 Tingting Li
Tingting Li Wenqiu Pan2†
Wenqiu Pan2† Ying Liu
Ying Liu Fei Wang
Fei Wang Licao Cui
Licao Cui- 1College of Bioscience and Engineering, Jiangxi Agricultural University, Nanchang, China
- 2State Key Laboratory of Crop Stress Biology in Arid Areas and College of Agronomy, Northwest A&F University, Yangling, China
Plant mitochondrial transcription termination factor (mTERF) family regulates organellar gene expression (OGE) and is functionally characterized in diverse species. However, limited data are available about its functions in the agriculturally important cereal barley (Hordeum vulgare L.). In this study, we identified 60 mTERFs in the barley genome (HvmTERFs) through a comprehensive search against the most updated barley reference genome, Morex V2. Then, phylogenetic analysis categorized these genes into nine subfamilies, with approximately half of the HvmTERFs belonging to subfamily IX. Members within the same subfamily generally possessed conserved motif composition and exon-intron structure. Both segmental and tandem duplication contributed to the expansion of HvmTERFs, and the duplicated gene pairs were subjected to strong purifying selection. Expression analysis suggested that many HvmTERFs may play important roles in barley development (e.g., seedlings, leaves, and developing inflorescences) and abiotic stresses (e.g., cold, salt, and metal ion), and HvmTERF21 and HvmTERF23 were significant induced by various abiotic stresses and/or phytohormone treatment. Finally, the nucleotide diversity was decreased by only 4.5% for HvmTERFs during the process of barley domestication. Collectively, this is the first report to characterize HvmTERFs, which will not only provide important insights into further evolutionary studies but also contribute to a better understanding of the potential functions of HvmTERFs and ultimately will be useful in future gene functional studies.
Introduction
One of the major differences between eukaryotes and prokaryotes is that the former has organelles, while the latter does not (Quesada, 2016). Due to endosymbiotic evolution from their cyanobacterial ancestors, most of the organellar genes within chloroplasts and mitochondria have been either lost or transferred to the nucleus (Gray, 2012). Current chloroplast and mitochondrial genomes retain only a tiny fraction of the genes, which are required for photosynthesis, gene expression, and electron transport chains (Lang et al., 1999; Yagi and Shiina, 2012; Robles and Quesada, 2021). Nevertheless, thousands of proteins have been predicted to be localized in plant mitochondria and chloroplasts according to bioinformatics analysis, most of which are encoded by the nuclear genome (Binder and Brennicke, 2003; Huang et al., 2013; Lee et al., 2013). Thus, the organellar gene expression (OGE) apparatus is a precisely coordinated system that largely depends on a great many of proteins encoded by nuclear genes (Pfannschmidt et al., 2015; Quesada, 2016). In the mitochondria and chloroplasts of higher plants, several transcriptional components of the functional OGE system have been reported. Three different polymerases involved in the transcriptional machinery have been demonstrated, including a multi-subunit plastid-encoded RNA polymerase (PEP) and two single-subunit nucleus-encoded RNA polymerases (NEPs) (Pfannschmidt et al., 2015). However, currently known auxiliary factors can only partially explain the transcriptional machinery, suggesting the existence of additional unidentified regulatory factors that are required for organellar gene transcription (Kühn et al., 2007; Liere et al., 2011).
Among the nucleus-encoded OGE factors, a novel protein family has received increasing concerns, namely, the mitochondrial transcription termination factor (mTERF) family. mTERF proteins, firstly characterized in animal mitochondria, were involved in mitochondrial transcription, translation, and DNA replication (Quesada, 2016). These proteins possess a variable number of ~30 amino acid “mTERF” motifs and comprise three leucine zipper-like elements separated by loops (Roberti et al., 2006), which are believed to confer the ability to recognize and bind to the typical mTERF motif on mitochondrial genome (Roberti et al., 2009). To date, four members, mTERF1 to mTERF4, have been described in vertebrates (Linder et al., 2005; Roberti et al., 2009). mTERF1, the founding member of this family, was originally considered to promote transcription termination of the heavy strand genes tRNA-Ler and 16S rRNA (Kruse et al., 1989). However, more recent studies proposed that mTERF1 only partially terminated heavy strand transcription, whereas its major function was to completely block transcriptional interference at the opposite light strand of the ribosomal RNA genes from which they originated (Terzioglu et al., 2013). Although mTERF2 is a non-specific mitochondrial DNA binding protein and works as a negative regulator of mitochondrial gene expression, the function of mTERF2 is largely unknown (Pellegrini et al., 2009; Huang et al., 2011). Similarly, mTERF3 has been demonstrated as a specific repressor of mammalian mitochondrial transcription initiation, and therefore slowing down cell metabolism (Park et al., 2007). Meanwhile, other studies also revealed that mTERF3 was essential for ribosome biogenesis, mitochondrial protein transcription, and translation (Andersson et al., 2011; Wredenberg et al., 2013). mTERF4 can directly regulate mitochondrial ribosomal biogenesis and protein translation by targeting to the ribosomal RNA methyltransferase NSUN4 (a 5-methylcytosine RNA methyltransferase) (Cámara et al., 2011; Spåhr et al., 2012; Yakubovskaya et al., 2012).
By contrast, the mTERF gene family has expanded to approximately 30 members during the evolutionary process of land plants (Quesada, 2016; Leister and Kleine, 2020). For example, there are 35 mTERFs in Arabidopsis thaliana, 33 in rice (Oryza sativa) (Kleine, 2012), 31 in maize (Zea mays) (Zhao et al., 2014), 25 in grape (Vitis vinifera) (Yin et al., 2021), and 35 in Capsicum annuum (Tang et al., 2019). The substantial expansion in the number of mTERF genes was accompanied by their increased involvement in diverse RNA metabolism processes, with the majority being involved in rRNA maturation and intron splicing in organelles (Méteignier et al., 2020). For instance, Arabidopsis mTERF8 mediates preferential transcription termination of the chloroplast gene psbJ by preferentially binding to the 3′-terminus (Xiong et al., 2020). Arabidopsis mTERF15 acts as an RNA binding protein that is required for mitochondrial nad2 intron 3 splicing and functional complex I activity, which is indispensable for plant growth and development (Hsu et al., 2014). mTERF6 is required for the maturation of the chloroplast Ile transfer RNA gene trnI.2 and regulates transcription termination of the PEP core subunit rpoA poly-cistron, thus further demonstrating the essential roles of mTERFs in leaf organogenesis and patterning in Arabidopsis (Romani et al., 2015; Robles et al., 2018b; Zhang et al., 2018). A more recent study reported that mTERF2 is implicated in the splicing of the group IIB introns of ycf3 (intron 1) and rps12 in Arabidopsis. Knock-down mTERF2 resulted in delayed flowering time and knock-out mTERF2 mutants were embryo lethal (Lee et al., 2021). In maize, ZmmTERF4 is involved in plastid ribosome accumulation and promote group II intron splicing of trnI.2, trnA, rpl2, atpF, and ycf3-2 in chloroplasts (Hammani and Barkan, 2014). Zmsmk3 affects complex I assembly by modulating nad4 intron 1 and nad1 intron 4 splicing, seedling growth, and kernel development (Pan et al., 2019). Moreover, recent studies have also proposed the importance of mTERF genes associated with a variety of abiotic stress responses, including heat, salt, and osmotic stresses. For instance, Arabidopsis SHOT1 (SUPPRESSOR OF HOT1-4 1) can indirectly increase thermotolerance by reducing reactive oxygen species (ROS) accumulation and increasing the expression of heat shock proteins (HSPs), particularly those localized to mitochondria (Kim et al., 2012). mTERF5/MDA1 (mTERF DEFECTIVE IN Arabidopsis1) not only has a dual function in the transcription and stabilization of specific chloroplast transcripts but also responds to salt, osmotic, and sugar stresses through perturbed abscisic acid (ABA) retrograde signaling during seedling establishment in Arabidopsis (Robles et al., 2012; Ding et al., 2019; Méteignier et al., 2020). Arabidopsis mTERF9 regulates chloroplast gene expression and development and responds to sugar, ABA, salt, and osmotic stresses (Robles et al., 2015). Similar to mTERF5 and mTERF9, loss of Arabidopsis mTREF27 resulted in mitochondria developmental defects and altered response to salt stress (Jiang et al., 2021). Arabidopsis mTERF10 and mTERF11 are involved in the response to salt stress, possibly through the ABA-mediated pathway (Xu et al., 2017). Recently, a new role was demonstrated for mTERF6 in response to adverse environmental stresses, such as ABA, ionic, and osmotic stresses (Robles et al., 2018a). Arabidopsis SOLDAT10 (SINGLET OXYGEN-LINKED DEATH ACTIVATOR10) controls plastid-specific rRNA expression and protein synthesis in plastids and is well known for its roles in the response to mild photooxidative stress (Meskauskiene et al., 2009). Collectively, mTERFs are essential for the regulation of OGE and play crucial roles in plant growth and development and in response to diverse abiotic stresses, at least in Arabidopsis and possibly in other higher plants. Nevertheless, detailed information about the molecular mechanisms of mTERFs is still rather limited in diverse plants, especially crop plants (Zhao et al., 2014).
As one of the earliest domesticated crops of ancient civilizations, barley (Hordeum vulgare L.) currently ranks as the fourth most abundant crop in terms of both area and tonnage harvested (Mayer et al., 2012). Barley is more adaptable to a wide range of agroclimatic conditions than its relative wheat and, as a result, is of high importance for human food, animal feed, and malt brewing (Jayakodi et al., 2020). The first draft sequence assembly of barley (Mayer et al., 2012) and its subsequent improved versions (Mascher et al., 2017; Monat et al., 2019) lay the foundation for the comprehensive identification and characterization of gene families at the genome-wide level. Here, the protein sequences of barley mTERFs were identified through a comprehensive search. The physicochemical properties, phylogenetic relationships, exon-intron gene structure, conserved motifs, expression profiles, and preliminary functions were systematically analyzed. Moreover, the single-nucleotide polymorphism (SNP) variation atlas of mTERFs for wild and landrace barley accessions was profiled. This study will not only shed light on the evolutionary mechanism of barley mTERFs, but also pave the way for their functional characterization in barley and beyond.
Materials and Methods
Identification of mTERF Gene Family Members in Barley
The protein sequences of barley Morex V2 were downloaded from the IPK database (https://doi.org/10.5447/ipk/2019/8), and the hidden Markov model (HMM) file of the mTERF domain (PF02536) was retrieved from the Pfam database. HMMER v2.41.1 was employed to search for the mTERF domain against the barley genome with the default inclusion threshold. The candidate sequences were further confirmed by using the NCBI-CDD (National Coalition Building Institute, Conserved Domains Database) (https://www.ncbi.nlm.nih.gov/cdd/), SMART (Simple Modular Architecture Research Tool) (http://smart.embl-heidelberg.de/), HMMER (https://www.ebi.ac.uk/Tools/hmmer/), and InterPro (http://www.ebi.ac.uk/interpro/search/sequence/) online tools. Subsequently, a BLAST (Basic Local Alignment Search Tool) search against barley ESTs (Expressed Sequence Tag) was conducted to determine the existence of putative mTERF genes. The molecular weight (MW), number of amino acids, theoretical isoelectric point (pI), and grand average of hydropathicity (GRAVY) were evaluated using the online tool ExPASy (http://web.expasy.org/protparam/). The subcellular localization was predicted using the TargetP online tools (http://www.cbs.dtu.dk/services/TargetP/).
Phylogenetic Relationship, Gene Structure, and Conserved Motif Analysis
Multiple sequence alignment of full-length proteins of HvmTERF genes was performed using the Clustal X program. An unrooted neighbor-joining (NJ) phylogenetic tree was constructed using MEGA X with 1,000 bootstrap replicates. The exon-intron gene structure was visualized using the Gene Structure Display Sever (GSDS) (http://gsds.cbi.pku.edu.cn/) based on the gene annotation GTF (Gene Transfer Format) file. The conserved protein motifs were obtained using online MEME (Multiple Em for Motif Elicitation) tools (https://meme-suite.org/meme/) with the following parameters: the maximum number of motifs was set to 10, any number of repetitions was allowed, and the optimum width ranged from 6 to 250. The 1.5 kb genomic sequences upstream of the coding regions were extracted and submitted to the PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) to identify the putative cis-acting regulatory elements in the promoter region. The transcripts of HvmTERFs were extracted and submitted to the psRNATarget online server (http://plantgrn.noble.org/psRNATarget/) to detect the candidate miRNA targets with the following parameters: published miRNAs from Brachypodium, barley, and wheat were chosen, with a maximum expectation = 4.
Gene Duplication and Comparative Genomics Analysis of Barley, Arabidopsis, Brachypodium, Rice, Grape, and Maize mTERFs
To reveal the duplication events of HvmTERF during barley evolution, an integrated method was employed to identify the duplicated pairs. First, MCScanX software was used to detect duplication events. Second, the following criteria were used as described by Chen et al. (1) the alignment of shorter genes covered ≥70% of longer genes; (2) the aligned region possessed an identity ≥70%; and (3) only one duplication event was counted for tightly linked genes (Gu et al., 2002; Chen et al., 2012). The duplicated events were manually combined into a non-redundant dataset to determine the orthologous relationships between barley and other species. The orthologs of mTERF genes in A. thaliana, Brachypodium distachyon, O. sativa, V. vinifera, and Z. mays were identified using InParanoid V4.1. The syntenic blocks within and among species were detected by MCscanX. To evaluate the evolutionary rate of the duplicated and syntenic genes, PAML (Phylogenetic Analysis by Maximum Likelihood) v4.3 software was utilized to calculate the non-synonymous (Ka) and synonymous (Ks) substitution ratios. The duplicated pairs were visualized using Circos v0.67 software.
Expression Analysis of HvmTERF Genes
To estimate the gene expression profile of HvmTERFs, RNA-seq samples from different tissues and developmental stages as well as plants responding to various biotic and abiotic stresses were retrieved from the NCBI Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/). The sample information and accession numbers are shown in Supplementary Table 1. The Hisat2 v2.1.0 and StringTie v1.3.5 pipelines were employed to calculate the fragments per kilobase of transcript per million fragments mapped (FPKM) value. The R package Ballgown was used to identify the differentially expressed genes. The differentially expressed genes were identified as having a false discovery rate (FDR) ≤ 0.05 and fold change ≥2. The heatmap and hierarchical clustering were generated using the pheatmap package embedded in R with the log2 transformed FPKM values. To determine the co-expressed genes with HvmTERFs, a co-expression network was constructed by weighted gene co-expression network analysis (WGCNA) in R. Here, a convenient one-step method was employed for network construction, and genes with the top 5% weighted values associated with HvmTERFs were categorized for further analysis. A BLAST search against Arabidopsis and rice proteins was performed to determine the potential functions of the co-expressed genes. Cytoscape v3.8.0 was implemented to display the co-expression networks.
Plant Materials, Treatment, and qRT-PCR Analysis
Seeds of the barley cultivar Morex were sterilized with 5% sodium hypochlorite for 10 min, rinsed with distilled water, and then germinated on wet filter paper at 25°C for 5 days. The germinated seeds were hydroponically cultured in a greenhouse under the following conditions: 20°C day/15°C night, 16 h light/8 h dark cycle, and 50% relative humidity. Three-leaf-stage seedlings were exposed to 150 mM NaCl, 20% PEG, 4°C, or 100 μM ABA for 0, 1, 6, 12, 24, and 48 h. Seedlings without any treatment at the same time point were used as the control. Leaves and roots were collected from three plants at each time point and promptly frozen in liquid nitrogen for RNA extraction with three biological replicates.
To further investigate the possible functions of HvmTERFs, a total of 25 HvmTERFs were randomly selected to detect their expression patterns through qRT-PCR (Quantitative Real-time PCR) analysis. The primers used in this study are listed in Supplementary Table 2. Total RNA was isolated using a Plant RNA extraction kit (Omega Biotek, USA), and cDNA was synthesized using 5X All-in-one RT MasterMix (ABM, Canada) following the manufacturer's instructions. HvACTIN2 (GenBank accession no. AY145451.1) was used as the internal control. The TB-Green® Premix Ex Taq™ II kit (Takara, Dalian, China) was used for qRT-PCR amplification in a QuantStudio™ Real-Time PCR system (Thermo Fisher, USA). The reaction protocol was as follows: 95°C for 30 s, followed by 40 cycles at 95°C for 3 s and 60°C for 30 s. The relative expression levels of candidate genes were calculated using the 2−ΔΔCT method. Three technical replicates were applied for each treatment (Livak and Schmittgen, 2001). Student's t-test was employed for statistical analysis by R software. The histogram was drawn using the ggplot package in R software. One asterisk (*) and double asterisk (**) indicate 0.05 and 0.01 significance level, respectively.
Nucleotide Variation, Population Structure, and Haplotype Analysis of HvmTERFs
To acquire the candidate HvmTERFs, a total of 220 barley resequencing samples were downloaded from the SRA database (Russell et al., 2016). The geographic distribution is presented in Supplementary Figure 1. The detailed material information is listed in Supplementary Table 3. BWA-MEM v0.7.13r1126 was used to map the clean reads against the barley reference genome. The PICARD-GATK pipeline was employed to generate single nucleotide polymorphisms (SNPs) with default parameters. The genomic distribution and potential function of the SNPs were annotated by SnpEFF v4.3. The SNPs located within the HvmTERF genes were retained for subsequent analysis. To further reveal the population structure of barley samples based on HvmTERF sequences, population structure analysis, phylogenetic tree analysis, and principal component analysis (PCA) were performed. ADMIXTURE v1.3.0 was used to infer the population structure with predefined K-values ranging from 2 to 5. The phylogenetic tree was constructed using Treebest v1.9.2. The Smartpca toolkit implemented in EIGENSOFT v4.2 was employed to conduct the PCA. Median-joining haplotype networks were constructed using the software programs DnaSP v5.10.01, Alignment v1.3.1.1, and Network v4.6.1.1. The network was visualized using Cytoscape v3.8.0. The nucleotide diversity (π) and Wright's F-statistic (Fst) were calculated using vcftools v0.1.16.
Results
Identification of mTERF Gene Family Members in Barley
The updated reference genome of barley, Morex v2, provided invaluable resources for HvmTERF identification, and a total of 60 mTERF genes were identified in barley using a combined method (Supplementary Tables 4, 5). Since there was no standard nomenclature, the barley mTERFs were designated as HvmTERF1 to HvmTERF60 according to their chromosome numbers and physical positions. The physicochemical properties of the HvmTERFs were further characterized. In detail, the mTERFs encoded proteins ranging from 105 (HvmTERF41) to 632 (HvmTERF59) amino acids in length, with pIs ranging from 5.41 (HvmTERF2) to 10.78 (HvmTERF10), and MWs ranging from 12.01 (HvmTERF41) to 71.82 kDa (HvmTERF59). The GRAVY values ranged from 0.259 (HvmTERF25) to −0.502 (HvmTERF7), with an average of −1.005. Most (61.67%) of the HvmTERFs displayed positive GRAVY values, suggesting hydrophobic characteristics. Subcellular location prediction revealed that most (78.33%) HvmTERFs were localized to mitochondria (39 HvmTERFs, 65%) or chloroplasts (8 HvmTERFs, 13.3%), and the remaining 13 HvmTERFs were targeted to other locations. To confirm the existence of HvmTERFs, a BLAST search against barley ESTs was performed. In total, 48 members of the HvmTERF gene family had EST records, whereas the remaining 12 mTERFs had no EST support, suggesting their stage- or tissue-specific expression profile or undetectable expression level.
Phylogenetic and Structural Domain Analysis of HvmTERFs
We examined the amino acid sequence features of the mTERF domain by multiple sequence alignment. The conserved mTERF motifs spanned approximately 30 amino acids in length, have been characterized in other plants and are believed to act as DNA-binding modules (Zhao et al., 2014) (Supplementary Figure 2; Supplementary Table 6). The sequence conservation percentages for each amino acid residue were calculated, and 15 amino acid sites were highly conserved with a consensus sequence percentage >60%. Consistent with previous studies (Zhao et al., 2014), three repeats of the leucine zipper-like heptad X3LX3 were identified in barley mTERF motifs, of which the conservation percentages were 62.71, 46.67, and 36.67% for Leu-8, Leu-16, and Leu-23, respectively. These results revealed that HvmTERFs possessed well-characterized mTERF motifs with conserved leucine residues like those in other plants, indicating the conserved evolutionary process of plant mTERF proteins. Surprisingly, the conservation percentages of Ile-1 and Tyr-20 in barley were significantly higher than those in Arabidopsis, rice, and maize, suggesting that these residues may play essential roles in the evolutionary history of HvmTERFs.
To further elucidate the evolutionary relationship of HvmTERFs, we constructed a phylogenetic tree based on the alignment of 128 mTERF protein sequences from Arabidopsis (35), rice (33), and barley (60) (Figure 1). These mTERF proteins were divided into nine monophyletic clades according to the classification given by Zhao (Zhao et al., 2014). The number of proteins assigned to different subfamilies varied greatly, of which subfamily IX contained 39 members, whereas subfamilies I, III, V, and VII possessed only one member.
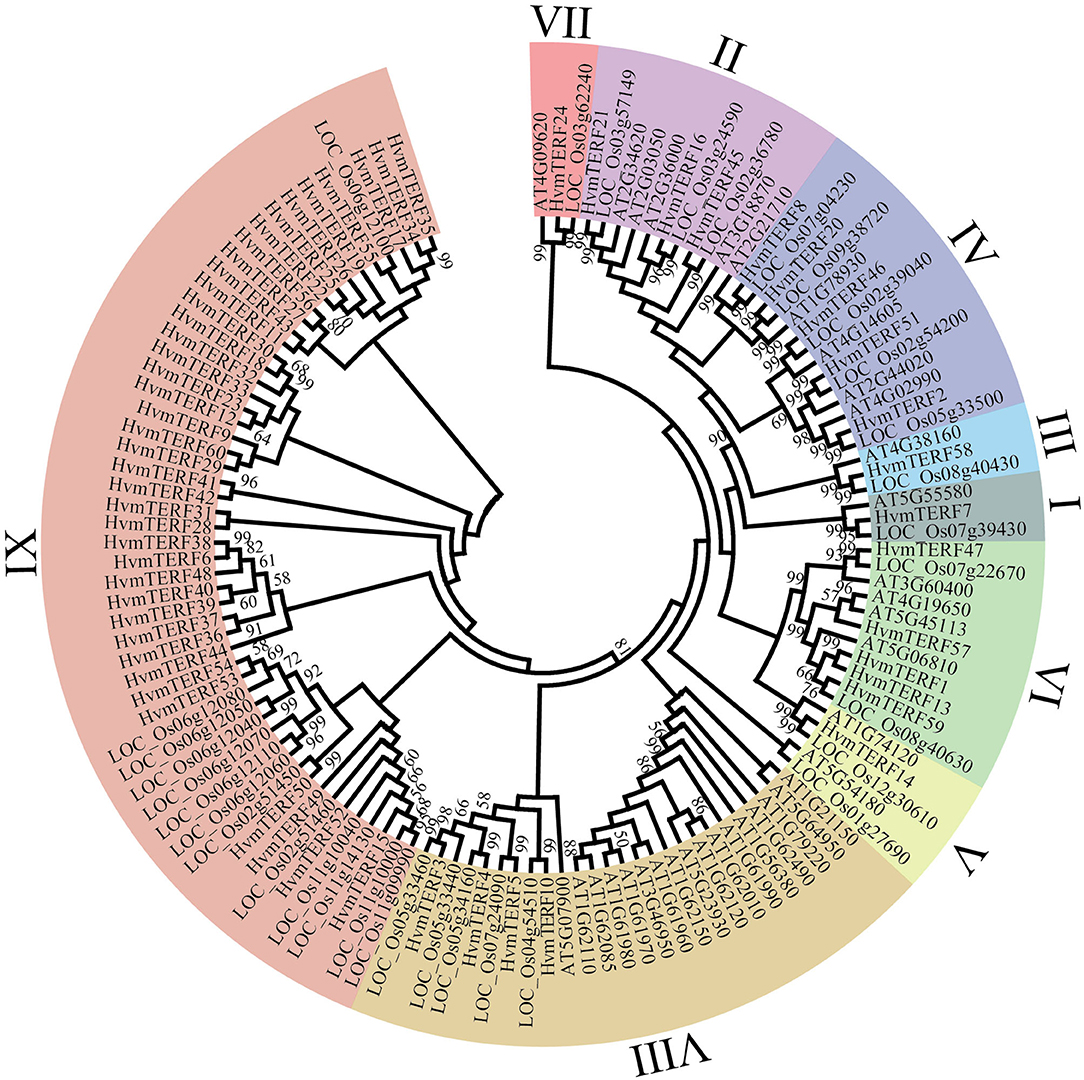
Figure 1. Phylogenetic analysis of mTERF proteins from Arabidopsis, rice, and barley. The phylogenetic tree was constructed using the Neighbor-joining method with 1,000 bootstrap replications. The nine subfamilies are marked with different colors.
The exon–intron structure not only provides additional evidence to support the phylogenetic topology but also increases the understanding of the functional diversification within a gene family. Therefore, the exon-intron structure of HvmTERFs was analyzed to obtain their evolutionary relationships (Figure 2). A solid correlation between gene structures and their phylogeny was observed. Genes clustered within the same subfamily displayed a similar exon-intron structure. Indeed, HvmTERFs within subfamilies II, VI, and VIII showed nearly identical exon lengths and tended to be intron-less. Nonetheless, we pinpointed the exon/intron gain/loss event within several clusters. For example, 37 out of 39 HvmTERFs within subfamily IX possessed only 1 exon, whereas HvmTERF29 and HvmTERF44 had 2 and 3 exons, indicating that they may have acquired additional exons during the evolutionary history of the mTERF gene family.
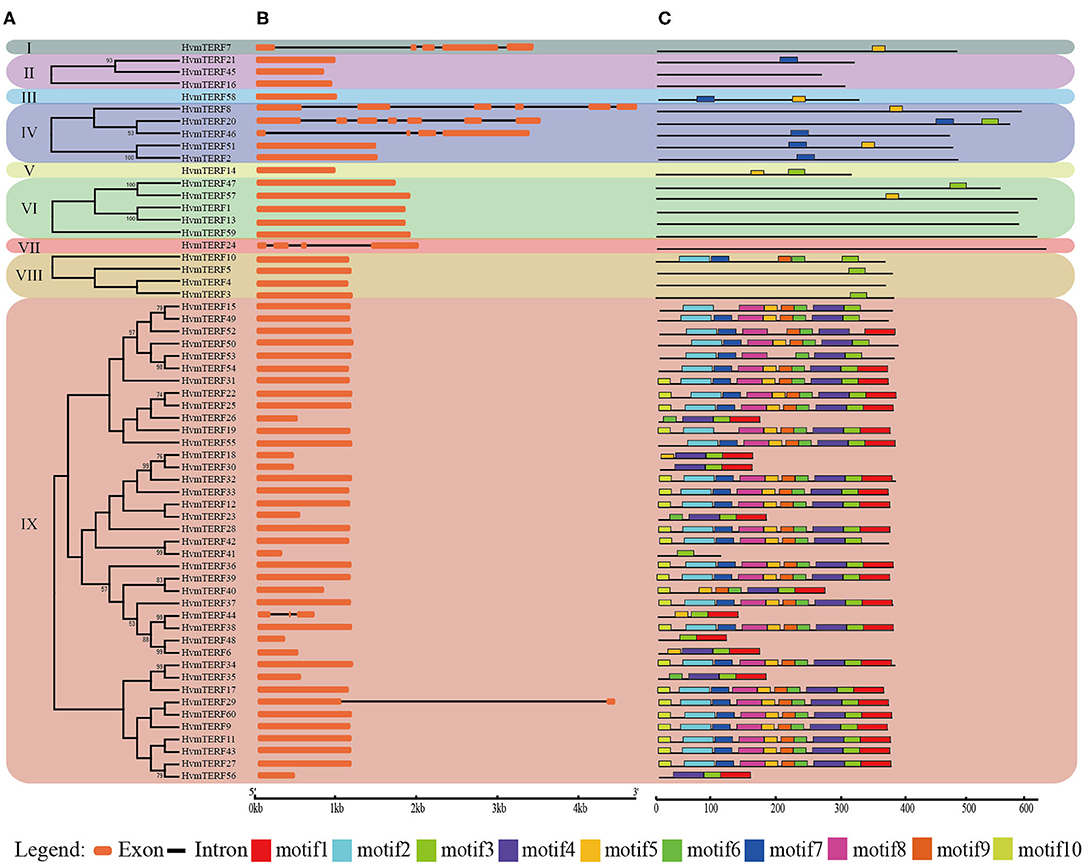
Figure 2. Phylogenetic tree, conserved motifs, and exon-intron structure analysis of HvmTERFs. (A) Phylogenetic tree for each subfamily. (B) Gene structure of HvmTERF genes. Exons are indicated as orange boxes. Black lines represent introns. (C) The motif composition of HvmTERFs. Motifs are designated as 1–10 and represented by different colors.
To further gain insight into the evolutionary relationships and functional regions among the HvmTERFs, the distribution pattern of the conserved motifs was also visualized (Figure 2). We identified 10 motifs and designated them as motif 1 to motif 10. Notably, no motif was identified within HvmTERFs 1, 4, 13, 16, 24, 45, and 59, possibly because the consensus sequence failed to reach the threshold in the MEME software. The HvmTERFs within the same clade showed similar motif numbers and distribution patterns. Motif 3 was shared by 44 members, ranking as the most abundant motif, followed by motif 4 (36) and motif 5 (36). Except for motifs 2, 5, 6, 7, and 9, the remaining motifs were specific to subfamily IX. We also observed a certain order of the identified motifs. For example, motif 2 tended to be tightly connect with motif 7, motif 5 was linked to motif 8, motif 6 was linked to motif 9, and motifs 1, 3, and 4 were linked.
Duplication Events and Orthologous Analysis of HvmTERFs
The HvmTERFs were unevenly located across the seven barley chromosomes in accordance with the barley genome annotation, of which 27 HvmTERFs were located on chromosome 6, ranking as the most populated chromosome, whereas the other six chromosomes had only 9 (chromosome 7H) to 3 (chromosome 3H) HvmTERF genes (Supplementary Figure 3). Interestingly, there was no positive correlation between chromosome length and the number of HvmTERFs (Pearson correlation r = −0.2994, p-value = 0.5141), indicating that longer chromosomes do not necessarily contain more HvmTERF genes.
In order to elucidate the expansion mechanism of HvmTERFs, tandem, and segmental duplication event analyses were performed using an integrated method (Figure 3). The results showed that 10 HvmTERFs (HvmTERFs 25 and 26, HvmTERFs 29 and 32, HvmTERFs 49 and 50, and HvmTERFs 36, 37, 38, and 39) were clustered into four tandemly duplicated regions on chromosome 6, indicating a gene distribution hot spot of HvmTERFs. It is noteworthy that all tandemly duplicated genes belonged to subfamily IX. Generally, it is difficult to segregate this kind of tightly linked gene arrangement through recombination in breeding or research. Furthermore, seven duplicated pairs composed of 13 HvmTERF genes were identified as segmental duplications. Except for HvmTERF1 and HvmTERF13, the remaining duplicated genes were clustered in subfamily IX. Remarkably, six segmentally duplicated gene pairs were associated with chromosome 6. Taken together, both tandem and segmental duplication events contributed to the expansion of HvmTERFs, mainly in subfamily IX.
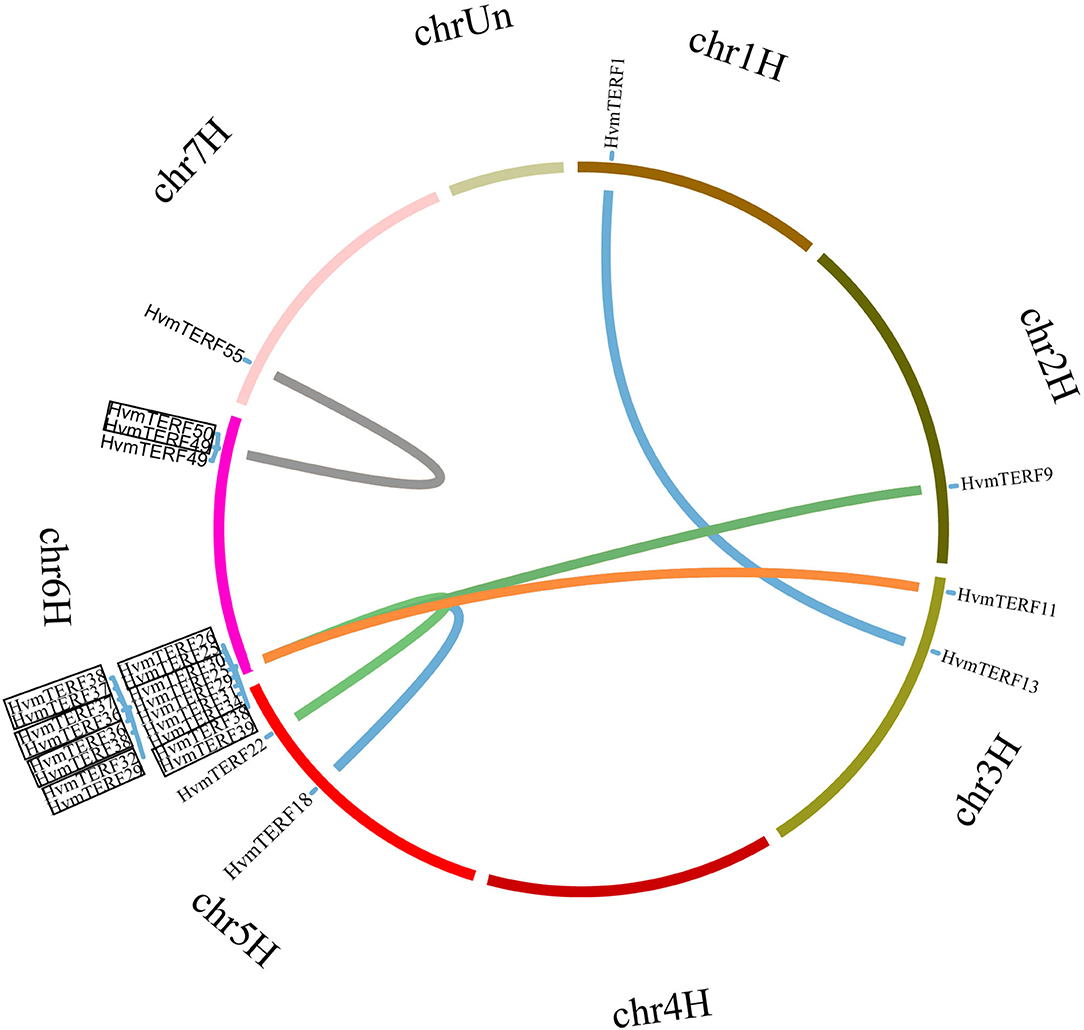
Figure 3. Chromosomal location and gene duplication of HvmTERFs. Tandemly duplicated gene pairs are highlighted with black boxes.
The non-synonymous (Ka) vs. synonymous (Ks) substitution ratios for duplicated gene pairs were further calculated to estimate the evolutionary constraints acting on HvmTERFs. Ka/Ks >1, =1, and <1 indicate positive, neutral and purifying selection, respectively (Lynch and Conery, 2000). In this study, the Ka/Ks ratio for duplicated gene pairs ranged from 0.1516 to 0.5662, with an average value of 0.3845, suggesting that these gene pairs have experienced strong purifying selective pressure during their expansion process (Supplementary Table 7).
To further infer the evolutionary mechanisms of HvmTERFs, we conducted comparative ortholog analysis with five representative species, including two dicots (A. thaliana and V. vinifera) and three monocots (B. distachyon, O. sativa, and Z. mays) (Figure 4). The ortholog analysis resulted in 16, 18, 39, 22, and 20 gene pairs between barley and the other five species (A. thaliana, V. vinifera, B. distachyon, O. sativa, and Z. mays), respectively. A total of 29 HvmTERF genes held orthologous relationships with those in B. distachyon, followed by O. sativa (22), Z. mays (19), V. vinifera (18), and A. thaliana (16). Nine HvmTERFs (HvmTERFs 2, 3, 5, 8, 14, 16, 46, 58, and 59) were found to possess one-to-one relationships among the five representative species. We thus proposed that these evolutionarily conserved genes may have essential roles during plant evolution. Interestingly, 21 gene pairs composed of 12 HvmTERFs were only identified between barley and B. distachyon, O. sativa, and Z. mays, suggesting that these orthologous pairs formed after the divergence between monocotyledonous and dicotyledonous plants. The Ka/Ks ratios of the mTERF gene pairs were also calculated. All orthologous mTERF gene pairs showed Ka/Ks <1, suggesting that these HvmTERFs might have been subjected to extensive purifying selective pressure (Supplementary Table 8).
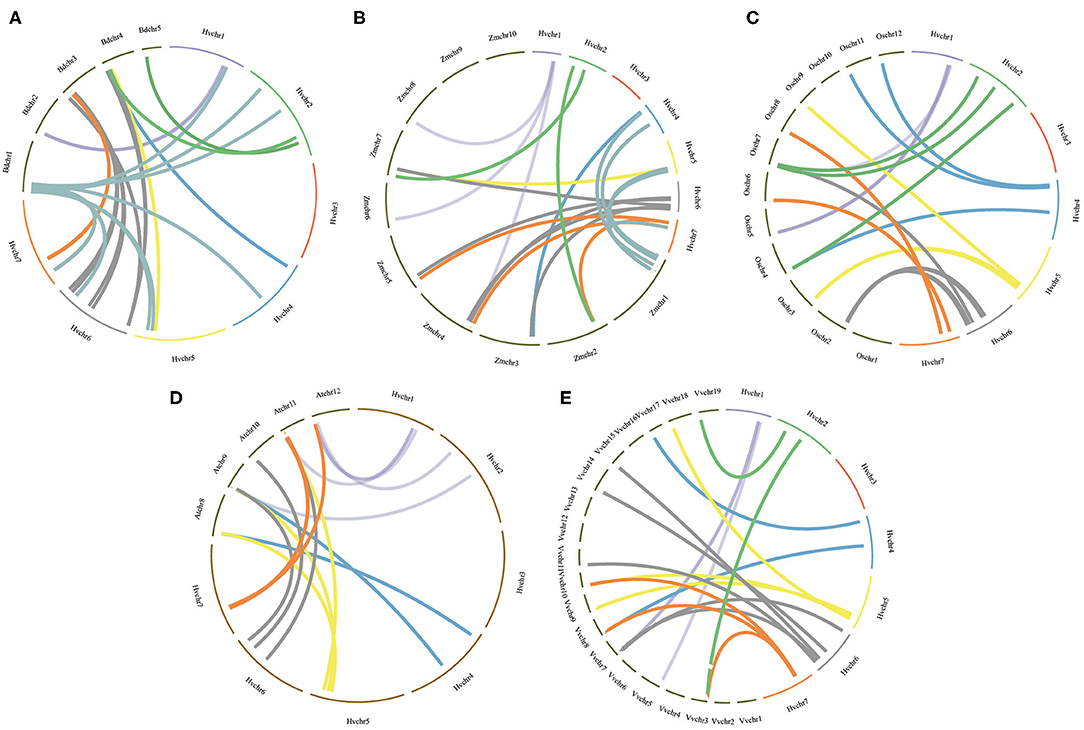
Figure 4. Orthologous analysis of HvmTERFs between barley and other five representative plant species. (A) Brachypodium distachyon, (B) Zea mays, (C) Oryza sativa, (D) Arabidopsis thaliana, (E) Vitis vinifera.
Analysis of cis-elements and miRNA Target Sites of HvmTERFs
Cis-elements play vital roles in the transcriptional regulation of genes during plant growth and development as well as the plant response to biotic and abiotic stresses. By searching the PlantCARE database, a total of 40 cis-elements were identified and further classified into four categories. As shown in Supplementary Table 9, a total of 12 kinds of light-responsive elements were observed, accounting for the majority of the putative cis-elements. There were 10 hormone-responsive regulatory elements present in the HvmTERF promoters, such as ABRE (ABA-Responsive Elements), CGTCA-motif/TGACG-motif, TGA-element, ERE (Estrogen Response Element), and TCA-element, which were associated with ABA, methyl MeJA (Methyl Jasmonate), auxin, ethylene, and SA (Salicylic Acid), respectively. We also identified several organogenesis-related cis-elements, such as MSA-like (Mitosis-Specific Activator) (cell cycle regulation, 4 genes), GCN4 (General Control Non-repressible-4) (endosperm expression, 3 genes), CAT-box (meristem expression, 32 genes), AC-I/II (xylem expression, 2 genes), and circadian (circadian control, 11 genes). Notably, five kinds of biotic and abiotic stress-related regulatory elements, including MBS (Myeloblastosis Binding Site), GC-motif, ARE (Anaerobic Response Element), LTR (Long Terminal Repeat), and Wun-motif, which responded to drought, anoxic-specific inducibility, anaerobic induction, low temperature, and wound damage, respectively, were identified in the HvmTERF promoter regions. Therefore, the variety and quantity of regulatory elements were present in distinct HvmTERF promoters, suggesting their potential functions in diverse signal transduction pathways and various stress adaptations in barley.
To obtain preliminary insight into the miRNA-mediated posttranscriptional regulatory mechanisms, the CDSs (Coding Sequence) of HvmTERFs were extracted to search for miRNA target sites. The results showed that a total of 12 mTERF-miRNA pairs were identified, referring to five miRNAs targeting 10 HvmTERFs (Supplementary Table 10). Most of the miRNAs controlled the expression of HvmTERFs by guiding mRNA cleavage, whereas HvmTERF34 and HvmTERF35 were regulated by translation inhibition. HvmTERF22 and HvmTERF46 were targeted by miRNA6192 upstream of the mTERF domain, whereas a total of six HvmTERFs were targeted by miRNA9662a-3p within the mTERF domain. miRNA7717b-5p and miRNA9962a-3p both targeted HvmTERF19 through transcript cleavage. Our findings suggested that miRNA was involved in the posttranscriptional regulation of HvmTERF, but the actual regulatory mechanism should be validated in future molecular biology experiments.
Expression Profile Analysis of HvmTERF Genes
To obtain preliminary insight into tissue- and stage-specific expression profiles and elucidate the potential roles of mTERFs in tissue development, the transcript abundances of HvmTERFs in 16 different tissues or stages were obtained using Illumina RNA-seq data. As shown in Figure 5, HvmTERFs were expressed in all barley RNA-seq samples studied. The mTERFs were highly expressed in seedlings, leaves and developing inflorescences. HvmTERFs 2, 7, 8, 16, 24, 45, 46, and 58 exhibited relatively high expression levels in most of the studied tissues and stages, suggesting these genes may play an important role in barley growth and development. It is noteworthy that HvmTERF24 ranked the most highly expressed gene with an average FPKM value of 28.58. We also identified tissue- and stage-specific HvmTERFs. HvmTERF3, HvmTERF5, HvmTERF30, HvmTERF35, and HvmTERF50 exhibited preferential expression in young inflorescences, senescing leaves, epidermis, lodicules, and senescing leaves, respectively. HvmTERF20 was predominantly expressed in seedlings and senescing leaves, whereas HvmTERF21 was mostly expressed in the epidermis and senescing leaves, suggesting that these genes were involved in tissue- or stage-specific development in barley. Interestingly, four HvmTERF genes (HvmTERFs 6, 26, 40, and 54) in subfamily IX exhibited almost no expression in any of the tissues and stages.
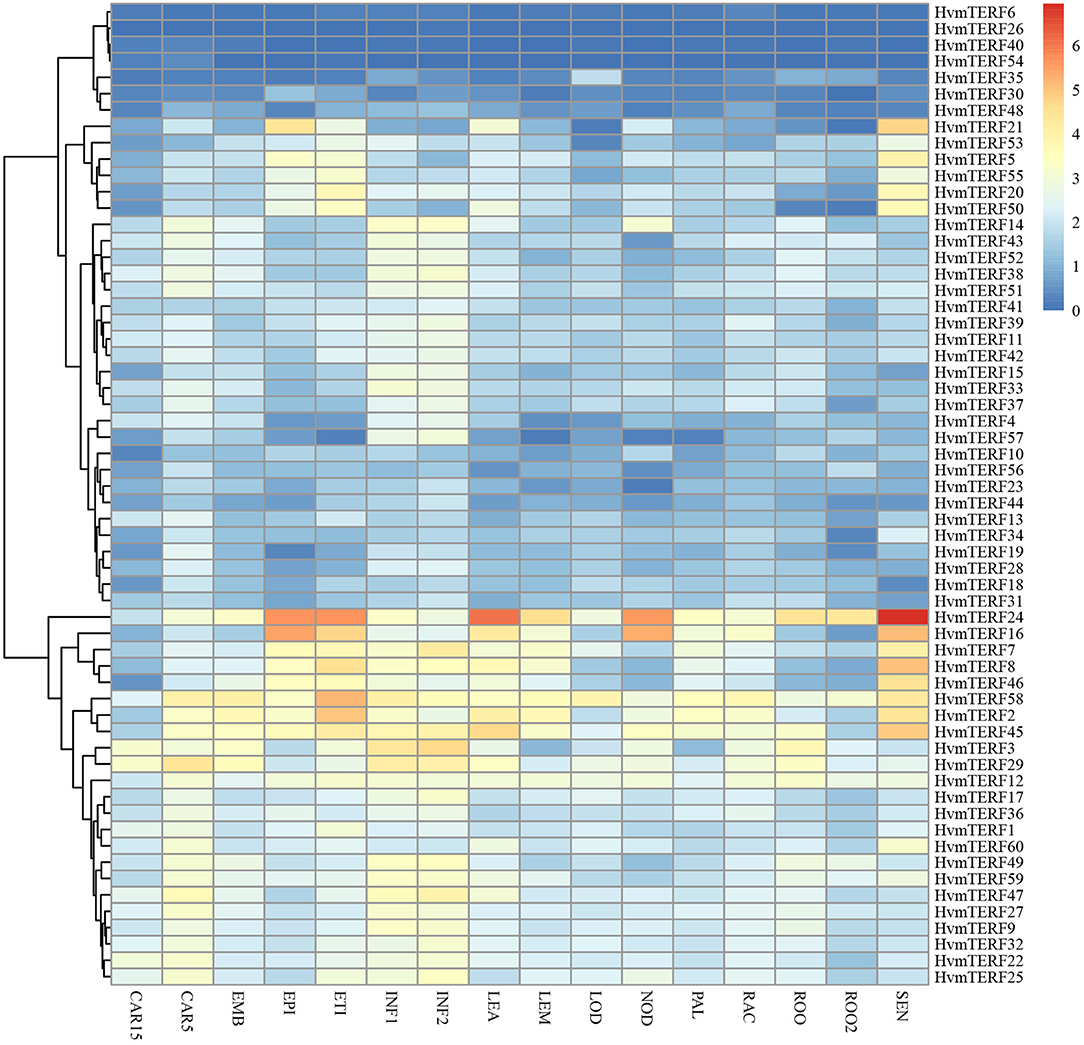
Figure 5. The expression profile of HvmTERF genes at different tissues or stage of barley. FPKM values were normalized by log2(FPKM+1) transform to represent color scores. CAR15/CAR5, developing grain (15/5 day after pollination); EMB, embryonic tissue (4 days); EPI, epidermal strips (4 weeks after pollination); ETI, etiolated seedling with 10 days old after planting; INF1, Young developing inflorescences with 5 mm; INF2, developing inflorescences with 1 cm; LEA, 10 cm shoots from seedlings; LEM, inflorescences, lemma(6 weeks after pollination); LOD, inflorescences, lodicule (6 weeks after pollination); NOD, developing tillers at third stem internode (6 weeks after pollination); PAL, dissected inflorescences, palea (6 weeks after pollination); RAC, inflorescences, rachis (5 weeks after pollination); ROO, roots from the seedlings with 17 and 28 days old after planting; ROO2, roots (4 weeks after pollination); SEN, senescing leaves (8 weeks after pollination).
To gain comprehensive information about the functions of HvmTERFs in response to abiotic stresses, the expression profiles of HvmTERFs under cold, salt, and metal ion stresses were further investigated. The results revealed that HvmTERFs 15, 23, and 33 were found to be upregulated under cold treatment, whereas seven HvmTERF genes were downregulated (Figure 6A). Notably, the tissue-specific gene HvmTERF16 (mTERF3/SL1, SEEDLING LETHAL 1) was downregulated with 2.65-fold change compared with the control. Since limited studies have been conducted on mTERF genes (Jiang et al., 2020), the biological function of HvmTERFs still need more experimental verification.
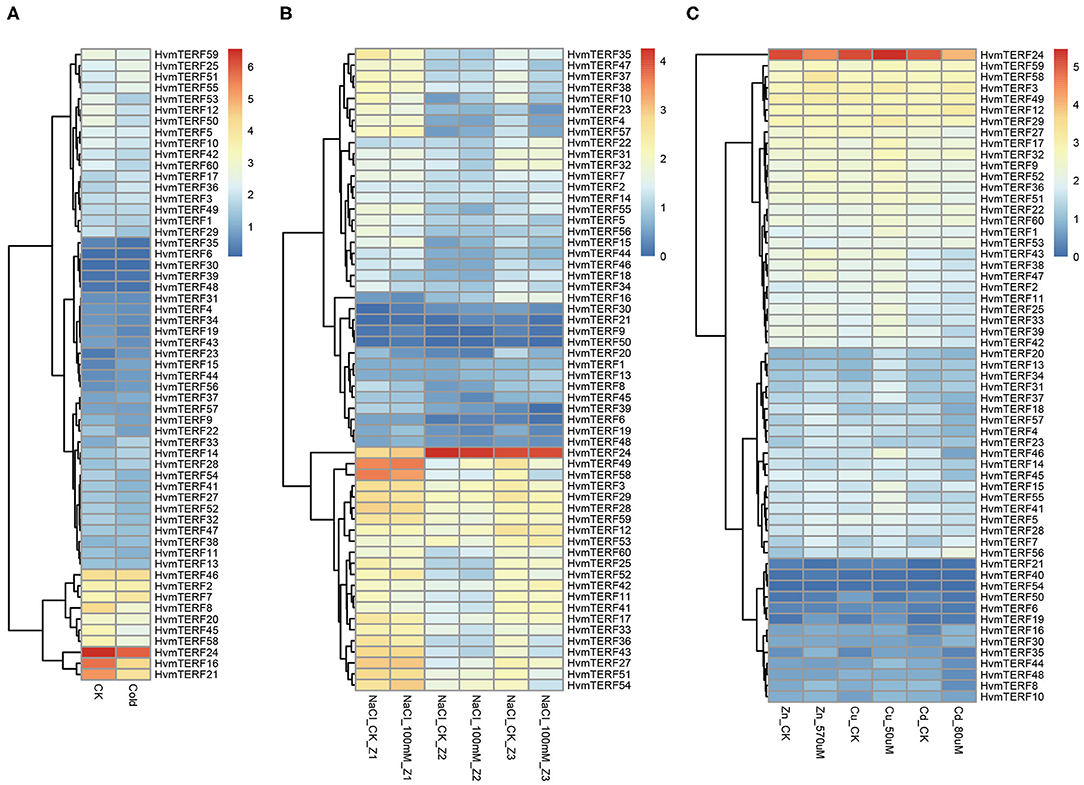
Figure 6. Expression profiles of mTERF genes under five stress conditions. (A) cold stress; (B) salt stress; (C) zinc, copper, and cadmium stress.
The expression patterns of HvmTERFs under salt treatment were further analyzed. A total of 5, 2, and 15 HvmTERF genes were differentially expressed in the root meristematic zone, elongation zone, and maturation zone, respectively (Figure 6B). Consistent with the expression pattern under cold treatment, most (72.72%) of the differentially expressed genes, including 1 in the meristematic zone and 15 in the maturation zone, were downregulated. Furthermore, HvmTERF50 was upregulated in the meristematic zone and downregulated in the mature zone. HvmTERF10 was upregulated in the elongation zone and downregulated in the mature zone. Notably, HvmTERF21 was upregulated in both the meristematic zone (3.49-fold) and elongation zone (4.01-fold) and downregulated in the maturation zone (2.95-fold).
We finally analyzed the expression profiles of HvmTERFs under metal ion toxicity stress. Among them, 4, 5, and 2 upregulated HvmTERFs were found to be zinc-, copper- and cadmium toxicity-responsive genes, and 2, 1, and 9 downregulated genes were also identified (Figure 6C). Remarkably, the expression levels of HvmTERF19 under zinc and copper treatment were 4.14- and 2.45-fold higher than those of the control. HvmTERF35 was significantly upregulated under zinc and copper treatment and downregulated under cadmium treatment. However, HvmTERF50 was upregulated under zinc treatment and downregulated under copper and cadmium treatment. HvmTERF21 was upregulated under cadmium treatment and downregulated under zinc treatment.
Co-expression Network Analysis Between HvmTERFs and Other Genes in Barley
Co-expression analysis has become an effective methodology for gene functional annotation (Wei and Chen, 2018). By using a large dataset of 148 RNA-seq samples, we attempted to construct a co-expression network of mTERF genes. A total of 162,373 interactions, composed of 27 HvmTERFs and 778 other co-expressed genes, were detected (Figure 7). In detail, 595 (76.48%), 260 (33.42%), 178 (22.88%), and 167 (21.47%) genes were predicted to be co-expressed with HvmTERF57, HvmTERF3, HvmTERF15, and HvmTERF33, respectively. The results suggested that these HvmTERFs may play central regulatory roles in the co-expression network. Interestingly, nine HvmTERFs were co-expressed with multiple transcription factors. For instance, 5 B3, 4 GRF (Growth-Regulating Factor), 3 C3H (Cysteine3Histidine), and 3 MYB (Myeloblastosis) family genes were co-expressed with 4, 4, 7, and 3 HvmTERFs. Transcription factors are essential regulators to repress or activate the expression of their target genes by binding to specific upstream elements (Jin et al., 2017). These results suggested that multiple transcription factors may interact with HvmTERFs, and further to control multitudinous growth and development processes, and response to environmental stressors in barley. Furthermore, 11 HvmTERFs were predicted to be co-expressed with SPLICEOSOME-ASSOCIATED PROTEIN 130 (SAP130a), a key gene that is required for specific spatiotemporal events during reproduction in Arabidopsis (Aki et al., 2011).
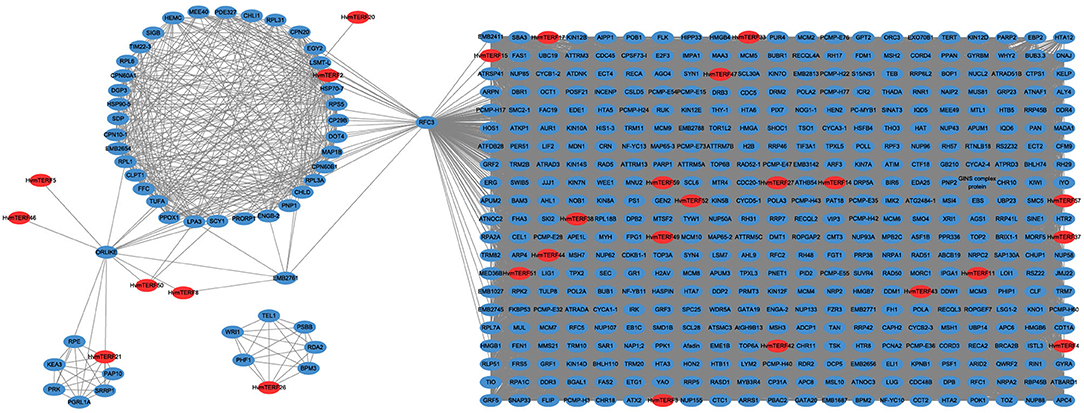
Figure 7. The co-expression network analysis of HvmTERFs with other barley genes. Only annotated genes are represented.
Gene Ontology (GO) enrichment analysis was further performed to determine the potential function of the mTERF co-expressed genes. The mTERF co-expressed genes were enriched in 155 GO terms (FDR <0.05), including 66 biological processes, 41 cellular components, and 48 molecular functions (Supplementary Figures 4–6; Supplementary Table 11). In the molecular function category, microtubule binding (GO:0008017), nucleoside-triphosphatase activity (GO:0017111), and tubulin binding (GO:0015631) ranked as the top three enriched terms, whereas intracellular non-membrane-bound organelle (GO:0043232), non-membrane-bound organelle (GO:0043228), and nucleus (GO:0005634) were the most common terms in the cellular components category. In the biological process category, the mTERF co-expressed genes were implicated not only in various biological functions (e.g., GO:0006260 DNA replication metabolic process; GO:0006259 DNA metabolic process; GO:0042254 ribosome biogenesis) but also in response to diverse stresses (e.g., GO:0033554 cellular response to stress; GO:0006974 cellular response to DNA damage stimulus).
Expression of HvmTERF Genes in Response to Salt, Drought, Cold, and ABA Treatment via qRT-PCR
Although differentially expressed HvmTERFs under different stresses were obtained based on RNA-seq data, comprehensive expression patterns of HvmTERFs in response to various stresses and phytohormones have not been reported. To better understand the expression patterns in response to diverse stress treatments (cold, salt, heat, and ABA), 25 HvmTERFs were randomly selected for qRT-PCR analysis. Under salt stress, all the candidate HvmTERFs were downregulated after 1, 3, and 12 h of treatment. HvmTERF23 was the most downregulated gene at the 1 h (5.93-fold), 3 h (12.89-fold), and 12 h (6.63-fold) time points (Supplementary Figure 7). Under 6 h salt treatment, nine HvmTERFs were found to be upregulated. Among them, the expression level of HvmTERF46 was dramatically increased with a 2.28-fold change value. Moreover, three upregulated HvmTERF genes were found at 24 h. Notably, HvmTERF21 exhibited significantly upregulated expression levels at 6 and 24 h.
Under drought treatment, a total of 1, 1, 5, and 5 HvmTERF genes were upregulated at 3, 6, 12, and 24 h (Supplementary Figure 8). Among them, HvmTERF21 showed 1.21-, 1.36-, 1.24-, and 2.58-fold changes at the 3, 6, 12, and 24 h time points, whereas HvmTERF52 displayed 1.99- and 2.00-fold changes at the 12 and 24 h time points, respectively. The MBS cis-acting element involved in drought inducibility was also detected within the promoter regions of these genes (Urao et al., 1993). For example, HvmTERFs52 possessed 2 MBS cis-acting elements. There were some exceptions, however, no MBS-acting element was detected in the promoter regions of HvmTERF21, implying this gene may have unknown elements acting in response to drought stress. Furthermore, HvmTERFs 2, 8, 19, 29, 43, and 49 were downregulated after drought treatment at all-time points.
Under cold treatment, we identified more upregulated HvmTERFs than those identified in response to salt and drought treatment, with 17, 19, 18, 17, and 20 upregulated genes after 1, 3, 6, 12, and 24 h of treatment, respectively (Supplementary Figure 9). Notably, HvmTERFs 9, 16, 21, 24, 25, 45, 49, and 51 were upregulated at all treated time points. The expression level of HvmTERFs dramatically decreased over time. The average fold change was 4.40 after 1 h of cold treatment, whereas it decreased to 1.24 after 48 h of stress, suggesting that HvmTERFs may mainly function in the initial response to cold injury.
The plant hormone ABA has been demonstrated to play important roles in improving the tolerance of plants to diverse stresses (Shinozaki and Yamaguchi-Shinozaki, 1997). qRT-PCR was also carried out to analyze the expression profiles of the 25 selected HvmTERFs after ABA treatment (Supplementary Figure 10). The heatmap revealed that HvmTERFs 9, 16, 21, 24, 28, and 29 exhibited upregulated expression patterns at all time points. Meanwhile, abundant ABRE cis-acting elements, the major cis-element for ABA-responsive gene expression, were identified in the promoter regions, such as five ABRE cis-element for HvmTERF28, three for HvmTERF16, and three for HvmTERF24. The expression level of HvmTERF46 displayed upregulated expression with a 10.16-fold change after 6 h of treatment, whereas HvmTERF53 showed 3.89- and 3.84-fold changes after 1 and 6 h of treatment, respectively.
Nucleotide Variation, Population Structure, and Haplotype Analysis of HvmTERF Genes
To reveal the variation landscape of HvmTERFs, public resequencing data of barley were employed to detect HvmTERF-related SNPs. The SNP calling pipeline yielded a total of 481 HvmTERF-related SNPs or approximately 8.01 SNPs per gene, representing the most comprehensive variation dataset of HvmTERFs to date (Supplementary Table 12). The majority of HvmTERF-related SNPs (70.68%) were located in the genic region, including 159 synonymous, 133 missense, 44 intron, 3 splice region, and 1 stop-gain variant (HvmTERF42) (Supplementary Table 13). The overall transition/transversion (Ts/Tv) ratio was 2.317, with A/G (35.55%) and C/T (34.30%) ranking as the most popular allelic substitution patterns. These results indicated that there was fewer purine to purine or pyrimidine to pyrimidine mutation than there was pyrimidine to purine or purine to pyrimidine mutations.
To further investigate the genetic relationship between wild and landrace barley populations, the PCA was conducted using HvmTERF-related SNPs. The first eigenvector explained 23.11% of the total genetic variance and captured the biological differentiation that separated wild barley from landrace barley. The second and third eigenvectors explained 12.16 and 11.10% of the variance, respectively, and distinguished the accessions geographically (Figures 8A,B; Supplementary Table 14). The same population affinities were recovered in the phylogenetic tree with a precise accession relationship (Figure 8C). ADMIXTURE analysis also recapitulated these findings (Figure 8D). When K = 2, a clear separation was observed in accordance with biological differentiation between wild and landrace barley. As K increased to 5, a definite separation was presented in accordance with the geographical source. Remarkably, a certain proportion of genetic admixture between wild and landrace barley was observed, suggesting the potential domestication origin of cultivated barley and ongoing gene flow between wild and landrace barley.
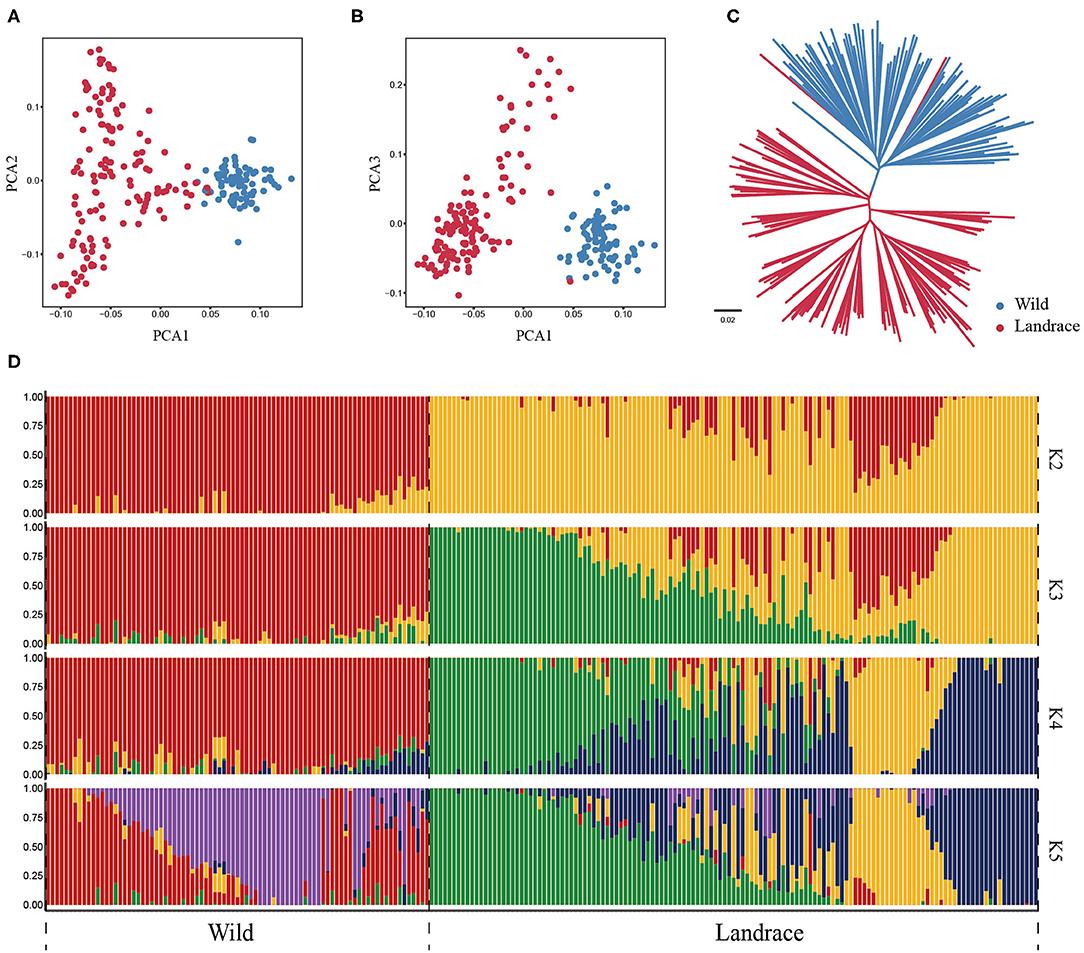
Figure 8. Population structure of wild barley accessions and landraces based on HvmTERF-related SNPs. (A) Principal component analysis PC1 vs. PC2, (B) Principal component analysis PC1 vs. PC3, (C) The NJ phylogenetic tree, (D) Population structure with K ranging from 2 to 5.
Population-based nucleotide diversity (π) was estimated based on HvmTERF-related SNPs. The nucleotide diversity decreased only 4.5% from wild barley (0.2491) to landrace barley (0.2380), indicating that this gene family suffered a weak genetic bottleneck in the process of domestication (Supplementary Figure 11). Wright's F-statistic is an informative indicator that measures population differentiation and gene flow intensity (Wright, 1951). Populations with Fst values >0.25 are considered highly differentiated (Fong et al., 2016). A relatively low Fst index (0.1310) was obtained between wild and landrace barley in accordance with the HvmTERF-related SNPs, indicating that this gene family was not subjected to strong selective pressure during barley domestication.
Haplotype dissection and comparison provide invaluable resources for understanding the evolutionary and domestication processes of important traits (Jan et al., 2019). To acquire a more precise depiction of the haplotype network, we constructed the complete haplotypes for the 220 accessions using HvmTERF-related SNPs. The median-joining method network generated a total of 481 HvmTERF haplotypes (one haplotype per accession) consisting of distinct wild and landrace groups (Figure 9). No shared haplotype between wild and landrace barley was observed in the network. The highly diverse wild accessions displayed geographical clustering patterns in terms of the Southern Levant (such as Israel and Jordan), Northern Levant (such as Syria and Turkey), and East of Levant (such as Iraq, Iran, and Central Asian counties). For landrace haplotypes, a geographical clustering pattern was obtained. However, a certain portion of accessions displayed no geographical clustering of haplotypes; for example, many shared haplotypes from Central Asia and Europe were observed in the median-joining network, suggesting a complex domestication process of HvmTERF in barley.
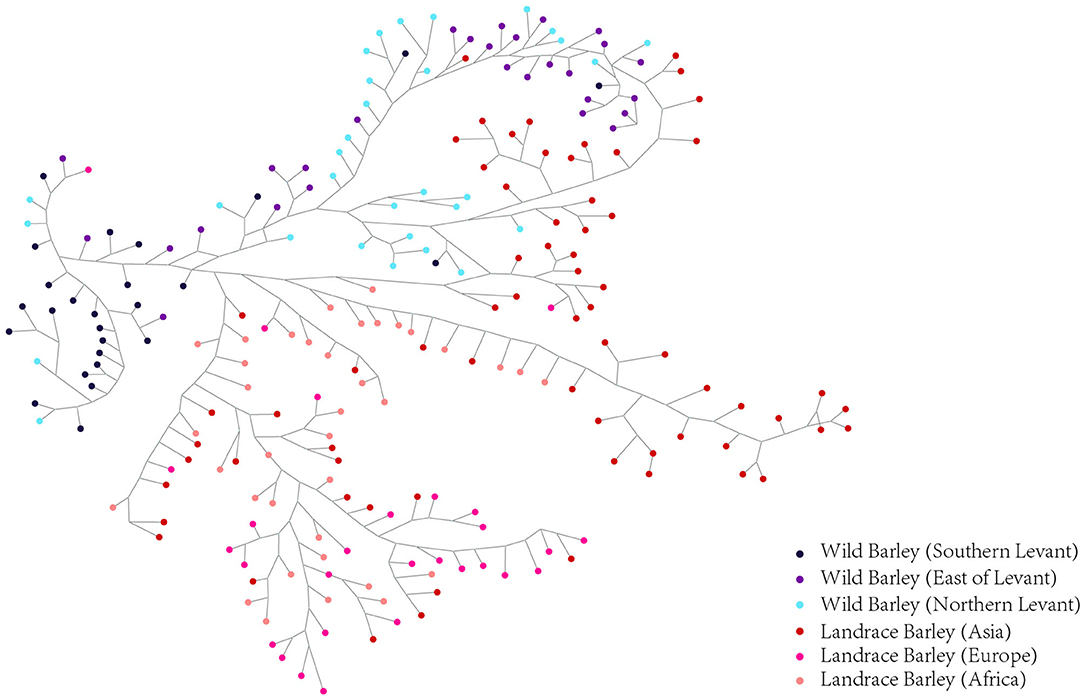
Figure 9. Median-Joining network analysis of wild barley accessions and landraces based on HvmTERF-related SNPs. Wild barley accessions were divided into Southern Levant, Northern Levant, and East of Levant groups. Landraces were divided into Asia, Europe, and Africa groups.
Discussion
Characterization of the mTERF Gene Family in Barley
The mTERF family, firstly identified in vertebrate mitochondria, is implicated in the regulation of organellar transcription, translation, and DNA replication (Quesada, 2016; Xiong et al., 2020). With the accomplishment of various whole genome sequencing projects, the identification and characterization of mTERF genes have been widely conducted in diverse plant species (Tang et al., 2019). As an inbreeding, diploid and temperate cereal crop, barley is well-studied in terms of cytology, genetics and molecular biology. However, its large genome size and high transposon content have long hindered barley genome assembly projects (Schulte et al., 2009). In recent years, the first physical sequence assembly for barley and its subsequent chromosome-scale reference sequence assembly (Morex V1), as well as the improved annotated reference genome assembly (Morex V2), have formed the basis for the identification and characterization of related gene families (Jayakodi et al., 2020). In this study, we carried out a comprehensive search for putative HvmTERFs, and a total of 60 HvmTERFs were identified in the barley Morex V2 genome assembly. Compared with its previous versions, the numbers of identified HvmTERFs were only 40 and 51 for the barley physical sequence assembly and Morex V1 assembly, respectively (Supplementary Table 15). In addition, in the Morex V1 assembly, two HvmTERF members named HORVU0Hr1G013680 and HORVU0Hr1G017090 were not mapped to the reference genome. However, they were anchored to chromosome 5 (HORVU.MOREX.r2.5HG0420050, HvmTERF20) and chromosome 6 (HORVU.MOREX.r2.6HG0509710, HvmTERF49) in the Morex V2 assembly. Each of the HvmTERFs contains the notable conserved mTERF domain. No premature termination codon was found in the coding region of HvmTERF genes, and most of them were supported by barley ESTs, ensuring the authenticity of gene family identification. Thus, this is the first gene family identification using the Morex V2 assembly at the whole genome-wide level, and the most updated and comprehensive information on the mTERF gene family in barley has been obtained to date.
The improved genome assembly also provided the definite physical locations of HvmTERFs. The HvmTERFs were distributed unevenly across seven chromosomes. The maximum number of HvmTERFs was located on the long arm end of chromosome 6, with a total of 19 HvmTERFs. Most of the HvmTERFs were located at the distal ends of the chromosome, but they were absent from the short arm of chromosome 3 and the long arm of chromosome 5. Similar findings were reported in other barley gene families, such as the non-specific lipid transfer protein (LTP) gene family and auxin/indole-3-acetic acid (Aux/IAA) gene family (Zhang et al., 2019; Shi et al., 2020). A possible cause might be that the distal and proximal ends of chromosomes are more gene-rich than the middle regions of chromosomes in barley (Mayer et al., 2012). In cereals, meiotic homologous chromosome recombination is skewed toward the distal regions of the chromosomes, leading to the biased distribution of genes that are concentrated in the distal regions (Higgins et al., 2012). The uneven distribution of recombination-rich regions ensures that there is abundant genetic diversity available to respond to various stressful conditions and dynamic environmental changes (Zhang et al., 2019).
In metazoans, the mTERF gene family contains four to five members (Roberti et al., 2009). In contrast, the mTERF gene family has expanded to approximately 30 members in land plants (Quesada, 2016). For example, there are 35 mTERFs in Arabidopsis, 33 in rice, 31 in maize, and 25 in grape. In this study, a total of 60 HvmTERF genes were identified in the barley reference genome, approximately two times as many as other higher plants. Given the complicated regulation of organellar genome transcription in land plants, the expansion of the mTERF gene family in barley could be induced by a complex mechanism. To gain insights into the evolutionary relationship within the mTERF gene family, we first constructed a phylogenetic tree using the mTERF proteins from barley, rice, and Arabidopsis. The mTERF proteins were categorized into nine subfamilies based on the classification set forth by Zhao (Zhao et al., 2014). Within the same subfamily, the gene structure and protein motif organization were highly conserved, supporting the phylogenetic analysis, and classification results. The phylogenetic tree further showed that eight subfamilies (subfamilies I–VIII) contained the mTERF proteins from these three species (barley, rice, and Arabidopsis), suggesting that these mTERF proteins evolved from common ancestors and then expanded independently in each species. Most of the subfamilies possessed comparable numbers of mTERF proteins, whereas monocot-specific subfamily IX does not contain any mTERF proteins from Arabidopsis, suggesting that subfamily IX formed after the divergence of monocots and dicots (Zhao et al., 2014). Furthermore, only 13 rice mTERF proteins were clustered in Group IX. In contrast, 39 mTERF proteins, more than half of the total mTERFs in barley, were assigned to this subfamily. Thus, we speculated that HvmTERFs within subfamily IX may have experienced noticeable expansion.
Gene duplication contributes to the expansion and evolution of gene families (Shi et al., 2020). To reveal the expansion mechanism of HvmTERFs, segmental and tandem duplication events were investigated. Fourteen pairs of HvmTERFs underwent gene duplication, including seven segmental and seven tandem duplication events. Remarkably, 17 mTERF genes consisting of 13 duplicated pairs contributed to the expansion of subfamily IX. Collectively, both segmental and tandem duplication contributed to the expansion of the HvmTERF gene family, mainly in subfamily IX, and further led to twice as many mTERF genes in barley as in other species. Moreover, the Ka/Ks values of all the paralogous gene pairs were <1, suggesting that they all underwent purifying selection.
HvmTERF Genes May Play Important Roles During Plant Growth, Abiotic Stress, and Phytohormone Responses
To obtain preliminary insight into the biological function of mTERFs, we checked the cis-elements in the promoter regions of HvmTERFs. The promoter regions contained various cis-elements associated with development/tissue specificity, promoter/enhancer elements, light responses, circadian control rhythms, and external stimuli and hormone responses, suggesting that HvmTERFs are involved in multiple biological processes. Since cis-element prediction was carried out based on a bioinformatics approach, further experimental validation is also required.
In vertebrates, the biological function of mTERFs in the regulation of mitochondrial transcription, replication, and translation has been well-documented (Castillo et al., 2019). Although land plants possess more mTERFs than mammals, the functions of mTERFs in plants are rather limited (Kleine and Leister, 2015; Quesada, 2016). Based on their loss-of-function phenotypes, which have mainly been characterized in Arabidopsis and maize, mTERFs in land plants are required for OGE and play essential roles in plant growth and development (Ding et al., 2019). In this study, the specific spatiotemporal expression of HvmTERFs in different developmental stages and tissues suggested that HvmTERFs might potentially play a vital role in various plant growth and developmental processes. HvmTERF2, HvmTERF16, and HvmTERF58 (orthologous to RUGOSA2, SL1/mTERF3, and mTERF6 in Arabidopsis, respectively) displayed high expression levels in the studied tissues and stages. In Arabidopsis, these orthologous genes are essential for plant photosynthesis, mitochondrial, and chloroplastic gene expression and development, and leaf patterning and organogenesis (Quesada et al., 2011; Jiang et al., 2020). HvmTERF24 was the most highly expressed gene in different organs. However, there is rather limited information on the functions of its orthologous gene in Arabidopsis (AtmTERF12). Recent research has only demonstrated that AtmTERF12 is not involved in the response to salt stress (Xu et al., 2017). Several tissue- and stage-specific genes were also identified. For instance, HvmTERF14 showed preferential expression in young inflorescences, whereas its ortholog mTERF15 is required for the cis-splicing of mitochondrial nad2 intron 3 in both Arabidopsis and maize and further regulates the small kernel phenotype in maize, implying that HvmTERF14 may achieve different functions in barley compared with other species (Hsu et al., 2014; Yang et al., 2020). Homologous analysis might provide information on the role of HvmTERFs. However, to ascertain HvmTERF function still requires further detailed and extensive experimental work (Yin et al., 2021).
In contrast to animals, plants are sessile organisms that are continuously exposed to and cannot escape environmental stresses. During evolution, the expansion and diversification of gene families played important roles in plant adaptation or tolerance to environmental extremes (Xu et al., 2017). A wide range of mechanisms have evolved in plants to cope with adverse environmental stresses at the molecular level. Compared with the control, a total of three and four upregulated genes were identified under cold and salt treatment, whereas under metal poisoning stresses, a total of four zinc, five copper, and two cadmium toxicity stress-related HvmTERFs were identified. Since similar studies are rather scarce, further experimental validation is required. Therefore, the expression patterns of HvmTERFs in response to various stresses were further investigated by qRT-PCR. Most of the qRT-PCR results were consistent with the RNA-seq results. For example, both the RNA-seq and qRT-PCR results demonstrated that HvmTERFs 19, 23, and 58 were downregulated in response to salt stress. By contrast, several upregulated HvmTERFs were also detected in response to various stresses. For instance, HvmTERF21 was upregulated under salt and cold stress based on RNA-seq, while this gene was significantly upregulated by cold, salt, drought and ABA stress via qRT-PCR analysis. Homology analysis revealed the involvement of its orthologous gene AtmTERF10 in salt tolerance, possibly through an ABA-mediated mechanism (Xu et al., 2017). Nonetheless, certain inconsistent expression patterns were also observed. RNA-seq data revealed that HvmTERF7 and HvmTERF46 were not induced by salt stress at the three root zones, while qRT-PCR analysis showed that these genes were significantly downregulated at 1, 3, 12, and 24 h and significantly upregulated at 6 h under salt treatment. Previous studies reported that mTERF9 (orthologous to HvmTERF7) and MAD1/mTERF5 (orthologous to HvmTERF46) contributed to salt tolerance in Arabidopsis (Robles et al., 2012, 2015; Núñez-Delegido et al., 2020). The inconsistent results between RNA-seq and qRT-PCR may be due to several putative reasons. First, the different barley varieties were used in the two experiments. The barley cultivar Clipper was used in RNA-seq, whereas the cultivar Morex was the experimental materials in qRT-PCR. Second, the plant materials were not exactly the same. The materials for qRT-PCR were roots, whereas the materials for RNA-seq were both roots and leaves. Third, the expression level of qRT-PCR was calculated based on the 2−ΔΔCT method, and the expression level of RNA-seq was estimated by FPKM. These two different calculation methods could not ensure that all the results are consistent. In brief, these results provide candidates for further functional investigation of mTERF genes in barley as well as in other cereal crops.
In addition, there was no correlation between expression patterns and phylogenetic relationships. The fate of HvmTERF genes from the same gene family could be described as neofunctionalization during expansion. For example, in subfamily IV, HvmTERFs 2, 8, and 46 showed relatively high expression in various tissues and developmental stages, whereas HvmTERF20 exhibited preferential expression in developing inflorescences and senescing leaves, and HvmTERF51 was not expressed in most of the developmental stages and tissues. Highly diverse expression patterns were also observed in subfamily IX, the most expanded subfamily. In addition, a divergent expression profile was investigated even for duplicated gene pairs. The duplicated genes HvmTERF9 and 34 showed divergent spatiotemporal expression patterns. HvmTERF34 was induced by copper, whereas its paralog, HvmTERF9, was significantly upregulated in the root meristematic zone under salt treatment. Similar patterns were also observed in other phylogenetic groups. These results suggested that close phylogenetic relationships are not essential for similar expression profiles, which was consistent with previous reports on other barley gene families (Li et al., 2019).
Nucleotide Diversity Analysis Indicated That HvmTERF Genes Experienced a Weak Bottleneck During Barley Domestication
Domestication is a plant-animal coevolution process that occurs when wild species are exposed to new selective environments associated with human cultivation and use, leading to morphological and physical changes that distinguish domesticated taxa from their wild ancestors (Purugganan and Fuller, 2009; Purugganan, 2019). Cultivated barley, domesticated from its progenitor wild barley (Hordeum vulgare ssp. spontaneum), has experienced a series of genetic changes that have caused differences in plant architecture and growth habits, collectively called the domestication syndrome (Hammer, 1984; Doebley et al., 2006). Domestication of barley resulted in a concomitant bottleneck that reduced nucleotide diversity in alleles (Haas et al., 2020). However, little is known about the changes in mTERFs resulting from domestication in barley. In this study, 481 SNPs were identified from 60 HvmTERF genes across 220 wild and landrace barley accessions. The SNPs were distributed unevenly along the genomic sequence, including a total of 292 exon and 44 intron variations, which was consistent with a previous study showing higher polymorphism of SNPs in exon regions than in intron regions (Lu et al., 2019) but opposite to observations in other studies (Uçarli et al., 2016; Xia et al., 2017). The PCA, admixture, and phylogenetic analyses clearly divided all the accessions into landraces and wild barley accessions and further distinguished them geographically. We further examined the geographical distribution of these haplotypes and found that the wild barley populations from the Northern Levant and East of Levant regions appeared to contribute most directly to the genetic composition of Asian landraces, while Southern Levant barley populations made a great contribution to African and European landraces. The genetic constitution of barley landraces indicated multiple origins from wild progenitor populations that resulted in the initial domestication and subsequent migration of early agriculturalists along the axes of the Afro-Eurasian world (Poets et al., 2015). Although multiple wild populations provided the basis for the genetic constitution of landraces, the broad geographic range of landraces also showed various regional correlations.
Domestication also resulted in a concomitant bottleneck that reduced sequence diversity across many genes (Haas et al., 2020). The nucleotide diversity of wild accessions was relatively higher than that of landrace accessions, with a total decrease of ~4.5%. Compared with the previous study, the average reduction in nucleotide diversity was 27% from wild barley accessions to landraces across the genome (Russell et al., 2016). The significantly lower nucleotide diversity loss passing from wild barley accessions to landraces in this study indicated that the HvmTERF gene family might have suffered simple bottleneck effects, rather than selection, in the process of barley domestication. This result was also verified by the relatively low Fst index. No significant divergence occurred between wild barley accessions and landraces regarding HvmTERFs. One plausible reason is that the hitchhiking effect reduced the nucleotide diversity of the linked loci associated with domestication (Kilian et al., 2006).
As in other plants, mTERFs are characterized as evolutionarily conserved and fundamentally universal signaling pathways. However, the comprehensive characterization of barley mTERF gene family remains to be elucidated in detail. Our data on the physicochemical properties, phylogenetic relationships, gene structures, conserved motifs, expansion patterns, expression profiles, and genetic variations will provide essential clues for investigating the biological functions and evolutionary history of mTERF gene family in barley.
Conclusion
In this study, a total of 60 mTERF genes were identified in barley, about two times as many as those in Arabidopsis and rice. Phylogenetic analysis categorized these genes into nine subfamilies, with approximately half of them assigned to subfamily IX, which was supported by exon-intron structure and conserved motif analyses. Both segmental and tandem duplications contributed significantly to the expansion of HvmTERFs, mainly in subfamily IX. Cis-acting regulatory element, expression profile and co-expression network analyses suggested that HvmTERFs might be involved in the development process, tolerance to diverse stresses and response to plant hormones. qRT-PCR analysis also revealed that HvmTERF21 and HvmTERF23 were significant induced by several abiotic stresses and/or phytohormone treatment, and these genes could be considered candidates for further functional studies. Finally, genetic variation analysis demonstrated that HvmTERFs may have experienced a weak genetic bottleneck, rather than selection, during the domestication process from wild to landrace barley. Taken together, our findings will not only provide a solid foundation for further evolutionary analysis but also facilitate the functional study of HvmTERFs and the molecular breeding of barley.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
XW, FW, and LC designed and supervised the project. TL, WP, YY, and YLiu performed the data analysis. WP and YLiu performed the experiments. TL, YLi, and LC drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Natural Science Foundation of China (Grant No. 32060458), Jiangxi Natural Science Foundation (Grant No. 20202BAB215002), and the Science and Technology Research Project of Jiangxi Provincial Department of Education (Grant No. GJJ180241). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.684619/full#supplementary-material
Supplementary Figure 1. Schematic geographic distribution of 220 wild barley and landraces accessions from different regions.
Supplementary Figure 2. Sequence characteristics and conserved amino acids ratio of mTERF proteins in four species. (A) Barley, (B) Rice, (C) Arabidopsis, (D) Maize.
Supplementary Figure 3. Distribution of HvmTERF genes on barley chromosomes. Chromosome numbers are shown at the top of each chromosome. The scale (Mb) is indicated on the left.
Supplementary Figure 4. GO enrichment (biological process) of HvmTERF co-expressed genes in the WGCNA network.
Supplementary Figure 5. GO enrichment (cellular component) of HvmTERF co-expressed genes in the WGCNA network.
Supplementary Figure 6. GO enrichment (molecular function) of HvmTERF co-expressed genes in the WGCNA network.
Supplementary Figure 7. The expression analysis of HvmTERF genes in response to salt. Error bar represent the standard error of the mean. One asterisk (*) indicates 0.05 significance level. Double asterisk (**) indicates 0.01 significance level.
Supplementary Figure 8. The expression analysis of HvmTERF genes in response to drought. Error bar represent the standard error of the mean. One asterisk (*) indicates 0.05 significance level. Double asterisk (**) indicates 0.01 significance level.
Supplementary Figure 9. The expression analysis of HvmTERF genes in response to cold. Error bar represent the standard error of the mean. One asterisk (*) indicates 0.05 significance level. Double asterisk (**) indicates 0.01 significance level.
Supplementary Figure 10. The expression analysis of HvmTERF genes in response to ABA. Error bar represent the standard error of the mean. One asterisk (*) indicates 0.05 significance level. Double asterisk (**) indicates 0.01 significance level.
Supplementary Figure 11. Nucleotide diversity of wild barley and landraces accessions. The horizontal line inside the box corresponds to the median of this distribution, the bottom and top of the box are the first and third quartiles and data points outside the whiskers can be considered as outliers. The nucleotide diversity was calculated based on HvmTERF-related SNPs.
Supplementary Table 1. Accession number and samples information of RNA-seq data used in this study.
Supplementary Table 2. The primers used in the qRT-PCR.
Supplementary Table 3. Summary information of the 220 wild barley accessions and landraces.
Supplementary Table 4. Characteristic features of mTERF gene family in barley.
Supplementary Table 5. Verification of the mTERF domains using the NCBI-CDD, SMART, HMMER, and InterPro online tools.
Supplementary Table 6. Consensus motifs analysis of mTERF domains in different species.
Supplementary Table 7. The segmental and tandem duplication events of HvmTERFs.
Supplementary Table 8. The Ka/Ks ratios and estimated divergence time between barley and other five species (A. thaliana, B. distachyon, O. sativa, Vitis vinifera, Zea mays).
Supplementary Table 9. Characteristics of cis-acting regulatory elements in the promoter regions of the HvmTERF genes.
Supplementary Table 10. List of putative miRNAs targeted to HvmTERF genes identified by psRNATarget online tool.
Supplementary Table 11. GO enrichment of HvmTERF co-expressed genes.
Supplementary Table 12. The genetic variation information (VCF format) of HvmTERF-related SNPs across the 220 wild barley accessions and landraces.
Supplementary Table 13. Distribution of SNPs within various genomic regions.
Supplementary Table 14. Tracy-Widom (TW) statistics and P-values for the first 10 eigenvalues in the PCA of HvmTERF-related SNPs.
Supplementary Table 15. The number of HvmTERFs from different barley assemblies.
Abbreviations
ABA, abscisic acid; ABRE, ABA-responsive elements; Aux/IAA, auxin/indole-3-acetic acid; BLAST, basic local alignment search tool; C3H, cysteine3histidine; CDD, conserved domains database; CDS, coding sequence; EST, expressed sequence tag; FDR, false discovery rate; FPKM, fragments per kilobase per million; ERE, estrogen response element; Fst, Wright's F-statistic; GCN4, general control non-repressible-4; GO, gene ontology; GRAVY, grand average of hydropathicity; GRF, growth-regulating factor; GSDS, Gene Structure Display Sever; GTF, Gene Transfer Format; HMM, hidden Markov model; HSP, heat shock protein; Ka, non-synonymous substitution rate; Ks, synonymous substitution rate; LTP, lipid transfer protein; LTR, long terminal repeat; MBS, myeloblastosis binding site; MDA1, mTERF DEFECTIVE IN Arabidopsis1; MeJA, methyl jasmonate; MEME, Multiple Em for Motif Elicitation; MSA-like, mitosis-specific activator; MW, molecular weight; MYB, myeloblastosis; NCBI, National Coalition Building Institute; NEP, nucleus-encoded RNA polymerase; NJ, neighbor-joining; OGE, organellar gene expression; PAML, Phylogenetic Analysis by Maximum Likelihood; PCA, principal component analysis; PEP, plastid-encoded RNA polymerase; pI, theoretical isoelectric point; qRT-PCR, Quantitative Real-time PCR; ROS, reactive oxygen species; SA, salicylic acid; SHOT1, SUPPRESSOR OF HOT1-4 1; SL1, SEEDLING LETHAL 1; SMART, simple modular architecture research tool; SNP, single nucleotide polymorphism; SOLDAT10, SINGLET OXYGEN-LINKED DEATH ACTIVATOR10; SRA, sequence read archive; Ts, transition; Tv, transversion; WGCNA, weighted gene co-expression network analysis.
References
Aki, S., Nakai, H., Aoyama, T., Oka, A., and Tsuge, T. (2011). AtSAP130/AtSF3b-3 function is required for reproduction in Arabidopsis thaliana. Plant Cell Physiol. 52, 1330–1339. doi: 10.1093/pcp/pcr077
Andersson, D. C., Fauconnier, J., Park, C. B., Zhang, S. J., Thireau, J., Ivarsson, N., et al. (2011). Enhanced cardiomyocyte Ca(2+) cycling precedes terminal AV-block in mitochondrial cardiomyopathy Mterf3 KO mice. Antioxid. Redox Signal. 15, 2455–2464. doi: 10.1089/ars.2011.3915
Binder, S., and Brennicke, A. (2003). Gene expression in plant mitochondria: transcriptional and post-transcriptional control. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 181–188; discussion 188–189. doi: 10.1098/rstb.2002.1179
Cámara, Y., Asin-Cayuela, J., Park, C. B., Metodiev, M. D., Shi, Y., Ruzzenente, B., et al. (2011). MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 13, 527–539. doi: 10.1016/j.cmet.2011.04.002
Castillo, A., Vilà, M., Pedriza, I., Pardo, R., Cámara, Y., Martín, E., et al. (2019). Adipocyte MTERF4 regulates non-shivering adaptive thermogenesis and sympathetic-dependent glucose homeostasis. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1298–1312. doi: 10.1016/j.bbadis.2019.01.025
Chen, L., Hu, W., Tan, S., Wang, M., Ma, Z., Zhou, S., et al. (2012). Genome-wide identification and analysis of MAPK and MAPKK gene families in Brachypodium distachyon. PLoS ONE 7:e46744. doi: 10.1371/journal.pone.0046744
Ding, S., Zhang, Y., Hu, Z., Huang, X., Zhang, B., Lu, Q., et al. (2019). mTERF5 acts as a transcriptional pausing factor to positively regulate transcription of chloroplast psbEFLJ. Mol. Plant 12, 1259–1277. doi: 10.1016/j.molp.2019.05.007
Doebley, J. F., Gaut, B. S., and Smith, B. D. (2006). The molecular genetics of crop domestication. Cell 127, 1309–1321. doi: 10.1016/j.cell.2006.12.006
Fong, M. Y., Rashdi, S. A., Yusof, R., and Lau, Y. L. (2016). Genetic diversity, natural selection and haplotype grouping of Plasmodium knowlesi gamma protein region II (PkγRII): comparison with the duffy binding protein (PkDBPαRII). PLoS ONE 11:e0155627. doi: 10.1371/journal.pone.0155627
Gray, M. W. (2012). Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 4:a011403. doi: 10.1101/cshperspect.a011403
Gu, Z., Cavalcanti, A., Chen, F. C., Bouman, P., and Li, W. H. (2002). Extent of gene duplication in the genomes of Drosophila, nematode, and yeast. Mol. Biol. Evol. 19, 256–262. doi: 10.1093/oxfordjournals.molbev.a004079
Haas, M., Himmelbach, A., and Mascher, M. (2020). The contribution of cis- and trans-acting variants to gene regulation in wild and domesticated barley under cold stress and control conditions. J. Exp. Bot. 71, 2573–2584. doi: 10.1093/jxb/eraa036
Hammani, K., and Barkan, A. (2014). An mTERF domain protein functions in group II intron splicing in maize chloroplasts. Nucleic Acids Res. 42, 5033–5042. doi: 10.1093/nar/gku112
Higgins, J. D., Perry, R. M., Barakate, A., Ramsay, L., Waugh, R., Halpin, C., et al. (2012). Spatiotemporal asymmetry of the meiotic program underlies the predominantly distal distribution of meiotic crossovers in barley. Plant Cell 24, 4096–4109. doi: 10.1105/tpc.112.102483
Hsu, Y. W., Wang, H. J., Hsieh, M. H., Hsieh, H. L., and Jauh, G. Y. (2014). Arabidopsis mTERF15 is required for mitochondrial nad2 intron 3 splicing and functional complex I activity. PLoS ONE 9:e112360. doi: 10.1371/journal.pone.0112360
Huang, M., Friso, G., Nishimura, K., Qu, X., Olinares, P. D., Majeran, W., et al. (2013). Construction of plastid reference proteomes for maize and Arabidopsis and evaluation of their orthologous relationships; the concept of orthoproteomics. J. Proteome Res. 12, 491–504. doi: 10.1021/pr300952g
Huang, W., Yu, M., Jiao, Y., Ma, J., Ma, M., Wang, Z., et al. (2011). Mitochondrial transcription termination factor 2 binds to entire mitochondrial DNA and negatively regulates mitochondrial gene expression. Acta Biochim. Biophys. Sin. 43, 472–479. doi: 10.1093/abbs/gmr035
Jan, H. U., Guan, M., Yao, M., Liu, W., Wei, D., Abbadi, A., et al. (2019). Genome-wide haplotype analysis improves trait predictions in Brassica napus hybrids. Plant Sci. 283, 157–164. doi: 10.1016/j.plantsci.2019.02.007
Jayakodi, M., Padmarasu, S., Haberer, G., Bonthala, V. S., Gundlach, H., Monat, C., et al. (2020). The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 588, 284–289. doi: 10.1038/s41586-020-2947-8
Jiang, D., Chen, J., Zhang, Z., and Hou, X. (2021). mitochondrial transcription termination factor 27 is required for salt tolerance in Arabidopsis thaliana. Int. J. Mol. Sci. 22. doi: 10.3390/ijms22031466
Jiang, D., Tang, R., Shi, Y., Ke, X., Wang, Y., Che, Y., et al. (2020). Arabidopsis seedling lethal 1 interacting with plastid-encoded RNA polymerase complex proteins is essential for chloroplast development. Front. Plant Sci. 11:602782. doi: 10.3389/fpls.2020.602782
Jin, J., Tian, F., Yang, D. C., Meng, Y. Q., Kong, L., Luo, J., et al. (2017). PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D1040–D1045. doi: 10.1093/nar/gkw982
Kilian, B., Ozkan, H., Kohl, J., von Haeseler, A., Barale, F., Deusch, O., et al. (2006). Haplotype structure at seven barley genes: relevance to gene pool bottlenecks, phylogeny of ear type and site of barley domestication. Mol. Genet. Genom. 276, 230–241. doi: 10.1007/s00438-006-0136-6
Kim, M., Lee, U., Small, I., des Francs-Small, C. C., and Vierling, E. (2012). Mutations in an Arabidopsis mitochondrial transcription termination factor-related protein enhance thermotolerance in the absence of the major molecular chaperone HSP101. Plant Cell 24, 3349–3365. doi: 10.1105/tpc.112.101006
Kleine, T. (2012). Arabidopsis thaliana mTERF proteins: evolution and functional classification. Front. Plant Sci. 3:233. doi: 10.3389/fpls.2012.00233
Kleine, T., and Leister, D. (2015). Emerging functions of mammalian and plant mTERFs. Biochim. Biophys. Acta 1847, 786–797. doi: 10.1016/j.bbabio.2014.12.009
Kruse, B., Narasimhan, N., and Attardi, G. (1989). Termination of transcription in human mitochondria: identification and purification of a DNA binding protein factor that promotes termination. Cell 58, 391–397. doi: 10.1016/0092-8674(89)90853-2
Kühn, K., Bohne, A. V., Liere, K., Weihe, A., and Börner, T. (2007). Arabidopsis phage-type RNA polymerases: accurate in vitro transcription of organellar genes. Plant Cell 19, 959–971. doi: 10.1105/tpc.106.046839
Lang, B. F., Gray, M. W., and Burger, G. (1999). Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 33, 351–397. doi: 10.1146/annurev.genet.33.1.351
Lee, C. P., Taylor, N. L., and Millar, A. H. (2013). Recent advances in the composition and heterogeneity of the Arabidopsis mitochondrial proteome. Front. Plant Sci. 4:4. doi: 10.3389/fpls.2013.00004
Lee, K., Leister, D., and Kleine, T. (2021). Arabidopsis mitochondrial transcription termination factor mTERF2 promotes splicing of group IIB introns. Cells 10:315. doi: 10.3390/cells10020315
Leister, D., and Kleine, T. (2020). Extending the repertoire of mTERF proteins with functions in organellar gene expression. Mol. Plant 13, 817–819. doi: 10.1016/j.molp.2020.04.003
Li, Y., Xiong, H., Cuo, D., Wu, X., and Duan, R. (2019). Genome-wide characterization and expression profiling of the relation of the HD-Zip gene family to abiotic stress in barley (Hordeum vulgare L.). Plant Physiol. Biochem. 141, 250–258. doi: 10.1016/j.plaphy.2019.05.026
Liere, K., Weihe, A., and Börner, T. (2011). The transcription machineries of plant mitochondria and chloroplasts: composition, function, and regulation. J. Plant Physiol. 168, 1345–1360. doi: 10.1016/j.jplph.2011.01.005
Linder, T., Park, C. B., Asin-Cayuela, J., Pellegrini, M., Larsson, N. G., Falkenberg, M., et al. (2005). A family of putative transcription termination factors shared amongst metazoans and plants. Curr. Genet. 48, 265–269. doi: 10.1007/s00294-005-0022-5
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, X., Fang, Y., Tian, B., Tong, T., Wang, J., Wang, H., et al. (2019). Genetic variation of HvXYN1 associated with endoxylanase activity and TAX content in barley (Hordeum vulgare L.). BMC Plant Biol. 19:170. doi: 10.1186/s12870-019-1747-5
Lynch, M., and Conery, J. S. (2000). The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155. doi: 10.1126/science.290.5494.1151
Mascher, M., Gundlach, H., Himmelbach, A., Beier, S., Twardziok, S. O., Wicker, T., et al. (2017). A chromosome conformation capture ordered sequence of the barley genome. Nature 544, 427–433. doi: 10.1038/nature22043
Mayer, K. F., Waugh, R., Brown, J. W., Schulman, A., Langridge, P., Platzer, M., et al. (2012). A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716. doi: 10.1038/nature11543
Meskauskiene, R., Würsch, M., Laloi, C., Vidi, P. A., Coll, N. S., Kessler, F., et al. (2009). A mutation in the Arabidopsis mTERF-related plastid protein SOLDAT10 activates retrograde signaling and suppresses (1)O(2)-induced cell death. Plant J. 60, 399–410. doi: 10.1111/j.1365-313X.2009.03965.x
Méteignier, L. V., Ghandour, R., Meierhoff, K., Zimmerman, A., Chicher, J., Baumberger, N., et al. (2020). The Arabidopsis mTERF-repeat MDA1 protein plays a dual function in transcription and stabilization of specific chloroplast transcripts within the psbE and ndhH operons. New Phytol. 227, 1376–1391. doi: 10.1111/nph.16625
Monat, C., Padmarasu, S., Lux, T., Wicker, T., Gundlach, H., Himmelbach, A., et al. (2019). TRITEX: chromosome-scale sequence assembly of Triticeae genomes with open-source tools. Genome Biol. 20:284. doi: 10.1186/s13059-019-1899-5
Núñez-Delegido, E., Robles, P., Ferrández-Ayela, A., and Quesada, V. (2020). Functional analysis of mTERF5 and mTERF9 contribution to salt tolerance, plastid gene expression and retrograde signalling in Arabidopsis thaliana. Plant Biol. 22, 459–471. doi: 10.1111/plb.13084
Pan, Z., Ren, X., Zhao, H., Liu, L., Tan, Z., and Qiu, F. (2019). A mitochondrial transcription termination factor, ZmSmk3, is required for nad1 Intron4 and nad4 intron1 splicing and kernel development in maize. G3 9, 2677–2686. doi: 10.1534/g3.119.400265
Park, C. B., Asin-Cayuela, J., Cámara, Y., Shi, Y., Pellegrini, M., Gaspari, M., et al. (2007). MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell 130, 273–285. doi: 10.1016/j.cell.2007.05.046
Pellegrini, M., Asin-Cayuela, J., Erdjument-Bromage, H., Tempst, P., Larsson, N. G., and Gustafsson, C. M. (2009). MTERF2 is a nucleoid component in mammalian mitochondria. Biochim. Biophys. Acta 1787, 296–302. doi: 10.1016/j.bbabio.2009.01.018
Pfannschmidt, T., Blanvillain, R., Merendino, L., Courtois, F., Chevalier, F., Liebers, M., et al. (2015). Plastid RNA polymerases: orchestration of enzymes with different evolutionary origins controls chloroplast biogenesis during the plant life cycle. J. Exp. Bot. 66, 6957–6973. doi: 10.1093/jxb/erv415
Poets, A. M., Fang, Z., Clegg, M. T., and Morrell, P. L. (2015). Barley landraces are characterized by geographically heterogeneous genomic origins. Genome Biol. 16:173. doi: 10.1186/s13059-015-0712-3
Purugganan, M. D. (2019). Evolutionary insights into the nature of plant domestication. Curr. Biol. 29, R705–R714. doi: 10.1016/j.cub.2019.05.053
Purugganan, M. D., and Fuller, D. Q. (2009). The nature of selection during plant domestication. Nature 457, 843–848. doi: 10.1038/nature07895
Quesada, V. (2016). The roles of mitochondrial transcription termination factors (MTERFs) in plants. Physiol. Plant. 157, 389–399. doi: 10.1111/ppl.12416
Quesada, V., Sarmiento-Mañús, R., González-Bayón, R., Hricová, A., Pérez-Marcos, R., Graciá-Martínez, E., et al. (2011). Arabidopsis RUGOSA2 encodes an mTERF family member required for mitochondrion, chloroplast and leaf development. Plant J. 68, 738–753. doi: 10.1111/j.1365-313X.2011.04726.x
Roberti, M., Bruni, F., Polosa, P. L., Gadaleta, M. N., and Cantatore, P. (2006). The Drosophila termination factor DmTTF regulates in vivo mitochondrial transcription. Nucleic Acids Res. 34, 2109–2116. doi: 10.1093/nar/gkl181
Roberti, M., Polosa, P. L., Bruni, F., Manzari, C., Deceglie, S., Gadaleta, M. N., et al. (2009). The MTERF family proteins: mitochondrial transcription regulators and beyond. Biochim. Biophys. Acta 1787, 303–311. doi: 10.1016/j.bbabio.2009.01.013
Robles, P., Micol, J. L., and Quesada, V. (2012). Arabidopsis MDA1, a nuclear-encoded protein, functions in chloroplast development and abiotic stress responses. PLoS ONE 7:e42924. doi: 10.1371/journal.pone.0042924
Robles, P., Micol, J. L., and Quesada, V. (2015). Mutations in the plant-conserved MTERF9 alter chloroplast gene expression, development and tolerance to abiotic stress in Arabidopsis thaliana. Physiol. Plant. 154, 297–313. doi: 10.1111/ppl.12307
Robles, P., Navarro-Cartagena, S., Ferrández-Ayela, A., Núñez-Delegido, E., and Quesada, V. (2018a). The characterization of Arabidopsis mterf6 mutants reveals a new role for mTERF6 in tolerance to abiotic stress. Int. J. Mol. Sci. 19:2388. doi: 10.3390/ijms19082388
Robles, P., Núñez-Delegido, E., Ferrández-Ayela, A., Sarmiento-Mañús, R., Micol, J. L., and Quesada, V. (2018b). Arabidopsis mTERF6 is required for leaf patterning. Plant Sci. 266, 117–129. doi: 10.1016/j.plantsci.2017.11.003
Robles, P., and Quesada, V. (2021). Research progress in the molecular functions of plant mTERF proteins. Cells 10:205. doi: 10.3390/cells10020205
Romani, I., Manavski, N., Morosetti, A., Tadini, L., Maier, S., Kühn, K., et al. (2015). A member of the arabidopsis mitochondrial transcription termination factor family is required for maturation of chloroplast transfer RNAIle(GAU). Plant Physiol. 169, 627–646. doi: 10.1104/pp.15.00964
Russell, J., Mascher, M., Dawson, I. K., Kyriakidis, S., Calixto, C., Freund, F., et al. (2016). Exome sequencing of geographically diverse barley landraces and wild relatives gives insights into environmental adaptation. Nat. Genet. 48, 1024–1030. doi: 10.1038/ng.3612
Schulte, D., Close, T. J., Graner, A., Langridge, P., Matsumoto, T., Muehlbauer, G., et al. (2009). The international barley sequencing consortium–at the threshold of efficient access to the barley genome. Plant Physiol. 149, 142–147. doi: 10.1104/pp.108.128967
Shi, Q., Zhang, Y., To, V. T., Shi, J., Zhang, D., and Cai, W. (2020). Genome-wide characterization and expression analyses of the auxin/indole-3-acetic acid (Aux/IAA) gene family in barley (Hordeum vulgare L.). Sci. Rep. 10:10242. doi: 10.1038/s41598-020-66860-7
Shinozaki, K., and Yamaguchi-Shinozaki, K. (1997). Gene expression and signal transduction in water-stress response. Plant Physiol. 115, 327–334. doi: 10.1104/pp.115.2.327
Spåhr, H., Habermann, B., Gustafsson, C. M., Larsson, N. G., and Hallberg, B. M. (2012). Structure of the human MTERF4-NSUN4 protein complex that regulates mitochondrial ribosome biogenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 15253–15258. doi: 10.1073/pnas.1210688109
Tang, B., Xie, L., Yi, T., Lv, J., Yang, H., Cheng, X., et al. (2019). Genome-wide identification and characterization of the mitochondrial transcription termination factors (mTERFs) in Capsicum annuum L. Int. J. Mol. Sci. 21:269. doi: 10.3390/ijms21010269
Terzioglu, M., Ruzzenente, B., Harmel, J., Mourier, A., Jemt, E., López, M. D., et al. (2013). MTERF1 binds mtDNA to prevent transcriptional interference at the light-strand promoter but is dispensable for rRNA gene transcription regulation. Cell Metab. 17, 618–626. doi: 10.1016/j.cmet.2013.03.006
Uçarli, C., McGuffin, L. J., Çaputlu, S., Aravena, A., and Gürel, F. (2016). Genetic diversity at the Dhn3 locus in Turkish Hordeum spontaneum populations with comparative structural analyses. Sci. Rep. 6:20966. doi: 10.1038/srep20966
Urao, T., Yamaguchi-Shinozaki, K., Urao, S., and Shinozaki, K. (1993). An Arabidopsis myb homolog is induced by dehydration stress and its gene product binds to the conserved MYB recognition sequence. Plant Cell 5, 1529–1539. doi: 10.1105/tpc.5.11.1529
Wei, K., and Chen, H. (2018). Comparative functional genomics analysis of bHLH gene family in rice, maize and wheat. BMC Plant Biol. 18:309. doi: 10.1186/s12870-018-1529-5
Wredenberg, A., Lagouge, M., Bratic, A., Metodiev, M. D., Spåhr, H., Mourier, A., et al. (2013). MTERF3 regulates mitochondrial ribosome biogenesis in invertebrates and mammals. PLoS Genet. 9:e1003178. doi: 10.1371/journal.pgen.1003178
Wright, S. (1951). The genetical structure of populations. Ann. Eugen. 15, 323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x
Xia, Y., Li, R., Bai, G., Siddique, K. H. M., Varshney, R. K., Baum, M., et al. (2017). Genetic variations of HvP5CS1 and their association with drought tolerance related traits in barley (Hordeum vulgare L.). Sci. Rep. 7:7870. doi: 10.1038/s41598-017-08393-0
Xiong, H. B., Wang, J., Huang, C., Rochaix, J. D., Lin, F. M., Zhang, J. X., et al. (2020). mTERF8, a member of the mitochondrial transcription termination factor family, is involved in the transcription termination of chloroplast gene psbJ. Plant Physiol. 182, 408–423. doi: 10.1104/pp.19.00906
Xu, D., Leister, D., and Kleine, T. (2017). Arabidopsis thaliana mTERF10 and mTERF11, but Not mTERF12, are involved in the response to salt stress. Front. Plant Sci. 8:1213. doi: 10.3389/fpls.2017.01213
Yagi, Y., and Shiina, T. (2012). Evolutionary aspects of plastid proteins involved in transcription: the transcription of a tiny genome is mediated by a complicated machinery. Transcription 3, 290–294. doi: 10.4161/trns.21810
Yakubovskaya, E., Guja, K. E., Mejia, E., Castano, S., Hambardjieva, E., Choi, W. S., et al. (2012). Structure of the essential MTERF4:NSUN4 protein complex reveals how an MTERF protein collaborates to facilitate rRNA modification. Structure 20, 1940–1947. doi: 10.1016/j.str.2012.08.027
Yang, Y. Z., Ding, S., Wang, Y., Wang, H. C., Liu, X. Y., Sun, F., et al. (2020). PPR20 is required for the cis-splicing of mitochondrial nad2 intron 3 and seed development in maize. Plant Cell Physiol. 61, 370–380. doi: 10.1093/pcp/pcz204
Yin, X., Gao, Y., Song, S., Hassani, D., and Lu, J. (2021). Identification, characterization and functional analysis of grape (Vitis vinifera L.) mitochondrial transcription termination factor (mTERF) genes in responding to biotic stress and exogenous phytohormone. BMC Genomics 22:136. doi: 10.1186/s12864-021-07446-z
Zhang, M., Kim, Y., Zong, J., Lin, H., Dievart, A., Li, H., et al. (2019). Genome-wide analysis of the barley non-specific lipid transfer protein gene family. Crop J. 7, 65–76. doi: 10.1016/j.cj.2018.07.009
Zhang, Y., Cui, Y. L., Zhang, X. L., Yu, Q. B., Wang, X., Yuan, X. B., et al. (2018). A nuclear-encoded protein, mTERF6, mediates transcription termination of rpoA polycistron for plastid-encoded RNA polymerase-dependent chloroplast gene expression and chloroplast development. Sci. Rep. 8:11929. doi: 10.1038/s41598-018-30166-6
Keywords: barley, mTERF gene family, duplication, expression profile, qRT-PCR, genetic variation
Citation: Li T, Pan W, Yuan Y, Liu Y, Li Y, Wu X, Wang F and Cui L (2021) Identification, Characterization, and Expression Profile Analysis of the mTERF Gene Family and Its Role in the Response to Abiotic Stress in Barley (Hordeum vulgare L.). Front. Plant Sci. 12:684619. doi: 10.3389/fpls.2021.684619
Received: 09 April 2021; Accepted: 23 June 2021;
Published: 15 July 2021.
Edited by:
Hakan Ozkan, Çukurova University, TurkeyReviewed by:
Huseyin Tombuloglu, Imam Abdulrahman Bin Faisal University, Saudi ArabiaVictor Quesada, Miguel Hernández University of Elche, Spain
Copyright © 2021 Li, Pan, Yuan, Liu, Li, Wu, Wang and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Licao Cui, Y3VpbGljYW9AanhhdS5lZHUuY24=
†These authors have contributed equally to this work