 Natascha D. Wagner
Natascha D. Wagner Martin Volf
Martin Volf Elvira Hörandl
Elvira Hörandl- 1Department of Systematics, Biodiversity and Evolution of Plants (With Herbarium), University of Goettingen, Göttingen, Germany
- 2Biology Centre of the Czech Academy of Sciences, Institute of Entomology, Ceske Budejovice, Czechia
Plastome phylogenomics is used in a broad range of studies where single markers do not bear enough information. Phylogenetic reconstruction in the genus Salix is difficult due to the lack of informative characters and reticulate evolution. Here, we use a genome skimming approach to reconstruct 41 complete plastomes of 32 Eurasian and North American Salix species representing different lineages, different ploidy levels, and separate geographic regions. We combined our plastomes with published data from Genbank to build a comprehensive phylogeny of 61 samples (50 species) using RAxML (Randomized Axelerated Maximum Likelihood). Additionally, haplotype networks for two observed subclades were calculated, and 72 genes were tested to be under selection. The results revealed a highly conserved structure of the observed plastomes. Within the genus, we observed a variation of 1.68%, most of which separated subg. Salix from the subgeneric Chamaetia/Vetrix clade. Our data generally confirm previous plastid phylogenies, however, within Chamaetia/Vetrix phylogenetic results represented neither taxonomical classifications nor geographical regions. Non-coding DNA regions were responsible for most of the observed variation within subclades and 5.6% of the analyzed genes showed signals of diversifying selection. A comparison of nuclear restriction site associated DNA (RAD) sequencing and plastome data on a subset of 10 species showed discrepancies in topology and resolution. We assume that a combination of (i) a very low mutation rate due to efficient mechanisms preventing mutagenesis, (ii) reticulate evolution, including ancient and ongoing hybridization, and (iii) homoplasy has shaped plastome evolution in willows.
Introduction
Plastid markers are frequently used in plant phylogenetics because they possess several advantages over nuclear markers (Taberlet et al., 1991; Gitzendanner et al., 2018). They are haploid but occur in high copy number, which simplifies the sequencing process. Additionally, the availability of various plastid markers with different levels of molecular evolution combined with the conserved structure of the plastome makes them a popular choice for molecular systematic studies on different levels of divergence (Shaw et al., 2005, 2007; Wicke and Schneeweiss, 2015). Plastomes are usually inherited uniparentally and only rarely show recombination between differentiated plastid genomes (Wolfe and Randle, 2004; Bock et al., 2014). In combination with nuclear markers, this makes plastomes useful for the analysis of introgression, hybridization, and polyploidy. In addition, the dispersion of maternally inherited genomes occurs at shorter geographic distances than for nuclear genomes. The consequence of a reduced gene dispersal and high genetic drift in organelle genomes is a generally pronounced geographic structure (Besnard et al., 2011). However, despite all these advantages, single plastid markers have not been able to resolve phylogenetic relationships in some lineages due to a lack of informative sites (e.g., Percy et al., 2014). The advent of next generation sequencing techniques has enabled researchers to overcome this lack of information by analyzing complete plastomes at moderate costs, e.g., via genome skimming (Straub et al., 2012; Wicke and Schneeweiss, 2015). The number of available plastomes in databases that potentially might serve as reference for read mapping has drastically increased over the last years. Thus, plastome phylogenomics has been used in a broad range of studies, e.g., in rapidly radiating groups (Barrett et al., 2014; Straub et al., 2014) and lineages with high species diversity (Huang et al., 2014; Givnish et al., 2015; Nargar et al., 2018).
The genus Salix L. (Salicaceae) comprises about 400–450 species of trees and shrubs mainly occurring in the Northern Hemisphere (Fang et al., 1999; Skvortsov, 1999; Argus, 2010). Willows are ecologically and economically important, e.g., for biomass production (Smart et al., 2005; Karp et al., 2011), and they are considered as keystone plants for insect diversity (Narango et al., 2020). The reconstruction of the willow phylogeny has proven to be difficult based on traditional Sanger sequencing markers, which have failed to resolve interspecific relationships (Leskinen and Alström-Rapaport, 1999; Azuma et al., 2000; Chen et al., 2010; Savage and Cavender-Bares, 2012; Barcaccia et al., 2014; Percy et al., 2014; Lauron-Moreau et al., 2015; Wu et al., 2015). Based on morphological characters, the genus is divided into three (or five) subgenera: subgenus Salix s.l. (including subgenera Salix L., Longifoliae (ANDERSSON) ARGUS, Protitae KIMURA, or excluding the latter two), subgenus Chamaetia (DUMORT) NASAROV in KOM., and subgenus Vetrix DUMORT (Skvortsov, 1999; Argus, 2010; Lauron-Moreau et al., 2015; Wu et al., 2015). Recent studies have recommended that the latter two subgenera be merged into the Chamaetia/Vetrix clade (Wu et al., 2015; Wagner et al., 2018, 2020, 2021). This clade comprises about three quarters of the described species diversity in Salix containing more than 300 species classified in about 40 sections. Previous molecular studies based on traditional markers were able to confirm the monophyly of the genus and to separate a small, basal clade of subtropical to temperate trees (subg. Salix s.l.) (Leskinen and Alström-Rapaport, 1999; Azuma et al., 2000; Chen et al., 2010; Savage and Cavender-Bares, 2012; Barcaccia et al., 2014; Percy et al., 2014; Lauron-Moreau et al., 2015; Wu et al., 2015). Nevertheless, they failed to resolve the relationships among species of the diverse Chamaetia/Vetrix clade of shrub willows due to a lack of informative sites. Percy et al. (2014) tried to interpret the lack of variation in plastid barcoding markers with either coalescence failure and incomplete lineage sorting, or a selective, trans-specific sweep for a certain haplotype. The latter idea was supported by the observation of a non-random distribution of haplotypes and of polymorphisms within genes (Percy et al., 2014). However, the authors included only four plastid loci and focused mainly North American species. Selective sweeps were also hypothesized by Huang et al. (2014) to occur in plastomes of a few tested willow species. We aim to test if this pattern can be confirmed for complete plastome data in a more comprehensive sampling of species.
While single plastid or nuclear markers have failed to resolve relationships, recently, restriction site associated DNA (RAD) sequencing has been used to resolve relationships within the Chamaetia/Vetrix clade, rendering all taxonomic species as distinct monophyletic lineages (Wagner et al., 2018, 2020; He et al., 2021b). However, the data contained exclusively nuclear information. The availability of additional whole plastome data would increase our understanding of reticulate evolution within the genus. Reticulate evolution could involve several processes: ancient incomplete lineage sorting, horizontal gene transfer, and/or interspecific hybridization. In case of hybridization, including the hybrid origin of allopolyploids, the position of a species will differ between phylogenies that are based on plastid data representing the maternal lineage and nuclear data reflecting biparental inheritance. By analyzing plastomes in combination with nuclear data, it is thus possible to test hypotheses on reticulate evolution (Wicke and Schneeweiss, 2015) and to gain insight into the mode of origin for polyploids. Extant hybridization and introgression is an extensively reported and studied phenomenon in Salix and occurs even between distantly related species (Skvortsov, 1999; Argus, 2010; Hörandl et al., 2012; Gramlich et al., 2018). Additionally, ancient hybrid origin via allopolyploidy has been demonstrated for several European species (Wagner et al., 2020). Therefore, with the incorporation of plastid data, we may gain insights into whether frequent hybridization and chloroplast capture are leading to a spread of a few dominant plastid haplotypes as assumed among subg. Chamaetia/Vetrix (Percy et al., 2014; Lauron-Moreau et al., 2015). Chloroplast capture (Rieseberg and Soltis, 1991) often leads to a geographic clustering of haplotypes rather than a species-specific clustering. This is especially frequent in taxa with known hybridization and introgression (e.g., Quercus, Pham et al., 2017).
Recently, several single Salix plastomes were published (e.g., Lu et al., 2019; Wu et al., 2019; Chen, 2020) and the method of complete plastome sequencing was applied to the family Salicaceae s.l. to study phylogenetic relationships and diversification of five genera with a special focus on Salix and Populus (Huang et al., 2017; Zhang et al., 2018). This set was expanded by Li et al. (2019) to 24 species representing 18 genera. However, the authors focused on higher taxonomic levels and not on subgeneric relationships. Furthermore, few of the previous accessions covered the Chamaetia/Vetrix clade that contains most of the willow species. In this study, we present 41 complete plastomes of 32 Salix species, representing 19 out of circa 40 sections, with a specific focus on Eurasian species of the diverse Chamaetia/Vetrix clade to analyze plastome structure and variability. We combine the data with available Salix plastomes from Genbank to determine the utility of complete plastomes for phylogenetic analyses. The reconstructed relationships of the genus are used to examine if the taxonomical classification and/or biogeographical distribution are reflected by plastome data. We test whether selective sweeps could have shaped the plastome diversity of willows. Furthermore, we compare the plastome to nuclear RAD sequencing data in order to discuss ancient and recent hybridization and introgression in our target group. Finally, we discuss possible reasons that can explain the observed level of plastome variability within the genus Salix.
Materials and Methods
Plant Material
For this study, we sampled 32 species (41 accessions) representing 19 sections sensu Skvortsov (1999) (Table 1, Supplementary Table 1). Four species belonged to Salix subg. Salix s.l., (s. Skvortsov) and 28 species belonged to the shrub willow clade Chamaetia/Vetrix. Next to sectional representation, we covered several ploidy levels. In total, we included 21 diploid, one triploid, five tetraploid, four hexaploid, and one octoploid species. The samples were collected mainly in Central Europe, however, additional samples from Spain, United Kingdom, Northern Europe, as well as the United States were included. Species were determined after Skvortsov (1999), Argus (2010), and Hörandl et al. (2012). Leaves were dried in silica gel and herbarium voucher specimens were deposited in the herbarium of the University of Goettingen (GOET) and the University of South Bohemia. For phylogenetic analyses, we integrated 20 available plastomes from Genbank (Supplementary Table 1).

Table 1. Plant material including taxonomic classification and origin.
Genome Skimming and Reference-Based Mapping
The DNA of all samples was extracted using the Qiagen DNeasy Plant Mini Kit following instructions from the manufacturer (Valencia, CA). After quality check, the DNA of 12 samples was sent to NIG - NGS Integrative Genomics Core Unit of the University Medical Center Göttingen (UMG) (https://www.humangenetik-umg.de/en/research/nig/) for library preparation and sequencing. About 1 μg DNA of each sample was used for library preparation using the PCR FreeDNA Sample Prep Kit (Illumina) followed by the Illumina TruSeq PE Cluster Kit. The 12 samples were barcoded and multiplexed. Whole genome shotgun sequencing was performed on one lane of an Illumina HiSeq 2500 platform producing 2 × 150 bp paired end reads. With this initial test set, we wanted to assess the utility of whole genome skimming for Salix plastome reconstruction. The sequencing libraries for the remaining samples were generated using the NEBNext® DNA Library Prep Kit following recommendations of manufacturer. The NEBNext® Multiplex Oligos for Illumina kit was used to add indices to each sample and to enrich the libraries via PCR using P5 and indexed P7 oligos. The PCR products were then purified (AMPure XP system). Whole genome shotgun sequencing was carried out on a Novaseq 6000 platform producing 2 × 150 bp paired end reads. The quality of the resulting sequencing reads was checked with FastQC v.0.10.1 (Andrews, 2010), and the reads were assembled de novo for a total of 36 samples using the software Fast-Plast v.1.2.8 (McKain and Wilson, 2017) under its default settings. The minimum coverage was 0.25 of the average coverage across the respective plastome. For five samples, Fast-Plast was not able to assemble the complete plastome. It is known that fragments of the plastome were transferred to the nuclear genome in Salix (see Huang et al., 2014). These pseudo-copies might cause problems in a de novo plastome assembly approach based on deep sequencing data. To obtain plastomes for the five samples, we utilized a “mapping-to-reference” approach to receive the respective plastomes. For the reference-based assembly, we used Geneious vR11 2020.2.4 (http://www.geneious.com, Kearse et al., 2012) as described in Ripma et al. (2014). The reads were mapped to the plastome of S. purpurea [Genbank accession NC026722].
The annotation of plastomes was done using CPGAVAS2 (Shi et al., 2019) with default settings applying the dataset containing 2,544 reference plastomes. The results were checked and edited with Geneious R11 2020.2.4 (www.geneious.com).
Phylogenetic Analyses
For final phylogenetic analyses, the sequences of the 41 produced plastomes were combined with 20 available Salix plastomes from Genbank resulting in a dataset comprising 61 samples (for details, see Supplementary Table 1). Complete plastid genomes were aligned as a single sequence with MAFFT v3 (as implemented in Geneious R11) by applying the automatic algorithm selection with a gap open penalty of 1.53 and an offset value of 0.123. One inverted repeat (IR) copy was excluded from the alignment to avoid double weighting of identical information. Because the overall variation was low, especially within the Chamaetia/Vetrix clade, the effects of misaligned regions in the subsequent tree topology could be pronounced (Parks et al., 2012; Duvall et al., 2020). Therefore, the alignment was optimized using Gblocks 0.91b (Castresana, 2000) with default settings (minimum number of sequences for a conserved position set to 25, minimum number of sequences for a flank position set to 40, maximum number of contiguous non-conserved positions set to 8, and minimum length of a block set to 10). The allowed gap position was set to “none” and “all,” respectively, and the results of both approaches were compared. The resulting alignments were extracted in PHYLIP format and used as input for Maximum Likelihood analysis using the general time-reversible (GTR)+Γ model of nucleotide substitution implemented in RAxML (Randomized Axelerated Maximum Likelihood) v.8.2.4 (Stamatakis, 2014). We conducted for each ML analysis a rapid bootstrapping (BS) analysis with 100 replicates. Resulting trees were obtained in FigTree v1.4.3 (Rambaut, 2014).
Haplotype Networks
Next to phylogenetic tree reconstruction, we used the plastome data to calculate haplotype networks with TCS v1.21 (Clement et al., 2000). Due to the large genetic distance between the subclades, we conducted two separate analyses without out-group: one for the closely related Chamaetia/Vetrix clade and one for subgenus Salix s.l. We used only coding regions to avoid homoplasy in the data set. Gaps were treated as missing data.
Statistical Tests
A geographic clustering rather than a taxonomic clustering is frequently observed in plastid-based studies (Gitzendanner et al., 2018). To test for this trend in our data, we correlated the genetic distance with the geographic distance of the included samples. We derived the genetic distance matrix from the branch lengths in the observed RAxML tree of the complete plastome dataset using the R package ape 5.0 (Paradis and Schliep, 2019). We calculated the geographic distance matrix based on the global positioning system (GPS) coordinates of our samples. For Genbank samples without detailed information on sampling localities, we used the distribution center of the species or the location of the institute that performed the analyses instead. We then correlated the two matrices using a Mantel test based on Pearson's product-moment correlation with 999 permutations in the R package vegan 2.5 (Oksanen et al., 2019). We performed the analysis with the full dataset and with subg. Salix and subg. Chamaetia/Vetrix clades separately. Furthermore, in the case of subg. Chamaetia/Vetrix, we also performed an analysis excluding its basal members (S. arbutifolia, S. rorida, S. magnifica, and S. oreinoma) that showed plastomes most divergent from the rest of the subgenus. All analyses were performed in R 3.6.1 (R Core Team, 2019).
To analyze if plastid genes are under selection, we calculated gene-wise ω (dN/dS ratios = non-synonymus vs. synonymous substitutions) with codeML implemented in paml version 4.8 (Yang, 2007). We used the model = 0 option, i.e., a single omega for the whole tree. We extracted the coding sequences (=CDS) of 72 genes (Supplementary Table 2) out of 78 genes in total and used the alignments and the RAxML tree of the complete sample set as input. Annotations of Genbank accessions were not complete in all cases and for statistical reasons we included only genes that were present/annotated in at least 60 of the 61 samples for this test.
Comparison to Nuclear RAD Sequencing Data of a Comparative Subset
To evaluate the phylogenetic resolution and topology of the plastome phylogeny, we compared 10 samples of our plastome data to a comparative sampling of already published RAD sequencing data. The RAD sequencing data are available at Genbank (Bioproject PRJNA433286). Previous RAD sequencing studies included two to four individuals per species and rendered species as monophyletic lineages (Wagner et al., 2018, 2020), and hence we used only one representative sample per species for the RAD sequencing analysis here. Salix triandra was used as an out-group in both datasets. For the comparison, we used a subset of 10 representative plastomes of the Chamaetia/Vetrix clade. We used the same accessions in both datasets whenever possible. However, in two cases, due to the low amount of extracted DNA, we substituted the species in the RAD analysis with another individual of the same species. The reduced RAD sequencing set was analyzed with ipyrad v7.24 (Eaton and Overcast, 2016) using the same settings as described in Wagner et al. (2018). The minimum number of samples sharing a locus was set to 4, the maximum number of single nucleotide polymorphism(s) SNP(s) per locus was set to 20, and the maximum number of indels per locus was set to 8. With respect to the mixed ploidy of the dataset, we used a maximum of four alleles in the settings of the ipyrad pipeline (for more details see Wagner et al., 2020). Maximum Likelihood analyses for both the concatenated RAD loci as well as the plastomes were performed as described above.
Results
Plastome Reconstruction
The shotgun sequencing revealed an average of 71.65 Mio raw paired reads per sample. An average of 6.86 Mio paired reads mapped to the plastome. The average coverage was 7,733 reads. The plastome lengths varied between 155,414 bp (S. mielichhoferi) and 160,386 bp (S. myrtilloides). Length variation was due to one large insertion at the margin of the inverted repeat (IRb) that was observed in 21 samples, two large indels (>200 bp) in S. triandra and species of subg. Salix, several smaller indels (2–80 bp), and repetitive motifs (SSRs, tandem repeats). All obtained plastomes showed the typical tetrapartite structure of two IRs separating the small single-copy (SSC) region from the large single-copy (LSC) region. They contained 78 protein coding genes, 30 tRNAs, and 3 rRNAs. The order of genes was identical in all newly assembled plastomes. The annotated plastomes were uploaded to Genbank, their respective accession numbers (MW435413–MW435453) are provided in Supplementary Table 1.
Phylogenetic Reconstructions
The plastome sequences presented here were aligned together with 20 available Salix plastomes from Genbank. The initial alignment of the complete plastomes of 61 samples had a length of 141,081 bp and after trimming of one IR and alignment optimization with Gblocks retained a length of 129,052 bp. The concatenated alignment of coding regions (CDS) had a length of 68,211 bp. The length of the edited alignment for the Chamaetia/Vetrix clade was 128,608 bp and 68,009 bp for the extracted coding regions. The lengths were 128,403 bp and 68,311 bp for subgenus Salix, respectively. The variation observed in the complete alignment was 1.68 and 0.72% in coding regions. Within the Chamaetia/Vetrix clade, 0.74% of sites were variable and we observed 0.41% variability in the alignment of extracted coding regions. Within subgenus Salix, we observed 0.64% variability and 0.35% of variable sites for CDS, respectively. A statistical comparison of the different alignment editing approaches is provided in Supplementary Table 3.
Relationships of Genus Salix Based on Plastome Data
The observed phylogenetic tree based on complete plastomes showed a clear separation of the subgenus Salix s.l. (tree willows) (BS 100) and the Chamaetia/Vetrix clade (shrub willows) plus S. triandra (BS 100) (Figure 1). Both accessions of S. triandra (BS 99) were found to be sister to the well-supported Chamaetia/Vetrix clade (BS 98). Within the Chamaetia/Vetrix clade, the observed resolution was low, indicated by short branches and no or low BS support for most branches. Salix arbutifolia was in sister position to the remaining samples of Chamaetia/Vetrix (BS 100), followed by the Asian species S. rorida, and a clade comprising S. magnifica and S. oreinoma (BS 71). The remaining samples formed a well-supported clade (BS 100) with Salix retusa (RET6) at an early diverging position. The two accessions of S. myrsinifolia grouped together with high support (BS 100), while all other species with more than one sample appeared polyphyletic. Additionally, the sectional classification was not reflected by the phylogeny. No geographical pattern was observed, e.g., Salix sitchensis from California was shown to be closely related to European S. myrsinifolia and S. caesia (BS 92). Asian S. gracilistyla appeared in close relationship to S. waldsteiniana and S. breviserrata, both occurring in the European Alps. However, resolution within this clade was extremely low. Within the subg. /Salix/ clade, S. babylonica was in sister relationship to a subclade (BS 100) containing the European tree species (S. alba, S. pentandra, and S. fragilis), and S. paraplesia from China. The North American willow S. interior was shown to be situated on a long branch and in sister position to the remaining samples of subg. Salix.
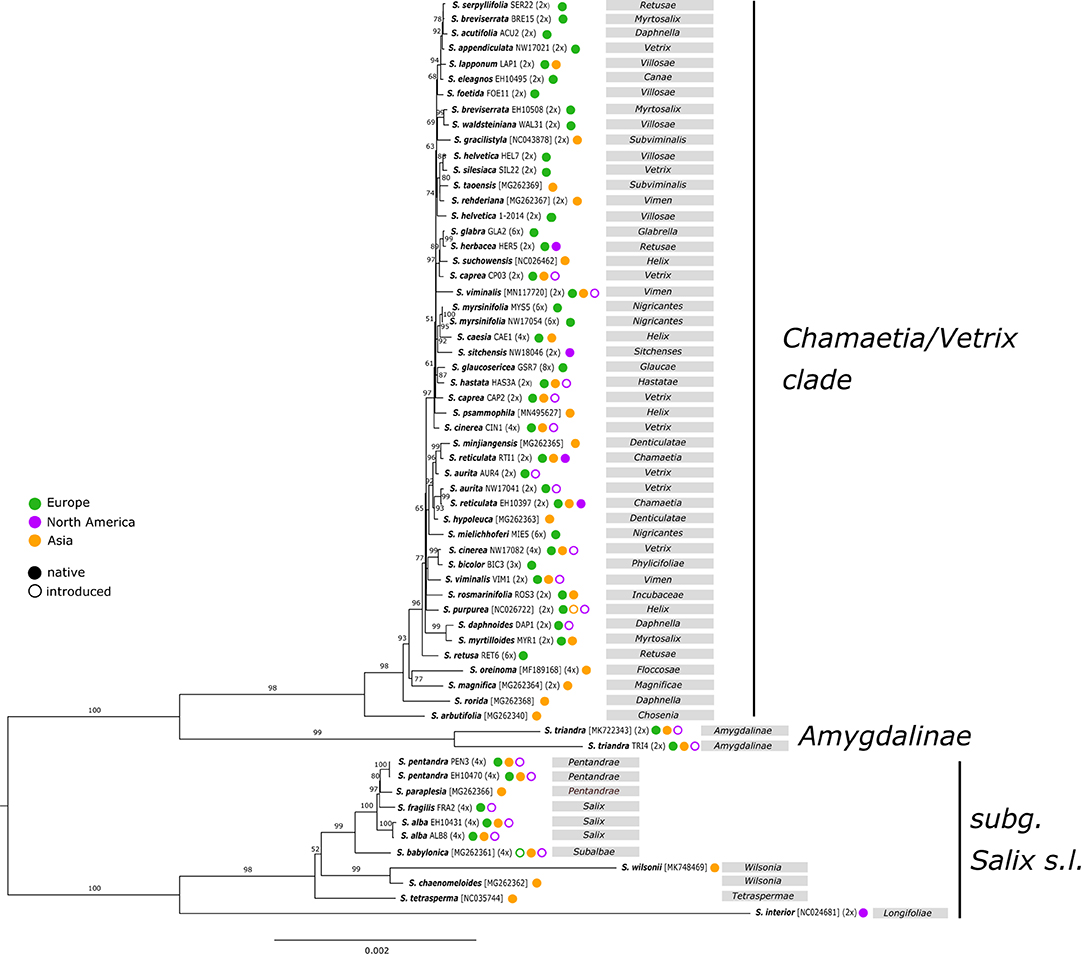
Figure 1. RAxML phylogeny based on complete plastomes of 61 samples representing 50 Salix species. Sample IDs or Genbank accession numbers for Genbank samples as well as ploidy levels are indicated behind species names. Bootstrap support values are given above branches. Geographic distribution of species is indicated behind sample names according to color coding. The taxonomical affiliation into sections is given in gray boxes. The two main clades are illustrated according to their subgeneric classification.
Haplotype Networks
We calculated haplotype networks of the two subclades based on coding regions (CDS). For subg. Salix, the haplotype network displayed the same clades as the RAxML tree (Supplementary Figure 1). The close relationships of tree willows collected in Europe were reflected by a central empty haplotype that was not occupied by any included species. Both S. alba accessions were only one mutational step apart from each other, both accessions of S. pentandra shared an identical haplotype. Salix paraplesia showed close relationships to S. alba, S. fragilis, and S. pentandra. The remaining Asian species were up to 92 mutational steps distinct from the central haplotype connecting the European tree willows plus S. paraplesia.
The haplotype network for the Chamaetia/Vetrix clade based on coding regions revealed that both accessions of S. myrsinifolia share the same haplotype. All other included samples showed a unique haplotype. Samples of the same species but from different localities did not group together, e.g., S. caprea from the United Kingdom was three mutational steps apart from S. caprea collected in the Czech Republic. Overall, the network showed three main groups that were connected via central haplotypes that were not occupied by included samples (Figure 2). Neither a geographical nor a taxonomical pattern was reflected.
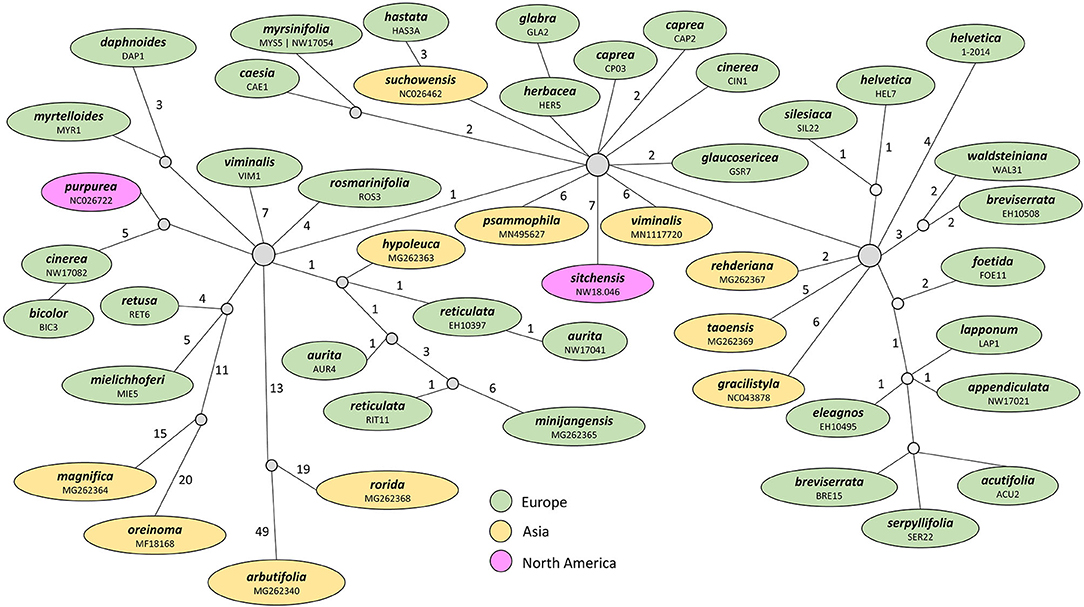
Figure 2. Haplotype network of 48 samples representing 42 species of the Chamaetia/Vetrix clade based on CDS regions. Each circle represents a unique haplotype, the number of mutational steps between haplotypes are indicated by numbers above branches. The applied color-coding reflects the geographic origin of samples. Both accessions of S. myrsinifolia share an identical haplotype. The colors highlight central haplotypes and closely related groups.
Statistical Tests
The Mantel tests revealed correlation of geographic and genetic distance in the case of the whole dataset (r = 0.1705 p = 0.018), for the subgenus Salix (r = 0.5025, p = 0.001), and for the whole subgenus Chamaetia/Vetrix clade (r = 0.2739, p = 0.018). When we excluded the early branching lineages of the Chamaetia/Vetrix clade, the significant correlation disappeared (r = 0.0619, p = 0.259).
The dN/dS ratios (non-synonymous vs. synonymous substitutions) revealed purifying selection for the majority of genes (ω values <1). Four genes showed ratios of positive, i.e., diversifying selection (rpl2, rpl16, rps15, and ycf 1). A complete list of gene-wise statistics and ω values is given in Supplementary Table 2.
Comparison of Plastome and RAD Sequencing Data
We compared the resulting phylogeny of 10 plastomes of the Chamaetia/Vetrix clade to published nuclear RAD sequencing data of the same subset of species. Salix triandra was used as an outgroup. The resulting plastome alignment of 130 kbp showed 0.5% variable sites. The RAD sequencing alignment of 57,084 concatenated RAD sequencing loci had a length of 4,669,722 bp and showed 8.05% variable sites. The alignment contained 26.3% missing data. The observed phylogeny based on RAD sequencing data was in accordance with formerly published data based on more samples (Wagner et al., 2018, 2020, 2021) (Figure 3). Salix reticulata was in sister position to the remaining species. Species belonging to section Vetrix formed a well-supported monophyletic group. The North American species S. sitchensis was situated in sister relationship to S. helvetica. The plastid phylogeny of the same subset showed tetraploid S. cinerea in sister position to the remaining species (Figure 3). The members of section Vetrix did not form a monophylum but occurred at different positions in the tree (highlighted in Figure 3). Salix sitchensis was in sister position to hexaploid S. myrsinifolia. The dwarf shrub S. breviserrata was found to be closely related to the Swiss willow S. helvetica. Overall, the branches were very short, however, the bootstrap values showed moderate to good support for the observed topology.
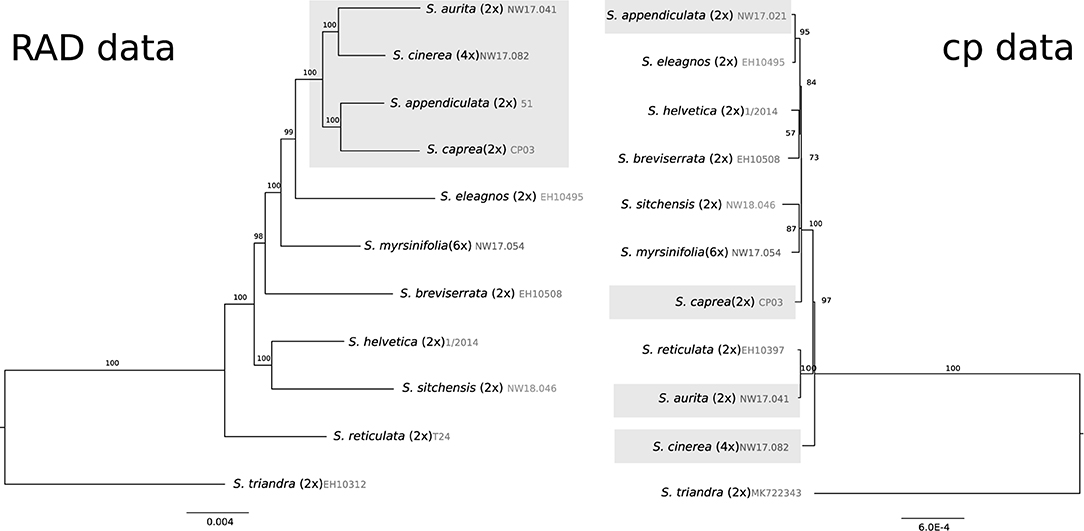
Figure 3. Comparison of Maximum Likelihood phylogenies of a subset of 10 species of the Chamaetia/Vetrix clade with S. triandra as out-group based on 57,084 RAD sequencing loci (left) and whole plastome data (right). RAD sequencing data from Wagner et al. (2018, 2020). Bootstrap support values are given above branches. The ploidy level and accession numbers are indicated behind species names, the members of section Vetrix are highlighted in gray.
Discussion
In 2014, Percy et al. proclaimed the “spectacular failure of barcodes for willows.” In contrast to single markers, we sought to analyze the variability of complete plastomes and their utility for phylogenetic analyses. We included 41 newly assembled plastomes and present here the first comprehensive study on plastome evolution on a subgeneric level within Salix. Compared to other angiosperms, our results reveal that the sequence variation in genus Salix is very low. Wu et al. (2015) observed 7.8% variable sites in their combined four-cp-marker set. However, the authors included next to Salix also samples of Populus and Dovyales as outgroups, which were responsible for most of the observed variation. To compare the results in detail, we extracted the four specific loci (matK, rbcL, atpB-rbcL, and trnD-trnT) of our dataset and revealed 3.2% variable sites for the combined loci and complete sampling, but only 0.4% for the Chamaetia/Vetrix clade and 2.4% for subg. Salix, respectively (see Supplementary Table 4).
Low Mutation Rates and Effective Repair Mechanisms in Coding Regions
Our alignment of the complete plastomes shows only 1.64% variable characters. Our alignment of all extracted coding regions shows only a variation of 0.72%. Genic regions of plastomes evolve with only about a third or half the rate of the nuclear genome (Wolfe et al., 1987). It is still unknown, why plant organellar genes have lower mutation rates than nuclear genes, but possible explanations include differences in replication enzymes, replication fidelities, mismatch repair, and low rates of genetic exchange (Gaut et al., 2011). Our results show a high degree of purifying selection in the protein coding genes of the plastome (Supplementary Table 2). The elimination of deleterious mutations might also affect linked sites and thus decrease the overall genetic variability (Charlesworth et al., 1993). Because the complete plastome can be treated as one single haplotype, this might be an explanation of the observed low variability. Next, recombination is a driver of purifying selection and can also happen between and among organelles (Bock et al., 2014). However, we assume that gene conversion might be a strong mechanism acting toward purifying selection in our dataset (Wolfe and Randle, 2004). Gene conversion is known from non-recombining systems, e.g., mitochondrial genomes (Mower et al., 2010) and nuclear genomes of ancient asexual animals (Flot et al., 2013). However, the elimination of deleterious mutations by gene conversion has also been proposed in plastid genomes (Khakhlova and Bock, 2006).
An overall low genetic divergence also occurs in the nuclear genomes of willows. Single barcoding regions like internal transcribed spacer (ITS) or single genes like rbcL and matK have failed to resolve interspecific relationships (Leskinen and Alström-Rapaport, 1999; Lauron-Moreau et al., 2015). Instead, thousands of nuclear RAD sequencing loci were required to resolve species-level relationships in the Chamaetia/Vetrix clade (Wagner et al., 2018, 2020, 2021; He et al., 2021b). Based on our results, as well as on previous studies, we infer generally low mutation rates in willow genomes, considering the relatively high age of the genus (up to 43.8 Ma; Wu et al., 2015). Efficient regulation of intracellular oxidative stress resulting from photosynthesis and respiration might avoid DNA damage and reduce frequencies of non-homologous DNA repair processes, which is generally a major source for mutagenesis (Friedberg and Meira, 2006). Willows are rich in antioxidants, especially in phenolics and other typical chemical compounds known for the regulation of redox homeostasis (Hörandl et al., 2012; Jia et al., 2020; Piatczak et al., 2020). Their hypothetical role in the observed low mutation rates would need to be tested. However, it is remarkable that a low mutation rate (c. one-sixth of Arabidopsis) has also been observed in nuclear, plastid, and mitochondrial genomes of poplar (Tuskan et al., 2006), the sister genus of Salix, which is similarly rich in phenolics, such as salicylates, tannins, or flavonoids (Palo, 1984).
Variable but Not Informative: Rapidly Evolving Non-coding Regions
In our dataset, we observed some length variation based on insertions/deletions resulting from sequence duplications in non-coding regions of the plastome, which is in the range of similar studies (Huang et al., 2017; Zhang et al., 2018; Li et al., 2019). Most of the observed variable characters occurred in rapidly evolving, non-coding parts of the plastome, as SSRs and other repetitive regions (Zhang et al., 2018; Li et al., 2019). Next to that, the haplotype network of the Chamaetia/Vetrix clade revealed mainly synonymous and non-directional mutations in coding regions (Figure 2). Homoplasy might be introduced by plastid haplotype polymorphism within and among individuals, resulting in paralogous copies (Wolfe and Randle, 2004). Further, the non-directional signal of mutations might lead to conflicting signals in the phylogeny (Parks et al., 2012; Duvall et al., 2020). Because the overall variability is very low, this effect might be even stronger within shrub willows. However, both the effects, low variation and non-directional signal, lead in combination to a non-resolved tree, especially in the Chamaetia/Vetrix clade. Interestingly, the effects in subgenus Salix seem to be less significant. Despite similar levels of variability, the topology of the subclade is much better resolved. This is in accordance with previous phylogenetic studies (Percy et al., 2014; Lauron-Moreau et al., 2015; Wu et al., 2015). Gene transfer from the plastome to the nucleus might give some additional explanation to the observed low variability (Bock and Timmis, 2008). For Populus trichocarpa, the transfer of the whole plastome to the nuclear genome was reported, while hints of transfer of single loci were also found in some Salix species (Huang et al., 2014). However, due to the lack of suitable genomes, we did not test for any transfer of plastid genes or larger portions of the plastome to the nuclear genome in our dataset. Nevertheless, the de novo assembly problems for five samples may have occurred due to the transfer of large portions of the plastome to the nucleus.
Molecular Dating Opposed Hypotheses of Rapid Radiation or Rapid Range Expansion From Refugia
Another explanation for the observed low plastome variation, especially within the Chamaetia/Vetrix clade, might be a large radiation or a rapid range expansion from refugia after the last glacial maximum (Percy et al., 2014; Lauron-Moreau et al., 2015). In this scenario, recently evolved species would share identical haplotypes. Our data on shrub willows revealed a low amount of variation, but almost no identical haplotypes were observed. Additionally, lineage diversification clearly predates the Pleistocene glaciations; the age of genus Salix was estimated as 43.8 Ma, and that of the Chamaetia/Vetrix clade as 23 Ma, respectively (Wu et al., 2015). Next to the diversification time, the distinctiveness of the species based on morphology and nuclear phylogenies also oppose a postglacial rapid radiation as an explanation for low plastome variability (Wagner et al., 2018, 2020; He et al., 2021b). However, an older radiation followed by fragmentation and genetic drift cannot be ruled out completely.
Differences Between Tree Willows (subg. Salix) and Shrub Willows (subg. Chamaetia/Vetrix)
Our comprehensive plastome phylogeny confirmed the differentiation into two distinct subgeneric clades (Wu et al., 2015; Huang et al., 2017; Zhang et al., 2018). An explanation for the split into two clades might be that species within subgenera and within sections hybridize more frequently than species between different subgenera (Hörandl, 1992). A recent study analyzed differences in sex determination systems in subg. Salix and the Chamaetia/Vetrix clade, which might be responsible for incompatibilities between the two subgeneric clades (He et al., 2021a). This would support our conclusion that the plastomes of the subgenera evolved more independently. Our data confirm the monophyly of species as well as the split of a New World and an Old World clade within subgenus Salix (Chen et al., 2010; Percy et al., 2014; Wu et al., 2015). The geographic pattern was further supported by the results of the Mantel test. Within the shrub willows, the somewhat isolated position of S. arbutifolia is in accordance with previous studies (Lauron-Moreau et al., 2015; Wu et al., 2015). The early branching Asian lineages corresponded to the Hengduan Mountain clade described in He et al. (2021b). Within the core Chamaetia/Vetrix clade, neither species-specific patterns nor support for previous sectional classification were found. The polyphyly of four species might be explained by homoplasy of plastid polymorphisms (see above).
Comparison of Nuclear and Plastid Data
We compared nuclear RAD sequencing data with plastome data for a subset of 10 samples. So far, few studies have performed a statistical comparison of the two reduced representation methods we used here (Pham et al., 2017; Mu et al., 2020). Our comparison clearly shows that RAD sequencing is much more efficient in resolving relationships within Salix than plastome data. The included members of the section Vetrix showed a well-supported monophyletic group in the RAD sequencing dataset, which is in accordance with previously published data (Wagner et al., 2018, 2020, 2021). However, the same species were scattered over the non-resolved tree in the plastome phylogeny (Figure 3). Maternal inheritance of plastomes might explain the observed incongruence of nuclear (RAD sequencing) and plastome phylogenies. These discrepancies could be further explained by chloroplast capture, and in the case of polyploid S. cinerea, by an allopolyploid origin (Wagner et al., 2020). In willows c. 40% of species are polyploid (Suda and Argus, 1968), which means that frequent allopolyploidy could have a major impact on phylogenetic relationships. Different ancient hybrid origins will influence the backbone of the plastome tree and thus explain discordance between nuclear and plastid phylogenies. Further, more recent hybridization or introgression events, even if infrequent, could occur between distantly related species, transferring the few and randomly appearing plastome polymorphisms to different genomic backgrounds of species.
The Lack of Any Biogeographical Pattern
In the presence of frequent hybridization as well as chloroplast capture, we would expect a biogeographic signal in the phylogeny and/or haplotype networks. Although our sampling represented mainly European species, it also included samples from several parts of Eurasia and North America. Based on our data, species of geographical proximity show distinct haplotypes while some species from separate continents share similar plastomes. For example, S. sitchensis from the West Coast of the United States shared a similar plastome with S. caesia and S. myrsinifolia from Europe. Over these huge distances, extant hybridization and frequent maternal gene flow via seeds is unlikely. However, within Eurasia, distribution areas of widespread species are often overlapping (see Skvortsov, 1999), and hybridization appears possible. Interestingly, the Mantel test revealed significant correlation of geographical and genetic distance in our dataset. This can be explained by the early branching lineages from China, which show quite distinct plastomes and might influence the results. When analyzing only the core clade of shrub willows, no correlation could be observed. This is in accordance with former results of Percy et al. (2014) who found correlation of geographical and genetic distance within the overall dataset, but no correlation within a large clade of shrub willows. The close relationship of Eurasian and North American shrub willow species in plastid-based phylogenies was also reported in Lauron-Moreau et al. (2015), who observed a large clade comprising boreo-arctic and montane to alpine species of both Eurasia and North America.
Plastid Genes Under Selection
Percy et al. (2014) assumed that hybridization/ introgression alone could not explain the small number of shared haplotypes between a large number of distinct willow morphospecies. They assumed a trans-specific selective sweep as a potential reason for one dominant haplotype. The positively selected plastome would have been able to spread rapidly, probably aided by widespread species hybridizing with the local ones. Our haplotype network of the Chamaetia/Vetrix clade, however, does not support the predominance of one certain haplotype. Further, most of the observed variation occurred in non-coding regions, and within genes, mostly in synonymous sites. However, the scenario of a selective sweep would require a positive selection of plastid genes. Indeed, Huang et al. (2017) tested plastid coding regions and found seven genes under selection in Salicaceae. However, this is not reflected in our results. Most tested protein coding genes showed purifying selection (dN/dS <1). Only four genes (5.6%) showed signals of positive selection (rpl2, rpl16, rps15, and ycf 1). Interestingly, they differed from the selective genes found by Huang et al. (2017). The genes analyzed by Percy et al. (2014) (matK, rbcL, rpoB, and rpoC1) were all under purifying selection with ω values far below one. Thus, our results based on more species strongly contradict the hypothesis of a selective sweep. In rpl2 and rps15, slight signals of positive selection were detected, but both are ribosomal genes. The signal of positive selection was strong (dN/dS > 1) only in the case of ycf 1. This large open reading frame was for a long time enigmatic and its function unknown. The gene ycf 1 has been predicted to have the highest nucleotide diversity (π) at the species level within angiosperm plastid genomes (Dong et al., 2012, 2015). More recently, it was shown that ycf 1 encodes for Tic214, a vital component of the Arabidopsis translocon on the inner chloroplast (TIC) membrane complex that is essential for plant viability (Kikuchi et al., 2013). However, in comparison to other plant genera, ycf 1 is relatively conserved and showed only 1.5% variability on the genus level within Salix. The lack of a predominant haplotype as well as the low number of genes under selection argue against the hypotheses of a selective sweep in willows.
Conclusions
The observed plastome variation in willows is much lower than in other angiosperm lineages. Thus, even complete plastome data are unsuitable for phylogenetic reconstruction, DNA barcoding, and analyses of biogeographical history in shrub willows. Usual explanations for plastome evolution patterns do not fit our data. Instead, the willow plastomes seem to have been shaped by extremely low mutation rates due to efficient mechanisms preventing mutagenesis, and further, by reticulate evolution and non-specific, rather random polymorphisms resulting in homoplasy. Consequently, the observed plastomes are neither species-specific nor reflect geographical patterns. Our results provide a caveat on relying solely on plastid phylogenies, a common practice in plant systematics. Our study demonstrates the importance of examining the evolution of plastid genomes thoroughly before applying them to questions of plant systematics, especially in cases of widespread, hybridizing taxa with low evolutionary rates.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, MW435413 - MW435453.
Author Contributions
NW planned and designed research. NW, MV, and EH conducted fieldwork. NW and MV performed experiments and analyzed data. NW wrote the manuscript. MV and EH contributed to the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was financially supported by the German research foundation Deutsche Forschungsgemeinschaft, DFG (Ho 5462/7-1 to EH and priority program Taxon-Omics SPP1991, project Wa3684/2-1 to NW). MV acknowledges funding by Czech Academy of Sciences, Programme for Research and Mobility Support of Starting Researchers (MSM200962004), and Grant Agency of the Czech Republic (20-10543Y).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RS declared a shared affiliation, with no collaboration, with one of the authors, MV, to the handling editor at the time of the review.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Bruce Baldwin, Susanne Gramlich, Andrea Danler, Katrin Scheufler, Claudia Pätzold, Carlo L. Seifert, Jan Michálek, Petr Kozel, Nela Nováková, and Tereza Holicová for collecting plant material and field assistance, and Salvatore Tomasello for help with data analysis. We thank John Bradican for his help with English editing of the manuscript. We thank four reviewers for their supportive comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.662715/full#supplementary-material
References
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Argus, G. W. (2010). “Salix,” in Flora of North America, Vol. 7 Magnoliophyta: Salicaceae to Brassicaceae, ed E. Flora of North America Editorial Committee (New York, NY: Oxford University Press), 23–51.
Azuma, T., Kajita, T., Yokoyama, J., and Ohashi, H. (2000). Phylogenetic relationships of Salix (Salicaceae) based on rbcL sequence data. Am. J. Bot. 87, 67–75. doi: 10.2307/2656686
Barcaccia, G., Meneghetti, S., Lucchin, M., and de Jong, H. (2014). Genetic segregation and genomic hybridization patterns support an allotetraploid structure and disomic inheritance for Salix species. Diversity 6, 633–651. doi: 10.3390/d6040633
Barrett, C. F., Specht, C. D., Leebens-Mack, J., Stevenson, D. W., Zomlefer, W. B., and Davis, J. I. (2014). Resolving ancient radiations: can complete plastid gene sets elucidate deep relationships among the tropical gingers (Zingiberales)? Ann. Bot. 113, 119–133. doi: 10.1093/aob/mct264
Besnard, G., Hernández, P., Khadari, B., Dorado, G., and Savolainen, V. (2011). Genomic profiling of plastid DNA variation in the Mediterranean olive tree. BMC Plant Biol. 11:80. doi: 10.1186/1471-2229-11-80
Bock, D. G., Andrew, R. L., and Rieseberg, L. H. (2014). On the adaptive value of cytoplasmic genomes in plants. Mol. Ecol. 23, 4899–4911. doi: 10.1111/mec.12920
Bock, R., and Timmis, J. N. (2008). Reconstructing evolution: gene transfer from plastids to the nucleus. BioEssays 30, 556–566. doi: 10.1002/bies.20761
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Charlesworth, B., Morgan, M. T., and Charlesworth, D. (1993). The effect of deleterious mutations on neutral molecular variation. Genetics 134, 1289–1303. doi: 10.1093/genetics/134.4.1289
Chen, A. J., Sun, H., Wen, J., Yang, Y., Chen, J., Sun, H., et al (2010). Molecular Phylogeny of Salix L. (Salicaceae) Inferred From Three Chloroplast Datasets and Its Systematic Implications. International Association for Plant Taxonomy (IAPT) Stable, 29–37. Available online at: http://www.jstor.org/stable/27757048
Chen, J. (2020). Characterization of the complete chloroplast genome of Salix variegata (Salicaceae). Mitochondrial DNA Part B Resour. 5, 196–197. doi: 10.1080/23802359.2019.1698989
Clement, M., Posada, D., and Crandall, K. A. (2000). TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x
Dong, W., Liu, J., Yu, J., Wang, L., and Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 7:e35071. doi: 10.1371/journal.pone.0035071
Dong, W., Xu, C., Li, C., Sun, J., Zuo, Y., Shi, S., et al. (2015). ycf 1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5:8348. doi: 10.1038/srep08348
Duvall, M. R., Burke, S. V., and Clark, D. C. (2020). Plastome phylogenomics of Poaceae: alternate topologies depend on alignment gaps. Bot. J. Linn. Soc. 192, 9–20. doi: 10.1093/botlinnean/boz060
Eaton, D. A. R., and Overcast, I. (2016). iPYRAD: Interactive Assembly and Analysis of RADseq Data Sets. Available online at: available: http://ipyrad.readthedocs.io/
Fang, C., Zhao, S., and Skvortsov, A. (1999). “Salicaceae,” in Flora of China, Vol. 4, eds Z. Y. Wu and P. H. Raven (St. Louis, MI: Science Press, Beijing and Missouri Botanical Garden Press), 139–274.
Flot, J. F., Hespeels, B., Li, X., Noel, B., Arkhipova, I., Danchin, E. G. J., et al. (2013). Genomic evidence for ameiotic evolution in the bdelloid rotifer Adineta vaga. Nature 500, 453–457. doi: 10.1038/nature12326
Friedberg, E. C., and Meira, L. B. (2006). Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage Version 7. DNA Repair 5, 189–209. doi: 10.1016/j.dnarep.2005.09.009
Gaut, B., Yang, L., Takuno, S., and Eguiarte, L. E. (2011). The patterns and causes of variation in plant nucleotide substitution rates. Annu. Rev. Ecol. Evol. Syst. 42, 245–266. doi: 10.1146/annurev-ecolsys-102710-145119
Gitzendanner, M. A., Soltis, P. S., Wong, G. K. S., Ruhfel, B. R., and Soltis, D. E. (2018). Plastid phylogenomic analysis of green plants: a billion years of evolutionary history. Am. J. Bot. 105, 291–301. doi: 10.1002/ajb2.1048
Givnish, T. J., Spalink, D., Ames, M., Lyon, S. P., Hunter, S. J., Zuluaga, A., et al. (2015). Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proc. R. Soc. B Biol. Sci. 282, 171–180. doi: 10.1098/rspb.2015.1553
Gramlich, S., Wagner, N. D., and Hörandl, E. (2018). RAD-seq reveals genetic structure of the F2-generation of natural willow hybrids (Salix L.) and a great potential for interspecific introgression. BMC Plant Biol. 18:317. doi: 10.1186/s12870-018-1552-6
He, L., Jia, K. H., Zhang, R. G., Wang, Y., Shi, T., Le, L.i, Z. C., et al. (2021a). Chromosome-scale assembly of the genome of Salix dunnii reveals a male-heterogametic sex determination system on chromosome 7. Mol. Ecol. Resour. 21, 1966–1982. doi: 10.1101/2020.10.09.333229
He, L., Wagner, N. D., and Hörandl, E. (2021b). Restriction-site associated DNA sequencing data reveal a radiation of willow species (Salix L., Salicaceae) in the Hengduan Mountains and adjacent areas. J. Syst. Evol. 59, 44–57. doi: 10.1111/jse.12593
Hörandl, E. (1992). Die Gattung Salix in Österreich (mit Berücksichtigung angrenzender Gebiete). Abh. Zool.-Bot. Ges. Österreich 27, 1–170.
Hörandl, E., Florineth, F., and Hadacek, F. (2012). Weiden in Österreich und Angrenzenden Gebieten (Willows in Austria and Adjacent Regions), 2nd ed. Vienna: University of Agriculture.
Huang, D. I., Hefer, C. A., Kolosova, N., Douglas, C. J., and Cronk, Q. C. B. (2014). Whole plastome sequencing reveals deep plastid divergence and cytonuclear discordance between closely related balsam poplars, Populus balsamifera and P. trichocarpa (Salicaceae). New Phytol. 204, 693–703. doi: 10.1111/nph.12956
Huang, Y., Wang, J., Yang, Y., Fan, C., and Chen, J. (2017). Phylogenomic analysis and dynamic evolution of chloroplast genomes in Salicaceae. Front. Plant Sci. 8, 1–13. doi: 10.3389/fpls.2017.01050
Jia, H., Wang, L., Li, J., Sun, P., Lu, M., and Hu, J. (2020). Physiological and metabolic responses of Salix sinopurpurea and Salix suchowensis to drought stress. Trees Struct. Funct. 34, 563–577. doi: 10.1007/s00468-019-01937-z
Karp, A., Hanley, S. J., Trybush, S. O., Macalpine, W., Pei, M., and Shield, I. (2011). Genetic improvement of willow for bioenergy and biofuels. J. Integr. Plant Biol. 53, 151–165. doi: 10.1111/j.1744-7909.2010.01015.x
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Khakhlova, O., and Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j.1365-313X.2006.02673.x
Kikuchi, S., Bédard, J., Hirano, M., Hirabayashi, Y., Oishi, M., Imai, M., et al. (2013). Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 339, 571–574. doi: 10.1126/science.1229262
Lauron-Moreau, A., Pitre, F. E., Argus, G. W., Labrecque, M., and Brouillet, L. (2015). Phylogenetic relationships of American willows (Salix L., Salicaceae). PLoS ONE 10:e0121965. doi: 10.1371/journal.pone.0121965
Leskinen, E., and Alström-Rapaport, C. (1999). Molecular phylogeny of Salicaceae and closely related Flacourtiaceae: evidence from 5.8 S, ITS 1 and ITS 2 of the rDNA. Plant Syst. Evol. 215, 209–227. doi: 10.1007/BF00984656
Li, M. M., Wang, D. Y., Zhang, L., Kang, M. H., Lu, Z. Q., Zhu, R., et al. (2019). Intergeneric relationships within the family Salicaceae s.l. based on plastid phylogenomics. Int. J. Mol. Sci. 20:3788. doi: 10.3390/ijms20153788
Lu, D., Hao, L., Huang, H., and Zhang, G. (2019). The complete chloroplast genome of Salix psamaphila, a desert shrub in northwest China. Mitochondrial DNA Part B Resour. 4, 3432–3433. doi: 10.1080/23802359.2019.1675485
McKain, M. R., and Wilson, M. (2017). Fast-Plast: Rapid de novo assembly and finishing for whole chloroplast genomes. Available online at: https://github.com/mrmckain/Fast-Plast
Mower, J. P., Stefanovi,ć, S., Hao, W., Gummow, J. S., Jain, K., Ahmed, D., et al. (2010). Horizontal acquisition of multiple mitochondrial genes from a parasitic plant followed by gene conversion with host mitochondrial genes. BMC Biol. 8:150. doi: 10.1186/1741-7007-8-150
Mu, X. Y., Tong, L., Sun, M., Zhu, Y. X., Wen, J., Lin, Q. W., et al. (2020). Phylogeny and divergence time estimation of the walnut family (Juglandaceae) based on nuclear RAD-Seq and chloroplast genome data. Mol. Phylogenet. Evol. 147:106802. doi: 10.1016/j.ympev.2020.106802
Narango, D. L., Tallamy, D. W., and Shropshire, K. J. (2020). Few keystone plant genera support the majority of Lepidoptera species. Nat. Commun. 11, 1–8. doi: 10.1038/s41467-020-19565-4
Nargar, K., Molina, S., Wagner, N., Nauheimer, L., Micheneau, C., and Clements, M. A. (2018). Australasian orchid diversification in time and space: molecular phylogenetic insights from the beard orchids (Calochilus, Diurideae). Aust. Syst. Bot. 31, 389–408. doi: 10.1071/SB18027
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, R., McGlinn, D., et al (2019). Vegan: Community Ecology Package. R package version 2.5-6. Available online at: https://CRAN.Rproject.org/package=vegan
Palo, R. T. (1984). Distribution of birch (Betula SPP.), willow (Salix SPP.), and poplar (Populus SPP.) secondary metabolites and their potential role as chemical defense against herbivores. J. Chem. Ecol. 10, 499–520. doi: 10.1007/BF00988096
Paradis, E., and Schliep, K. (2019). Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Parks, M., Cronn, R., and Liston, A. (2012). Separating the wheat from the chaff: mitigating the effects of noise in a plastome phylogenomic data set from Pinus L. (Pinaceae). BMC Evol. Biol. 12:100. doi: 10.1186/1471-2148-12-100
Percy, D. M., Argus, G. W., Cronk, Q. C., Fazekas, A. J., Kesanakurti, P. R., Burgess, K. S., et al. (2014). Understanding the spectacular failure of DNA barcoding in willows (Salix): does this result from a trans-specific selective sweep? Mol. Ecol. 23, 4737–4756. doi: 10.1111/mec.12837
Pham, K. K., Hipp, A. L., Manos, P. S., and Cronn, R. C. (2017). A time and a place for everything: phylogenetic history and geography as joint predictors of oak plastome phylogeny. Genome 60, 720–732. doi: 10.1139/gen-2016-0191
Piatczak, E., Dybowska, M., Płuciennik, E., Kośla, K., Kolniak-ostek, J., and Kalinowska-lis, U. (2020). Identification and accumulation of phenolic compounds in the leaves and bark of Salix alba (L.) and their biological potential. Biomolecules 10, 1–17. doi: 10.3390/biom10101391
R Core Team. (2019). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: https://www.R-project.org/
Rambaut, A. (2014). Figtree, A Graphical Viewer of Phylogenetic Trees. Available online at: http://tree.bio.ed.ac.uk/software/figtree
Rieseberg, L. H., and Soltis, D. E. (1991). Phylogenetic consequences of cytoplasmic geneflow in plants. Evol. Trends Plants 5, 65–84.
Ripma, L. A., Simpson, M. G., and Hasenstab-Lehman, K. (2014). Geneious! Simplified genome skimming methods for phylogenetic systematic studies: a case study in Oreocarya (Boraginaceae). Appl. Plant Sci. 2:1400062. doi: 10.3732/apps.1400062
Savage, J. A., and Cavender-Bares, J. (2012). Habitat specialization and the role of trait lability in structuring diverse willow (genus Salix) communities. Ecology 93, 138–150. doi: 10.1890/11-0406.1
Shaw, J., Lickey, E. B., Beck, J. T., Farmer, S. B., Liu, W., Miller, J., et al. (2005). The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 92, 142–166. doi: 10.3732/ajb.92.1.142
Shaw, J., Lickey, E. B., Schilling, E. E., and Small, R. L. (2007). Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the Tortoise and the hare III. Am. J. Bot. 94, 275–288. doi: 10.3732/ajb.94.3.275
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi: 10.1093/nar/gkz345
Skvortsov, A. K. (1999). “Willows of Russia and adjacent countries,” in Taxonomical and Geographical Revision, eds A. G. Zinovjev, G. W. Argus, J. Tahvanainen, and H. Roininen (Finland: Joensuu).
Smart, L. B., Volk, T. A., Lin, J., Kopp, R. F., Phillips, I. S., Cameron, K. D., et al. (2005). Genetic improvement of shrub willow (Salix spp.) crops for bioenergy and environmental applications in the United States. Unasylva 56, 51–55.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Straub, S. C. K., Moore, M. J., Soltis, P. S., Soltis, D. E., Liston, A., and Livshultz, T. (2014). Phylogenetic signal detection from an ancient rapid radiation: effects of noise reduction, long-branch attraction, and model selection in crown clade Apocynaceae. Mol. Phylogenet. Evol. 80, 169–185. doi: 10.1016/j.ympev.2014.07.020
Straub, S. C. K., Parks, M., Weitemier, K., Fishbein, M., Cronn, R. C., and Liston, A. (2012). Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am. J. Bot. 99, 349–364. doi: 10.3732/ajb.1100335
Suda, Y., and Argus, G. W. (1968). Chromosome numbers of some North American Salix. Brittonia 20, 191–197. doi: 10.2307/2805440
Taberlet, P., Gielly, L., Pautou, G., and Bouvet, J. (1991). Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol. Biol. 17, 1105–1109. doi: 10.1007/BF00037152
Tuskan, G. A., DiFazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. and Gray). Science 313, 1596–604. doi: 10.1126/science.1128691
Wagner, N. D., Gramlich, S., and Hörandl, E. (2018). RAD sequencing resolved phylogenetic relationships in European shrub willows (Salix L. subg. Chamaetia and subg. Vetrix) and revealed multiple evolution of dwarf shrubs. Ecol. Evol. 8, 8243–8255. doi: 10.1002/ece3.4360
Wagner, N. D., He, L., and Hörandl, E. (2020). Phylogenomic relationships and evolution of polyploid Salix species revealed by RAD sequencing data. Front. Plant Sci. 11, 1–38. doi: 10.3389/fpls.2020.01077
Wagner, N. D., He, L., and Hörandl, E. (2021). The evolutionary history, diversity, and ecology of willows (Salix l.) in the European alps. Diversity 13, 1–16. doi: 10.3390/d13040146
Wicke, S., and Schneeweiss, G. M. (2015). “Next-generation organellar genomics: potentials and pitfalls of highthroughput technologies for molecular evolutionary studies and plant systematics,” in Next Generation Sequencing in Plant Systematics. Regnum; Vegetabile Book Series of the IAPT (International Association of Plant Taxonomy), eds E. Hörandl and M. Appelhans (Königstein: Koeltz Scientific Books). doi: 10.14630/000002
Wolfe, A. D., and Randle, C. P. (2004). Recombination, heteroplasmy, haplotype polymorphism, and paralogy in plastid genes: implications for plant molecular systematics. Syst. Bot. 29, 1011–1020. doi: 10.1600/0363644042451008
Wolfe, K. H., Li, W. H., and Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. U.S.A. 84, 9054–9058. doi: 10.1073/pnas.84.24.9054
Wu, D., Wang, Y., and Zhang, L. (2019). The complete chloroplast genome sequence of an economic plant Salix wilsonii. Mitochondrial DNA Part B 4, 3560–3562. doi: 10.1080/23802359.2019.1668311
Wu, J., Nyman, T., Wang, D.-C., Argus, G. W., Yang, Y.-P., and Chen, J.-H. (2015). Phylogeny of Salix subgenus Salix s.l. (Salicaceae): delimitation, biogeography, and reticulate evolution. BMC Evol. Biol. 15:31. doi: 10.1186/s12862-015-0311-7
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Keywords: Chamaetia/Vetrix clade, Eurasia, genome skimming, North America, phylogenomics, plastome evolution
Citation: Wagner ND, Volf M and Hörandl E (2021) Highly Diverse Shrub Willows (Salix L.) Share Highly Similar Plastomes. Front. Plant Sci. 12:662715. doi: 10.3389/fpls.2021.662715
Received: 01 February 2021; Accepted: 23 July 2021;
Published: 03 September 2021.
Edited by:
Susann Wicke, Humboldt University of Berlin, GermanyReviewed by:
Roswitha Schmickl, Academy of Sciences of the Czech Republic (ASCR), CzechiaJulia Bechteler, University of Bonn, Germany
Doerte Harpke, Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Germany
Michael Gruenstaeudl, Freie Universität Berlin, Germany
Copyright © 2021 Wagner, Volf and Hörandl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natascha D. Wagner, bmF0YXNjaGEud2FnbmVyQHVuaS1nb2V0dGluZ2VuLmRl