 Nilsa Emilia Munguambe1
Nilsa Emilia Munguambe1 Shouta Inoue1Zita Demeter1
Shouta Inoue1Zita Demeter1 Yoshiyuki Yamagata2
Yoshiyuki Yamagata2 Hideshi Yasui2Shao-Hui Zheng1
Hideshi Yasui2Shao-Hui Zheng1 Daisuke Fujita1*
Daisuke Fujita1*- 1Tropical Crop Improvement Laboratory, Faculty of Agriculture, Saga University, Saga, Japan
- 2Plant Breeding Laboratory, Faculty of Agriculture, Kyushu University, Fukuoka, Japan
Hybrid breakdown, a form of postzygotic reproductive barrier, has been reported to hinder gene flow in many crosses between wild and cultivated rice. Here, the phenomenon of hybrid breakdown was observed as low-tillering (i.e., low tiller number) in some progeny of an interspecific cross produced in an attempt to introduce Oryza meridionalis Ng (W1625) chromosomal segments into Oryza sativa L. ssp. japonica “Taichung 65” (T65). Low-tillering lines were obtained in BC4-derived progeny from a cross between W1625 and “Taichung 65,” but the locus for low-tillering could not be mapped in segregating populations. As a second approach to map the locus for low-tillering, we analyzed an F2 population derived from a cross between the low-tillering lines and a high-yielding indica cultivar, “Takanari.” A major QTL for low-tillering, qLTN4, was detected between PCR-based markers MS10 and RM307 on the long arm of chromosome 4, with a LOD score of 15.6. The low-tillering phenotype was associated with weak growth and pale yellow phenotype; however, low-tillering plant had less reduction of grain fertility. In an F4 population (4896 plants), 563 recombinant plants were identified and the low-tillering locus was delimited to a 4.6-Mbp region between markers W1 and C5-indel3729. This region could not be further delimited because recombination is restricted in this region of qLTN4, which is near the centromere. Understanding the genetic basis of hybrid breakdown, including the low-tillering habit, will be important for improving varieties in rice breeding.
Introduction
Panicle number in rice (Oryza sativa L.) is a yield component that is directly influenced by tillering ability during the growth period. Tillers are additional culms (stems) that develop from the main culm and are similar to branches (Smith and Dilday, 2003). Tiller number is an important trait for improving rice varieties in breeding programs. In the late 1980s, new plant type (NPT) breeding was launched at the International Rice Research Institute (IRRI, Los Banos, Philippines) to increase rice yield potential. One of the target traits for the NPT was the low-tillering trait (i.e., low number of tillers) because low-tillering varieties have fewer unproductive tillers (Janoria, 1989). Rice varieties with high tiller number are generally suitable under some conditions; however, excessive tillering can lead to yield reductions due to an increased number of unproductive tillers. Under wet direct-seeding conditions or in locations where drought is expected to occur episodically, desirable genotypes are expected to have few but vigorous tillers (Fujita et al., 2010). Therefore, the development of rice varieties with optimal tiller number for a specific environment could play an important role in increasing production (Kebrom et al., 2012).
Numerous studies have reported that tiller and panicle number (PN) is controlled by multiple genes (Zhu et al., 2011; Hussien et al., 2014). Across these studies, several major genes for tiller number, initially identified through mutant phenotypes, have been isolated and validated. The gene TEOSINTE BRANCHED-1, which negatively regulated lateral branching, was detected on chromosome 3 (Takeda et al., 2003). MONOCULM 1, which encodes a putative GRAS family nuclear protein, was located on chromosome 6 and promotes the formation of axillary buds (Li et al., 2003). Two genes for reduced culm number, RCN8 and RCN9, have been mapped on the long arm of chromosome 1 and the long arm of chromosome 6, respectively (Jiang et al., 2006). The Ltn (low tiller number) gene from “Aikawa 1” was detected on the long arm of chromosome 8 (Fujita et al., 2010), and ltn2 from NPT rice was identified on chromosome 7 at a distance of 2.1 cM from the SSR marker RM21950 (Uddin et al., 2016).
The use of exotic germplasm, including wild rice species, is expected to extend the available genetic variation in cultivated rice. To facilitate improvement of disease and insect resistance, resistance genes from wild rice species have been introduced into cultivated rice varieties (Khush, 1997). However, the identification of genes from wild rice species is difficult because of reproductive barriers between rice cultivars and wild rice species. Thus, there are few reports on exploitation of genes from wild rice species related to agronomic traits such as tiller number. To identify potentially useful genetic factors in wild rice species, Yoshimura et al. (2010) developed a set of introgression lines (ILs) carrying wild donor chromosomes segments in an uniform genetic background. The ILs are important materials for precisely evaluating the phenotype of each plant and fine-mapping genes as single Mendelian factors.
As mentioned above, the introgression of agronomically useful genes from wild rice into cultivated rice is often prevented by reproductive barriers. Hybrid breakdown, a type of postzygotic reproductive barrier, is defined as weakness or sterility in the F2 or later generations (Fukuoka et al., 1998; Yamamoto et al., 2007, 2010; Miura et al., 2008; Kubo, 2013). In previous studies, genes inducing hybrid breakdown have been reported in both inter- and intraspecific crosses of AA genome species. The AA genome wild rice is considered to be the direct ancestor of cultivated rice. The hybrid weakness f-1 gene from Oryza glumaepatula was located on the short arm of chromosome 4 and found to induce hybrid breakdown in the genetic background of japonica rice variety “Taichung 65” (T65) (Sobrizal and Yoshimura, 2009). The hbd1(t) gene from Oryza nivara was located on the short arm of chromosome 2 and produced a similar hybrid breakdown phenotype in the “Koshihikari” genetic background (Miura et al., 2008). To our knowledge there are no other reports of hybrid breakdown genes involving other AA-genome species.
To broaden the available rice genetic resources from wild species, O. meridionalis (AA genome), a wild rice that is endemic to Oceania, New Guinea, and Australia (Sotowa et al., 2013) was used to develop 36 ILs with introduced chromosome segments of O. meridionalis in the genetic background of T65 (Yoshimura et al., 2010). Among these ILs, we identified one line with yellow leaves and greatly reduced tiller number. Through analysis of a segregating population derived from a cross between the low-tillering IL and T65, we identified a locus related to low-tillering but could not map it in that population. By crossing the low-tillering line to another cultivar (“Takanari”), producing segregating populations, and performing substitution mapping, we delimited a major QTL for low-tillering.
Materials and Methods
Plant Materials Containing Low-Tillering Trait in T65 Genetic Background
In a previous study O. meridionalis (W1625) was crossed with O. sativa ssp. japonica “Taichung 65”, and the F1 plants were repeatedly backcrossed to T65 as the recurrent parent (Figure 1; Yoshimura et al., 2010). A set of W1625 ILs (36 lines) in BC4F4 was selected by marker-assisted selection (MAS) (Yoshimura et al., 2010). One line among the W1625 ILs showed segregation of a particular phenotype with weaker growth (low-tillering and yellow leaves) compared with normal plants. To identify the locus controlling low tiller number, a BC4F5 segregating population (derived from a single self-pollinated BC4F4 plant) was used for genetic analysis. To confirm the genotype at the low-tillering locus for each BC4F5 plant, the corresponding BC4F6 lines were used for progeny tests. A set of 119 BC4F5-derived lines was used for analyzing the introgressed chromosomal segments with SSR markers. BC5F1 plants derived from a cross between T65 and low-tillering plant (BC4F5) were used for confirming the effect of the low-tillering trait on other agronomic traits. The BC5F2 plants were evaluated for tillering number and panicle structure.
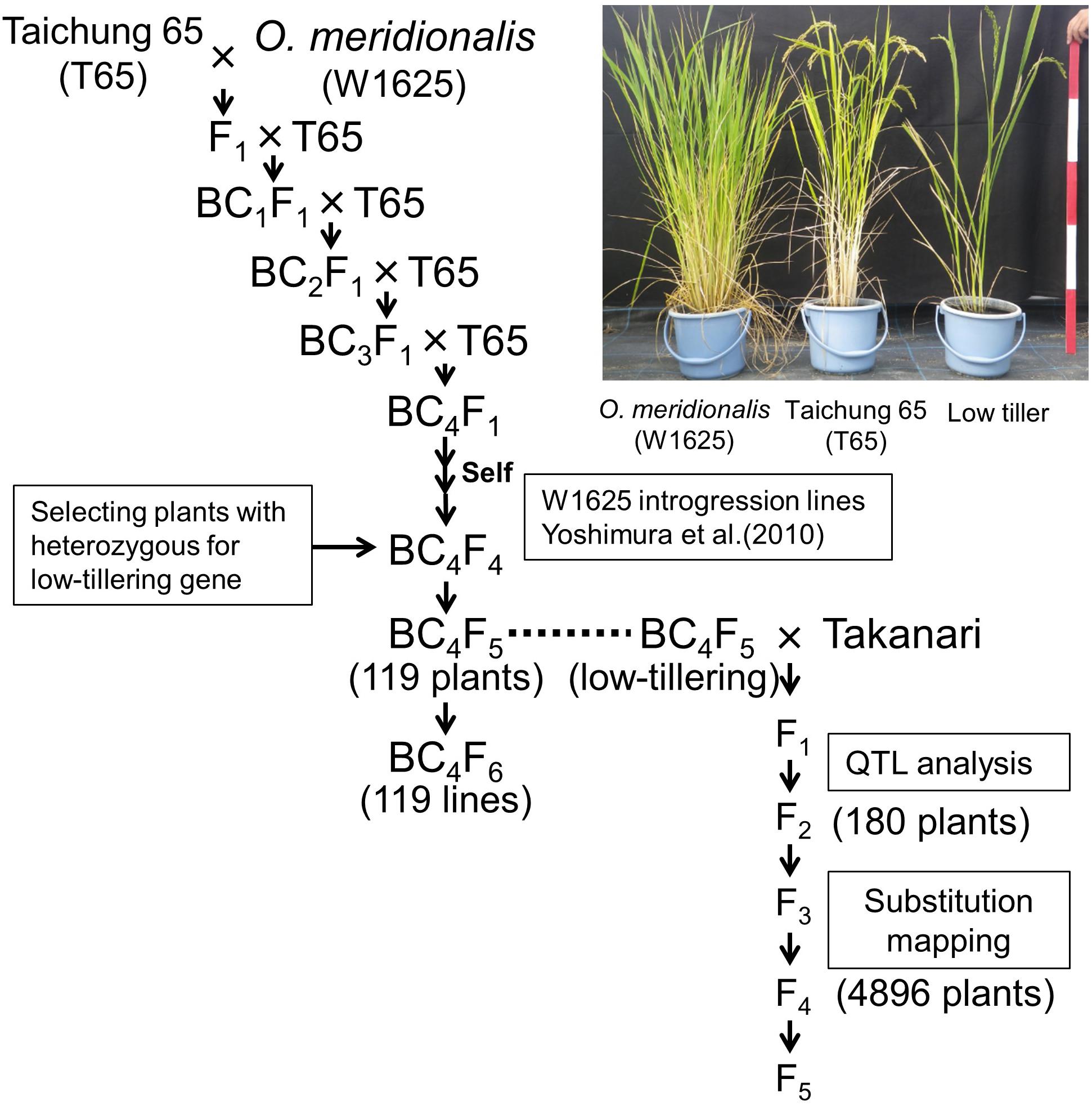
Figure 1. Breeding scheme for developing lines with low-tillering. The BC4F5 and BC4F6 populations were derived from a cross between “Taichung 65” (T65) and O. meridionalis. The F2 population was derived from a cross between low-tillering BC4F5 plants and “Takanari.”
Plant Materials for Mapping the QTL
To map the locus for low-tillering, we developed an F2 population derived from a cross between the low-tillering plant (BC4F5) and a high-yielding indica cultivar, “Takanari.” The F2 plants were self-pollinated to generate F3, F4, and F5 lines for substitution mapping of the low-tillering locus. F2:3 and F4:5 lines were used for progeny tests to confirm the genotypes at the low-tillering locus of the F2 and F4 plants, respectively.
Characterization of Agronomic Traits in Normal- and Low-Tillering Lines
All seeds were incubated at 25°C for 3 days to induce germination and then sown in seeding trays. The seedlings were transplanted to the paddy field at Saga University (Saga, Japan) at 30 days after sowing and planted one plant per hill. The BC4F5 population was planted with 18 cm between plants within a row and 30 cm between rows in an experimental field at Kyushu University (Kasuya, Fukuoka, Japan). The other materials (BC4F6, BC5F1, BC5F2, F2, F3, F4, and F5) were planted with 20 cm between plants within a row and 25 cm between rows in an experimental field at Saga University (Saga, Japan). The PNs per plant were observed at the maturity stage and measured in all segregating populations. In the F3, and F5 generations, which were used for progeny tests (using 20 plants of each entry), plants with fewer than 5 panicles were considered as low-tillering. To examine tiller number after transplanting, 10 plants per entry were transplanted to 5-L pots and tiller numbers were counted every week. To understand the effects on other agronomic traits caused by low-tillering locus, the BC4F6 and BC5F1 plants (10 plants of each entry) were evaluated for culm length (CL), panicle length (PL), PN, leaf length (LL), leaf width (LW), number of primary branches (NPB), number of secondary branches (NSB), total spikelet number per panicle (TSN), grain fertility per panicle (GF), total number of spikelets on the primary branch (TSPB), and total number of spikelet on the secondary branch (TSSB).
DNA Extraction and Genotyping
The genomic DNA of parents and segregating populations was extracted from freeze-dried leaves by using the potassium acetate protocol (Dellaporta et al., 1983). In preparation for extraction, the leaves were collected in 96-well deep-well plates and crushed using a FastPrep 96 (MP Biomedicals, United States). DNA marker genotypes were determined by PCR with a Gene Atlas thermal cycler (ASTEC, Japan). PCR amplification conditions consisted of 96°C for 5 min; followed by 35 cycles of 96°C for 5 min, 55°C for 30 s, and 72°C for 30 s; followed by a final extension of 25°C for 1 min. The PCR products were separated by electrophoresis in 4% agarose gel, stained with ethidium bromide, and viewed under ultraviolet light.
Construction of the Linkage Map and QTL Analysis
The DNA markers were first screened for polymorphism between the low-tillering line and “Takanari” and then used for genotyping the F2 populations. The genetic map for the F2 population was constructed using 113 markers that were polymorphic between the parents and distributed across all 12 chromosomes. In this map, genetic distances were determined using the Kosambi function. The QTL analysis was performed by Windows QTL Cartographer software version 2.5 and conducted using data for PN and DNA marker genotypes (Wang et al., 2012). On the basis of a 1000-permutation test, a LOD score greater than 3.6 was considered as the threshold at a 0.05 level of significance.
Substitution Mapping of the Low-Tillering Locus
To determine the precise location of the low-tillering locus, the tillering phenotypes and genotypes of DNA markers around the low-tillering locus in 180 F3 lines were used to confirm the genotypes at that locus in the F2 population. To further delimit the location of the low-tillering locus, a large segregating F4 population (4896 plants) derived from F3 lines heterozygous at the low-tillering locus were screened with markers RM8213 and RM307 to select recombinant plants. The genotypes of recombinant plants were analyzed using nine SSR markers (RM16459, RM16502, RM16535, RM16550, RM16605, RM16626, and RM307) and three Indel markers (RH7, W1, C5-indel3729, C5-indel3743, and C5-indel3757) around the low-tillering locus (Supplementary Table 1). Finally, an F5 progeny test was performed to confirm the genotype of the low-tillering locus for each F4 individual.
Results
Identification of the Low-Tillering Locus
The tiller numbers per plant of W1625, T65, and a low-tillering line (BC4F6 generation which seems to have had homozygous low-tillering lines) were evaluated every week during the tillering stage (Figure 2). Throughout the tiller development stage, the low-tillering line continually showed few tillers. The PNs at maturity of W1625, T65, and the low-tillering line were 44, 13, and 4, respectively. The number of panicles per plant of W1625 was higher than that of T65 at the maturing and greatly reduced in the low-tillering line compared with both parents. These data suggested that the total number of panicles in the low-tillering line (with T65 genetic background) was affected by one or more introgressed segments from W1625.
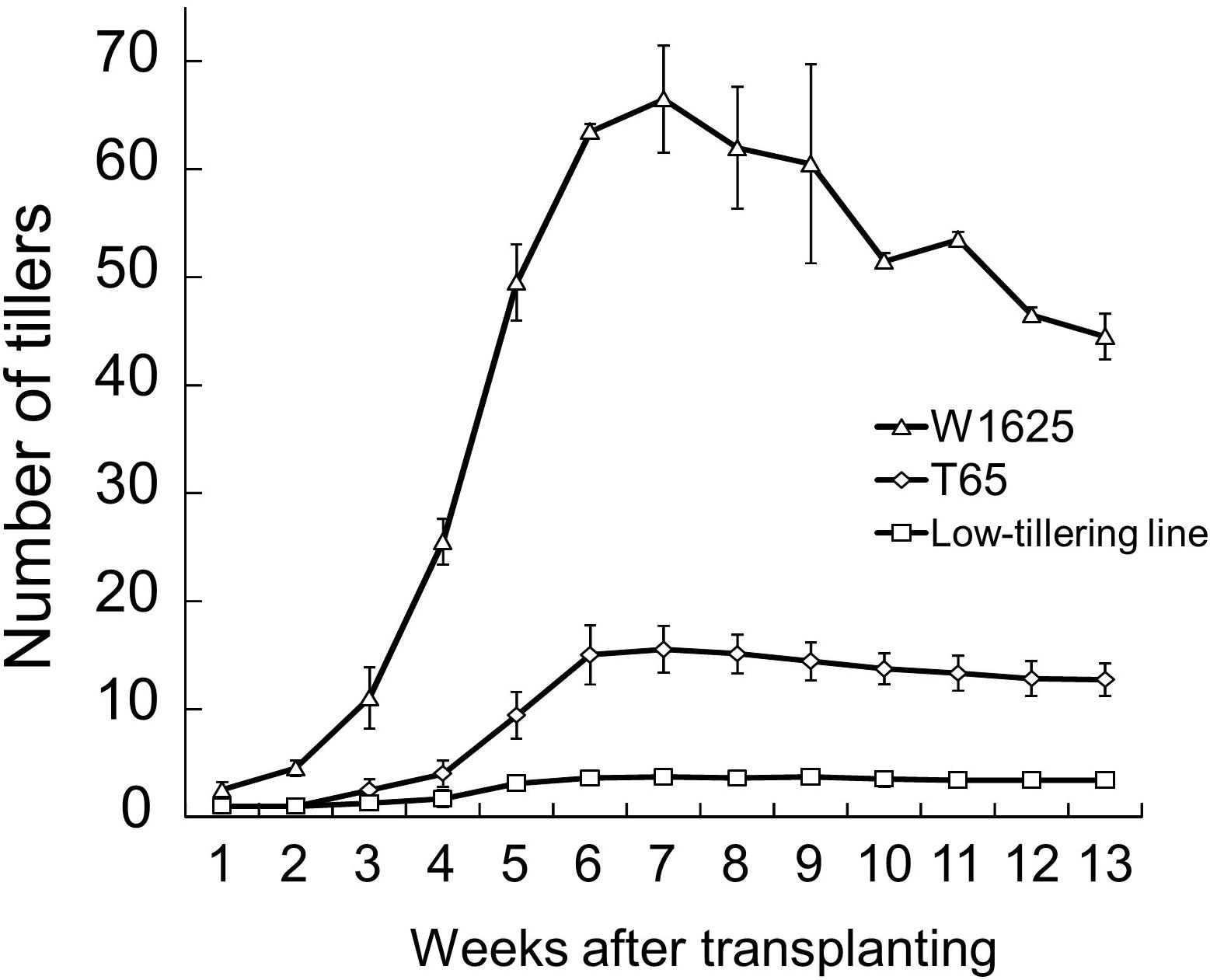
Figure 2. Fluctuation in tiller number during plant development in W1625, T65, and the low-tillering line. Data are means ± SD (n = 10).
To identify the locus for low-tillering, the BC4F5 segregating population was analyzed. The frequency distribution of PN in that population showed a bimodal distribution: 15 plants with low-tillering (one panicle per plant) and 104 plants with PN ranging from 4 to 10 (Figure 3A). To elucidate the genotypes at the low-tillering locus in the BC4F5 segregating population, the PNs in BC4F6 lines were observed as a progeny test. Among the 119 BC4F5 plants, 15 plants were homozygous for the W1625 allele at the low-tillering locus (progeny were all low-tillering), 60 plants were heterozygous (progeny segregated for low-tillering phenotype), and 44 plants were homozygous for the T65 allele (progeny were all normal tillering). The segregation ratio in the BC4F5 population (15:60:44) did not fit the 1:2:1 expected ratio (χ2 = 14.1, P = 0.001).
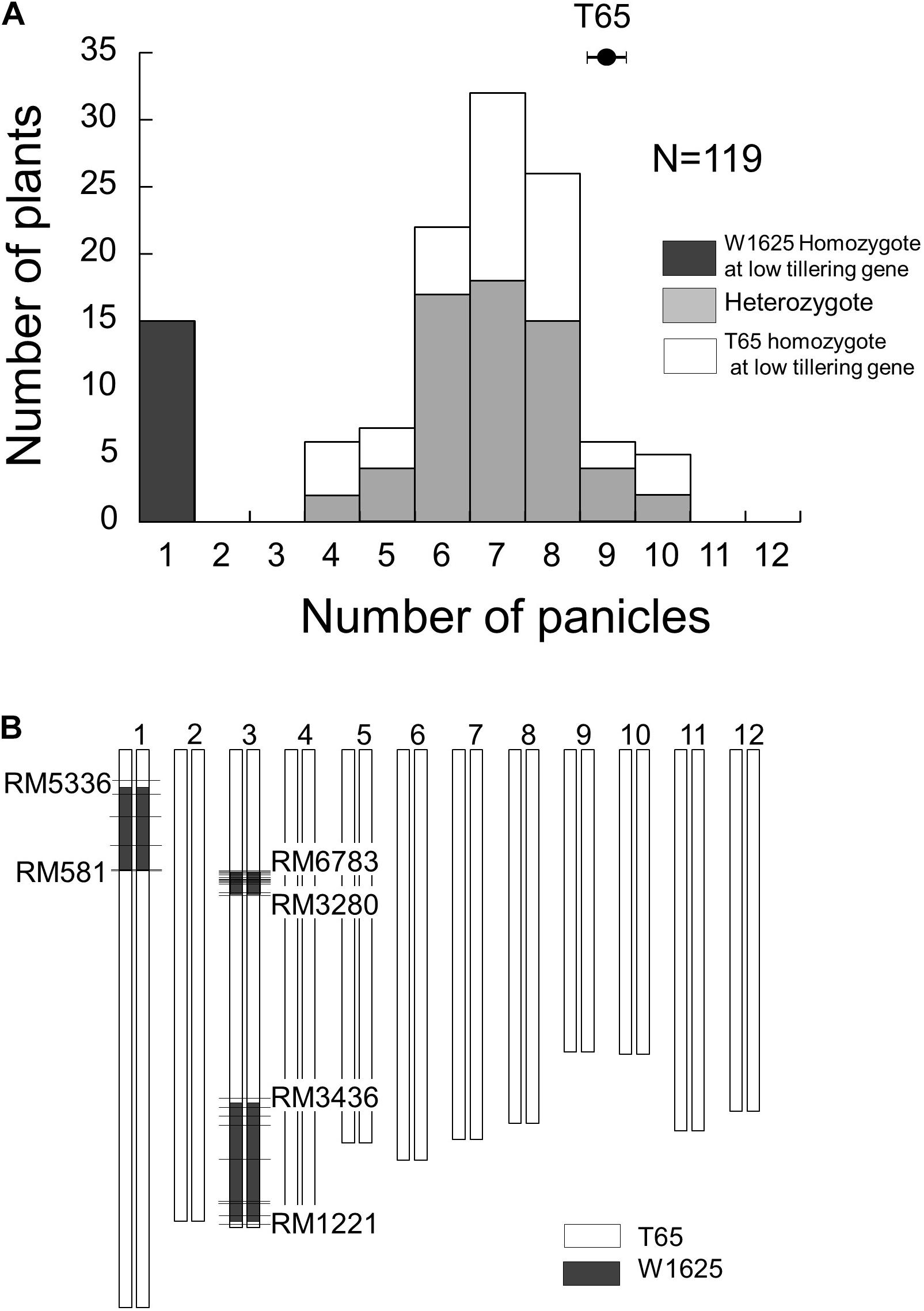
Figure 3. Frequency distribution of panicle number (A) and graphical genotype of low-tillering line (B) in BC4F5 population derived from a cross between T65 and W1625. Black, gray, and white colors in (A) indicate W1625 homozygote, heterozygote, and T65 homozygote for low-tillering locus through scored based on segregation of phenotypes in the next generation. The horizontal bars in (B) show the location of polymorphic SSR markers.
Identification of W1625 Chromosomal Segments Retained in the BC4F5 Population
The chromosomal constitution of the low-tillering line was assessed by using 492 SSR markers (Figure 3B). There were three introgressed segments from W1625 detected in the low-tillering line: one on the short arm of chromosome 1 and two on chromosome 3. Additionally, through genotyping by sequencing, three large introgressed segments from W1625 on the low-tillering line were identified at same regions using 5445 SNPs and there are three small regions introgressed segment from W1625 on chromosomes 4, 10, and 12 (Supplementary Figure 1). There was no association between tiller number and genotype of these three large W1625 segments in the BC4F5 segregating population. In BC5F2 segregating population, there are no association between agronomic traits (PN, TSN, and SNB) and genotype of these three large W1625 segments and the TSN and SNB of BC5F2 plants with low-tillering showed lower than that of T65 (Supplementary Figure 2). These results suggested that other chromosomes might harbor small introgressed segments from W1625 that are related to the low-tillering phenotype, TSN and SNB.
Detection of a QTL for Panicle Number
Because we were unable to detect the location of the low-tillering locus in the BC4F5 population, we developed an F2 population from a cross between the low-tillering line and “Takanari” to conduct QTL analysis. The PNs of the parents were 4 for the low-tillering line and 11 for “Takanari” (Figure 4). The frequency distribution for PN per plant in the F2 population showed a continuous distribution with approximately 60% of the individuals having a higher PN than the parents. To estimate the location of the QTL for PN, a QTL analysis was conducted using this F2 population. A QTL designated qLTN4 (QTL for low tiller number on chromosome 4) was detected between MS10 and RM307 on the short arm of chromosome 4 (Table 1). This QTL had a LOD score of 15.6 and explained 30.4% of the phenotypic variation for PN. The allele from the low-tillering line at qLTN4 was associated with decreased PN per plant. Additionally, five regions on chromosomes 5, 6, 8, 10, and 11 had LOD peaks and LOD score is less than threshold of LOD score at 5% level. The alleles from the low-tillering line at two QTLs, qLTN5, and qLTN6, showed increasing PN per plant, while the alleles from the low-tillering line at three QTLs, qLTN8, qLTN10, and qLTN11, showed decreasing PN per plant.
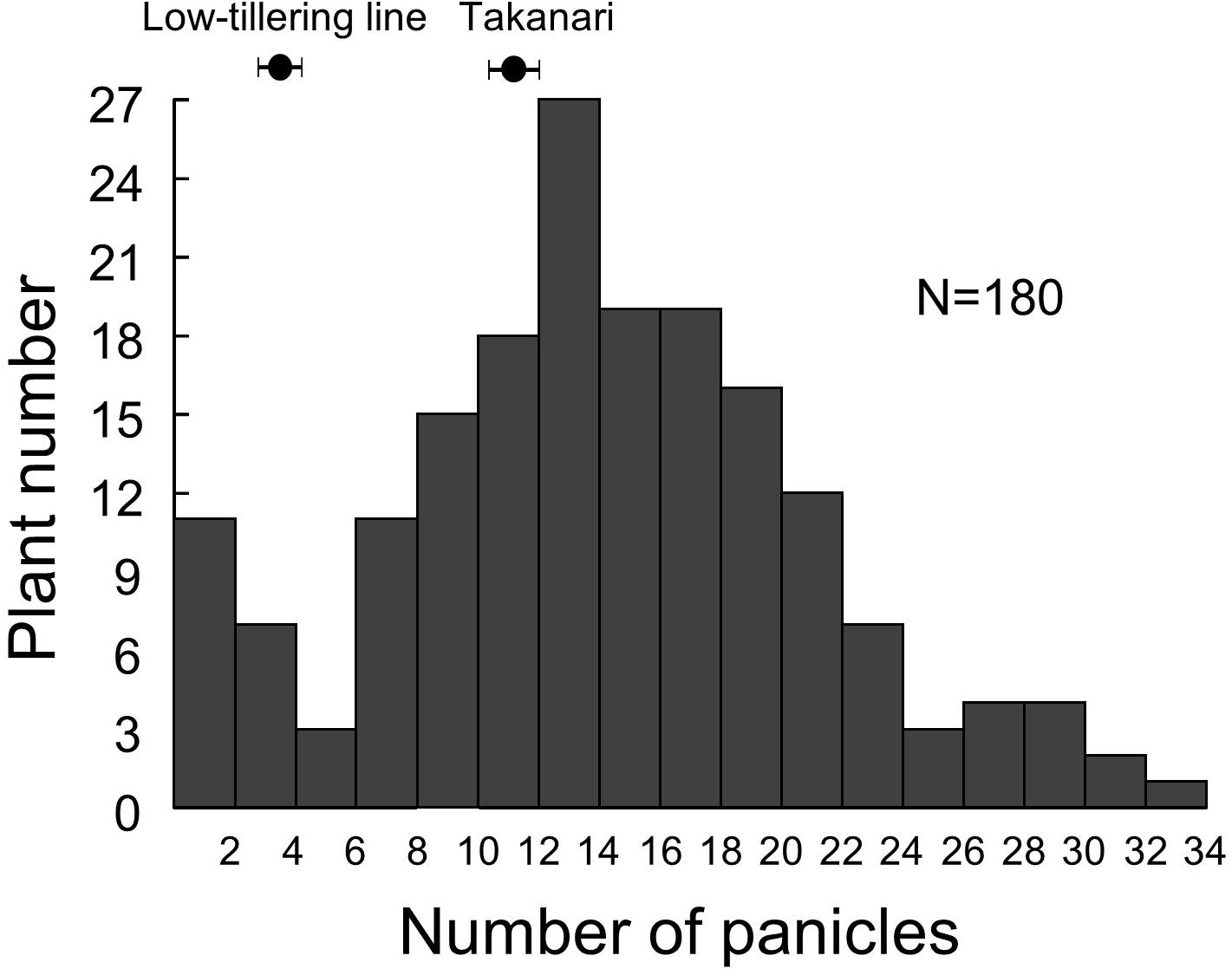
Figure 4. Frequency distribution of panicle number in F2 population derived from a cross between low-tillering plants and “Takanari.”
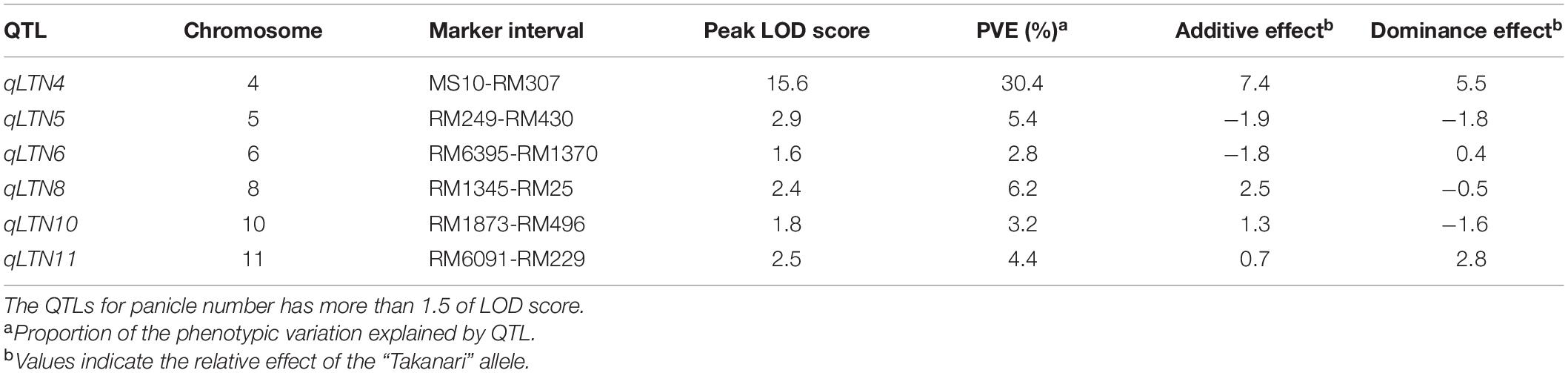
Table 1. The QTL for tiller number identified in an F2 population derived from a cross between “Takanari” and low-tillering line.
Substitution Mapping of qLTN4
To delimit the location of qLTN4, a progeny test was performed using F3 lines. These lines, derived from F2 plants with recombination between markers RM8213 and RM307, were classified into three phenotypes: low-tillering fixed, segregating, and normal-tillering fixed. By comparing F2 genotypes and F3 phenotypes, qLTN4 was located within an 8.8-Mbp interval between markers RM8213 and RM307 (Figure 5A). To further refine the location of qLTN4, recombinant plants were selected from 13 F4 families with heterozygosity at qLTN4. Among a large segregating population (4896 F4 plants), 563 plants with a recombination event between markers RM8213 and RM307 were selected. These 563 plants were evaluated for PN and genotyped using PCR-based markers. Based on the phenotypes and genotypes at each marker for 13 recombinant plants, lines ISC11-3 and ISC8-18, with recombination that took place between markers W1 and C5-indel3729, delimited the low-tillering locus to a 4.6-Mbp region (Figure 5B).
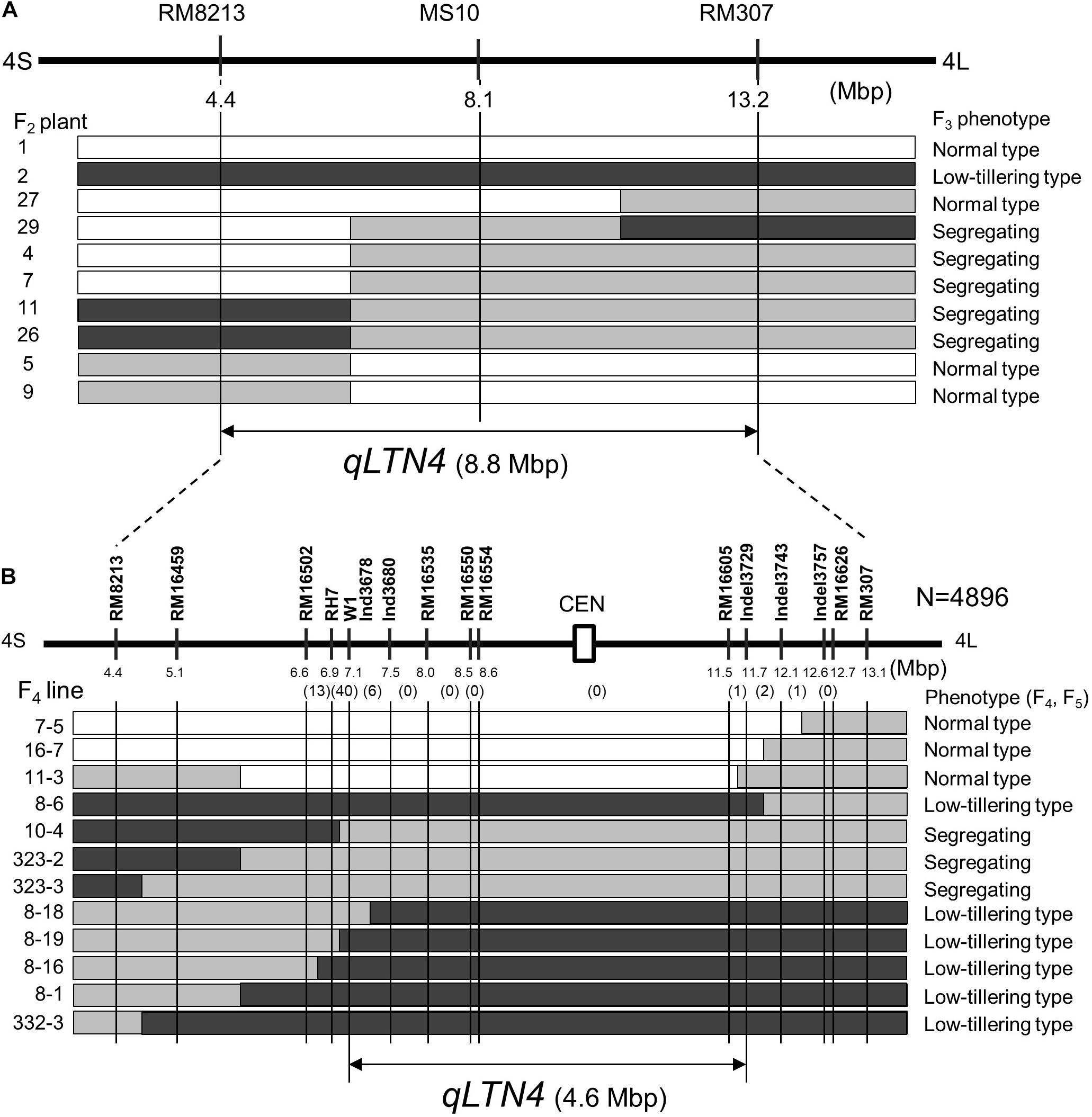
Figure 5. Substitution map of locus for low tiller number. (A) Substitution mapping of qLTN4 in F2 plants and F3 lines derived from a cross between “Takanari” and the low-tillering line. (B) Substitution mapping of qLTN4 in F4 and F5 lines. Black, gray, and white indicate regions homozygous for marker allele from low-tillering line, heterozygous, and homozygous for marker allele from “Takanari.” The white rectangle with CEN indicate centromere region of chromosome 4.
Characterization of Agronomic Traits Affected by qLTN4 on Low-Tillering Line
To determine the influence of qLTN4 on agronomic traits, we characterized panicle architecture and other traits in the low-tillering line (BC4F6). CL and PN, of the low-tillering line were significantly lower than those of T65 despite their similar genetic background (Table 2). On the other hand, LW was significantly larger in the low-tillering line than in T65 or the BC5F1. BC5F1 plants derived from a cross between T65 and the low-tillering line had similar agronomic traits to T65 with the exception of PL, which was significantly larger in the BC5F1. There were no significant differences for CL, PN, LL, and LW between T65 and BC5F1 plants. Several traits related to panicle architecture, i.e., TSPB, TSSB, and TSN, were significantly lower in the low-tillering line than in T65. The grain fertility of the low-tillering line was 75.4%, significantly lower than that of T65 (94.5%; Table 3). However, NPB, NSB, and TSPB of the BC5F1 were not significantly different from those of the low-tillering line.
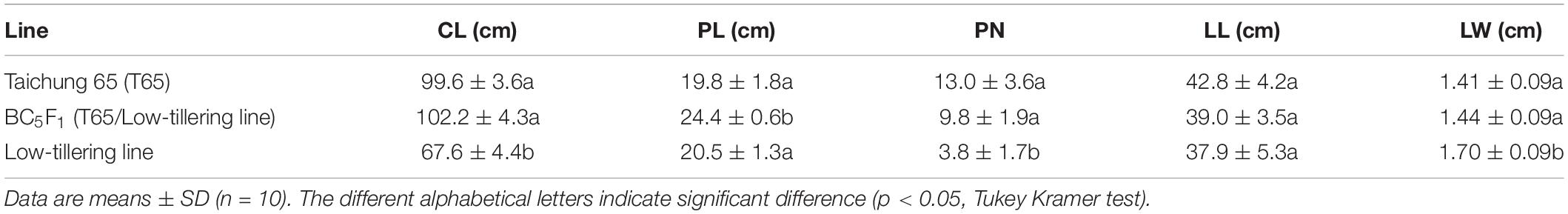
Table 2. Agronomic traits of “Taichung 65” (T65), BC5F1 (T65/low-tillering line), and low-tillering line.
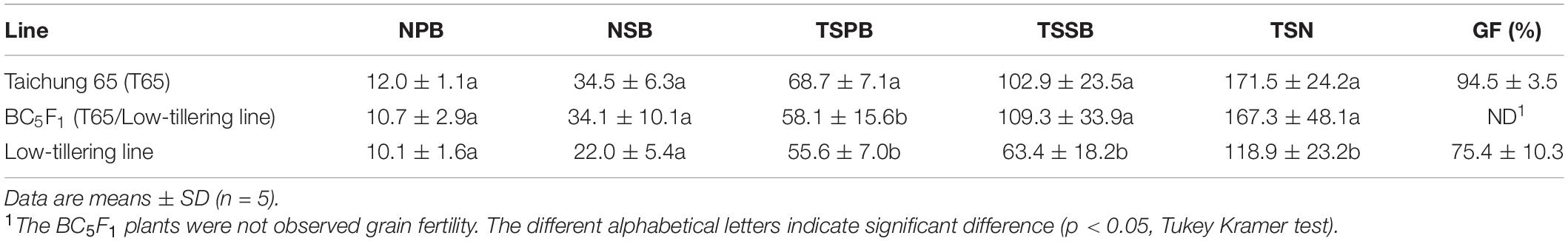
Table 3. Panicle structure of “Taichung 65” (T65), BC5F1 (T65/low-tillering line), and low-tillering line.
Discussion
Tillering, i.e., the production of multiple stems, is an important agronomic trait that has been widely studied in cereals. In previous studies, genes and QTLs with large effects on tiller number were mapped in various regions of the rice genome. MONOCULM 1 was located on chromosome 6 (Li et al., 2003), Ltn from “Aiwaka 1” on chromosome 8 (Fujita et al., 2010), and ltn2 from an NPT variety on chromosome 7 (Uddin et al., 2016). Among the QTLs and genes for tiller number on chromosome 4, tn4-1 was located between markers RG190 and RG908 and was detected by the conditional QTL method (Yan et al., 1998). The QTL detected in the present study, qLTN4, was detected on chromosome 4 and had a large contribution to the phenotypic variation (30.4%) and high LOD score (15.6). The chromosomal region of qLTN4 overlapped with that of tn4-1 (Yan et al., 1998), suggesting that qLTN4 might be same QTL.
In a previous study, segregation distortion as a consequence of nuclear and cytoplasmic factors and zygotic selection was observed in two reciprocal F2 crosses and a BC1 population (Reflinur et al., 2014). Another report mentioned that segregation distortion was caused by other factors such as population size, genotyping errors, and some non-biological factors (Falconer and Mackay, 1996). Furthermore, among different cross combination, different factors may contribute to segregation distortion (Xu et al., 1997). In the present study, the distribution of PN in the BC4F5 segregating population showed a slightly bimodal distribution but did not fit the expected 3:1 ratio. In the F2 population, significant segregation distortion was observed at MS10 (near qLTN4), reducing the frequency of low-tillering plants. Similarly, the low-tillering gene locus segregated in the BC4F5 population as 44 homozygous for the T65 allele, 60 heterozygous and 15 homozygous for the W1625 allele. This same tendency for segregation distortion around qLTN4 in both F2 and BC4F5 populations is evidence for segregation distortion in this region.
The low-tillering lines were characterized in different growth conditions. In BC4F5 population, the plants with 18 cm between plants within a row and 30 cm were grown in Fukuoka, JAPAN. In BC4F6, BC5F1, F2, F3, F4, and F5, the plants with 20 cm between plants within a row and 25 cm were grown in Saga, JAPAN. The PN of low-tillering plant is one in BC4F5 population (Figure 3A), while the PN of low-tillering line of BC4F6 is 3.8 (Table 2). The difference suggested that the PN of low-tillering line is influenced by environmental factors such as transplanting interval. Therefore, we defined plants with fewer than 5 panicles as low-tillering in progeny test (F3 and F5 those were grown in Saga, JAPAN)
The low-tillering line was analyzed for detecting introgression segments from W1625. Based on SSR markers, three segments were identified on chromosomes 1 and 3. However, the chromosome segment from W1625 around the qLTN4 region on the low-tillering line was not detected. Therefore, through genotyping by sequencing, 5445 SNPs was used for detecting W1625 chromosomal segment on low-tillering line. Three large segments on chromosomes 1 and 3 and three additional small segment on chromosomes 4, 10, and 12 have been detected. However, the location of small chromosomal segment of W1625 on chromosome 4 is outside qLTN4 region. Furthermore, we read whole genome sequence of low-tillering lines and several SNPs from W1625 detected around qLTN4 region (Supplementary Figure 3). However, we cannot find exact evidence of association between low-tillering and W1625 chromosomal segment in this area. Therefore, these results suggested that there are possibilities for small introgression segment of W1625 locating on this region or natural mutation of low tiller line. The SNPs and NGS analysis were conducted based on Nipponbare genome sequence, therefore, we might not detect W1625 chromosomal segments that are absent in Nipponbare genome. In future study, de novo sequence of W1625 would be necessary for detecting W1625 chromosomal segments. Also, the detection of qLTN4 on earlier generation of segregation population derived from cross between T65 and W1625 would be possible to confirm association between low-tillering and W1625 chromosomal segments.
Meiotic recombination, which is the exchange of DNA between homologous chromosomes, is a fundamental process in eukaryotic reproduction (Dreissig et al., 2019). Previous studies have reported that recombination rates vary along chromosomes in most organisms with large genomes. High recombination rates were found near the distal ends of the chromosomes while low recombination rates were found in the region surrounding the centromere (Stapley et al., 2017; Haenel et al., 2018). Here, qLTN4 was detected near the centromeric region of chromosome 4. In substitution mapping of qLTN4, we failed to narrow down the candidate region due to difficulty in finding recombinant plants in the F3:4 generation. This difficulty might have been caused by recombination restriction around the centromeric region of chromosome 4. In 4896 plants those were segregated at qLTN4, there is region for recombination restriction between RM16535 (8.0-Mbp) and RM16605 (11.5-Mbp) and we cannot obtain any recombinant plants between RM16535 and RM16605. Additionally, the genotypes of qLTN4 through progeny test were completely linked to those of RM16535 and RM16605. To further delimit the qLTN4 genomic region and identify candidate genes, expanding segregating population will be essential in a future study.
Hybrid breakdown has been described as sterility and weakness in F2 or later generations resulting from inter- or intraspecific crosses and can affect all aspects of plant development. Several studies have reported plants expressing various phenotypes such as sterility; severe reduction in plant height, tiller number, root number, and root length; or even plant death before heading time (Sato and Morishima, 1988; Kubo and Yoshimura, 2002; Miura et al., 2008). Yamamoto et al. (2010) observed two types of weak plants, severe and mild, in the F2 progeny of a cross between “Koshihikari” and “Habataki.” In the present study, the low-tillering plants (BC4F5) derived from a cross between T65 and O. meridionalis showed severe reduction in PN but only moderate reduction in other panicle traits, CL, and grain fertility compared with the recurrent parent. The phenotypes of the low-tillering line were considered to be caused by hybrid breakdown because they were observed in later generations derived from crosses between normal parental lines. We consider the type of hybrid breakdown that occurred in this study as the mild type described by Yamamoto et al. (2010). The low-tillering locus on the short arm of chromosome 4 was associated with reduction in CL, PL, PN, LW, NSB, and TSN. In a previous study by Sobrizal and Yoshimura (2009), the hybrid weakness f-1 gene, associated with hybrid breakdown in a cross between T65 and O. glumaepatula, was located between G3006 and C933 (12.7-Mbp) on the short arm chromosome 4 and was completely linked to C708 (6.3-Mbp), C802 (6.9-Mbp), and R288 (8.2-Mbp). The hybrid weakness f-1 gene showed a similar phenotype to qLTN4 in the genetic background of T65. The qLTN4 was located between W1 (7.1-Mbp) to Indel3729 (11.7-Mbp) and was overlapped with the regions of hybrid weakness f-1 gene. Thus, we infer that the locus identified in our study might be the same locus that was previously identified by Sobrizal and Yoshimura (2009). Several studies have reported distant relationships between O. meridionalis and other AA-genome rice species (Second, 1986; Noredo et al., 1998). These results suggest that various AA-genome rice species may share a conserved region around qLTN4 that is related to hybrid breakdown.
Several studies have proposed that interactions between complementary genes derived from each parent are the cause of hybrid weakness and hybrid breakdown (Fukuoka et al., 1998; Kubo and Yoshimura, 2002; Ichitani et al., 2007, 2011). In our study, only one locus involved in hybrid breakdown was detected. Assuming that hybrid breakdown in this case was also caused by the interaction of two complementary genes, the hybrid breakdown gene from T65 remains to be identified. Miura et al. (2008) were able to identify the gene responsible for hybrid breakdown in O. nivara; however, the complementary gene for hybrid breakdown from “Koshihikari” was not identified, suggesting a possible complex mechanism controlling this trait.
Hybrid breakdown has been reported as a hindrance to gene flow in many plant crosses. In this study, we observed a beneficial trait, low-tillering frequency, as a result of hybrid breakdown. Because the effects on other agronomic traits were mild, the allele with low-tillering on chromosome 4 might be useful for rice genetic improvement via production of low-tillering lines in breeding programs.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
NM, SI, and DF designed the research. DF, NM, SI, and ZD performed the research. S-HZ, YY, and HY provided advice on the experiments. NM and DF wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully thank Atsushi Yoshimura from Kyushu University for the kind provision of introgression lines with low-tillering.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.633247/full#supplementary-material
References
Dellaporta, S. L., Wood, J., and Hicks, J. B. (1983). A plant DNA minipreparation: version II. Plant Mol. Biol Rep. 1, 19–21. doi: 10.1007/BF02712670
Dreissig, S., Mascher, M., and Heckmann, S. (2019). Variation in recombination rate is shaped by domestication and environmental conditions in barley. Mol. Biol. Evol. 36, 2029–2039. doi: 10.1093/molbev/msz141
Falconer, D. S., and Mackay, T. F. C. (1996). Introduction to Quantitative Genetics. London: Longmans Green.
Fujita, D., Ebron, L. A., Araki, E., Kato, H., Khush, G. S., Sheehy, J. E., et al. (2010). Fine mapping of a gene for low-tiller number, Ltn, in japonica rice (Oryza sativa L.) variety Aikawa 1. Theor. Appl. Genet. 120, 1233–1240. doi: 10.1007/s00122-009-1251-7
Fukuoka, S., Namai, H., and Okuno, K. (1998). RFLP mapping of the genes controlling hybrid breakdown in rice (Oryza sativa L.). Theor. Appl. Genet. 97, 446–449. doi: 10.1007/s001220050915
Haenel, Q., Laurentino, T. G., Roesti, M., and Berner, D. (2018). Meta-analysis of chromosome-scale crossover rate variation in eukaryotes and its significance to evolutionary genomics. Mol. Ecol. 27, 2477–2497. doi: 10.1111/mec.14699
Hussien, A., Tavakol, E., Horner, D. S., Muñoz-Amatriaín, M., Muehlbauer, G. J., and Rossini, L. (2014). Genetics of tillering in rice and barley. Plant Genome 7:plantgenome2013.10.0032. doi: 10.3835/plantgenome2013.10.0032
Ichitani, K., Namigoshi, K., Sato, M., Taura, S., Aoki, M., Matsumoto, Y., et al. (2007). Fine mapping and allelic dosage effect of Hwc1, a complementary hybrid weakness gene in rice. Theor. Appl. Genet. 114, 1407–1415. doi: 10.1007/s00122-007-0526-0
Ichitani, K., Taura, S., Tezuka, T., Okiyama, Y., and Kuboyama, T. (2011). Chromosomal location of HWA1 and HWA2, complementary hybrid weakness genes in rice. Rice 4, 29–38. doi: 10.1007/s12284-011-9062-2
International Rice Genome Sequencing Project (2005). The map-based sequence of the rice genome. Nature 436, 793–800. doi: 10.1038/nature03895
Jiang, H., Guo, L. B., Xue, D. W., Zeng, D. L., Zhang, G. H., Dong, G. J., et al. (2006). Genetic analysis and gene-mapping of two reduced-culm-number mutants in rice. J. Integr. Plant Biol. 48, 341–347. doi: 10.1111/j.1744-7909.2006.00224.x
Kebrom, T. H., Chandler, P. M., Swain, S. M., King, R. W., Richards, R. A., and Spielmeyer, W. (2012). Inhibition of tiller bud outgrowth in the tin mutant of wheat is associated with precocious internode development. Plant Physiol. 160, 308–318. doi: 10.1104/pp.112.197954
Khush, G. S. (1997). Origin, dispersal, cultivation and variation of rice. Plant Mol. Biol. 35, 25–34. doi: 10.1007/978-94-011-5794-0_3
Kubo, T. (2013). Genetic mechanisms of postzygotic reproductive isolation: an epistatic network in rice. Breed Sci. 63, 359–366. doi: 10.1270/jsbbs.63.359
Kubo, T., and Yoshimura, A. (2002). Genetic basis of hybrid breakdown in a Japonica/Indica cross of rice, Oryza sativa L. Theor. Appl. Genet. 105, 906–911. doi: 10.1007/s00122-002-1059-1
Li, X., Qian, Q., Fu, Z., Wang, Y., Xiong, G., Zeng, D., et al. (2003). Control of tillering in rice. Nature 422, 618–621. doi: 10.1038/nature01518
Liu, Y., Wu, H., Chen, H., Liu, Y., He, J., Kang, H., et al. (2015). A gene cluster encoding lectin receptor kinases confers broad-spectrum and durable insect resistance in rice. Nat. Biotech. 33, 301–305. doi: 10.1038/nbt.3069
McCouch, S. R., Teytelman, L., Xu, Y., Lobos, K. B., Clare, K., Walton, M., et al. (2002). Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res. 9, 199–207. doi: 10.1093/dnares/9.6.199
Miura, K., Yamamoto, E., Morinaka, Y., Takashi, T., Kitano, H., Matsuoka, M., et al. (2008). The hybrid breakdown 1(t) locus induces interspecific hybrid breakdown between rice Oryza sativa cv. Koshihikari and its wild relative O. nivara. Breed Sci. 58, 99–105. doi: 10.1270/jsbbs.58.99
Noredo, M. E. B., Juliano, A. B., Lu, B. R., and Jackson, M. T. (1998). Taxonomic status of Oryza glumaepatula Steud. II. Hybridization between new world diploids and AA-genome species from Asia and Australia. Genet Resour. Crop Evol. 45, 205–214. doi: 10.1023/A:1008634400519
Reflinur, Kim, B., Jang, S. M., Chu, S. H., Bordiya, Y., Akter, M. B., et al. (2014). Analysis of segregation distortion and its relationship to hybrid barriers in rice. Rice 7:3. doi: 10.1186/s12284-014-0003-8
Sato, Y. I., and Morishima, H. (1988). Distribution of the genes causing F2 chlorosis in rice cultivars of the Indica and Japonica types. Theor. Appl. Genet. 75, 723–727. doi: 10.1007/BF00265594
Second, G. (1986). “Isozymes and phylogenetic relationship in Oryza,” in Proceedings of the Rice Genetics Proceedings of the International Rice Genetics Symposium, (Philippines: IRRI), 27–39. doi: 10.1142/9789812814265_0003
Smith, C. W., and Dilday, R. H. (2003). Rice: Origin, History, Technology and Production. New Jersey: John Wiley & Sons, Inc.
Sobrizal, and Yoshimura, A. (2009). Hybrid weakness found in the backcross progeny of an inter-specific cross of Oryza sativa / O. glumaepatula. Atom Indonesia 35, 49–56. doi: 10.17146/aij.2009.47
Sotowa, M., Ootsuka, K., Kobayashi, Y., Hao, Y., Tanaka, K., Ichitani, K., et al. (2013). Molecular relationships between Australian annual wild rice, Oryza meridionalis, and two related perennial forms. Rice 6:26. doi: 10.1186/1939-8433-6-26
Stapley, J., Feulner, P. G. D., Johnston, S. E., Santure, A. W., and Smadja, C. M. (2017). Variation in recombination frequency and distribution across eukaryotes: patterns and processes. Philos. Trans. R. Soc. B Biol. Sci. 372:20160455. doi: 10.1098/rstb.2016.0455
Takeda, T., Suwa, Y., Suzuki, M., Kitano, H., Ueguchi-Tanaka, M., Ashikari, M., et al. (2003). The OsTB1 gene negatively regulates lateral branching in rice. Plant J. 33, 513–520. doi: 10.1046/j.1365-313X.2003.01648.x
Uddin, M. N., Tomita, A., Obara, M., Yanagihara, S., and Fukuta, Y. (2016). Identification of a low tiller gene from a new plant type cultivar in rice (Oryza sativa L.). Breed Sci. 66, 790–796. doi: 10.1270/jsbbs.16143
Wang, S., Basten, C. J., Gaffney, P., and Zeng, Z. B. (2012). Windows QTL Cartographer 2.5. User Manual. Raleigh, NC: Bioinformatics Research Centre, North Carolina State Univ.
Xu, Y., Zhu, L., Xiao, J., Huang, N., and McCouch, S. R. (1997). Chromosomal regions associated with segregation distortion of molecular markers in F2, backcross, doubled haploid, and recombinant inbred populations in rice (Oryza sativa L.). Mol. Gen. Genet. 253, 535–545. doi: 10.1007/s004380050355
Yamamoto, E., Takashi, T., Morinaka, Y., Lin, S., Kitano, H., Matsuoka, M., et al. (2007). Interaction of two recessive genes, hbd2 and hbd3, induces hybrid breakdown in rice. Theor. Appl. Genet. 115, 187–194. doi: 10.1007/s00122-007-0554-9
Yamamoto, E., Takashi, T., Morinaka, Y., Lin, S., Wu, J., Matsumoto, T., et al. (2010). Gain of deleterious function causes an autoimmune response and Bateson-Dobzhansky-Muller incompatibility in rice. Mol. Genet. Genomics 283, 305–315. doi: 10.1007/s00438-010-0514-y
Yan, J. Q., Zhu, J., He, C. X., Benmoussa, M., and Wu, P. (1998). Quantitative trait loci analysis for the developmental behavior of tiller number in rice (Oryza sativa L.). Theor. Appl. Genet. 97, 267–274. doi: 10.1007/s001220050895
Yang, H., You, A., Yang, Z., Zhang, F., He, R., Zhu, L., et al. (2004). High-resolution genetic mapping at the Bph15 locus for brown planthopper resistance in rice (Oryza sativa L.). Theor. Appl. Genet. 110, 182–191. doi: 10.1007/s00122-004-1844-0
Yonemaru, J., Choi, S. H., Sakai, H., Ando, T., Shomura, A., Yano, M., et al. (2015). Genome-wide indel markers shared by diverse Asian rice cultivars compared to Japanese rice cultivar ‘Koshihikari’. Breed Sci. 65, 249–256. doi: 10.1270/jsbbs.65.249
Yoshimura, A., Nagayama, H., Sobrizal, Kurakazu, T., Sanchez, P. L., Doi, K., et al. (2010). Introgression lines of rice (Oryza sativa L.) carrying a donor genome from the wild species, O. glumaepatula Steud. and O. meridionalis Ng. Breed Sci. 60, 597–603. doi: 10.1093/nar/gkh340
Keywords: rice, Oryza meridionalis, tiller number, substitution mapping, hybrid breakdown
Citation: Munguambe NE, Inoue S, Demeter Z, Yamagata Y, Yasui H, Zheng S-H and Fujita D (2021) Substitution Mapping of a Locus Responsible for Hybrid Breakdown in Populations Derived From Interspecific Introgression Line. Front. Plant Sci. 12:633247. doi: 10.3389/fpls.2021.633247
Received: 25 November 2020; Accepted: 22 March 2021;
Published: 21 April 2021.
Edited by:
Kazuki Matsubara, Institute of Crop Science (NARO), JapanReviewed by:
Gang Zhi Zhao, Nanjing Agricultural University, ChinaJiawu Zhou, Food Crops Research Institute, Yunnan Academy of Agricultural Sciences, China
Copyright © 2021 Munguambe, Inoue, Demeter, Yamagata, Yasui, Zheng and Fujita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daisuke Fujita, ZGZ1aml0YUBjYy5zYWdhLXUuYWMuanA=