 Wei Chen1†
Wei Chen1† Yan Li1†Shouhong Zhu1Shengtao Fang1Lanjie Zhao1Yan Guo1Junyi Wang1Li Yuan1
Yan Li1†Shouhong Zhu1Shengtao Fang1Lanjie Zhao1Yan Guo1Junyi Wang1Li Yuan1 Youjun Lu2
Youjun Lu2 Fang Liu1
Fang Liu1 Jinbo Yao1*Yongshan Zhang1*
Jinbo Yao1*Yongshan Zhang1*- 1State Key Laboratory of Cotton Biology, Institute of Cotton Research, Chinese Academy of Agricultural Sciences, Anyang, China
- 2School of Biological Science and Food Engineering, Anyang Institute of Technology, Anyang, China
Cotton (Gossypium) seed fibers can be divided into lint (long) or fuzz (very short). Using fiberless (fuzzless-lintless) mutants, the lint initiation gene Li3 was identified by map-based cloning. The gene is an R2R3-MYB transcription factor located on chromosome D12 (GhMML3_D12). Sequence analysis revealed that li3 is a loss-of-function allele containing a retrotransposon insertion in the second exon that completely blocks the gene’s expression. The genetic loci n2 and n3 underlying the recessive fuzzless phenotype in Gossypium hirsutum were also mapped. The genomic location of n3 overlapped with that of the dominant fuzzless locus N1, and n3 appeared to be a loss-of-function allele caused by a single nucleotide polymorphism (SNP) mutation in the coding region of GhMML3_A12. The n2 allele was found to be co-located with li3 and originated from G. babardense. n2 and li3 are possibly the multiple alleles of the GhMML3_D12 gene. Genetic analysis showed that Li3 and N3 are a pair of homologs with additive effects for the initiation of fibers (fuzz or lint). In addition, the presence of another locus was speculated, and it appeared to show an inhibitory effect on the expression of GhMML3. These findings provide new information about the genetic factors affecting the initiation of fibers in cotton.
Introduction
The cotton genus (Gossypium) includes approximately 50 species, and the diploid (n = 13) species can be divided into eight diploid genome groups (A–G and K; Wendel and Cronn, 2003). Allotetraploid cotton resulted from the merger of two formerly isolated diploid genomes (A- and D-genome ancestors; Wendel and Cronn, 2003). Nearly all cotton species have epidermal seed trichomes (fibers), though in some D-genome species, the fibers are relatively sparse. Seed fibers are usually divided into lint and fuzz according to mature fiber length. Lint fibers are spinnable (generally <25 mm) and are initiated −1 to 0 days post-anthesis (DPA), while fuzz fibers are much shorter (<5 mm) and are initiated 3–5 DPA (Lang, 1938; Stewart, 1975).
Upland cotton (Gossypium hirsutum, AD1) is the most widely cultivated allotetraploid species. Many “naked seed” (fuzzless-linted) mutants have been identified in G. hirsutum (Gh) cultivars, and these can be divided into dominant and recessive types based on genetic analyses (Kearney and Harrison, 1927; Carver, 1929; Ware, 1929; Ware et al., 1947). A dominant allele N1 and a recessive allele n2 have been proposed to be responsible for the fuzzless phenotype in some mutant lines (Endrizzi et al., 1985). Recently, two new recessive loci (n3 and n4t) were identified, and an N3 allele was thought to have epistatic effects on the expression of the n2 locus (Turley and Kloth, 2002; Bechere et al., 2012). In addition, the N1 and n2 alleles were found to significantly reduce lint percentage (Turley and Kloth, 2002, 2008). All Gossypium barbadense (Gb; another cultivated allotetraploid species, AD2) lines display the recessive fuzzless phenotype, although the amount of fuzz is affected by environmental factors (Kearney and Harrison, 1928).
Various fiberless (fuzzless-lintless) mutant lines have also been reported in Gh. These include Line L40 (Musaev and Abzalov, 1972), Mcu5 (Nadarajan and Rangasamy, 1988), Xu142 fl (Zhang and Pan, 1991), and SL1-7-1 (Turley and Ferguson, 1996). However, the genotypes of these lines and the relationships among them remained to be clearly elucidated (Turley and Kloth, 2008). For example, three genotypes have been proposed for Xu142 fl: a three-locus type (n1n1n2n2li3li3) proposed by Zhang and Pan (1991), a four-locus type (n1n1n2n2li3li3li4li4) proposed by Du et al. (2001), and another four-locus type (n1n1n2n2li3li3n3n3) proposed by Turley and Kloth (2008). Xu142 fl was thought to differ from the original wild-type Xu142 (n1n1N2N2li3li3) at the N2 locus (Zhang and Pan, 1991). Interestingly, a fiberless line, MD17 (N1N1n2n2n3n3), was developed from the cross between dominant (N1N1N2N2n3n3) and recessive (n1n1n2n2n3n3) naked seed lines, meaning that the interaction of fuzzless loci (N1, n2, and n3) could also produce the fiberless phenotype (Turley, 2002). Besides these major loci, there should be other genes modifying the amount of fuzz or lint on the seed, because considerable variation in the fuzzless/fiberless phenotype has been observed in many uncharacterized fiber mutants (Turley and Kloth, 2008).
The N1 and the n2 loci were mapped to chromosomes 12 (A12) and 26 (D12), respectively (Endrizzi and Ramsay, 1980; Samora et al., 1994). However, in a later study, both loci were genetically mapped to A12 by molecular markers (Rong et al., 2005). Recently, the N1 gene was identified by map-based cloning and found to be an MYBMIXTA-like (MML) transcription factor 3 on chromosome A12 (GhMML3_A12; Wan et al., 2016). MML3_A12 promotes the initiation of fibers, but its expression is suppressed by a natural antisense transcript (NAT) in dominant naked seed mutants. MML3 was also named GhMYB25-like in an earlier study, and its reduced expression was also found in Xu142 fl (Walford et al., 2011). Suppression of GhMYB25-like resulted in fiberless seeds (Walford et al., 2011). Another MML transcription factor, GhMML4_D12, was thought to be the li3 locus responsible for the initiation of lint fibers in Xu142 fl (Wu et al., 2018). Interestingly, MML3 and MML4 are coterminous on chromosomes A12 and D12. A recent study proposed that MML3_D12 also is a major contributing locus for the recessive fuzzless trait of Gb (Zhu et al., 2018). Interestingly, besides these protein-coding genes, some long noncoding RNAs were found to be associated with fiber initiation in the comparison of the transcriptomic differences between Xu142 fl and normal lines (Wang et al., 2015; Hu et al., 2018). Huang et al. (2013) identified an R2R3-MYB gene GbMYB2 involved in fiber initiation and elongation, and this gene also showed a higher expression level in Xu142 compared with Xu142 fl. Another R2R3-MYB gene, GhMYB212, was also found to regulate fiber initiation and elongation (Sun et al., 2019). In Gossypium arboreum (A2-genome), many fuzzless mutants have been found, and they share a common dominant repressor that has been speculated to be the GLABRA2-interacting repressor (GIR1) gene on chromosome 8 (Du et al., 2018; Feng et al., 2019). Recently, a homeodomain-leucine zipper gene GaHD-1 on Chr06 was found to be the likely causative gene for the lintless trait in G. arboreum (Ding et al., 2020; Liu et al., 2020).
In this study, the li3 locus of Xu142 fl was identified by map-based cloning, and the possible variations of DNA sequences in this locus likely responsible for the lintless phenotype were detected. Primary mapping of n2 and n3 loci in n2 was conducted, and a single nucleotide polymorphism (SNP) potentially underlying the n3 mutation was also revealed. Based on the genotyping of F2 plants derived from the cross TM-1 × Xu142 fl, a new genotype was proposed for the fiberless phenotype of Xu142 fl.
Materials and Methods
Plant Materials
An F2 population derived from Xu142 fl × n2 was used for segregation analysis and mapping of the Li3 locus. A BC1 mapping population was also developed for fine mapping. Xu142 fl and n2 are naturally occurring fiberless and fuzzless-linted mutants, respectively (Figure 1). Xu142 fl was found in a commercial Gh variety Xu142 (with normal fuzz and lint) in 1987. For mapping of the N2 and N3 loci, an F2 population derived from n2 × TM-1 was used. TM-1 is a standard G. hirsutum line with a normal seed phenotype (Kohel et al., 1970). For mapping of the fuzzless loci of Gb, an F2 population derived from TM-1 × Hai7124 was used. An F2 population derived from TM-1 × Xu142 fl was used for the segregation analysis of the Li3 and N3 loci. In addition, 6 fiberless, 25 dominant fuzzless, and 26 recessive fuzzless lines were used for genotyping of Li3 and N3 loci (Supplementary Table S1). For association analysis of Li3 and N3 loci, 387 Gh accessions with normal fiber phenotype were used (including Xu142). All cotton plants were grown in the field at the Experimental Station of the Cotton Research Institute of the Chinese Academy of Agricultural Sciences (Anyang, China). Seed phenotypes were scored based on visual inspection at maturity of seeds, and at least five bolls in a single plant were investigated for each material. Lint percentage data were collected and evaluated following the conventional cotton breeding methods using at least 1,000 seeds and the formula (weight of ginned fibers/weight of seeds before ginning).

Figure 1. Fuzz and lint phenotypes of TM-1[wild type (WT)] and representative fiber mutants. For linted materials, the left panel show seeds with lint (before ginning); the right panel show seeds with fuzz (after ginning). Very sparse lint could be found for some fiberless (lintless-fuzzless) mutants.
Bulk Segregant Analysis and Mapping-by-Sequencing
For bulk construction, genomic DNA was isolated from young leaves of the 30 selected plants (assumed to have homozygous recessive genotype) and bulked in an equal ratio to form the pool for each segregating population. Pools and DNA of corresponding parents were subjected to sequencing on the Illumina HiSeq 2,500 platform with 100-bp paired-end reads generated. Identification of SNPs was performed according to the procedure described by Zhu et al. (2017). Briefly, after quality check using FastQC, the clean reads were aligned to the TM-1 reference genome (NAU-NBI Assembly v1.1) using BWA software (Li et al., 2009). The GATK Toolkit was used for calling of SNPs (McKenna et al., 2010), and the SNP filtering was performed according to the methods reported by Reumers et al. (2012). Minimum read coverage of SNPs was set to 5 and 10 for parent and pool, respectively. The sequencing depth and coverage for each mapping population are summarized in Supplementary Table S2. Association mapping was conducted using the method proposed by Abe et al. (2012) and Zhu et al. (2018). Only homozygous SNPs between the parents of the population were used for association mapping. For each SNP site, the SNP index was calculated as follows: SNP index = R/T, where R is the depth of reads of recessive parental types in the pool, and T is the total depth of reads in the pool. For the SNP responsible for the recessive genotype or its closely linked SNPs, the SNP indices should be or close to 1. Average SNP indices were calculated using a sliding-window-based approach (1.0 Mb window with 100 kb increments) and plotted for each chromosome. If there are less than five SNPs in a given 1.0 Mb window, the average SNP index value for this window will be ignored.
Genotyping and Linkage Analysis
The SNPs in the association region of Li3 were converted to PCR-based markers using the modified allele-specific PCR method (Hayashi et al., 2006; Chen et al., 2015), and these markers were used to genotype the plants from the F2 population of Xu142 fl × n2. The linkage map was constructed with MapMaker v 3.0 (Lander et al., 1987). Recombination frequency was transformed into the genetic distance (centimorgans, cM) using Kosambi’s mapping function (Kosambi, 1944).
Sequence Comparison
The candidate gene was amplified from the genomic DNA of the TM-1 and other materials using the primers described in Supplementary Table S3. In addition, the full-length coding fragments of MML3 were also amplified from the −1 DPA-ovule cDNA of TM-1 and Xu142 fl. PrimeSTAR GXL high fidelity DNA polymerase (Takara, Dalian, China) was used for PCR. The PCR products were subcloned into a pEASY-Blunt cloning vector (Transgen, Beijing, China), and no fewer than eight clones were sequenced for each PCR product. The cloned sequences were assigned to the At- or Dt-subgenome of the reference genome sequence (Yuan et al., 2015; Zhang et al., 2015). Sequence alignment was performed using ClustalW2.1
Gene Expression Analysis
The expression levels of MML3_D12 and MML4_D12 were analyzed by quantitative real-time PCR (qRT-PCR) in ABI Prism 7,500 system according to the manufacturer’s protocol. Developing ovules at −3, −1, 0, 1, 3, and 5 DPA were harvested and immediately frozen in liquid nitrogen. Total RNA was isolated from the whole ovules using the RNAprep Pure Plant Kit (Tiangen, Beijing, China). The total RNA was reverse transcribed to cDNA using a PrimeScript® RT reagent kit with a gDNA Eraser (Takara, Dalian, China). Quantitative PCR (qPCR) was performed using SYBR® Premix Ex Taq™ (Tli RNaseH Plus; Takara, Dalian, China). Cotton ACTIN14 (GenBank accession number: AY305733) was used as an internal control. Relative expression levels were determined by the ∆Ct method. For each set of materials, three biological and two technical replicates were used. RT-PCR primers are listed in Supplementary Table S3. PCR efficiency of these primers was determined by LinRegPCR (Ruijter et al., 2009). All qPCR primer pairs had a similar amplification efficiency (91.7–97.3%).
Results
Map-Based Cloning of li3
Previous studies concerning the inheritance of the fuzzless/fiberless seed phenotypes showed that Xu142 fl differed from n2 only at the Li3 locus (Turley and Kloth, 2008). Thus, the cross Xu142 fl × n2 was made to generate a segregating population for the mapping of the Li3 locus. Seeds of F1 plants were fuzzless but linted, and segregation of linted and lintless seed phenotypes in the F2 and BC1 populations both fit the expected ratios (3:1 in F2; 1:1 in BC1; Supplementary Table S4). The DNA pool was generated by bulking 30 lintless individuals in the F2 population, and these were subsequently subjected to high-throughput sequencing. A single unique genomic region was found on chromosome D12 (GhMML3_D12; Figure 2A). Linkage analysis using two flanking SNP markers showed that the li3 locus was located in the region of 44.06–48.97 Mb (Figure 2B). Further fine-mapping using 1,805 F2 and 2,967 BC1 plants narrowed down the li3 locus to a ~270 kb region that contained 16 predicted open reading frames (ORFs; Figure 2C, Supplementary Table S5). Out of these ORFs, two genes coding AtMYB16/AmMIXTA orthologs (GhMML3_D12 and GhMML4_D12) appear to be the candidates for Li3, as they are the only genes shown to be preferentially expressed in ovules during the fiber initiation stage (−3 to 5 DPA) in the previous analyses (Bedon et al., 2014; Zhang et al., 2015; Wu et al., 2018; Supplementary Table S6). However, for GhMML4_D12, sequence alignment did not find any likely causal variation for the li3 mutation (please see detailed description in the later section “li3 and n2 might be the multiple alleles of GhMML3_D12”).
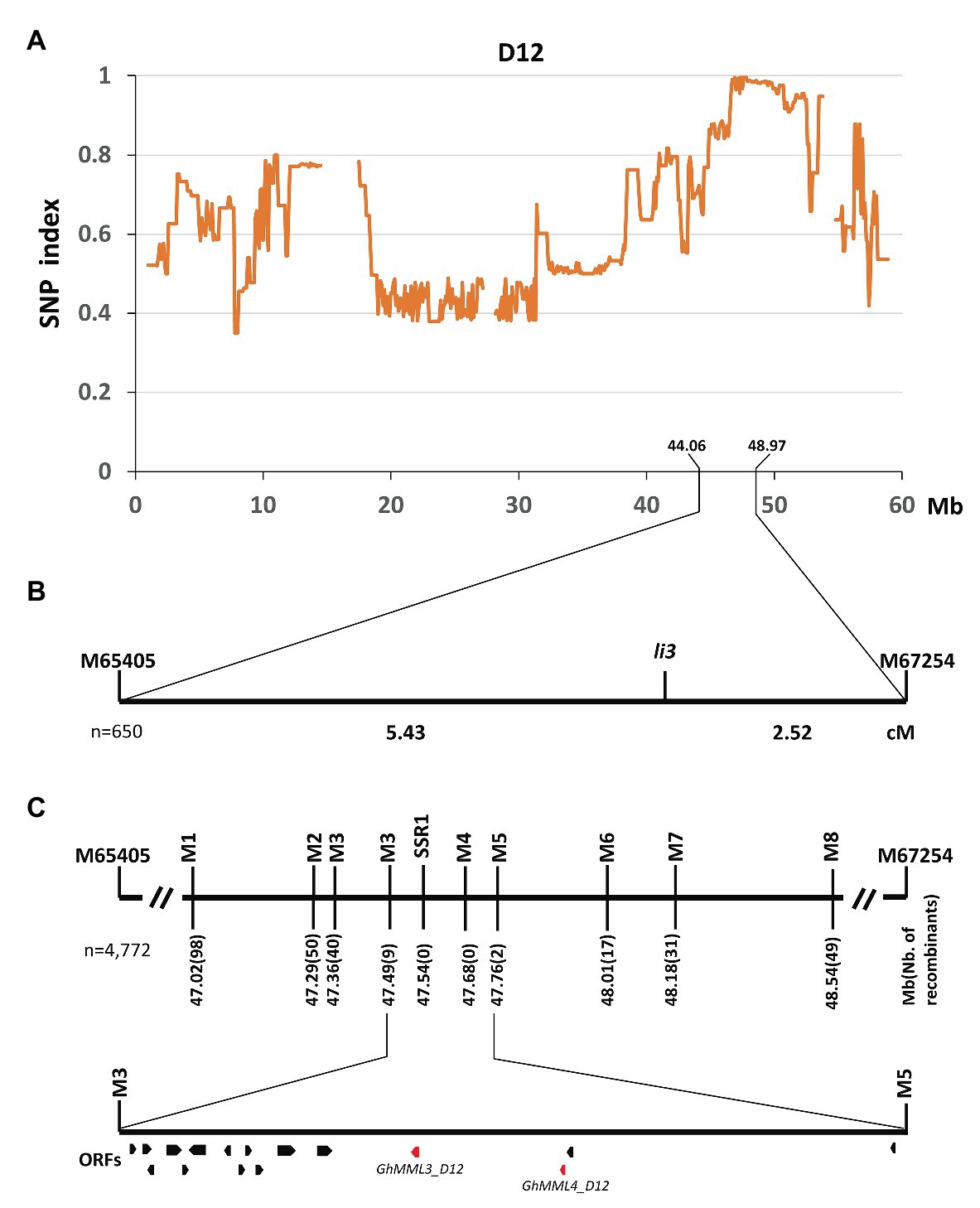
Figure 2. Fine mapping of the Li3 gene. (A) The SNP index plot on chromosome D12 (GhMML3_D12). Averaged SNP index values in a moving window of 1.0 Mb with 100 kb increments were calculated and plotted. (B) Primary mapping of Li3. cM, centiMorgan. (C) Fine mapping of Li3. Open reading frames (ORFs) are indicated by boxes.
When amplified by subgenome-specific primers for GhMML3_D12, genomic DNA of Xu142 fl displayed a longer PCR product compared with those of expected length in normal and fuzzless-mutant lines, and the same size products were also found in three other fiberless mutants (Figure 3A). However, the other two fiberless lines, MD17 and SL1-7-1, displayed a normal-length amplification. The full-length genomic sequences of GhMML3_D12 of Xu142 fl and three other fiberless mutants showed that they all carried a 3,635-bp insertion fragment in the second exon (Figure 3B). Further sequence analyses indicated that the insertion fragment is a putative Ty1/copia long terminal repeat (LTR) retrotransposon (named Ghli3_ret hereafter). The components of a typical Ty1/copia element, such as LTRs, gag, and pol, could be found in Ghli3_ret (Supplementary Figure S1). The retrotransposon consists of a 427-bp 5′-LTR, a 2,781-bp internal region, and a 427-bp 3′-LTR. The sequences of the two LTRs are entirely the same, which suggests that this insertion event is relatively recent. A further screening using li3-specific primers (forward primer in Intron 1 and reverse primer in 5′LTR; Supplementary Table S3) in 387 Gh and 373 Gb cultivars showed that the Ghli3_ret insertion only existed in these four fiberless mutants (Supplementary Tables S7, S8). The marker assay also showed that this insertion was absent in Xu142.
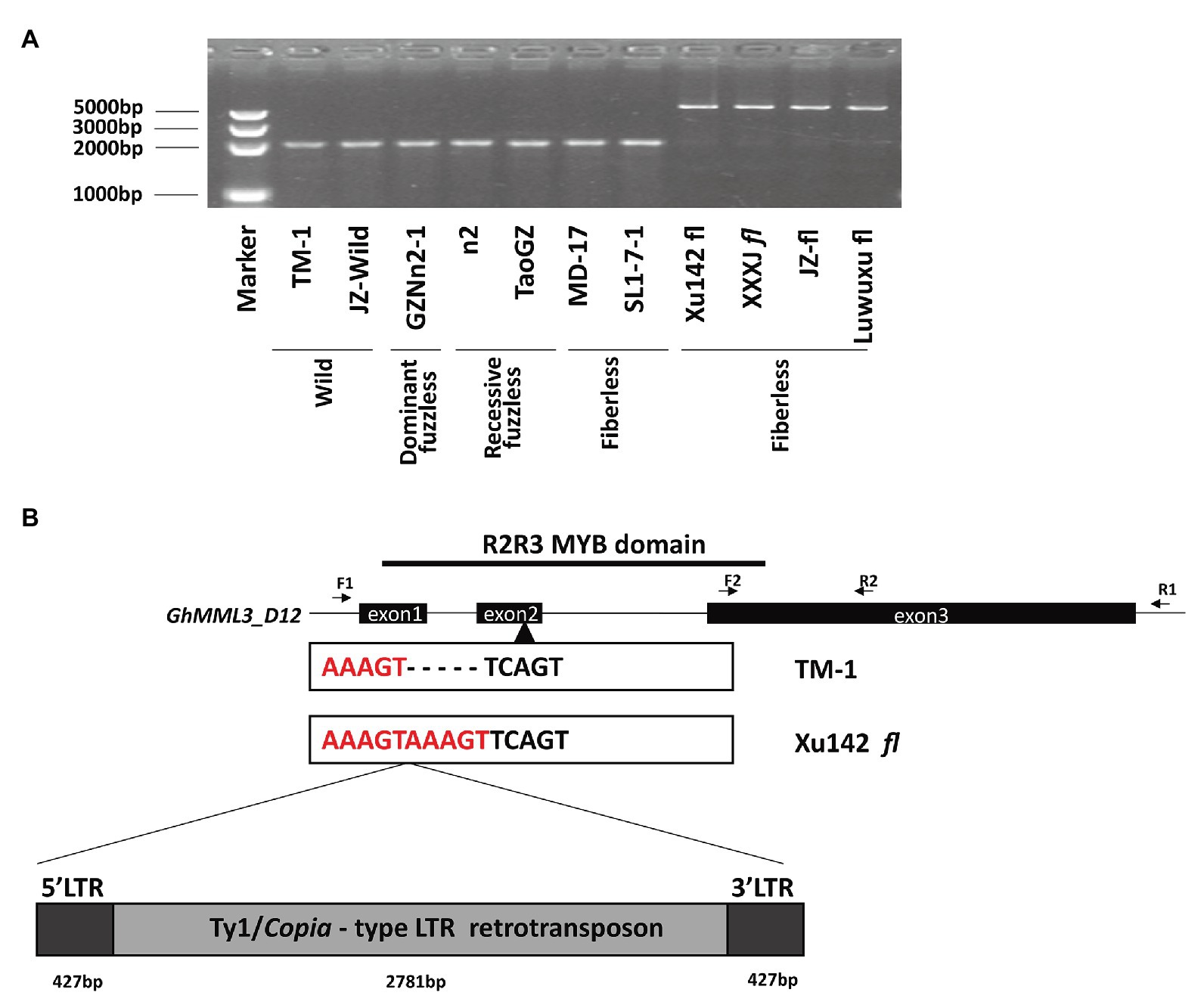
Figure 3. The sequence variation between TM-1 and Xu142 fl. (A) Detection of retrotransposon insertion using F1/R1 primer pair. (B) Structure of the GhMML3_D12. F1/R1 primer pair was used for the amplification of the full DNA sequence of GhMML3_D12. The F2/R2 primer pair was used for quantitative real-time PCR (qRT-PCR).
The Ghli3_ret insertion in GhMML3_D12 might block the expression of the gene. qRT-PCR was used to analyze the expression level of the GhMML3_D12 transcript at different development stages of ovules. Compared with TM-1, the transcript abundance of GhMML3_D12 was significantly reduced in Xu142 fl at −1 and 0 DPA stages (Figure 4). Sequencing of cDNA using primers universal to the At- and Dt-subgenome alleles also indicated that GhMML3_D12 in Xu142 fl might not be expressed, because all sequenced clones were GhMML3_A12 type (15 out of 15). Actually, Walford et al. (2011) also reported the absence of GhMML3_D12 mRNA in Xu142 fl. These results indicated that the insertion of Ghli3_ret interrupted the transcription of GhMML3_D12, the best candidate for Li3.
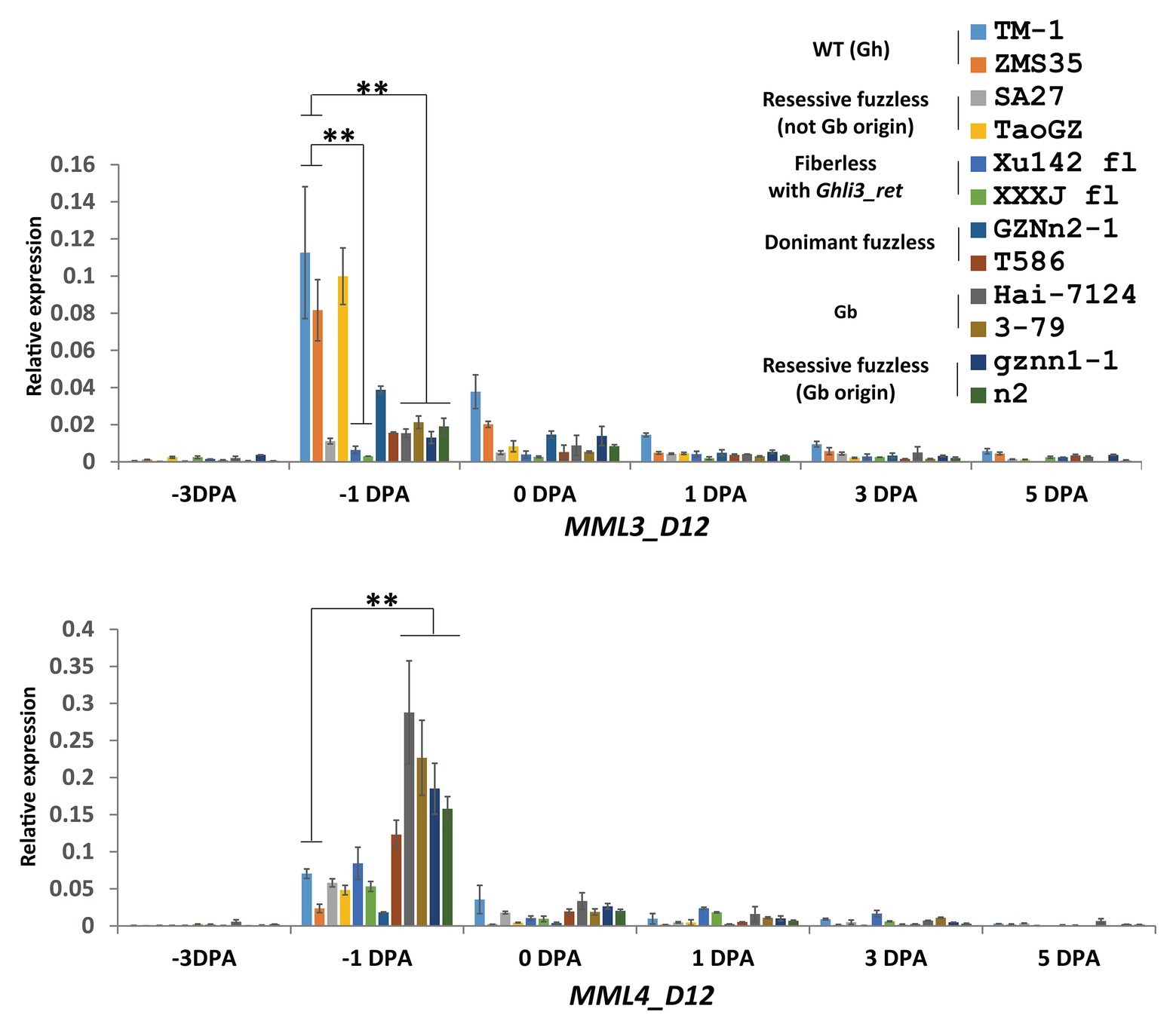
Figure 4. Quantitative real-time PCR measurements of MML3_D12 and MML4_D12 in ovule development. Error bars indicate the standard deviation of three biological replicates. Significant differences between groups are indicated by an asterisk (**p < 0.01) according to Student’s t-tests.
Primary Mapping of n2 and n3
Previous studies showed that the recessive fuzzless mutant n2 might differ from normal cotton at the N2 and N3 loci (Turley and Kloth, 2002). Thus, the cross n2 (n1n1n2n2n3n3) × TM-1 (n1n1N2N2N3N3) was made to generate a segregating population for mapping of these two loci. F1 plants show the fuzzy phenotype, though the fuzz density of seeds is lower compared with that of TM-1 (Figure 1). The fuzz phenotype of F2 plants was investigated by visual inspection. The results showed that segregation of fuzzy and fuzzless phenotypes fit the expected ratio (15:1; Supplementary Table S4). However, it should be noted that there was a broad variation in fuzz density for those fuzzy plants, and only a fraction showed similar density with that of TM-1. Again, the strategy of bulked segregant analysis (BSA) combined with whole-genome resequencing was used. The DNA pool was generated by bulking 30 fuzzless individuals (with genotype assumed to be n1n1n2n2n3n3) in the F2 population with subsequent high-throughput sequencing (Illumina HiSeq × Ten platform). Two candidate genomic regions for n2 and n3 were found in BSA analysis: ~47–48 Mb on chromosome D12 and ~74–75 Mb on chromosome A12 (Figure 5A). Interestingly, the region on D12 overlapped with the location of Li3, and the region on A12 overlapped with that of N1. GhMML3 or GhMML4 again was considered to be a good candidate for N2 or N3, because in these regions, no other gene showed preferential expression in ovules during the fiber initiation stage (−3 to 5 DPA) in the previous RNA-Seq data (Zhang et al., 2015; Supplementary Table S6).
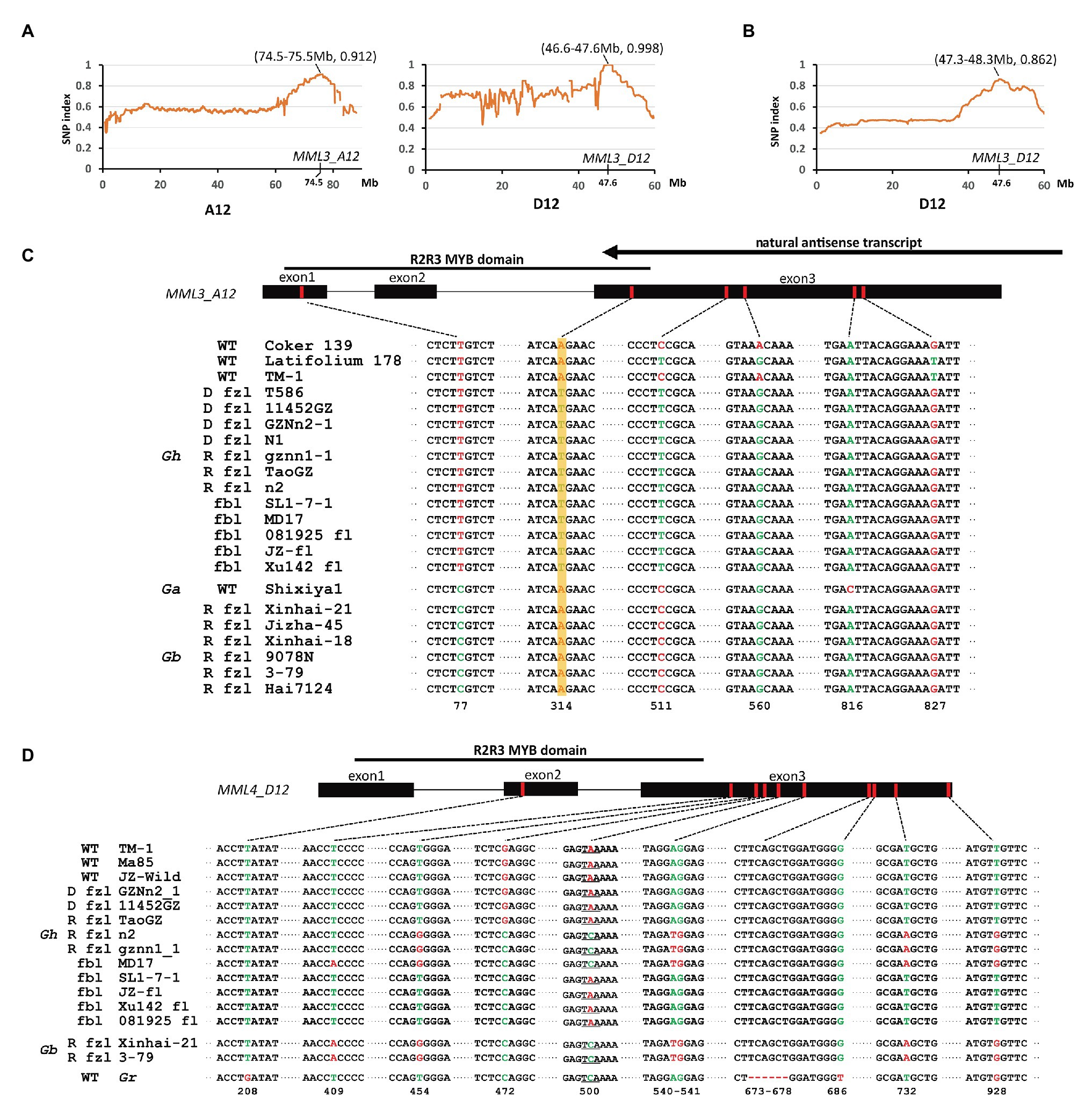
Figure 5. Mapping of the recessive fuzzless genes. (A) The SNP index plot on chromosomes A12 and D12 using the F2 population of n2 × TM-1. (B) The SNP index plot on GhMML3_D12 using the F2 population of TM-1 × Hai7124. Averaged SNP index values in a moving window of 1.0 Mb with 100 kb increments were calculated and plotted. The window with the largest index value is shown in brackets. (C) Non-synonymous mutations in MML3_A12. D fzl, dominant fuzzless; R fzl, recessive fuzzless; and fbl, fiberless. The arrow indicates the NAT of GhMML3_A12 found by Wan et al. (2016). (D) Non-synonymous mutations in MML4_D12.
n3 Is One of the GhMML3_A12 Alleles
GhMML4_A12 was found to maintain a very low expression level in ovules throughout different developmental stages in previous and the present studies (Zhang et al., 2015; Supplementary Figure S2), and no sequence variation in the coding sequence (CDS) region was found between mutants and the wild type (WT; Supplementary Figure S3), so it is unlikely to be the candidate gene for these two loci (N2 and N3). In GhMML3_A12, sequence alignment found an SNP (A314T) common among n2, Xu142 fl, and GZNn2-1 that caused a non-synonymous coding mutation (K105M) in the DNA-binding domain of this gene (Figure 5C; Supplementary Figures S4, S5). The screening of 387 normal Gh lines and 57 fiber mutants using a diagnostic marker based on this SNP showed that this mutation was found in all fiber mutant lines, no matter whether they were fuzzless (dominant or recessive) or fiberless (Supplementary Tables S1, S7). In previous studies (Turley and Kloth, 2002, 2008), the dominant fuzzless mutants have been shown to contain the N2 allele, so this SNP should be the underlying mutation for the n3 locus. That is, if this SNP is the causal mutation for the n2 locus, those dominant fuzzless mutants would contain the n2 allele, which is contradictory to the results of the previous studies. Interestingly, Wan et al. (2016) proposed that the extremely low expression of GhMML3_A12 in N1 is associated with NAT production (Figure 5C). This NAT is highly active in the dominant fuzzless mutants and suppresses many MML genes besides MML3. In normal and recessive fuzzless lines, this NAT is weak or absent (Wan et al., 2016). Thus, the n3 and N1 loci are actually different mutation sites in and around GhMML3_A12, and both disrupt the normal function of GhMML3_A12.
The marker assay showed that only six normal lines contained the n3 allele (Supplementary Table S9), so it appears to be a rare allele in normal lines (allele frequency was 0.015). The screening in 373 Gb accessions showed that this allele was also missing in Gb lines (Supplementary Table S8). Thus, all Gb lines seem to contain the N3 locus, though they are all fuzzless.
Another SNP (C511T) in GhMML3_A12 was also common among n2, Xu142 fl, and GZNn2-1, and this SNP also caused a non-synonymous coding mutation (P171S; Figure 5C; Supplementary Figures S4, S5). However, this SNP was found in many normal lines in the previous genome-wide association study (GWAS) analyses, and no significant association was found with any surveyed traits (Wang et al., 2017; Ma et al., 2018). Furthermore, this SNP is located outside the MYB domain of GhMML3_A12, so it should be excluded as the causal mutation for n3.
To trace the origin of the n3 locus, 51 fuzzless and 405 normal Gh wild accessions were surveyed using the diagnostic marker based on the A314T mutation (Supplementary Table S10). The n3 mutation was found in only two fuzzless accessions, marie-galante 38 (TX-881) and marie-galante 39A (TX-882), and it was absent in all normal wild accessions.
Interestingly, Xu142 was found to contain the N3 allele in the marker assay. That is, it contained wild-type genotypes (Li3Li3N3N3) for both li3 and n3 loci. Thus, it is possible that Xu142 used in the present study is not the original line (Xu142 fl derived). Actually, Xu142 was also found to differ from Xu42 fl at more than two loci in another study (Hu et al., 2018). In future study, attention should be called to the truth of Xu142, considering that the original line might be lost because of the low probability of opening pollination during germplasm conservation.
li3 and n2 Might Be the Multiple Alleles of GhMML3_D12
Because n3 is located in GhMML3_A12, the n2 locus should be in the other candidate genomic region on D12 that overlaps with the location of li3 (Figure 5A). The sequence alignment showed that genomic sequences of GhMML3_D12 and GhMML4_D12 of n2, gznn1-1, and MD17 were the most similar to those of Gb reference genomes (Liu et al., 2015; Yuan et al., 2015). Considering that nearly all Gb accessions in nature are fuzzless, the n2 allele of these mutants might have been transferred from fuzzless Gb lines. For GhMML3_D12, no sequence variation in CDS was found to be specific for mutants proposed to contain the n2 allele (Supplementary Figure S6). In the promoter region of GhMML3_D12, a 480-bp insertion at the −252 site (the star codon set as the zero site) was found when compared with GhMML3_A12 and other homologs in Gossypium raimondii (D5-genome) and G. arboreum (Supplementary Figure S7). This insertion exists in both Gh and Gb. A 7 bp target site duplication sequence (ACATAGT) was found beside the insertion. Alignment analyses showed that sequences similar to this insertion widely distributed in the genomes of Gossypium species, but were missing in other organisms (Supplementary Table S11). Thus, this insertion appears to be a transposon specific to cotton. Interestingly, in the insertion, an 80-bp deletion was found in some Gh lines, differing from the 22-bp deletion in Gb, n2, gznn1-1, and MD17. Several other sequence variants were found between Gh and Gb in the promoter region. When using a diagnostic marker based on the differences in the promoter region between Gh and Gb lines, 9 out of 24 recessive fuzzless lines were found to be of Gb origin (Supplementary Table S1).
For GhMML4_D12, sequence alignment found that a non-synonymous coding mutation (G472C cause E158Q) was common for n2, gznn1-1, Xu142 fl, MD17, and 2 Gb reference lines (Figure 5D; Supplementary Figure S8), so this appears to be a candidate variation for the n2 locus, considering that all these lines were proposed to contain the n2n2 genotype in previous studies (Turley and Kloth, 2002, 2008). However, this variation was also found in some of the dominant fuzzless lines (14 out of 23) that were thought to contain the N2 allele (Supplementary Table S1). This SNP variation was also found in the GWAS analysis, and it appeared to be arranged randomly in the 352 cotton accessions (Wang et al., 2017). Another SNP (C500A) that resulted in the early termination of GhMML4_D12 translation because of the formation of a TAA stop codon was found in all Gh lines surveyed, except for n2, gznn1-1, MD17, and other Gb origin recessive fuzzless lines. Thus, this SNP should not be the causal mutation of the n2 locus, because it was also found in the normal Gh lines that were thought to contain the N2 allele. In addition, this mutation was located outside the MYB domain of GhMML4_D12, and it might not affect its DNA-binding function. Interestingly, the expression level of GhMML4_D12 was much higher for −1 DPA-ovules in Gb and Gb origin recessive fuzzless lines compared with those with the TAA mutation (Figure 4), further suggesting that this gene might not be the candidate for N2.
Overall, no mutation was found common among lines containing the n2 allele (based on previously proposed genetic models) in the genomic sequences of GhMML3_D12 and GhMML4_D12 in the present study. However, the expression level of GhMML3_D12 was much lower for −1 DPA-ovules in lines assumed to contain the n2 allele compared with the WT (Figure 4). Considering that transcription of GhMML3_D12 was also completely interrupted in the li3 mutants, li3 and n2 might be multiple alleles of the GhMML3_D12 gene (so li3 was renamed as n2Xu).
Mapping of Fuzzless Loci in Gb
Considering that all Gb cultivars are fuzzless and that some of the recessive fuzzless Gh lines were of Gb origin, it was essential to map the fuzzless loci in Gb. An F2 population derived from the cross TM-1 × Hai7124 was developed, and the fuzz phenotype was investigated by visual inspection. Fuzz density of seeds in the population seemed to display a continuous distribution, from TM-1-like to Hai7124-like. Of the 1,117 F2 plants, only 39 plants displayed naked seeds and the fuzz density of those seeds was even lower than that of Hai7124, indicating that the fuzzless seed trait of Hai7124 might be regulated by multiple loci. The DNA pool was generated by bulking these fuzzless individuals and was subsequently subjected to high-throughput sequencing (Illumina HiSeq × Ten platform). Only a single genomic region on D12 was found significant in association mapping, and this region overlapped with those of the Li3/N2 loci (Figure 5B). This location is also the same as the major locus mapped in Gb using another cross (Zhu et al., 2018), though another four minor loci were also identified in their study. These results indicate that the n2 allele originated from Gb.
Genetic Analysis of Lintless/Fuzzless Loci in Xu142 fl
To further dissect the inheritance model for the fiberless trait of Xu142 fl, the genotypes of Li3/N2 and N3 loci of F2 plants from the cross TM-1 × Xu142 fl were surveyed by PCR markers based on the mutations responsible for these two loci in GhMML3_D12 (Ghli3_ret insertion) and GhMML3_A12 (A314T), respectively (Figure 6A). In the background of N2N2 (homozygous Li3 allele), two groups of fuzz density were observed in each genotype of the N3 locus. It seems that there was a third locus (named N5) displaying an inhibitory effect on the expression of Li3 (GhMML3_D12) because of the ratio of lower fuzz density plants: higher fuzz density plants was close to 3:1 in each genotype of the N3 locus. The combination of N2N2n3n3N5_ resulted in the fuzzless phenotype. In the background of N2n2Xu, the N5 locus still displayed a suppression effect on the Li3 locus, because two groups of fuzz density were still observed in each genotype of the N3 locus. The combination of N2n2Xun3n3N5_ resulted in the fuzzless phenotype. In the background of n2Xun2Xu, only a single fuzz density was observed for each genotype of the N3 locus. Thus, the N5 locus seems to show no inhibitory effect on the expression of N3 (GhMML3_A12). The combination of n2Xun2XuN3n3_ _ resulted in the fuzzless phenotype, and the double homozygous recessive genotype (nli3nli3n3n3_ _) displayed the fiberless phenotype.
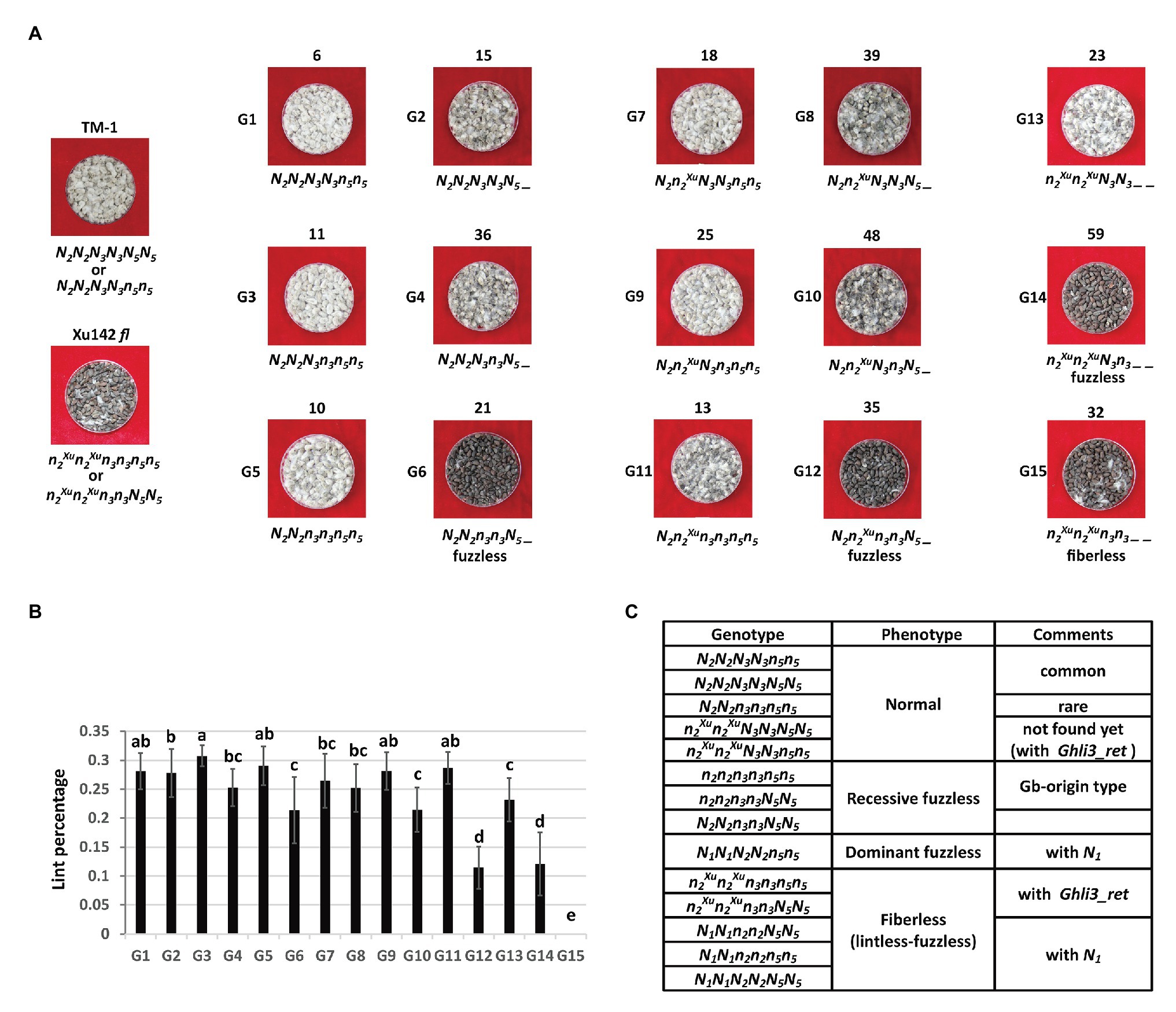
Figure 6. Segregation of the fiber traits by N2/N3/N5 combination in the F2 population of TM-1 × Xu142 fl. (A) Fuzz phenotype of parents and representative F2 plants. The number on the top is the number of plants in each group. The proposed genotype is on the bottom. N2, n2Xu are WT and Xu142 fl alleles in the N2 locus, respectively. (B) The lint percentage of each group. Error bars indicate standard deviations. Multiple comparisons were conducted by Duncan’s new multiple comparisons test. (C) Proposed genotypes for WT cotton and fiber mutants by N1/N2/N3/N5 combination.
The N5 locus might also repress the initiation of lint. The average lint percentage of those plants with the dominant N5_ genotype was usually lower than that of plants with n5n5 in the same genotype combination of Li3/N2 and N3 loci (Figure 6B). Interestingly, the fuzzless N2N2n3n3N5_ plants showed a much higher lint percentage than the other two types of fuzzless plants (N2n2Xun3n3N5_ and n2Xun2XuN3n3_ _).
Based on these results, a four-locus genetic model could be proposed for Xu142 fl and other fiber mutants when taking into account the N1 locus (Figure 6C). In this model, only dominant fuzzless mutants have a single genotype.
Discussion
For the fine-mapping of the li3 (n2Xu) locus, the same parents and types of mapping population were used between this study and that of Wu et al. (2018), and the same mapping region was also obtained. The SNP site (C500A) that resulted in the early termination of GhMML4_D12 was regarded as the causative mutation for li3 in their study. However, our results showed that this mutation was universal in Gh lines, and native MML4_D12 was only found in Gb and recessive fuzzless Gh lines with n2 allele (Gb origin). Like Xu142 fl, the wild-type Gh lines also displayed a much lower expression level of GhMML4_D12 compared with Gb and recessive fuzzless lines (with n2 allele). Furthermore, the RNA interference suppression of GhMML4 did not result in the lintless phenotype in their study. These results indicated that GhMML4_D12 should not be the candidate gene for li3. In this study, a retrotransposon insertion in GhMML3_D12 was found, and the expression of this transcript was accordingly disrupted. The insertion was only found in four fiberless mutants. Furthermore, Walford et al. (2011) found that the reduced expression of GhMML3 resulted in a fiberless phenotype. Thus, the insertion in GhMML3_D12 is likely the real underlying mutation for li3.
In the present research, the fiberless mutant Xu142 fl was found to be a double mutant line (n2Xun2Xun3n3), and the mutations in the copies of GhMML3 on A12 and D12 were sufficient to explain the fiberless phenotype in the F2 population. GhMML3_D12 was found to be a good candidate for the recessive fuzzless n2 locus in Gh, and it also appeared to be a major locus underlying the fuzzless seed trait in Gb. Furthermore, the n3 locus appeared to be a prerequisite for all the naked or fiberless seed mutants in cultivated Gh lines. The RNA interference suppression of MML3 resulted in fiberless seed phenotypes similar to that of Xu142 fl and reduced expression of other fiber-expressed MYBs (such as GhMML7/GhMYB25, GhMML8, and GhMML9) was also found in these silenced plants (Walford et al., 2011; Bedon et al., 2014). In a transcriptome profiling study of five fiber mutants, a number of transcription factor families (such as MYB, WRKY, bZIP, and bHLH) were found to be differently modulated between WT and mutants (Wan et al., 2014). Thus, the present and previous results imply that GhMML3 is a key factor having a prominent effect on fiber initiation and that it may function as an upstream gene in a regulatory cascade for fiber initiation (Hu et al., 2016). However, the discovery of the N1 and N5 locus shows that there are some upstream regulatory factors modulating the expression of GhMML3.
The K105M in the DNA-binding domain of MML3_A12 appeared to be the best candidate for n3 in the present study. This residue (K105) is located in the DNA-binding motif of the MYB domain, and no variation of this site has been found among different plant species (Supplementary Figure S5). The mutation might disrupt the DNA-binding activity of MML3, so n3 should be a loss-of-function allele. The genetic analyses in the F2 population of the cross TM-1 × Xu142 fl also indicated that n3 was a recessive allele. A recent GWAS (using 352 cotton accessions) showed that this SNP allele was missing in all of the normal cotton except for one line, 611Bo (Wang et al., 2017). In another GWAS (using 419 cotton accessions), only 10 out of 335 normal-seed lines were found to contain this allele (Ma et al., 2018; Supplementary Table S9). In both GWAS, all of the fiber mutants contained this SNP site. Thus, this mutation appears to be a prerequisite for the naked or fiberless seed in Gh. It appears to be a rare allele in normal lines, which is also reasonable based on the four-locus genetic model deduced in the present study. In the background of N2N2n5n5, the plants with the n3n3 genotype could still display normal fuzz phenotype. In addition, this SNP site was also found to be significantly associated with lint percentage in the GWAS analysis of Gh lines (Ma et al., 2018).
There are different mapping results for the n2 locus in previous studies. In Endrizzi and Ramsay (1980) n2 was mapped to D12, but in later studies, it was mapped to A12 (Rong et al., 2005; Li et al., 2019). Song et al. (2010) found that the n2 locus could be mapped to A12 or D12, depending on the mapping population used. Actually, some of these studies may have ignored the segregation of the n3 locus in mapping populations, thus leading to the confusing mapping results for n2. Because of the broad variation in the fuzz phenotype in some mapping populations, some fuzzy plants with low fuzz density would be classified into the fuzzless group. Thus, at times, the fuzz phenotype from some mapping populations would fit the expected ratios based on monogenic inheritance in segregation analyses, even though n2 and n3 were both segregating.
In this study, all four fiberless mutants from China were found to contain the retrotransposon Ghli3_ret in GhMML3_D12, and the sequences of these Ghli3_ret were all the same. The Ghli3_ret insertion in these mutants might have the same origin, though the pedigree of those lines is missing. However, the n3 allele was found in all fiber mutants of cultivated Gh, and it would be a low probability event if all of these mutations happened independently. The genetic analyses in previous and the present studies indicated that the fuzzless/fiberless traits of all fiber mutants in Gh have polygenic inheritance. Thus, it is possible that these mutants actually are natural combinations of some rare mutations of different gene loci existing in normal lines. Actually, for the cross TM-1 × Xu142 fl used in the present study, all F2 plants with n2Xun2XuN3N3 and some plants with N2N2n3n3 genotypes displayed the normal seed phenotype. Although the n2Xu allele was not found outside the fiberless mutants used in the present study, it might exist in some normal lines currently not surveyed.
The locations of loci mapped in the present study indicated that the initiation of fuzz and lint seems to be under the same (or partly overlapping) gene regulatory network. The genetic analysis using the F2 plants of TM-1 × Xu142 fl indicated that the lintless phenotype resulted from the additive effects of the two fuzzless loci. The fiberless phenotype of the synthetic line MD17 is an example of this kind of additive effect (Turley, 2002). Actually, the lower lint percentage had been observed in most of the fuzzless mutants (Supplementary Table S1). These results are not surprising considering MML3_A12 and MML3_D12 affecting the initiation of fiber with an additive manner. However, no fuzz-lintless mutant has been found in Gh or Gb so far. Thus, some key genetic loci (currently unknown) responsible for lint initiation might be at the upstream position of the regulatory network that also controls the initiation of fuzz.
The variation responsible for the n2 allele still needs to be examined in further studies. Only one SNP (C76T) in the CDS region of MML3_D12 was found to be specific to materials containing the n2 allele. However, this SNP is a synonymous mutation and is located outside the DNA-binding motif. There are several sequence variants in the promoter region of MML3_D12 between Gh and Gb, and some of them might be responsible for the lower expression level of GbMML3_D12. A promoter activity assay with different combinations of promoter sequences is necessary to find the key region regulating the expression of MML3_D12.
Data Availability Statement
The sequences of MML4_A12 (GenBank accession number: MN510689–MN510696), MML3_D12 (MN510697–MN510712), MML3_A12 (MN510713–MN510729), MML4_D12 (MN510730–MN510742), MML3_D12 promoters (MN510743–MN510757), and Ghli3_ret (MN486221) have been deposited in the GenBank database.
Author Contributions
WC, YLi, SZ, JY, and YZ designed the research, performed most of experiments, and analyzed the data. SF, LZ, YG, JW, LY, FL, and YLu performed part of the experiments. WC and YZ interpreted the results and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Natural Science Foundation of China (31621005).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the national mid-term cotton gene bank of ICR-CAAS and national wild cotton nursery for providing the cotton materials.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.593679/full#supplementary-material
Footnotes
References
Abe, A., Kosugi, S., Yoshida, K., Natsume, S., Takagi, H., Kanzaki, H., et al. (2012). Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 30, 174–178. doi: 10.1038/nbt.2095
Bechere, E., Turley, R. B., Auld, D. L., and Zeng, L. (2012). A new fuzzless seed locus in an upland cotton (Gossypium hirsutum L.) mutant. Am. J. Plant Sci. 3, 799–804. doi: 10.4236/ajps.2012.36096
Bedon, F., Ziolkowski, L., Walford, S. A., Dennis, E. S., and Llewellyn, D. J. (2014). Members of the mybmixta-like transcription factors may orchestrate the initiation of fiber development in cotton seeds. Front. Plant Sci. 5:179. doi: 10.3389/fpls.2014.00179
Carver, W. A. (1929). The inheritance of certain seed, leaf, and flower characters in Gossypium hirsutum and some of their genetic interrelations1. J. Am. Soc. Agron. 21, 467–480.
Chen, W., Yao, J., Chu, L., Yuan, Z., Li, Y., and Zhang, Y. (2015). Genetic mapping of the nulliplex-branch gene (gb_nb1) in cotton using next-generation sequencing. Theor. Appl. Genet. 128, 539–547. doi: 10.1007/s00122-014-2452-2
Ding, M., Cao, Y., He, S., Sun, J., Dai, H., Zhang, H., et al. (2020). GaHD1, a candidate gene for the Gossypium arboreum SMA-4 mutant, promotes trichome and fiber initiation by cellular H2O2 and Ca2+ signals. Plant Mol. Biol. 103, 409–423. doi: 10.1007/s11103-020-01000-3
Du, X., Huang, G., He, S., Yang, Z., Sun, G., Ma, X., et al. (2018). Resequencing of 243 diploid cotton accessions based on an updated a genome identifies the genetic basis of key agronomic traits. Nat. Genet. 50, 796–802. doi: 10.1038/s41588-018-0116-x
Du, X. M., Pan, J. J., Wang, R. H., Zhang, T. Z., and Shi, Y. Z. (2001). Genetic analysis of presence and absence of lint and fuzz in cotton. Plant Breed. 120, 519–522. doi: 10.1046/j.1439-0523.2001.00643.x
Endrizzi, J. E., and Ramsay, G. (1980). Identification of ten chromosome deficiencies in cotton. J. Hered. 71, 45–48.
Endrizzi, J. E., Turcotte, E. L., and Kohel, R. J. (1985). Genetics, cytology, and evolution of Gossypium. Adv. Genet. 23, 271–375.
Feng, X., Cheng, H., Zuo, D., Zhang, Y., Wang, Q., Liu, K., et al. (2019). Fine mapping and identification of the fuzzless gene GaFzl in DPL972 (Gossypium arboreum). Theor. Appl. Genet. 132, 2169–2179. doi: 10.1007/s00122-019-03330-3
Hayashi, K., Yoshida, H., and Ashikawa, I. (2006). Development of PCR-based allele-specific and InDel marker sets for nine rice blast resistance genes. Theor. Appl. Genet. 113, 251–260. doi: 10.1007/s00122-006-0290-6
Hu, H., He, X., Tu, L., Zhu, L., Zhu, S., Ge, Z., et al. (2016). GhJAZ2 negatively regulates cotton fiber initiation by interacting with the R2R3-MYB transcription factor GhMYB25-like. Plant J. 88, 921–935. doi: 10.1111/tpj.13273
Hu, H., Wang, M., Ding, Y., Zhu, S., Zhao, G., Tu, L., et al. (2018). Transcriptomic repertoires depict the initiation of lint and fuzz fibres in cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 16, 1002–1012. doi: 10.1111/pbi.12844
Huang, Y., Liu, X., Tang, K., and Zuo, K. (2013). Functional analysis of the seed coat-specific gene GbMYB2 from cotton. Plant Physiol. Biochem. 73, 16–22. doi: 10.1016/j.plaphy.2013.08.004
Kearney, T. H., and Harrison, G. J. (1927). The inheritance of smoothness seeds in cotton. J. Agric. Res. 35, 193–217.
Kearney, T. H., and Harrison, G. J. (1928). Variation in seed fuzziness on individual plants of Pima cotton. J. Agric. Res. 37, 465–472.
Kohel, R. J., Richmond, T. R., and Lewis, C. F. (1970). Texas marker-1. Description of a genetic standard for Gossypium hirsutum L. Crop Sci. 10, 670–671.
Kosambi, D. D. (1944). The estimation of map distance from recombination values. Ann. Hum. Genet. 12, 172–175.
Lander, E. S., Green, P., Abrahamson, J., Barlow, A., Daly, M. J., Lincoln, S. E., et al. (1987). MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1, 174–181. doi: 10.1016/0888-7543(87)90010-3
Li, H., Handsaker, B., Wysoker, A., Fennell, T., and Ruan, J. (2009). The sequence alignment-map format and samtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, S., Zuo, D., Cheng, H., Zhang, Y., Wang, Q., Liu, K., et al. (2019). Fine mapping of the fuzzless gene n2 in cotton. Cotton Sci. 31, 114–120. doi: 10.11963/1002-7807.lsmsgl.20190325
Liu, X., Moncuquet, P., Zhu, Q. H., Stiller, W., and Wilson, I. (2020). Genetic identification and transcriptome analysis of lintless and fuzzless traits in Gossypium arboreum L. Int. J. Mol. Sci. 21:1675. doi: 10.3390/ijms21051675
Liu, X., Zhao, B., Zheng, H., Hu, Y., Lu, G., Yang, C., et al. (2015). Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci. Rep. 5:14139. doi: 10.1038/srep14139
Ma, Z., He, S., Wang, X., Sun, J., Zhang, Y., Zhang, G., et al. (2018). Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 50, 803–813. doi: 10.1038/s41588-018-0119-7
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation dna sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Musaev, D. A., and Abzalov, M. M. (1972). Some questions concerning the inheritance of fuzzy in cotton seeds (G. hirsutum L.) (in Russian). Genetika 8, 7–16.
Nadarajan, N., and Rangasamy, S. R. (1988). Inheritance of the fuzzless-lintless character in cotton (Gossypium hirsutum). Theor. Appl. Genet. 75, 728–730.
Reumers, J., Rijk, P. D., Hui, Z., Liekens, A., Smeets, D., Cleary, J., et al. (2012). Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat. Biotechnol. 30, 61–68. doi: 10.1038/nbt.2053
Rong, J., Pierce, G. J., Waghmare, V. N., Rogers, C. J., Desai, A., Chee, P. W., et al. (2005). Genetic mapping and comparative analysis of seven mutants related to seed fiber development in cotton. Theor. Appl. Genet. 111, 1137–1146. doi: 10.1007/s00122-005-0041-0
Ruijter, J. M., Ramakers, C., Hoogaars, W. M. H., Karlen, Y., Bakker, O., van den Hoff, M. J. B., et al. (2009). Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37:e45. doi: 10.1093/nar/gkp045
Samora, P. J., Stelly, D. M., and Kohel, R. J. (1994). Localization and mapping of the Le1, and Gl2 loci of cotton (Gossypium hirsutum L.). J. Hered. 85, 152–157.
Song, L., Guo, W. Z., Qin, H. D., Ding, Y. Z., and Zhang, T. Z. (2010). Genetic analysis and molecular validation of chromosome assignment for fuzzless genes N1 and n2 in cotton. J. Nanjing Agric. Univ. 33, 21–26. doi: 10.7685/j.issn.1000-2030.2010.01.005
Stewart, J. M. (1975). Fiber initiation on the cotton ovule (Gossypium hirsutum). Am. J. Bot. 62, 723–730.
Sun, W., Gao, Z., Wang, J., Huang, Y., Chen, Y., Li, J., et al. (2019). Cotton fiber elongation requires the transcription factor GhMYB212 to regulate sucrose transportation into expanding fibers. New Phytol. 222, 864–881. doi: 10.1111/nph.15620
Turley, R. B. (2002). Registration of MD 17 fiberless upland cotton as a genetic stock. Crop Sci. 42, 994–995. doi: 10.2135/cropsci2002.994a
Turley, R. B., and Ferguson, D. L. (1996). Changes in ovule proteins during early fiber development in a normal and a fiberless line of cotton (Gossypium hirsutum L.). J. Plant Physiol. 149, 695–702.
Turley, R. B., and Kloth, R. H. (2002). Identification of a third fuzzless seed locus in upland cotton (Gossypium hirsutum L.). J. Hered. 93, 359–364. doi: 10.1093/jhered/93.5.359
Turley, R. B., and Kloth, R. H. (2008). The inheritance model for the fiberless trait in upland cotton (Gossypium hirsutum L.) line SL1-7-1: variation on a theme. Euphytica 164, 123–132. doi: 10.1007/s10681-008-9670-0
Walford, S. A., Wu, Y., Llewellyn, D. J., and Dennis, E. S. (2011). GhMYB25-like: a key factor in early cotton fibre development. Plant J. 65, 785–797. doi: 10.1111/j.1365-313X.2010.04464.x
Wan, Q., Guan, X., Yang, N., Wu, H., Pan, M., Liu, B., et al. (2016). Small interfering RNAs from bidirectional transcripts of GhMML3_A12 regulate cotton fiber development. New Phytol. 210, 1298–1310. doi: 10.1111/nph.13860
Wan, Q., Zhang, H., Ye, W., Wu, H., and Zhang, T. (2014). Genome-wide transcriptome profiling revealed cotton fuzz fiber development having a similar molecular model as Arabidopsis trichome. PLoS One 9:e97313. doi: 10.1371/journal.pone.0097313
Wang, M., Tu, L., Lin, M., Lin, Z., Wang, P., Yang, Q., et al. (2017). Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 49, 579–587. doi: 10.1038/ng.3807
Wang, M., Yuan, D., Tu, L., Gao, W., He, Y., Hu, H., et al. (2015). Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 207, 1181–1197. doi: 10.1111/nph.13429
Ware, J. O., Benedict, L. I., and Rolfe, W. H. (1947). A recessive naked-seed character in upland cotton. J. Hered. 38, 313–320.
Wendel, J. F., and Cronn, R. (2003). Polyploidy and the evolutionary history of cotton. Adv. Agron. 78, 139–186. doi: 10.1016/S0065-2113(02)78004-8
Wu, H., Tian, Y., Wan, Q., Fang, L., Guan, X., Chen, J., et al. (2018). Genetics and evolution of MIXTA genes regulating cotton lint fiber development. New Phytol. 217, 883–895. doi: 10.1111/nph.14844
Yuan, D., Tang, Z., Wang, M., Gao, W., Tu, L., Jin, X., et al. (2015). The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci. Rep. 5:17662. doi: 10.1038/srep17662
Zhang, T., Hu, Y., Jiang, W., Fang, L., Guan, X., Chen, J., et al. (2015). Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. doi: 10.1038/nbt.3207
Zhang, T. Z., and Pan, J. J. (1991). Genetic analysis of a fuzzless-lintless mutant in Gossypium hirsutum L. Jiangsu J. Agric. Sci. 7, 13–16.
Zhu, J., Chen, J., Gao, F., Xu, C., Wu, H., Chen, K., et al. (2017). Rapid mapping and cloning of the virescent-1 gene in cotton by bulked segregant analysis-next generation sequencing and virus-induced gene silencing strategies. J. Exp. Bot. 68, 4125–4135. doi: 10.1093/jxb/erx240
Keywords: fiber initiation, fiberless mutants, fuzzless mutants, MYBMIXTA-like transcription factors, cotton (Gossypium spp.)
Citation: Chen W, Li Y, Zhu S, Fang S, Zhao L, Guo Y, Wang J, Yuan L, Lu Y, Liu F, Yao J and Zhang Y (2020) A Retrotransposon Insertion in GhMML3_D12 Is Likely Responsible for the Lintless Locus li3 of Tetraploid Cotton. Front. Plant Sci. 11:593679. doi: 10.3389/fpls.2020.593679
Edited by:
Keiichi Okazaki, Niigata University, JapanReviewed by:
Iain Wilson, Commonwealth Scientific and Industrial Research Organisation (CSIRO), AustraliaKaijing Zuo, Shanghai Jiao Tong University, China
Copyright © 2020 Chen, Li, Zhu, Fang, Zhao, Guo, Wang, Yuan, Lu, Liu, Yao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongshan Zhang, MTM5Mzg2OTgyOTlAMTYzLmNvbQ==; Jinbo Yao, eWFvamJAY3JpY2Fhcy5jb20uY24=
†These authors have contributed equally to this work