 Kazuki Fujiwara1,2*
Kazuki Fujiwara1,2* Yuki O. Kobayashi3Manami Usui4Kazuya Nishioka5Misa Nakamura6Shinji Kawano6Yoshihiro Okada7Akira Kobayashi3Atsushi Miyasaka2Kazuyuki Hirayae2Yoshiyuki Kushima4Yatsuka Nishi5Hiroyoshi Inoue2
Yuki O. Kobayashi3Manami Usui4Kazuya Nishioka5Misa Nakamura6Shinji Kawano6Yoshihiro Okada7Akira Kobayashi3Atsushi Miyasaka2Kazuyuki Hirayae2Yoshiyuki Kushima4Yatsuka Nishi5Hiroyoshi Inoue2- 1Institute for Plant Protection, National Agriculture and Food Research Organization (NARO), Tsukuba, Japan
- 2Division of Agro-Environment Research, Kyushu Okinawa Agricultural Research Center, NARO, Koshi, Japan
- 3Division of Upland Farming Research, Kyushu Okinawa Agricultural Research Center, NARO, Miyakonojo, Japan
- 4Miyazaki Agricultural Experiment Station, Miyazaki, Japan
- 5Kagoshima Prefectural Institute for Agricultural Development, Kagoshima, Japan
- 6Okinawa Prefectural Agricultural Research Center, Itoman, Japan
- 7Division of Agro-Environment Research, Kyushu Okinawa Agricultural Research Center, NARO, Itoman, Japan
Foot rot disease caused by Diaporthe destruens (formerly Plenodomus destruens) has become a major concern for the production of sweet potato [Ipomoea batatas (L.) Lam.] in Japan. A related fungus Diaporthe batatas, which causes dry rot disease of sweet potato, is native and is widespread in fields in Japan. The similar characteristics of these two pathogens pose a challenge for conventional disease diagnosis. Currently, there are no effective molecular measures for identifying and distinguishing D. destruens and D. batatas. Here, we demonstrate a real-time PCR assay that distinguishes and quantifies D. batatas and D. destruens from co-infected sweet potato. The assay was performed with various simulated DNA combinations of D. batatas and D. destruens ranging from 1:1 to 1:100000. The assay was also used with the ratios of D. batatas: D. destruens: sweet potato DNA ranging from 1:1:1 to 1:1:100000. These assays produced a specific amplification product for each of the pathogens, and quantified the fungal biomass over the entire range tested without detecting false positives. The assay was validated by using infected sweet potato collected from various fields; it showed sufficient sensitivity and specificity to quantify and distinguish D. batatas and D. destruens from these field samples. Thus, our real-time PCR assay would be a useful tool for diagnosis of D. batatas and D. destruens and is expected to provide the foundation for the design of integrated disease management strategies for foot rot disease in sweet potato.
Introduction
Foot rot is an important fungal disease in sweet potato [Ipomoea batatas (L.) Lam.]. The disease was first observed in Virginia, United States (Harter,1913a,b; Harter and Weimer, 1929) and is widespread in some countries in North and South America (Lopes et al., 1994; Clark et al., 2013), eastern Africa (Steinbauer and Kushman, 1971; Skoglund and Smit, 1994), Asia (Huang et al., 2012; Gai et al., 2016; Paul et al., 2019), and New Zealand (Liu et al., 2014). The causal pathogen is Diaporthe destruens (Harter) Hirooka, Minosh, and Rossman [formerly Plenodomus destruens Harter; syn. Phomopsis destruens (Harter) Boerema, Loerakker, and Hamers). Foot rot infection in sweet potato causes black stem, wilting, and plant death due to stem girdling. Root rot is often developed in storage roots. Severe infection involves hardening of the black stem lesion and rotten tubers in the soil. In Japan, foot rot disease was first observed in Southern Kyushu and Okinawa regions (Kobayashi, 2019) including Okinawa and Kagoshima Prefectures in 2018, and Miyazaki Prefecture in 2019. In sweet potato cultivation, development of foot rot disease is often indistinguishable from that of other soil-borne diseases including dry rot disease caused by Diaporthe batatas Harter & E.C. Field (Udayanga et al., 2015), Fusarium wilt of sweet potato caused by several Fusarium spp. such F. acuminatum, F. incarnatum, F. oxysporum, F. proliferatum, and F. solani (Scruggs and Quesada-Ocampo, 2016), and bacterial wilt and root rot of sweet potato caused by Dicheya spp. (syn. Erwinia chrysanthemi; Schaad and Brenner, 1977). The complex of symptoms often masks the emergence of foot rot disease. Of the above pathogens, D. batatas rarely causes serious damage to sweet potatoes in the field; however, even if the storage roots look healthy at harvesting, the infection triggers further disease development, similar to that caused by D. destruens, in the roots during storage. Thus, whereas foot rot disease manifests in the field, the damage increases post-harvesting in cases of co-infection with D. batatas and D. destruens. This co-infection makes it more difficult to identify a causative pathogen, and hinders the preparation of seed tubers for planting in the following year. Thus, we need a novel molecular measure to effectively identify and distinguish D. batatas and D. destruens.
Molecular methods for rapid detection and identification of causative pathogens are powerful tools in disease diagnosis. The internal transcribed spacer (ITS) regions of ribosomal DNA (rDNA) are commonly used in the identification and phylogenetic study of fungi (Chase and Fay, 2009; Seifert, 2009). PCR-based methods using the ITS sequences have been successfully used to identify Diaporthe spp. and Phomopsis spp. (Baumgartner et al., 2013; Udayanga et al., 2014), which are difficult to discriminate by morphological features alone (Hosseini et al., 2020). Currently, conventional PCR (Lin et al., 2017) and loop-mediated isothermal amplification (LAMP; Maruyama et al., 2020) are the only techniques available for diagnosis of D. destruens, but there are technical concerns about their sensitivity and specificity for assessing infection with D. destruens. There is currently no robust method for disease management of D. destruens, and the disease management relies on pathogen diagnostic measures to determine the cause of disease and degree of infection for informing the decision-making in countermeasures. Therefore, development of an efficient molecular diagnostic tool for foot rot disease is essential. To this end, we have established a real-time PCR technique for reliable evaluation of both D. batatas and D. destruens in co-infected sweet potato tissues.
Materials and Methods
Pathogens Used in This Study
Sweet potato seedlings and storage roots with appearance of foot rot symptoms were collected from several local fields in the Miyazaki, Kagoshima, and Okinawa Prefectures, Japan, in 2018. Infected plant materials were washed with sterilized distilled water, and 1-cm3 pieces of the materials were cut and plated onto potato dextrose agar (PDA) plates (BD, NJ, United States) with 50 μg mL–1 of ampicillin (Sigma, Munich, Germany) or sweet potato dextrose agar (SPDA) plates [50% sweet potato filtrate obtained by boiling 500 g diced sweet potato in 1,000 mL distilled water and filtering, 20 g glucose (FUJIFILM Wako, Tokyo, Japan), and 15 g agar (FUJIFILM Wako, Tokyo, Japan)]. After 2 weeks, small fungal colonies were picked with a sterilized toothpick and transferred to fresh PDA or SPDA plates. Fungal colonies were subjected to Diaporthe species identification based on morphological characteristics and DNA sequencing. Single spores of D. destruens and D. batatas were isolated under a light microscope, and isolates were cultured as described above, providing 11 D. batatas isolates and 17 D. destruens isolates (Supplementary Table 1). We also used the following Diaporthe and Phomopsis species provided from the National Agriculture and Food Research Organization (NARO) Genebank1 : D. gardeniae (MAFF 410870), D. nobilis (MAFF 245916), D. santonensis (MAFF 410114), P. asparagi (MAFF 150068), P. cucurbitae (MAFF 410452), P. fukushii (MAFF 625033), P. macrospora (MAFF 410314), P. velata (MAFF 410118), and P. vexans (MAFF 150147). These fungal species were maintained on PDA or SPDA plates until use. To prepare culture plate samples for DNA extraction, single fungal disks were taken from the culture plates by a 5-mm cork borer, and then suspended in AP1 buffer [a component of the DNeasy Plant Mini Kit (Qiagen, Venlo, Netherlands)]. The samples were then stored at −20°C until use.
Isolates of D. destruens were cultured on SPDA plates at 25°C in dark for 5 days and then exposed to black light radiation for 2 weeks by using an FL20S black light (Toshiba, Tokyo, Japan) to induce sporulation. Alpha-conidia were collected from the culture plates and suspended in sterilized distilled water. The conidia were adjusted to a density of ∼1 × 107 cells mL–1 by using a hemocytometer under a light microscope. Collection of D. batatas conidia was performed as described for D. destruens conidia. The conidial suspensions were stored at −20°C until use.
DNA Extraction
All prepared samples were disrupted in a Multi-Bead Shocker (Yasui Kikai, Osaka, Japan) with 1 mm diameter zirconia beads (Sarstedt, Nümbrecht, Germany) at 3,000 rpm for 45 s, and the disrupted samples were chilled on ice for 3 min. This procedure was performed three times. DNA from conidial suspensions, culture plates, and plant materials were extracted with a DNeasy Plant Mini Kit or DNeasy Plant Maxi Kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s instructions. In conidial and culture plate samples, three and five biological replicates, respectively, were used to prepare DNA templates. Each of the DNA templates was adjusted to 5 ng μL–1. The DNA templates from conidial samples were used to prepare a 10-fold dilution series of DNA from 5 to 0.00005 ng μL–1.
To assess detection efficiency in co-infection of D. batatas and D. destruens, a fixed concentration of fungal DNA of one pathogen (5 ng μL–1) was mixed with a progressively decreasing DNA concentration of the other pathogen (5, 0.5, 0.05, 0.005, 0.0005, and 0.00005 ng μL–1). This provided a ratio of DNA concentrations in the range from 1:1 down to 1:100000. To simulate co-infection with sweet potato, an increasing concentration of sweet potato DNA (5 to 50 ng μL–1) was mixed with a decreasing concentration of D. batatas and D. destruens DNA (5 to 0.0005 ng μL–1 each). This generated a ratio of DNA concentrations of D. batatas: D. destruens: sweet potato from 1:1:1 to 1:1:100000. For preparation of DNA templates, D. batatas strain MOKM-3S-B and D. destruens strain KTJ-1R-a were used as representative strains. A sweet potato DNA template was prepared from healthy tubers.
Development and Specificity of Primers
We designed new primer sets Db ITS and Dd ITS (Table 1) from ITS1 and ITS2 of rDNA by comparison of the DNA sequences of D. batatas (KU577616, MG827239, and NR_152456) and D. destruens (JN848791, MH465671, MH465672, and MH465673) obtained from the NCBI database and the DNA sequences of 11 D. batatas isolates and 17 D. destruens isolates. The primer design was carried out using Primer-BLAST2.

Table 1. Primers used in this study.
Primer specificity was tested in both conventional PCR and real-time PCR. Db ITS and Dd ITS primers were tested against closely related Diaporthe species and Phomopsis species as well as D. batatas and D. destruens. Template DNA was prepared from culture plates of each fungal species as described above. In conventional PCR, a single PCR mixture was prepared by mixing three biological replicates of each of fungal pathogen DNA (5 ng μL–1). Real-time PCR reactions were prepared from three biological replicates of each of fungal pathogen DNA.
Conventional PCR and Real-Time PCR Conditions
For conventional PCR with Db ITS or Dd ITS primer sets, we used TaKaRa Ex Taq Hot Start Version (Takara, Tokyo, Japan) according to the manufacturer’s instructions. Reaction mixtures contained 0.1 μL of TaKaRa Ex Taq HS, 2 μL of 10 × Ex Taq Buffer, 1.6 μL of dNTP Mixture, 0.5 μM of each primer (Forward/Reverse), and 2 μL of DNA template in a final volume of 20 μL. The conventional PCR protocol consisted of initial denaturation at 94°C for 2 min, and 32 cycles of denaturation at 98°C for 10 s, annealing at 55°C for 30 s, and extension at 72°C for 1 min. The PCR products were detected by 1% agarose gel electrophoresis. We also used a SPPD primer set (Table 1) for conventional PCR as previously reported (Lin et al., 2017); these PCR products were also detected by 1% agarose gel electrophoresis.
Real-time PCR was performed in a QuantStudio 5 real-time PCR system (Thermo Fisher Scientific, MA, United States) using SYBR Premix ExTaq II (Takara, Tokyo, Japan) according to the manufacturer’s protocol. Reaction mixtures contained 1 × TB Green Premix Ex Taq II, 1 × ROX Reference Dye II, 0.4 μM of each primer (Forward/Reverse), and 2 μL of DNA template in a final volume of 20 μL. The real-time PCR protocol consisted of initial denaturation at 94°C for 1 min, and 32 cycles of denaturation at 96°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s. Further denaturation at 96°C for 15 s, holding at 55°C for 1 min, and heating from 55 to 96°C for 15 s were carried out for melting curve analysis. In both conventional and real-time PCR, stem and tuberous DNA of healthy sweet potato plants as well as no template were used as negative controls.
Validation of Real-Time PCR Assay
The simulation of standard curve using a 10-fold dilution series of DNA from 5 to 0.00005 ng μL–1 was carried out. The regression equations and R2 coefficient of determination for quantification cycle (Cq) versus the natural logarithm of concentration of amplified genomic DNA (gDNA) were calculated. The amplification efficiency (%) was calculated by the following equation: [10(–1/(slope)) − 1] × 100 based on logarithm of gDNA concentration.
DNA Sequencing
Fungal isolates were subjected to DNA sequencing analysis. Culture plates of each isolate were used for DNA extraction as described above. Conventional PCR was performed using a universal ITS1 and ITS4 primer set (White et al., 1990; Table 1), which amplifies the ITS regions in fungi; the PCR protocol was initial denaturation at 95°C for 4 min, followed by 35 cycles of denaturation at 95°C for 1 min, annealing at 56°C for 1 min, and extension at 72°C for 2 min, with a final extension for 10 min at 72°C. Each conventional PCR product was purified with the NucleoSpin Gel and PCR Clean-up (Takara, Tokyo, Japan). DNA sequencing was performed in an ABI PRISM 3100-Avant Genetic Analyzer (Thermo Fisher Scientific, MA, United States) using the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, MA, United States). Unincorporated dye terminators were removed with NucleoSEQ (Takara, Tokyo, Japan). Sequences were analyzed by BLAST3.
Collection of Sweet Potato Seedlings in the Field
Sweet potato plants were collected from 37 sites in the Kyushu and Okinawa regions where foot rot disease appeared under natural conditions: two from Fukuoka Prefecture, 17 from Kagoshima Prefecture, four from Kumamoto Prefecture, three from Nagasaki Prefecture, six from Miyazaki Prefecture, and five from Okinawa Prefecture. Stems and tubers with or without symptoms were harvested from June to October in 2019 and 2020. In total, 55 seedlings and 35 tubers from the above regions were obtained. We generated 77 samples from stems and 83 samples from tubers of the obtained plants. For processing, stems and tubers were washed with distilled water, and their surfaces were sterilized by wiping with a Kimwipe paper soaked with 70% ethanol before use. The stem tissues were cut into small pieces of ∼5-mm width. The tubers were cut into 1-cm3 pieces including the skin tissue. Each processed sample was kept in a separate plastic bag. For DNA extraction, 50 mg of plant material was ground in AP1 buffer with a mortar and pestle. The collected samples and homogenized samples were stored at −20°C until use. For both stem and tubers, the diagnostic index was scored on a scale of 0 to 2 by real-time PCR detection using the primer sets Db ITS and Dd ITS (0, no symptoms with no pathogen detected; 1, symptomless but pathogen detected; and 2, symptomatic with pathogen detected).
Statistical Analysis
The differences in relative frequency of pathogen detection among symptomless and symptomatic sweet potato plants were assessed by the Fisher’s exact test. The differences in pathogen concentration measured as Cq value among various categories in the diagnostic index were analyzed by Steel–Dwass test. Statistical analysis was performed with R v. 3.5.0 software, and P values < 0.01 were classified as statistically significant.
Results
Primer Specificity and Validation of Real-Time PCR
We tested the primer specificity against D. batatas and D. destruens in conventional PCR and real-time PCR. Our newly developed primers for D. batatas and D. destruens (Supplementary Figure 1) yielded different products for each pathogen in conventional PCR, i.e., a 317 bp fragment of Db ITS and a 258 bp fragment of Dd ITS (Figure 1). Conventional PCR amplifications with Db ITS and Dd ITS primers were successful for conidial and culture plate DNA of all D. batatas and D. destruens isolates that we obtained (K. Fujiwara, unpublished data). In contrast, the previously reported SPPD primer set (Lin et al., 2017) did not distinguish D. batatas from D. destruens (Figure 1). Among the tested species of the family Diaporthaceae, the Db ITS and Dd ITS primers were specific to D. batatas and D. destruens, respectively (Supplementary Figure 2). In real-time PCR, both Db ITS and Dd ITS primers yielded amplicons specific to D. batatas and D. destruens, respectively (Supplementary Figure 3). A single melting temperature (Tm) peak indicated that D. batatas–specific amplicons were detected at 89.3 (±0.4) Tm value by Db ITS and D. destruens–specific amplicons were detected at 86.9 (±0.4) Tm value by Dd ITS. Amplification of a 10-fold DNA dilution series (5 to 0.00005 ng DNA μL–1) showed a linear relationship between DNA concentration and Cq value for both Db ITS and Dd ITS primers, but the quantification was limited to concentrations of <0.0005 ng μL–1 in both pathogens (Figure 2). In the detection of D. batatas, Db ITS produced specific amplicons from 13.7 (±0.2) to 28.2 (±0.2) cycles, and Dd ITS produced specific amplicons from 16.6 (±0.3) to 31.4 (±0.1) cycles in the dilution series. The amplification efficiency was found to be 87.4% in Db ITS and 84.3% in Dd ITS. Both primers produced no false positives.
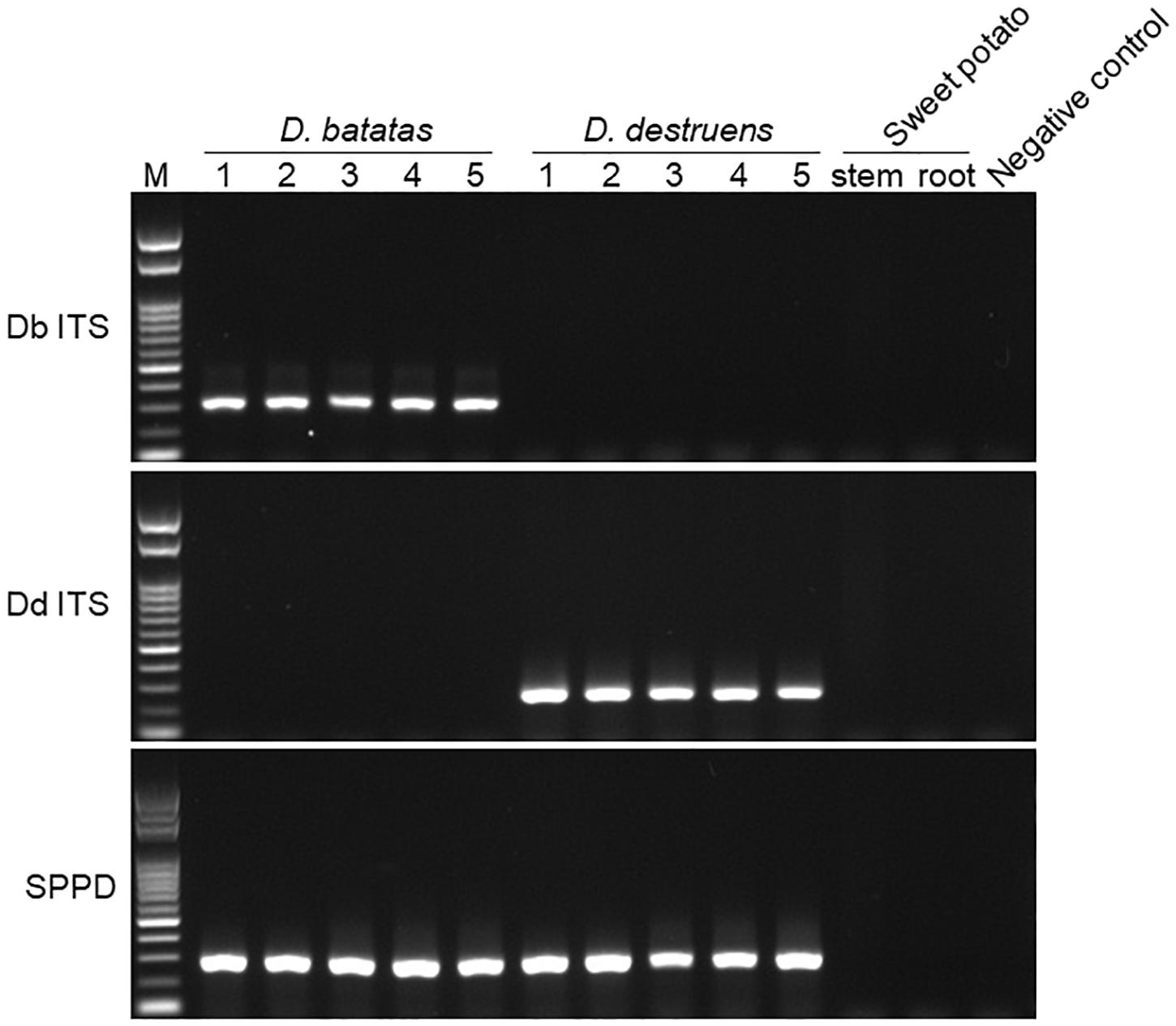
Figure 1. Amplification of Diaporthe batatas and Diaporthe destruens DNA by conventional PCR. Newly developed primer sets Db ITS and Dd ITS were tested against D. batatas and D. destruens DNA, respectively, in five individual PCR amplifications. The SPPD primer set previously reported was used for comparison. Template DNA (5 ng μL–1) were prepared from culture plates of D. batatas strain MOKM-3S-B and D. destruens strain KTJ-1R-a, respectively. Stem and tuberous DNA of healthy sweet potato plants as well as no template were used as negative controls. Amplicons produced by Db ITS, Dd ITS, and SPPD primer sets were 317, 258, and 289 bp, respectively. M, marker.
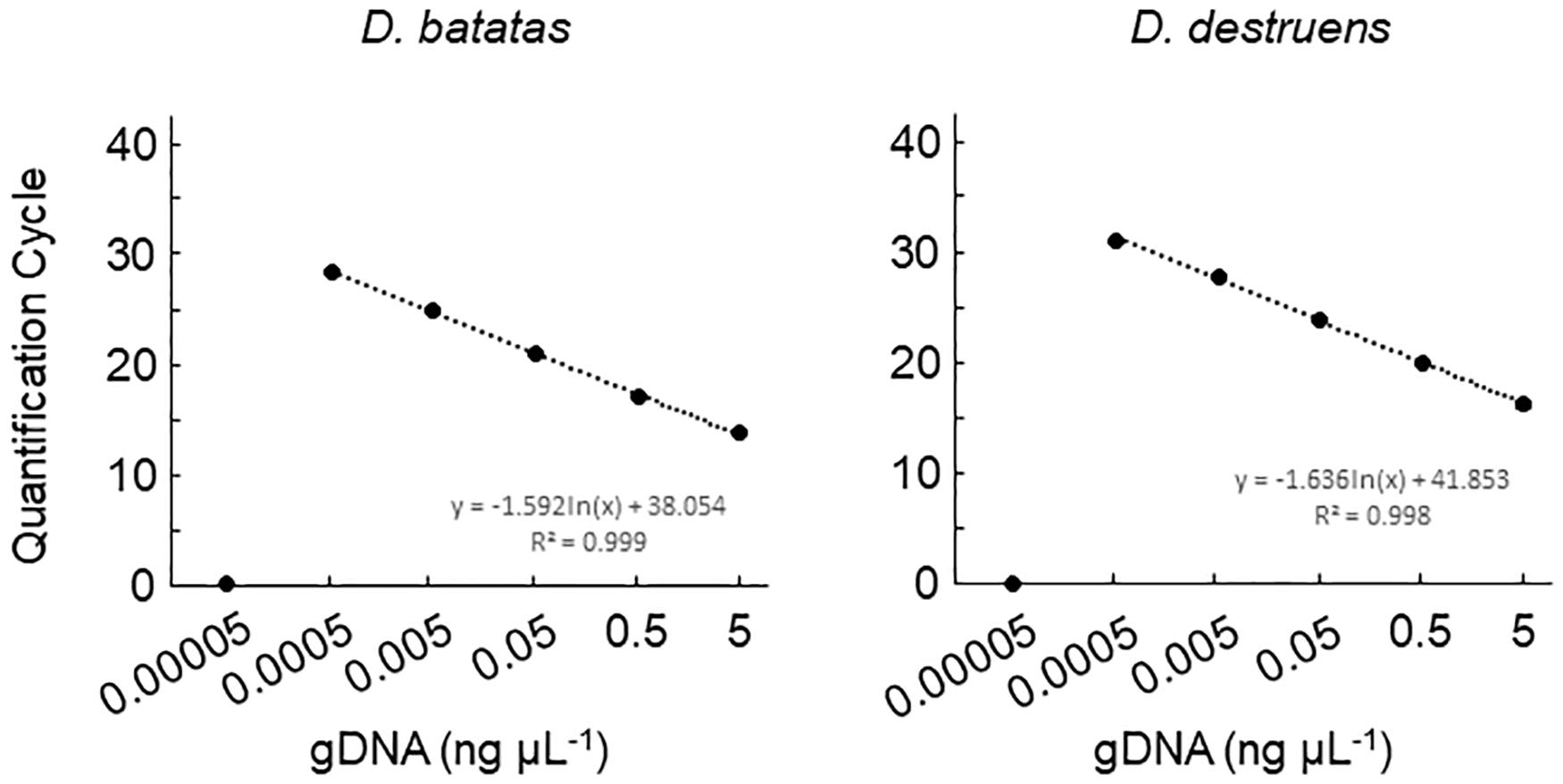
Figure 2. Amplification of Diaporthe batatas and Diaporthe destruens DNA by real-time PCR with Db ITS and Dd ITS primers. Template DNA was prepared from conidial samples separately from D. batatas strain MOKM-3S-B and D. destruens strain KTJ-1R-a, and were used to prepare 10-fold DNA dilution series of DNA (5.0, 0.5, 0.05, 0.005, 0.0005, and 0.00005 ng μL–1). Three biological replicates of each dilution were prepared. The regression equations and R2 coefficient of determination for Cq value versus the natural logarithm of concentration of amplified gDNA are shown on the chart. Data represent means ± standard deviation where visible.
Detection Efficiencies of D. batatas and D. destruens DNA in a Simulated Co-infection
We next investigated the detection efficiencies of Db ITS and Dd ITS primers against the DNA of D. batatas and D. destruens under different DNA mixing ratios by decreasing the DNA concentration of one species while keeping the DNA concentration of the other species constant. Both Db ITS and Dd ITS primers were capable of quantifying each of their target pathogen DNA down to a ratio of 1:10000 (Figure 3A). Db ITS amplified D. batatas DNA fragments from 13.9 (±0.3) to 30.2 (±2.0) cycles and Dd ITS yielded amplicons from 16.5 (±0.2) to 31.3 (±0.5) cycles in the range of 1:1 to 1:10000. On the other hand, quantification beyond 1:10000 was limited in both primer sets. There were no false positives for either Db ITS or Dd ITS primers against the mixed DNA.
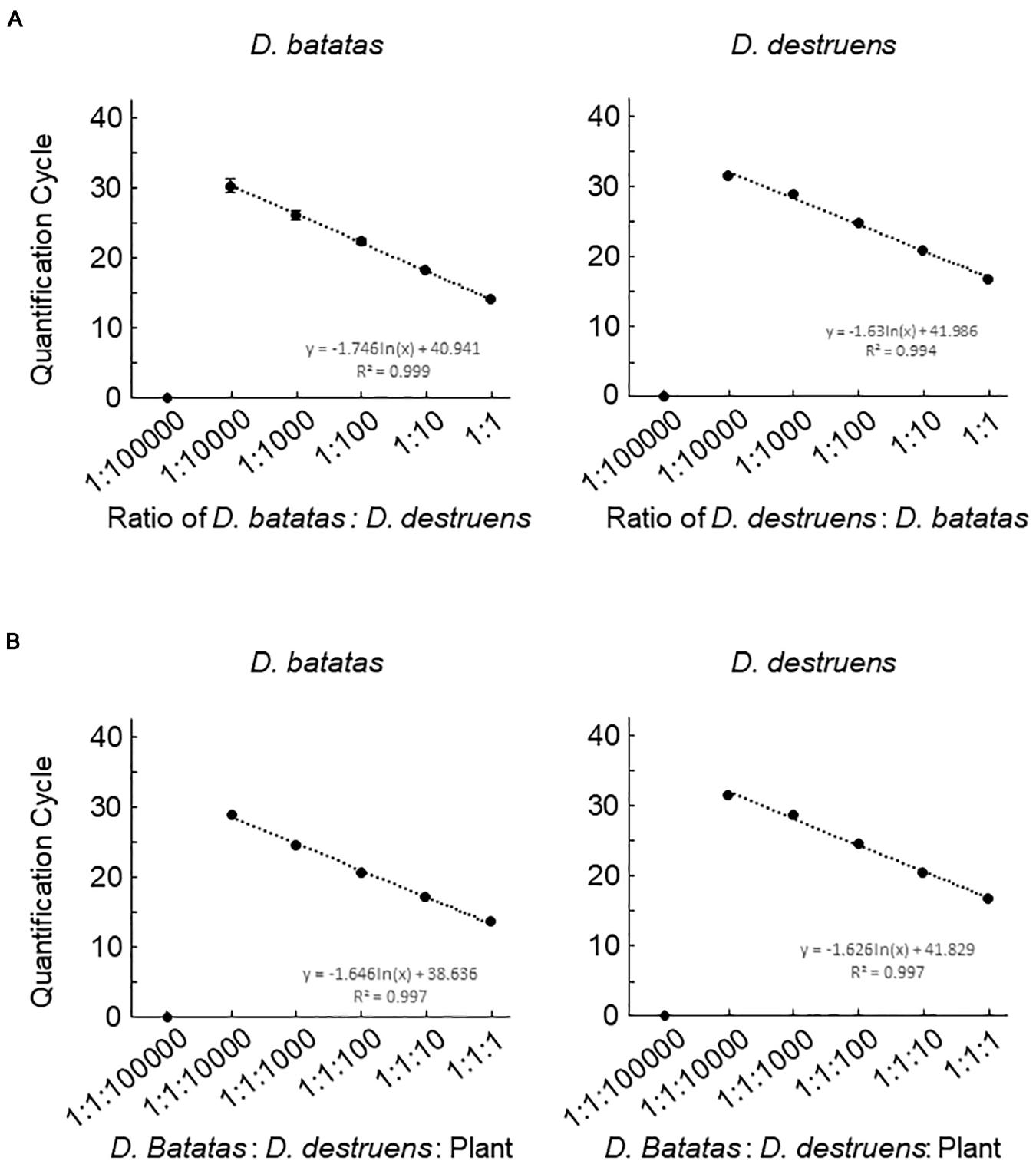
Figure 3. Detection efficiencies of the assay for Diaporthe batatas and Diaporthe destruens DNA in a simulated co-infection. Coinfection was simulated using mixtures of template DNA at various ratios. (A) mixtures of D. batatas and D. destruens DNA and (B) mixtures of fungal DNA and background sweet potato DNA. In the left panel of (A), D. destruens DNA (5 ng μL–1) was mixed with a 10-fold dilution series of D. batatas DNA (5, 0.5, 0.05, 0.005, 0.0005, and 0.00005 ng DNA μL–1), providing ratios of D. batatas to D. destruens DNA in the range of 1:1 to 1:100000. In the right panel of (A) D. batatas and D. destruens were exchanged and tested at the same DNA mixing ratios. In (B), ratios of D. batatas: D. destruens: sweet potato DNA from 1:1:1 to 1:1:100000 were prepared by decreasing the concentrations of D. batatas and D. destruens DNA (5 to 0.0005 ng μL–1) while increasing the sweet potato DNA concentration (5 to 50 ng μL–1). The DNA concentrations in each reaction were 5:5:5, 5:5:50, 0.5:0.5:50, 0.05:0.05:50, 0.005:0.005:50, and 0.0005:0.0005:50 ng μL–1. Three biological replicates of each dilution were prepared. The regression equations and R2 coefficient of determination for Cq value versus the natural logarithm of the ratio of the various DNA indicated are shown on the charts. Data represent means ± standard deviation where visible.
We assessed the ability of the newly developed primers to quantify fungal DNA mixed with abundant sweet potato DNA. Db ITS and Dd ITS primers produced amplicons specific to target fungal pathogen DNA from a D. batatas: D. destruens: sweet potato DNA ratio of 1:1:1 to 1:1:10000 without false positives (Figure 3B). Recovery of both D. batatas and D. destruens DNA in a mixed sample was not achieved beyond the detection limit being 1:1:10000, resulting in quantification from 13.6 (±0.2) to 28.8 (±0.3) cycles by Db ITS and 16.7 (±0.3) to 31.4 (±0.4) cycles by Dd ITS. These results demonstrated that D. batatas and D. destruens DNA present in a background of sweet potato DNA was detectable by using Db ITS and Dd ITS primers.
Evaluation of Naturally Infected Sweet Potato Plants in the Field
We collected 55 sweet potato plants and 35 tubers with or without symptoms from 37 fields where foot rot disease appears in the Kyushu and Okinawa regions. The symptomatic specimens showed foot rot disease–like symptoms: i.e., black lesions with wilted tissues in the stems and discoloration with soft or rotten tissues in the tubers. From the collected seedlings and tubers, we generated stem samples (N = 77) and tuber samples (N = 83), respectively. For each sample, we applied a diagnostic index scored on a scale of 0 to 2 (see section “Materials and Methods”), based on the real-time PCR assay with the new primer sets. Overall, for both stem (Figure 4 and Supplementary Table 2) and tuber tissues (Figure 5 and Supplementary Table 3), D. destruens was found predominantly in symptomatic tissues rather than symptomless tissues; D. batatas was not as frequent as D. destruens in either symptomatic or symptomless tissues. The differences in relative frequency and concentration of D. batatas and D. destruens between symptomless and symptomatic sweet potato plants were highly significant in both stems and tubers (P < 0.01; Figures 4, 5). For symptomatic stem tissues (diagnostic index 2), specific amplicons of D. destruens were generated from all 38 samples: eight of these samples were also positive for D. batatas, indicating co-infection of D. batatas and D. destruens (Figure 4B). On the other hand, for symptomless stem tissues (diagnostic indexes 0 and 1), nine of 39 samples were positive for D. destruens, including one sample that displayed co-infection with D. batatas; one sample was positive for D. batatas alone. In symptomatic tuber tissues (diagnostic index 2), 37 out of 39 samples were positive for D. destruens, including five samples that displayed co-infection with D. batatas, and two samples were positive for D. batatas alone (Figure 5B). Specific amplicons of D. destruens, but not D. batatas, were also detected in three samples of symptomless tubers with pathogen (diagnostic index 1). The remaining 41 samples of symptomless tubers had no detectable pathogen (diagnostic index 0). In short, these results demonstrated that co-infection of D. destruens and D. batatas was detectable and distinguishable in real-time PCR diagnosis for foot rot disease.
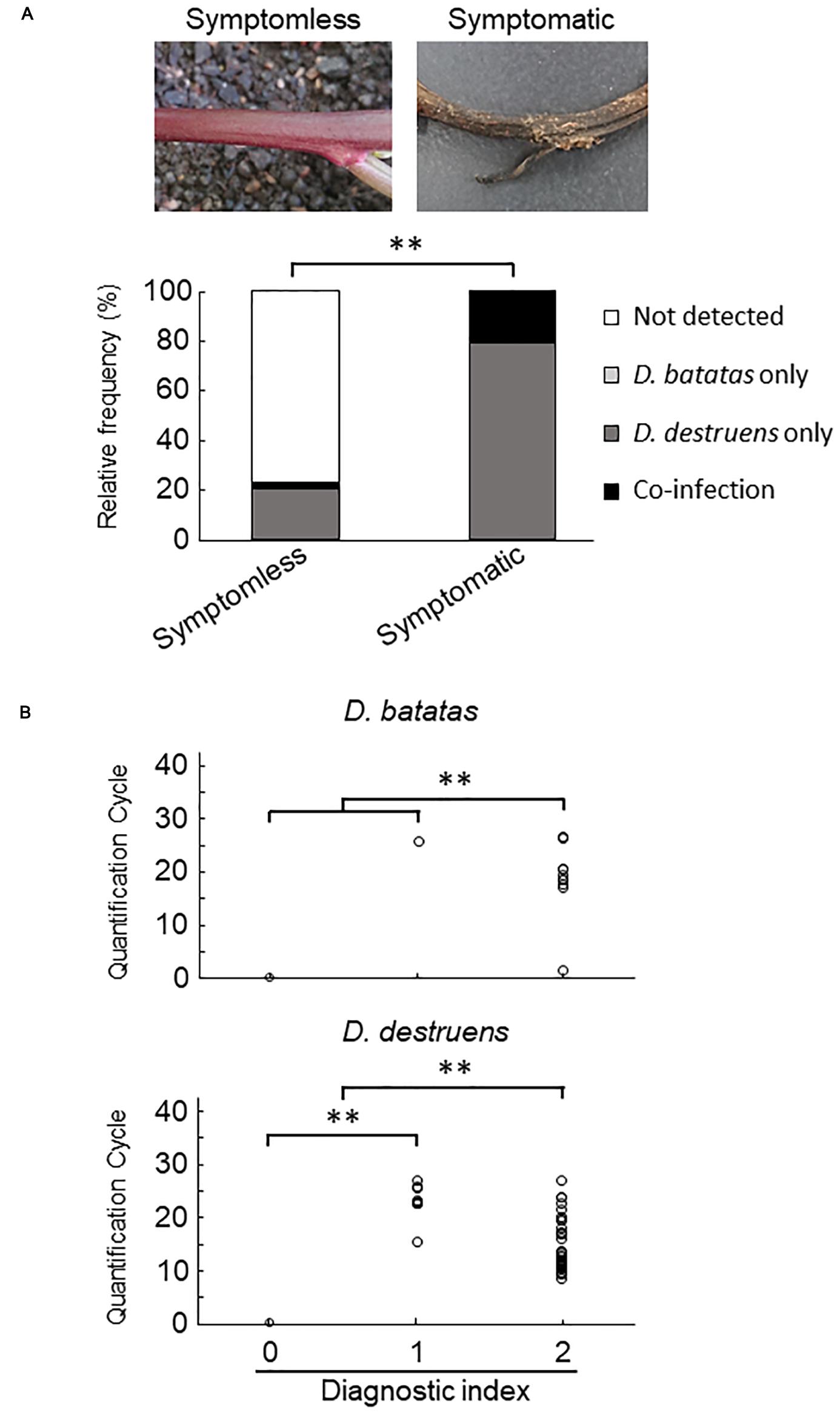
Figure 4. Frequency of amplification of D. batatas and D. destruens DNA from naturally infected sweet potato stems. (A) Relative frequency of D. batatas and D. destruens detection in sweet potato stems with or without symptomatic disorder. The relative frequency of detection of the two pathogens was significantly different between symptomless and symptomatic stem tissues (n = 77, ∗∗P < 0.01, Fisher’s exact test). (B) Real-time PCR detection of D. batatas and D. destruens DNA from naturally infected sweet potato seedlings in the field. Quantification cycle using the new primers is graphed against diagnostic index of stem (0, symptomless with no pathogen detected; 1, symptomless but pathogen detected; and 2, symptomatic disorder with pathogen detected). The concentrations of D. destruens and D. batatas in stems with diagnostic index 2 were significantly higher than in those with index 0 or 1, and the concentration of D. destruens in stems with diagnostic index 1 was significantly higher than in those with index 0 (n = 77, ∗∗P < 0.01, Steel–Dwass test).
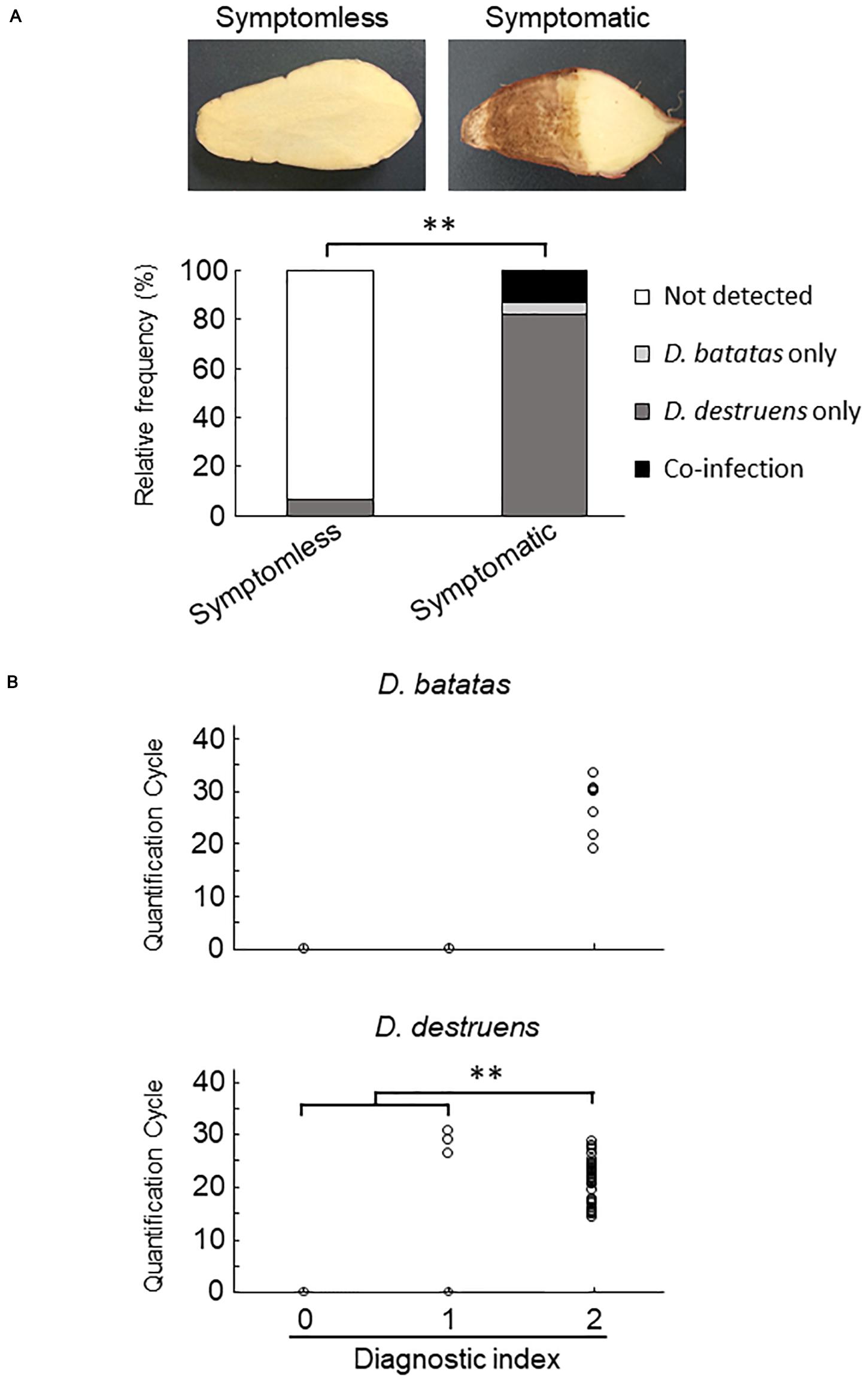
Figure 5. Frequency of amplification of D. batatas and D. destruens DNA from naturally infected sweet potato tubers. (A) Relative frequency of D. batatas and D. destruens detection in sweet potato tubers with or without symptomatic disorder. Detection frequency of the two pathogens among symptomless and symptomatic tuber tissues was highly differentiated (n = 83, ∗∗P < 0.01, Fisher’s exact test). (B) Amplification of D. batatas and D. destruens DNA from naturally infected sweet potato tubers in the field. Diagnostic index of sweet potato tubers scored on a scale of 0 to 2 along with real-time PCR detection using the new primer sets (0, symptomless with no pathogen detected; 1, symptomless but pathogen detected; and 2, symptomatic disorder with pathogen detected). The concentration of D. destruens in tubers with diagnostic index 2 was significantly higher than in those with the index 1 (n = 83, ∗∗P < 0.01, Steel–Dwass test), while there was no statistical significance in D. batatas.
Discussion
This study demonstrates that co-infection of D. batatas and D. destruens in sweet potato can be detected and distinguished by real-time PCR using the new primer sets Db ITS and Dd ITS. The technical reliability of our new PCR-based method is expected to be sufficient for diagnostic use. Our finding that D. destruens is frequently presented in symptomless sweet potatoes as well as symptomatic plants indicates that, for practical diagnostic evaluation of field plants, it is necessary to assess the presence of D. batatas and D. destruens in stems and tubers of both symptomless and symptomatic tissues. Also, the findings highlight the potential risks of further distribution of the fungal pathogens through symptomless seedlings or seed potatoes. Further studies are required to establish a molecular-based diagnosis process for foot rot disease based on our real-time PCR technique. This process would be a powerful tool for field evaluation of foot rot disease in sweet potato, even in cases of co-infection with other soil-borne pathogens that have been difficult to diagnose until now.
To ensure the accuracy and reliability of our real-time PCR technique in different laboratories, it is important to evaluate various combinations of real-time PCR thermal cyclers and reagents (Borst et al., 2004; Schoder et al., 2005; Bacich et al., 2011). In addition, different approaches for DNA extraction may influence the sensitivity of PCR detection of D. batatas and D. destruens, as previously reported for several fungal pathogens (Fredricks et al., 2005; Kidd et al., 2020). We performed an interlaboratory test in four experienced PCR laboratories in our project, all of whom successfully applied the approach demonstrated in this study (K. Fujiwara, unpublished data). The detection sensitivity for D. batatas and D. destruens varied slightly in the interlaboratory studies when different real-time PCR reagents were used. Thus, it is necessary to optimize the PCR conditions and operation process when performing conventional PCR and real-time PCR for diagnosis targeting D. batatas and D. destruens.
In general, the specificity and quantitative nature of PCR amplification in detection of D. batatas and D. destruens is negatively impacted by the generation of false positives from a non-target DNA (Grosdidier et al., 2016). Our study demonstrates that our real-time PCR technique for distinguishing D. batatas and D. destruens was specific and generated no false positives for up to 32 cycles, which is within the detection limit of the primers. However, a potential drawback in that Db ITS and Dd ITS primers yield false positives to other closely-related Diaporthe and Phomopsis species (Rossman et al., 2014; Gao et al., 2016) might arise. Post-PCR approaches to screen for non-target amplification may need to be employed. For instance, in this study, amplicons generated from closely-related Diaporthe and Phomopsis species beyond the detection limit were distinguishable from D. batatas by DNA sequencing (K. Fujiwara, unpublished data). The evaluation of potential false positive results could also be achieved by other methods such as restriction fragment length polymorphism or microsatellite analysis (Koreth et al., 1996; Borst et al., 2004). Understanding the potential risks of obtaining false positives in PCR would allow field evaluation of foot rot disease in naturally infected sweet potato.
The use of real-time PCR assays may provide a new approach for managing foot rot disease of sweet potato. Currently, there are no chemicals that can be effectively used to control the disease (Kobayashi, 2019). However, the heavy use of fungicides is not advisable for sweet potato production because of food safety regulation in Japan. Also, development of fungicide-resistant D. destruens due to the heavy use of fungicides might be a major concern (Batzer and Mueller, 2020). In this study, D. destruens in symptomless sweet potato seedlings was frequently detected by real-time PCR using Dd ITS primers. Thus, our methodology can be used to screen for disease-free seedlings and prevent the use of infected seedlings or nurseries in the preparation of disease-free seedlings and seed tubers. Future use of the real-time PCR assays to monitor D. destruens in soil could potentially identify the risk of foot rot disease and guide control decisions before disease development. Thus, the PCR approach could contribute to the development of countermeasures and could help establish a scheme to control foot rot disease of sweet potato by evaluating seedlings and/or soils before planting, to identify fields where foot rot disease occurs.
Conclusion
Our study provides a novel PCR method to identify D. batatas, which causes dry rot disease, and D. destruens, which causes foot rot disease, of sweet potato. The real-time PCR assay is specific for each of the fungal pathogens and can distinguish them in co-infected sweet potato. Currently, there is no effective cure or control measure for D. destruens in sweet potato production in Japan. We expect that our PCR assay will provide a useful tool for diagnosis of D. destruens and will form the foundation of the design of integrated disease management strategies for foot rot disease.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
KF and HI conducted the experiments and drafted the manuscript. MU, MN, SK, and YN assisted in evaluating PCR results and provided critical advice on PCR. YOK, KN, YO, AK, AM, KH, and YK contributed to the collection of fungal isolates, field samples, and diagnosis of infected materials. YOK also contributed to the design and the management of a project including this work. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Project of the NARO Bio-oriented Technology Research Advancement Institution (Research Program on Development of Innovative Technology; Grant No. 01020C).
Conflict of Interest
The authors declare one relevant Japanese patent application, 2020-140356.
Acknowledgments
We appreciate the support of the Pest and Crop Disease Prevention Office in the Fukuoka Agriculture and Forestry Research Center, the Kumamoto Plant Protection Office, and the Nagasaki Prefectural Government Pest Control Office in collecting infected sweet potato plants. We thank Michiyo Teramoto, Noriko Hidaka, and Yuko Fujii for their support in plant material processing and DNA extraction.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.694053/full#supplementary-material
Footnotes
- ^ https://www.gene.affrc.go.jp/index_en.php
- ^ https://www.ncbi.nlm.nih.gov/tools/primer-blast/
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi
References
Bacich, D. J., Sobek, K. M., Cummings, J. L., Atwood, A. A., and O’Keefe, D. S. (2011). False negative results from using common PCR reagents. BMC Res. Notes 4:457. doi: 10.1186/1756-0500-4-457
Batzer, J. C., and Mueller, D. S. (2020). Soybean fungal endophytes Alternaria and Diaporthe spp. are differentially impacted by fungicide application. Plant Dis. 104, 52–59. doi: 10.1094/PDIS-05-19-1001-RE
Baumgartner, K., Fujiyoshi, P. T., Travadon, R., Castlebury, L. A., Wilcox, W. F., and Rolshausen, P. E. (2013). Characterization of species of Diaporthe from wood cankers of grape in eastern North American vineyards. Plant Dis. 97, 912–920. doi: 10.1094/PDIS-04-12-0357-RE
Borst, A., Box, A. T. A., and Fluit, A. C. (2004). False-positive results and contamination in nucleic acid amplification assays: suggestions for a prevent and destroy strategy. Eur. J. Clin. Microbiol. Infect. Dis. 23, 289–299. doi: 10.1007/s10096-004-1100-1
Chase, M. W., and Fay, M. F. (2009). Barcoding of plants and fungi. Science 325, 682–683. doi: 10.1126/science.1176906
Clark, C. A., Ferrin, D. M., Smith, T. P., and Gerald, J. H. (2013). Compendium of Sweetpotato Diseases, Pests, and Disorders. Saint Paul, MIN: The American Phytopathological Society Press.
Fredricks, D. N., Smith, C., and Meier, A. (2005). Comparison of six DNA extraction methods for recovery of fungal DNA as assessed by quantitative PCR. J. Clin. Microbiol. 43, 5122–5128. doi: 10.1128/JCM.43.10.5122-5128.2005
Gai, Y., Ma, H., Chen, X., Zheng, J., Chen, H., and Li, H. (2016). Stem blight, foot rot and storage tuber rot of sweet potato caused by Plenodomus destruens in China. J. Gen. Plant Pathol. 82, 181–185. doi: 10.1007/s10327-016-0661-z
Gao, Y. H., Liu, F., and Cai, L. (2016). Unravelling Diaporthe species associated with Camellia. Syst. Biodivers. 14, 102–117. doi: 10.1080/14772000.2015.1101027
Grosdidier, M., Aguayo, J., Marçais, B., and Ioos, R. (2016). Detection of plant pathogens using real−time PCR: how reliable are late Ct values? Plant Pathol. 66, 359–367. doi: 10.1111/ppa.12591
Harter, L. L., and Weimer, J. L. (1929). A Monographic Study of Sweet Potato Diseases and Their Control, Technical Bulletin. Washington, DC: U.S. Department of Agriculture, 1–118.
Hosseini, B., Ei-Hasan, A., Link, T., and Voegele, R. T. (2020). Analysis of the species spectrum of the Diaporthe/Phomopsis complex in European soybean seeds. Mycol. Prog. 19, 455–469. doi: 10.1007/s11557-020-01570-y
Huang, C. W., Chuang, M. F., Tzean, S. S., Yang, H. R., and Ni, H. F. (2012). Occurrence of foot rot disease of sweet potato caused by Phomopsis destruens in Tai-wan. Plant Pathol. Bull. 21, 47–52. [In Chinese],Google Scholar
Kidd, S. E., Chen, S. A. C., Meyer, W., and Halliday, C. L. (2020). A new age in molecular diagnostics for invasive fungal disease: are we ready? Front. Microbiol. 10:2903. doi: 10.3389/fmicb.2019.02903
Kobayashi, Y. O. (2019). Outbreak and management of foot rot of sweetpotato. Plant Prot. 73, 501–505. [In Japanese],Google Scholar
Koreth, J., O’Leary, J. J., and O’D McGee, J. (1996). Microsatellites and PCR genomic analysis. J. Pathol. 178, 239–248. doi: 10.1002/(SICI)1096-9896(199603)178:3<239::AID-PATH506<3.0.CO;2-5
Lin, C. Y., Huang, C. W., Yang, H. R., Lai, S. Y., and Ni, H. F. (2017). A method for the specific detection of Phomopsis destruens in sweet potato by PCR. J. Taiwan Agric. Res. 66, 276–285. doi: 10.6156/JTAR/2017.06604.02
Liu, Q. C., Liu, J., Zhang, P., and He, S. Z. (2014). “Root and tuber crops,” in Encyclopedia of Agriculture and Food Systems, ed. N. Van Alfen (San Diego, CA: Elsevier), 46–61.
Lopes, C. A., Boff, P., and Duarte, V. (1994). Foot rot of sweet potato in Brazil. Pesqui. Agropecu. Bras. 29, 1407–1410.
Maruyama, M., Tokuda, R., Okano, Y., Miyazaki, A., Sato, H., Maejima, K., et al. (2020). “Development of a specific detection method for sweet potato foot rot fungus (Plenodomus destruens) based on LAMP,” in Japanese Journal of Phytopathology: Abstracts of the Papers Presented at the Annual Meeting of the Society, 2020 March 19-21, Kagoshima, Japan, ed. Y. Ishinose (Tokyo: The Phytopathological Society of Japan), 157–247.
Paul, N. C., Nam, S. S., Park, W., Yang, J. W., and Kachroo, A. (2019). First report of storage tuber rot in sweetpotato (Ipomoea batatas) caused by Plenodomus destruens in Korea. Plant Dis. 103:1020. doi: 10.1094/PDIS-10-18-1776-PDN
Rossman, A., Udayanga, D., Castlebury, L. A., and Hyde, K. D. (2014). (2304) Proposal to conserve the name Diaporthe eres against twenty-one competing names (Ascomycota: Diaporthales: Diaporthaceae). Taxon 63, 934–935. doi: 10.12705/634.23
Schaad, N. W., and Brenner, D. (1977). A bacterial wilt and root rot of sweet potato caused by Erwinia chrysanthemi. Phytopathology 67, 302–308. doi: 10.1094/phyto-67-302
Schoder, D., Schmalwieser, A., Schauberger, G., Hoorfar, J., Kuhn, M., and Wagner, M. (2005). Novel approach for assessing performance of PCR cyclers used for diagnostic testing. J. Clin. Microbiol. 43, 2724–2728. doi: 10.1128/JCM.43.6.2724-2728.2005
Scruggs, A. C., and Quesada-Ocampo, L. M. (2016). Etiology and epidemiological conditions promoting Fusarium root rot in sweetpotato. Phytopathology 106, 909–919. doi: 10.1094/PHYTO-01-16-0009-R
Seifert, K. (2009). Progress toward DNA barcoding of fungi. Mol. Ecol. Resour. 9, 83–89. doi: 10.1111/j.1755-0998.2009.02635.x
Skoglund, L. G., and Smit, N. E. J. M. (1994). Major Diseases and Pests of Sweetpotatoes in Eastern Africa. Lima: International Potato Center.
Steinbauer, C. E., and Kushman, L. J. (1971). Sweetpotato Culture and Diseases, Agriculture Handbook No. 388. Washington, DC: United States Department of Agriculture, 49–68.
Udayanga, D., Castlebury, L. A., Rossman, A. Y., Chukeatirote, E., and Hyde, K. D. (2014). Insights into the genus Diaporthe: phylogenetic species delimitation in the D. eres species complex. Fungal Divers. 67, 203–229. doi: 10.1007/s13225-014-0297-2
Udayanga, D., Castlebury, L. A., Rossman, A. Y., Chukeatirote, E., and Hyde, K. D. (2015). The Diaporthe sojae species complex: phylogenetic re-assessment of pathogens associated with soybean, cucurbits and other field crops. Fungal Biol. 5, 383–407. doi: 10.1016/j.funbio.2014.10.009
White, T. J., Bruns, T., Lee, S., and Taylor, J. W. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: A Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (New York, NY: Academic Press, Inc.), 315–322. doi: 10.1016/b978-0-12-372180-8.50042-1
Keywords: Diaporthe batatas, Diaporthe destruens, foot rot disease, real-time PCR, sweet potato
Citation: Fujiwara K, Kobayashi YO, Usui M, Nishioka K, Nakamura M, Kawano S, Okada Y, Kobayashi A, Miyasaka A, Hirayae K, Kushima Y, Nishi Y and Inoue H (2021) Real-Time PCR Assay for the Diagnosis and Quantification of Co-infections by Diaporthe batatas and Diaporthe destruens in Sweet Potato. Front. Plant Sci. 12:694053. doi: 10.3389/fpls.2021.694053
Received: 12 April 2021; Accepted: 21 May 2021;
Published: 22 June 2021.
Edited by:
Christina Cowger, Plant Science Research Unit, Agricultural Research Service, United States Department of Agriculture, United StatesReviewed by:
Narayan Chandra Paul, Chonnam National University, South KoreaKandeeparoopan Prasannath, Eastern University, Sri Lanka, Sri Lanka
Copyright © 2021 Fujiwara, Kobayashi, Usui, Nishioka, Nakamura, Kawano, Okada, Kobayashi, Miyasaka, Hirayae, Kushima, Nishi and Inoue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuki Fujiwara, a3owODE0QGFmZnJjLmdvLmpw