 Yangyun Zhou1,2†
Yangyun Zhou1,2† Jingxian Feng3†
Jingxian Feng3† Qing Li1Doudou Huang1Xiao Chen3Zenan Du3
Qing Li1Doudou Huang1Xiao Chen3Zenan Du3 Zongyou Lv3Ying Xiao3
Zongyou Lv3Ying Xiao3 Yonglong Han2
Yonglong Han2 Junfeng Chen3*Wansheng Chen1,3*
Junfeng Chen3*Wansheng Chen1,3*- 1Department of Pharmacy, Changzheng Hospital, Naval Medical University (Second Military Medical University), Shanghai, China
- 2Department of Pharmacy, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai, China
- 3Research and Development Center of Chinese Medicine Resources and Biotechnology, Institute of Chinese Materia Medica, Shanghai University of Traditional Chinese Medicine, Shanghai, China
Salvia miltiorrhiza Bunge (Lamiaceae) is an economically important medicinal plant as well as an emerging model plant. Our previous studies indicate that SmMYC2b is a positive transcription factor that can affect the biosynthesis of phenolic acids and tanshinones in S. miltiorrhiza. Moreover, MYC2s are well known to induce the development of lateral roots. As tanshinones are mainly distributed in the periderm, the promotion of lateral root development probably leads to increased accumulation of tanshinones. In this paper, we firstly discovered that SmMYC2b played a dual regulatory role in effectively enhancing the tanshinone accumulation by activating tanshinone biosynthetic pathway and promoting lateral root development. The expression levels of the previously studied pathway genes SmCPS1, SmKSL1, SmCYP76AH1, SmCYP76AH3, and SmCYP76AK1 dramatically increased. In addition, SmMYC2b was proved to exhibit a similar function as other homologs in promoting lateral root development, which increased the tanshinone produced tissue and further enhanced the biosynthesis of tanshinones. RNA-seq assays revealed that SmMYC2b-regulated genes comprised 30.6% (1,901 of 6,210) of JA-responsive genes, confirming that SmMYC2b played a crucial role in transcriptional regulation of JA-regulated genes. Overall, we concluded that SmMYC2b could enhance tanshinone accumulation by activating the tanshinone biosynthetic pathway and promoting lateral root development. Our study provides an effective approach to enhance the production of desired tanshinones and enriches our knowledge of the related regulatory network.
Introduction
Salvia miltiorrhiza Bunge (Lamiaceae) is a perennial herb that is a source of the famous traditional Chinese medicine (TCM) Danshen. Cultivated in East Asia, the dried roots and rhizomes of S. miltiorrhiza are mainly used in treating coronary heart disease, angina, acute ischemic stroke, and other cardiovascular diseases (Lin and Hsieh, 2010). The bioactive components in S. miltiorrhiza are mainly lipophilic diterpenoids (including tanshinone IIA, tanshinone I, tanshinone IIB, and cryptotanshinone) and hydrophilic phenolics (such as salvianolic acids and rosmarinic acid) (Gong et al., 2011; Su et al., 2015). Based on previous studies, the biosynthesis of terpenoids consists of three stages: isoprene precursor biosynthesis, direct skeleton precursor generation, and the biosynthesis of various tanshinones (Chen et al., 2011; Chang et al., 2019; Xue et al., 2019). The first stage mainly generates isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) derived from the mevalonate (MVA) pathway and the methylerythritol phosphate (MEP) pathway (Vranová et al., 2013; Zhao et al., 2013). The second stage is the generation of farnesyl diphosphate (FPP), geranyl diphosphate (GPP) and geranylgeranyl diphosphate (GGPP), which further form the precursors of monoterpenes, sesquiterpenes, and diterpenes respectively (Liang et al., 2002; Szkopińska and Płochocka, 2005). At present, the first two stages have been studied clearly and are shared by all terpenes. The third stage involves a variety of terpenoid synthases and modifying enzymes, which determine the structural diversity of terpenoids and are species specific (Keeling et al., 2008; Degenhardt et al., 2009).
The metabolic engineering of one or more rate-limiting enzymes is an effective strategy to increase the metabolic flux towards the desirable compounds. Significant progress has been made in increasing the production of the bioactive compounds by overexpression of the key enzymes (Shi et al., 2016; Shi et al., 2020; Zhang et al., 2020). A large number of bioactive compounds are distributed in the specific tissues, such as the antitumor drug paclitaxel and the antimalarial drug quinine, which are mainly distributed in the bark. To reach high yield of the bioactive components, increasing the producing tissues can be an ideal strategy. The β-glucosidase offers a possibility to increase artemisinin accumulation by promoting gland density in Artemisia annua (Singh et al., 2015).
Tanshinone pigments accumulate in the periderm (Xu et al., 2015), while salvianolic acid B (Sal B) is mainly detected in the phloem and xylem of roots (Xu et al., 2016b). As tanshinones are mainly distributed in the periderm, the rhizomes/roots of S. miltiorrhiza are typically reddish brown. This is also consistent with the classification and screening of TCM Danshen. During the screening of Danshen materials, roots with different morphologies are often used in four applications. It is generally accepted that the tanshinone content is higher in the dark reddish brown and deeply branched S. miltiorrhiza roots. This kind of material is generally used to extract tanshinones. Phenolic acids are usually extracted from coarser roots. Materials with good shape and pharmacopoeia standard content are used for slicing. Others are used in the extraction of tanshinones and phenolic acids together.
It will be exciting to realize the dual activation of the tanshinone biosynthetic pathway and the specific tissue development. The biosynthesis of the tanshinones and the development of specific tissues are regulated by many factors, among which transcription factors (TFs) that control the abundance or activity of multiple enzymes have become promising regulators (Zhang et al., 2015; Deng et al., 2020a). In our previous studies, the TAR1 TF is demonstrated to play a dual regulatory role in the artemisinin biosynthesis by controlling trichome development and promoting artemisinin pathway in A. annua (Tan et al., 2015). In S. miltiorrhiza, it is not clear whether there is an ideal TF with similar dual function. The MYC2 TF is widely known to induce the formation of lateral roots (Yadav, 2005; Sun et al., 2009). As tanshinones are mainly distributed in the periderm, overexpressing MYC2b may promote the development of lateral roots to enhance the content of tanshinones. Our previous studies indicate that SmMYC2b is a positive JA-responsive TF that facilitates the biosynthesis of tanshinone IIA and salvianolic acid B (Zhou et al., 2016). Tanshinones and phenolic acids may also accumulate following overexpressing of MYC2b. Overexpressing MYC2b could be a potential strategy to increase tanshinones in one step by promoting tanshinone pathway gene expression and lateral root development.
In the present paper, we implemented transcriptome profiling by RNA sequencing (RNA-seq) to mine MYC2-targeted genes. We showed that SmMYC2b could promote the biosynthesis of tanshinone biosynthesis genes (SmCPS1, SmKSL1, SmCYP76AH1, SmCYP76AH3, and SmCYP76AK1). SmMYC2b mainly modulated the downstream tanshinone pathway, instead of the nonspecific upstream pathway. The content of tanshinones dramatically increased with the exceptional decrease in carnosic acid (CA), suggesting its role as an important tanshinone precursor. MYC2b was also proposed to positively regulate lateral root development. These data suggest that SmMYC2b dramatically enhances the accumulation of tanshinones by activating the tanshinone pathway and promoting lateral root development in Salvia miltiorrhiza.
Materials and Methods
Plant Materials and Growth Conditions
Danshen (Salvia miltiorrhiza Bunge) plants were cultivated in the botanical garden of the Second Military Medical University, Shanghai, China. To obtain sterile S. miltiorrhiza seedlings, seeds were immersed in 75% alcohol for 1 min and afterwards were washed three times with distilled water. After subsequent treatment with 0.1% HgCl2 for 5 min, the seeds were rinsed three times for 3 min each with sterile distilled water and incubated on Murashige and Skoog (MS) solid medium plus 3% sucrose with an appropriate pH value of 5.6. The sterilized seedlings were grown in a growth room at 24 ± 2°C under a 16-h light/8-h dark photoperiod for propagation.
Seeds of an Arabidopsis thaliana (L.) mutant were sown in a soil matrix. PCR identified approximately 1-month-old mutant plants with good growth status that were transformed by the soaking method. The aboveground parts of the A. thaliana plants were immersed in transformation buffer for 5–15 s to ensure that all buds were immersed. Then, the plants were laid flat and covered to keep out light. The next day, the cover was removed, and the plants were raised and transferred to normal conditions for growth. Their seeds were collected and stored.
S. miltiorrhiza Bunge plants were also cultivated in the botanical garden of the Second Military Medical University, Shanghai, China. To examine the expression patterns of SmMYC2b, we collected roots of S. miltiorrhiza plants at different times (5, 15, 30, 60, 90, 120, 150, 180, 210, 240, 270, and 300 d). There were three biological replicates for every time point.
Vector Construction and Plant Transformation
A modified pHB-flag vector that could express C-terminally Flag-tagged fusion protein was chosen as an overexpression vector. The full-length open reading frame (ORF) of SmMYC2b was amplified with primers 5’-TGATCAATGGGGGTTGTTGGTTGG-3’ and 5’-ACTAGTCCCGAGAGATAACTGATG-3’, and inserted into the BamH I and Spe I sites of the pHB-flag vector. The constructed pHB-flag-MYC2b vector was introduced into the disarmed Agrobacterium tumefaciens EHA105 strain. The pHB-flag vector was also transformed with the same method to serve as a control.
Agrobacterium-mediated construction of transgenic plants was performed according to previous protocols (Yan and Wang, 2007). Generally, 1-month-old S. miltiorrhiza plants with seven to eight leaves were chosen for genetic transformation. The leaves were cut into 0.5 × 0.5 cm discs. All explants were cocultured on MS basal medium with A. tumefaciens EHA105 at 25°C for 2 d in the dark. Afterwards, the explants were transformed to selective media supplemented with different concentrations of cefotaxime (from 500 mg/L gradually reduced to 0) and hygromycin (2 mg/L). The identification of MYC2b transgenic seedlings was conducted by western blot (WB) assay. Total soluble protein was extracted by adding Plant Protein Extraction Reagent (CWBIO, Beijing, China) supplemented with Protease Inhibitor Cocktail. The samples were denatured for 5 min in boiling water with protein loading buffer. After separation on an 8% SDS-PAGE mini-gel, the targeted proteins were blotted to a nitrocellulose membrane. The blots were probed with the anti-FLAG mouse antibody (1:1,000, Sigma). The corresponding HRP-labelled goat anti-mouse IgG(H+L) antibody (Beyotime, China) was used as a secondary antibody at a final dilution of 1:1,000. A Chemiscope 3000 mini was used to capture chemiluminescence according to the manufacturer’s instructions (Clinx, Shanghai, China). The fresh roots from five selected lines were subjected to root imaging analysis as biological replicates. The roots were weighed and collected for root imaging analysis using WinRHIZO software (WinRhizo, Regent Instruments, Canada).
Changes in Root Morphological Caused by SmMYC2b in A. thaliana
The constructed pHB-flag-MYC2b vector was introduced into A. thaliana MYC2 mutant (jin1-9) lines to generate complementary mutant SmMYC2b lines. Different genotypes of A. thaliana seeds (Col-0, jin1-9) were sown on 0.5×MS plates for germination. The SmMYC2b seeds were sown on hygromycin B selective medium. Then, after 7 d, the seedlings were plated on 0.5×MS with 50 μM methyl jasmonate (Sigma) or a solvent control on square plates and were finally transferred to a growth room in a vertical position. Root growth was monitored regularly, plates were photographed and root length was measured after 7 d of vertical cultivation.
MeJA Treatment and Construction of RNA-Seq Libraries
Two-month-old CK and MYC2bOE sterile seedlings grown on 0.5×MS medium were treated with either 100 μM MeJA or mock solution (ethanol). After treatment for 1 h, roots from the treated seedlings were collected from three biological replicates. Total RNA was extracted using an RNeasy Plant Mini Kit (Qiagen, German) following the user manual. The extracted RNA was quantified using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, America) and assessed using an Agilent 2100 Bioanalyser (Agilent Technologies, America). Total RNA (3 μg for each sample) was used to construct mRNA libraries according to the manufacturer’s instructions. Sequencing was conducted on an Illumina HiSeq 2500 platform.
Analysis of RNA-Seq Data
The S. miltiorrhiza reference genome and corresponding gene model annotation files were downloaded directly from public genome websites (Xu et al., 2016a). More than three biological replicates (six MYC2bOE lines, four CK lines, three MeJA-treated MYC2bOE lines, and three MeJA-treated CK lines) were sequenced on an Illumina HiSeq 2500. Bowtie 2 v2.1.0 (Langmead and Salzberg, 2012) was used to map the clean reads from each sample to reference sequence transcripts. The differentially expressed genes (DEGs) were screened using the DEGseq v1.20.0 (Wang et al., 2010) package with the MARS (MA-plot-based method with random sampling model) model. When the following conditions were all satisfied: |log2Fold change| ≥ 1, FDR (q value) < 0.001 and RPKM of at least one sample >20, the genes were considered differentially expressed. To clarify differences in gene function, gene ontology (GO) enrichment analysis was implemented using GOseq (Young et al., 2010) based on the Wallenius noncentral hypergeometric distribution. GO terms with corrected P-values <0.05 were considered significantly enriched in the DEGs. The GO terms with FDR value (q value) <0.05 were considered strikingly enriched in the DEGs.
Quantitative Real-Time PCR (qRT-PCR)
The relative expression analysis of S. miltiorrhiza genes was verified by qRT-PCR. Total RNA was isolated using a commercial kit (TIANGEN, Beijing, China). One microgram of the extracted total RNA was reverse transcribed to generate first strand cDNA using a TransScript® First-Strand cDNA Synthesis SuperMix Kit (TransGen Biotech, Beijing, China). qRT-PCR assays were conducted on a Termal Cycler Dice Real Time System TP800 according to the manufacturer’s instructions (Takara Bio Inc., Dalian, China). Expression levels were normalized to the corresponding housekeeping gene (18S RNA gene). All qRT-PCR experiments were carried out with three independent replicates. Gene-specific primers were designed and are listed in Table S1.
Chromatin Immunoprecipitation Assay
Chromatin immunoprecipitation (ChIP) assays were conducted according to previously published protocols (Nelson et al., 2006; Abdelaty et al., 2008). Briefly, 1 g of fresh root tissues was collected and cross-linked using 1% formaldehyde under vacuum for 15 min. The reaction was stopped by adding glycine to a final concentration of 0.125 M. Then, the roots were ground to a fine powder in liquid nitrogen. The chromatin was extracted, sonicated, and immunoprecipitated with ChIP grade rabbit anti-FLAG antibodies (CST #14793S). The protein-DNA complex was reverse cross-linked, and the protein was digested. Subsequently, the ChIP-DNA was purified and subjected to ChIP-qPCR. The gene-specific primers are listed in Table S2.
Compound Extraction and Analysis
Positive MYC2bOE seedlings were confirmed by western blot assay. The roots of six transgenic and four independent control seedlings were subjected to compound analysis. The harvested roots were dried at 40°C overnight to a constant dry weight and milled to homogeneous powders that could sieve through No. 100 mesh. Active compounds containing lipophilic tanshinones and hydrophilic phenolic acids were extracted following previously reported methods (Zhu et al., 2007; Gu et al., 2012). A 0.02 g powder sample was precisely weighed and extracted in 4 ml 70% methanol under sonication for 30 min. After centrifugation at 10,000 rpm for 5 min, the supernatant was collected and filtered through a 0.22 μm organic membrane. Afterwards, the samples were injected into the HPLC system for content analysis by applying the standard curve method. TT was designated as the summed content of DTI, TI, CT, and TIIA.
CA Treatment Assay
Based on the above research, a proposed biosynthetic route from CA to tanshinones was presumed. Wild-type hairy roots of S. miltiorrhiza from the same line were grown in specialized MSOH liquid culture. Plants with uniform growth were randomly divided into two groups: the CA treatment group and the control group. CA dissolved in 70% ethanol was added into the liquid medium to reach a final concentration of 50 μg/ml, while controls were treated with 70% ethanol. The hairy roots were harvested for tanshinone content determination after treatment for 0, 1, 2, 4, and 8 d. The collected hairy roots were dried at 50°C and the determination was consistent with the above method. The experiment was performed with three independent replicates.
Data Availability
The raw RNA-seq read data are publicly available at the NCBI Sequence Read Archive with the accession number PRJNA623221 (http://www.ncbi.nlm.nih.gov/sra/).
Results
Overexpression of SmMYC2b in Transgenic S. miltiorrhiza Plants Causes Morphological Changes
To further investigate the regulatory effect of SmMYC2b on active compounds, we constructed the SmMYC2b overexpression vector pHB-flag-MYC2 (Figure 1A) to generate transgenic plants. The growth process of the transgenic plants is shown in Figure S1. Positive MYC2b overexpression (MYC2bOE) plants were identified using the WB method (Figure 1B). Compared with CK, we surprisingly found that MYC2bOE plants exhibited a much more developed root systems (Figures 1C, D). Notably, obvious lateral root phenotypes were observed in MYC2bOE plants. Then, WinRHIZO software was applied to capture root morphological trait data including length, surface area (SurfArea), root volume (RootVolume), average diameter (AvgDiam), tips, forks, and crossings. Principal component analysis (PCA) was applied as a powerful tool to reduce the complexity of the data. As shown in Figure 1E, the OPLS-DA plot denoted that the samples could be classified into two groups, MYC2bOE and CK. A heatmap (Figure 1F) was generated and coupled with hierarchical clustering analysis to further understand the difference in root characteristics among the transgenic and CK plants. The X-axis shows the clustering of the two groups, including MYC2bOE and CK. Root tips, forks, and crossings on the Y-axis were clustered into one group as they were correlated with each other. RootVolume, length, and SurfArea were correlated with each other. Volcano plots are a powerful tool to screen different root traits between two groups and display two important indicators, fold change (FC) and p-value, in one image. The root length, tips, and SurfArea were differential root indexes based on their fold changes and P-values (Figure 1G). The forks and crossings of MYC2bOE were more than 2-fold those of CK. However, the results of the t-test did not show a significant difference due to the excessive intragroup differences.
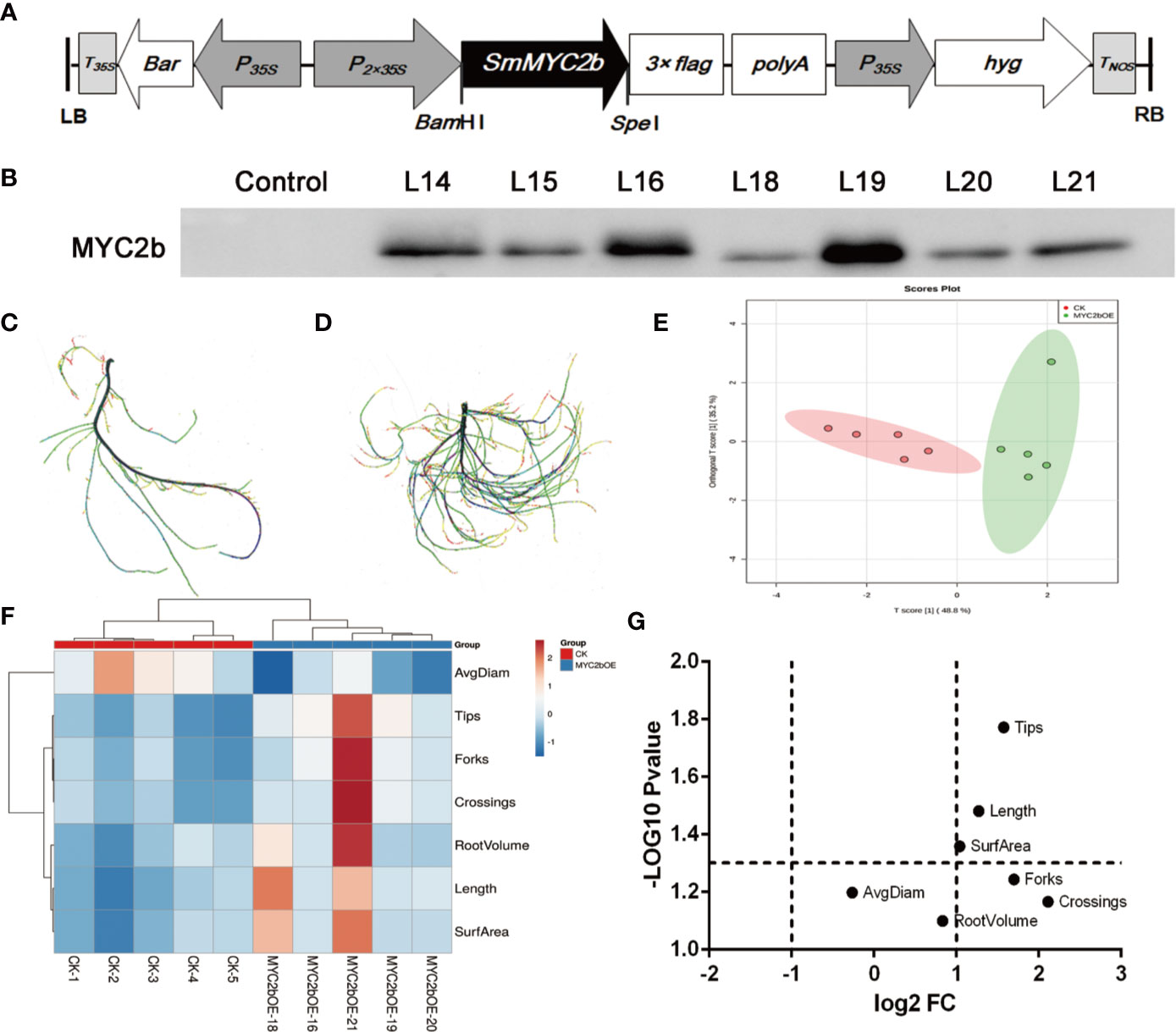
Figure 1 SmMYC2b promotes the development of lateral roots in S. miltiorrhiza. (A) Schematic diagram of the constructed plant overexpression vector pHB-flag-MYC2b. (B) Western blot analysis of the SmMYC2b protein in transgenic S. miltiorrhiza plants. L14, L15, L16, L18, L19, L20, and L21 denote different MYC2b overexpression lines. (C, D) show CK and MYC2bOE sterilized plants, respectively, with different root morphological characteristics. (E) PCA analysis, (F) heatmap, and (G) volcano plot of roots from CK and SmMYC2b transgenic plants. The analysis was performed with five independent replicates.
SmMYC2b Rescues the Phenotype of the A. thaliana MYC2 Mutant
Previous studies indicate that MYC2 positively promoted lateral root formation and inhibited primary root elongation. To investigate whether SmMYC2b exhibited similar functions in roots, transgenic plants were produced. Different genotypes of A. thaliana seeds (Col-0, jin1-9 and SmMYC2b) were harvested, sown, and treated with 100 μM MeJA or not treated (mock group). As shown in Figure 2, the roots with different genotypes or treatments exhibited different phenotypes. Two-way ANOVA with genotype and treatment as factors was performed to examine significant differences between groups. Genotype (wild type versus jin1-9 versus SmMYC2b) and treatment (mock versus MeJA treatment) caused differences in primary root length and lateral root density, respectively (Table S3). Moreover, there was no interaction between the two factors. Overall, JA was discovered to promote the development of lateral roots and reduce the elongation of primary roots. Unlike JA, SmMYC2b dramatically promoted lateral root formation without inhibiting the primary root elongation.
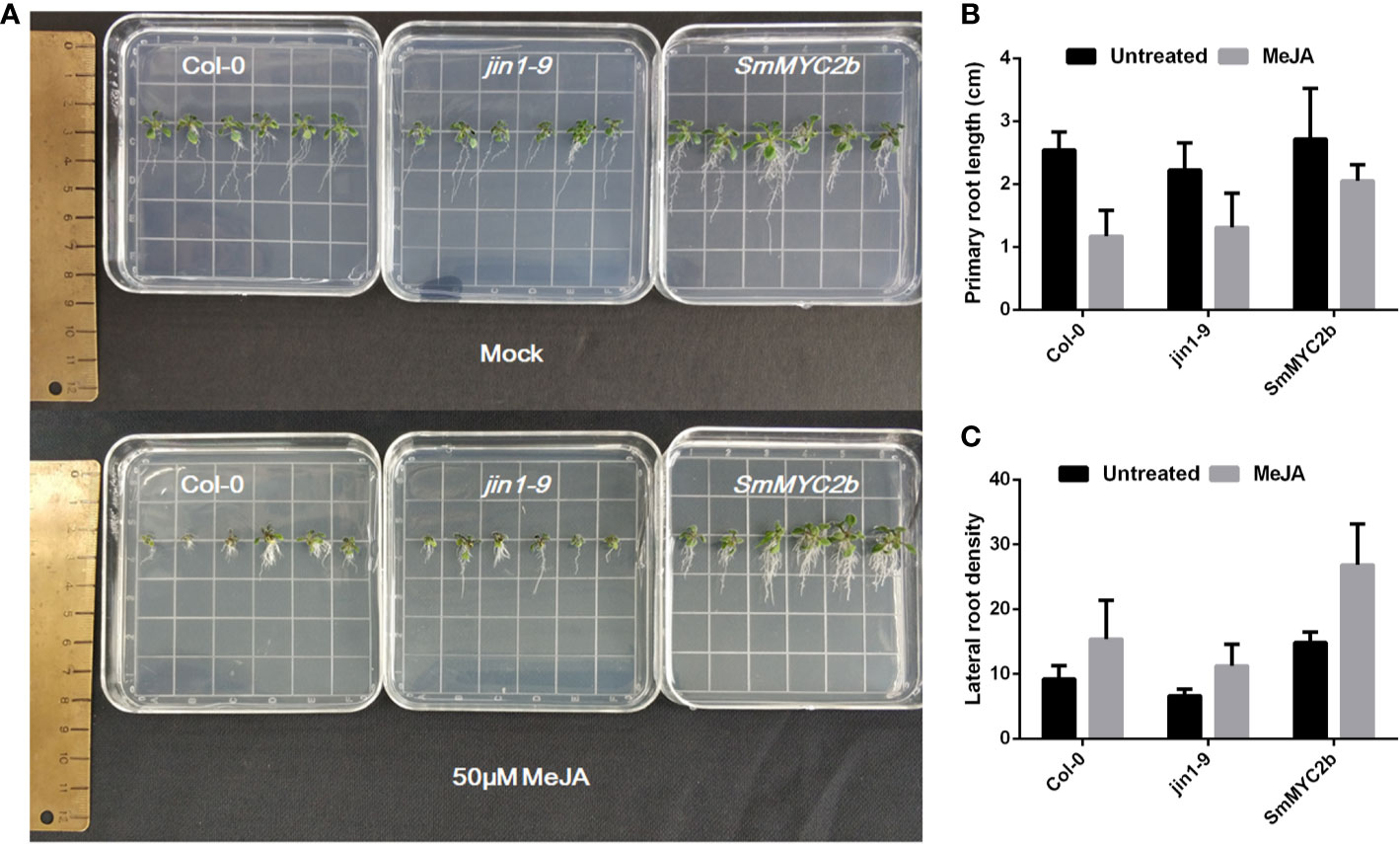
Figure 2 MYC2b facilitates the development of lateral roots without inhibition of primary roots. (A) Photographs of Col-0 (wild type), jin1-9 (MYC2 mutant type), and SmMYC2b (SmMYC2b overexpressed in jin1-9) A. thaliana plants with or without MeJA treatment. (B) Primary root length of different groups in (A); Values are the means with SE (n = 6). (C) Lateral root density of different groups in (A). Lateral root density was given as the number of lateral roots calculated per centimeter of primary root. Values are the means with SE (n = 6).
SmMYC2b Promotes the Biosynthesis of Tanshinones
To investigate the effect of SmMYC2b on phenolic acids and tanshinones, we determined the active compounds of transgenic MYC2bOE and CK plants. Among the six determined MYC2b overexpression lines, three lines that exhibited extremely significant enhancement in tanshinone content were subjected to content analysis. A significant increase in tanshinones (15.74-fold TI and 10.89-fold TT) is shown in Figure 3. The DTI, CT, and TIIA contents respectively increased 5.63-, 8.01-, and 3.13-fold, respectively, in OE compared with CK plants. However, t-test results did not exhibit significant differences owing to excessive intragroup differences. MYC2bOE-19 contained 9.46-fold DTI, 15.40-fold CT, and 8.22-fold TIIA contents compared with those of the CK. However, the other MYC2bOE lines harbored much higher contents. In addition, the content of CA, which was proposed to be an upstream compound of tanshinones according to its molecular structure, dramatically decreased. There was no obvious change in the content of the compounds (Phe, Tyr, CinA, 4-HPPA, DSS, HGA, CafA, RA, SAA, and SAB) in the SAB pathway (Figure S2). ChIP-qPCR assays were conducted to validate the RNA-seq results. SmCPS1 and SmKSL1, which are involved in the tanshinone pathway, were discovered to be SmMYC2b-regulated genes whose promoter sequences were downloaded from the GenBank database with the accession numbers KY937191 and KY937192, respectively (Figure S3).
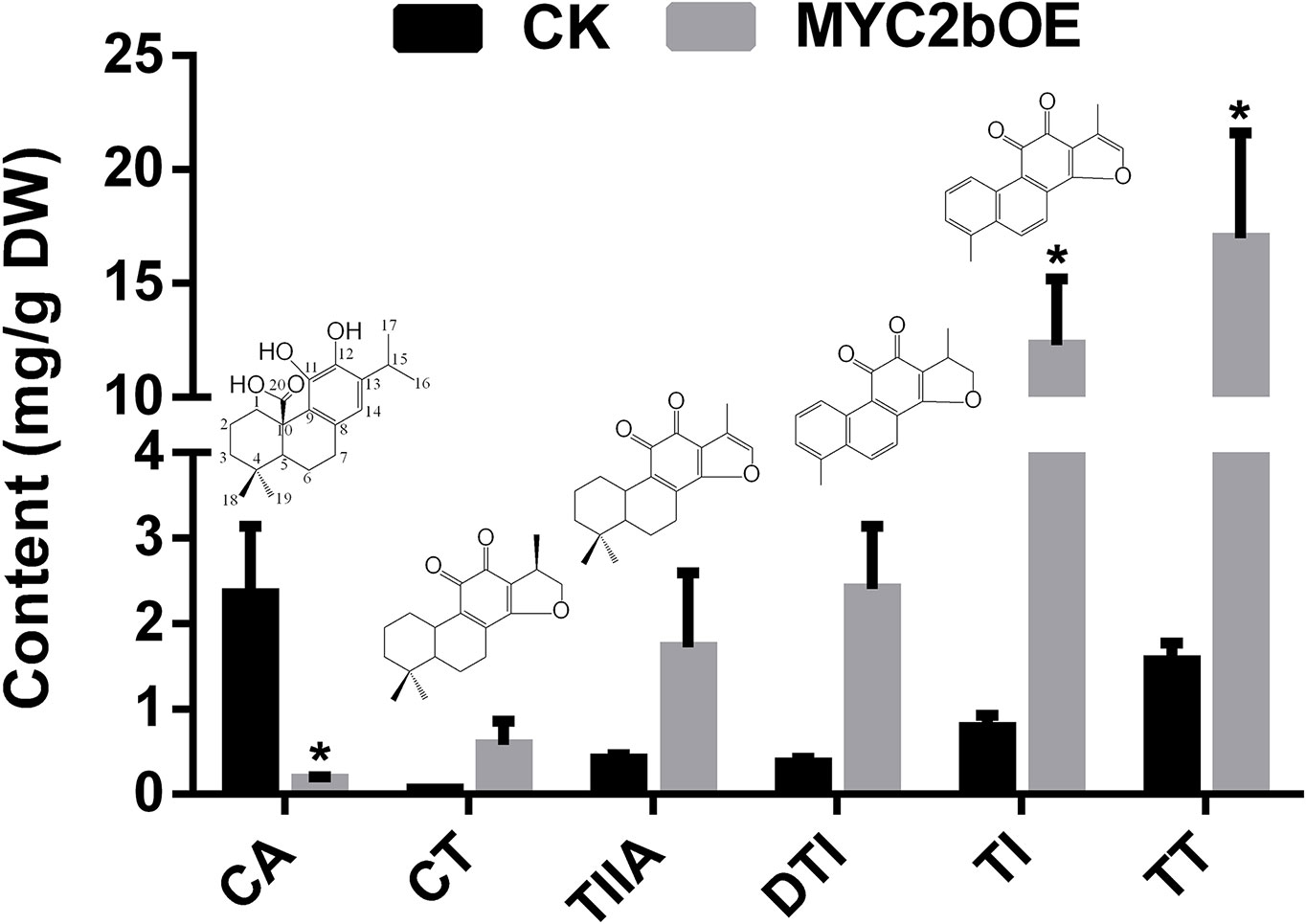
Figure 3 Overexpression of SmMYC2b enhances the biosynthesis of tanshinones. Values are the means with SE (n = 3–4). Asterisks suggest significant differences at P < 0.05; CA, carnosic acid; DTI, dihydrotanshinone I; TI, tanshinone I; CT, cryptotanshinone; TIIA, tanshinone IIA; TT, total tanshinone.
Tanshinones predominantly accumulated in the roots of S. miltiorrhiza, especially in the rhizome. To provide more evidence supporting the physiological roles of SmMYC2b in tanshinone biosynthesis, we examined the expression pattern of SmMYC2b. In general, the expression level of SmMYC2b increased with time (Figure S4).
SmMYC2b Positively Regulates Tanshinone Biosynthesis Genes to Enhance the Biosynthesis of Tanshinones
Because the mechanism by which how MYC2b affects the biosynthesis of tanshinones is unclear, RNA-seq analysis was conducted to mine potential tanshinone biosynthesis genes. The tanshinones were derived from the cytosol-localized mevalonic acid (MVA) pathway and plastid-localized methylerythritol phosphate (MEP) pathway. The expression levels of genes involved in the MVA pathway were almost unchanged (Figure 4). There was no obvious change in upstream genes in the MEP pathway. However, downstream genes in the MEP pathway significantly increased, suggesting that MYC2b mainly targets relative downstream biosynthesis genes to increase the content of tanshinones. SmCPS1 and SmCPS2 were proposed to be involved in distinct tanshinone pathways in the root periderm and aerial tissues respectively. The expression level of SmCPS1 was discovered to be increased while that of SmCPS2 decreased, collectively leading to an increase in the tanshinone content. SmKSL1 was discovered to be quite specific in its production of miltiradiene from CPP. CYP76AH1 was able to efficiently catalyze the conversion of miltiradiene to ferruginol in the tanshinone pathway. CYP76AH3 and CYP76AK1 formed a bifurcating pathway for the biosynthesis of tanshinones. The increase in the expression levels of SmCPS1, SmKSL1, SmCYP76AH1, SmCYP76AH3, and SmCYP76AK1 collectively enhanced tanshinone production. The expression levels of genes in the SAB pathway did not exhibit significant changes, which is consistent with the content determination results (Figure S5).
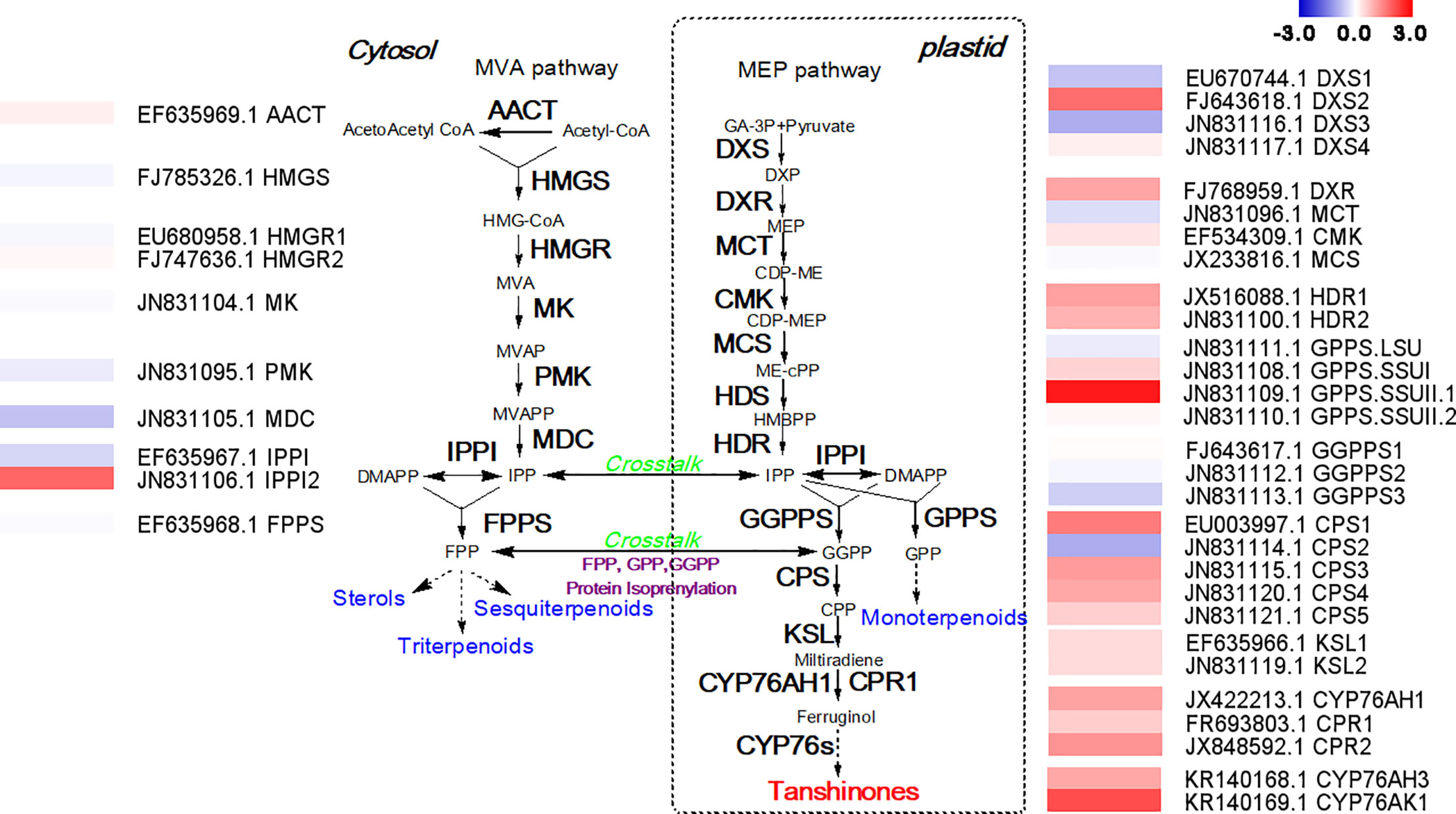
Figure 4 Overexpression of SmMYC2b promotes the expression of tanshinone biosynthetic genes. AACT, acetoacetyl-CoA thiolase; CDP-ME, 4-(cytidine 5’-diphospho)-2-C-methyl- D-erythritol; CDP-MEP, 2-phospho-4-(cytidine 5’-diphospho)-2-C-methyl-D-erythritol; CMK, 4-diphosphocytidyl-2C-methyl-D-erythritol kinase; CPP, copalyl diphosphate; Cprimary root, cytochrome p450 reductase; CPS, copalyl diphosphate synthase; CYP, cytochrome p450; DMAPP, dimethylallyl pyrophosphate; DXP, 1-deoxy-D-xylulose 5-phosphate; DXR, 1-deoxy-D-xylulose 5- phosphate reductoisomerase; DXS, 1-deoxy-D-xylulose-5-phosphate synthase; FPP, farnesyldiphosphate; FPPS, farnesyl diphosphate synthase; GA-3P, glyceraldehyde-3-phosphate; GGPP, geranylgeranyl diphosphate; GGPP, geranylgeranyl diphosphate; GGPPS, geranylgeranyl diphosphate synthase; GPP, geranyl diphosphate; GPPS, geranyl diphosphate synthase; HDR, 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase; HDS, 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate synthase; HMBPP, 4-hydroxy-3-methylbut-2-enyl diphosphate; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; HMGR, 3-hydroxy-3-methylglutaryl-CoA reductase; HMGS, 3-hydroxy-3-methylglutaryl-CoA synthase; IDS, isoprenyl diphosphate synthase; IPP, isopentenyl pyrophosphate; IPPI, isopentenyl diphosphate isomerase; KSL, kaurene synthase-like; MCS, 2C-methyl-D-erythritol 2,4-cyclodiphosphate synthase; MCT, 2C-methyl-D-erythritol 4-phosphate cytidyl transferase; MDC, mevalonate diphosphate decarboxylase; ME-cPP, 2-C-methyl-D-erythritol-2,4-cyclodiphosphate; MEP, 2-C-methyl-d-erythritol 4-phosphate; MK, Mevalonate kinase; MVA, mevalonate; MVAP, mevalonate 5-phosphate; MVAPP, mevalonate 5-diphosphate; PMK, phosphomevalonate kinase. The fold change of gene expression levels measured in RPKM is shown in a log2 scale. Red represents upregulated genes and blue indicates downregulated genes.
Transcriptome Analysis of MYC2b-Regulated Genes During JA Signaling
Based on its efficient and extensive regulatory characteristics, MYC2 is considered a regulatory hub within the JA signaling pathway (Kazan and Manners, 2012; Du et al., 2017). The role of MYC2 orthologues in the regulation of JA-mediated biosynthesis of secondary metabolites has been well established. To investigate the role of MYC2b in the JA pathway and screen MYC2b-regulated genes, we performed RNA-seq analysis. Transcriptome profiles of four groups including CK and MYC2bOE plants with or without MeJA treatment, were compared by a pairwise comparison method to screen MYC2b-regulated genes (MYC2bOE vs CK), JA-regulated genes (MeJA-CK vs CK) and interacting genes (shown in Dataset 1). We identified 6,210 JA-regulated genes, 3,118 MYC2b-regulated genes, and 1,901 interacting genes (Figure 5). GO analysis of JA and MYC2b co-regulated genes was performed and is shown in Figures 5C, D. The co-regulated genes were mainly involved in metabolic processes, cellular processes, single-organism processes, biological regulation, and localization. The majority of these genes exerted binding and catalytic functions. JA-regulated genes and MYC2b-regulated genes exhibited similar gene functions (Figure S6). JA-regulated tanshinone and SAB pathway genes are also displayed in Figure 5E. As the group of JA-regulated genes included MYC2b-regulated genes and non-MYC2b-regulated genes, 4,309 non-MYC2b-regulated genes were discovered. To further narrow MYC2b-regulated genes, we compared DEGs of MeJA-MYC2bOE vs CK plants with MeJA-CK vs CK plants. The considerably upregulated or downregulated genes were designated MYC2b-regulated genes (2,018). The number of interacting genes between the 2,018 MYC2b-regulated genes and the above 3,118 MYC2b-regulated genes was narrowed to 666 (shown in Dataset 2). Sixteen lateral root development genes regulated by SmMYC2b are listed in Table S4.
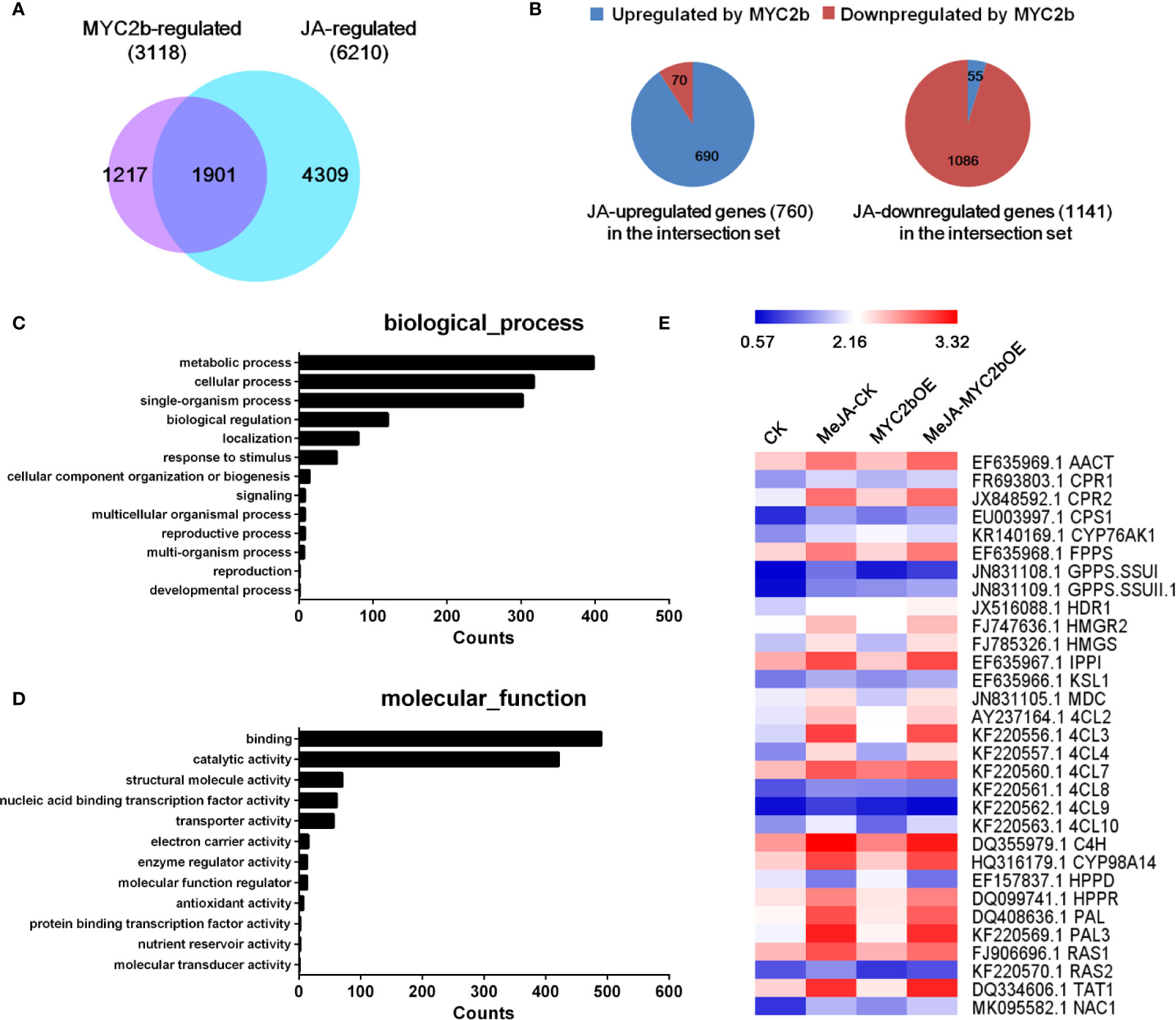
Figure 5 SmMYC2b plays a vital role in the JA pathway. (A) Venn diagram illustrating the overlap of JA-regulated genes and MYC2b-regulated genes. (B) Distribution of MYC2b-upregulated and MYC2b-downregulated genes among the 1901 JA and MYC2b co-regulated genes. (C, D) GO analysis of JA and MYC2b co-regulated genes, respectively. (E) Expression of typical genes regulated by JA. The average fragments per kilobase of exon per million fragments mapped (RPKM, log10 scale) of each gene is shown.
CA Was Presumed to Be an Important Precursor of Tanshinones
Based on the above results, CA was presumed to be a potential precursor of tanshinones. To further explain the relationship between CA and tanshinones, CA feeding studies were carried out. S. miltiorrhiza hairy root cultures were used as materials, which were characterized by a fast growth rate and were genetically stable (Guillon et al., 2006; Miklaszewska et al., 2017). As shown in Figure 6, the color of the liquid medium in which S. miltiorrhiza hairy roots were growing surprisingly turned yellow after treatment for 1 d, faded slowly after 4 d, and returned to transparent after 8 d. At the same time, the content of CA, CT, TIIA, DTI, and TI increased significantly after CA treatment, reached the maximum values at 4 d and then decreasing. On the 4th day, the content of CA, CT, TIIA, DTI, and TI reached 95.50, 4.12, 2.39, 5.90, and 3.26 times those of the control, respectively. The exogenous addition of CA greatly increased the content of CA, CT, TIIA, DTI, and TI, suggesting that CA was probably an important precursor of tanshinone components. More direct evidence should be provided to explain the relationship between CA and tanshinones.
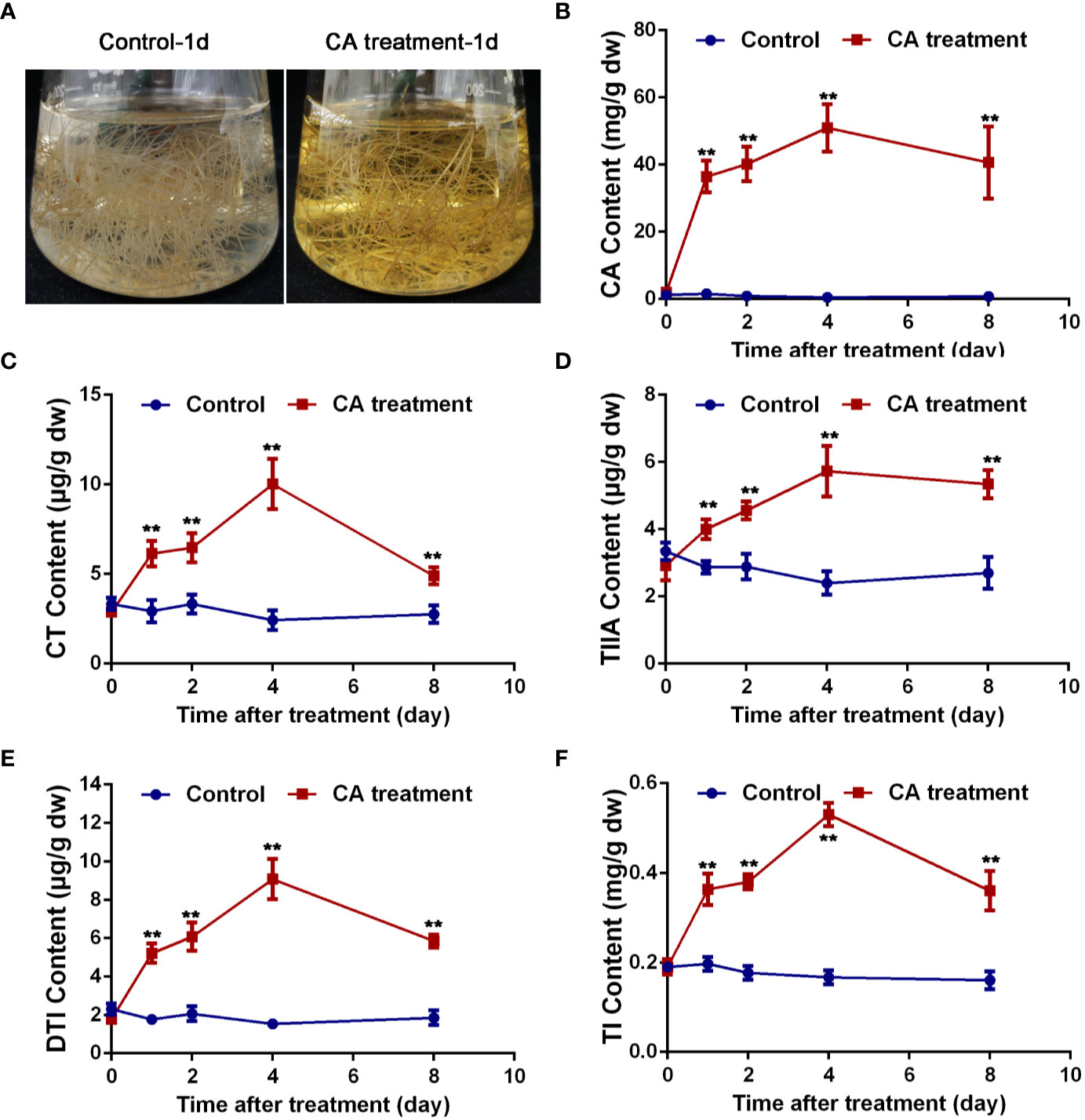
Figure 6 Exogenous CA treatment caused changes in tanshinone accumulation. (A) The hairy root medium treated with CA for 1 d turned yellow. (B–F) exhibited content changes in CA, CT, TIIA, DTI, and TI, respectively. Asterisks denote significant differences at P < 0.05. Values are the means with SE (n = 3).
Discussion
To improve plant survival, plant terpenoids serve as phytoalexins that can effectively resist pest and pathogen attack (Schmelz et al., 2011; Schmelz et al., 2014). Previous reports indicate that the terpenoid contents change during plant development in many plant species (Niu et al., 2015; Aizpurua-Olaizola et al., 2016). Thus, to characterize the secondary metabolism in plants, plant growth and development should be taken into consideration. MYC2 was discovered to be an efficient regulator in the modulation of secondary metabolites. However, there is still limited understanding of the regulatory effects of SmMYC2b on bioactive compounds. To investigate the expression level of SmMYC2b throughout the plant life cycle, we collected S. miltiorrhiza roots at various time points. Overall, the expression level of MYC2b increases with plant aging.
Lateral roots help anchor the plant securely in the soil as well as engage in water and nutrient uptake. In the present study, SmMYC2b promoted lateral root development. This result is consistent with previous findings in other species (Yadav, 2005). A previous investigation demonstrates that JA represses the expression of PLT1 and PLT2 transcription factors to inhibit primary root elongation, and this effect is executed by MYC2 (Shen et al., 2016; Ishimaru et al., 2018). However, SmMYC2b exerts opposite functions in regulating primary root growth. SmMYC2b stimulates root growth by promoting lateral root development and primary root elongation. As tanshinone pigments are mainly distributed in the periderm (Xu et al., 2015), the promotion of root development is accompanied by an increase in tanshinone producing tissues, leading to enhanced accumulation of tanshinones.
The plant hormone JA plays noteworthy roles in multiple plant processes, including plant resistance, growth, and development. MYC2 is known as a vitally important TF involved in most aspects of the JA signaling pathway (Kazan and Manners, 2012; Du et al., 2017). Here, RNA-seq assays revealed that SmMYC2b-regulated genes comprised 30.6% (1,901 of 6,210) of JA-responsive genes. Moreover, the interacting genes (the MYC2b and JA co-regulated genes) comprised 60.9% (1,901 of 3,118) of the MYC2b-regulated genes. As SmMYC2a was formerly designated as a similar TF of SmMYC2b (Zhou et al., 2016), SmMYC2a and SmMYC2b are proposed to exert the most influence on JA function.
The metabolic engineering of key TFs provides a feasible strategy for improving the content of tanshinones and other bioactive compounds (Sun et al., 2019; Deng et al., 2020b; Hao et al., 2020). As MYC2 orthologs are master regulators of JA-mediated secondary metabolite biosynthesis, metabolic engineering using SmMYC2b as candidate can be a promising strategy to achieve high yield of target compounds in S. miltiorrhiza. Previous knockdown of SmMYC2b causes a significant decrease in tanshinone and SAB biosynthesis, indicating that SmMYC2b is a positive regulator performing similar functions as other MYC2 homologs (Dombrecht et al., 2007; Zhang et al., 2011; Shen et al., 2016). More efforts should be made to explain the related complicated regulatory networks. In this study, RNA-seq data were sufficiently analyzed to mine clues supporting the above deduction. SmMYC2b specifically enhances the expression of genes involved in the downstream pathway of tanshinones, including SmCPS1, SmKSL1, SmKSL2, SmCYP76AH1, SmCYP76AH3, and SmCYP76AK1. Among the characterized SmCPS1-5 genes, only SmCPS1 exhibits catalytic activity in tanshinone biosynthesis in the roots (Cui et al., 2015). SmKSL1 was discovered to specifically catalyze the next reaction step in the production of miltiradiene (Gao et al., 2009). Miltiradiene can be effectively converted into ferruginol by SmCYP76AH1 (Guo et al., 2013). SmCYP76AH3 and SmCYP76AK1 both exhibit promiscuity, leading to a diversity of tanshinones (Guo et al., 2016).
Overexpression of SmMYC2b also leads to activation of the tanshinone pathway. Compared with CK, the contents of DTI, TI, CT, and TIIA exhibited different degrees of increase. In contrast, the content of CA dramatically decreased. By far, the tanshinone biosynthetic pathway hasn’t been completely elucidated, extremely the specific downstream pathway. The latest work on S. miltiorrhiza characterized CYP76AH3 which catalyzes ferruginol to 11-hydroxy ferruginol (Guo et al., 2016). In other species including Rosmarinus officinalis, Salvia fruticose, and Salvia pomifera, 11-hydroxyferruginol is converted to CA by CYP76AK6-8 (Ignea et al., 2016; Scheler et al., 2016). In S. miltiorrhiza, whether CYP76AK6-8 homologs are involved in similar processes has not yet been reported. CA belongs to the group of labdane-type diterpene, and is synthesized from geranylgeranyl diphosphate (GGPP) (Ignea et al., 2016; Scheler et al., 2016). Based on the structural characteristics, it is reasonable to deduce that CA can be converted to CT by decarboxylation reaction and cyclization reaction. Afterwards, the metabolic flux is proposed to occur successively from CT, TIIA, DTI, to TI by dehydrogenation reactions. Coincidentally, the contents of CT, TIIA, DTI, and TI were discovered to successively increase, leading to the intensive accumulation of terminal products. However, the content of CA dramatically decreased. As the expression levels of CA upstream pathway genes, including SmCPS1, SmKSL1, SmCYP76AH1, and SmCYP76AH3, consistently increase, CA accumulation should increase, in contrast with the content determination results. It is reasonable to deduce that the decrease in CA content leads to an increase in tanshinones, indicating that CA is a crucial precursor of tanshinones. However, more evidence is required to support this hypothesis.
To further explore the function of CA, CA feeding studies were carried out. After treatment for 4 d, the contents of CA, CT, TIIA, DTI, and TI reached their maximum values. The contents of CA, CT, TIIA, DTI, and TI reached 95.50, 4.12, 2.39, 5.90, and 3.26 times those of the control, respectively. The exogenous addition of CA greatly increased the contents of CA, CT, TIIA, DTI, and TI, suggesting that CA is an essential precursor of tanshinone components that provide raw material for the biosynthesis and regulation of tanshinone components. MYC2b promotes the transformation of the CA precursor to tanshinones to realize extremely high accumulation of end products. Moreover, SmMYC2b mainly modulates the downstream genes of the tanshinone pathway instead of the upstream genes, which are shared with other pathways, such as monoterpene and sesquiterpene pathways. Our previous studies indicate that SmMYC2b is a positive transcription factor that can affect the biosynthesis of phenolic acids and tanshinones in S. miltiorrhiza hairy roots. Phenolic acids and tanshinones are supposed to be increased by overexpression of SmMYC2b. However, there was no significant up-regulating of SAB pathway, suggesting the role of endogenous SmMYC2b is sufficient for the induction of SAB biosynthesis genes (Zhang et al., 2011).
The extensive roles of JA in the regulation of pathway genes involved in bioactive compounds are well established (Wasternack and Hause, 2013). MYC2 is a master regulator that modulates wide range of functions triggered by JA signaling pathway (Kazan and Manners, 2012; Du et al., 2017; Wasternack and Song, 2017). As JA-induced biosynthesis of many secondary metabolites is executed by MYC2 (Zhou and Memelink, 2016), overexpression of SmMYC2b can be a promising method to increase the tanshinone yield in S. miltiorrhiza. In this paper, SmMYC2b is proved to promote two physiological aspects of S. miltiorrhiza roots, tanshinone biosynthesis and lateral roots development, which are both decisive factors of yield and quality of this medicinal material. Thus, SmMYC2 might provide an efficient regulatory target for breeding of S. miltiorrhiza, as well as a basis for exploring the mechanism behind the extensive roles of MYC2 in planta.
Data Availability Statement
The raw RNA-seq read data are publicly available at the NCBI Sequence Read Archive with the accession number PRJNA623221 (http://www.ncbi.nlm.nih.gov/sra/).
Author Contributions
YZ, JC, and WC planned and designed the research. YZ, JF, QL, DH, XC, ZD, JC, ZL, YX performed experiments. YZ, JF, DH, YH, JC, and WC analyzed data. YZ and JC wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
We are grateful to Shujuan Zhao and Ronghui Tan for preparing materials. This work was financially supported by the National Natural Science Foundation of China (81603220, 81673550, 31970325, and 31770329), National Key R&D Program of China (2019YFC1711100) and Shanghai Rising-Star Program (18QB1402700, China).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.559438/full#supplementary-material
References
Abdelaty, S., Raúl, A. V., Zoya, A. (2008). An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in Arabidopsis plants. Nat. Protoc. 3, 1018–1025. doi: 10.1038/nprot.2008.66
Aizpurua-Olaizola, O., Soydaner, U., Öztürk, E., Schibano, D., Simsir, Y., Navarro, P., et al. (2016). Evolution of the cannabinoid and terpene content during the growth of Cannabis sativa plants from different chemotypes. J. Nat. Prod. 79, 324–331. doi: 10.1021/acs.jnatprod.5b00949
Chang, Y., Wang, M., Li, J., Lu, S. (2019). Transcriptomic analysis reveals potential genes involved in tanshinone biosynthesis in Salvia miltiorrhiza. Sci. Rep. 9, 14929. doi: 10.1038/s41598-019-51535-9
Chen, F., Tholl, D., Bohlmann, J., Pichersky, E. (2011). The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229. doi: 10.1111/j.1365-313X.2011.04520.x
Cui, G., Duan, L., Jin, B., Qian, J., Xue, Z., Shen, G., et al. (2015). Functional divergence of diterpene syntheses in the medicinal plant Salvia miltiorrhiza. Plant Physiol. 169, 1607–1618. doi: 10.1104/pp.15.00695
Degenhardt, J., Köllner, T. G., Gershenzon, J. (2009). Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 70, 1621–1637. doi: 10.1016/j.phytochem.2009.07.030
Deng, C., Shi, M., Fu, R., Zhang, Y., Wang, Q., Zhou, Y., et al. (2020a). An ABA-responsive SmbZIP1 is involved in modulating biosynthesis of phenolic acids and tanshinones in Salvia miltiorrhiza. J. Exp. Bot. doi: 10.1093/jxb/eraa295
Deng, C., Wang, Y., Huang, F., Lu, S., Zhao, L., Ma, X., et al. (2020b). SmMYB2 promotes salvianolic acid biosynthesis in the medicinal herb Salvia miltiorrhiza. doi: 10.1111/jipb.12943
Dombrecht, B., Xue, G. P., Sprague, S. J., Kirkegaard, J. A., Ross, J. J., Reid, J. B., et al. (2007). MYC2 differentially modulates diverse jasmonate-dependent functions in Arabidopsis. Plant Cell 19, 2225–2245. doi: 10.1105/tpc.106.048017
Du, M., Zhao, J., Tzeng, D. T., Liu, Y., Deng, L., Yang, T., et al. (2017). MYC2 orchestrates a hierarchical transcriptional cascade that regulates jasmonate-mediated plant immunity in tomato. Plant Cell 29, 1883–1906. doi: 10.1105/tpc.16.00953
Gao, W., Hillwig, M. L., Huang, L., Cui, G., Wang, X., Kong, J., et al. (2009). A functional genomics approach to tanshinone biosynthesis provides stereochemical insights. Org. Lett. 11, 5170–5173. doi: 10.1021/ol902051v
Gong, Y., Li, Y., Lu, Y., Li, L., Abdolmaleky, H., Blackburn, G. L., et al. (2011). Bioactive tanshinones in Salvia miltiorrhiza inhibit the growth of prostate cancer cells in vitro and in mice. Int. J. Cancer 129, 1042–1052. doi: 10.1002/ijc.25678
Gu, X., Chen, J., Xiao, Y., Di, P., Xuan, H., Zhou, X., et al. (2012). Overexpression of allene oxide cyclase promoted tanshinone/phenolic acid production in Salvia miltiorrhiza. Plant Cell Rep. 31, 2247–2259. doi: 10.1007/s00299-012-1334-9
Guillon, S., Trémouillaux-Guiller, J., Pati, P. K., Rideau, M., Gantet, P. (2006). Harnessing the potential of hairy roots: dawn of a new era. Trends Biotechnol. 24, 403–409. doi: 10.1016/j.tibtech.2006.07.002
Guo, J., Zhou, Y. J., Hillwig, M. L., Shen, Y., Yang, L., Wang, Y., et al. (2013). CYP76AH1 catalyzes turnover of miltiradiene in tanshinones biosynthesis and enables heterologous production of ferruginol in yeasts. Proc. Natl. Acad. Sci. U.S.A. 110, 12108–12113. doi: 10.1073/pnas.1218061110
Guo, J., Ma, X., Cai, Y., Ma, Y., Zhan, Z., Zhou, Y. J., et al. (2016). Cytochrome P450 promiscuity leads to a bifurcating biosynthetic pathway for tanshinones. New Phytol. 210, 525–534. doi: 10.1111/nph.13790
Hao, X., Pu, Z., Cao, G., You, D., Zhou, Y., Deng, C., et al. (2020). Tanshinone and salvianolic acid biosynthesis are regulated by SmMYB98 in Salvia miltiorrhiza hairy roots. J. Adv. Res. 23, 1–12. doi: 10.1016/j.jare.2020.01.012
Ignea, C., Athanasakoglou, A., Ioannou, E., Georgantea, P., Trikka, F. A., Loupassaki, S., et al. (2016). Carnosic acid biosynthesis elucidated by a synthetic biology platform. Proc. Natl. Acad. Sci. U.S.A. 113, 3681–3686. doi: 10.1073/pnas.1523787113
Ishimaru, Y., Hayashi, K., Suzuki, T., Fukaki, H., Prusinska, J., Meesters, C., et al. (2018). Jasmonic acid inhibits auxin-induced lateral rooting independently of the CORONATINE INSENSITIVE 1 receptor. Plant Physiol. 177, 1704–1716. doi: 10.1104/pp.18.00357
Kazan, K., Manners, J. M. (2012). MYC2: the master in action. Mol. Plant 6, 686–703. doi: 10.1093/mp/sss128
Keeling, C. I., Weisshaar, S., Lin, R. P., Bohlmann, J. (2008). Functional plasticity of paralogous diterpene synthases involved in conifer defense. Proc. Natl. Acad. Sci. U.S.A. 105, 1085–1090. doi: 10.1073/pnas.0709466105
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Liang, P., Ko, T., Wang, A. H. (2002). Structure, mechanism and function of prenyltransferases. Eur. J. Biochem. 269, 3339–3354. doi: 10.1046/j.1432-1033.2002.03014.x
Lin, T., Hsieh, C. (2010). Pharmacological effects of Salvia miltiorrhiza (Danshen) on cerebral infarction. Chin. Med. 5:22. doi: 10.1186/1749-8546-5-22
Miklaszewska, M., Banaś, A., Królicka, A. (2017). Metabolic engineering of fatty alcohol production in transgenic hairy roots of Crambe abyssinica. Biotechnol. Bioeng. 114, 1275–1282. doi: 10.1002/bit.26234
Nelson, J. D., Denisenko, O., Bomsztyk, K. (2006). Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 1, 179–185. doi: 10.1038/nprot.2006.27
Niu, J., Chen, Y., An, J., Hou, X., Cai, J., Wang, J., et al. (2015). Integrated transcriptome sequencing and dynamic analysis reveal carbon source partitioning between terpenoid and oil accumulation in developing Lindera glauca fruits. Sci. Rep. 5, 15017. doi: 10.1038/srep15017
Scheler, U., Brandt, W., Porzel, A., Rothe, K., Manzano, D., Božić, D., et al. (2016). Elucidation of the biosynthesis of carnosic acid and its reconstitution in yeast. Nat. Commun. 7, 12942. doi: 10.1038/ncomms12942
Schmelz, E. A., Kaplan, F., Huffaker, A., Dafoe, N. J., Vaughan, M. M., Ni, X., et al. (2011). Identity, regulation, and activity of inducible diterpenoid phytoalexins in maize. Proc. Natl. Acad. Sci. U. S. A. 108, 5455–5460. doi: 10.1073/pnas.1014714108
Schmelz, E. A., Huffaker, A., Sims, J. W., Christensen, S. A., Lu, X., Okada, K., et al. (2014). Biosynthesis, elicitation and roles of monocot terpenoid phytoalexins. Plant J. 79, 659–678. doi: 10.1111/tpj.12436
Shen, Q., Lu, X., Yan, T., Fu, X., Lv, Z., Zhang, F., et al. (2016). The jasmonate-responsive AaMYC2 transcription factor positively regulates artemisinin biosynthesis in Artemisia annua. New Phytol. 210, 1269–1281. doi: 10.1111/nph.13874
Shi, M., Luo, X., Ju, G., Li, L., Huang, S., Zhang, T., et al. (2016). Enhanced diterpene tanshinone accumulation and bioactivity of transgenic Salvia miltiorrhiza hairy roots by pathway engineering. J. Agric. Food Chem. 64, 2523–2530. doi: 10.1021/acs.jafc.5b04697
Shi, M., Gong, H., Cui, L., Wang, Q., Wang, C., Wang, Y., et al. (2020). Targeted metabolic engineering of committed steps improves anti-cancer drug camptothecin production in Ophiorrhiza pumila hairy roots. Ind. Crops Prod. 148, 112277. doi: 10.1016/j.indcrop.2020.112277
Singh, N. D., Kumar, S., Daniell, H. (2015). Expression of β-glucosidase increases trichome density and artemisinin content in transgenic Artemisia annua plants. Plant Biotechnol. J. 14, 1034–1045. doi: 10.1111/pbi.12476
Su, C., Ming, Q., Khalid, R., Han, T., Qin, L. (2015). Salvia miltiorrhiza: Traditional medicinal uses, chemistry, and pharmacology. Chin. J. Nat. Med. 13, 163–182. doi: 10.1016/S1875-5364(15)30002-9
Sun, J., Xu, Y., Ye, S., Jiang, H., Chen, Q., Liu, F., et al. (2009). Arabidopsis ASA1 is important for jasmonate-mediated regulation of auxin biosynthesis and transport during lateral root formation. Plant Cell 21, 1495–1511. doi: 10.1105/tpc.108.064303
Sun, M., Shi, M., Wang, Y., Huang, Q., Yuan, T., Wang, Q., et al. (2019). The biosynthesis of phenolic acids is positively regulated by the JA-responsive transcription factor ERF115 in Salvia miltiorrhiza. J. Exp. Bot. 70, 243–254. doi: 10.1093/jxb/ery349
Szkopińska, A., Płochocka, D. (2005). Farnesyl diphosphate synthase; regulation of product specificity. Acta Biochim. Pol. 52, 45–55. doi: 10.18388/abp.2005_3485
Tan, H., Xiao, L., Gao, S., Li, Q., Chen, J., Xiao, Y., et al. (2015). TRICHOME ARTEMISININ REGULATOR 1 is required for trichome development and artemisinin biosynthesis in Artemisia annua. Mol. Plant 8, 1396–1411. doi: 10.1016/j.molp.2015.04.002
Vranová, E., Coman, D., Gruissem, W. (2013). Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 64, 665–700. doi: 10.1146/annurev-arplant-050312-120116
Wang, L., Feng, Z., Wang, X., Wang, X., Zhang, X. (2010). DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138. doi: 10.1093/bioinformatics/btp612
Wasternack, C., Hause, B. (2013). Jasmonates: biosynthesis, perception, signal transduction and action in plant stress response, growth and development. An update to the 2007 review in Annals of Botany. Ann. Bot. 111, 1021–1058. doi: 10.1093/aob/mct067
Wasternack, C., Song, S. (2017). Jasmonates: biosynthesis, metabolism, and signaling by proteins activating and repressing transcription. J. Exp. Bot. 68, 1303–1321. doi: 10.1093/jxb/erw443
Xu, Z., Peters, R. J., Weirather, J., Luo, H., Liao, B., Zhang, X., et al. (2015). Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 82, 951–961. doi: 10.1111/tpj.12865
Xu, H., Song, J., Luo, H., Zhang, Y., Li, Q., Zhu, Y., et al. (2016a). Analysis of the genome sequence of the medicinal plant Salvia miltiorrhiza. Mol. Plant 9, 949–952. doi: 10.1016/j.molp.2016.03.010
Xu, Z., Luo, H., Ji, A., Zhang, X., Song, J., Chen, S. (2016b). Global identification of the full-length transcripts and alternative splicing related to phenolic acid biosynthetic genes in Salvia miltiorrhiza. Fron. Plant Sci. 7, 100. doi: 10.3389/fpls.2016.00100
Xue, L., He, Z., Bi, X., Xu, W., Wei, T., Wu, S., et al. (2019). Transcriptomic profiling reveals MEP pathway contributing to ginsenoside biosynthesis in Panax ginseng. BMC Genomics 20, 383. doi: 10.1186/s12864-019-5718-x
Yadav, V. (2005). A basic helix-loop-helix transcription factor in Arabidopsis, MYC2, acts as a repressor of blue light-mediated photomorphogenic growth. Plant Cell 17, 1953–1966. doi: 10.1105/tpc.105.032060
Yan, Y., Wang, Z. (2007). Genetic transformation of the medicinal plant Salvia miltiorrhiza by Agrobacterium tumefaciens-mediated method. Plant Cell Tiss. Org. 88, 175–184. doi: 10.1007/s11240-006-9187-y
Young, M. D., Wakefield, M. J., Smyth, G. K., Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14. doi: 10.1186/gb-2010-11-2-r14
Zhang, H., Bokowiec, M. T., Rushton, P. J., Han, S., Timko, M. P. (2011). Tobacco transcription factors NtMYC2a and NtMYC2b form nuclear complexes with the NtJAZ1 repressor and regulate multiple jasmonate-inducible steps in nicotine biosynthesis. Mol. Plant 5, 73–84. doi: 10.1093/mp/ssr056
Zhang, Y., Butelli, E., Alseekh, S., Tohge, T., Rallapalli, G., Luo, J., et al. (2015). Multi-level engineering facilitates the production of phenylpropanoid compounds in tomato. Nat. Commun. 6, 219–246. doi: 10.1038/ncomms9635
Zhang, P., Du, H., Wang, J., Pu, Y., Yang, C., Yan, R., et al. (2020). Multiplex CRISPR/Cas9-mediated metabolic engineering increases soya bean isoflavone content and resistance to soya bean mosaic virus. Plant Biotechnol. J. 18, 1384–1395. doi: 10.1111/pbi.13302
Zhao, L., Chang, W., Xiao, Y., Liu, H., Liu, P. (2013). Methylerythritol phosphate pathway of isoprenoid biosynthesis. Annu. Rev. Biochem. 82, 497–530. doi: 10.1146/annurev-biochem-052010-100934
Zhou, M., Memelink, J. (2016). Jasmonate-responsive transcription factors regulating plant secondary metabolism. Biotechnol. Adv. 34, 441–449. doi: 10.1016/j.biotechadv.2016.02.004
Zhou, Y., Sun, W., Chen, J., Tan, H., Xiao, Y., Li, Q., et al. (2016). SmMYC2a and SmMYC2b played similar but irreplacable roles in regulating the biosynthesis of tanshinones and phenolic acids in Salvia miltiorrhiza. Sci. Rep. 6, 22852. doi: 10.1038/srep22852
Zhu, Z., Zhang, H., Zhao, L., Dong, X., Li, X., Chai, Y., et al. (2007). Rapid separation and identification of phenolic and diterpenoid constituents from radix Salvia miltiorrhiza by high-performance liquid chromatography diode-array detection, electrospray ionization time-of-flight mass spectrometry and electrospray ionization quadrupole ion trap mass spectrometry. Rapid Commu. Mass Sp. 21, 1855–1865. doi: 10.1002/rcm.3023
Keywords: Salvia miltiorrhiza, SmMYC2b, tanshinone biosynthesis, lateral root development, transcriptome
Citation: Zhou Y, Feng J, Li Q, Huang D, Chen X, Du Z, Lv Z, Xiao Y, Han Y, Chen J and Chen W (2020) SmMYC2b Enhances Tanshinone Accumulation in Salvia miltiorrhiza by Activating Pathway Genes and Promoting Lateral Root Development. Front. Plant Sci. 11:559438. doi: 10.3389/fpls.2020.559438
Received: 06 May 2020; Accepted: 27 August 2020;
Published: 11 September 2020.
Edited by:
Goetz Hensel, Heinrich Heine University Düsseldorf, GermanyReviewed by:
Guoyin Kai, Zhejiang Chinese Medical University, ChinaDongfeng Yang, Zhejiang Sci-Tech University, China
Copyright © 2020 Zhou, Feng, Li, Huang, Chen, Du, Lv, Xiao, Han, Chen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junfeng Chen, MTIzNDc4MzFAcXEuY29t; Wansheng Chen, Y2hlbndhbnNoZW5nQHNtbXUuZWR1LmNu
†These authors have contributed equally to this work