 Neha Agrawal
Neha Agrawal Mehak Gupta
Mehak Gupta Surinder S. Banga
Surinder S. Banga JS (Pat) Heslop-Harrison
JS (Pat) Heslop-Harrison- 1Department of Plant Breeding and Genetics, Punjab Agricultural University, Ludhiana, India
- 2Department of Genetics and Genome Biology, University of Leicester, Leicester, United Kingdom
- 3South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China
Crop brassicas include three diploid [Brassica rapa (AA; 2n = 2x = 16), B. nigra (BB; 2n = 2x = 18), and B. oleracea (CC; 2n = 2x = 20)] and three derived allotetraploid species. It is difficult to distinguish Brassica chromosomes as they are small and morphologically similar. We aimed to develop a genome-sequence based cytogenetic toolkit for reproducible identification of Brassica chromosomes and their structural variations. A bioinformatic pipeline was used to extract repeat-free sequences from the whole genome assembly of B. rapa. Identified sequences were subsequently used to develop four c. 47-mer oligonucleotide libraries comprising 27,100, 11,084, 9,291, and 16,312 oligonucleotides. We selected these oligonucleotides after removing repeats from 18 identified sites (500–1,000 kb) with 1,997–5,420 oligonucleotides localized at each site in B. rapa. For one set of probes, a new method for amplification or immortalization of the library is described. oligonucleotide probes produced specific and reproducible in situ hybridization patterns for all chromosomes belonging to A, B, C, and R (Raphanus sativus) genomes. The probes were able to identify structural changes between the genomes, including translocations, fusions, and deletions. Furthermore, the probes were able to identify a structural translocation between a pak choi and turnip cultivar of B. rapa. Overall, the comparative chromosomal mapping helps understand the role of chromosome structural changes during genome evolution and speciation in the family Brassicaceae. The probes can also be used to identify chromosomes in aneuploids such as addition lines used for gene mapping, and to track transfer of chromosomes in hybridization and breeding programs.
Introduction
The genus Brassica (family Brassicaceae, with some 37 species) includes six major vegetable or oil crops: three diploid [B. rapa (AA genome composition, 2n = 20), B. nigra (BB, 2n = 16), and B. oleracea (CC, 2n = 18)] and three allotetraploid species [B. juncea (AABB, 2n = 36), B. napus (AACC, 2n = 38), and B. carinata (BBCC, 2n = 34)]. The allotetraploid brassicas evolved from pair-wise natural hybridizations between the three basic diploids. Although monophyletic, evolution of diploid Brassica genomes (A, B, and C) is complex (Lagercrantz and Lydiate, 1996; Lagercrantz, 1998). Inferences from comparative genome biology and phylogenetic reconstructions from whole genome sequences of brassica diploids are consistent with their common origin from an ancient paleohexaploid (γ event), followed by two whole-genome duplications (Wang et al., 2011; Zhang et al., 2018; Sun et al., 2019) and an additional whole-genome triplication (WGT), leaving extant brassica genomes as massively rearranged versions of an ancestral paleo-hexaploid genome. The chromosome structural changes resulted from chromosome breakages, fusions, inversions, and deletions after each cycle of polyploidy and diploidization (Schranz et al., 2006; Mandakova and Lysak, 2008; Schubert and Lysak, 2011). Despite the erosion of collinearity, high synteny and DNA sequence homologies continue to exist among Brassicaceae genomes (Tang et al., 2008; Cheng et al., 2012), representing regions with more conserved gene order or synteny blocks. There are 24 (A–X) conserved genome blocks (GBs) or ancestral karyotypes (AK) in the family Brassicaceae (Schranz et al., 2006). Each Brassica genome has three or six regions that are orthologous to Arabidopsis thaliana (Lysak et al., 2005, 2007; Cheng et al., 2014). These also harbor highly repeated sequences and complicated centromeric regions relative to A. thaliana (Lagercrantz and Lydiate, 1996; Lagercrantz, 1998; Lan et al., 2000; Chalhoub et al., 2014; Liu et al., 2014; Yang et al., 2016; Zhang et al., 2018). Though gene content evolution mirrored genome changes (Cheng et al., 2012; Tang et al., 2012), orthologs in the syntenic regions retained their functionality. Knowledge of syntenic genes and genomic regions among closely related species is important to explain genome diversification (Lyons et al., 2008). Furthermore, genetic exchanges in the regions of shared synteny are vital for mobilizing genes of interest across species domains, without precipitating non-compensating translocations. In silico analysis of DNA sequence data has been vital for the understanding of evolutionary mechanisms that framed structure of existing plant genomes (Salse and Feuillet, 2011).
Fluorescent in situ hybridization (FISH) is a powerful molecular cytogenetic technique to characterize karyotype variation at chromosome level by direct localization of repetitive DNA sequences on plant chromosomes (Jiang and Gill, 2006; Patokar et al., 2016; Song et al., 2020), enabling identification of chromosomes even in species with small and morphologically indistinguishable chromosomes, and comparison of chromosomal organization between species. However, such probes can be inconsistent in chromosome identification for multiplicity of repeat sequences and variations in their genomic locations (Mukai et al., 1993; Fransz et al., 1998; Kato et al., 2004; Danilova et al., 2012; Komuro et al., 2013; Koo et al., 2016; Amosova et al., 2017; Krivankova et al., 2017; Hou et al., 2018; Said et al., 2018), and repetitive DNA sequences with suitable genomic locations may not exist. BAC-based chromosome painting techniques have been used to construct high-resolution karyotypes (Kulikova et al., 2001; Pecinka et al., 2004; Zhang et al., 2006; Xiong and Pires, 2011; Wang et al., 2012) and identify chromosome structural variations (Lysak et al., 2005, 2006; Mandakova and Lysak, 2008; Idziak et al., 2011, 2014; Peters et al., 2012; Szinay et al., 2012; Mandakova et al., 2013). In a remarkable experiment, Lysak et al. (2006) explained the origin of each chromosome of A. thaliana relative to the ancestral n = 8 karyotype. These involved four chromosomal inversions, two translocations and three chromosome fusion events based on ordered BAC pools. Mandakova and Lysak (2008) also used multiple selected BACs as probes to explain monophyletic origin of the x = 7 tribes in Brassicaceae family through reduction of chromosome number from n = 8 in ancestral karyotype to n = 7, with different fusion and intrachromosomal inversion events. Mandakova et al. (2019) combined BAC-based chromosome painting, genomic in situ hybridization (GISH) and multi-gene phylogenetics to explain the role of post polyploidy chromosome structural variation in the origin and evolution of the Camelina sativa polyploid complex. However, identifying genetically mapped BACs with complete genome coverage is a challenge in most species; repetitive DNA sequences in the target DNA and BAC probes can cause non-specific hybridization. Development of repeat-free probes has proved difficult in some species (Bertioli et al., 2013), although in some cases repetitive DNA, particularly derived from repetitive DNA, may be valuable to identify different genomes in hybrids (Santos et al., 2015; Huang et al., 2020).
Use of massive pools of short synthetic oligonucleotides as probes for chromosomal in situ hybridization can allow design of probes to label any part of a chromosome as a band, or be designed to label (“paint”) a complete chromosome (Beliveau et al., 2015; Braz et al., 2018; Šimoníková et al., 2019). The oligonucleotide libraries use a defined set of unique sequences, selected in silico out of assembled genome sequences with chromosomal region specificity. These are highly sensitive and provide consistent chromosome labeling and signal intensity. Synthesis and labeling of massive oligonucleotide pools typically require thousands of oligonucleotides, 20–100 bp long. Their synthesis is now possible with a range of newly available commercial sources (Affymetrix, Combimatrix, Twist, inkjet printing/Agilent, and Mycroarray/Arbor Biosciences), using whole genome draft assemblies to anchor genome sequence information directly to chromosome topographies for getting a phylogenetic view of species. We expect this approach to provide a correct view of evolutionary relationships among species as single copy genomic regions are used to develop oligonucleotide pools. oligonucleotide libraries have been used as robust FISH probes in many plant species to construct molecular cytogenetic karyotypes (Braz et al., 2018; Meng et al., 2018), characterize chromosomal rearrangements and visualize homoeologous pairing among related species (Han et al., 2015; Qu et al., 2017; Xin et al., 2018) and integrate pseudomolecules of reference genome sequence of Musa acuminata spp. malaccensis “DH Pahang” to individual chromosomes in situ (Šimoníková et al., 2019).
Here, we aimed to develop an easy, robust and efficient oligonucleotide based cytogenetic toolkit for consistent and reproducible characterization of chromosomes or their structural variants in Brassica. Karyotype construction in Brassicaceae family has been challenging because of small chromosome size and the absence of cytological landmarks. The probes are useful for determining chromosome evolution or homology in the family Brassicaceae and applicable in genomic studies and in plant breeding aiming to exploit the germplasm pool. These probes allow comparative studies using oligonucleotides from conserved DNA sequences from one species in other genetic related species. We constructed four oligonucleotide libraries from 18 identified regions of B. rapa assembled genome sequence and tested them to identify all chromosomes of A, B, C, and R (Raphanus sativus) genomes. Our probes could also differentiate chromosome arms and pre-existing translocations in a commercial genotype of B. rapa. We also report an improved method for immortalization of oligonucleotide libraries to optimize the cost of oligonucleotide paints, which can otherwise be expensive or require demanding optimization.
Materials and Methods
Plant Materials
Brassica rapa, B. nigra, B. oleracea, and Raphanus sativus were used for comparative FISH analysis. Seeds of B. rapa ssp. chinensis cv. Chiifu-401 (pak choi or Chinese cabbage) were obtained from the University of Warwick, United Kingdom and B. rapa ssp. rapa cv. Turnip Purple Top Milan was sourced from Mr. Fothergill’s Seeds, United Kingdom. Other seeds of B. nigra, B. oleracea, and R. sativus were from the germplasm collections maintained at Punjab Agricultural University, Ludhiana, India.
Design of Oligonucleotide Pools
Four different sets of oligonucleotide pools were designed, each one labeled with a different fluorophore to create a multi-color barcode for identification of individual B. rapa chromosomes (Supplementary Table 1). One to three ranges of 0.5–1 MB in size were selected from different regions (sub-telomeric, intercalary, or sub-centromeric) of the DNA sequences of all of the 10 chromosomes of B. rapa (2n = 2x = 20) to create unique chromosome specific hybridization patterns upon simultaneous hybridization with four oligonucleotide sets. We downloaded chromosome assemblies of B. rapa cv. Chiifu-401 (synonyms Chiffu and Chifu) V_2.5, B. oleracea V_1, Raphanus sativus V_1 from the public Brassica database (BRAD1). The B. nigra chromosome assembly was kindly provided by Isobel Parkin (Saskatoon; Perumal et al., 2020). Linux command lines were used to split the target regions for probe design into 47 bp fragments with a 3 bp gap to prevent potential steric interference by adjacent oligonucleotide probes during in situ hybridization. We retained fragments with 30–66% GC content. oligonucleotides were further screened in sequential steps against all known repetitive sequences, including rDNA, chloroplast, published repeats (pBo, pBc families; Harrison and Heslop-Harrison, 1995) and a new repetitive motif library developed from unassembled, raw Illumina reads. Frequencies of all 32-mers (k-mer) were calculated, and the most abundant 5,000 were concatenated. Any 47-mer oligonucleotides mapping to approximately 28 of more bases of the concatenated sequence were discarded. We also tested against highly repetitive motifs from graph-based clustering of the raw reads using RepeatExplorer (Novak et al., 2013) to remove further repeats. The depleted libraries were then mapped back to published whole genome sequences (Bowtie2). Firstly, in B. rapa, any oligonucleotides mapping outside the target region were discarded (Figure 1) and primers added for PCR amplification for some pools (Figure 2). Reads from the target region were then mapped to reference with B. nigra chromosome to select sequence(s) common between the species occurring only in homoeologous and syntenic genome regions (Figure 3). Final oligonucleotides sets were also mapped to B. oleracea and R. sativus and in some cases screened against inclusion of repetitive sequences from these species that were less abundant in the source B. rapa genome. In silico hybridization simulations showed characteristic binding patterns of four oligonucleotide libraries to the genomes A, B, C, and R. Dot-plots were constructed among various chromosomes belonging to different genomes around the regions where probes were hybridizing to estimate degree of similarity and identify any rearrangements or major sequence insertions between the species (Figure 3).

Figure 1. Pipeline for designing of synthetic oligonucleotide libraries (Table 1) to chromosome loci. After generating 47 bp oligonucleotides (“oligos”) from the target regions, those with ambiguities or extreme GC-content, or that hybridize (assessed by mapping in silico) to repetitive sequences and non-target chromosomes, are discarded. The final pool has oligonucleotides hybridizing specifically to the target chromosomal regions.
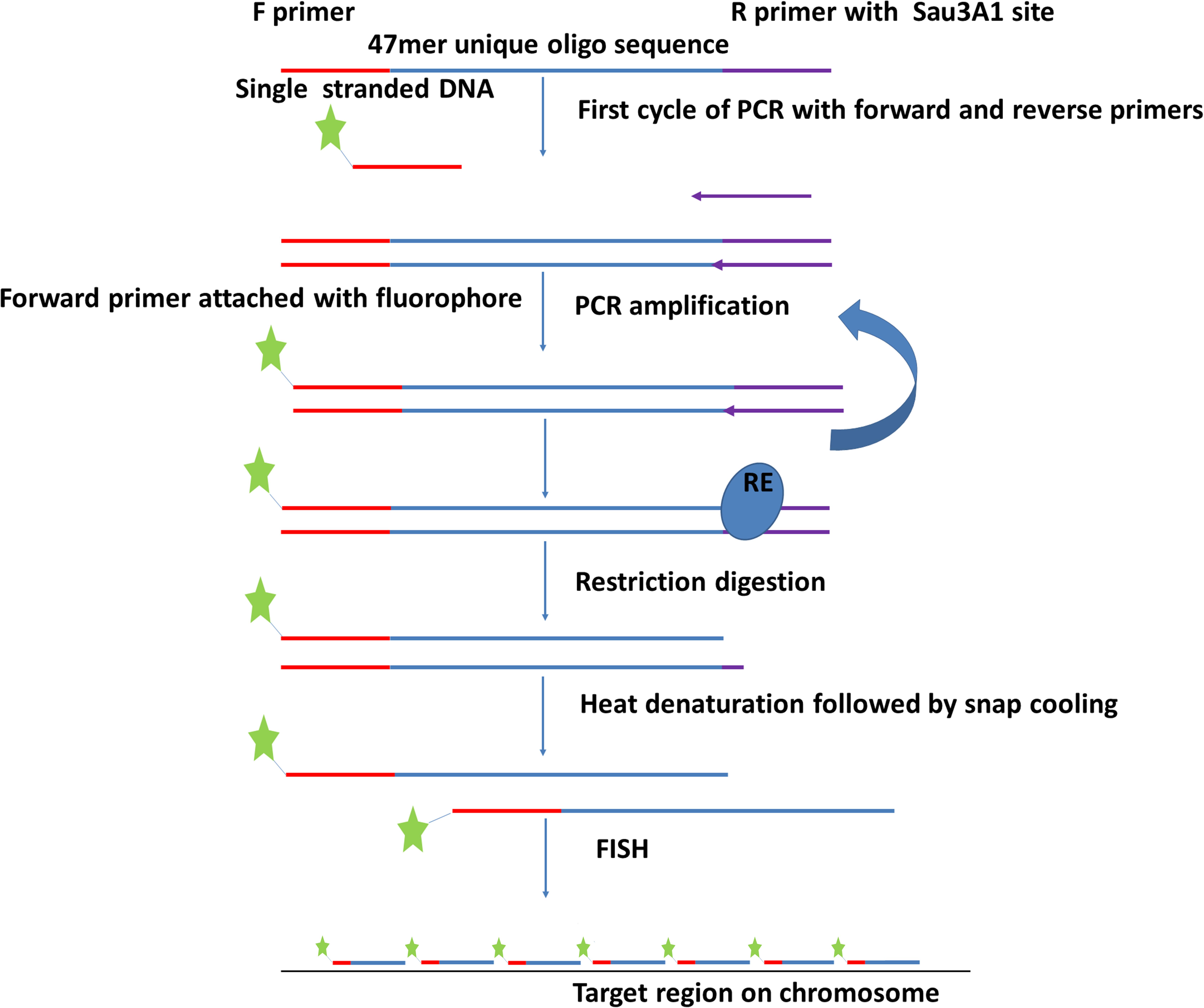
Figure 2. Flowchart depicting procedure for amplification of oligonucleotide libraries (immortalization). The oligonucleotides (Figure 1) are synthesized with 5′ and 3′ primers and amplified by PCR using one labeled and one unlabeled primer. After restriction enzyme digestion to remove the unlabeled end complementary to the reverse primer, the labeled product is hybridized to chromosome preparations.
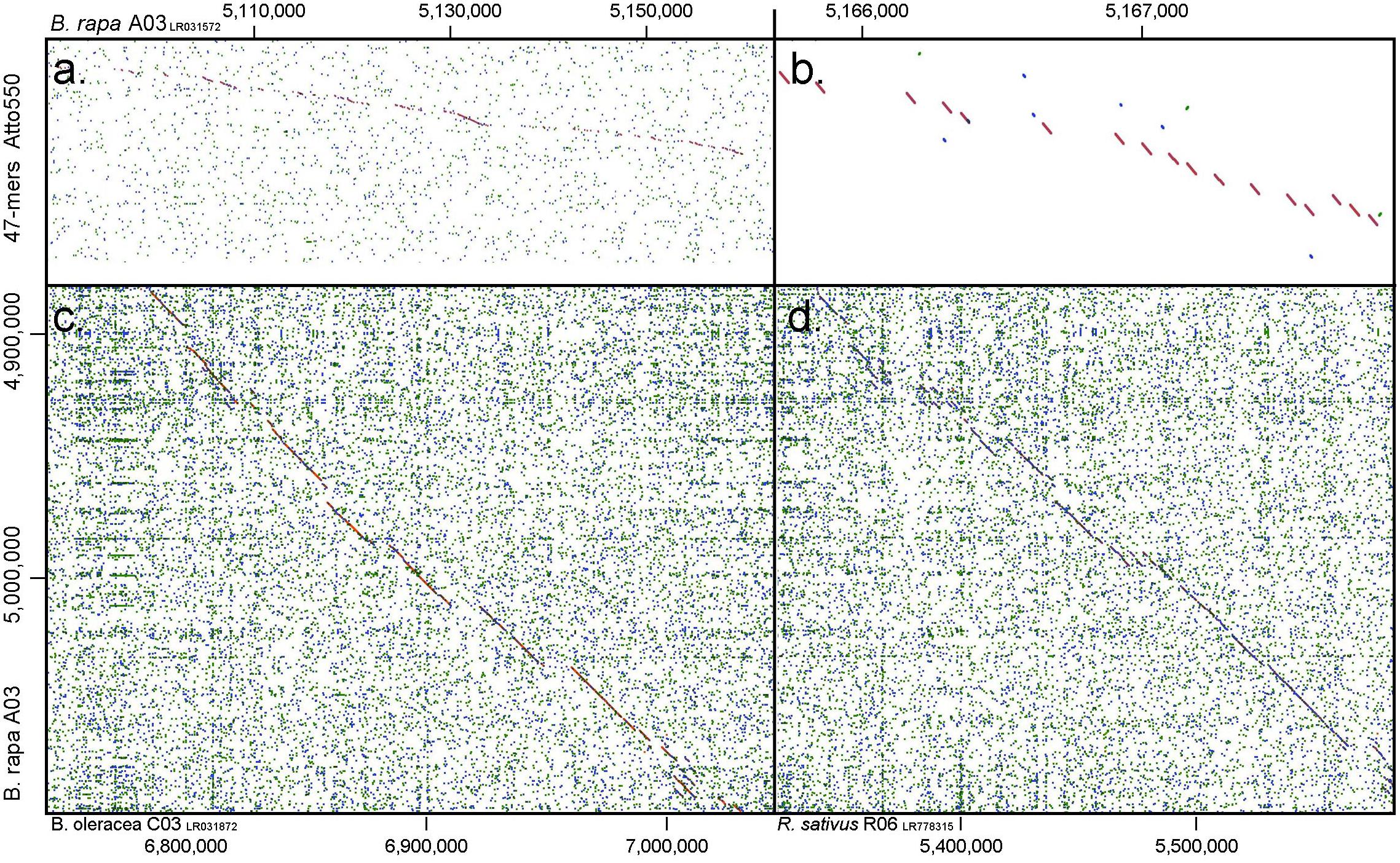
Figure 3. Dot plots depicting similarity between (a,b) oligonucleotide pool of 47-mers labeled with Atto550 and B. rapa chromosome A03 (1.0 gramene.org, y-axis): gaps show regions where nucleotides have been deleted by the selection procedure; and comparing A03 with (c) B. oleracea C03 and (d) Raphanus sativus R06. Diagonal lines show regions of high similarity where oligonucleotide probes would hybridize to both genomes, while gaps indicate insertions in one of the genomes or regions of lower homology.
Immortalization of Oligonucleotide Libraries
Oligonucleotides were designed with addition of a 20 bp 5′ primer annealing site (complementary to T7 primer), 47 bp of the unique oligo, and a 20 nt 3′ primer annealing site containing Sau3AI restriction site (GATCTCTGCATCTAGTAATG) (Figure 2). Unlabeled oligo libraries were ordered from Arbor Biosciences (Ann Arbor, MI). Each synthesized library contained 100 ng of DNA. These libraries were amplified and labeled simultaneously using PCR. Briefly, the PCR mixture of 50 μl reaction included 1 pmol DNA from the oligo library pool, 25 μM each of F (T7 primer 5′end labeled with the fluorochrome Cy5) and R (CTAGAAGTTACTGAGAGATC) primers, (underlined sequence depicts Sau3AI restriction site), 40 mM dNTPs, 1 unit of Platinum SuperFi DNA Polymerase in 5X high fidelity (HF) buffer and enhancer. The reactions were cycled as: 98°C for 30 s, 2x (98°C for 30 s, 59°C for 10 s, and 72°C for 10 s), continuing with a 2°C reduction each cycle till 53°C, 15–20x (98°C for 10 s, 53°C for 10 s, and 72°C for 10 s), 72°C for 1 min then held at 15°C. After amplification, the PCR product was digested with Sau3AI (Figure 2) to remove 3′ primers. Digested product was then purified using commercially available cycle purification kit of DNA from Omega and used as a probe for in situ hybridization.
Chromosome Preparations and in situ Hybridization Protocol
Metaphase chromosome preparations and in situ hybridization was performed according to Schwarzacher and Heslop-Harrison (2000) with minor modifications. The most stringent post-hybridization washes were carried out in 0.1X SSC at 42°C. The custom synthesized and labeled oligonucleotide library pools were directly used as FISH probes. Preparations were counterstained with DAPI in VectaShield antifade solution. The slides were examined and FISH images were captured using a Nikon Eclipse N80i fluorescent microscope equipped with a DS-QiMc monochromatic camera (Nikon, Japan). Raw images were processed with Adobe Photoshop using only functions that affect the whole image equally.
Results
Development of Oligonucleotide-Based Probes for Chromosome Identification in Brassica
We designed oligonucleotides from 18 different regions of B. rapa genome (BRAD B. rapa Version 2.5) using the strategy outlined in Figure 1. Genome coordinates for 18 identified regions are available in table 1. Each chromosomal region comprised 1,997–5,420 oligonucleotides, spanning over 500–1,000 kb (Table 1). Three libraries were custom synthesized and labeled with dyes Atto488-Green, Atto550-Red and Atto594-Yellow for use as robust probes. We also amplified an unlabeled library and labeled it with Cy5-Far Red, using standard PCR techniques (without an RNA intermediate) to develop an immortal probe. We retained 63,787 oligonucleotides (47 bp long) in four libraries (27,100, 11,084, 9,291, and 16,312 oligonucleotides, chromosomal locations and sequences shown in Supplementary Table 1). Simulated hybridization of oligonucleotides with whole genome sequence of B. rapa produced results in form of 18 intense peaks on ten chromosomes of B. rapa. The designed oligonucleotide-pools generated 18 distinct FISH signals on 10 chromosomes of B. rapa (chromosomes A01–A10) and the characteristic hybridization patterns identified all individual B. rapa chromosomes. Importantly, all the regions used for probe design gave signals. We also tested transferability of A genome probes to other brassica and radish genomes (Figures 3a–d). Simulations had earlier shown that the same oligonucleotide sets could depict 16, 18, and 18 sites in B (B. nigra), C (B. oleracea), and R (R. sativus) genomes respectively. The position (along the chromosome between centromere and telomere), intensity (number of probes with high homology) and width (region showing many probes hybridizing) of the respective peaks differed from those observed for A-genome. As an example, chromosome C01 showed the same two peaks as recorded in A01, in the same order between telomere and centromere (Figures 4A,B). The first peak, observed by hybridization of probes, complemented a region 4 Mbp from the start of chromosome A01 and 4.8 Mbp from start of C01. Hybridization of the probes produced the second peak on A01, with a region initializing at 29.5 Mbp from the start; C01 produced this peak at 34.5 Mbp. The number of probes predicted to hybridize
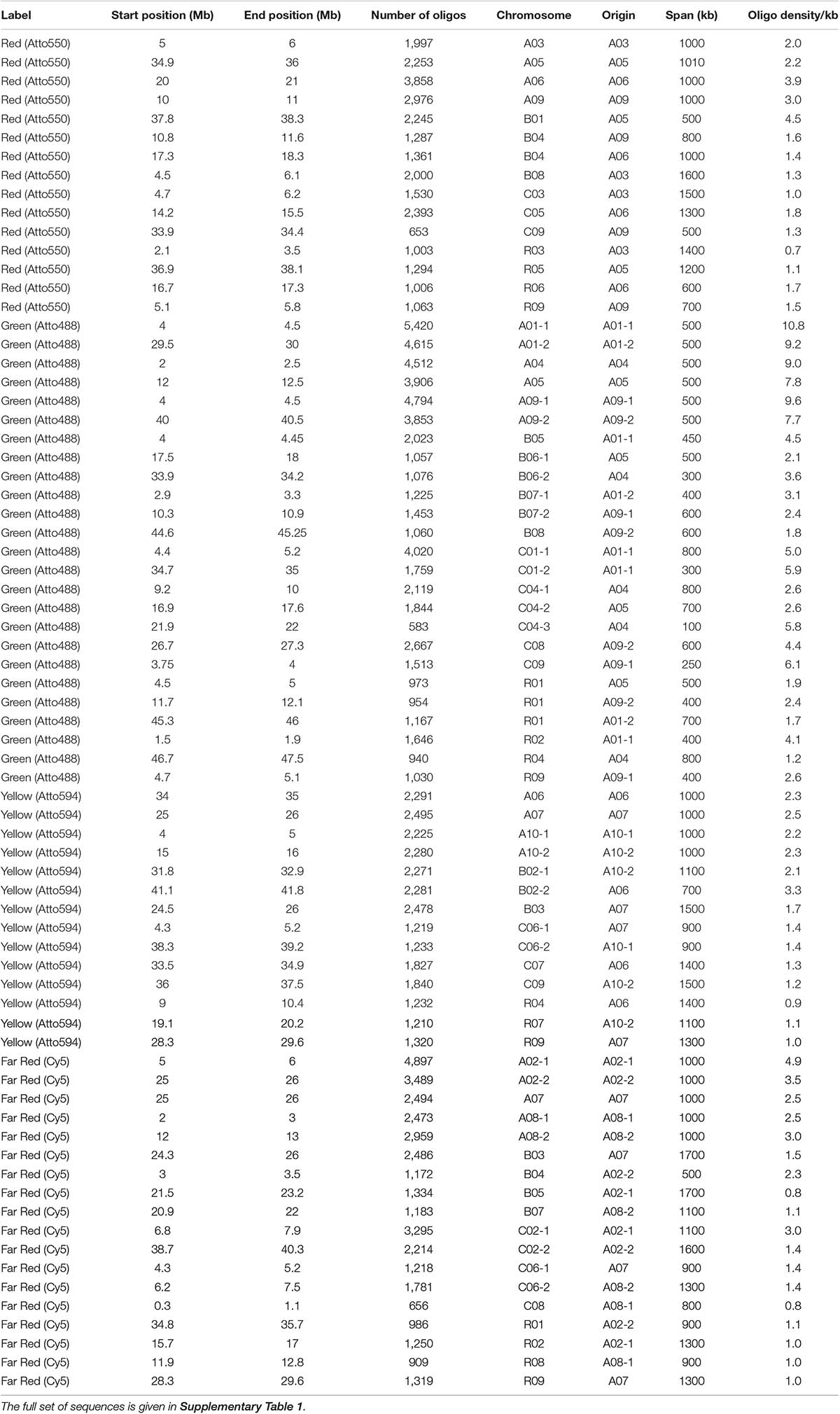
Table 1. Details of design of synthetic massive oligonucleotide pools at each chromosomal locus (A, B, and C genomes and chromosomes number), with start and end position along the sequence and this span, the number of oligonucleotides (oligos) designed, density of oligonucleotides over the region, and fluorochrome label used.
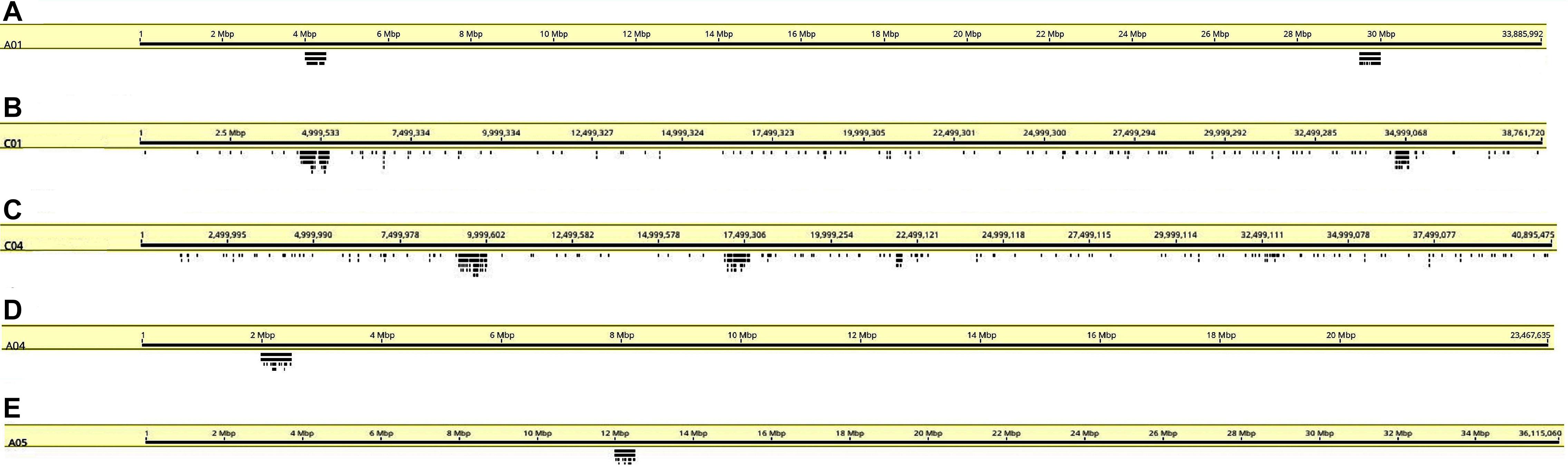
Figure 4. Coverage graphs (stacked bars below chromosomes) depicting hybridization simulations of oligonucleotide libraries made from the B. rapa A genome on chromosomes (A) A01, (B) C01, (C) C04, (D) A04, and (E) A05. Sites are only within target regions on the A genome chromosomes as expected from the design strategy, but a few hybridization sites are seen elsewhere on C genome chromosomes (B,C).
at the peaks in C01 was lower than those observed in A01 by around a factor of two, presumably because of divergence of the low-copy sequences in the region. Chromosome C01 is slightly longer than A01 (by about 5 Mbp). The first C01 signal peak was wider than the A01 equivalent, suggesting multiple insertions in C01 regarding A01. However, the second C01 peak was narrower, indicating sequence expansion of A01 relative to C01. C04 peaks shared some similarities with peaks of both A04 and A05, although the relationship between these chromosomes was not as strong as observed between A01 and C01 (Figures 4C–E). In both A04 and C04 the first peak hybridized with the same oligonucleotides, although the hybridization occurred in C04 further into the chromosome by approximately 7.4 Mbp. The sequences on the A05 peak were also present in C04, in a different location and in reverse orientation. Both C04 peaks had low signal intensity, showing divergence. The homoeologous chromosomes have evolved through insertions, deletions and translocations (and also through repetitive sequence homogenization, although this evolutionary mechanism would not be detected by the low-copy oligonucleotides). A dot-plot shows the locations of the 47 bp oligonucleotides on the chromosome-of-origin for a section of B. rapa chromosome A03 (Figures 3a,b) with gaps showing oligonucleotides that were deleted by the selection procedure in Figure 1. Additional dot-plot analyses show the comparison of the A03 chromosomes, with insertions (gaps) and regions of weaker and strong homology with (diagonal lines) (Figures 3c,d) with B. oleracea C03 and Raphanus sativus R06. Differences in the location, size and intensities of the peak signals allowed identification of all chromosomes of A, B, C, and R genomes (Figures 5A–D). In silico developed ideotypes also revealed chromosomal rearrangements such as translocations and fusion events for genome coordinates used for probe synthesis (Figures 6A–Q). As allotetraploid Brassica species evolved from the direct pairwise hybridizations between three diploid species, so the synthesized probes can be efficiently used to identify all chromosomes of B. juncea, B. carinata, and B. napus.
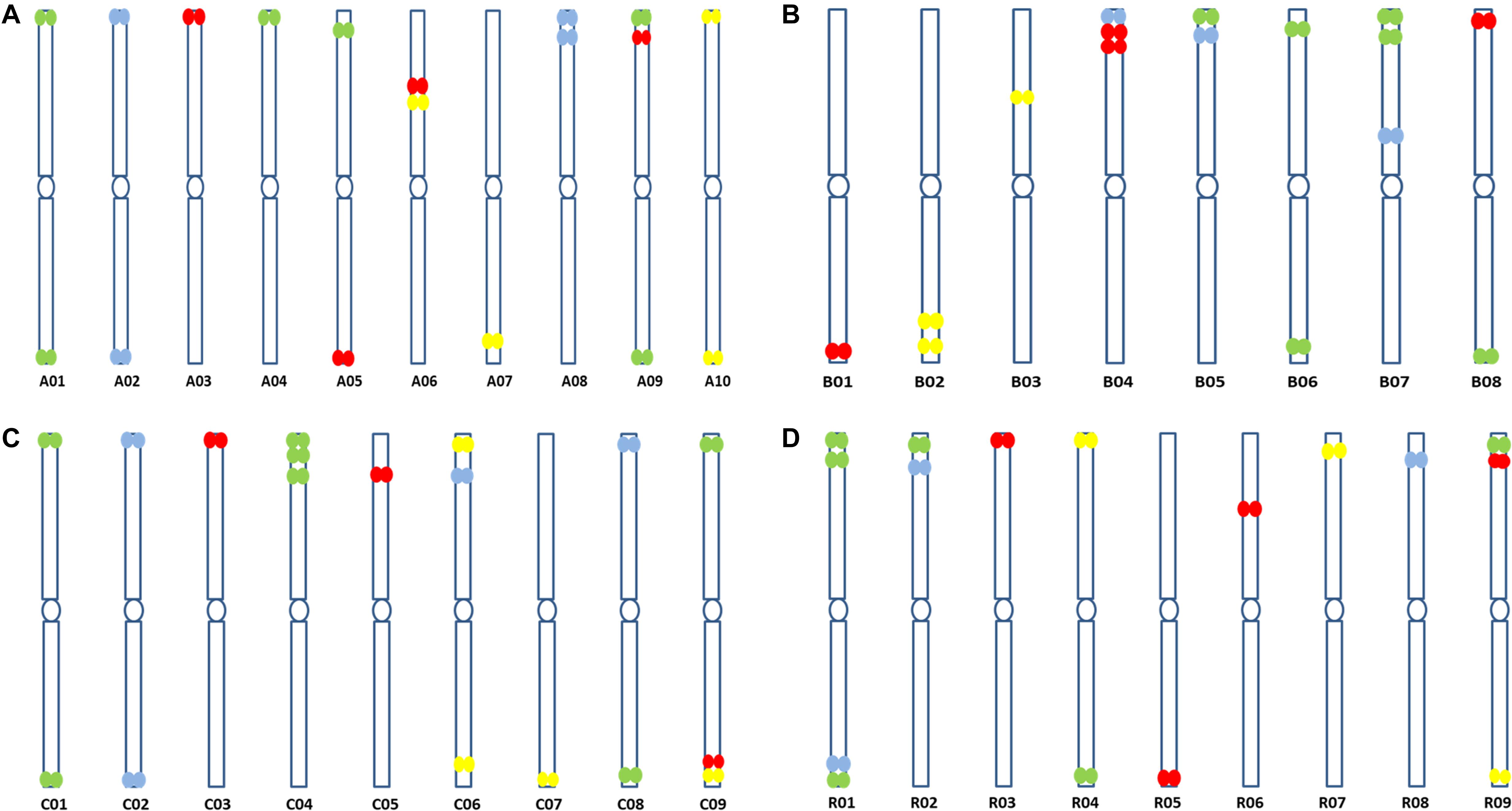
Figure 5. Predicted ideotypes of (A) B. rapa (A genome, 2n = 2x = 20) (B) B. nigra (B genome, 2n = 2x = 16), (C) B. oleracea (C genome, 2n = 2x = 18), and (D) R. sativus (R genome, 2n = 2x = 18) showing sites of hybridization of the four oligonucleotide libraries (red, yellow, green, and cyan) on diagrammatic chromosomes.

Figure 6. (A–Q) Predicted structural chromosomal rearrangements among cultivated genomes of Brassica based on sequence data and locations of hybridization of the four oligonucleotide libraries.
Comparative in situ Studies Using Developed Oligonucleotide-Based FISH Probes
Chromosomal in situ hybridization of the oligonucleotide probe pools on four diploid species B. rapa (cultivars Chiffu-401 and Purple Top Milan), B. nigra, B. oleracea, and Raphanus sativus validated the predicted outcomes. FISH signals derived from the four probes matched exactly to the patterns predicted in B. rapa Chiffu-401, B. nigra, and R. sativus (Figures 7a–d, 8a–h). With B. oleracea, one library (Atto 550, red) produced a pair of extra signals (Figure 8e). This might result from a regional duplication in the genotype used for validation of oligonucleotide probes, based on publicly available genome sequence of B. oleracea, or possible a small duplication that was not assembled. Notably, one commercial cultivar of B. rapa (turnip Purple Top Milan) exhibited a translocation with the yellow-colored library for Chiffu-401 (Figure 8h). Thus, these oligonucleotide libraries detect chromosomal translocations and duplications in different cultivars belonging to the same species.
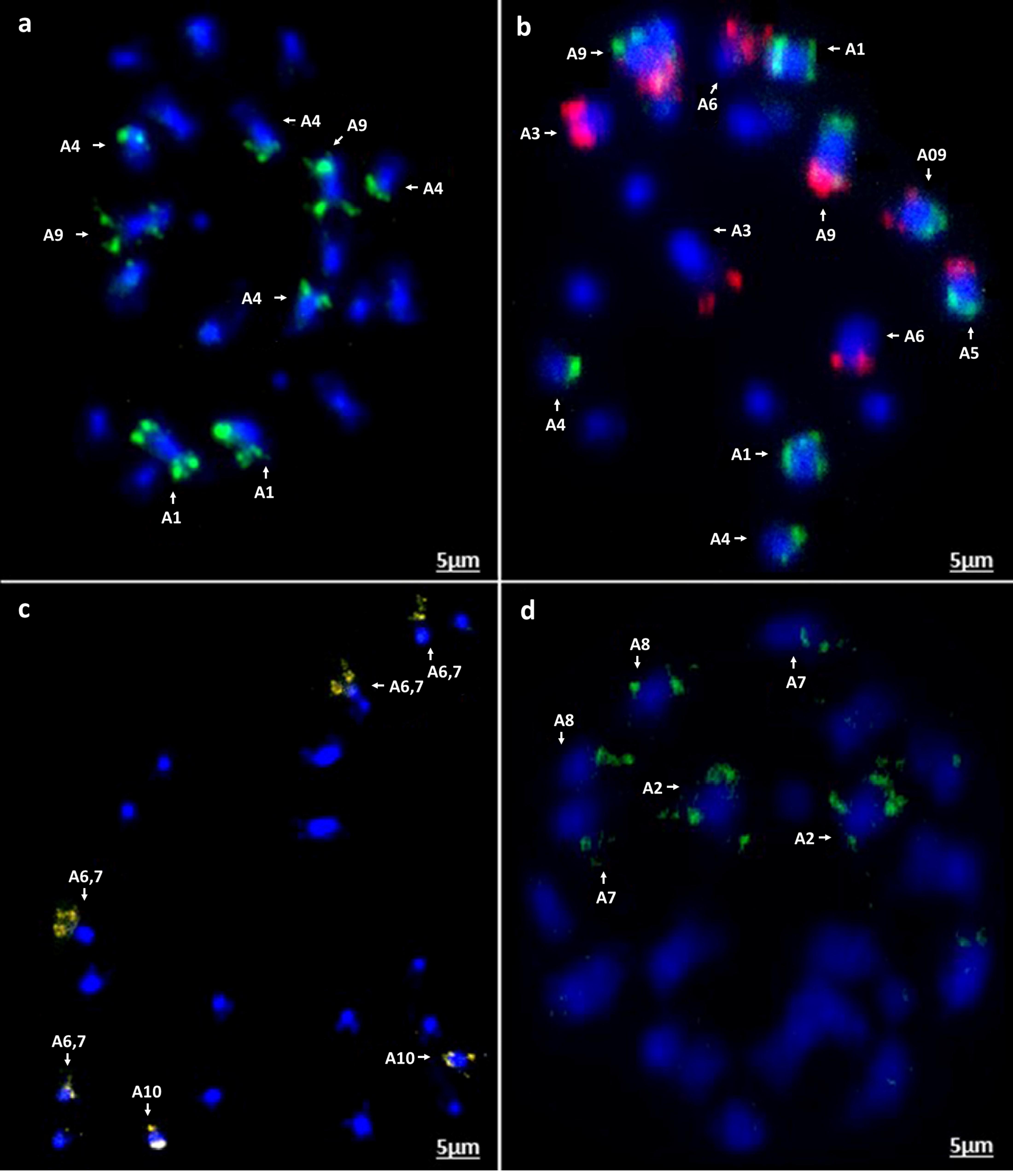
Figure 7. In situ hybridization on mitotic chromosome spreads of B. rapa cv. Chiffu-401 using four oligonucleotide libraries. Chromosomes are stained blue with DAPI and oligonucleotide probe hybridization sites are seen in the other colors (a) green-Atto488, (b) red-Atto550 and green-Atto488 as dual color in situ, (c) yellow-Atto594, and (d) green-Cy5. Scale bars = 5 μm.
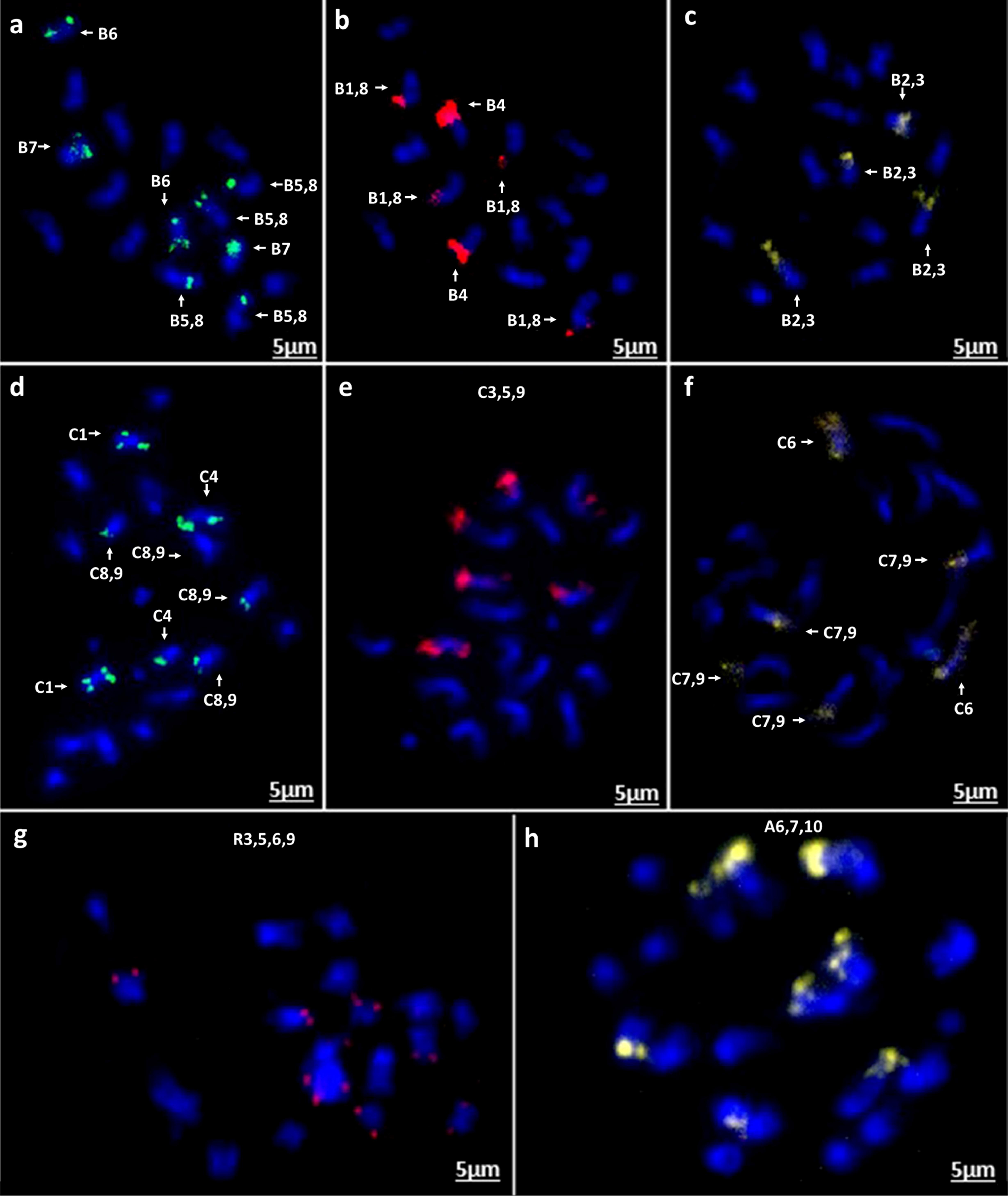
Figure 8. In situ hybridization on mitotic chromosome spreads of B. nigra using four oligonucleotide libraries as Figure 7. (a) green-Atto488, (b) red-Atto550, (c) yellow-Atto594; B. oleracea (d) green-Atto488, (e) red-Atto550 (f) yellow-Atto594; R. sativus (g) red-Atto550; (h) B. rapa (cv. Turnip Purple Top Milan) yellow-Atto594 demonstrating presence of an intraspecific chromosomal translocation compared to B. rapa cv. Chiffu-401 (Figure 7c).
Discussion
We were able to exploit the reference genome of B. rapa to develop massive oligonucleotide pools from 18 chromosomal regions containing only single-copy sequences (Figure 1 and Table 1). These probe pools can be used to identify unambiguously all chromosomes and chromosome arms of crop brassica (A, B, and C) and radish (R) genomes in a single in situ hybridization experiment (Figures 7a–d, 8a–h). Our probes recognized corresponding homoeologous chromosomes and regions across these species and thus are able to anchor sequence maps, check sequence assemblies, and identify chromosomal rearrangements including translocations, inversions and fusion/fission events occurring both within and between species. Our probes designed to specific regions are specific, robust, and identify all chromosomes, unlike repetitive DNA probes developed using repetitive DNA sequences (e.g., 5S, 45S, Cent Br1 and Cent Br2, PBrSTR, PBnSTR, PBoSTR, PBnBH35, and pBcKB4; Harrison and Heslop-Harrison, 1995; Snowdon et al., 1997; Fukui et al., 1998; Kulak et al., 2002; Lim et al., 2005, 2007; He et al., 2015; Xu et al., 2016; Wang et al., 2017; Sun et al., 2019). The in situ hybridization mapping results are largely in concordance with physical genome sequence maps of the four species, achieved by high-coverage (deep) sequencing including use of long-molecule and mate-pair approaches. In the future, the high-quality reference genome sequence will be used to identify genetic polymorphisms including SNPs (single nucleotide polymorphisms) by re-sequencing, Genotyping-by-Sequencing (GBS) or RNA-seq approaches with relatively low coverage. However, chromosomal rearrangements are unlikely to be detected by either shallow or selective sequencing approaches. Using the oligonucleotide pools, we were able to detect an intra-specific rearrangement between the reference sequence of B. rapa Chiffu-401 (pak choi) and turnip Purple Top Milan (Figure 8h). The presence of such a translocation would restrict the ability to exchange genetic materials between the B. rapa varieties by making hybrids to exploit the diversity present. Our probes also detect chromosome rearrangements that precipitated differentiation of Brassica species from the ancestral crucifer karyotype. Oligonucleotide based karyotypes facilitate aligning and numbering of chromosomes in integration with the linkage maps or physical maps, and will also help to improve the quality of genome sequence assembly via identification of gaps or duplications in assembled plant genomes.
The strength and distribution of oligonucleotide signals varied between four species depending upon differences in genetic relationships and genomic sequences, and the results were robust between experiments. Short probes such as the oligonucleotides are valuable in combination with immunolabeling of chromosomal proteins in recombination or chromatin studies (Sepsi et al., 2018). As per phylogenetic reconstructions, A and C genomes of Brassica are more closely related to each other than to B and R genomes (Prakash et al., 2009). In our results, we also found that the C genome chromosomes generated the more similar signals to corresponding A genome chromosomes suggesting greater sequences or genetic similarity. More dispersed or dissimilar signals were obtained on the chromosomes of B and R genomes. The developed probes will also facilitate development of cytogenetic stocks, especially chromosome addition and substitution lines by identifying the chromosomes. Such chromosome stocks are eminently viable in brassicas as these crops can tolerate chromatin gain or loss because of the buffering provided by their palaeopolyploid nature (Cheng et al., 2014; Alix et al., 2017). Random chromosome addition (Prakash et al., 2009) or substitution lines developed in brassicas are stable (Banga, 1988; Gupta et al., 2016), and likely to be of increasing value for breeding as they allow characterization of genes of agronomic and quality relevance.
Oligonucleotide pools will be of significant value for identifying and tracing alien introgressions in brassicas, since the conservation of the low copy sequences, unlike many repetitive DNA elements, is relatively conserved between the sequenced crops and wild species. The genome sequences and gene diversity of many wild Brassicaceae genomes are being studied to expand the genepool available to breeders. The use of alien introgressions and the characterization of recombinant chromosomes is known to be of value in wheat, using probes that label whole alien chromosomes (Patokar et al., 2016), not available in brassicas. In some cases, BAC (Bacterial Artificial Chromosome) probes carrying the genes of interest for introgression in the Brassicaceae can be used (Niemela et al., 2012), but few new BAC libraries are being characterized or even maintained in the 21st century, and the hybridization is difficult to optimize. Because the oligonucleotides are designed in largely single-copy regions of the genome, there is a relatively high conservation with well-studied Brassica genomes. The computational pipelines as described in Figure 1 would be able to identify oligonucleotides with homology to regions assembled from low-coverage reads by mapping the wild genome reads to a phylogenetically distant reference genome, while excluding repetitive genome regions from the wild genomes, without having a high-quality assembly. Furthermore, the strategy would enable design of probes related to regions of interest, for example to track introgression of regions carrying genes of interest in a breeding program involving backcrossing.
Use of synthetic labeled oligonucleotides is convenient and efficient in ensuring all the probes are similarly labeled. However, it is relatively high-cost, requiring some 10 pmol of each probe, so for four labels costing more than United States $100 per slide. Amplification of the oligonucleotides via PCR makes the cost of probes, once synthesized, less prohibitive. Previous approaches have recommended oligonucleotide amplification via an RNA intermediate and extensive optimization of emulsion PCR steps which were not required with the two-step cycles and high denaturation temperature used here. We could show amplification of DNA probes via a robust PCR method to generate hundreds of ng of product from a few picomoles of oligonucleotide-pool. Theoretically, amplification can be selective and there may be self-priming products, but our results here showed no obvious differences using probes labeled during synthesis.
In conclusion, we could identify all Brassica chromosomes in the major A, B, and C genomes, with the use of massive pools of designed synthetic oligonucleotide probes. Following the design strategy including screening against new genome-wide, unbiased repetitive DNA sequence motifs, libraries can be made to target any appropriate chromosomal region. Appropriate designing of probes is critical as even a few repetitive motifs in the oligonucleotides, if not filtered out by the bioinformatic analysis can make the whole library less efficient.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
JH-H, SB, and NA designed the experiments. NA performed the experiments. NA, MG, and JH-H performed the data analysis. NA, MG, SB, and JH-H wrote the manuscript. All authors approved the submission.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported under the Newton-Bhabha Fund United Kingdom-India Pulses and Oilseeds Research Initiative, with funding from United Kingdom’s Official Development Assistance Newton Fund awarded by United Kingdom Biotechnology and Biological Sciences Research Council (BB/R019819/1). The Indian component of the studies was conducted with financial support from Department of Biotechnology, under the project “Germplasm enhancement for crop architecture and defensive traits in Brassica juncea L. Czern. and Coss.” NA acknowledges financial support from DFID (Department for International Development), United Kingdom for the award of Commonwealth split site fellowship during one year of the studies. SB is also grateful to Indian Council of Agricultural Research for salary support from ICAR National Professor Project “Broadening the genetic base of Indian mustard (Brassica juncea) through alien introgressions and germplasm enhancement.”
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.598039/full#supplementary-material
Footnotes
References
Alix, K., Gérard, P. R., Schwarzacher, T., and Heslop-Harrison, J. S. (2017). Polyploidy and interspecific hybridization: partners for adaptation, speciation and evolution in plants. Ann. Bot. 120, 183–194. doi: 10.1093/aob/mcx079
Amosova, A. V., Bolsheva, N. L., Zoshchuk, S. A., Twardovska, M. O., Yurkevich, O. Y., Andreev, I. O., et al. (2017). Comparative molecular cytogenetic characterization of seven Deschampsia (Poaceae) species. PLoS One 12:e0175760. doi: 10.1371/journal.pone.0175760
Banga, S. S. (1988). C-genome chromosome substitution lines in Brassica juncea (L.) Coss. Genetica 77, 81–84. doi: 10.1007/bf00057756
Beliveau, B. J., Boettiger, A. N., Avendaño, M. S., Jungmann, R., McCole, R. B., Joyce, E. F., et al. (2015). Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat. Commun. 12, 1–3. doi: 10.1111/mmi.12942
Bertioli, D. J., Vidigal, B., Nielen, S., Ratnaparkhe, M. B., Lee, T. H., Leal-Bertioli, S. C., et al. (2013). The repetitive component of the A genome of peanut (Arachis hypogaea) and its role in remodelling intergenic sequence space since its evolutionary divergence from the B genome. Ann. Bot. 112, 545–559. doi: 10.1093/aob/mct128
Braz, G. T., He, L., Zhao, H., Zhang, T., Semrau, K., Rouillard, J. M., et al. (2018). Comparative oligo-FISH mapping: an efficient and powerful methodology to reveal karyotypic and chromosomal evolution. Genetics 208, 513–523. doi: 10.1534/genetics.117.300344
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953.
Cheng, F., Wu, J., Fang, L., Sun, S., Liu, B., Lin, K., et al. (2012). Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PLoS One 7:e36442. doi: 10.1371/journal.pone.0036442
Cheng, F., Wu, J., and Wang, X. (2014). Genome triplication drove the diversification of Brassica plants. Hortic. Res. 15, 14024–14030.
Danilova, T. V., Friebe, B., and Gill, B. S. (2012). Single-copy gene fluorescence in situ hybridization and genome analysis: Acc-2 loci mark evolutionary chromosomal rearrangements in wheat. Chromosoma 121, 597–611. doi: 10.1007/s00412-012-0384-7
Fransz, P., Armstrong, S., Alonso-blanco, C., Fischer, T. C., Torres-Ruiz, R. A., and Jones, G. (1998). Cytogenetics for the model system Arabidopsis thaliana. Plant J. 13, 867–876.
Fukui, K., Nakayama, S., Ohmido, N., Yoshiaki, H., and Yamabe, M. (1998). Quantitative karyotyping of three diploid Brassica species by imaging methods and localization of 45S rDNA loci on the identified chromosomes. Theor. Appl. Genet. 96, 325–330. doi: 10.1007/s001220050744
Gupta, M., Mason, A. S., Batley, J., Bharti, S., Banga, S., and Banga, S. S. (2016). Molecular-cytogenetic characterization of C-genome chromosome substitution lines in Brassica juncea (L.) Czern and Coss. Theor. Appl. Genet. 129, 1153–1166. doi: 10.1007/s00122-016-2692-4
Han, Y., Zhang, T., Thammapichai, P., Weng, Y., and Jiang, J. (2015). Chromosome-specific painting in Cucumis species using bulked oligonucleotides. Genetics 200, 771–779. doi: 10.1534/genetics.115.177642
Harrison, G. E., and Heslop-Harrison, J. S. (1995). Centromeric repetitive DNA sequences in the genus Brassica. Theor. Appl. Genet. 90, 157–165. doi: 10.1007/bf00222197
He, Q., Cai, Z., Hu, T., Liu, H., Bao, C., Mao, W., et al. (2015). Repetitive sequence analysis and karyotyping reveals centromere-associated DNA sequences in radish (Raphanus sativus L.). BMC Plant Biol. 15:105. doi: 10.1186/s12870-015-0480-y
Hou, L., Xu, M., Zhang, T., Xu, Z., Wang, W., Zhang, J., et al. (2018). Chromosome painting and its applications in cultivated and wild rice. BMC Plant Biol. 18:110. doi: 10.1186/s12870-018-1325-2
Huang, Y., Chen, H., Han, J., Zhang, Y., Ma, S., Yu, G., et al. (2020). Species-specific abundant retrotransposons elucidate the genomic composition of modern sugarcane cultivars. Chromosoma 129, 45–55. doi: 10.1007/s00412-019-00729-1
Idziak, D., Betekhtin, A., Wolny, E., Lesniewska, K., Wright, J., Febrer, M., et al. (2011). Painting the chromosomes of Brachypodium-current status and future prospects. Chromosoma 120, 469–479. doi: 10.1007/s00412-011-0326-9
Idziak, D., Hazuka, I., Poliwczak, B., Wiszynska, A., Wolny, E., and Hasterok, R. (2014). Insight into the karyotype evolution of Brachypodium species using comparative chromosome barcoding. PLoS One 9:e93503. doi: 10.1371/journal.pone.0093503
Jiang, J., and Gill, B. S. (2006). Current status and the future of fluorescence in situ hybridization (FISH) in plant genome research. Genome 49, 1057–1068. doi: 10.1139/g06-076
Kato, A., Lamb, J. C., and Birchler, J. A. (2004). Chromosome painting using repetitive DNA sequences as probes for somatic chromosome identi?cation in maize. Proc. Natl. Acad. Sci. U.S.A. 101, 13554–13559. doi: 10.1073/pnas.0403659101
Komuro, S., Endo, R., Shikata, K., and Kato, A. (2013). Genomic and chromosomal distribution patterns of various repeated DNA sequences in wheat revealed by a fluorescence in situ hybridization procedure. Genome 56, 131–137. doi: 10.1139/gen-2013-0003
Koo, D. H., Zhao, H., and Jiang, J. (2016). Chromatin-associated transcripts of tandemly repetitive DNA sequences revealed by RNA-FISH. Chromosome Res. 24, 467–480. doi: 10.1007/s10577-016-9537-5
Krivankova, A., Kopecky, D., Stoces, S., Dolezel, J., and Hribova, E. (2017). Repetitive DNA: a versatile tool for karyotyping in Festuca pratensis huds. Cyto Genome Res. 151, 96–105. doi: 10.1159/000462915
Kulak, S., Hasterok, R., and Maluszynska, J. (2002). Karyotyping of Brassica amphidiploids using 5S and 25S rDNA as chromosome markers. Hereditas 137, 79–80.
Kulikova, O., Gualtieri, G., Geurts, R., Kim, D. J., Cook, D., Huguet, T., et al. (2001). Integration of the FISH pachytene and genetic maps of Medicago truncatula. Plant J. 27, 49–58. doi: 10.1046/j.1365-313x.2001.01057.x
Lagercrantz, U. (1998). Comparative mapping between Arabidopsis thaliana and Brassica nigra indicates that Brassica genomes have evolved through extensive genome replication accompanied by chromosome fusions and frequent rearrangements. Genetics 150, 1217–1228.
Lagercrantz, U., and Lydiate, D. J. (1996). Comparative genome mapping in Brassica. Genetics 144, 1903–1910.
Lan, T. H., DelMonte, T. A., Reischmann, K. P., Hyman, J., Kowalski, S. P., McFerson, J., et al. (2000). An EST-enriched comparative map of Brassica oleracea and Arabidopsis thaliana. Genome Res. 10, 776–788. doi: 10.1101/gr.10.6.776
Lim, K. B., De Jong, H., Yang, T. J., Park, J. Y., Kwon, S. J., Kim, J. S., et al. (2005). Characterization of rDNAs and tandem repeats in the heterochromatin of Brassica rapa. Mol. Cell 19, 41–55.
Lim, K. B., Yang, T. J., Hwang, Y. J., Kim, J. S., Park, J. Y., Kwon, S. J., et al. (2007). Characterization of the centromere and peri−centromere retrotransposons in Brassica rapa and their distribution in related Brassica species. Plant J. 49, 173–183. doi: 10.1111/j.1365-313x.2006.02952.x
Liu, S., Liu, Y., Yang, X., Tong, C., Edwards, D., Parkin, I. A., et al. (2014). The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 23:3930.
Lyons, E., Pedersen, B., Kane, J., Alma, M., Ming, R., Tang, H., et al. (2008). Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol. 148, 1772–1781. doi: 10.1104/pp.108.124867
Lysak, M. A., Berr, A., Pecinka, A., Schmidt, R., McBreen, K., and Schubert, I. (2006). Mechanisms of chromosome number reduction in Arabidopsis thaliana and related Brassicaceae species. Proc. Natl. Acad. Sci. U.S.A. 103, 5224–5229. doi: 10.1073/pnas.0510791103
Lysak, M. A., Cheung, K., Kitschke, M., and Bureš, P. (2007). Ancestral chromosomal blocks are triplicated in Brassiceae species with varying chromosome number and genome size. Plant Physiol. 145, 402–410. doi: 10.1104/pp.107.104380
Lysak, M. A., Koch, M. A., Pecinka, A., and Schubert, I. (2005). Chromosome triplication found across the tribe Brassicae. Genome Res. 15, 516–525. doi: 10.1101/gr.3531105
Mandakova, T., and Lysak, M. A. (2008). Chromosomal phylogeny and karyotype evolution in x= 7 crucifer species (Brassicaceae). Plant Cell 20, 2559–2570. doi: 10.1105/tpc.108.062166
Mandakova, T., Marhold, K., and Lysak, M. A. (2013). The widespread crucifer species Cardamine flexulosa is an allotetraploid with a conserved subgenomic structure. New Phytol. 201, 982–992. doi: 10.1111/nph.12567
Mandakova, T., Pouch, M., Brock, J. R., Al-Shehbaz, I. A., and Lysak, M. A. (2019). Origin and evolution of diploid and allopolyploid camelina genomes were accompanied by chromosome shattering. Plant Cell 31, 2596–2612.
Meng, Z., Zhang, Z., Yan, T., Lin, Q., Wang, Y., Huang, W., et al. (2018). Comprehensively characterizing the cytological features of Saccharum spontaneum by the development of a complete set of chromosome-specific oligo probes. Front. Plant Sci. 9:1624.
Mukai, Y., Nakahara, Y., and Yamamoto, M. (1993). Simultaneous discrimination of the three genomes in hexaploid wheat by multicolor ?uorescence in situ hybridization using total genomic and highly repeated DNA probes. Genome 36, 489–494. doi: 10.1139/g93-067
Niemela, T., Seppänen, M., Badakshi, F., Rokka, V. M., and Heslop-Harrison, J. P. (2012). Size and location of radish chromosome regions carrying the fertility restorer Rfk1 gene in spring turnip rape. Chromosome Res. 20, 353–361. doi: 10.1007/s10577-012-9280-5
Novak, P., Neumann, P., Pech, J., Steinhaisl, J., and Macas, J. (2013). RepeatExplorer: a galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29, 792–803. doi: 10.1093/bioinformatics/btt054
Patokar, C., Sepsi, A., Schwarzacher, T., Kishii, M., and Heslop-Harrison, J. S. (2016). Molecular cytogenetic characterization of novel wheat-Thinopyrum bessarabicum recombinant lines carrying intercalary translocations. Chromosoma 125, 163–172. doi: 10.1007/s00412-015-0537-6
Pecinka, A., Schubert, V., Meister, A., Kreth, G., Klatte, M., Lysak, M. A., et al. (2004). Chromosome territory arrangement and homologous pairing in nuclei of Arabidopsis thaliana are predominantly random except for NOR-bearing chromosomes. Chromosoma 113, 258–269. doi: 10.1007/s00412-004-0316-2
Perumal, S., Koh, C. S., Jin, L., Buchwaldt, M., Higgins, E., Zheng, C., et al. (2020). High contiguity long read assembly of Brassica nigra allows localization of active centromeres and provides insights into the ancestral Brassica genome. bioRxiv doi: 10.1101/2020.02.03.932665
Peters, S. A., Bargsten, J. W., Szinay, D., van de Belt, J., Visser, R. G., Bai, Y., et al. (2012). Structural homology in the Solanaceae: analysis of genomic regions in support of synteny studies in tomato, potato and pepper. Plant J. 71, 602–614. doi: 10.1111/j.1365-313x.2012.05012.x
Prakash, S., Bhat, S. R., Quiros, C. F., Kirti, P. B., and Chopra, V. L. (2009). Brassica and Its Close allies: cytogenetics and evolution. Plant Breed. Rev. 31, 21–187. doi: 10.1002/9780470593783.ch2
Qu, M., Li, K., Han, Y., Chen, L., Li, Z., and Han, Y. (2017). Integrated karyotyping of woodland strawberry (Fragaria vesca) with oligopaint FISH probes. Cytogenet. Genome Res. 153, 158–164. doi: 10.1159/000485283
Said, M., Hribova, E., Danilova, T. V., Karafiatova, M., Cizkova, J., Friebe, B., et al. (2018). The Agropyron cristatum karyotype, chromosome structure and cross-genome homoeology as revealed by fluorescence in situ hybridization with tandem repeats and wheat single-gene probes. Theor. Appl. Genet. 131, 2213–2227. doi: 10.1007/s00122-018-3148-9
Salse, J., and Feuillet, C. (2011). Palaeogenomics in cereals: modeling of ancestors for modern species improvement. Comptes. Rendus. Biol. 334, 205–211. doi: 10.1016/j.crvi.2010.12.014
Santos, F. C., Guyot, R., do Valle, C. B., Chiari, L., Techio, V. H., Heslop-Harrison, P., et al. (2015). Chromosomal distribution and evolution of abundant retrotransposons in plants: gypsy elements in diploid and polyploid Brachiaria forage grasses. Chromosome Res. 23, 571–582. doi: 10.1007/s10577-015-9492-6
Schranz, M. E., Lysak, M. A., and Mitchell-Olds, T. (2006). The ABC’s of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends Plant Sci. 11, 535–542. doi: 10.1016/j.tplants.2006.09.002
Schubert, I., and Lysak, M. A. (2011). Interpretation of karyotype evolution should consider chromosome structural constraints. Trends Genet. 27, 207–216. doi: 10.1016/j.tig.2011.03.004
Schwarzacher, T., and Heslop-Harrison, P. (2000). Practical in situ Hybridization. Milton Park: BIOS Scientific Publishers, UK.
Sepsi, A., Fábián, A., Jäger, K., Heslop-Harrison, J. S., and Schwarzacher, T. (2018). ImmunoFISH: simultaneous visualisation of proteins and DNA sequences gives insight into meiotic processes in nuclei of grasses. Front. Plant Sci. 14:1193.
Šimoníková, D., Nìmeèková, A., Karafiátová, M., Uwimana, B., Swennen, R., Doležel, J., et al. (2019). Chromosome painting facilitates anchoring reference genome sequence to chromosomes in situ and integrated karyotyping in banana (Musa spp.). Front. Plant Sci. 10:1503.
Snowdon, R. J., Kohler, W., and Kohler, A. (1997). Chromosomal localization and characterization of rDNA loci in the Brassica A and C genomes. Genome 40, 582–587. doi: 10.1139/g97-076
Song, Z., Dai, S., Bao, T., Zuo, Y., Xiang, Q., Li, J., et al. (2020). Analysis of structural genomic diversity in Aegilops umbellulata, Ae. markgrafii, Ae. comosa, and Ae. uniaristata by fluorescence in situ hybridization karyotyping. Front. Plant Sci. 9:710.
Sun, D., Wang, C., Zhang, X., Zhang, W., Jiang, H., Yao, X., et al. (2019). Draft genome sequence of cauliflower (Brassica oleracea L. var. botrytis) provides new insights into the C genome in Brassica species. Horticulture Res. 6:82.
Szinay, D., Wijnker, E., van den Berg, R., Visser, R. G., de Jong, H., and Bai, Y. (2012). Chromosome evolution in Solanum traced by cross−species BAC−FISH. New Phytol. 195, 688–698. doi: 10.1111/j.1469-8137.2012.04195.x
Tang, H., Woodhouse, M. R., Cheng, F., Schnable, J. C., Pedersen, B. S., Conant, G., et al. (2012). Altered patterns of fractionation and exon deletions in Brassica rapa support a two-step model of paleohexaploidy. Genetics 190, 1563–1574. doi: 10.1534/genetics.111.137349
Tang, X., Szinay, D., and Lang, C. (2008). Cross-species bacterial artificial chromosome-fluorescence in situ hybridization painting of the tomato and potato chromosome 6 reveals undescribed chromosomal rearrangements. Genetics 180, 1319–1328. doi: 10.1534/genetics.108.093211
Wang, G. X., He, Q. Y., Macas, J., Novak, P., Neumann, P., Meng, D. X., et al. (2017). Karyotypes and distribution of tandem repeat sequences in Brassica nigra determined by fluorescence in situ hybridization. Cytogenet. Genome Res. 152, 158–165. doi: 10.1159/000479179
Wang, J., Lydiate, D. J., Parkin, I. A., Falentin, C., Delourme, R., Carion, P. W., et al. (2011). Integration of linkage maps for the amphidiploid Brassica napus and comparative mapping with Arabidopsis and Brassica rapa. BMC Genomics 12:101.
Wang, W., Huang, S., Liu, Y., Fang, Z., Yang, L., Hua, W., et al. (2012). Construction and analysis of a high-density genetic linkage map in cabbage (Brassica oleracea L. var. capitata). BMC Genomics 13:523. doi: 10.1186/1471-2164-13-523
Xin, H., Zhang, T., Han, Y., Wu, Y., Shi, J., Xi, M., et al. (2018). Chromosome painting and comparative physical mapping of the sex chromosomes in Populus tomentosa and Populus deltoides. Chromosoma 127, 313–321. doi: 10.1007/s00412-018-0664-y
Xiong, Z., and Pires, J. C. (2011). Karyotype and identification of all homoeologous chromosomes of allopolyploid Brassica napus and its diploid progenitors. Genet 187, 37–49. doi: 10.1534/genetics.110.122473
Xu, Z., Xie, B., Wu, T., Xin, X., Man, L., Tan, G., et al. (2016). Karyotyping and identifying all of the chromosomes of allopolyploid Brassica juncea using multicolor FISH. Crop J. 4, 266–274. doi: 10.1016/j.cj.2016.05.006
Yang, J., Liu, D., Wang, X., Ji, C., Cheng, F., Liu, B., et al. (2016). The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 48, 1225–1232. doi: 10.1038/ng.3657
Zhang, X., Scheuring, C., Tripathy, S., Xu, Z., Wu, C., Ko, A., et al. (2006). An integrated BAC and genome sequence physical map of Phytophthora sojae. Mol. Plant Microbe Interact. 19, 1302–1310. doi: 10.1094/mpmi-19-1302
Keywords: Oligo-FISH, chromosomes, translocations, Brassica, karyotypes, oligonucleotides, evolution, genomics
Citation: Agrawal N, Gupta M, Banga SS and Heslop-Harrison JSP (2020) Identification of Chromosomes and Chromosome Rearrangements in Crop Brassicas and Raphanus sativus: A Cytogenetic Toolkit Using Synthesized Massive Oligonucleotide Libraries. Front. Plant Sci. 11:598039. doi: 10.3389/fpls.2020.598039
Received: 23 August 2020; Accepted: 30 November 2020;
Published: 23 December 2020.
Edited by:
Ryo Fujimoto, Kobe University, JapanReviewed by:
Isabelle Colas, The James Hutton Institute, United KingdomByoung-Cheorl Kang, Seoul National University, South Korea
Copyright © 2020 Agrawal, Gupta, Banga and Heslop-Harrison. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: JS (Pat) Heslop-Harrison, cGhoNEBsZWljZXN0ZXIuYWMudWs=; cGhoNEBsZS5hYy51aw==; orcid.org/0000-0002-3105-2167