 Xu Wang1†
Xu Wang1† Xinlei Yang1,2†
Xinlei Yang1,2† Yucheng Feng1Phat Dang3Wenwen Wang1
Yucheng Feng1Phat Dang3Wenwen Wang1 Rita Graze4
Rita Graze4 Josh P. Clevenger5
Josh P. Clevenger5 Ye Chu6
Ye Chu6 Peggy Ozias-Akins6Corley Holbrook7
Peggy Ozias-Akins6Corley Holbrook7 Charles Chen1*
Charles Chen1*- 1Department of Crop, Soil and Environmental Sciences, Auburn University, Auburn, AL, United States
- 2State Key Laboratory of North China Crop Improvement and Regulation, Laboratory of Crop Germplasm Resources of Hebei, Hebei Agricultural University, Baoding, China
- 3United States Department of Agriculture–Agricultural Research Service National Peanut Research Laboratory, Dawson, GA, United States
- 4Department of Biology, Auburn University, Auburn, AL, United States
- 5HudsonAlpha Institute for Biotechnology, Huntsville, AL, United States
- 6Center for Applied Genetic Technologies, University of Georgia, Tifton, GA, United States
- 7United States Department of Agriculture–Agricultural Research Service Crop Genetics and Breeding Research, Tifton, GA, United States
Cultivated peanut (Arachis hypogaea) is one of the most widely grown food legumes in the world, being valued for its high protein and unsaturated oil contents. Drought stress is one of the major constraints that limit peanut production. This study’s objective was to identify the drought-responsive genes preferentially expressed under drought stress in different peanut genotypes. To accomplish this, four genotypes (drought tolerant: C76-16 and 587; drought susceptible: Tifrunner and 506) subjected to drought stress in a rainout shelter experiment were examined. Transcriptome sequencing analysis identified that all four genotypes shared a total of 2,457 differentially expressed genes (DEGs). A total of 139 enriched gene ontology terms consisting of 86 biological processes and 53 molecular functions, with defense response, reproductive process, and signaling pathways, were significantly enriched in the common DEGs. In addition, 3,576 DEGs were identified only in drought-tolerant lines in which a total of 74 gene ontology terms were identified, including 55 biological processes and 19 molecular functions, mainly related to protein modification process, pollination, and metabolic process. These terms were also found in shared genes in four genotypes, indicating that tolerant lines adjusted more related genes to respond to drought. Forty-three significantly enriched Kyoto Encyclopedia of Genes and Genomes pathways were also identified, and the most enriched pathways were those processes involved in metabolic pathways, biosynthesis of secondary metabolites, plant circadian rhythm, phenylpropanoid biosynthesis, and starch and sucrose metabolism. This research expands our current understanding of the mechanisms that facilitate peanut drought tolerance and shed light on breeding advanced peanut lines to combat drought stress.
Introduction
Cultivated peanut (Arachis hypogaea L.) is an important legume that is grown mainly on arid and semiarid regions where peanut productivity is usually limited by water deficit (Pimratch et al., 2008, 2009; Songsri et al., 2008; Balota et al., 2012; Dinh et al., 2013; Brasileiro et al., 2015). Drought stress during the mid-growing season from flowering to pod development leads to a severe reduction in peanut pod yield due to the highest water requirement during this period (Stansell and Pallas, 1985; Sterling et al., 1989). How to sustain and even increase peanut production to meet growing population needs, whereas environmental conditions are deteriorating, is a major challenge that the peanut industry faces. Developing drought-tolerant varieties adapted to various levels of drought stress is a priority for many peanut breeding programs (Zhao et al., 2018). Unfortunately, traditional breeding approaches achieve little progress because drought-stress tolerance is a polygenic trait, and little is known about the molecular signaling and regulatory mechanisms of peanuts under drought stress.
The vulnerability of peanut to drought stress depends on genotypic variability (Stansell and Pallas, 1985; Greenberg et al., 1992; Puangbut et al., 2009; Devi et al., 2010; Dinh et al., 2013). Genotypic variations in several physiological characteristics associated with drought tolerance, including transpiration and photosynthesis rate, have been identified and provide opportunities to breed high-yielding drought-tolerant genotypes (Pimratch et al., 2008; Balota et al., 2012). RNA-sequencing (RNA-Seq), a technique for genome-wide gene expression analysis, provides a powerful alternative to facilitate the development of drought-tolerant genotypes (Li et al., 2014; Lu et al., 2014; Zhao et al., 2018). Recently, candidate genes and expression profiles in many crops, including wheat, corn, soybean, and peanut, evaluating plant response to environmental stress conditions were determined with RNA-Seq technology (Mathioni et al., 2011; Petre et al., 2012; Chen et al., 2013; Li et al., 2014; Brasileiro et al., 2015; Ruan et al., 2018; Zhao et al., 2018; Long et al., 2019). Large-scale screening of peanut had identified some drought-related candidate genes such as basic leucine zipper (bZIP) transcription factor genes that were observed from the wild relative of cultivated peanut, Arachis duranensis, when the plant was subjected to drought with 18% soil water content (Guimarães et al., 2012). Likewise, Li et al. (2014) reported 621 genes that were rapidly induced under water deficit conditions and the key drought response mechanisms in peanut function through the abscisic acid (ABA)-dependent pathway. In addition, more than 4,000 genes were identified to be associated with drought stress, in which 224 transcription factors and genes were involved in photosynthesis-antenna proteins, carbon metabolism, and the citrate cycle (Zhao et al., 2018).
As the genome sequence of the cultivated peanut cultivar Tifrunner was released (Bertioli et al., 2019), a more accurate transcriptome assembly can be obtained by mapping to the reference genome. Thus, the present study’s objective was to discover drought-induced genes by comparing drought-tolerant with drought-susceptible lines under drought stress by mapping RNA-Seq data to the cultivated peanut reference genome. The data generated in this research will provide insights into molecular mechanisms that underlie drought tolerance and provide a novel resource to further advance molecular breeding research in peanut.
Materials and Methods
Plant Materials and Experimental Design
The experiments were performed using four cultivated peanut genotypes, Tifrunner (susceptible), C76-16 (tolerant), 587 (tolerant), and 506 (susceptible), which were selected based on the drought study conducted in 2015 and 2016. The genotypes 587 and 506 are two recombinant inbred lines (RILs) derived from the cross “Tifrunner × C76-16,” representing the highest and lowest drought-tolerant level. The “Tifrunner × C76-16” RIL population was one of the 16 nested mapping populations (Holbrook et al., 2013). A split-plot design with randomized complete block design within was adopted in this study. All seeds were planted in a single-row (15 × 120 cm) at a rate of 10 seeds m–1 under rainout shelters at the United States Department of Agriculture (USDA) Agricultural Research Service National Peanut Research Laboratory in Dawson, GA, United States, to create artificial drought stress. Two rainout shelters were designated with two treatments, including full irrigation and middle-season drought, and each shelter consists of three blocks. Irrigation was provided for both treatments right after seed sowing to encourage uniform germination. The irrigated treatment (designated as “irrigated control”) received a full irrigation schedule throughout the growing season based on the evapotranspiration replacement described by Stansell et al. (1976). The drought treatment (designated as “treatment”) was fully irrigated at the beginning of the growing season until 61 days after planting. Water-deficit stress was applied at 61 days after planting by withheld water for four consecutive weeks starting at −10 kPa of soil water potential at 20 cm depth and progressively advanced to −700 kPa after 1 week of treatment and reached −1,050 to −1,200 kPa in the second week and stabilized for another 2 weeks. Specific leaf area, 15N and 13C natural abundance were determined to reflect physiological responses to drought stress based on the method described by Dang et al. (2012). Besides the water treatment, all other agronomic management practices were applied according to the University of Georgia’s best management practices for peanuts.
RNA Extraction and Library Construction
Fully expanded leaves (second nodal) were randomly collected from each genotype at the end of the drought period in 2016. Leaf samples of each genotype were flash-frozen using liquid nitrogen and stored at −80°C until RNA extraction. Three leaflets randomly collected from each biological replication were pooled, and approximately 0.2-g pooled leaf samples were ground in liquid nitrogen for RNA extraction. Total RNA was extracted using a modified cetyltrimethylammonium bromide method (Yin et al., 2011) and purified using a Direct-Zol RNA MiniPrep Kit (Zymo Research, Irvine, CA, United States). The purity and integrity of RNA were analyzed using NanoDrop ND-1000 UV/Vis spectrophotometer (Thermo Scientific, Wilmington, DE, United States) and Agilent 2100 Bioanalyzer (Agilent, United States), respectively. A total of 24 libraries of complementary DNA (4 genotypes × 2 treatments × 3 replicates) were constructed and subsequently sequenced using an Illumina HiSeq 4000 instrument at the Beijing Genomics Institute.
Quantitative Reverse Transcription-Polymerase Chain Reaction Validation of Differentially Expressed Genes
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was applied to verify the transcription levels of 14 randomly selected genes. The same RNA samples in high-throughput sequencing were used for qRT-PCR. Gene-specific primers were designed by Primer Premier 3.0 software (Supplementary Table 1). Each 10-μl qRT-PCR reaction mixture contained 1 μl of 10-fold diluted first-strand complementary DNA, 0.3 μl of each primer (10 μM), and 5-μl 2 × PowerUPTM SYBRTM Green Master Mix (Applied Biosystems, Carlsbad, CA, United States). A Bio-Rad CFX96 real-time PCR system was used under the following conditions: 50°C for 2 min, 95°C for 2 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. For normalizing expression levels, yellow-leaf-specific protein 8 (NM_120912) was used as a reference gene. Non-specific products were identified by melting-curve analysis (17 genes reduced to 14 genes due to elimination of non-specific amplification). The quantification cycle value of each gene and RNA-seq results are listed in Supplementary Table 2. Relative gene expression levels were calculated using the 2–△△Ct method (Livak and Schmittgen, 2001).
Bioinformatics Analysis
Quality Control, Alignment, and Genome-Guided Assembly
The raw reads from RNA-seq were trimmed with Trimmomatic (Bolger et al., 2014). Clean reads were obtained by removing the adaptor sequences, ambiguous “N” nucleotides, and low-quality reads from the raw data. The read quality was assessed using FastQC (Andrews, 2010) before and after trimming. High-quality clean data were subjected to the downstream analyses. The RNA-seq data analysis pipeline followed the protocol described by Trapnell et al. (2012). Each sample was mapped to the reference genome by Tophat2 (Kim et al., 2013), with all the parameters setting to default. The cultivated peanut genome and the annotation file (Bertioli et al., 2019) were used as a reference for alignment. The alignment files of the 24 samples from Tophat2 were input into Cufflinks (Trapnell et al., 2012) for transcripts reconstruction.
To identify the novel transcript sequences, all the assemblies were compared with the reference annotation using Cuffcompare. Novel transcript sequences were then compared with the “Nr” database at National Center for Biotechnology Information by BLASTX to achieve gene functional annotation.
Identification of Differentially Expressed Genes
The expected number of fragments per kilobase of transcript sequence per millions of base pairs sequenced was used to represent the gene expression levels based on the length of the gene and reads count mapped to this gene. The differentially expressed genes (DEGs) analysis was performed using Cuffdiff [false discovery rate (FDR) < 0.05] (Trapnell et al., 2012). By comprising gene expression between “irrigated control” and “drought treatment” samples for each genotype, the DEGs were identified. The calculated P-value was then adjusted through FDR correction. Genes with adjusted P-values < 0.05 were considered as significantly differentially expressed.
Gene Ontology Enrichment Analysis and Kyoto Encyclopedia of Genes and Genomes Pathway Analysis
Gene ontology (GO) terms for gene models accessible in genome annotation were directly retrieved from the “GFF” file downloaded at PeanutBase1. GO terms for the novel transcripts were assigned using Blast2Go (Conesa et al., 2005). To combine the GO terms of the annotated genes and novel genes, the GO enrichment analysis for DEGs was performed using AgriGO2 (Tian et al., 2017). GO terms with FDR-adjusted P-value < 0.05 were considered as significantly enriched by DEGs. The enriched GO terms were subsequently visualized using REVIGO (Supek et al., 2011). To identify important pathways involved by the DEGs, the transcripts were assigned to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways using the webserver3 against the Arabidopsis thaliana, Glycine max, A. duranensis, and Arachis ipaensis gene datasets using the bidirectional best hit method. KEGG enrichment analysis was conducted on the KOBAS 3.0 webserver4 (Wu et al., 2006).
Results
We have observed progressive changes of soil water potential at 20 cm depth starting at −10 kPa advanced to −700 kPa after 1-week treatment and reached −1,050 to −1,200 kPa in the second week and stabilized for another 2 weeks. After 4 weeks of middle-season drought stress treatment, specific leaf area for these four genotypes were 34.85 ± 2.90 for C76-16, 33.41 ± 3.17 for 587, 29.40 ± 4.95 for Tifrunner, and 28.89 ± 2.97 for 506 compared with irrigated treatment at 30.31 ± 4.6 for C76-16, 32.22 ± 0.65 for 587, 31.89 ± 1.45 for Tifrunner, and 40.65 ± 0.64 for 506, indicating that these genotypes have different levels of physiological response to drought stress. There are significant differences found among the four genotypes under drought stress vs. non-differences under irrigation for 15N and 13C natural abundances (Wang et al., 2021 under review).
Genome-Guided Assembly and Annotation of Novel Transcripts
To assess the global transcriptome profile of peanut leaf samples in response to drought stress, RNA-Seq analysis was performed using peanut leaves under drought treatments. RNA-seq of 24 samples of the four genotypes with three replicates under “irrigated control” or “treatment” generated a total of 1,059,869,097 pairs of 100-bp cleaned reads (197.41 Gb) with an average of 44.16 million read pairs per library representing coverage of 77.27 times. After trimming, 87.81% of the raw reads, including 930,991,527 paired reads and 77,462,493 unpaired reads, high-quality and vector-trimmed sequences were retained (Table 1). The cleaned reads were mapped to the cultivated peanut genome, and the overall mapping rate per library ranged from 62.20 to 79.30%, with an average mapping rate of 73.42% (Table 1).
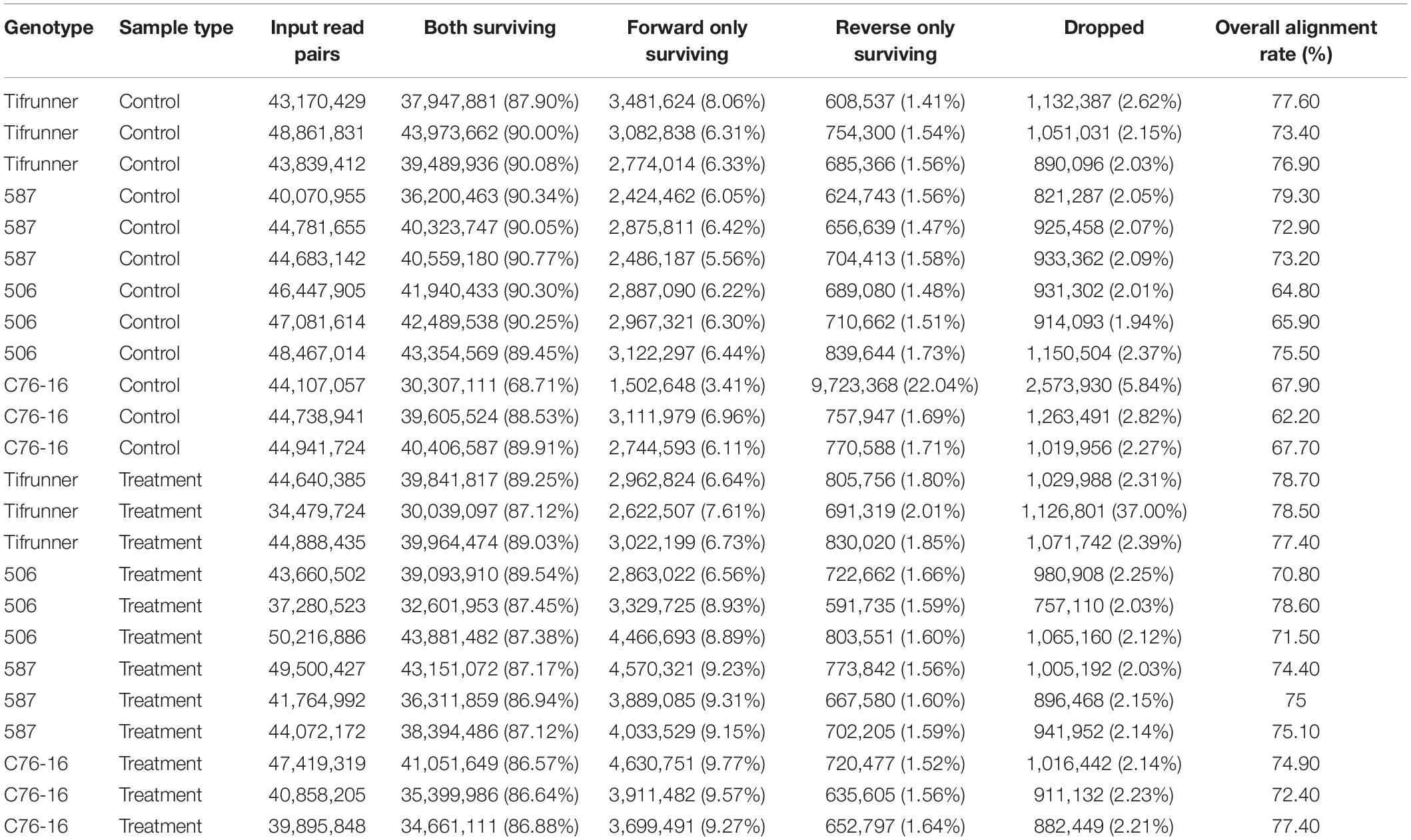
Table 1. Summary of library, trimming, and alignment of reads to A. hypogaea genome in each library.
Through the genome-guided assembly, a total of 73,575 genes were assembled for Tifrunner, 73,610 genes were assembled for C76-16, 73,898 genes were assembled for 587, and 73,900 genes were assembled for 506. There were 66,437 (90.30%), 66,445 (90.27%), 66,373 (89.82%), and 66,378 (89.82%) assembled genes matched to genes annotated from the cultivated peanut reference genome in Tifrunner, C76-16, 587, and 506, respectively (Table 2), resulting in 7,138, 7,165, 7,525, and 7,522 novel genes that were identified in Tifrunner, C76-16, 587, and 506, respectively.
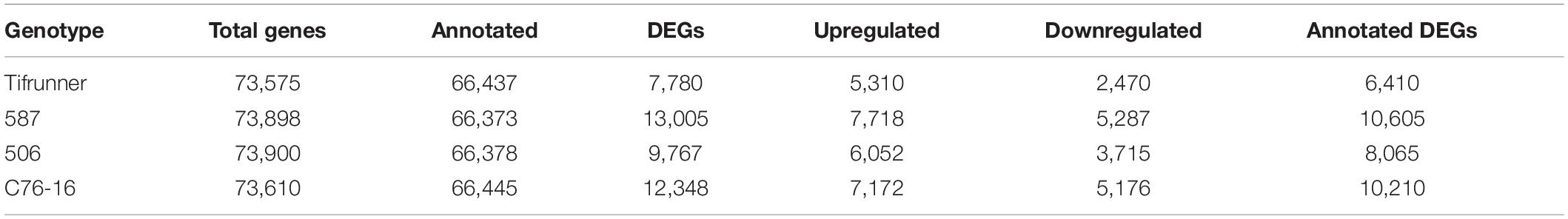
Table 2. Summary of library and alignment of reads to A. hypogaea genome in each genotype.
Differentially Expressed Genes
The DEGs were determined between “irrigated control” and “treatment” samples of each genotype. For the drought-susceptible genotypes, there were 7,780 genes differentially expressed in Tifrunner and 9,767 differentially expressed in 506 (Table 2). Of the 7,780 DEGs in Tifrunner, the levels of gene expression of 5,310 genes were increased, and 2,470 genes were decreased. For genotype 506, 6,052 genes were upregulated, and 3,715 genes were downregulated in the drought treatment (Table 2). For the drought-tolerant genotypes, 12,348 DEGs were identified in C76-16, including 7,172 upregulated genes and 5,176 downregulated genes. In addition, a total of 13,005 DEGs were identified in 587 to be upregulated (7,718 genes) or downregulated (5,287 genes). Among the DEGs identified, 6,410, 10,210, 10,605, and 8,065 DEGs were annotated with the reference genome in Tifrunner, C76-16, 587, and 506, respectively (Table 2).
Pairwise comparison of the DEGs from the four genotypes was performed to investigate which genes failed to respond to drought stress in drought-susceptible genotypes and in drought-tolerant genotypes (Figure 1A). A total of 5,703 DEGs, including 3,668 upregulated genes and 2,035 downregulated genes, were shared by 506 and two drought-tolerant genotypes (Figure 1A). Tifrunner shared 4,611 DEGs (3,246 upregulated and 1,365 downregulated) with the drought-tolerant genotypes (Figure 1A). Among the identified DEGs in the drought-tolerant lines, 3,860 genes were shared between 587 and C76-16, with 10,315 DEGs exclusively detected in the two drought-tolerant genotypes. Moreover, there were 2,457 DEGs identified in all four genotypes, and these genes were used in the subsequent GO and KEGG enrichment analysis.
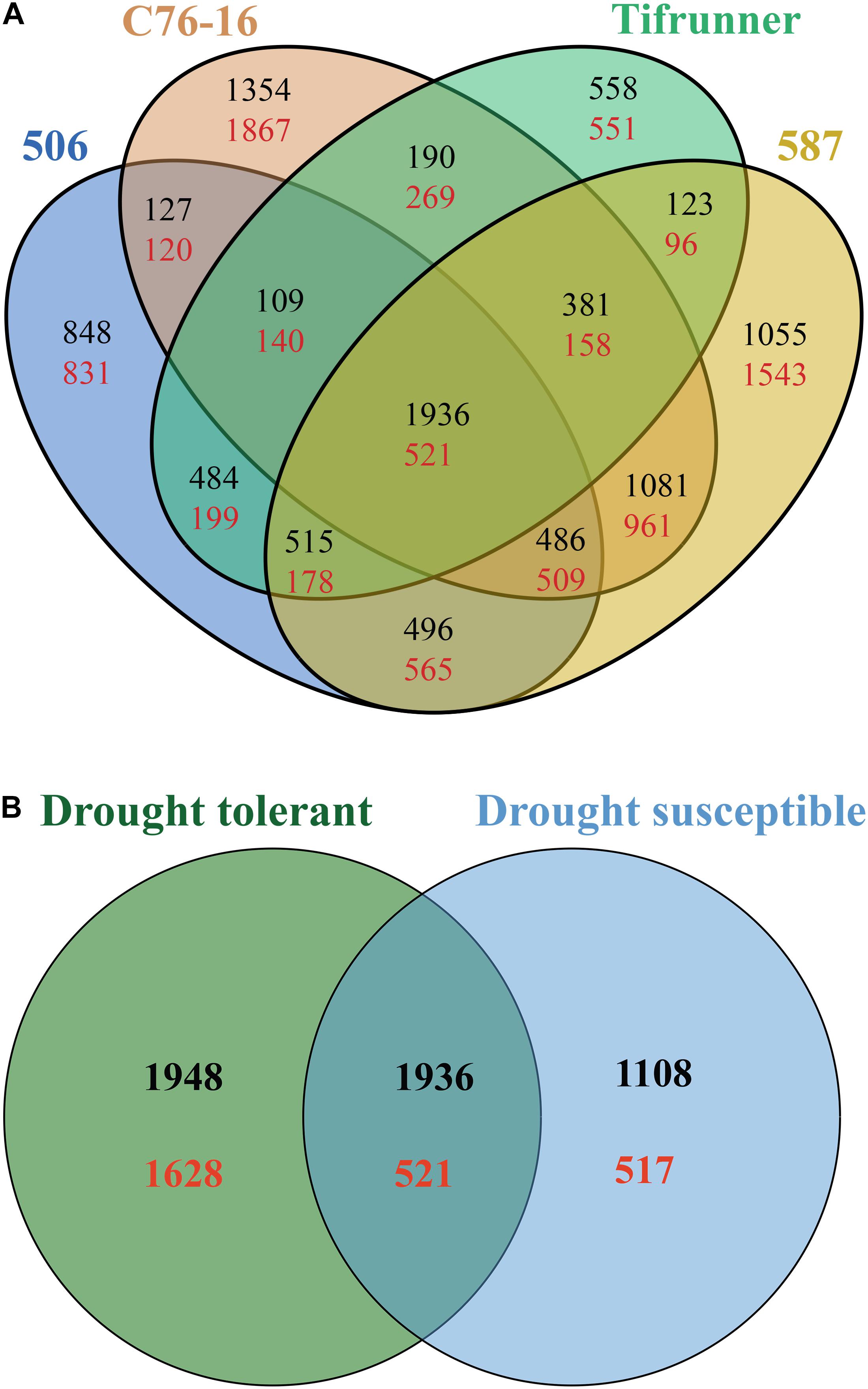
Figure 1. Comparison of the annotated DEGs among the four genotypes (A) and among the drought-susceptible genotypes, Tifrunner, and 506, and among drought-tolerant genotypes, C76-16, and 587 (B). Black color numbers present upregulated genes, and red color numbers present downregulated genes.
Among the identified DEGs in the drought-tolerant lines, 6,033 genes (3,884 upregulated genes and 2,149 downregulated genes) were shared between 587 and C76-16. A total of 4,082 shared DEGs with 3,044 upregulated and 1,038 downregulated genes in drought-susceptible lines (Tifrunner and 506). A total of 2,457 DEGs were shared between drought-tolerant lines and drought-susceptible lines with 1,936 upregulated and 521 downregulated genes. In addition, 3,576 DEGs were identified in both drought-tolerant lines but were not identified in drought-susceptible lines. Also, 1,625 genes were differentially expressed in drought-susceptible lines, which were not in drought-tolerant lines (Figure 1B).
Among the 2,457 DEGs shared by all four genotypes, a log2-fold change of >2 and <−2 thresholds was used to select the most significant DEGs. After filtering, only 250 genes were determined as significant DEGs resulting in 78 unique genes with their expression profiles shown in Figures 2, 3. Among these 78 genes, 76 were upregulated under drought stress, whereas 2 were downregulated.
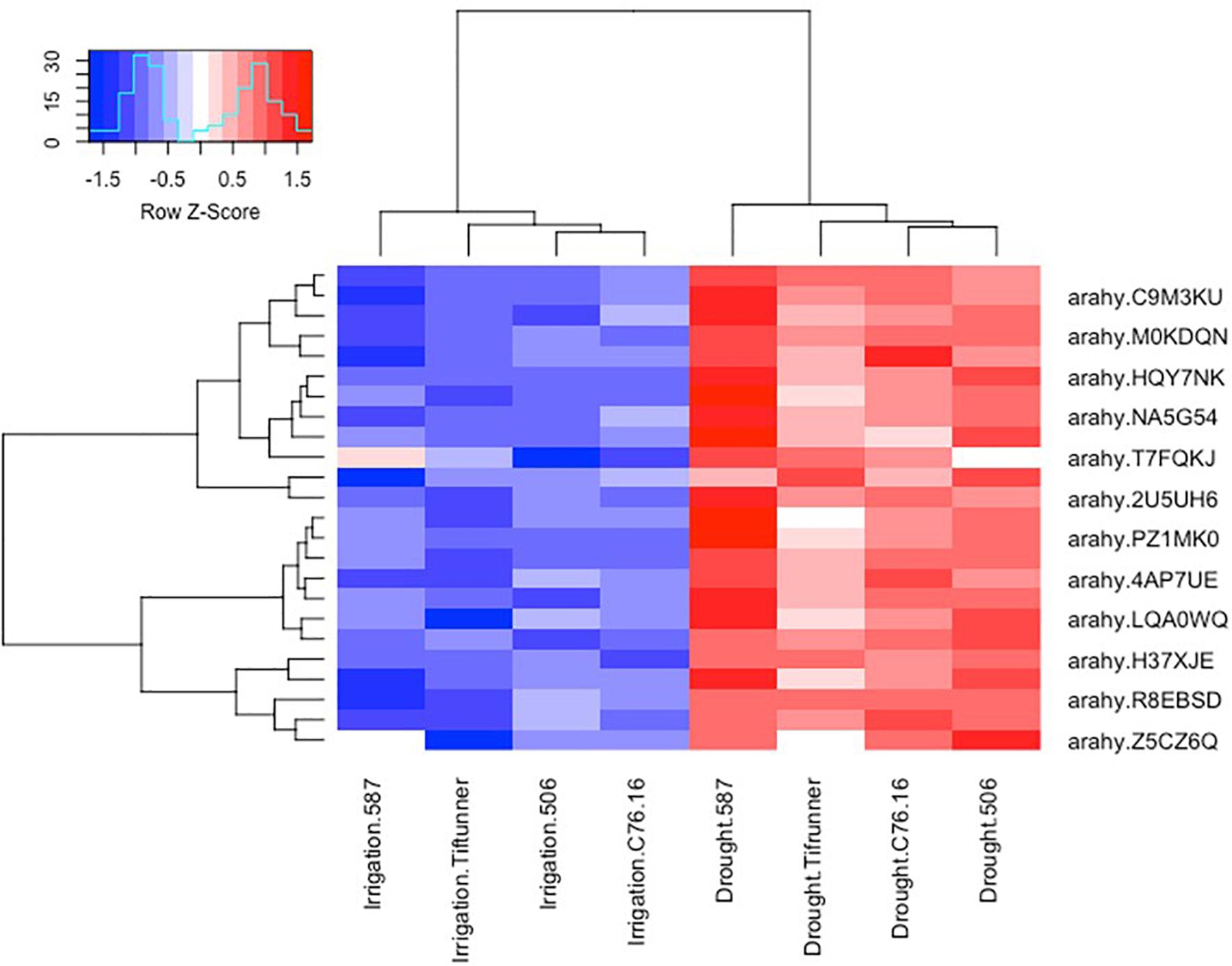
Figure 2. Expression profiles of the ABA-related differentially expressed genes shared by all four genotypes under irrigated and drought treatments. Log10 transformed FPKM values were used. “Blue” color indicates no expression or low expression level, and “red” color indicates high expression level. arahy.C9M3KU, myb transcription factor; arahy.M0KDQN, F-box family protein; arahy.HQY7NK, glucan endo-1,3-beta-glucosidase-like; arahy.NA5G54, WRKY family transcription factor; arahy.T7FQKJ, asparagine synthetase 3; arahy.2U5UH6, subtilisin-like serine protease; arahy.PZ1MK0, myb transcription factor; arahy.4AP7UE, U-box domain-containing protein 21-like; arahy.LQA0WQ, glutathione S-transferase family protein; arahy.H37XJE, calcium-dependent lipid-binding (CaLB domain) family protein; arahy.R8EBSD, RING-H2 finger protein 2B; IPR013083 (link is external) (zinc finger, RING/FYVE/PHD-type); arahy.Z5CZ6Q (link is external) glutathione S-transferase family protein.
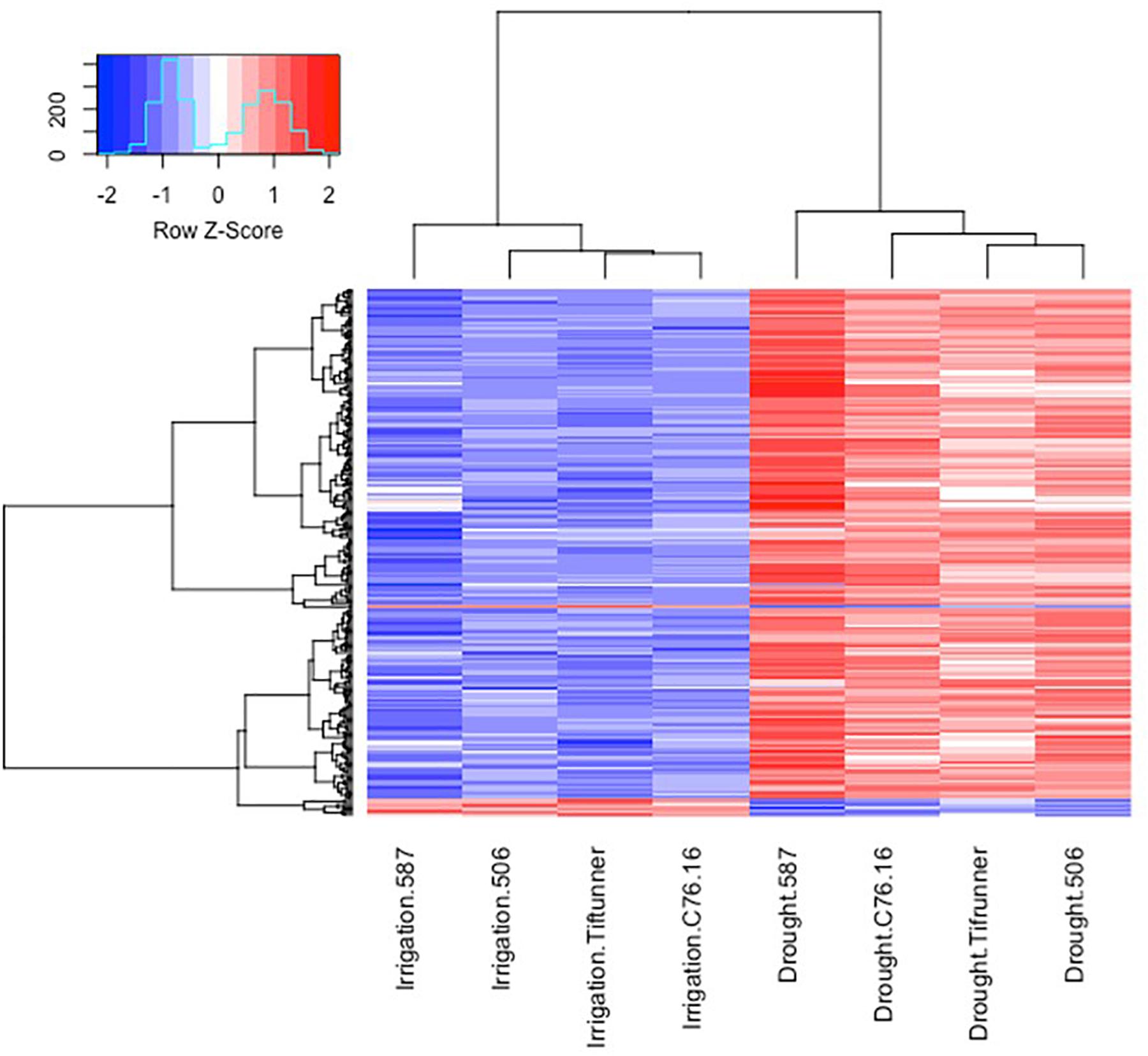
Figure 3. Expression profiles of the differentially expressed genes shared by all four genotypes under irrigated and drought treatments. Log10 transformed FPKM values were used. “Blue” color indicates no expression or low expression level, and “red” color indicates high expression level
Gene Ontology Enrichment and Functional Classification of Differentially Expressed Genes
Gene ontology assignments were used to classify the functions of DEGs. GO enrichment analysis was performed on the 2,457 genes to identify processes and functions overrepresented in the DEGs. The 2,457 drought-responsive DEGs were assigned into 139 enriched GO terms consisting of 86 biological processes and 53 molecular functions (Table 3). In addition, a total of 74 GO terms were identified in the DEGs expressed only in drought-tolerant lines, including 55 biological processes and 19 molecular functions, mainly related to protein modification process, pollination, and metabolic process (Supplementary Table 3). These GO terms were also identified in DEGs shared in both drought-tolerant lines and drought-susceptible lines, indicating that drought-tolerant lines adjusted more genes to respond more positively to drought. We also analyzed the enrichment of DEGs expressed only in drought-susceptible lines and identified 32 GO terms (Supplementary Table 3).
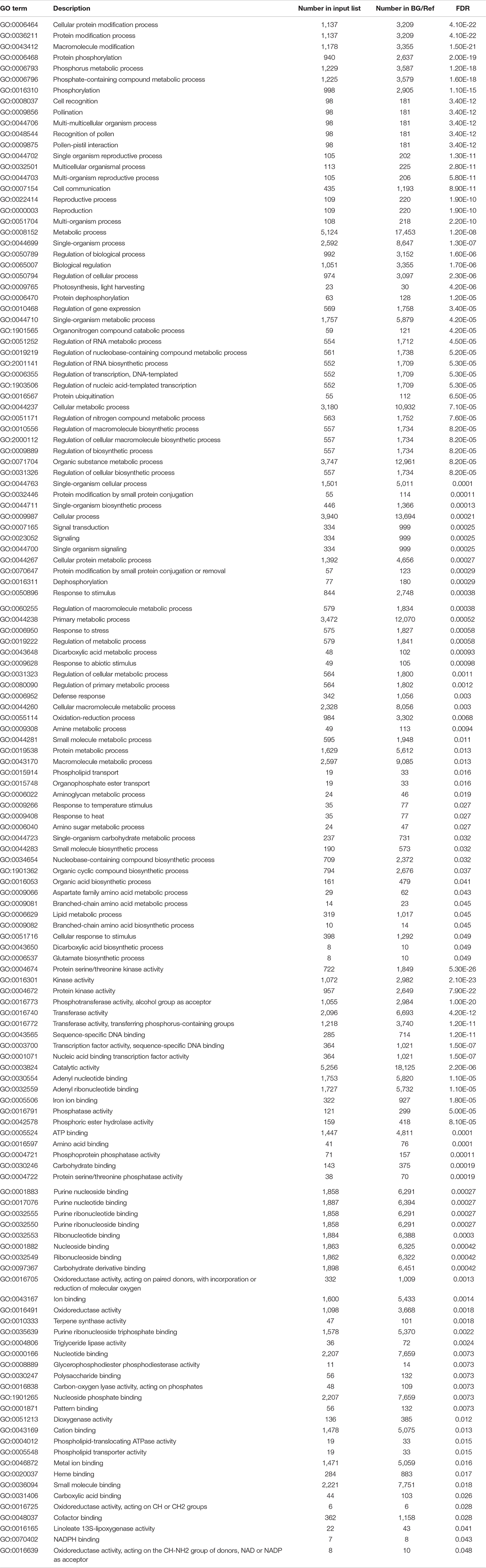
Table 3. Enriched GO terms of the DEGs common in drought-tolerant genotypes and drought-susceptible genotypes.
The most significantly enriched GO term in biological processes was the cellular protein modification process, followed by the protein modification process. Several other protein modification-related processes include protein dephosphorylation, protein ubiquitination, protein modification by small protein conjugation, protein metabolic process, and protein serine/threonine kinase activity, indicating the importance of protein modification in drought response. In addition, a cluster of GO terms related to defense response, such as response to stimulus, heat, abiotic variations, and temperature, were also observed. Furthermore, many reproductive-related GO terms, which included pollination, pollen–pistil interaction, reproduction, and reproductive process, were highly enriched, indicating the effects of drought stress on plant reproduction processes. There were also GO terms related to signaling (signaling and signal transduction) and regulation (regulation of transcription, RNA biosynthetic, and RNA metabolic processes) that were enriched. Additionally, kinase-related GO terms were observed, including protein serine/threonine kinase activity, kinase activity, and protein kinase activity in molecular function (Table 3). There were also many GO terms related to the oxidation-reduction processes, such as oxygen, oxidoreductase activity, and dioxygenase activity.
Kyoto Encyclopedia of Genes and Genomes Pathway Enrichment Analysis of Differentially Expressed Genes
A total of 741 KEGG Ontology terms were assigned to those 2,457 DEGs common in both drought-tolerant genotypes and drought-susceptible genotypes. The KEGG enrichment analysis was conducted against the Arabidopsis thaliana gene dataset using KOBAS 3.0 (Table 4). Forty-three pathways were identified to be significantly enriched (FDR-adjusted P-value < 0.05) pathways, including metabolic pathways, biosynthesis of secondary metabolites, circadian rhythm-plant, phenylpropanoid biosynthesis, starch and sucrose metabolism, etc. (Figure 4). Photosynthesis-related KEGG pathways such as photosynthesis-antenna proteins and carbon fixation in photosynthetic organisms were observed. Proline, arginine, and proline synthesis and metabolism pathways were enriched, which have been reported to be associated with drought tolerance.
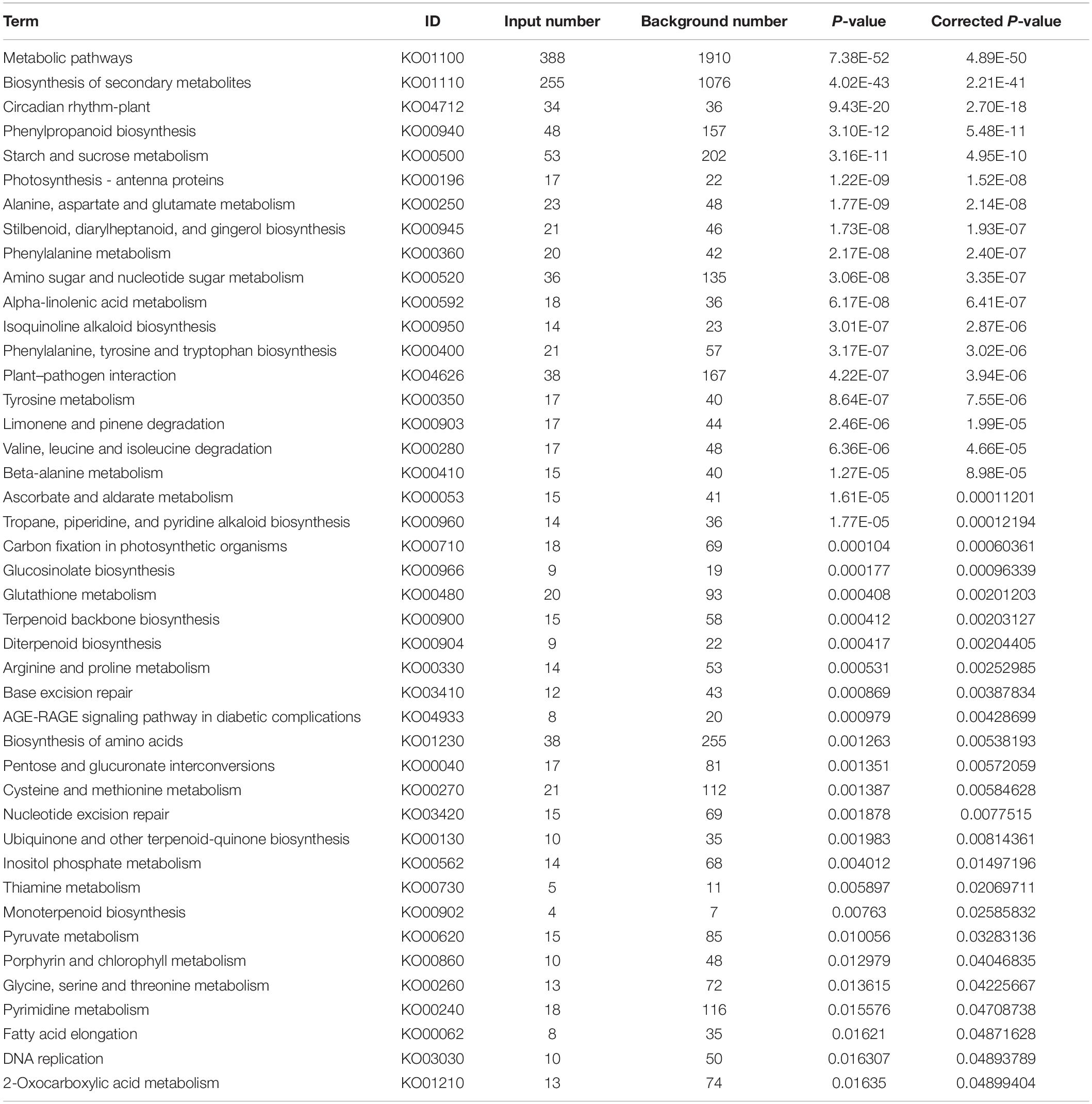
Table 4. Enriched KEGG ontology terms of the DEGs common in drought-tolerant genotypes and drought-susceptible genotypes.
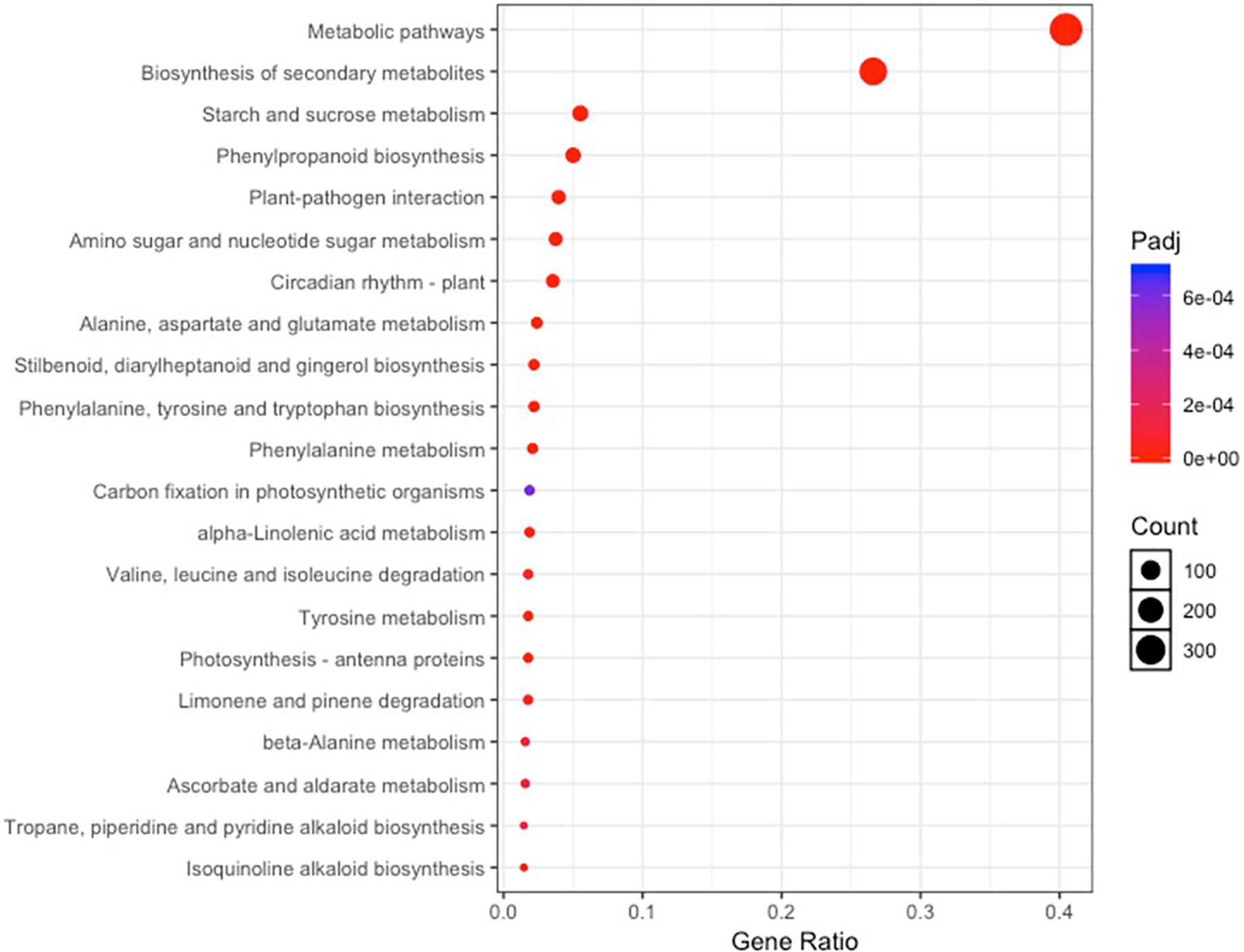
Figure 4. KEGG pathway enrichment analysis based on the differentially expressed genes shared by all four genotypes under irrigated and drought treatments.
Validation of RNA Sequencing Using Quantitative Reverse Transcription-Polymerase Chain Reaction
To validate the gene expression levels of the DEGs, 14 DEGs shared by all four genotypes under irrigated and drought treatments were selected for validation using qRT-PCR (Supplementary Table 2). All randomly selected 14 DEGs were confirmed to be upregulated in all four genotypes (Supplementary Table 2). Among these genes, three genes (“Arahy.JL3DG7,” “Arahy.Z1CJI9,” and “Arahy.UKNP9M”) were confirmed to be upregulated in all genotypes but significantly less than the expression level determined by RNA-seq. The correlation coefficient (R) between log2 (fold change) from RNAseq and ΔΔCq is 0.87, indicating that overall, the results of qRT-PCR agreed well with most findings from RNA-seq analysis.
Discussion
This research aims to define potential drought tolerance mechanism(s) by defining biochemical and molecular processes between drought-tolerant peanut versus drought-susceptible lines. A progressive drought treatment approach, in contrast to intermittent drought, was chosen to observe a linear relationship between plant response and increasing levels of water deficit. In contrast to the survival mechanism that plants may face in severe drought and high-heat environments, the peanuts grown in the many production areas around the world look to minimize yield loss or even to increase yield under increasing drought incidence. The selection of middle season drought was chosen to observe yield differences, and relatively short drought treatment (4 weeks) provides a fine separation of peanut response comparing drought-tolerant with drought-susceptible genotypes.
Transcriptome data are valuable resources for discovering gene expression levels, characterizing new alleles, and developing molecular markers associated with drought responses by investigating plants under abiotic or biotic stress. However, studies focusing on the transcriptome of peanuts under drought stress are limited. Li et al. (2014) demonstrated 47,842 unigenes with 621 induced DEGs (≥1.5 fold change compared with control) in the seedlings of peanut (Arachis hypogaea L.) cultivar Yueyou7 in South China under water deficit condition, and 22 putative transcription factor (TF) genes were reported as drought-responsive. They concluded that the main drought response mechanism in peanut function was through the ABA-dependent pathway. RNA-Seq analyses on two wild relatives of cultivated peanut under drought conditions, Arachis stenosperma (7,722 contigs) and A. duranensis (12,792 contigs), classified TF transcripts into 25 and 20 families, respectively (Guimarães et al., 2012). A more recent study assembled 51,554 genes in cultivated peanut root samples under drought conditions, where 4,648 DEGs were identified by comparing the irrigation with drought treatment (Zhao et al., 2018). In contrast, the present study analyzed the transcriptome of four peanut lines by mapping the sequenced library to the cultivated peanut reference genome and thus provided a more thorough dataset showing gene regulations under drought stress. We reported 73,575, 73,898, 73,900, and 73,610 genes for Tifrunner, 587, 506, and C76-16 with 7,780, 13,005, 9,767, and 12,348 DEGs in each genotype, respectively.
The majority of the DEGs were involved in secondary metabolite biosynthesis, photosynthesis, and response to heat and abiotic stimulus. This is supported by several studies reporting plant stress resistance systems (Farooq et al., 2009; Zhao et al., 2018). Some of the DEGs in response to drought stress were involved in 11 enriched KEGG pathways (carbon fixation in photosynthetic organisms; starch and sucrose metabolism; photosynthesis-antenna proteins; porphyrin and chlorophyll metabolism; cysteine and methionine metabolism; circadian rhythm-plant; pyruvate metabolism, amino sugar, and nucleotide sugar metabolism, glycine, serine, and threonine metabolism; phenylpropanoid biosynthesis; and phenylalanine metabolism), which is consistent with the results demonstrated in a recent study on peanut (Zhao et al., 2018). DEGs enriched in carbon metabolism pathway, starch and sucrose metabolism pathway, and photosynthesis-antenna proteins suggesting plant photosynthesis were affected due to decreasing carbon dioxide assimilation rate under mid-season drought stress (Farooq et al., 2009). Similarly, several genes involved in ascorbate and aldarate metabolism, carbon fixation, and photosynthesis were also reported to drought stimuli in other plants, including Boehmeria nivea and Chrysanthemum morifolium (Liu et al., 2013; Xu et al., 2013). However, our data indicated that ribosome and plant hormone signal transduction were not enriched in peanut in response to mid-season drought, suggesting plant host, growth stages, sampling dates, and treatments might play roles in the demonstrated variability.
Drought stress significantly affected the transcripts of some key genes related to secondary metabolism. For example, protein ubiquitination is demonstrated to regulate plant drought stress response (Liu et al., 2013; Zhang et al., 2013). Our GO enrichment analysis indicated peanut ubiquitin-related genes were highly enriched in both drought-tolerant and drought-susceptible genotypes. Fifty DEGs were identified to correlate with protein ubiquitination. For example, Arahy.4AP7UE played a role in protein ubiquitination that may negatively regulate ABA and drought response. In support of our data, the ubiquitin-related gene AhUBC2 was shown to enhance drought tolerance by regulating the expression of a stress-responsive gene (Wan et al., 2011).
Among the enriched KEGG and GO, many signaling-related GO terms were enriched indicating the potential importance of related pathways in peanut plants under drought stress. Interestingly, many ABA pathways related to DEGs were significantly induced by drought stress, including Arahy.UN6GTT, Arahy.RRZ6LI, Arahy.KS1HEQ, Arahy.H5H05M, and Arahy.KLG2UC. Plant hormones play significant roles in maintaining plants alive such as growth, under environmental stress, and senescence (Davies, 2013). ABA, as an important plant hormone being produced in the roots in response to drought, was widely studied for its role in regulating guard cell movement to close stomata (Li et al., 2014). ABA functions in plants under drought by regulating the development of reproductive tissues through massive transcriptional reprogramming events under long-term drought stress, which further reduces the plant growth and crop yield (Degenkolbe et al., 2009; Sreenivasulu et al., 2012). The previous study in peanut identified 279 DEGs that were significantly overlap in expression between the water deficit only and water deficit + ABA treatment groups, indicating the significant role of ABA in signaling under drought (Li et al., 2014). Furthermore, combining the reproduction-related pathway enriched in this study, we speculated that the induced ABA-related DEGs might further affect the reproduction process of peanuts under drought stress due to the 4-week drought period. This confirms the previous finding that ABA regulates the development of reproductive tissues under long-term drought (Degenkolbe et al., 2009; Sreenivasulu et al., 2012). Besides the ABA pathway-related genes, we also found a range of ethylene-related and auxin signaling pathway-related genes, and these genes can differentially express in the peanut under drought-stress conditions.
Late embryogenesis abundant (LEA) proteins are mainly low molecular weight (10–30 kDa) proteins, which are involved in defending higher plants from damage triggered by abiotic stresses, especially drought (dehydration). The present study identified two LEA genes (arachy. RD0T5B and arachy. XWYH2Z) that are shared by all four genotypes, five LEA genes (arachy.3IB3IU, arachy.Q07BGG, arachy.R9W6MW, arachy.B0SKQG, and arachy.FY9BZZ) shared uniquely in drought-tolerant genotypes, and only one LEA gene (arachy.P4KHGY) shared in drought-susceptible genotypes (Table 5). In our study, more LEA genes were upregulated in drought-tolerant genotypes than in drought-susceptible genotypes. This indicated the essential role of LEA proteins in peanuts under drought stress, especially in drought-tolerant genotypes. Accumulation of LEA proteins has also been found to occur in peanut roots when peanut plants under drought stress (Zhao et al., 2018). Regarding the many peanut LEA gene subfamilies, the precise functions are still enigmatic, and further research should be performed to elucidate the possible roles of these genes in peanut stress tolerance.
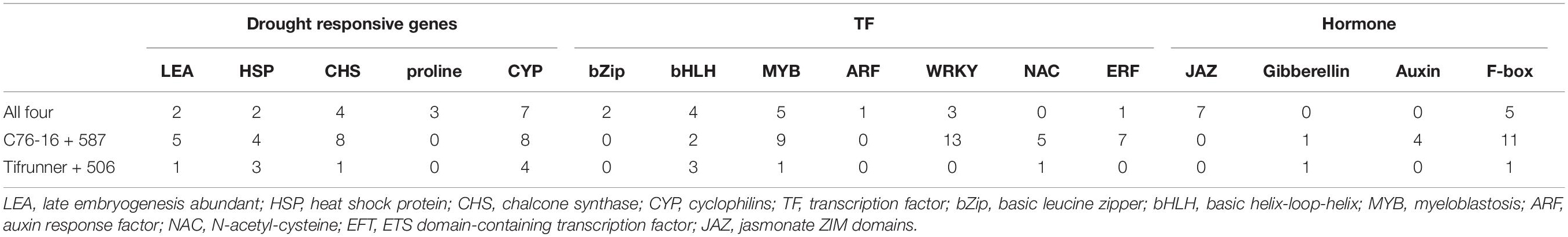
Table 5. Drought responsive genes, transcription factors family, and hormones may be involved in peanut tolerance to dehydration stress.
Transcription factors (sequence-specific DNA-binding factors) are proteins that bind to specific DNA sequences, thus manipulating the RNA transcription rate for genes (Latchman, 1997). TFs may perform their functions alone or with other proteins in a complex via promoting as an activator or blocking as a repressor the recruitment of RNA polymerase to specific genes. In legumes, different TF subfamilies might show different regulation under stress (Udvardi et al., 2007). In our study, many TFs families have been identified, and many of them have been reported to be involved in the plant drought-tolerance system (Table 5). In the present study, most of the TFs were enriched in MYB, WRKY, and ERF. In the present study, five genes from the MYB family were highly induced under drought stress in all four genotypes. In addition, nine MYB TFs were highly induced, particularly in the two drought-tolerant genotypes, and only one MYB TF was induced only in the two drought-susceptible genotypes. This indicated the significance of the MYB family in drought stress, especially in drought-tolerant genotypes. The MYB family has been documented to act through the ABA signaling cascade to regulate stomatal movement and as a result of water loss regulation in Arabidopsis and rice (Yanhui et al., 2006; Dai et al., 2007).
The present study demonstrated that mid-season drought alters the transcriptome profile in four peanut genotypes with varying drought-tolerant levels. Thousands of novel genes of cultivated peanuts were identified and annotated. The DEGs involved in circadian rhythm-plant, phenylpropanoid biosynthesis, starch and sucrose metabolism, photosynthesis-antenna proteins, etc., were enriched. In addition, the ABA-related pathway was considered as one of the most important mechanisms underlying drought tolerance in peanuts. This study provided insights into putative peanut response against drought stress.
Genetic variability between drought-tolerant and drought-susceptible genotypes evaluated is extremely low, suggesting that biochemical and molecular differences in drought treatment may be qualitative and subtle. The general conclusion is that the peanut drought response in this study has many similarities with other drought studies but with additive novel observations. A higher number of DEGs were observed in drought-tolerant compared with drought-susceptible lines, and there were more upregulated genes than downregulated genes in both (Supplementary Table 3). Comparing DEGs, 3,576 DEGs were observed only in drought-tolerant lines, and 1,625 DEGs were specific to drought-susceptible lines. GO terms were defined for these genes showing 74 for drought-tolerant lines highly enriched for cellular processes, protein modification, and gene-regulation and 32 for drought-susceptible lines enriched for catalytic activity, ion binding, and carbon/oxygen-binding activity. These gene activities highlight the subtleties of gene regulation demonstrated in our previous work showing drought-regulated gene expression evaluating a subset of transcription factors (Dang et al., 2012). In summary, these results showed that the mechanism underlying drought tolerance in peanuts involves a complex network of multiple hormones and a variety of molecular responses. However, the underlying regulatory mechanisms still need to be further studied. Therefore, studying plant hormone signaling pathways will be crucial in understanding the regulatory mechanism in peanut drought tolerance.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm. nih.gov/BioSample, accessions: SAMN17150141–SAMN17150164.
Author Contributions
CC is a director of the project. He supervised and coordinated the experiments and revised the manuscript. XW and XY conducted the experiments and wrote draft of manuscript. YF supervised RT-PCR experiments in her lab. PD performed rainout shelter drought treatment, collected leaf samples, and revised the manuscript. WW conducted data analysis for reversion. RG, JC, YC, PO-A, and CH revised the manuscript. All authors contributed to the manuscript and approved the final version of the manuscript to be published.
Funding
This work was supported by funding from the USDA-NIFA fund 2017-68008-26305 and in part by the National Peanut Board and Alabama Agricultural Experiment Station.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to Sam Hilton, Joseph Powell, Dr. Marshall Lamb, Dr. Ron Sorenson, Dr. Chris Butts, and other personnel from the USDA Agricultural Research Service National Peanut Research Lab at Dawson, GA, United States for assistance in this research.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.645291/full#supplementary-material
Supplementary Table 1 | qPCR primer design.
Supplementary Table 2 | Quantification calculation and comparison with RNAseq.
Supplementary Table 3 | Supplemental table GO of DEGs expressed in drought tolerant genotypes and susceptible genotypes.
Footnotes
- ^ http://peanutbase.org
- ^ http://systemsbiology.cau.edu.cn/agriGOv2/
- ^ http://www.genome.jp/kaas-bin/kaas_main
- ^ http://kobas.cbi.pku.edu.cn/index.php
References
Andrews, S. (2010). FASTQC. A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed September 29, 2014).
Balota, M., Isleib, T. G., and Tallury, S. (2012). Variability for drought related traits of virginia-type peanut cultivars and advanced breeding lines. Crop Sci. 52, 2702–2713. doi: 10.2135/cropsci2012.03.0207
Bertioli, D. J., Jenkins, J., Clevenger, J., Dudchenko, O., Gao, D., Seijo, G., et al. (2019). The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 51:877. doi: 10.1038/s41588-019-0405-z
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brasileiro, A. C. M., Morgante, C. V., Araujo, A. C. G., Leal-Bertioli, S. C. M., Silva, A. K., Martins, A. C. Q., et al. (2015). Transcriptome profiling of wild Arachis from water-limited environments uncovers drought tolerance candidate genes. Plant Mol. Biol. Rep. 33, 1876–1892. doi: 10.1007/s11105-015-0882-x
Chen, T., Lv, Y., Zhao, T., Li, N., Yang, Y., and Yu, W. G. (2013). Comparative transcriptome profiling of a resistant vs. susceptible tomato (Solanum lycopersicum) cultivar in response to infection by tomato yellow leaf curl virus. PLoS One 8:e80816. doi: 10.1371/journal.pone.0080816
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Dai, X., Xu, Y., Ma, Q., Xu, W., Wang, T., Xue, Y., et al. (2007). Overexpression of an R1R2R3 MYB gene, OsMYB3R-2, increases tolerance to freezing, drought, and salt stress in transgenic Arabidopsis. Plant Physiol. 143, 1739–1751. doi: 10.1104/pp.106.094532
Dang, P. M., Chen, C. Y., and Holbrook, C. C. (2012). Identification of drought-induced transcription factors in peanut (Arachis hypogaea L.). J. Mol. Biochem. 1:3.
Davies, P. J. (2013). Plant Hormones: Physiology, Biochemistry and Molecular Biology. Berlin: Springer Science & Business Media.
Degenkolbe, T., Do, P. T., Zuther, E., Repsilber, D., Walther, D., Hincha, D. K., et al. (2009). Expression profiling of rice cultivars differing in their tolerance to long-term drought stress. Plant Mol. Biol. 69, 133–153. doi: 10.1007/s11103-008-9412-7
Devi, M. J., Sinclair, T. R., and Vadez, V. (2010). Genotypic variability among peanut (Arachis hypogea L.) in sensitivity of nitrogen fixation to soil drying. Plant Soil 330, 139–148. doi: 10.1007/s11104-009-0185-9
Dinh, H. T., Kaewpradit, W., Jogloy, S., Vorasoot, N., and Patanothai, A. (2013). Biological nitrogen fixation of peanut genotypes with different levels of drought tolerance under mid-season drought. SABRAO J. Breed. Genet. 45, 491–503.
Farooq, M., Wahid, A., Kobayashi, N., Fujita, D., and Basra, S. M. A. (2009). “Plant drought stress: effects, mechanisms and management,” in Sustainable Agriculture, eds E. Lichtfouse, M. Navarrete, P. Debaeke, S. Véronique, and C. Alberola (Dordrecht: Springer Netherlands), 153–188. doi: 10.1007/978-90-481-2666-8_12
Greenberg, D. C., Williams, J. H., and Ndunguru, B. J. (1992). Differences in yield determining processes of groundnut (Arachis hypogaea L.) genotypes in varied drought environments1. Ann. Appl. Biol. 120, 557–566. doi: 10.1111/j.1744-7348.1992.tb04915.x
Guimarães, P. M., Brasileiro, A. C., Morgante, C. V., Martins, A. C., Pappas, G., Silva, O. B. Jr., et al. (2012). Global transcriptome analysis of two wild relatives of peanut under drought and fungi infection. BMC Genomics 13:387. doi: 10.1186/1471-2164-13-387
Holbrook, C. C., Isleib, T. G., Ozias-Akins, P., Chu, Y., Knapp, S., Tillman, B., et al. (2013). Development and phenotyping of recombinant inbred line (RIL) populations for peanut (Arachis hypogaea). Peanut Sci. 40, 89–94. doi: 10.3146/ps13-5.1
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, A. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Latchman, D. S. (1997). Transcription factors: an overview. Int. J. Biochem. Cell Biol. 29, 25435–25442. doi: 10.1016/s1357-2725(97)00085-x
Li, X. Y., Lu, J. B., Liu, S., Liu, X., Lin, Y. Y., and Li, L. (2014). Identification of rapidly induced genes in the response of peanut (Arachis hypogaea) to water deficit and abscisic acid. BMC Biotechnol. 14:58. doi: 10.1186/1472-6750-14-58
Liu, J., Xia, Z., Wang, M., Zhang, X., Yang, T., and Wu, J. (2013). Overexpression of a maize E3 ubiquitin ligase gene enhances drought tolerance through regulating stomatal aperture and antioxidant system in transgenic tobacco. Plant Physiol. Biochem. 73, 114–120. doi: 10.1016/j.plaphy.2013.09.006
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Long, H., Zheng, Z., Zhang, Y., Xing, P., Wan, X., Zheng, Y., et al. (2019). An abscisic acid (ABA) homeostasis regulated by its production, catabolism and transport in peanut leaves in response to drought stress. bioRxiv [Preprint]. doi: 10.1101/569848 bioRxiv: 569848,
Lu, X. Y., Li, J. Q., Liu, X. N., and Ma, J. (2014). De novo transcriptome of the desert beetle Microdera punctipennis (Coleoptera: Tenebrionidae) using illumine RNA-seq technology. Mol. Biol. Rep. 41, 7293–7303. doi: 10.1007/s11033-014-3615-6
Mathioni, M. S., Belo, A., Rizzo, C. J., and Dean, R. A. (2011). Transcriptome profiling of the rice blast fungus during invasive plant infection and in vitro stresses. BMC Genomics 12:49. doi: 10.1186/1471-2164-12-49
Petre, B., Morin, E., Tisserant, E., and Hacquard, S. (2012). RNA-seq of early infected poplar leaves by the rust pathogen Melampsora laricipopulina uncovers PtSultr3; 5, a fungal-induced host sulfate transporter. PLoS One 7:e44408. doi: 10.1371/journal.pone.0044408
Pimratch, S., Jogloy, S., Vorasoot, N., Toomsan, B., Kesmala, T., Patanothai, A., et al. (2008). Effect of drought stress on traits related to N2 fixation in eleven peanut (Arachis hypogaea L.) genotypes differing in degrees of resistance to drought. Asian J. Plant Sci. 7, 334–342. doi: 10.3923/ajps.2008.334.342
Pimratch, S., Jogloy, S., Vorasoot, N., Toomsan, B., Kesmala, T., Patanothai, A., et al. (2009). Heritability of N fixation traits, and phenotypic and genotypic correlations between N fixation traits with drought resistance traits and yield in Peanut. Crop Sci. 49, 791–800. doi: 10.2135/cropsci2008.06.0331
Puangbut, D., Jogloy, S., Vorasoot, N., Akkasaeng, C., Kesmalac, T., and Patanothai, A. (2009). Variability in yield responses of peanut (Arachis hypogaea L.) genotypes under early season drought. Asian J. Plant Sci. 8, 254–264. doi: 10.3923/ajps.2009.254.264
Ruan, J., Guo, F., Wang, Y., Li, X., Wan, S., Shan, L., et al. (2018). Transcriptome analysis of alternative splicing in peanut (Arachis hypogaea L.). BMC Plant Biol. 18:139. doi: 10.1186/s12870-018-1339-9
Songsri, P., Jogloy, S., Kesmala, T., Vorasoot, N., Akkasaeng, C., Patanothai, A., et al. (2008). Heritability of drought resistance traits and correlation of drought resistance and agronomic traits in peanut. Crop Sci. 48, 2245–2253. doi: 10.2135/cropsci2008.04.0228
Sreenivasulu, N., Harshavardhan, V. T., Govind, G., Seiler, C., and Kohli, A. (2012). Contrapuntal role of ABA: does it mediate stress tolerance or plant growth retardation under long-term drought stress? Gene 506, 265–273. doi: 10.1016/j.gene.2012.06.076
Stansell, J. R., and Pallas, J. E. (1985). Yield and quality response of Florunner peanut to applied drought at several growth stages. Peanut Sci. 12, 64–70. doi: 10.3146/pnut.12.2.0005
Stansell, J. R., shepherd, J. L., Pallas, J. E. Jr., Bruce, R. R., Minton, N. A., Bell, D. K., et al. (1976). Peanuts responses to soil water variables in the southeast. Peanut Sci. 3, 44–48.
Sterling, C. M., Ong, C. K., and Black, C. R. (1989). The response of groundnut (Arachis hypogaea L.) to timing of irrigation. J. Exp. Bot. 40, 1145–1153. doi: 10.1093/jxb/40.10.1145
Supek, F., Bošnjak, M., Škunca, N., and Šmuc, T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800. doi: 10.1371/journal.pone.0021800
Tian, T., Liu, Y., Yan, H., You, Q., Yi, X., Du, Z., et al. (2017). agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 45, W122–W129. doi: 10.1093/nar/gkx382
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Udvardi, M. K., Kakar, K., Wandrey, M., Montanari, O., Murray, J., Andriankaja, A., et al. (2007). Legume transcription factors: global regulators of plant development and response to the environment. Plant Physiol. 144:2007. doi: 10.1104/pp.107.098061
Wan, X., Mo, A., Liu, S., Yang, L., and Li, L. (2011). Constitutive expression of a peanut ubiquitin-conjugating enzyme gene in Arabidopsis confers improved water-stress tolerance through regulation of stress-responsive gene expression. J. Biosci. Bioeng. 111, 478–484. doi: 10.1016/j.jbiosc.2010.11.021
Wu, J., Mao, X., Cai, T., Luo, J., and Wei, L. (2006). KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 34, W720–W724. doi: 10.1093/nar/gkl167
Xu, Y., Gao, S., Yang, Y., Huang, M., Cheng, L., Wei, Q., et al. (2013). Transcriptome sequencing and whole genome expression profiling of chrysanthemum under dehydration stress. BMC Genomics 14:662. doi: 10.1186/1471-2164-14-662
Yanhui, C., Xiaoyuan, Y., Kun, H., Meihua, L., Jigang, L., Zhaofeng, G., et al. (2006). The MYB transcription factor superfamily of Arabidopsis: expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol. Biol. 60, 107–124.
Yin, D., Liu, H., Zhang, X., and Cui, D. (2011). A protocol for extraction of high-quality RNA and DNA from Peanut plant tissues. Mol. Biotechnol. 49, 187–191. doi: 10.1007/s12033-011-9391-9
Zhang, S., Qi, Y., Liu, M., and Yang, C. (2013). SUMO E3 Ligase AtMMS21 regulates drought tolerance in Arabidopsis thaliana F. J. Integr. Plant Biol. 55, 83–95. doi: 10.1111/jipb.12024
Keywords: transcriptome, drought, induced genes, cultivated peanut, water deficit
Citation: Wang X, Yang X, Feng Y, Dang P, Wang W, Graze R, Clevenger JP, Chu Y, Ozias-Akins P, Holbrook C and Chen C (2021) Transcriptome Profile Reveals Drought-Induced Genes Preferentially Expressed in Response to Water Deficit in Cultivated Peanut (Arachis hypogaea L.). Front. Plant Sci. 12:645291. doi: 10.3389/fpls.2021.645291
Received: 23 December 2020; Accepted: 12 March 2021;
Published: 30 April 2021.
Edited by:
Alfonso Clemente, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Bishal Tamang, University of Illinois at Urbana-Champaign, United StatesXiao-Ren Chen, Yangzhou University, China
Copyright © 2021 Wang, Yang, Feng, Dang, Wang, Graze, Clevenger, Chu, Ozias-Akins, Holbrook and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles Chen, Y3ljMDAwMkBhdWJ1cm4uZWR1
†These authors have contributed equally to this work