We also detected elevated RN/RS ratios in some Callitris genes. For example, the RN/RS ratios of plastid translation-related genes (clpP, infA, rpl, and rps) and plastid translocon component genes (ycf1 and ycf2) were greater than 0.5. Two genes, infA (1.38) and ycf2 (1.08), had RN/RS ratios greater than 1 (Figure 5B). In contrast, the RN/RS ratio of other genes, such as photosynthesis- and transcription-related genes, were generally lower than 0.4 in Callitris. Collectively, these results may suggest that Callitris encountered positive selection or relaxed functional constraints in plastid genes related to translation and protein modification.
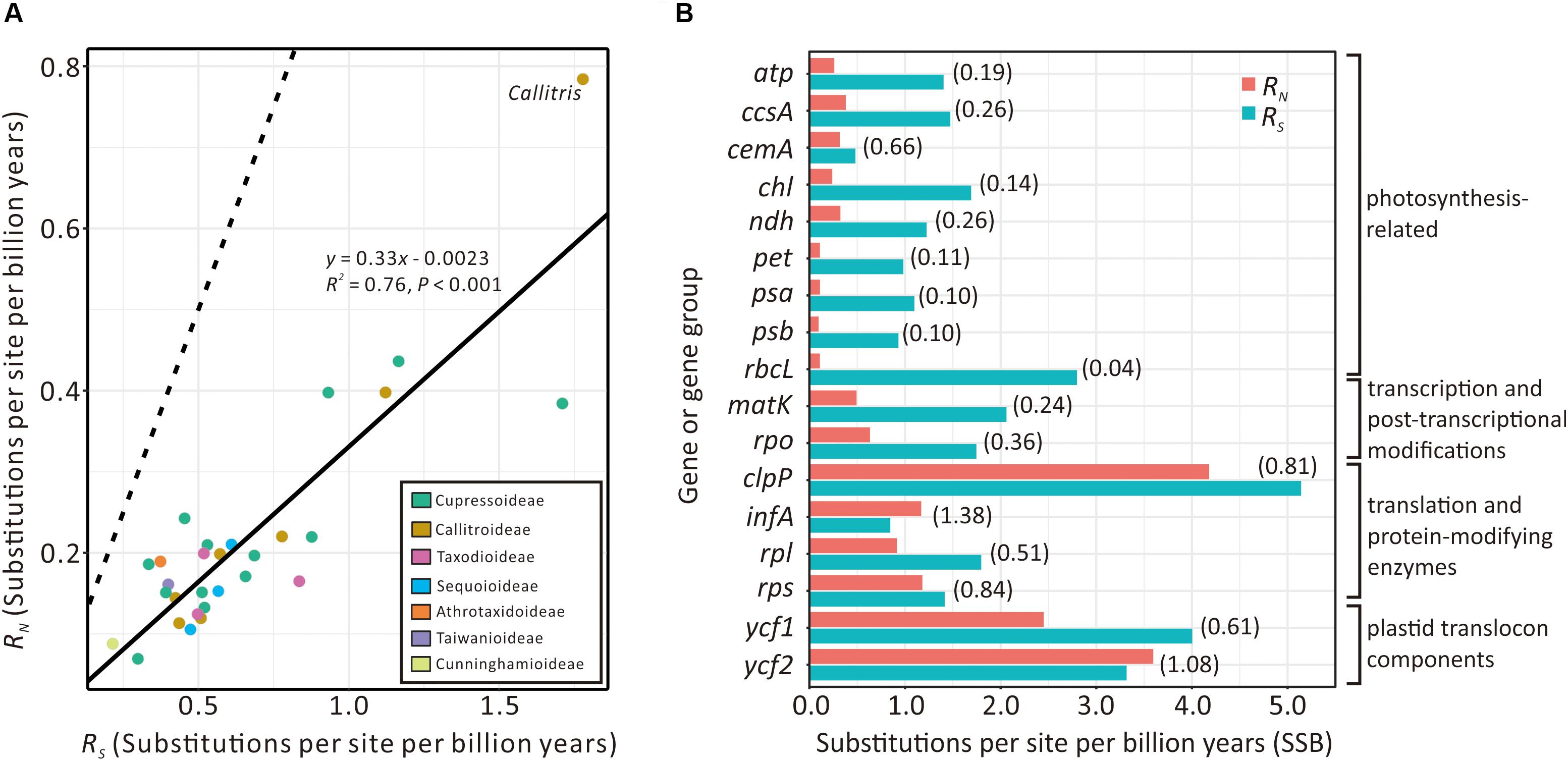
Figure 5. Comparison of plastid absolute non-synonymous (RN) and synonymous (RS) substitution rates across seven Cupressaceous subfamilies. (A) Regression analysis showing a strong positive correlation between RS and RN among the 29 sampled genera. The solid line depicts the linear regression line with its equation, R2, and P-value indicated. The dashed line indicates the expected pattern if RS = RN. (B) Bar plot showing RN vs. RS of plastid genes/groups in Callitris. RN/RS ratios are in parentheses.
Discussion
Filling the Gaps in Plastid Phylogenomics of Cupressaceae
Prior plastid phylogenomics of Cupressaceae focused on the taxa of Northern Hemispheric origin (Wu and Chaw, 2016; Qu et al., 2017a, b; Zhu et al., 2018) and only investigated one Southern Hemispheric taxon (Callitris rhomboidea; included in Wu and Chaw, 2016). The present study adds ten newly elucidated plastomes, including seven Southern Hemispheric taxa (six Callitroideae and one Athrotaxidoideae) and three Northern Hemispheric taxa (two Cupressoideae and one Sequoioideae). The addition of these ten genera enabled us to build the most comprehensive plastid phylogenomics of Cupressaceae to date – amounting to 29 of the 30–32 Cupressaceous genera recognized worldwide.
Both our ML and BI trees (Figure 1) support the monophyly of Callitroideae, with Austrocedrus sister to the remaining genera. This agrees with the views of Mao et al. (2012); Yang et al. (2012), and Leslie et al. (2018), but not Crisp et al. (2019) who placed the Pilgerondendron–Libocedrus clade as sister to the remaining Callitroideae genera. The phylogenetic positions of other Callitroideae genera are also congruent with the four above-mentioned studies. The intergeneric relationships within Cupressoideae and Sequoioideae agree well with those of Mao et al. (2012) and Leslie et al. (2018). Athrotaxis, the sole genus of Athrotaxidoideae, is supported as sister to a clade including Taxodioideae, Sequoioideae, Callitroideae, and Cupressoideae (Figure 1). This placement, however, contradicts those of Yang et al. (2012) and Lu et al. (2014), which placed Athrotaxis as sister to the Taxodioideae–Callitroideae–Cupressoideae clade and the Sequoioideae subfamily, respectively.
Numerous tRNA Gene Losses and IGS Reductions Contributed to Plastome Downsizing in Callitroideae
We show that plastome downsizing is especially pronounced in the Callitroideae genera. No plastome reduction was observed in the other Southern Hemispheric clade, Athrotaxidoideae. Callitroideae plastomes lost seven tRNA genes, several of which are dispensable. For example, trnP-GGG is frequently lost from the plastomes of major land plant lineages, such as mosses (Physcomitrella patens and Tortula ruralis; Oliver et al., 2010), liverworts (Marchantia polymorpha; Ohyama, 1996), lycophytes (Selaginella; Mower et al., 2019), gymnosperms (Sciadopitys verticillata; Hsu et al., 2016), and all angiosperms (Wicke et al., 2011). Two other genes (trnS-GGA and trnT-GGU) have been experimentally verified as non-essential plastid genes (Alkatib et al., 2012b). In contrast, the absence of any of the four remaining tRNA genes (trnG-UCC, trnM-CAU, trnT-UGU, and trnV-UAC) is lethal for tobacco (Alkatib et al., 2012a, b). It is interesting to note that some parasitic plants still retain ∼30 plastid tRNA genes despite having smaller plastomes than those of Callitroideae genera (see Wicke and Naumann, 2018).
Loss of these essential tRNA genes is unique to the Callitroideae, as no other Cupressaceous subfamilies have lost any of them (Figure 3). It was previously proposed that losses of tRNA genes might be compensated either by: (1) codon reassignment (Kollmar and Mühlhausen, 2017), (2) wobbling or superwobbling mechanisms (Alkatib et al., 2012b), or (3) cytosolic tRNA import (Maréchal-Drouard et al., 1999). Although codon reassignment has evidently rectified the missing tRNA in plastomes of several taxa (Su et al., 2019; Turmel et al., 2019), it was not detected in our data (Supplementary Table S3). Wobbling or superwobbling also could not compensate for these tRNA losses since the four missing tRNA genes are part of the minimum set of 25 tRNA species required to encode all codons (Alkatib et al., 2012b). Instead, the Callitroideae plastids may import cytosolic tRNAs to replace the tRNA defect, as previously proposed in various parasitic plants (Wolfe et al., 1992; Su et al., 2019). The exact mechanisms of essential tRNA compensation in Callitroideae plastids will be an interesting topic for further studies.
The mutational burden hypothesis posits that excess non-coding DNA is a liability because any mutation at the intronic splice sites, transcription factor binding sites, or core promoters will be deleterious to the genome (Lynch, 2006). Lineages with low mutation rates tend to accumulate non-coding DNA, leading to the assertion that elevated mutation rates enhance the degree of genome compaction in organelles (Lynch, 2006; Smith, 2016). Our results demonstrate signatures of both accelerated substitution rates and positive selection on the branch leading to Callitroideae (Figure 1). Conforming to the mutational burden hypothesis, this accelerated rate might have facilitated the removal of non-functional sequences, contributing to the low IGS content in Callitroideae (Table 1). Together, these data suggest that the plastome downsizing in the Callitroideae occurred prior to the subfamily diversification ca. 112–75 million years ago (Cretaceous Period; Supplementary Figure S4). This timeline coincides with the fragmentation of East and West Gondwana into smaller continents and the transition period from gymnosperm-dominated to angiosperm-dominated flora on the Gondwana (McLoughlin, 2001).
Potential Implications of Reduced and Compact Plastomes in Callitroideae
Given their small plastome size, Callitroideae genera might have selective advantages over other subfamilies containing larger plastomes. Streamlined genomes are hypothesized to have shorter replication times and consume fewer resources such as phosphorus and nitrogen (Hessen et al., 2010; Mann and Chen, 2010). The saved resources can be reallocated to RNA production and increasing the rate of protein synthesis, eventually leading to a higher growth rate (Hessen et al., 2010). Similarly, several studies also linked genome size to cell size, arguing that smaller genomes may facilitate cell size reduction, and consequently faster growth (Bennett, 1987; Hessen et al., 2010; Simonin and Roddy, 2018). However, the trend is less straightforward in algal plastomes, not all of which have a strong positive correlation between plastome size and cell size (Smith, 2017). As plastome downsizing has been commonly found in various lineages, it may be worthy to investigate the correlation among plastome size, cell size, and growth rate.
Callitroideae plastomes also contain the fewest GC-end codons among the Cupressaceae subfamilies (Figure 4 and Supplementary Figure S3). This bias in the base composition might stem from the fact that GC nucleotides are energetically more “expensive” than AT nucleotides (Rocha and Danchin, 2002). The AT base-pair also uses one fewer nitrogen atom than the GC base-pair (Dufresne et al., 2005). Collectively, these imply that the downsized plastomes of Callitroideae may have a competitive edge over other sympatric plants. These characteristics closely resemble the plastomes of another gymnosperm group, the gnetophytes, which grow well in arid or angiosperm-dominated environments (Wu et al., 2009).
Data Availability Statement
The complete plastid genomes of the ten Cupressaceous genera have been deposited in the DDBJ under the accession numbers LC500575–LC500584.
Author Contributions
ES performed the experiments, analyzed the data, and wrote the manuscript. S-MC collected the plant specimens and supervised the project. C-SW and S-MC critically revised and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by research grants from the Ministry of Science and Technology Taiwan (MOST 106-2311-B-001-005 to S-MC) and a postdoctoral fellowship from Academia Sinica (to ES).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Holly Forbes and Clare Loughran (UC Botanical Garden at Berkeley) and Dr. Sabine Etges and Lars Leonhard (Botanical Garden, Heinrich-Heine-University) for their kind permission to collect samples from their gardens.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00901/full#supplementary-material
References
Alkatib, S., Fleischmann, T. T., Scharff, L. B., and Bock, R. (2012a). Evolutionary constraints on the plastid tRNA set decoding methionine and isoleucine. Nucleic Acids Res. 40, 6713–6724. doi: 10.1093/nar/gks350
Alkatib, S., Scharff, L. B., Rogalski, M., Fleischmann, T. T., Matthes, A., Seeger, S., et al. (2012b). The contributions of wobbling and superwobbling to the reading of the genetic code. PLoS Genet. 8:e1003076. doi: 10.1371/journal.pgen.1003076
Andrew, S. (2011). FastQC: a Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk?/projects/fastqc/ (accessed October 6, 2011).
Bennett, M. D. (1987). Variation in genomic form in plants and its ecological implications. New Phytol. 106, 177–200. doi: 10.1111/j.1469-8137.1987.tb04689.x
Boisvert, S., Laviolette, F., and Corbeil, J. (2010). Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J. Comput. Biol. 17, 1519–1533. doi: 10.1089/cmb.2009.0238
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brodribb, T. J., Pittermann, J., and Coomes, D. A. (2012). Elegance versus speed: examining the competition between conifer and angiosperm trees. Int. J. Plant Sci. 173, 673–694. doi: 10.1086/666005
Chaw, S.-M., Wu, C.-S., and Sudianto, E. (2018). Evolution of gymnosperm plastid genomes: Plastid Genome Evolution Advances in botanical research. Amsterdam: Elsevier, 195–222. doi: 10.1016/bs.abr.2017.11.018
Choi, I.-S., Jansen, R., and Ruhlman, T. (2019). Lost and found: return of the inverted repeat in the legume clade defined by its absence. Genome Biol. Evol. 11, 1321–1333. doi: 10.1093/gbe/evz076
Crick, F. H. (1966). Codon–anticodon pairing: the wobble hypothesis. J. Mol. Biol. 19, 548–555. doi: 10.1016/S0022-2836(66)80022-0
Crisp, M. D., Cook, L. G., Bowman, D. M. J. S., Cosgrove, M., Isagi, Y., and Sakaguchi, S. (2019). Turnover of southern cypresses in the post-Gondwanan world: extinction, transoceanic dispersal, adaptation and rediversification. New Phytol. 221, 2308–2319. doi: 10.1111/nph.15561
Csurös, M. (2010). Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 26, 1910–1912. doi: 10.1093/bioinformatics/btq315
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
dos Reis, M., and Yang, Z. (2011). Approximate likelihood calculation on a phylogeny for Bayesian estimation of divergence times. Mol. Biol. Evol. 28, 2161–2172. doi: 10.1093/molbev/msr045
Dufresne, A., Garczarek, L., and Partensky, F. (2005). Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol. 6:R14. doi: 10.1186/gb-2005-6-2-r14
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Farjon, A., and Filer, D. (2013). An Atlas of the World’s Conifers: An Analysis of their Distribution, Biogeography, Diversity and Conservation Status. Leiden: BRILL.
Gadek, P. A., Alpers, D. L., Heslewood, M. M., and Quinn, C. J. (2000). Relationships within Cupressaceae sensu lato: a combined morphological and molecular approach. Am. J. Bot. 87, 1044–1057. doi: 10.2307/2657004
Gao, F., Chen, C., Arab, D. A., Du, Z., He, Y., and Ho, S. Y. W. (2019). EasyCodeML: a visual tool for analysis of selection using CodeML. Ecol. Evol. 9, 3891–3898. doi: 10.1002/ece3.5015
Hedges, S. B., Marin, J., Suleski, M., Paymer, M., and Kumar, S. (2015). Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol. 32, 835–845. doi: 10.1093/molbev/msv037
Hessen, D. O., Jeyasingh, P. D., Neiman, M., and Weider, L. J. (2010). Genome streamlining and the elemental costs of growth. Trends Ecol. Evol. (Amst.) 25, 75–80. doi: 10.1016/j.tree.2009.08.004
Hsu, C.-Y., Wu, C.-S., and Chaw, S.-M. (2016). Birth of four chimeric plastid gene clusters in japanese umbrella pine. Genome Biol. Evol. 8, 1776–1784. doi: 10.1093/gbe/evw109
Hu, F., Lin, Y., and Tang, J. (2014). MLGO: phylogeny reconstruction and ancestral inference from gene-order data. BMC Bioinformatics 15:354. doi: 10.1186/s12859-014-0354-6
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Jansen, R. K., and Ruhlman, T. A. (2012). “Plastid genomes of seed plants,” in Genomics of Chloroplasts and Mitochondria Advances in Photosynthesis and Respiration, eds R. Bock and V. Knoop (Dordrecht: Springer), 103–126. doi: 10.1007/978-94-007-2920-9_5
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kollmar, M., and Mühlhausen, S. (2017). How tRNAs dictate nuclear codon reassignments: only a few can capture non-cognate codons. RNA Biol. 14, 293–299. doi: 10.1080/15476286.2017.1279785
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lê, S., Josse, J., and Husson, F. (2008). factominer: anr package for multivariate analysis. J. Stat. Softw. 25, 1–18. doi: 10.18637/jss.v025.i01
Leslie, A. B., Beaulieu, J., Holman, G., Campbell, C. S., Mei, W., Raubeson, L. R., et al. (2018). An overview of extant conifer evolution from the perspective of the fossil record. Am. J. Bot. 105, 1531–1544. doi: 10.1002/ajb2.1143
Leslie, A. B., Beaulieu, J. M., Rai, H. S., Crane, P. R., Donoghue, M. J., and Mathews, S. (2012). Hemisphere-scale differences in conifer evolutionary dynamics. Proc. Natl. Acad. Sci. U.S.A. 109, 16217–16221. doi: 10.1073/pnas.1213621109
Lin, C.-P., Huang, J.-P., Wu, C.-S., Hsu, C.-Y., and Chaw, S.-M. (2010). Comparative chloroplast genomics reveals the evolution of Pinaceae genera and subfamilies. Genome Biol. Evol. 2, 504–517. doi: 10.1093/gbe/evq036
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Lu, Y., Ran, J.-H., Guo, D.-M., Yang, Z.-Y., and Wang, X.-Q. (2014). Phylogeny and divergence times of gymnosperms inferred from single-copy nuclear genes. PLoS One 9:e107679. doi: 10.1371/journal.pone.0107679
Lynch, M. (2006). Streamlining and simplification of microbial genome architecture. Annu. Rev. Microbiol. 60, 327–349. doi: 10.1146/annurev.micro.60.080805.142300
Mann, S., and Chen, Y.-P. P. (2010). Bacterial genomic G+C composition-eliciting environmental adaptation. Genomics 95, 7–15. doi: 10.1016/j.ygeno.2009.09.002
Mao, K., Milne, R. I., Zhang, L., Peng, Y., Liu, J., Thomas, P., et al. (2012). Distribution of living Cupressaceae reflects the breakup of Pangea. Proc. Natl. Acad. Sci. U.S.A. 109, 7793–7798. doi: 10.1073/pnas.1114319109
Maréchal-Drouard, L., Dietrich, A., Mireau, H., Peeters, N., and Small, I. (1999). “The import of cytosolic trna into plant mitochondria,” in Mitochondrial Diseases, ed. P. Lestienne (Berlin: Springer), 317–326. doi: 10.1007/978-3-642-59884-5_24
McLoughlin, S. (2001). The breakup history of Gondwana and its impact on pre-Cenozoic floristic provincialism. Aust. J. Bot. 49:271. doi: 10.1071/BT00023
Mower, J. P., Ma, P.-F., Grewe, F., Taylor, A., Michael, T. P., VanBuren, R., et al. (2019). Lycophyte plastid genomics: extreme variation in GC, gene and intron content and multiple inversions between a direct and inverted orientation of the rRNA repeat. New Phytol. 222, 1061–1075. doi: 10.1111/nph.15650
Nylander, J. A. A. (2004). MrModeltest Version 2. Uppsala: Evolutionary Biology Centre, Uppsala University.
Ohyama, K. (1996). Chloroplast and mitochondrial genomes from a liverwort, Marchantia polymorpha–gene organization and molecular evolution. Biosci. Biotechnol. Biochem. 60, 16–24. doi: 10.1271/bbb.60.16
Oliver, M. J., Murdock, A. G., Mishler, B. D., Kuehl, J. V., Boore, J. L., Mandoli, D. F., et al. (2010). Chloroplast genome sequence of the moss Tortula ruralis: gene content, polymorphism, and structural arrangement relative to other green plant chloroplast genomes. BMC Genomics 11:143. doi: 10.1186/1471-2164-11-143
Qu, X.-J., Jin, J.-J., Chaw, S.-M., Li, D.-Z., and Yi, T.-S. (2017a). Multiple measures could alleviate long-branch attraction in phylogenomic reconstruction of Cupressoideae (Cupressaceae). Sci. Rep. 7:41005. doi: 10.1038/srep41005
Qu, X.-J., Wu, C.-S., Chaw, S.-M., and Yi, T.-S. (2017b). Insights into the Existence of Isomeric Plastomes in Cupressoideae (Cupressaceae). Genome Biol. Evol. 9, 1110–1119. doi: 10.1093/gbe/evx071
R Core Team (2019). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Rocha, E. P. C., and Danchin, A. (2002). Base composition bias might result from competition for metabolic resources. Trends Genet. 18, 291–294. doi: 10.1016/S0168-9525(02)02690-2
Sanderson, M. J., Copetti, D., Búrquez, A., Bustamante, E., Charboneau, J. L. M., Eguiarte, L. E., et al. (2015). Exceptional reduction of the plastid genome of saguaro cactus (Carnegiea gigantea): Loss of the ndh gene suite and inverted repeat. Am. J. Bot. 102, 1115–1127. doi: 10.3732/ajb.1500184
Simonin, K. A., and Roddy, A. B. (2018). Genome downsizing, physiological novelty, and the global dominance of flowering plants. PLoS Biol. 16:e2003706. doi: 10.1371/journal.pbio.2003706
Smith, D. R. (2016). The mutational hazard hypothesis of organelle genome evolution: 10 years on. Mol. Ecol. 25, 3769–3775. doi: 10.1111/mec.13742
Smith, D. R. (2017). Does cell size impact chloroplast genome size? Front. Plant Sci. 8:2116. doi: 10.3389/fpls.2017.02116
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stewart, C. N., and Via, L. E. (1993). A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. BioTechniques 14, 748–750.
Su, H.-J., Barkman, T. J., Hao, W., Jones, S. S., Naumann, J., Skippington, E., et al. (2019). Novel genetic code and record-setting AT-richness in the highly reduced plastid genome of the holoparasitic plant Balanophora. Proc. Natl. Acad. Sci. U.S.A. 116, 934–943. doi: 10.1073/pnas.1816822116
Sudianto, E., Wu, C.-S., Lin, C.-P., and Chaw, S.-M. (2016). Revisiting the plastid phylogenomics of pinaceae with two complete plastomes of pseudolarix and tsuga. Genome Biol. Evol. 8, 1804–1811. doi: 10.1093/gbe/evw106
Tesler, G. (2002). GRIMM: genome rearrangements web server. Bioinformatics 18, 492–493. doi: 10.1093/bioinformatics/18.3.492
Turmel, M., Lopes Dos, Santos, A., Otis, C., Sergerie, R., and Lemieux, C. (2019). Tracing the evolution of the plastome and mitogenome in the chloropicophyceae uncovered convergent tRNA gene losses and a variant plastid genetic code. Genome Biol. Evol. 11, 1275–1292. doi: 10.1093/gbe/evz074
Vaidya, G., Lohman, D. J., and Meier, R. (2011). SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. doi: 10.1111/j.1096-0031.2010.00329.x
Wicke, S., and Naumann, J. (2018). Molecular Evolution of Plastid Genomes in Parasitic Flowering Plants: Plastid Genome Evolution Advances in Botanical Research. Amsterdam: Elsevier, 315–347. doi: 10.1016/bs.abr.2017.11.014
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wolfe, K. H., Morden, C. W., Ems, S. C., and Palmer, J. D. (1992). Rapid evolution of the plastid translational apparatus in a nonphotosynthetic plant: loss or accelerated sequence evolution of tRNA and ribosomal protein genes. J. Mol. Evol. 35, 304–317. doi: 10.1007/bf00161168
Wu, C.-S., and Chaw, S.-M. (2016). Large-scale comparative analysis reveals the mechanisms driving plastomic compaction, reduction, and inversions in conifers II (Cupressophytes). Genome Biol. Evol. 8, 3740–3750. doi: 10.1093/gbe/evw278
Wu, C.-S., Lai, Y.-T., Lin, C.-P., Wang, Y.-N., and Chaw, S.-M. (2009). Evolution of reduced and compact chloroplast genomes (cpDNAs) in gnetophytes: selection toward a lower-cost strategy. Mol. Phylogenet. Evol. 52, 115–124. doi: 10.1016/j.ympev.2008.12.026
Wu, C.-S., Wang, Y.-N., Hsu, C.-Y., Lin, C.-P., and Chaw, S.-M. (2011). Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 3, 1284–1295. doi: 10.1093/gbe/evr095
Xia, X. (2018). DAMBE7: new and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 35, 1550–1552. doi: 10.1093/molbev/msy073
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z.-Y., Ran, J.-H., and Wang, X.-Q. (2012). Three genome-based phylogeny of Cupressaceae s.l.: further evidence for the evolution of gymnosperms and Southern Hemisphere biogeography. Mol. Phylogenet. Evol. 64, 452–470. doi: 10.1016/j.ympev.2012.05.004
Keywords: callitroids, Callitroideae, cupressophytes, plastid genome, genome downsizing, Southern Hemisphere
Citation: Sudianto E, Wu C-S and Chaw S-M (2020) The Origin and Evolution of Plastid Genome Downsizing in Southern Hemispheric Cypresses (Cupressaceae). Front. Plant Sci. 11:901. doi: 10.3389/fpls.2020.00901
Received: 01 April 2020; Accepted: 02 June 2020;
Published: 23 June 2020.
Edited by:
Kathleen Pryer, Duke University, United StatesReviewed by:
Christopher Randle, Sam Houston State University, United StatesMaria D. Logacheva, Lomonosov Moscow State University, Russia
Copyright © 2020 Sudianto, Wu and Chaw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shu-Miaw Chaw, c21jaGF3QHNpbmljYS5lZHUudHc=
†ORCID: Edi Sudianto, orcid.org/0000-0002-0771-0385; Shu-Miaw Chaw, orcid.org/0000-0003-2499-7071