 Karol Krak
Karol Krak Petra Caklová1
Petra Caklová1 David Kopecký
David Kopecký Václav Mahelka
Václav Mahelka- 1Czech Academy of Sciences, Institute of Botany, Prùhonice, Czechia
- 2Faculty of Environmental Sciences, Czech University of Life Sciences Prague, Prague 6, Czechia
- 3Czech Academy of Sciences, Institute of Experimental Botany, Centre of the Region Haná for Biotechnological and Agricultural Research, Olomouc, Czechia
- 4Experimental Taxonomy, Leibniz Institute of Plant Genetics and Crop Plant Research, Gatersleben, Germany
- 5German Centre of Integrative Biodiversity Research (iDiv) Halle–Jena–Leipzig, Leipzig, Germany
Nuclear ribosomal DNA (nrDNA) has displayed extraordinary dynamics during the evolution of plant species. However, the patterns and evolutionary significance of nrDNA array expansion or contraction are still relatively unknown. Moreover, only little is known of the fate of minority nrDNA copies acquired between species via horizontal transfer. The barley genus Hordeum (Poaceae) represents a good model for such a study, as species of section Stenostachys acquired nrDNA via horizontal transfer from at least five different panicoid genera, causing long-term co-existence of native (Hordeum-like) and non-native (panicoid) nrDNAs. Using quantitative PCR, we investigated copy number variation (CNV) of nrDNA in the diploid representatives of the genus Hordeum. We estimated the copy number of the foreign, as well as of the native ITS types (ribotypes), and followed the pattern of their CNV in relation to the genus’ phylogeny, species’ genomes size and the number of nrDNA loci. For the native ribotype, we encountered an almost 19-fold variation in the mean copy number among the taxa analysed, ranging from 1689 copies (per 2C content) in H. patagonicum subsp. mustersii to 31342 copies in H. murinum subsp. glaucum. The copy numbers did not correlate with any of the genus’ phylogeny, the species’ genome size or the number of nrDNA loci. The CNV was high within the recognised groups (up to 13.2 × in the American I-genome species) as well as between accessions of the same species (up to 4×). Foreign ribotypes represent only a small fraction of the total number of nrDNA copies. Their copy numbers ranged from single units to tens and rarely hundreds of copies. They amounted, on average, to between 0.1% (Setaria ribotype) and 1.9% (Euclasta ribotype) of total nrDNA. None of the foreign ribotypes showed significant differences with respect to phylogenetic groups recognised within the sect. Stenostachys. Overall, no correlation was found between copy numbers of native and foreign nrDNAs suggesting the sequestration and independent evolution of native and non-native nrDNA arrays. Therefore, foreign nrDNA in Hordeum likely poses a dead-end by-product of horizontal gene transfer events.
Introduction
Ribosomal RNA (rRNA) is an essential structural component of ribosomes, the sites of protein synthesis. Ribosomes consist of two subunits, each of which is composed of several proteins and rRNA molecules. In eukaryotes, the large subunit consists of three rRNA molecules (25–26S, 5.8S and 5S), whereas the small subunit includes just one rRNA (18S) molecule (Ben-Shem et al., 2011). High demand for ribosomal RNA needed for ribosome assembly is satisfied through the transcription of numerous copies of the nuclear ribosomal (nrDNA) genes. Three of the four rRNA genes in eukaryotes, coding for 18S, 5.8S, and 26S (altogether referred to as 35S), are separated by two internal transcribed spacers (ITS) and together constitute a single transcription unit (Potapova and Gerton, 2019). Transcription units are separated from one another by intergenic spacers (IGS). Plant genomes harbour thousands of nrDNA transcription units, which are organised in large tandem arrays forming the so-called nucleolar organiser regions (NORs) located on a variable number of chromosomes (Srivastava and Schlessinger, 1991; Dubcovsky and Dvořák, 1995).
The number of nrDNA copies has been found to correlate with genome size at large scale in both plants and animals (Prokopowich et al., 2003). Nevertheless, at lower taxonomic levels this relationship is largely unexplored. The extent of the nrDNA copy number variation (CNV) depends on the group studied. It is somewhat conserved among liverworts, mosses and hornworts (Rosato et al., 2016). Conservation is highly variable among species (Cronn et al., 1996) as well as within species of seed plants (Govindaraju and Cullis, 1992; Malinská et al., 2010), fungi (Johnson et al., 2015) and vertebrates (Veiko et al., 2007). Despite the fact that nrDNA occupies a significant portion of the eukaryotic genome, the relationship between species-level phylogeny and nrDNA copy number has barely been investigated (Sproul et al., 2020).
Nuclear ribosomal DNA is an exemplar member of a multigene family and known for its ability to maintain sequence homogeneity. The nrDNA units change their sequences in a highly synchronised manner – described as concerted evolution (Arnheim et al., 1980). Notwithstanding, nrDNAs of different origin have been found to coexist within a single genome as a result of hybridisation and allopolyploidisation events (Álvarez and Wendel, 2003). The patterns of sequence evolution and array contraction and/or expansion of the putative parental copies, co-existing within the hybrid genomes, are well explored (Matyášek et al., 2007; Malinská et al., 2010). The fates of the parental ribotypes range from complete homogenisation (Wendel et al., 1995; Fuertes Aguilar et al., 1999) over independent evolution of parental sequences and their maintenance at different abundances (e.g., Malinská et al., 2010) to the occurrence of newly arising recombinant ribotypes (e.g., Ko and Jung, 2002). Horizontal gene transfer (HGT) represents another mechanism that can contribute to increased diversity of nrDNA within a single genome. However, the evolutionary dynamics of nrDNA sequences acquired via HGT has not been investigated.
Recently, Mahelka et al. (2017) described extensive HGT involving nrDNA. They found that wild barley (Hordeum, Pooideae) species possess, in addition to their native nrDNA copies, nrDNA sequences that correspond to grasses from the subfamily Panicoideae. The transferred nrDNAs occur only in the I-genome Hordeum taxa (= sect. Stenostachys; for infrageneric classification of Hordeum see Blattner, 2009; Brassac and Blattner, 2015), and certain wild barley species (and individuals) possess non-native nrDNAs from up to five panicoid donors (namely Arundinela, Euclasta, Panicum, Paspalum, and Setaria). Phylogenetic patterns suggest the acquisition of the panicoid DNA occurred via at least nine independent horizontal transfers within a timeframe of between 5 and 1 mya. Based on substitution patterns within the ITS region and the absence of mRNA expression, the authors considered the foreign ribotypes as pseudogenes (Mahelka et al., 2017). In a follow-up study, Mahelka et al. (2021) focused specifically on the transfer from a Panicum-like donor into Hordeum that is likely the oldest of the nine transfers between panicoid grasses and Hordeum. It predated the diversification of sect. Stenostachys, and occurred between 5 and 1.7 mya. Along with several protein-coding genes and transposable elements, these authors show that the Panicum-like ribotypes resided within a specific Panicum-derived chromosomal segment, which is located on a NOR-bearing chromosome, although on the opposite chromosome arm in various Hordeum species from sect. Stenostachys.
Mahelka et al. (2017) carried out a detailed characterisation of the non-native genetic material at the level of sequence variation in a phylogenetic context, but did not focus on quantification of the foreign nrDNAs in the Hordeum genomes. The number of particular foreign ribotypes in the Hordeum genomes – in terms of copy number, coupled with within – as well as between-species dynamics – remains an unanswered questions.
The objective of our study is to investigate the CNV of nrDNA in diploid representatives of the genus Hordeum. In the main, we ask what is the CNV of the foreign ribotypes in the I-genome species? Also, whether, and how, the CNV correlates with the CNV of native ribotypes in Hordeum? To answer these questions, we investigated the following: (i) the CNV of the native ribotype in diploid species representing the entire genus Hordeum; (ii) the CNV of particular foreign ribotypes in species of section Stenostachys; (iii) any correlations in the patterns of CNV between the native and foreign ribotypes; (iv) any correlation between the CNV of both native and foreign ribotypes and species-level phylogeny, genome size and the number of nrDNA loci. We hypothesise that a minor proportion of foreign ribotypes in the total nrDNA, coupled with independent evolution of foreign and native ribotypes, suggest the persistence of foreign nrDNAs within minor nrDNA loci aside from the main and active nrDNA loci (NORs). Together with other lines of evidence, such as non-functionality of the foreign nrDNAs and their confirmed occurrences in chromosomal parts located outside the NORs (Mahelka et al., 2017, 2021), the above scenario would constitute additional support for the status of foreign nrDNAs in Hordeum as dead-end by-products of HGT events.
Materials and Methods
Plant Material and DNA Quality Check
The plant material and high-quality DNA used in this study was available from a previous study (Mahelka et al., 2017). Details of the plant material is provided in Table 1 and Supplementary Table 1, for further details on the origins of particular accessions see Mahelka et al. (2017). The DNA of all samples was checked for integrity on agarose gel, and quantified using Qubit fluorometer (Thermo Fisher) with the Quibit dsDNA HS Assay kit (Thermo Fisher) according to the manufacturer’s instructions.
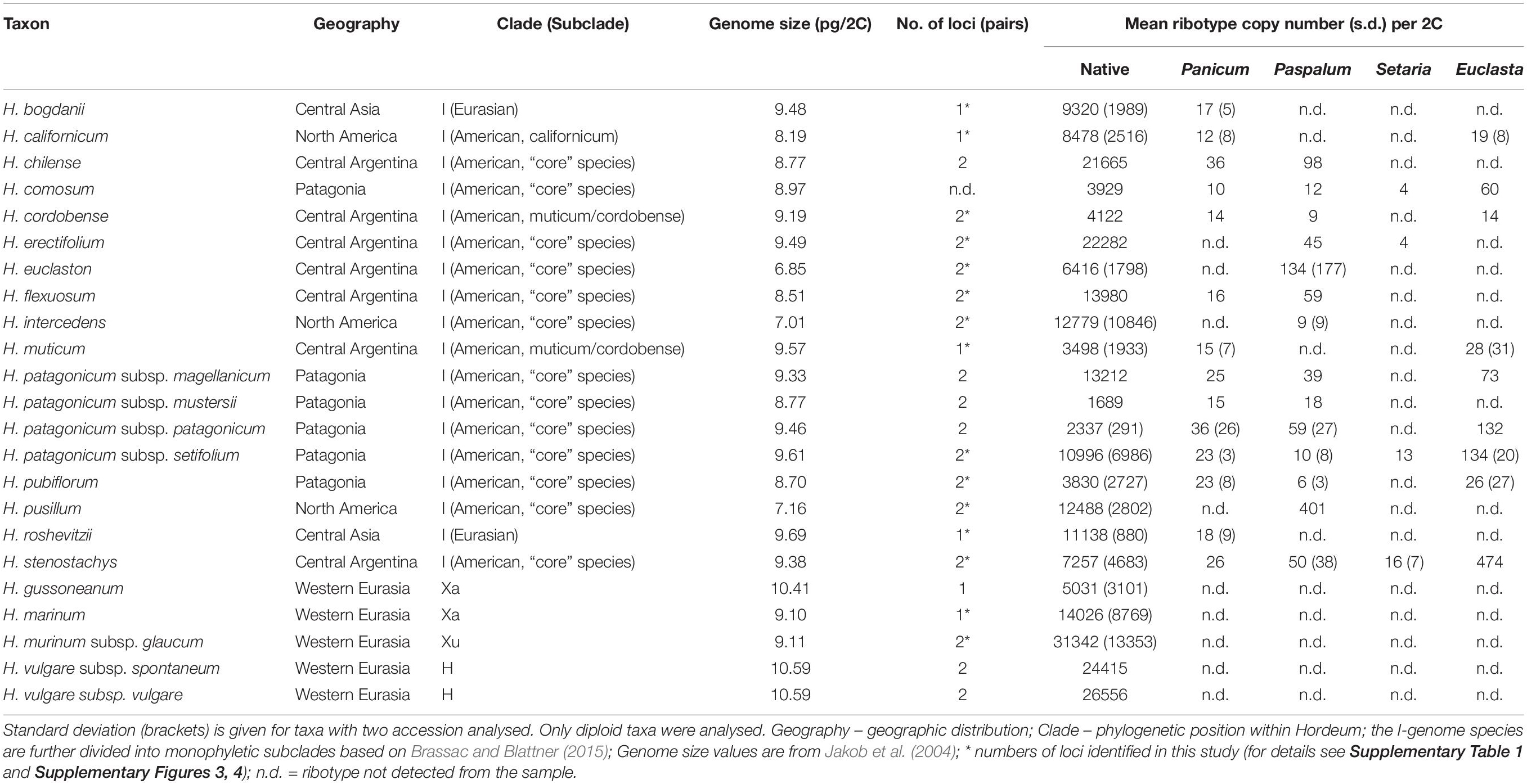
Table 1. Copy numbers of native and foreign ribotypes in Hordeum as estimated using qPCR.
Preparation of Standards for qPCR Estimation
Estimation of copy number using qPCR, requires the parallel measurement of standards to control qPCR efficiency. We employed results from our previous study (Mahelka et al., 2017), in which bacterial colonies containing all of the different ribotypes (both native and foreign) were stored as deep-frozen glycerol stocks. From this source we retrieved the relevant representative samples, and from these we isolated the plasmids using the Qiagen Plasmid Mini Kit (Qiagen) according to manufacturer’s instructions. The plasmids were linearised using an appropriate restriction enzyme with a single recognition site in the plasmid region. The linearised plasmids were purified using the Qiaquick PCR Purification Kit (Qiagen) and their concentrations were measured with a Qubit fluorometer using the Quibit dsDNA HS Assay kit (Thermo Fisher). The plasmids were then diluted to a concentration of 1 ng/μl. Copy numbers of the target ribotypes were calculated for the amplified fragments using the following equation: Copy number (ng–1) DNA = (6.022 × 1023)/(L × 10–9 × 660), where L is the length in bp of the amplified fragment (e.g., the insert), as implemented in the online tool1. Lengths of the inserts were as follows: native (Hordeum-like) 644 bp, Panicum-like ribotype 538 bp, Paspalum-like ribotype 537 bp, Setaria-like ribotype 539 bp, and Euclasta-like ribotype 536 bp. The plasmids were serially diluted to obtain a range of copies of the target ribotypes between 106 and 1 (in a 10-fold dilution series) and these dilution series were used for the qPCR efficiency estimates.
Development and Testing of qPCR Assays
The qPCR assays, targeting the ITS1-5.8S-ITS2 region of nrDNA, were developed based on the sequence variation available from Mahelka et al. (2017). We targeted the primers to the regions showing the highest specificity for each ribotype. At first, the specificity of the primers was tested in silico using Geneious 10.2.6 (Biomatters Ltd.), later, specificity was tested using the ribotype-specific plasmid standards. Each standard was amplified by all qPCR assays. Assays showing cross-amplification were further redesigned and tested, until only specific amplification of the target ribotype was obtained. Further, the reaction conditions of each assay were optimised using the serial dilutions of the respective plasmid standards in order to obtain maximal amplification efficiency.
Limitation of the qPCR Assays
Our initial intention was to develop seven qPCR assays: five targeting the foreign ribotypes as found in Hordeum, e.g., Panicum-, Paspalum-, Euclasta-, Setaria-, and Arundinella-like (Mahelka et al., 2017), one assay specific for the native Hordeum ribotype and a universal assay that would amplify the native as well as the foreign ribotypes (total nrDNA). Unfortunately, the overall pattern of sequence variation hindered the development of an assay specific for Arundinella and the assay targeting the native Hordeum ribotype. Hence, we use the universal assay not only to estimate the copy number of total nrDNAs but also to estimate the copy number of the native ribotype. Therefore, in species of sect. Stenostachys, an overestimate of native ribotype is likely. However, given the low proportion of foreign ribotypes among the total nrDNAs, the bias is considered unlikely to be serious. In the species outside this section (H. vulgare, H. marinum, H. murinum, H. gussoneanum) that lack the foreign ribotypes, the estimate is likely to be unbiased.
Estimation of nrDNA Copy Number Using qPCR
All qPCRs were carried out using the LightCycler 480 II Real-Time PCR instrument (Roche). To quantify the native ribotype, a TaqMan probe-based assay with the Light Cycler 480 Probes Master kit (Roche) was used, under the following cycling conditions: 10 min at 95°C, followed by 45 cycles of denaturation (95°C, 10 s), annealing (60°C, 30 s), and extension (72°C, 1 s).
The foreign ribotypes were quantified using SYBR Green I-based assays and the LightCycler 480 SYBR green I master kit (Roche), under the following cycling conditions: 5 min at 95°C, followed by 40 cycles of denaturation (95°C, 10 s), annealing (assay-specific Ta, 10 s; for specific conditions see Supplementary Table 2) and extension (72°C, 15 s). To detect the possible formation of primer dimers or unspecific amplification, the cycling was concluded with a standard melting curve analysis.
The efficiencies of the qPCR assays were estimated from standard calibration curves based on serial 10-fold dilutions of plasmid standards with specific ribotype sequence inserts, ranging from 106 to 1 copies of target nrDNA. The absolute quantification of the target sequences was carried out based on the standard calibration curves using the LightCycler 480 software, version 1.5 (Roche). The resulting concentrations of amplicon DNA, expressed as DNA copy number (ng–1), were further normalised to the genome sizes of the Hordeum species analysed (Jakob et al., 2004). For each quantification standard, three technical replicates were used, whereas for each analysed individual two technical replicates were included in the qPCR reactions.
Estimation of nrDNA Copy Number Using Illumina Data
In addition to the qPCR-based estimation of CNV, we estimated CNV of native ribotypes from whole-genome sequencing data. This analysis provides an alternative method to the qPCR-based estimation, enabling estimation of relative sensitivity of both methods. Therefore, only a subset of Hordeum samples was included in this analysis (specified below). In principal, the CNV was calculated from the number of Illumina reads mapped to a reference out of the total number of reads used for the mapping. The CN estimation was calculated as described in Wang et al. (2019), with two modifications. First, to avoid potential bias caused by uneven coverage within the rDNA region (genes vs. spacers), we used a complete repeat unit (18S-ITS1-5.8S-ITS2-26S) as a reference. Second, we used 2C DNA content as input data in the formula to conform with the qPCR-based CN estimation. A unique reference was used for each species: Each reference consisted of a species-specific ITS region (retrieved from Mahelka et al., 2017), which was surrounded by universal 18S and 26S rDNA genes. The genes were common to all species and have been derived from Hordeum bogdanii. The transcription unit of H. bogdanii was completed by mapping genomic reads of H. bogdanii against a Secale cereale reference (JF489233). The genes’ and spacers’ boundaries were then adjusted with the aid of an alignment consisting of multiple rDNA units of grasses including the newly assembled H. bogdanii. Lengths of the reference sequences varied slightly among the Hordeum samples (see Table 2). Mapping of reads was done using Bowtie2 (Langmead and Salzberg, 2012) implemented in Geneious version R10 (Biomatters Ltd.), using local alignment type and lowest sensitivity.
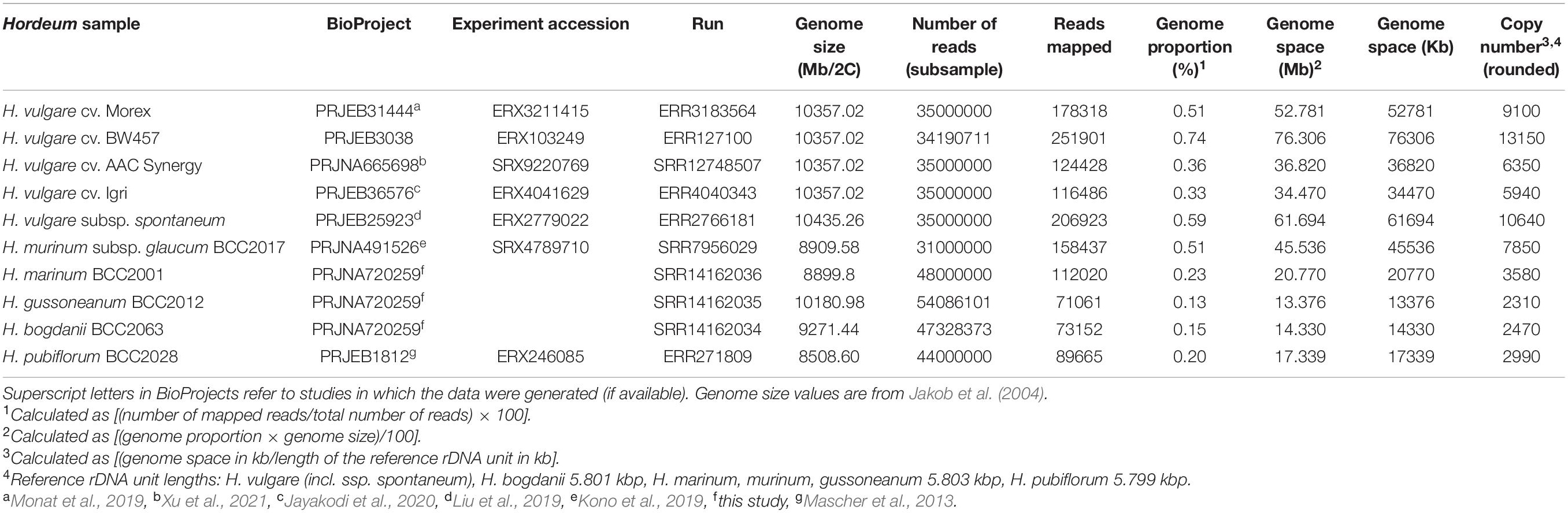
Table 2. Copy numbers of native ribotype in Hordeum as estimated using Illumina data.
We used for the analysis samples representing all major Hordeum lineages (genomes H, Xu, Xa, and I; Blattner, 2009): H. vulgare subsp. vulgare (cultivars Morex, BW457, AAC Synergy, Igri; genome H), H. vulgare subsp. spontaneum (H), H. murinum subsp. glaucum (Xu), H. marinum (Xa), H. gussoneanum (Xa), H. bogdanii (I), and H. pubiflorum (I). For H. murinum subsp. glaucum, H. marinum, H. gussoneanum, H. bogdanii, and H. pubiflorum, we used the same accessions (but not individuals), as we used for the qPCR-based estimation of CN (compare Table 2 and Supplementary Table 1). Short read archives were either downloaded from NCBI database, or we used our unpublished data (Table 2). For each species, the reads were subsampled to ca 1 × genomic coverage. Details on the accessions analysed and short read archives used for the analysis are provided in Table 2.
Identification of nrDNA Loci Number
The number of nrDNA loci was determined by fluorescent in situ hybridisation on metaphase chromosomes using the pTa71 probe as described in Mahelka and Kopecký (2010). The experiments were done under conditions of ∼77% stringency.
Data Analysis
We carried out a number of basic exploratory statistics to evaluate the variation of estimated nrDNA copy numbers. To determine the effects of variables, we used linear regression, ANOVA, Tukey’s HSD test and two-way ANOVA in R version 3.6.3 (R Core Team, 2020).
Results
We carried out a qPCR-based estimation of nrDNA copy number in the diploid representatives of the genus Hordeum. The main objective was to investigate the dynamics and evolution of the foreign ribotypes, in particular by estimating the CNV of the foreign ribotypes and comparing these with the CNV of native Hordeum ribotypes. For a subset of taxa, representing all major Hordeum clades (genomes), we performed an alternative estimation of CNV of native ribotype using low coverage Illumina sequencing.
Copy Number Estimates Based on qPCR and Illumina Read Mapping Are Correlated
The copy numbers of the native ribotype estimated using qPCR and Illumina read mapping differed in all analysed taxa. In all samples, mean qPCR-based values were higher than the values obtained from read mapping. The biggest difference was observed for H. murinum (4×), whereas in H. pubiflorum the difference was much smaller (1.3×, Supplementary Table 3). Overall, the copy numbers estimated using the two methods were correlated (adjusted R2 = 0.743, p = 0.0078; Supplementary Figure 1). Since we focus primarily on relative copy number differences, and a detailed sample including all diploid Hordeum species was used only to estimate CN based on qPCR, we consider qPCR as the default method. Therefore, CN estimates are hereafter qPCR-based, unless otherwise stated.
Copy Number of Native Ribotype Varies Between Species and Individuals
For the native Hordeum ribotype, we found an up to 19-fold difference in the mean copy number among the taxa analysed. The lowest mean value, 1689 copies per 2C content, was estimated for H. patagonicum subsp. mustersii and the highest, 31,342 copies, for H. murinum subsp. glaucum (Table 1). A high variation in copy number (up to fourfold) was also observed between different accessions of the same taxa (Supplementary Table 1). To determine whether the observed variation in nrDNA copy number was affected by variations in the DNA extraction efficiency (and therefore represents a methodological artefact), we carried out a regression analysis. We identified no correlation between DNA extract concentration (as a measure of DNA extraction efficiency) and nrDNA copy number (adjusted R2 = −0.025, p = 0.71; Supplementary Figure 2). This indicates that there is no significant effect of DNA extraction efficiency on the measured CN value. For H. vulgare subsp. vulgare, intraspecific CNV could be estimated also from Illumina read mapping. Here, the four accessions (different cultivars) showed up to a 2.21-fold variation in the copy number (Table 2).
Phylogeny, Genome Size, and Number of nrDNA Loci Have No Effect on CNV of the Native Ribotype
We assessed the effect of genome size, number of nrDNA loci and phylogeny on the observed patterns of CNV. There were either one or two pairs of nrDNA loci in the Hordeum samples analysed (Supplementary Table 1 and Supplementary Figures 3, 4). We found no significant relationship between the copy number of the native ribotype and genome size or the number of nrDNA loci (Supplementary Figure 5). Conversely, the nrDNA copy number was significantly affected by a group’s phylogeny (ANOVA, F = 3.396, p < 0.05). However, the post hoc comparisons (Tukey’s HSD test) revealed that the only significant difference was that between the Xu-genome and the American I-genome species (Figure 1).
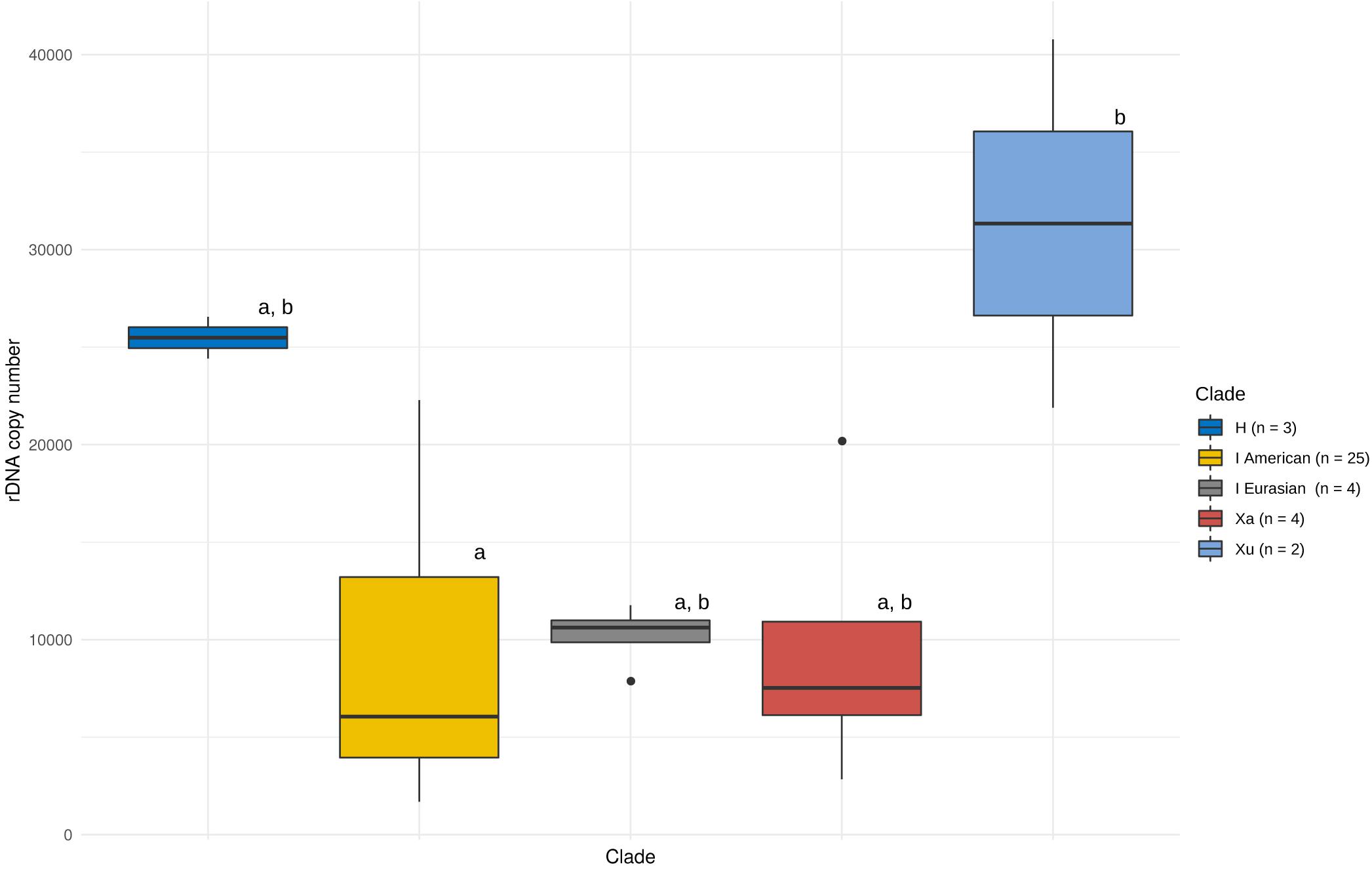
Figure 1. Copy number variation of the native ribotype in Hordeum. Variation within and between major phylogenetic groups is shown. The boxes represent the range between 25th and 75th percentil. The whiskers represent the distance between hinges to the 1.5 × IQR (IQR is the inter-quartile range, or distance between the first and third quartiles) to both sides (toward the maximum as well as minimum values). Vertical bars represent the median values and outlayers are dispalyed as individual points. Phylogenetic grouping is based on Brassac and Blattner (2015). Differences among means (solid lines in boxes) at P = 0.05 (as analysed by ANOVA) are indicated by letters above boxes (boxes with the same letters are not significantly different).
The CNV was high within the recognised phylogenetic groups (Table 3). The highest variation (in the ratio of maximal and minimal values, estimated within a lineage) was observed among the American I-genome species (13.2-fold). On the other hand, the H genome, represented by H. vulgare, was the most homogeneous group with respect to nrDNA copy number. Within this group, the two subspecies differed only by a factor of 1.1 (Table 3).
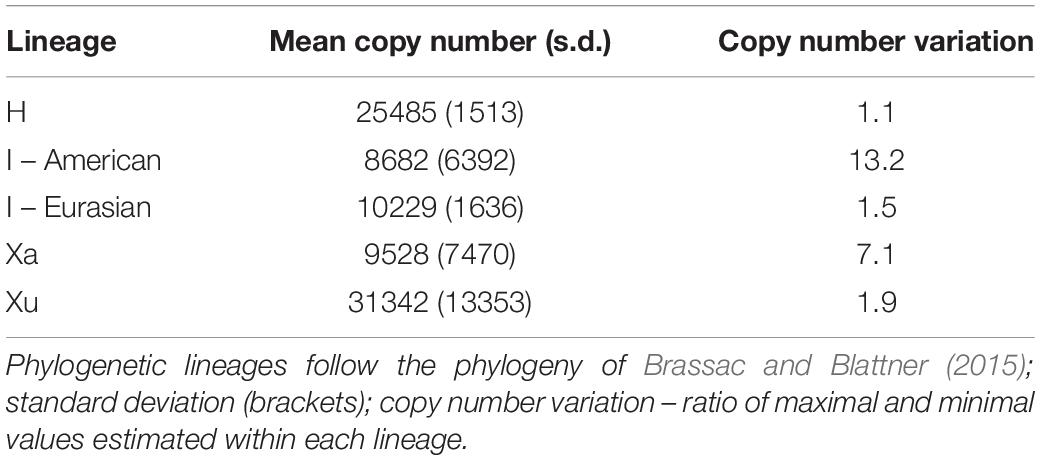
Table 3. Copy number variation of native Hordeum ribotype within the major phylogenetic lineages.
Foreign Ribotypes Represent a Minor Proportion of Total nrDNA
Apart from the native nrDNA, the I-genome Hordeum species contain foreign ribotypes acquired from panicoid grasses. The sequence divergence of particular ribotypes allowed us to design specific qPCR assays targeting four out of the five foreign ribotypes (namely Panicum-, Setaria-, Paspalum-, and Euclasta-like) and hence to estimate their CNV. The estimated copy number of the foreign ribotypes was remarkably low compared with that of the native ribotypes. The estimates ranged from a few copies to tens, solely hundreds of copies (Table 1). Individual foreign ribotypes represent only a small fraction of the estimated total number of nrDNA copies (0.01 – 12.02% depending on ribotype; Supplementary Table 1). Overall, the Euclasta-like ribotype was the most abundant and the Setaria-like ribotype was the least abundant (Figure 2). The two remaining ribotypes showed intermediate values. The differences among the abundances of the foreign ribotypes were not significant (ANOVA, F = 2.572, p = 0.06).
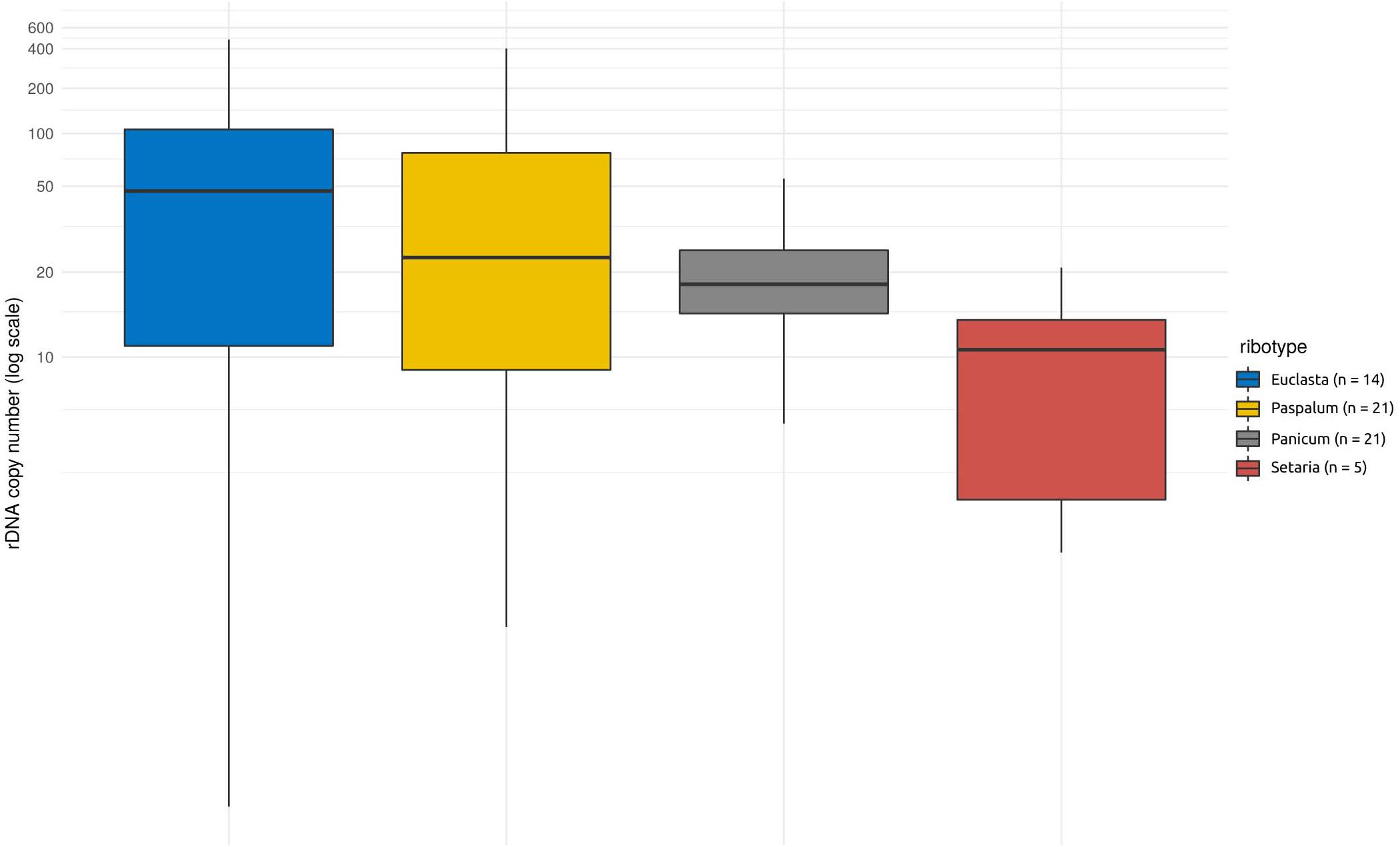
Figure 2. Copy number variation within and among the foreign ribotypes in Hordeum species of sect. Stenostachys. No significant differences at P = 0.05 were detected using ANOVA.
We carried out a regression analysis to test whether the copy numbers of the foreign ribotypes were affected by the copy number of the native ribotype. We found the estimated abundances of foreign ribotypes were independent of the abundance of the native ribotype (Supplementary Figure 6). This indicates the high specificity of the qPCR assays (indicating the absence of significant cross-amplification of the non-target template by the qPCR assays). It points to the independent evolution of the foreign and native ribotypes within the genomes studied.
Phylogeny and Geography Have No Effect on the Copy Numbers of Foreign Ribotypes
Next, we asked whether the abundances of the foreign ribotypes correlated with the phylogenetic relationships of the I-genome Hordeum species. For this purpose, we subdivided the I-genome group into subclades, which reflected the group’s phylogeny (Table 1). We found no significant relationship between the abundance of the foreign ribotypes and the phylogeny (ANOVA, F = 0.758, p = 0.52 for the differences between the subclades, F = 0.968, p = 0.43 for the interaction of the subclade and ribotype, Figure 3). We further investigated whether the CNV of the foreign ribotypes was affected by the geographic origins of the species analysed. Again, the copy number of the foreign ribotypes did not differ significantly among samples of different geographic origin (ANOVA, F = 1.077, p = 0.37) and the interactions between the foreign ribotype and geographic origin of the sample were not significant either (F = 0.551, p = 0.74).
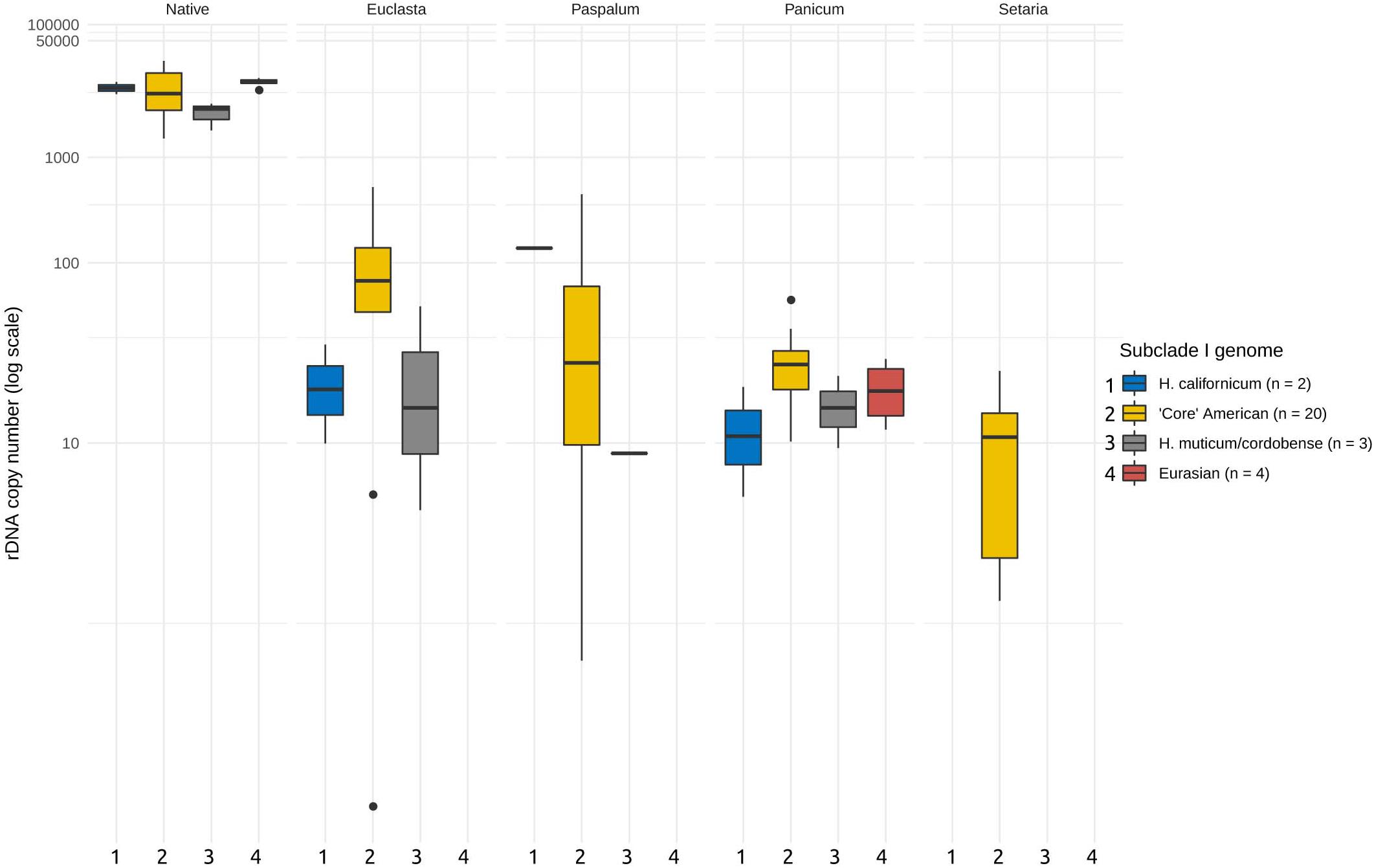
Figure 3. Copy number variation of the foreign ribotypes in relation to major phylogenetic groups. Phylogenetic grouping is based on Brassac and Blattner (2015). Foreign ribotypes only occur in Hordeum species of sect. Stenostachys. No significant differences at P = 0.05 were revealed by ANOVA. Copy numbers of native ribotype are given for comparison.
Discussion
Apart from their native nrDNA copies, species of section Stenostachys from the barley genus Hordeum harbour foreign nrDNA copies, presumably acquired after a series of HGT events from panicoid grasses (Mahelka et al., 2017). To investigate the evolution and dynamics of the foreign ribotypes, we estimated their CNV using specific qPCR assays, and related the values to the CNV of native Hordeum ribotypes. We hypothesised that the HGTs resulted in insertions of foreign nrDNA arrays at random places across Hordeum genomes, so that the foreign ribotypes represent non-functional entities, which reside apart from the NORs in the Hordeum species. Such a pattern would imply independent evolution of native and foreign ribotypes, which can be tested using relationships between CNV of foreign and native ribotypes.
The Usefulness of qPCR and Mapping of Short Reads for Quantification of nrDNA
To investigate the differences in CNV of individual ribotypes, we targeted our qPCR assays to ITS of nrDNA. These regions showed sufficient variation to discriminate between the foreign ribotypes, as well as between the foreign ribotypes and the native one. One of the assays was used to estimate the CNV of the native (Hordeum) ribotype across the genus Hordeum. We used this method mainly to seek for relative differences in CNV of particular ribotypes in the different species, or in the major phylognetic groups. For precise estimates of absolute copy numbers, one should target the rRNA genes (Long et al., 2013; Wang et al., 2019), ideally coupled with a normalisation using a well-defined and confirmed single-copy gene(s) (Long et al., 2013; Rosato et al., 2016; Rabanal et al., 2017). Conversely, specific qPCR assays targeting the ITS region, capture all target sequences irrespective of their genomic locations, and thus represent an efficient tool for detection of potentially pseudogenised, interspersed, non-native nrDNA copies. Thus, this approach is suitable for detection and quantification of minority acquired copies, for example via HGT.
To get insight into the sensitivity of the qPCR-based CN estimation, we implemented an alternative method based on Illumina read mapping. We thus obtained an independent estimate of CNV for a subset of taxa, representing all the major phylogenetic groups included in the study. We found considerable differences between the two methods, with qPCR-based CN estimates showing 1.3–4 × higher values (Supplementary Table 3). Despite the differences between the absolute values, the overall CN estimates were correlated. Hence, we believe that relative differences (between taxa and ribotypes), as inferred from qPCR here, are valid.
The consistently higher CN values estimated using qPCR, than those estimated by read mapping, may partly stem from different methodology of both approaches. While the qPCR targeted ITS region and therefore measured CN thereof, using Illumina data we estimated the number of complete rDNA units. It is likely that the ITS region outnumbers complete rDNA repeat units in plant genomes, because additive ITS copies are scattered across genomic locations outside the main NORs. We found that there are tens of ITS1 copies dispersed across all chromosomes except chr3 within the Morex V2 assembly of H. vulgare (Monat et al., 2019) (Supplementary File 1). In Hordeum samples analysed by us, a visual inspection of mapped reads suggests that there is no obvious increase in coverage within the ITS region. On the other hand, there is a decrease of reads, which mapped to the 26S gene in H. bogdanii, H. marinum, and H. vulgare cultivars BW457 and AAC Synergy. Thus, Illumina-based CNs can be underestimated in these cases. In any case, uneven coverage of a region of interest by reads is a potential source of error in CN estimation based on read mapping.
Potential discrepancies of relative values (e.g., the opposite ratios of CNs obtained for H. bogdanii accession BCC2063 and H. pubiflorum BCC2028; Table 2 and Supplementary Table 1), can be attributed to intraspecific variation of CN, when differences among individuals are commonplace (discussed below). In this particular case (H. bogdanii vs. H. pubiflorum), identical accessions were used, but not the same individuals. Even the use of plant material from a seed bank is not a guarantee of genotype homogeneity. For example, Jakob et al. (2014) studied genetic diversity in wild barley Hordeum vulgare subsp. spontaneum. Interestingly, ex situ samples of this taxon showed genetic admixture, as 22% of gene bank samples did not correspond with the geographic pattern found in wild barley accessions collected in the field (in which the incongruence was only found in 5% of samples). The authors speculated, that although H. vulgare is classified as an inbreeder, the admixture was likely caused by hybridisation during propagation and maintenance, and, possibly, handling of the ex situ gene bank samples. Thus, different individuals may actually represent diverse genotypes even if they are maintained under the same accession number. In this respect, revealing the net effect of particular methods investigated in the same individual plants (or even better in the same DNA extract), would be necessary.
CNV Among Ribotypes
One result of the multiple HGTs from panicoid grasses into Hordeum is that the I-genome species (and even some individual plants) harbour up to five foreign ribotypes. Moreover, these correspond to distinct panicoid genera, namely Panicum, Paspalum, Setaria, Euclasta, and Arundinella (Mahelka et al., 2017). In most cases, multiple foreign ribotypes were found in single individual plants. One individual of H. stenostachys even combined foreign ribotypes from all of the five panicoid genera. Although the distribution of foreign ribotypes among the I-genome species has been well described, information on their quantity, dynamics (variation among individuals) and evolution (variation among species) is largely lacking.
The CNV of individual foreign ribotypes provides valuable information, as it may indicate potential functionality and thus evolutionary potential in Hordeum genomes through their quantity and inferred chromosomal localisation. In another study, Mahelka et al. (2021) provide detailed characterisation of the Panicum-derived chromosomal segment in Hordeum species. The authors reconstructed the foreign DNA segment using sequencing of BAC clones in H. bogdanii (specimen BCC2063) and H. pubiflorum (BCC2028). There were two Panicum-like copies of the ITS region identified within the segment, and the copies occurred at two distinct sites. Thus, given the confirmed presence of a Panicum-derived segment on both chromosomal homologs, there are at least four Panicum-like copies in H. bogdanii and H. pubiflorum. The copy number of Panicum-like ribotypes as inferred from qPCR shows higher values (21 copies in H. bogdanii BCC2023 and 29 copies in H. pubiflorum BCC2028). This higher count can be explained by the presence of additional copies beyond the boundaries of the Panicum-derived segment, which would not have been detected by Mahelka et al. (2021). Furthermore, we still cannot rule out the possibility that some of the foreign ribotypes reside within the native nrDNA loci. The coexistence of different nrDNA variants within a single genome has been documented in allopolyploid and homoploid hybrids (e.g., Kovařík et al., 2008; Fehrer et al., 2009; Zozomová-Lihová et al., 2014) or even in presumed pure diploid species with different paralogous rDNA loci (Blattner, 2004). For these, several scenarios have been described, including maintenance of the different ribotypes within their original loci, formation of new recombinant ribotypes or gradual replacement of one ribotype by another due to interlocus recombination (reviewed in Álvarez and Wendel, 2003). We found only a small proportion of nrDNA is of foreign origin, supporting their presence solely in minor arrays, co-localised with other genetic material of foreign origin as expected under the HGT scenario (Mahelka et al., 2017, 2021). Due to their localisation, out of reach of homogenisation mechanisms, such minor arrays may remain conserved in plant genomes in relatively stable amounts over millions of years (Mahelka et al., 2021).
CNV of nrDNA Is High Both Within and Between Species, and Is Independent of Phylogeny, Genome Size, and Number of nrDNA Loci in Hordeum Species
A high level of CNV of the native ribotype detected between individuals of the same species (e.g., a 4-fold difference in H. intercedens) as well as between species within phylogenetic groups (e.g., a 13.2-fold difference within the American I-genome species) is not unusual. In other studies, high variation in nrDNA copies has been reported not only among closely related species and among individuals within a species but even between different tissues of the same individual. For example, Govindaraju and Cullis (1992) detected a 12-fold variation in nrDNA copy numbers among individuals within populations of Pinus rigida, whereas variation between populations reached values up to 21-fold. Populations of Arabidopsis thaliana from northern and southern Sweden differed in their genome size by more than 10%, mainly due to the variability in nrDNA copy number (Long et al., 2013). In Vicia faba, nrDNA copy number was shown to differ between individuals within a population (up to 95-fold differences) and between different tissues of the same individual, showing up to a 12-fold difference (Rogers and Bendich, 1987). Mechanisms underlying such differences still remain to be revealed. In Hordeum, the highest variation within phylogenetic groups was observed for the American I-genome species. In this case, the results may have been partly skewed by the number of samples, since the I-genome group is the most species-rich within Hordeum. Since we aimed to analyse two individuals per each species, some of the species-poor clades (H, Xu) were represented by only two individuals. Multiple samples from within other than I-genome group were only included for H. vulgare (H genome), in which CN of four cultivars was estimated using read mapping (Table 2). These four samples showed variation that was twice that of the two samples from the H group estimated using qPCR (H. vulgare subsp. vulgare and spontaneum). Since intra-specific variation of rDNA was described for both (Saghai-Maroof et al., 1984), it is likely that the inclusion of more wild barley individuals could further increase the CNV within the H-genome group.
In Hordeum, genome size is correlated with the genus’ phylogeny (Jakob et al., 2004). Our data on the CNV of nrDNA suggest that this correlation is not due to quantity of nrDNA. Despite the high level of CNV detected between Hordeum species, the only significant difference was that between the Xu- and the American I-genome species. We did not find a significant correlation between the nrDNA copy number and the genome size even when individual plants were examined (Supplementary Figure 5A). We further questioned whether the observed CNV could be an effect of the number of nrDNA loci. Since the number of nrDNA loci (particularly of the minor ones) varies in Hordeum (Taketa et al., 1999, 2001), using nrDNA-FISH we identified the number of loci in the same accessions, as used for the CNV analysis. The Hordeum species involved in this study carried one or two pairs of loci (Table 1). This is in accord with other studies, reporting for diploid species one or two pairs of major loci plus up to four pairs of minor loci (Leitch and Heslop-Harrison, 1992; Taketa et al., 1999, 2001). The absence of a significant relationship between CNV and the number of nrDNA loci suggests that the number of loci is not responsible for the variation in nrDNA copy number in Hordeum. Instead, within-loci dynamics (causing contraction or expansion of nrDNA arrays) seems to be the dominant mechanism underlying CNV in this group. A striking example fitting this hypothesis is H. murinum subsp. glaucum analysed here. The hybridisation signal after nrDNA-FISH in accession BCC2017 (Supplementary Figure 4B) is particularly stronger than that observed in the other accession of the same species (BCC2002, Supplementary Figure 4A). This pattern is consistent with copy number of nrDNA, when accession BCC2017 contains almost twice the number of copies. Although nrDNA-FISH is not a precise method to quantify copy number of rDNA, a correlation between copy number of nrDNA and intensity of hybridisation signal presumably exists.
At inter-specific level, the relationship between nrDNA copy number and phylogeny is still poorly explored. Inter-specific genome size evolution has been addressed for almost two decades, especially for plants (e.g., Albach and Greilhuber, 2004; Jakob et al., 2004; Weiss-Schneeweiss et al., 2006; Chrtek et al., 2009; Kang et al., 2014; Mandák et al., 2016). Besides investigating the patterns of genome size variation in a phylogenetic context, recent studies have focused on identification of particular genomic components as the main drivers of these changes (Zedek et al., 2010; Talla et al., 2017; Blommaert et al., 2019; Hloušková et al., 2019; Wong et al., 2019; McCann et al., 2020; Vitales et al., 2020). To date, nrDNA has been confirmed as being a substantial driver of interspecific genome size evolution only in a group of ground beetles (Sproul et al., 2020). Otherwise, no obvious relationship has been observed to enable generalisation of nrDNA evolution.
Conclusion
Here, we reveal the patterns of copy number variation of nrDNA in diploid species of the genus Hordeum. For this purpose, we categorised the nrDNA types as native or foreign ribotypes. While the native, Hordeum-like, ribotype is present in all Hordeum species, the occurrence of foreign ribotypes is restricted to the I-genome Hordeum species (Hordeum sect. Stenostachys), which acquired these ribotypes via a series of horizontal transfers. The foreign ribotypes were present in the respective genomes only at low copy numbers (a few copies to hundreds of copies) and represent a relatively minor fraction of the total nrDNA. We detected a high level of variation in the copy numbers of particular ribotypes at all hierarchical levels examined – between species groups, between species, and between individuals within a species. This variation did not correspond to any of the genus’ phylogeny, the species’ genome size, or the number of nrDNA loci. Overall, we can consider nrDNA copy number as a dynamic trait in Hordeum.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
KK, VM, and PC: conceived and designed the study. PC: conducted lab experiments. DK: provided in situ hybridisation experiments. FB: provided the Illumina data. KK and VM: analysed the data. KK: drafted the manuscript with contribution from VM. All co-authors contributed to the final version of the manuscript.
Funding
The research was financially supported by the Czech Science Foundation (Grant No. 17-06548S) and the long-term research development project RVO 67985939 of the Czech Academy of Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.672879/full#supplementary-material
Supplementary Figure 1 | Correlation between copy numbers of native ribotype estimated using qPCR and Illumina read mapping.
Supplementary Figure 2 | Correlation between the DNA extraction efficiency (expressed as DNA extract concentration) and the copy number of nrDNA, for both native (A) and foreign (B) ribotypes.
Supplementary Figure 3 | Molecular cytogenetic analysis of Hordeum species. Fluorescent signal of 45S-rDNA (labeled with biotin, arrows) has been observed after FISH on one or two pairs of chromosomes of H. bogdanii BCC2063 (A), H. bulbosum GRA970 (B), H. californicum BCC2057 (C), H. cordobense BCC2067 (D), H. erectofolium BCC2026 (E), H. euclaston BCC2022 (F), H. flexuosum BCC2023 (G), H. intercedens BCC2044 (H), and H. marinum BCC2001 (I). In H. cordobense (D) and H. erectifolium (E), minor loci of 45S-rDNA on one pair of chromosomes have been observed (arrowheads). Hordeum bulbosum GRA970 is tetraploid (2n = 28), hence it was excluded from qPCR analyses. Scale bar 10 μm.
Supplementary Figure 4 | Molecular cytogenetic analysis of Hordeum species. Fluorescent signal of 45S-rDNA (labeled with biotin, arrows) has been observed after FISH on one or two pairs of chromosomes of H. murinum BCC2002 (A), H. murinum BCC2017 (B), H. muticum BCC2042 (C), H. patagonicum subsp. setifolium BCC2032 (D), H. pubiflorum BCC2028 (E), H. pusillum BCC2049 (F), H. roshevitzii BCC2015 (G), H. stenostachys BCC2041 (H), and H. vulgare cv. Morex (I). In H. murinum BCC2017 (B) and H. vulgare cv. Morex (I), minor loci of 45S-rDNA on one pair of chromosomes have been observed (arrowheads). Scale bar 10 μm.
Supplementary Figure 5 | (A) Correlation between the genome size (2C value) and the copy number of native ribotype, (B) correlation between the number of nrDNA loci and the copy number of native ribotype.
Supplementary Figure 6 | Correlations between the copy numbers of native and foreign ribotypes. (A) Setaria-like ribotype; (B) Paspalum-like ribotype; (C) Panicum-like ribotype; (D) Euclasta-like ribotype.
Supplementary Table 1 | Characteristics of Hordeum samples used in the study.
Supplementary Table 2 | Detailed description of the qPCR assays used to quantify nrDNA in Hordeum.
Supplementary Table 3 | Comparison of copy numbers of native ribotype estimated using qPCR and Illumina read mapping.
Supplementary File 1 | The distribution of internal transcribed spacer 1 (ITS1) of nuclear ribosomal DNA in the genome of Hordeum vulgare.
Footnotes
References
Albach, D. C., and Greilhuber, J. (2004). Genome size variation and evolution in Veronica. Ann. Bot. 94, 897–911. doi: 10.1093/aob/mch219
Álvarez, I., and Wendel, J. F. (2003). Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 29, 417–434. doi: 10.1016/S1055-7903(03)00208-2
Arnheim, N., Krystal, M., Schmickel, R., Wilson, G., Ryder, O., and Zimmer, E. (1980). Molecular evidence for genetic exchanges among ribosomal genes on nonhomologous chromosomes in man and apes. Proc. Natl. Acad. Sci. U.S.A. 77, 7323–7327. doi: 10.1073/pnas.77.12.7323
Ben-Shem, A., de Loubresse, N., Melnikov, S., Jenner, L., Yusupova, G., and Yusupov, M. (2011). The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 334, 1524–1529. doi: 10.1126/science.1212642
Blattner, F. R. (2004). Phylogenetic analysis of Hordeum (Poaceae) as inferred by nuclear rDNA ITS sequences. Mol. Phylogenet. Evol. 33, 289–299. doi: 10.1016/j.ympev.2004.05.012
Blattner, F. R. (2009). Progress in phylogenetic analysis and a new infrageneric classification of the barley genus Hordeum (Poaceae: Triticeae). Breed. Sci. 59, 471–480. doi: 10.1270/jsbbs.59.471
Blommaert, J., Riss, S., Hecox-Lea, B., Mark Welch, D. B., and Stelzer, C. P. (2019). Small, but surprisingly repetitive genomes: transposon expansion and not polyploidy has driven a doubling in genome size in a metazoan species complex. BMC Genomics 20:466. doi: 10.1186/s12864-019-5859-y
Brassac, J., and Blattner, F. R. (2015). Species-level phylogeny and polyploid relationships in Hordeum (Poaceae) inferred by next-generation sequencing and in silico cloning of multiple nuclear loci. Syst. Biol. 64, 792–808. doi: 10.1093/sysbio/syv035
Chrtek, J., Zahradníček, J., Krak, K., and Fehrer, J. (2009). Genome size in Hieracium subgenus Hieracium (Asteraceae) is strongly correlated with major phylogenetic groups. Ann. Bot. 104, 161–178. doi: 10.1093/aob/mcp107
Cronn, R. C., Zhao, X., Paterson, A. H., and Wendel, J. F. (1996). Polymorphism and concerted evolution in a tandemly repeated gene family: 5S ribosomal DNA in diploid and allopolyploid cottons. J. Mol. Evol. 42, 685–705. doi: 10.1007/BF02338802
Dubcovsky, J., and Dvořák, J. (1995). Ribosomal RNA multigene loci: nomads of the Triticeae genomes. Genetics 140, 1367–1377. doi: 10.1093/genetics/140.4.1367
Fehrer, J., Krak, K., and Chrtek, J. (2009). Intra-individual polymorphism in diploid and apomictic polyploid hawkweeds (Hieracium, Lactuceae, Asteraceae): disentangling phylogenetic signal, reticulation, and noise. BMC Evol. Biol. 9:239. doi: 10.1186/1471-2148-9-239
Fuertes Aguilar, J., Rosselló, J. A., and Nieto Feliner, G. (1999). Nuclear ribosomal DNA (nrDNA) concerted evolution in natural and artificial hybrids of Armeria (Plumbaginaceae). Mol. Ecol. 8, 1341–1346. doi: 10.1046/j.1365-294X.1999.00690.x
Govindaraju, D. R., and Cullis, C. (1992). Ribosomal DNA variation among populations of a Pinus rigida Mill. (pitch pine) ecosystem: I. distribution of copy numbers. Heredity (Edinb) 69, 133–140. doi: 10.1038/hdy.1992.106
Hloušková, P., Mandáková, T., Pouch, M., Trávníček, P., and Lysák, M. A. (2019). The large genome size variation in the Hesperis clade was shaped by the prevalent proliferation of DNA repeats and rarer genome downsizing. Ann. Bot. 124, 103–120. doi: 10.1093/aob/mcz036
Jakob, S. S., Meister, A., and Blattner, F. R. (2004). The considerable genome size variation of Hordeum Species (Poaceae) is linked to phylogeny, life form, ecology, and speciation rates. Mol. Biol. Evol. 21, 860–869. doi: 10.1093/molbev/msh092
Jakob, S. S., Rödder, D., Engler, J. O., Shaaf, S., Özkan, H., Blattner, F. R., et al. (2014). Evolutionary history of wild barley (Hordeum vulgare subsp. spontaneum) analyzed using multilocus sequence data and paleodistribution modeling. Genome Biol. Evol. 6, 685–702. doi: 10.1093/gbe/evu047
Jayakodi, M., Padmarasu, S., Haberer, G., Bonthala, V. S., Gundlach, H., Monat, C., et al. (2020). The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 588, 284–289. doi: 10.1038/s41586-020-2947-8
Johnson, S. M., Carlson, E. L., and Pappagianis, D. (2015). Determination of ribosomal DNA copy number and comparison among strains of Coccidioides. Mycopathologia 179, 45–51. doi: 10.1007/s11046-014-9820-y
Kang, M., Tao, J., Wang, J., Ren, C., Qi, Q., Xiang, Q. Y., et al. (2014). Adaptive and nonadaptive genome size evolution in Karst endemic flora of China. New Phytol. 202, 1371–1381. doi: 10.1111/nph.12726
Ko, K. S., and Jung, H. S. (2002). Three nonorthologous ITS1 types are present in a polypore fungus Trichaptum abietinum. Mol. Phylogenet. Evol. 23, 112–122. doi: 10.1016/S1055-7903(02)00009-X
Kono, T. J. Y., Liu, C., Vonderharr, E. E., Koenig, D., Fay, J. C., Smith, K. P., et al. (2019). The fate of deleterious variants in a barley genomic prediction population. Genetics 213, 1531–1544. doi: 10.1534/genetics.119.302733
Kovařík, A., Dadejová, M., Lim, Y. K., Chase, M. W., Clarkson, J. J., Knapp, S., et al. (2008). Evolution of rDNA in Nicotiana allopolyploids: A potential link between rDNA homogenization and epigenetics. Ann. Bot. 101, 815–823. doi: 10.1093/aob/mcn019
Langmead, B., and Salzberg, S. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leitch, I. J., and Heslop-Harrison, J. S. (1992). Physical mapping of the 18S-5.8S-26S rRNA genes in barley by in situ hybridization. Genome 35, 1013–1018. doi: 10.1139/g92-155
Liu, M., Li, Y., Ma, Y., Zhao, Q., Stiller, J., Feng, Q., et al. (2019). The draft genome of a wild barley genotype reveals its enrichment in genes related to biotic and abiotic stresses compared to cultivated barley. Plant Biotechnol. J. 18, 443–456. doi: 10.1111/pbi.13210
Long, Q., Rabanal, F. A., Meng, D., Huber, C. D., Farlow, A., Platzer, A., et al. (2013). Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nat. Genet. 45, 884–890. doi: 10.1038/ng.2678
Mahelka, V., and Kopecký, D. (2010). Gene capture from across the grass family in the allohexaploid Elymus repens (L.) Gould (Poaceae, Triticeae) as evidenced by ITS, GBSSI, and molecular cytogenetics. Mol. Biol. Evol. 27, 1370–1390. doi: 10.1093/molbev/msq021
Mahelka, V., Krak, K., Fehrer, J., Caklová, P., Nagy Nejedlá, M., Čegan, R., et al. (2021). A Panicum-derived chromosomal segment captured by Hordeum a few million years ago preserves a set of stress-related genes. Plant J. 105, 1141–1164. doi: 10.1111/tpj.15167
Mahelka, V., Krak, K., Kopecký, D., Fehrer, J., Šafář, J., Bartoš, J., et al. (2017). Multiple horizontal transfers of nuclear ribosomal genes between phylogenetically distinct grass lineages. Proc. Natl. Acad. Sci. U.S.A. 114, 1726–1731. doi: 10.1073/pnas.1613375114
Malinská, H., Tate, J. A., Matyášek, R., Leitch, A. R., Soltis, D. E., Soltis, P. S., et al. (2010). Similar patterns of rDNA evolution in synthetic and recently formed natural populations of Tragopogon (Asteraceae) allotetraploids. BMC Evol. Biol. 10:291. doi: 10.1186/1471-2148-10-291
Mandák, B., Krak, K., Vít, P., Pavlíková, Z., Lomonosova, M. N., Habibi, F., et al. (2016). How genome size variation is linked with evolution within Chenopodium sensu lato. Perspect. Plant Ecol. Evol. Syst. 23, 18–32. doi: 10.1016/j.ppees.2016.09.004
Mascher, M., Richmond, T. A., Gerhardt, D. J., Himmelbach, A., Clissold, L., Sampath, D., et al. (2013). Barley whole exome capture: a tool for genomic research in the genus Hordeum and beyond. Plant J. 76, 494–505. doi: 10.1111/tpj.12294
Matyášek, R., Tate, J. A., Lim, Y. K., Šrubařová, H., Koh, J., Leitch, A. R., et al. (2007). Concerted evolution of rDNA in recently formed Tragopogon allotetraploids is typically associated with an inverse correlation between gene copy number and expression. Genetics 176, 2509–2519. doi: 10.1534/genetics.107.072751
McCann, J., Macas, J., Novák, P., Stuessy, T. F., Villaseñor, J. L., and Weiss-Schneeweiss, H. (2020). Differential genome size and repetitive DNA evolution in diploid species of Melampodium sect. Melampodium (Asteraceae). Front. Plant Sci. 11:362. doi: 10.3389/fpls.2020.00362
Monat, C., Padmarasu, S., Lux, T., Wicker, T., Gundlach, H., Himmelbach, A., et al. (2019). TRITEX: chromosome-scale sequence assembly of Triticeae genomes with open-source tools. Genome Biol. 20, 284. doi: 10.1186/s13059-019-1899-5
Potapova, T. A., and Gerton, J. L. (2019). Ribosomal DNA and the nucleolus in the context of genome organization. Chromosom. Res. 27, 109–127. doi: 10.1007/s10577-018-9600-5
Prokopowich, C. D., Gregory, T. R., and Crease, T. J. (2003). The correlation between rDNA copy number and genome size in eukaryotes. Genome 46, 48–50. doi: 10.1139/g02-103
R Core Team (2020). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rabanal, F. A., Nizhynska, V., Mandáková, T., Novikova, P. Y., Lysák, M. A., Mott, R., et al. (2017). Unstable inheritance of 45S rRNA genes in Arabidopsis thaliana. G3 Genes Genom. Genet. 7, 1201–1209. doi: 10.1534/g3.117.040204
Rogers, S. O., and Bendich, A. J. (1987). Heritability and variability in ribosomal RNA genes of Vicia faba. Genetics 117, 285–295.
Rosato, M., Kovařík, A., Garilleti, R., and Rosselló, J. A. (2016). Conserved organisation of 45S rDNA sites and rDNA gene copy number among major clades of early land plants. PLoS One 11:e0162544. doi: 10.1371/journal.pone.0162544
Saghai-Maroof, M. A., Soliman, K. M., Jorgensen, R. A., and Allard, R. W. (1984). Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc. Natl. Acad. Sci. U.S.A. 81, 8014–8018. doi: 10.1073/pnas.81.24.8014
Sproul, J. S., Barton, L. M., and Maddison, D. R. (2020). Repetitive DNA profiles reveal evidence of rapid genome evolution and reflect species boundaries in ground beetles. Syst. Biol. 69, 1137–1148. doi: 10.1093/sysbio/syaa030
Srivastava, A. K., and Schlessinger, D. (1991). Structure and organization of ribosomal DNA. Biochimie 73, 631–638. doi: 10.1016/0300-9084(91)90042-Y
Taketa, S., Ando, H., Takeda, K., and von Bothmer, R. (2001). Physical locations of 5S and 18S-25S rDNA in Asian and American diploid Hordeum species with the I-genome. Heredity (Edinb) 86, 522–530. doi: 10.1046/j.1365-2540.2001.00768.x
Taketa, S., Harrison, G. E., and Heslop-Harrison, J. S. (1999). Comparative physical mapping of the 5S and 18S-25S rDNA in nine wild Hordeum species and cytotypes. Theor. Appl. Genet. 98, 1–9. doi: 10.1007/s001220051033
Talla, V., Suh, A., Kalsoom, F., Dinca, V., Vila, R., Friberg, M., et al. (2017). Rapid increase in genome size as a consequence of transposable element hyperactivity in wood-white (Leptidea) butterflies. Genome Biol. Evol. 9, 2491–2505. doi: 10.1093/gbe/evx163
Veiko, N. N., Shubaeva, N. O., Malashenko, A. M., Beskova, T. B., Agapova, R. K., and Lyapunova, N. A. (2007). Ribosomal genes in inbred mouse strains: interstrain and intrastrain variation of copy number and extent of methylation. Russ. J. Genet. 43, 1021–1031. doi: 10.1134/S1022795407090086
Vitales, D., Álvarez, I., Garcia, S., Hidalgo, O., Nieto Feliner, G., Pellicer, J., et al. (2020). Genome size variation at constant chromosome number is not correlated with repetitive DNA dynamism in Anacyclus (Asteraceae). Ann. Bot. 125, 611–623. doi: 10.1093/aob/mcz183
Wang, W., Wan, T., Becher, H., Kuderová, A., Leitch, I. J., Garcia, S., et al. (2019). Remarkable variation of ribosomal DNA organization and copy number in gnetophytes, a distinct lineage of gymnosperms. Ann. Bot. 123, 767–781. doi: 10.1093/aob/mcy172
Weiss-Schneeweiss, H., Greilhuber, J., and Schneeweiss, G. M. (2006). Genome size evolution in holoparasitic Orobanche (Orobanchaceae) and related genera. Am. J. Bot. 93, 148–156. doi: 10.3732/ajb.93.1.148
Wendel, J. F., Schnabel, A., and Seelanan, T. (1995). Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc. Natl. Acad. Sci. U.S.A. 92, 280–284. doi: 10.1073/pnas.92.1.280
Wong, W. Y., Simakov, O., Bridge, D. M., Cartwright, P., Bellantuono, A. J., Kuhn, A., et al. (2019). Expansion of a single transposable element family is associated with genome-size increase and radiation in the genus Hydra. Proc. Natl. Acad. Sci. U.S.A. 116, 22915–22917. doi: 10.1073/pnas.1910106116
Xu, W., Tucker, J. R., Bekele, W. A., You, F. M., Fu, Y.-B., Khanal, R., et al. (2021). Genome assembly of the Canadian two-row malting barley cultivar AAC Synergy. G3 Genes Genom. Genet. 11:jkab031. doi: 10.1093/g3journal/jkab031
Zedek, F., Šmerda, J., Šmarda, P., and Bureš, P. (2010). Correlated evolution of LTR retrotransposons and genome size in the genus Eleocharis. BMC Plant Biol. 10:265. doi: 10.1186/1471-2229-10-265
Keywords: copy number variation (CNV), horizontal gene transfer (HGT), internal transcribed spacer (ITS), nuclear ribosomal DNA (nrDNA), fluorescent in situ hybridisation (FISH), phylogeny, qPCR (quantitative PCR), xenolog
Citation: Krak K, Caklová P, Kopecký D, Blattner FR and Mahelka V (2021) Horizontally Acquired nrDNAs Persist in Low Amounts in Host Hordeum Genomes and Evolve Independently of Native nrDNA. Front. Plant Sci. 12:672879. doi: 10.3389/fpls.2021.672879
Received: 26 February 2021; Accepted: 22 April 2021;
Published: 17 May 2021.
Edited by:
Ales Kovarik, Academy of Sciences of the Czech Republic (ASCR), CzechiaReviewed by:
Natalia Borowska-Zuchowska, University of Silesia in Katowice, PolandAnna Dimitrova, Institute of Plant Physiology and Genetics (BAS), Bulgaria
Copyright © 2021 Krak, Caklová, Kopecký, Blattner and Mahelka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Václav Mahelka, dmFjbGF2Lm1haGVsa2FAaWJvdC5jYXMuY3o=