 Chao Liu1
Chao Liu1 Jinpeng Wang1,2
Jinpeng Wang1,2 Pengchuan Sun1
Pengchuan Sun1 Jigao Yu1Fanbo Meng1,3Zhikang Zhang1,3He Guo1Chendan Wei1Xinyu Li1Shaoqi Shen1
Jigao Yu1Fanbo Meng1,3Zhikang Zhang1,3He Guo1Chendan Wei1Xinyu Li1Shaoqi Shen1 Xiyin Wang1,2,3*
Xiyin Wang1,2,3*- 1School of Life Sciences, North China University of Science and Technology, Tangshan, China
- 2National Key Laboratory for North China Crop Improvement and Regulation, Hebei Agriculture University, Baoding, China
- 3Institute for Genomics and Bio-Big-Data, Chengdu University of Traditional Chinese Medicine, Chengdu, China
Polyploidies produce a large number of duplicated regions and genes in genomes, which have a long-term impact and stimulate genetic innovation. The high similarity between homeologous chromosomes, forming different subgenomes, or homologous regions after genome repatterning, may permit illegitimate DNA recombination. Here, based on gene colinearity, we aligned the (sub)genomes of common wheat (Triticum aestivum, AABBDD genotype) and its relatives, including Triticum urartu (AA), Aegilops tauschii (DD), and T. turgidum ssp. dicoccoides (AABB) to detect the homeologous (paralogous or orthologous) colinear genes within and between (sub)genomes. Besides, we inferred more ancient paralogous regions produced by a much ancient grass-common tetraploidization. By comparing the sequence similarity between paralogous and orthologous genes, we assumed abnormality in the topology of constructed gene trees, which could be explained by gene conversion as a result of illegitimate recombination. We found large numbers of inferred converted genes (>2,000 gene pairs) suggested long-lasting genome instability of the hexaploid plant, and preferential donor roles by DD genes. Though illegitimate recombination was much restricted, duplicated genes produced by an ancient whole-genome duplication, which occurred millions of years ago, also showed evidence of likely gene conversion. As to biological function, we found that ~40% catalytic genes in colinearity, including those involved in starch biosynthesis, were likely affected by gene conversion. The present study will contribute to understanding the functional and structural innovation of the common wheat genome.
Highlights
Homeologous genes in common wheat, likely converted by one another, show long-lasting genome instability after polyploidization.
Introduction
Common wheat is one of the most widely grown cereal crops in the world and an essential source of food, its production affects the global economy, and failed harvests could lead to social unrest (Choulet et al., 2014; Wang et al., 2015; Appels et al., 2018). The sequencing of its relatives, the diploid Triticum urartu (genotype AdAd; hereafter capital letters to indicate genome types and subscripts to indicate ploidy of organisms carrying them, with d, t, or h to show diploid, tetraploid, or hexaploid, respectively), and Aegilops tauschii (genome DdDd), and the tetraploid wild emmer wheat (T. turgidum ssp. dicoccoides, genome AtAtBtBt) contributed to understanding their genome evolution and innovation (Middleton et al., 2014; Du et al., 2017; Zimin et al., 2017; Gardiner et al., 2019). Wheat, as a heterologous hexaploid (genotype AhAhBhBhDhDh), was possibly produced when T. urartu hybridized with an ear of wheat carrying a B genome, thereby forming a neo-tetraploid that hybridized with the A. tauschii. Two hybridization process made the common wheat genome complicated and made its genome even more complicated is that a grass-common whole-genome duplication (cWGD) having occurred ~100 million years ago produced thousands of duplicated genes in the extant grass genomes (Paterson et al., 2004; Wang et al., 2015; Du et al., 2017). The event played an important role in promoting the formation of new species of grasses (Sun et al., 2017).
Genetic recombination is the primary source of genetic novelty (Kurosawa and Ohta, 2011). In plants, meiotic and mitotic recombinations are reciprocal, involving asymmetric exchange of genetic information between the homologs (Jia et al., 2013; Gardiner et al., 2019). Whereas, irreversible recombination involves the transfer of information from one site to its homeolog, leading to gene transformation (Datta et al., 1997; Kurosawa and Ohta, 2011). Gene conversion involves the unidirectional transfer of genetic material from a ‘donor’ sequence to a homologous ‘acceptor.’ In eukaryotes, it constitutes the main form of homologous recombination initiated by DNA double-strand breaks, as reviewed previously (Chen et al., 2007).
Whole-genome duplication will result in a large number of paralogous genes on homeologous chromosomes or chromosomal regions, and substantial similarity between homeologous regions could permit the occurrence of illegitimate recombination (as compared to normal homologous recombination), often resulting in a pattern called gene conversion that could be explained by that a gene is replaced by its homologous genes (Wang et al., 2009). There were reports to show that gene conversion could have occurred between paralogs in grasses and other plants. In some cases, gene conversion could have been frequent and is even on-going between paralogs on the homeologous chromosomes, e.g., rice chromosome 11 and its cWGD homeolog, chromosome 12 (Wang et al., 2009; Kurosawa and Ohta, 2011; Wang et al., 2011; Wang et al., 2019).
Illegitimate recombination has profound consequences on the evolution of paralogous genes and the chromosomes that they reside in. While paralogous recombination elevates base mutation rates, evolutionary rate, and DNA sequence deletion, converted regions have highly similar paralogous DNA (Wang et al., 2009; Wang et al., 2011). This is a seemingly perplexing but an essence reasonable finding. Temporally segmental restriction of paralogous recombination along homeologous chromosomes has produced stepwise strata of increased similarity from their centromeres to chromosome ends (Wang and Paterson, 2011). Increased mutation rates may foster genetic innovation. A significant QTL (qS12) resulting in hybrid male sterility was mapped within ~400 kb region adjacent to the highly recombined areas on the short arm of chromosome 12 (Zhang et al., 2011). Moreover, two genes harboring the E3-ligase RING-C2 domain on the distal parts of rice chromosomes 11 and 12, were identified, and have possibly evolved in concert via gene conversion (Jung et al., 2012).
Despite thousands of paralogs in wheat genomes produced by recurrent polyploidies, a comprehensive analysis of gene conversion is lacking. Here, by aligning the wheat and its relatives’ genomes, we inferred gene colinearity within a genome and orthology between them, and eventually characterized the pattern of illegitimate recombination and gene conversion. The present work will contribute to the understanding of genetic and structural innovation of wheat genes and chromosomes.
Results
Alignment of Wheat and Relative Genomes
Chromosome homeology was revealed by inferring colinear genes using the software ColinearScan (Wang et al., 2006). We revealed 232 AhBh, 187 AhDh, and 154 BhDh homeologous regions between wheat subgenomes, involving 14,161, 15,376, and 15,553 homeologous genes, respectively (Table 1). This meant about 40% of their genes were involved in inter-subgenome colinearity. Actually, as to the numbers and percentages of colinear genes between genomes, BhDh shared better gene colinearity than the other pairs of subgenomes; while AhBh shared the least gene colinearity. As to each of the three subgenomes, Dh shared a better colinearity with the other two subgenomes. This seems to agree with the inference that Dh subgenome joined the latest to produce the hexaploid wheat and/or has a conservative nature, as discussed later.
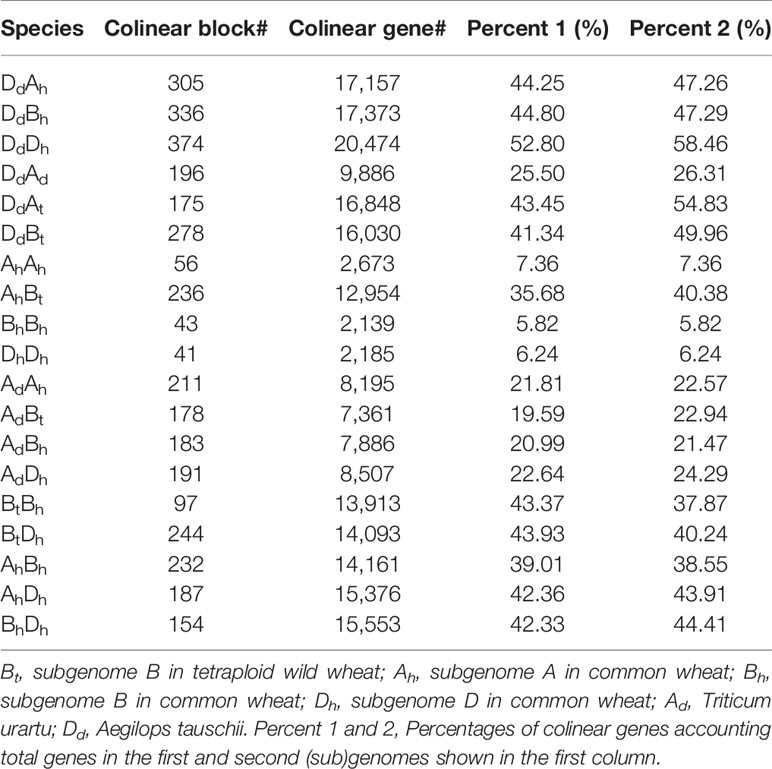
Table 1 The Number of colinear genes within and among the wheat and relative genomes.
Similarly, we detected orthologous regions and genes between the diploid wheat relative genomes (AdAd from T. urantu and DdDd from A. tauschii) and the corresponding common wheat subgenomes. Actually, with the same parameters, we inferred 211 AdAh and 374 DdDh orthologous regions, including 8,195 and 20,474 colinear genes, respectively. Notably, the Dd and Dh shared much better orthology than the Ad and Ah did, possibly due to much more genome fractionation in the Ad and Ah. This fact also supports Dh’s being a later player to form the hexaploid. We also revealed gene colinearity between the tetraploid wild wheat, T. turgidum (AtAtBtBt), with the common wheat genome. Mainly, the tetraploid Bt subgenome and hexaploid Bh subgenome shared 97 orthologous regions, involving 13,913 colinear genes and 80.0% of these colinear genes are supported by OrthorMCL (Table 2, Figure 1).
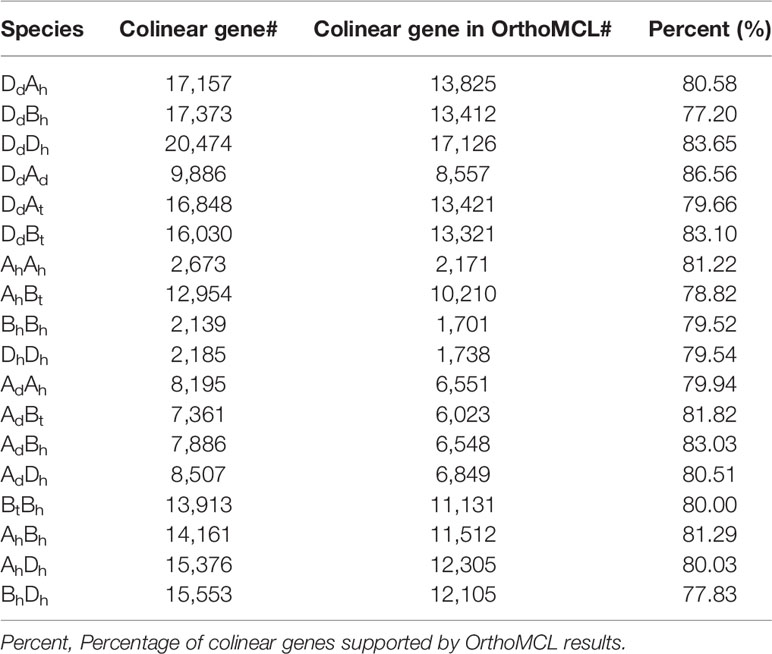
Table 2 Number of paralogous and orthologous genes supported by OrthoMCL within and among wheat and relative genomes.
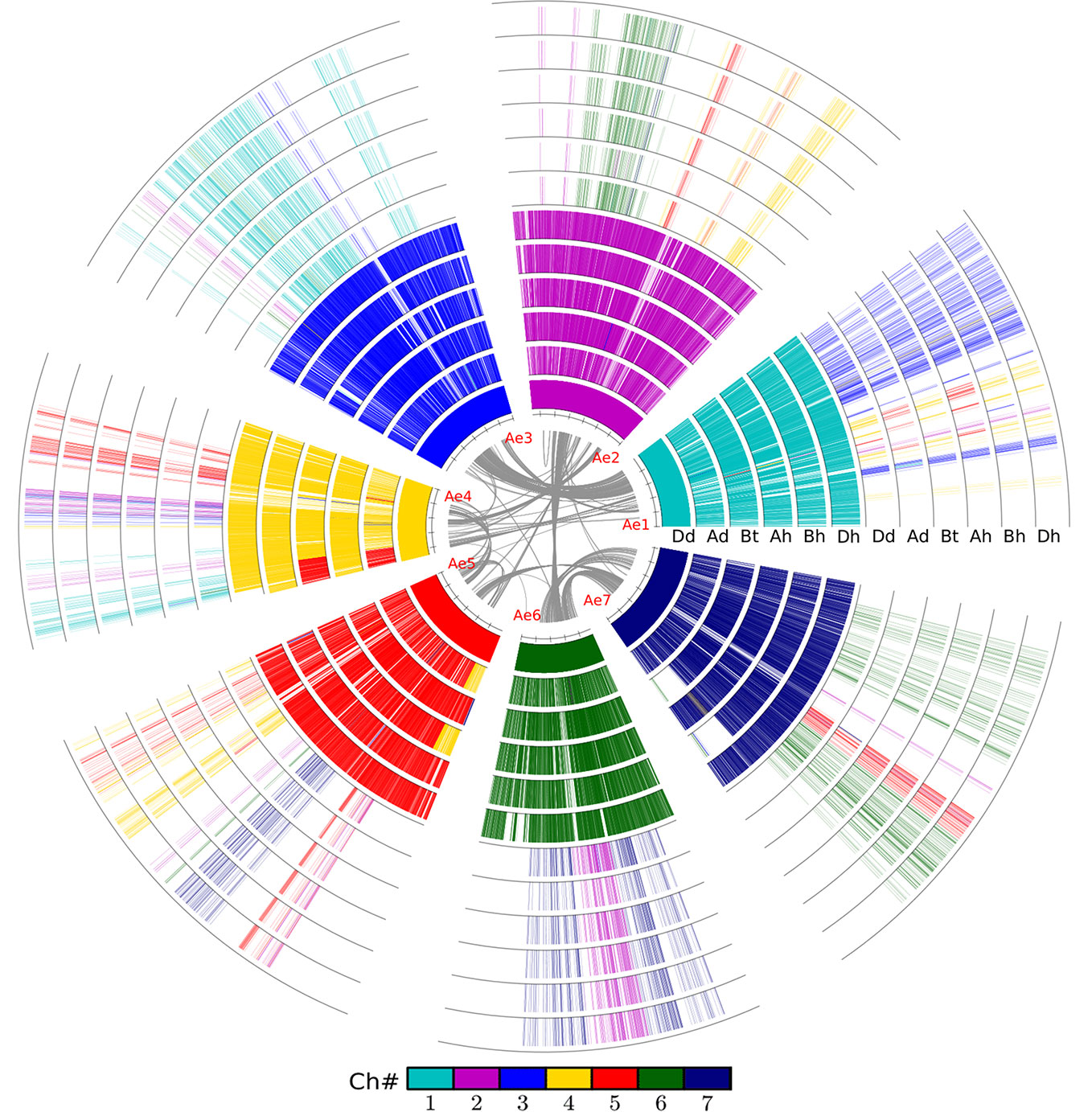
Figure 1 Alignment of the wheat and relative genomes with wheat DD as reference. The whole-genome duplication (WGD) in the common grass ancestor of plants caused them to have at least two circles of chromosomes. The hybridization event caused common wheat to have six such chromosomes. The innermost circle represents the seven chromosomes of the wheat DD genome (Dd) from Aegilops tauschii, and the gray lines linking paralogous genes. Ad, Triticum urartu; Bt, subgenome B in tetraploid wild wheat; Ah, Bh, and Dh, three subgenomes of common wheat.
Gene Conversion Between Common Wheat Subgenomes
To infer likely gene conversion, we retrieved homologous gene quartets from the above constructed multiple alignments. With Dd as the reference, we assumed 7,462 AdAhDdDh homeologous gene quartets, with each quartet containing a Dd gene, and its ortholog in common wheat Dh subgenome, and their corresponding orthologs in Ad genome and common wheat Ah genome (Table 1). This meant that about 42.36–43.91% of colinear genes in each genome were involved in the revealed quartets (Figure 2A). Theoretically, the common wheat AhDh homeologous genes were more diverged than each with their diploid orthologs. However, due to gene conversion following illegitimate recombination, the homeologs might be more similar to one another, resulting in an aberrant tree topology. The phylogenetic tree of the homologous quartet with gene conversion is shown in Figures 2C–H. By checking tree topology after aligning the gene sequence of each quartet, we found that 164 common wheat homeologs (~2.20% of all those involved in quartets) were likely converted.
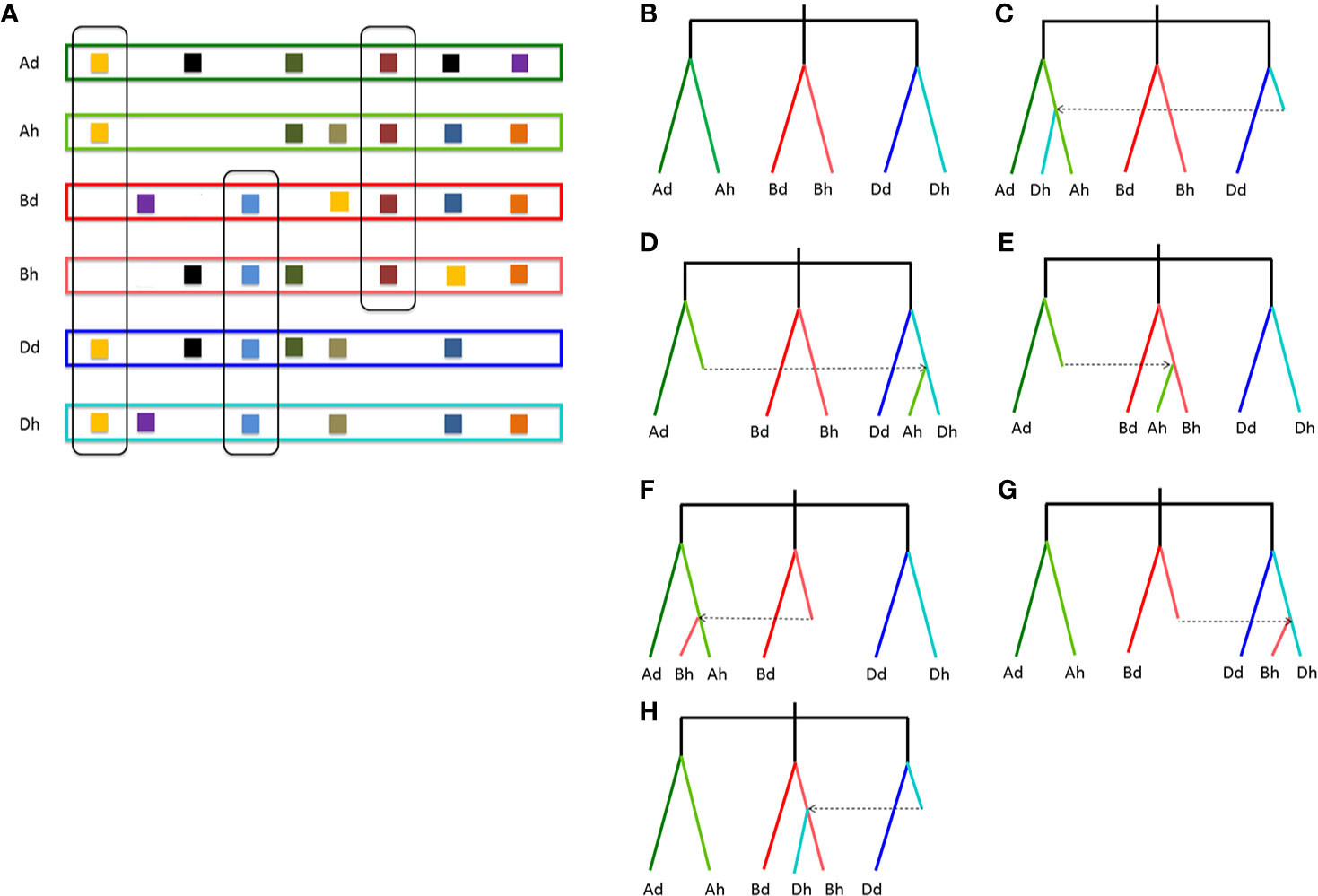
Figure 2 Homologous gene quartets and inference of conversion based on phylogenetic topology changes. (A) Colinear genes and quartets of homologous genes. Squares show genes, and the same color ones show homologous genes. If conversion does not occur, the expected phylogenetic relationship of a homologous quartet is shown in (B); (C) if a Dh gene is converted by an Ah paralog; (D) if an Ah gene is converted by a Dh paralog; (E) if an Ah gene is converted by a Bh paralog; (F) if a Bh gene is converted by an Ah paralog; (G) if a Bh gene is converted by a Dh paralog; (H) if a Dh gene is converted by a Bh paralog. Places without squares indicate gene loss.
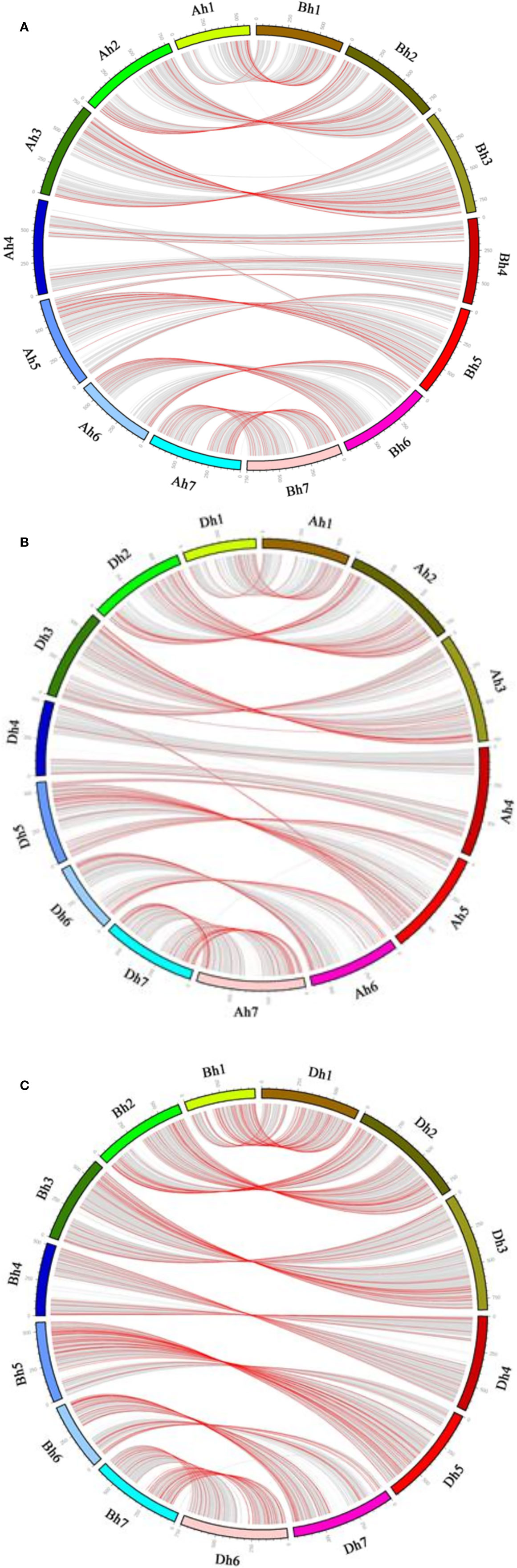
Figure 3 Gene conversion between wheat subgenomes. In each subfigure, the outer circle shows the seven chromosomes of each of wheat subgenome. Non-converted homeologous gene pairs between the subgenomes are connected by curvy gray lines, converted pairs by curvy red lines. (A) Ah-Bh; (B) Ah-Dh; (C) Bh-Dh.
For there was not a diploid B genome available, we took the Bt genome from the emmer wheat in the analysis and constructed AdAhBtBh and BtBhDdDh quartets (Table 3). We inferred 6059 AdAhBtBh quartets and found that 127 (or 2.10% of all those involved in quartets) AhBh homeologs were likely converted, showing much more conversion than the BhDh homeologs. Similarly, we inferred 12,770 Bh–Dh quartets, and found that 253 (or 1.99% of all those involved in quartets) were likely converted, showing similar gene conversion rates between subgenomes (Figure 3). With the converted homeologs, one copy might act as the donor, while the other as the acceptor. If one gene was taken as the donor, the sequence of the acceptor would be converted to be identical or highly similar, immediately after the conversion, to the donor’s ortholog in the diploid (tetraploid) plant. It seemed that in AhDh gene conversions, about 62.20% of the converted genes took the Dh homeologs as the donor. This showed that the subgenome D was more likely taken as the repairing template during recombination with the subgenome A. With AhBh and BhDh converted genes, the Bh homeologs were more often (59.84 and 66.40%) taken as the donors, respectively, showing the subgenome B was often taken as the repairing template when pairing with the subgenomes A or D (Table 3).
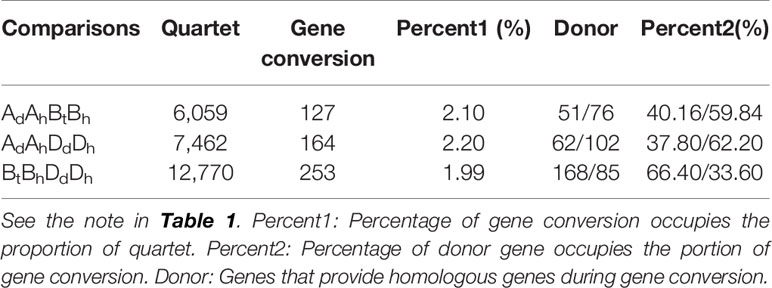
Table 3 Gene conversion between subgenomes in common wheat.
The Lower Gene Conversion Rate Between Ancient Paralogs Produced by the Grass Common Whole-Genome Duplication
We found likely gene conversion between ancient paralogs in each subgenome produced by the cWGD ~100 million years ago. To see gene conversions between ancient paralogs, such as those in the subgenome Ah, we first revealed the paralogs having preserved colineartiy resulting from the cWGD, inferred their Ad orthologs, and found the AdAhAdAh quartets. Then, we checked likely tree topology changes, being aberrant from the expected one. The orthologs were supposed to be more similar than each to the paralog in the same subgenome. However, if the paralogs, such as those Ah paralogs, were more similar, it showed likely gene conversion. Here, we found about 700 homologous quartets within each of the three subgenomes and showed that ancient gene conversion rates varied from 0.83 to 1.71% (Table 4), being lower (though not significantly) than those between these subgenomes, as estimated above.

Table 4 Gene conversion between paralogs in common wheat.
Gene Function Analysis
By performing Gene Ontology analysis, we found that, in the whole genome level, specific functional genes were enriched (Figure 4). The organelle-related functional genes were significantly more likely to be converted compared to all colinear genes (~3.32 to 1.89%, Fisher′s exact test p-value = 0.041). Similarly, the organelle extracellular region genes were significantly more likely to be converted compared to all colinear genes (~0.70% to 0, p-value = 0.004). Genes involved in the nutrient reservoir and molecular transduction were also significantly likely affected by conversion (Figure 4D). Comparatively, the membrane part genes and biological regulation genes showed substantially less likely to be involved in conversion as compared to all colinear genes (~4.71 and 7.63%, p-value = 0.0182; and ~9.95 and 15.45%, p-value = 0.002) (Figure 4A).
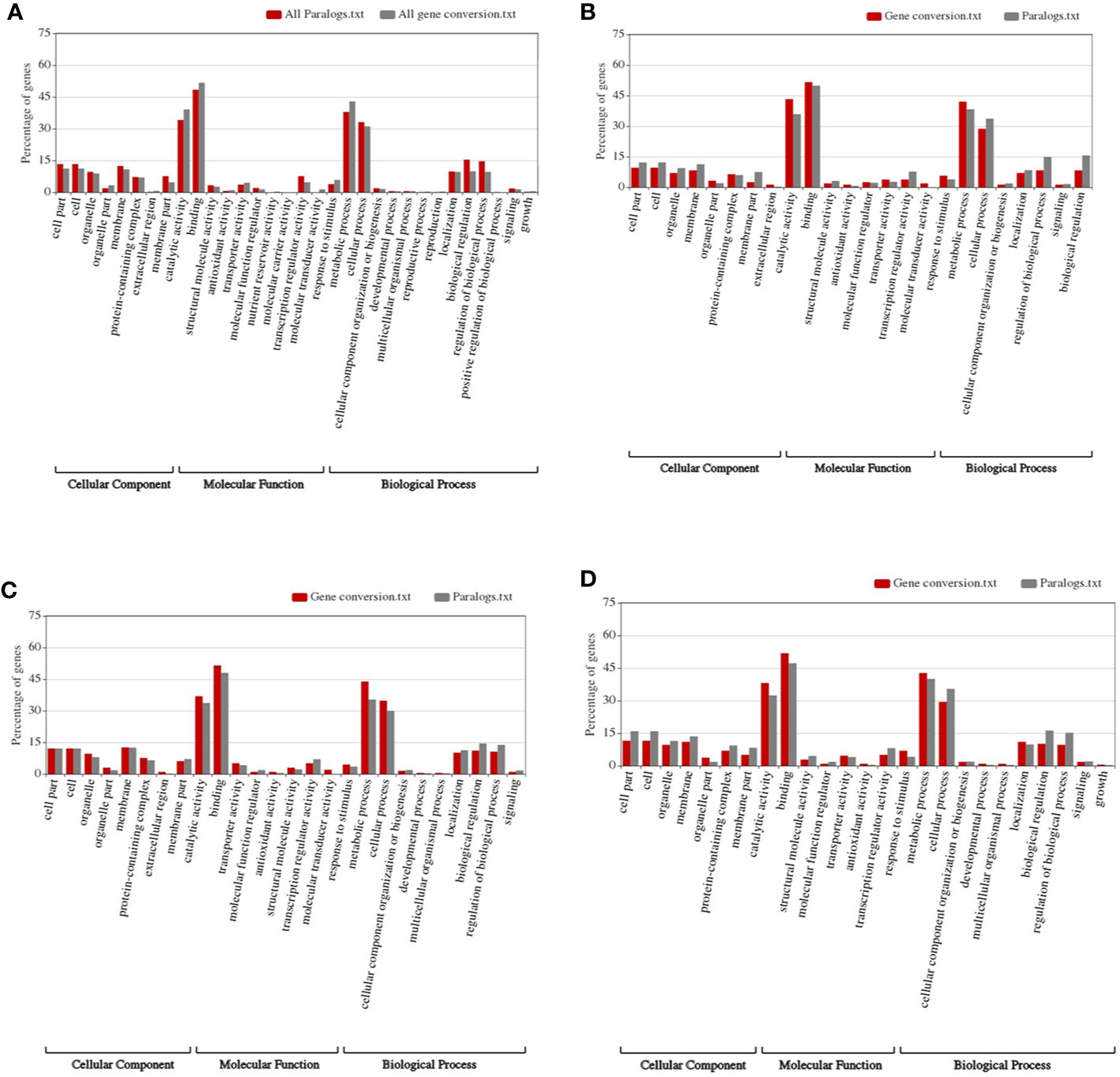
Figure 4 Construction of functional spectrum of subgenomes in wheat. (A) whole genome; (B) Ah subgenome; (C) Bh subgenome; (D) Dh subgenome.
As to each subgenome, specific genes were enriched in converted genes (Figures 4B–D). For subgenome B, the molecular transducer activity genes showed a significant increase in converted genes (~0 to 2.02%, p-value = 0.0004). As to the signaling genes in subgenome A, those in subgenome D showed a considerable increase (1.27 and 10.09%, p-value = 0.0002). The biological regulation genes in subgenome D showed a significant increase (~1.83 to 8.28%, p-value = 0.003).
Starch, one of the final products of photosynthesis, helps form the wheat seeds. About 65–80% of the grain composition of common wheat is starch. Here, we found that about 40% of starch biosynthesis catalytic genes were likely involved in gene conversion in the whole genome and each its subgenome. Therefore, we selected a set of catalytic genes regulating starch biosynthesis, the converted ones, and their close homologs in wheat and relative (sub)genomes to construct a phylogenetic tree (Figure 5). The converted genes shown here are Ah7g3962/Bh7g2982, Ah6g3817/Dh6g3664, Ah5g5014/Ah4g3299, Bh1g2423/Dh1g2297, each pair showing conversion between different subgenomes.
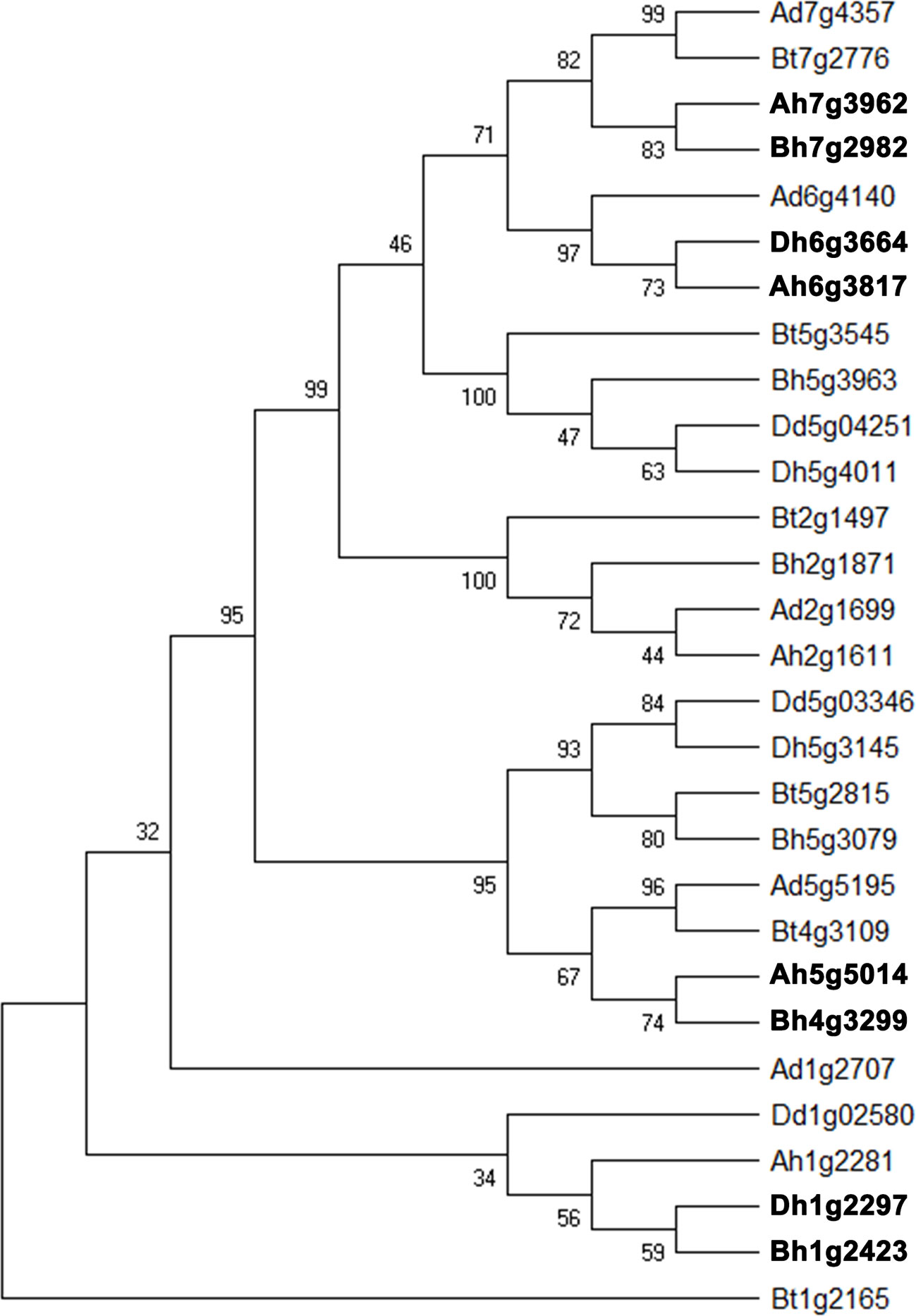
Figure 5 Phylogenetic tree of homologous gene quartet with catalytic activity in starch biosynthesis. Bt, subgenome B in tetraploid wild wheat; Dd, Aegilops tauschii; Ad, Triticum urartu; Ah, Bh, and Dh, three subgenomes of common wheat. Gene names in bold font show conversion affected ones.
Discussion
The doubling of the whole-genome often leads to an extremely unstable genome of the species (Freeling et al., 2012; Zhang et al., 2013; Wang et al., 2018). Illegitimate recombination, in contrast to recombination usually between homologous chromosomes, may occur between homeologous chromosomes (Wang et al., 2009; Woodhouse et al., 2010; Murat et al., 2014). Genome instability is often characterized by large-scale DNA losses, DNA rearrangement, even chromosome breakages, and fusions, which eventually contribute to regaining genome stability. After some time, which is difficult to define how long it is, the duplicated genome would come to a calm state. However, though at a much-lowered rate, illegitimate recombination may still occur between homeologous chromosomal regions. In rice, sorghum and other grasses, we found illegitimate recombination could last tens of millions of years, resulting in a particular stratum structure between two rice homeologous chromosomes, and between their sorghum counterparts, and many involved rice and sorghum genes might have been converted by their homeologous copies (Wang et al., 2009; Wang et al., 2011).
Here, we checked likely gene conversion in wheat, an important world-wide crop, and found hundreds of converted genes between the subgenomes. This shows that the wheat genome has not been quite stable after the formation of its hexaploid structure about 10,000 years ago (Feldman et al., 2012). There has been genetically illegitimate recombination between the subgenomes, contributing to the structural and functional changes of homeologous chromosomes and the genes residing on them. We also found likely gene conversion between more ancient duplicated genes produced by the cWGD having occurred ~100 million of years ago (Wang et al., 2015). Though the conversion scale of duplicated genes within each subgenome is lower than that between different subgenomes, it suggested the long-lasting lingering effect of illegitimate recombination after the whole-genome duplication, as reported previously in rice and its Oryza relatives, sorghum, and other grasses (Jacquemin et al., 2009; Wang et al., 2011; Ling et al., 2013).
Notably, we revealed that genes from different common wheat subgenomes tend to play different roles during the occurrence of conversion. The subgenome DD shares more colinear genes with the other two subgenomes and its genes act more likely as donors, showing likely preferential roles of DD chromosomes and the genes residing on them. This may be related to the fact that subgenome DD was the last one to be added to form the hexaploid plant, common wheat (Middleton et al., 2014; Du et al., 2017; Zimin et al., 2017; Gardiner et al., 2019). The initial formation of a tetraploid ancestor (AABB) may have been followed likely wide-spread DNA and gene losses, accompanying illegitimate recombination between homeologous chromosomes, due to genomic instability of the tetraploid. After this turmoil stage, the homeologous chromosomes became much different, and the tetraploid genome came to a relatively stable state. Then the third genome DD, contributed by an ancient plant that A. tauschii is derived from, hybridized with the tetraploid to form the hexaploid common wheat. The newly joined subgenome DD lives much at ease with the reduced DNA content of the subgenomes AA and BB. Though there should have been continual DNA losses and illegitimate recombination, the occurrence rates should have likely decreased. This eventually resulted in a better gene colinearity of the subgenome DD with the other two than the other ones with one another, as observed above. Furthermore, relatively innocent state of DD genes might render themselves privileges to act as the donors during gene conversion, to preserve ancestral gene structures and functions.
The above-inferred conversion involved homeologs produced by the formation of the hexaploid about 10,000 years ago, while the tetraploid progenitor (AABB) might have formed at least 0.5 million years ago (Marcussen et al., 2014; Brenchley et al., 2012). Comparatively, the divergence of their diploid ancestor occurred at least 2.5 to 7 million years ago (Marcussen et al., 2014; Cheng et al., 2019). With updated fossil evidence applying to colinear genes, a grass-family level estimation of their Triticeae common ancestor suggested its appearing more than 20 million years ago (Wang et al., 2015). These facts mean that the wheat homeologs have much diverged from one another and phylogenetic inference of gene conversion between homeologs by comparing their close orthologs in diploid or tetraploid progenitor would not likely result in high false positives. This supports the credibility of the present gene conversion inference. However, this could really be an underestimation of gene conversion. Thus, it is not easy of converted paralogs to be well supported at such rigorous criteria about gene tree topology change. The subgenome copies are quite similar to each other, and there may be no much time to gather mutations between the diploid-hexaploid orthologs to show likely occurrence of gene conversion between the hexaploid subgenome copies.
Notably, we found that around 40% of starch biosynthesis catalytic genes were involved in gene conversion, showing that conversion may favor certain types of functional genes, as shown with the above pilot characterization with GO. Actually, while characterizing the phylogeny of those catalytic genes, the constructed phylogenetic tree involved low Bootstrap values in some branches. However, the credibility of the tree was well supported by known or expected phylogenetic relationship, paralogy between homeologs from subgenomes, or orthology between homeologs from different organisms. Actually, the known or expected relationship between homeologs was further supported by inferred gene colinearity, which is a good criteria to explore the real phylogeny, especially when it was disturbed by non-uniform evolutionary rates of plant genes (Meng et al., 2020).
Materials and Methods
Materials
The genome-wide datasets, including whole-genome sequences, annotated genes, and their translated proteins, were retrieved from following public databases.
Tritium aestivum: URGI, https://wheat-urgi.versailles.inra.fr; Triticum urartu: MBK, http://www.mbkbase.org; T.turgidum ssp.dicoccoides: http://wewseq.wixsite.com; Aegilops tauschii: http://aegilops.wheat.ucdavis.edu/ATGSP.
Gene Colinearity Inference
By using the similarity comparison tool BLASTP compared the proteins within and between the genomes of wheat and related species, and scoring according to the similarity between the protein sequences, we inferred the relationship of gene homology according to the results of the comparison, and the expected value (E-value) is limited to 1e−5. Then the result of BLASTP was used for the software ColinearScan to infer colinear gene pairs. Colinear genes are those in a genomic region sharing locational orders with their homologs in another genomic region. The detection of genomic homologous colinear fragments was based on the principle of similarity between genes, which was combined with the position information of genes on the chromosomes. The colinearity analysis software ColinearScan was used to infer homologous regions (or blocks) containing five or more colinear genes (Wang et al., 2017). The maximal searching gap between colinear genes was set to be 50 intervening non-colinear genes(P <0.05), as often adopted in previous analyses (Sun et al., 2017). Besides, we used OrthoMCL (http://www.micans.org/mcl/src/mcl-latest.tar.gz) to identify orthologous and paralogous genes to support the credibility of the gene colinearity inferred above (Li et al., 2003; Brenchley et al., 2012).
Inference of Homologous Quartets and Gene Conversion
We used wheat A. tauschii genomes as a reference to construct multiple alignments. In details, each genome was compared and aligned to the referenced genome by inferring gene colinearity; any two genomes were compared to find inter-genome colinearity; and then, each genome was aligned to itself to find paralogous genes produced by cWGD (Wang et al., 2015; Ling et al., 2018; Liu et al., 2019). We used multiple alignments to construct homologous gene quartets based on the homologous relationships. By checking colinear genes in the multiple alignments, the paralogous gene pairs in each of the involved genomes and the orthologous gene pairs between genomes were obtained, and homologous gene quartets were retrieved. Using these quartets, we inferred gene conversion based on the aberrant topology of each homologous gene quartet. Aberrant tree topology is that the relationship between homeologous genes is different from the expected phylogeny (Figure 2A). With each gene quartet, multiple sequence alignment was performed using ClustalW (Thompson et al., 1994). If in the alignment, the number of gaped sites in any of the aligned sequences is more than 50% of the alignment length or the amino acid identity was less than 40%. Then the alignment was discarded for further inference (Wang et al., 2009) As to the conversion inference with each homologous quartet, phylogenetic gene trees were constructed by using Neighbor-joining approach implemented in MEGAX with encoded amino acid sequences as input, and default parameters were adopted (Tamura et al., 2007). Bootstrap of 1,000 iterations was performed to improve the reliability of the evolutionary tree. Then, with trees supported by bootstrap value >70%, we checked the tree topology to find whether there was likely gene conversion. Synonymous nucleotide substitutions on synonymous substitution sites (Ks) between homologous genes (Nei and Gojobori, 1986) were estimated to characterize the distance between homologs with the program implemented in Bioperl (Stajich et al., 2002). Whole-genome scale gene conversion was displayed by using Circos software (Krzywinski et al., 2009).
GO Enrichment Analysis
We performed GO enrichment analysis and used the domain similarity comparison software InterProScan to annotate genes with GO (Gaudet, 2019). The best international standard matching wheat genes was obtained through the BLASTP comparison between the international standard protein database and the whole wheat genomic protein file. Then, we used WEGO2.0 (http://wego.genomics.org.cn/) to generate the wheat gene function map. The log function standardized the number of genes involved in each category, and then the functional distribution and correlation were analyzed. The p-value was calculated using hypergeometric delivery to explain the significance of gene enrichment. Here we selected a set of starch catalytic activity function genes to build a phylogenetic tree. CLUSTALW was used to perform sequence alignment on the protein sequences (Thompson et al., 1994), and a phylogenetic tree was constructed using the Maximum Likelihood (Murshudov et al., 1997). The pilot value was based on 1,000 copies.
Data Availability Statement
The datasets generated for this study can be found in the URGI, https://wheat-urgi.versailles.inra.fr.
Author Contributions
XW conceived and led the research. CL, JW, PS, JY, FM, ZZ, HG, CW, and XL performed the analysis or contributed analysis tools. XW and CL wrote the paper. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors XW.
Acknowledgments
We appreciate financial support from the Ministry of Science and Technology of the People’s Republic of China (2016YFD0101001), China National Science Foundation (3117022), Tangshan Key Laboratory Project to XW. We thank the helpful discussion with researchers at the iGeno Co. Ltd, China.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.01076/full#supplementary-material
References
Appels, R., Eversole, K., Feuillet, C., Keller, B., Rogers, J., Stein, N., et al. (2018). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Sci. (N. Y. NY) 361, 6403. doi: 10.1126/science.aar7191
Brenchley, R., Spannagl, M., Pfeifer, M., Barker, G. L. A., D’Amore, R., Allen, A. M., et al. (2012). Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491, 705–710. doi: 10.1038/nature11650
Chen, J. M., Cooper, D. N., Chuzhanova, N., Férec, C., Patrinos, G. P. (2007). Gene conversion: mechanisms, evolution and human disease. Nat. Rev. Genet. 8, 762–775. doi: 10.1038/nrg2193
Cheng, H., Liu, J., Wen, J., Nie, X., Xu, L., Chen, N., et al. (2019). Frequent intra- and inter-species introgression shapes the landscape of genetic variation in bread wheat. Genome Biol. 20, 136. doi: 10.1186/s13059-019-1744-x
Choulet, F., Alberti, A., Theil, S., Glover, N., Barbe, V., Daron, J., et al. (2014). Structural and functional partitioning of bread wheat chromosome 3B. Science 345:1249721. doi: 10.1126/science.1249721
Datta, A., Hendrix, M., Lipsitch, M., Jinks-Robertson, S. (1997). Dual roles for DNA sequence identity and the mismatch repair system in the regulation of mitotic crossing-over in yeast. Proc. Natl. Acad. Sci. U. States A. 94, 9757–9762. doi: 10.1073/pnas.94.18.9757
Du, H., Yu, Y., Ma, Y., Gao, Q., Cao, Y., Chen, Z., et al. (2017). Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 8, 15324. doi: 10.1038/ncomms15324
Feldman, M., Levy, A. A., Fahima, T., Korol, A. (2012). Genomic asymmetry in allopolyploid plants: wheat as a model. J. Exp. Bot. 63, 5045–5059. doi: 10.1093/jxb/ers192
Freeling, M., Woodhouse, M. R., Subramaniam, S., Turco, G., Lisch, D., Schnable, J. C., et al. (2012). Fractionation mutagenesis and similar consequences of mechanisms removing dispensable or less-expressed DNA in plants. Curr. Opin. Plant Biol. 15, 131–139. doi: 10.1016/j.pbi.2012.01.015
Gardiner, L. J., Wingen, L. U., Bailey, P., Joynson, R., Brabbs, T., Wright, J., et al. (2019). Analysis of the recombination landscape of hexaploid bread wheat reveals genes controlling recombination and gene conversion frequency. Genome Biol. 20, 69. doi: 10.1186/s13059-019-1675-6
Gaudet, P. (2019). The Gene Ontology. Encyclopedia Bioinf. Comput. Biol. 2, 1–7. doi: 10.1016/B978-0-12-809633-8,20500-1
Jacquemin, J., Laudie, M., Cooke, R. (2009). A recent duplication revisited: phylogenetic analysis reveals an ancestral duplication highly-conserved throughout the Oryza genus and beyond. BMC Plant Biol. 9, 146. doi: 10.1186/1471-2229-9-146
Jia, J., Zhao, S., Kong, X., Li, Y., Zhao, G., He, W., et al. (2013). Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496, 91–95. doi: 10.1038/nature12028
Jung, C. G., Lim, S. D., Hwang, S. G., Jang, C. S. (2012). Molecular characterization and concerted evolution of two genes encoding RING-C2 type proteins in rice. Gene 505, 9–18. doi: 10.1016/j.gene.2012.05.060
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kurosawa, K., Ohta, K. (2011). Genetic diversification by somatic gene conversion. Genes 2, 48–58. doi: 10.3390/genes2010048
Li, L., Stoeckert, C. J., Roos, D. S. (2003). OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Ling, H., Zhao, S., Liu, D., Wang, J., Sun, H., Zhang, C., et al. (2013). Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496, 87–90. doi: 10.1038/nature11997
Ling, H., Ma, B., Shi, X., Liu, H., Dong, L., Sun, H., et al. (2018). Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 557, 424–428. doi: 10.1038/s41586-018-0108-0
Liu, M., Li, Y., Ma, Y., Zhao, Q., Stiller, J., Feng, Q., et al. (2019). The draft genome of a wild barley genotype reveals its enrichment in genes related to biotic and abiotic stresses compared to cultivated barley. Plant Biotechnol. J. 18, 443–456. doi: 10.1111/pbi.13210
Marcussen, T., Sandve, S. R., Heier, L., Spannagl, M., Pfeifer, M., Jakobsen, K. S., et al. (2014). Ancient hybridizations among the ancestral genomes of bread wheat. Science 345, 1250092. doi: 10.1126/science.1250092
Meng, F., Pan, Y., Wang, J., Yu, J., Liu, C., Zhang, Z., et al. (2020). Cotton duplicated genes produced by polyploidy show significantly elevated and unbalanced evolutionary rates, overwhelmingly perturbing gene topology. Front. Genet. 11, 239. doi: 10.3389/fgene.2020.00239
Middleton, C. P., Senerchia, N., Stein, N., Akhunov, E. D., Keller, B., Wicker, T., et al. (2014). Sequencing of chloroplast genomes from wheat, barley, rye and their relatives provides a detailed insight into the evolution of the Triticeae tribe. PloS One 9, e85761. doi: 10.1371/journal.pone.0085761
Murat, F., Zhang, R., Guizard, S., Flores, R., Armero, A., Pont, C., et al. (2014). Shared subgenome dominance following polyploidization explains grass genome evolutionary plasticity from a seven protochromosome ancestor with 16K protogenes. Genome Biol. Evol. 6, 12–33. doi: 10.1093/gbe/evt200
Murshudov, G. N., Vagin, A. A., Dodson, E. J. (1997). Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallographica D53, 240–255. doi: 10.1107/S0907444996012255
Nei, M., Gojobori, T. (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. MolBiolEvol 3, 418–426. doi: 10.1093/oxfordjournals.molbev.a040410
Paterson, A. H., Bowers, J. E., Chapman, B. A. (2004). Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. U. States A. 101, 9903–9908. doi: 10.1073/pnas.0307901101
Stajich, J. E., Block, D., Boulez, K., Brenner, S. E., Chervitz, S. A., Dagdigian, C., et al. (2002). The bioperl toolkit: Perl modules for the life sciences. Genome Res. 12, 1611–1618. doi: 10.1101/gr.361602
Sun, S., Wang, J., Yu, J., Meng, F., Xia, R., Wang, L., et al. (2017). Alignment of Common Wheat and Other Grass Genomes Establishes a Comparative Genomics Research Platform. Front. Plant Sci. 8, 1480. doi: 10.3389/fpls.2017.01480
Tamura, K., Dudley, J., Nei, M., Kumar, S. (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. doi: 10.1093/molbev/msm092
Thompson, J. D., Higgins, D. G., Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Wang, X., Paterson, A. H. (2011). Gene conversion in angiosperm genomes with an emphasis on genes duplicated by polyploidization. Genes 2, 1–20. doi: 10.3390/genes2010001
Wang, X., Shi, X., Li, Z., Zhu, Q., Kong, L., Tang, W., et al. (2006). Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice. BMC Bioinf. 7, 447. doi: 10.1186/1471-2105-7-447
Wang, X., Tang, H., Bowers, J. E., Paterson, A. H. (2009). Comparative inference of illegitimate recombination between rice and sorghum duplicated genes produced by polyploidization. Genome Res. 19, 1026–1032. doi: 10.1105/tpc.114.125583
Wang, X., Tang, H., Paterson, A. H. (2011). Seventy million years of concerted evolution of a homeologous chromosome pair, in parallel, in major Poaceae lineages. Plant Cell. 23, 27–37. doi: 10.1105/tpc.110.080622
Wang, X., Wang, J., Jin, D., Guo, H., Lee, T.,. H., Liu, T., et al. (2015). Genome Alignment Spanning Major Poaceae Lineages Reveals Heterogeneous Evolutionary Rates and Alters Inferred Dates for Key Evolutionary Events. Mol. Plant 8, 885–898. doi: 10.1016/j.molp.2015.04.004
Wang, J., Sun, P., Li, Y., Liu, Y., Yu, J., Ma, X., et al. (2017). Hierarchically Aligning 10 Legume Genomes Establishes a Family-Level Genomics Platform. Plant Physiol. 174, 284–300. doi: 10.1104/pp.16.01981
Wang, J., Yu, J., Li, J., Sun, P., Wang, L., Yuan, J., et al. (2018). Two Likely Auto-Tetraploidization Events Shaped Kiwifruit Genome and Contributed to Establishment of the Actinidiaceae Family. iScience 7, 230–240. doi: 10.1016/j.isci.2018.08.003
Wang, J., Yuan, J., Yu, J., Meng, F., Sun, P., Li, Y., et al. (2019). Recursive Paleohexaploidization Shaped the Durian Genome. Plant Physiol. 179, 209–219. doi: 10.1104/pp.18.00921
Woodhouse, M. R., Schnable, J. C., Pedersen, B. S., Lyons, E., Lisch, D., Subramaniam, S., et al. (2010). Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PloS Biol. 8, e1000409. doi: 10.1371/journal.pbio.1000409
Zhang, H., Zhang, C., Sun, Z., Yu, W., Gu, M., Liu, Q., et al. (2011). A major locus qS12, located in a duplicated segment of chromosome 12, causes spikelet sterility in an indica-japonica rice hybrid. TAG Theor. Appl. Genet. Theoretische Und Angewandte Genetik. 123, 1247–1256. doi: 10.1007/s00122-011-1663-z
Zhang, H., Bian, Y., Gou, X., Dong, Y., Rustgi, S., Zhang, B., et al. (2013). Intrinsic karyotype stability and gene copy number variations may have laid the foundation for tetraploid wheat formation. Proc. Natl. Acad. Sci. U. S A. 110, 19466–19471. doi: 10.1073/pnas.1319598110
Keywords: common wheat, gene colinearity, gene conversion, polyploidization, homeologous gene
Citation: Liu C, Wang J, Sun P, Yu J, Meng F, Zhang Z, Guo H, Wei C, Li X, Shen S and Wang X (2020) Illegitimate Recombination Between Homeologous Genes in Wheat Genome. Front. Plant Sci. 11:1076. doi: 10.3389/fpls.2020.01076
Received: 09 March 2020; Accepted: 30 June 2020;
Published: 21 July 2020.
Edited by:
Yuannian Jiao, Institute of Botany (CAS), ChinaReviewed by:
Liangsheng Zhang, Zhejiang University, ChinaMehboob-ur- Rahman, National Institute for Biotechnology and Genetic Engineering, Pakistan
Copyright © 2020 Liu, Wang, Sun, Yu, Meng, Zhang, Guo, Wei, Li, Shen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiyin Wang, d2FuZy54aXlpbkBnbWFpbC5jb20=