 Qian Yang
Qian Yang Chunyu Zhang
Chunyu Zhang Chao Liu
Chao Liu- College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, China
Crosses that lead to heterosis have been widely used in the rapeseed (Brassica napus L.) industry. Cytoplasmic male sterility (CMS)/restorer-of-fertility (Rf) systems represent one of the most useful tools for rapeseed production. Several CMS types and their restorer lines have been identified in rapeseed, but there are few studies on the mechanisms underlying fertility restoration. Here, we performed morphological observation, map-based cloning, and transcriptomic analysis of the F2 population developed by crossing the CMS line WNJ01A with its restorer line Hui01. Paraffin-embedded sections showed that the sporogenous cell stage was the critical pollen degeneration period, with major sporogenous cells displaying loose and irregular arrangement in sterile anthers. Most mitochondrial electron transport chain (mtETC) complex genes were upregulated in fertile compared to sterile buds. Using bulked segregant analysis (BSA)-seq to analyze mixed DNA pools from sterile and fertile F2 buds, respectively, we identified a 6.25 Mb candidate interval where Rfw is located. Using map-based cloning experiments combined with bacterial artificial chromosome (BAC) clone sequencing, the candidate interval was reduced to 99.75 kb and two pentatricopeptide repeat (PPR) genes were found among 28 predicted genes in this interval. Transcriptome sequencing showed that there were 1679 DEGs (1023 upregulated and 656 downregulated) in fertile compared to sterile F2 buds. The upregulated differentially expressed genes (DEGs) were enriched in the Kyoto Encyclopedia of Genes and Genomes (KEGG) lysine degradation pathway and phenylalanine metabolism, and the downregulated DEGs were enriched in cutin, suberine, and wax biosynthesis. Furthermore, 44 DEGs were involved in pollen and anther development, such as tapetum, microspores, and pollen wall development. All of them were upregulated except a few such as POE1 genes (which encode Pollen Ole e I allergen and extensin family proteins). There were 261 specifically expressed DEGs (9 and 252 in sterile and fertile buds, respectively). Regarding the fertile bud-specific upregulated DEGs, the ubiquitin–proteasome pathway was enriched. The top four hub genes in the protein–protein interaction network (BnaA09g56400D, BnaA10g18210D, BnaA10g18220D, and BnaC09g41740D) encode RAD23d proteins, which deliver ubiquitinated substrates to the 26S proteasome. These findings provide evidence on the pathways regulated by Rfw and improve our understanding of fertility restoration.
Introduction
Rapeseed (Brassica napus L.) is one of the most important oil crops worldwide. Crosses that lead to heterosis, involving significantly increased seed yield, have been widely used for rapeseed production in the past decades (Morrison et al., 2016; Shen et al., 2017). Cytoplasmic male sterility (CMS) combined with its maintainer line and restorer line is a highly valuable resource to produce hybrid seeds of rapeseed (Li et al., 2015).
Plant CMS is generally caused by new chimeric open reading frames (ORFs) in the mitochondrial genome, and they are usually generated through mitochondrial genome rearrangement (Tang H. et al., 2017). These novel ORFs are usually homologous or co-transcribed with the genes encoding proteins of the mitochondrial electron transport chain (mtETC) complexes or ATP synthase, so these systems do not function normally (Bohra et al., 2016). Restorer-of-fertility (Rf) genes in the nuclear genomes of the restorer lines can downregulate the CMS genes to reverse male sterility. Several Rf genes, which encode diverse functional proteins, have been identified. For instance, maize Rf2 and Rf4 encode an aldehyde dehydrogenase and a bHLH transcription factor (TF) for T-CMS and C-CMS, respectively (Liu and Schnable, 2002); rice RF17 and RF2 encode an acetyl-carrier protein and a glycine-rich protein for CW-CMS and LD-CMS, respectively (Fujii and Toriyama, 2009); and sugar beet Rf1 encodes a putative M48 family peptidase for Owen-CMS (Kitazaki et al., 2015). However, most cloned plant Rf genes encode pentatricopeptide repeat (PPR)-containing proteins (Kubo et al., 2020). These proteins are RNA-binding factors that participate in mRNA processing after transport to a mitochondrion (Schmitz-Linneweber and Small, 2008). During the fertility restoration of CMS, they downregulate sterility genes by cleaving their transcripts or inhibiting their translation (Gaborieau et al., 2016).
In B. napus, there are a variety of CMS systems, such as pol, ogu, nap, hau, Nsa, and inap. Among them, the pol and ogu CMS lines are the most widely used to produce hybrid seeds. Regarding pol CMS, a chimeric ORF, orf224, is co-transcribed with atp6 and causes male sterility (Singh and Brown, 1993; L’Homme et al., 1997; Liu et al., 2016). Its restorer gene, Rfp, encodes a mitochondria-targeted PPR protein that downregulates orf224 (Liu et al., 2016). Ogu CMS originated in Raphanus sativus and was introduced into B. napus. Regarding ogu CMS, orf138 is considered to be the gene that causes male sterility (Bonhomme et al., 1992). Its restorer gene Rfo from R. sativus encodes a PPR protein that downregulates orf138 protein at the translation level (Uyttewaal et al., 2008). Another B. napus CMS system, nap, is rarely used to produce hybrid seeds because the sterility is easily affected by temperature change in most cases. In this system, orf222 is the key sterility gene, and it is co-transcribed with both orf139 and an exon of a trans-spliced gene, nad5c (L’Homme et al., 1997). Its restorer gene, Rfn, on chromosome A09 of B. napus, encodes a PPR protein. However, the accumulation of orf222 transcripts was not obviously changed in the restorer line or in transgenic fertility-restored lines compared to the nap CMS line (Liu et al., 2017).
In recent years, new CMS systems from Brassica or related species have been discovered and introduced into B. napus. The hau CMS system originated from Brassica juncea and its sterility gene, orf288, encodes a cytotoxic protein that causes aborted pollen development (Wan et al., 2008; Jing et al., 2012). orf288 transcription was upregulated in the male-sterile line compared to the restorer line, but there was no significant difference at the protein level (Heng et al., 2018). A recent research reported that the restorer gene Rfh of hau CMS has been cloned (Wang H. et al., 2021). Nsa CMS is a novel B. napus CMS system derived from the cytoplasm of Sinapis arvensis, and its restorer line NR1 was developed from a disomic alien addition line of B. napus and S. arvensis (Wei et al., 2010). Three ORFs (orf224, orf309, and orf346), which possess chimeric and transmembrane structures, are the candidate sterility genes of Nsa CMS (Sang et al., 2019). It was further confirmed that ORF346 is the key gene for pollen abortion in Nsa CMS (Sang et al., 2021). Another CMS system, inap CMS, was obtained via backcross of the somatic hybrid of Isatis indigotica (Chinese woad) and B. napus, with B. napus as the recurrent parent (Kang et al., 2017). A restorer line carrying a dominant Rf gene was successfully developed for inap CMS (Li et al., 2019). Further research is required to isolate their restorer genes and use them in rapeseed breeding.
Anther and pollen development are extremely complex processes involving many genes and pathways. Therefore, transcriptome sequencing has been applied in many crops as an effective tool to study global transcription networks to elucidate underlying mechanisms. To elucidate the genome-wide molecular mechanisms underlying Brassica CMS and fertility restoration, transcriptomic profiling using RNA-seq has been used. For example, transcriptomic analysis of pol CMS indicated that the energy deficiency caused by orf224/atp6 may downregulate many genes involved in pollen development via nuclear–mitochondrial interactions (An et al., 2014). Through multi-omics joint analysis and yeast two-hybrid assay, seven Rfp interacting proteins related to RNA editing, anther and tapetum development, and five orf224 interacting proteins participating in the electronic respiratory transmission chain, anther development and oxidative phosphorylation were obtained and further verified (Wang B. et al., 2021). In ogu CMS, sterility may be caused by delayed tapetum degradation, as genes related to tapetum programmed cell death were downregulated compared to in the maintainer line. Additionally, sporopollenin biosynthesis and its key genes were both inhibited in ogu CMS (Xing et al., 2018; Lin et al., 2019). Another transcriptomics study on ogu CMS, pol CMS, and their shared maintainer line reported that most of the DEGs that were downregulated in both CMS lines were involved in pollen development, carbon metabolism, lipid metabolism, the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation (Wei et al., 2020).
Currently, many hybrid rapeseed cultivars are bred using the pol CMS system in China. Thus, it is necessary to develop new stable CMS systems (with restorer lines) to solve the issue of cytoplasmic simplification (i.e., most of the sterile cytoplasm of current commercial hybrids belong to a single type) to avoid risks to the rapeseed industry. We previously developed a novel type of CMS, WNJ01A, from a natural male-sterile B. juncea mutant. It was introduced into B. napus and displayed a stable and complete pollen-abortion phenotype. Recently, its restorer line, Hui01, was obtained through distant hybridization between WNJ01A CMS and B. rapa. Here, we aimed to finely map and isolate the candidate restorer gene, designated Restorer of fertility for WNJ01A (Rfw). We also aimed to conduct a transcriptomic analysis to identify the genes and pathways involved in male sterility and fertility restoration. The results will facilitate understanding of the molecular mechanisms of WNJ01A CMS and its fertility restoration, and they will also provide a foundation for its use in breeding.
Materials and Methods
Plant Materials
We previously developed the WJS1A CMS line from a natural B. juncea mutant. To obtain WNJ01A CMS, the sterile cytoplasm was introduced into B. napus cv. Huashuang4 by backcrossing for more than 10 generations. Subsequently, we crossed WNJ01A CMS (female parent) with B. rapa ssp. pekinensis via distant hybridization. We then searched for fertile plants in their progenies after continuous selfing, the fertile progeny lines were obtained and it was further confirmed that they could reverse the sterility in WNJ01A CMS by test crossing. One restorer line with the best ability of restoring fertility was designated Hui01.
Genetic Analysis of Fertility Restoration for WNJ01A
WNJ01A (female parent) was crossed with restorer line Hui01 (male parent) to produce the F1 population and then the F2 population was generated via self-pollination of F1 plants. The fertility of each plant was determined by assessing at least five flowers per plant a minimum of three difference times during the flowering period, following a previously described method (Liu et al., 2012).
Cytological Characterization of Fertility Restoration for WNJ01A
Fresh fertile and sterile floral buds (0–8 mm) at various developmental stages from F2 plants were collected and fixed in FAA fixative (3.7% formaldehyde, 50% ethanol, and 5.0% acetic acid) and processed as previously described (Ning et al., 2019). Paraffin-embedded sections were assessed using the Arabidopsis thaliana classification criteria for the anther development period (Chang F. et al., 2011).
Whole-Genome Resequencing of Bulked DNA Pools
To determine the genomic region containing Rfw, 37 extremely fertile plants and 38 extremely sterile plants were selected from the F2 population. Genomic DNA was extracted from 100 mg fresh leaves from each plant and purified using a DNA Secure Plant Kit (DP320; TIANGEN, China). A NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, United States) was used to determine the DNA concentration and quality. Next, 80 ng DNA per plant were mixed to construct the F- and S-pools. A HiSeq 3000 PE150 sequencing platform (Illumina, United States) was used to perform high-throughput library construction and sequencing for the S- and F-pools.
Bulked segregant analysis (BSA)-seq, involving the abovementioned clean genome resequencing data and using the reference genome B. napus cv. Darmor-bzh (Chalhoub et al., 2014), was conducted as previously described (Tang Q. et al., 2017; Su et al., 2020). To locate the Rfw gene, we calculated the proportion of reads in the F- and S-pools with single-nucleotide polymorphism (SNP)/insertion/deletion (InDel) sites that were distinct from the of Darmor-bzh reference reads (i.e., SNP/InDel-index) and the difference in this index between the F- and S-pools [i.e., Δ(SNP/InDel-index)]. To eliminate low-quality SNP/InDels, we discarded all loci with an SNP/InDel-index < 0.3 (Takagi et al., 2013). Using the sliding window method (with a 1 Mb window size and 100 kb increment), the average index values of loci were calculated. The SNP/InDel-index of the F- and S-pools and the corresponding Δ(SNP/InDel-index) in the sliding window were used to construct SNP/InDel-index plots.
Map-Based Cloning of Rfw
To develop the set of molecular markers, the InDel polymorphism sites detected during genome resequencing in the candidate interval, simple sequence repeats (SSR) sites in this interval, and publicly available molecular markers in this interval were used. The molecular markers were verified in a mixed DNA pool from fertile and sterile plants, with a small population being used to verify the linkage between the molecular markers and fertility restoration. All primer sequences of linked molecular markers are given in Supplementary Table 1. Next, 4454 sterile F2 plants were subjected to map-based cloning experiments to finely map Rfw, based on the numbers of recombinant plants and the known physical locations of a set of molecular markers.
Bacterial Artificial Chromosome (BAC) Screening, Sequencing, and Candidate Gene Prediction
To obtain the candidate region sequence, we constructed a BAC library (using CopyControlTM pCC1BACTM Vector; EPICENTRE, United States) for the restorer line Hui01. HindIII restriction enzyme was used to digest the genomic DNA and then 90–120 kb DNA fragments were selected by pulse-field electrophoresis and purified. The DNA fragments were ligated into the CopyControlTM pCCIBACTM Vectors in the library kit. The library contained about 40,000 clones with 90–120 kb DNA insert size, representing about 5 × B. napus genome. After batch transformation of EPI300 E. coli, the bacterial suspension was evenly smeared onto LB medium containing 12.5 μg/mL chloramphenicol. Thereafter, the strains were grown and then eluted in a sterile centrifuge tube containing chloramphenicol before further propagation. We mixed sets of 1000 single clones to create 40 pools. Next, the BAC plasmids in each pool were extracted using a Large-Construct Kit (QIAGEN, United States). A molecular marker (TY21) that co-segregated with fertility restoration was used to screen for pools containing a positive clone. Finally, the molecular markers flanking the finely mapped candidate interval were used to analyze the physical location of the interval in the positive clones, which underwent high-throughput sequencing using Illumina NovaSeq (Illumina, United States) and Oxford Nanopore Technologies (ONT; United Kingdom).
Four reference genomes containing A subgenomes (B. rapa cv. chiifu-401: AA; B. napus cv. Darmor-bzh and Zhongshuang11: AACC; B. juncea var. tumida: AABB) were used to conduct a collinearity analysis of the candidate Rfw genes in the finely mapped Rfw region. The genes in the candidate interval of Hui01 were analyzed and predicted through the Fgenesh gene-finder1. The gene coding sequence (CDS) was compared and analyzed with ClustalX.
Analysis of Mitochondrial Gene Transcripts Using RT-qPCR
Annotation information of the B. juncea var. tumida and jiangpu mitochondrial genomes (accession nos. KJ461445 and JF920288, respectively) were obtained from the National Center for Biotechnology Information (NCBI) (Chang S. et al., 2011; Zhao et al., 2016). We selected 32 known mitochondrial genes, including genes related to mtETC complex I (NADH dehydrogenase, nad), complex III (ubiquinol–cytochrome c reductase, cob), complex IV (cytochrome c oxidase, cox), complex V (ATP synthase, atp), cytochrome c biogenesis, ribosome small subunit, ribosome large subunit, twin-arginine translocation (tatC), and intron maturase (matR).
Transcriptional levels in the fertile and sterile buds were assessed using RT-qPCR. All genes and primer sequences are detailed in Supplementary Table 2. A RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, United States) was used to reverse transcribe the total RNA following the manufacturer’s protocol. A Hieff® qPCR SYBR Green Master Mix (No Rox) kit (YEASEN, China) and a CFX384 Touch Real-Time PCR Detection System (Bio-Rad, United States) were used for analysis of the transcription levels. Bna.actin7 was used for normalization in RT-qPCR (for normalization corresponding to total RNA levels).
Transcriptomic Sequencing
Floral buds (0–2 mm) were carefully harvested from fertile and sterile F2 plants. Three biological replicates (involving five buds each) were used for the fertile-pool (F-pool; Rfw1, Rfw2, and Rfw3) and three for the sterile-pool (S-pool; rfw1, rfw2, and rfw3). The buds were frozen in liquid nitrogen at once and then stored at –80°C until further processing. Total RNA was extracted using TRIzol (Invitrogen, United States) according to the manufacturer’s instructions. A HiSeq X Ten platform (Illumina, United States) was used for transcriptome sequencing (Biomarker Technologies, China).
Identification of DEGs and Enrichment Analyses
Differentially expressed genes (DEGs) between fertile and sterile buds were identified with the DESeq R package (1.10.1), using the Benjamini–Hochberg procedure to adjust the generated P-values to control the false discovery rate (FDR). DEGs were identified based on adjusted P < 0.05, Fragments Per Kilobase of transcript per Million mapped read (FPKM) fold change ≥ 2, and FDR < 0.01.
Next, we used the GOseq R package based on the Wallenius non-central hypergeometric distribution to perform Gene Ontology (GO) enrichment analysis on the DEGs (Young et al., 2010), which accounts for gene length bias in DEGs. We also performed a Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis using KOBAS software (Mao et al., 2005). The KEGG and GO analyses were performed via the Bioinformatics platform2.
Interaction Analysis
To predict the protein–protein interactions (PPIs) of the DEGs, the DEG sequences were used in a BLASTx search against the genomes of related species that had PPI information in the STRING database3. The PPI network (confidence score > 0.7), with KEGG annotations, was visualized using Cytoscape (Shannon et al., 2003). The hub genes were identified using the Cytoscape plugin CytoHubba (Song et al., 2020; Zuo et al., 2020).
Results
Morphological Characterization and Genetic Analysis of CMS and Restorer Lines
The cytoplasm of the CMS line WNJ01A originated from a natural male-sterile B. juncea mutant. We developed its restorer line Hui01 via distant hybridization between WNJ01A CMS and B. rapa. The sepals, petals, and pistils of both the WNJ01A and Hui01 lines exhibited normal development (Figure 1). However, the albino WNJ01A anthers were significantly smaller and their filaments were dramatically shorter (Figures 1A,C) compared to the Hui01 anthers (Figures 1B,D). Across more than 20 generations over 11 years (2010–2020) under various ecological conditions (in spring and winter) in China, WNJ01A maintained complete pollen abortion without being affected by temperature or photoperiod.

Figure 1. Phenotypic characterization of floral organs. (A,C) Sterile flower of CMS line WNJ01A. (B,D) Fertile flower of restorer line Hui01. Bars = 0.5 cm.
To elucidate the genetic characteristics of fertility restoration of WNJ01A CMS by Hui01, the fertility of the F1 and F2 populations of a WNJ01A CMS × Hui01 cross was evaluated. All F1 plants produced normally fertile pollen, indicating that Hui01 can completely restore the fertility of WNJ01A CMS. In the F2 population, there were 1606 fertile and 555 sterile plants, which fitted the expected 3:1 ratio (χc2 = 0.5012, P = 0.46). Thus, the fertility restoration of WNJ01A CMS is controlled by a dominant gene. It was designated Restorer of fertility for WNJ01A (Rfw).
Cytological Characterization of Anther Development
To accurately characterize pollen abortion, paraffin-embedded sections of the buds of fertile and sterile F2 plants were observed. At the anther primordia division stage, the cells of anther primordia were dividing, with almost no difference between fertile and sterile anthers (Figures 2A1,B1). However, at the sporogenous cell stage, major sporogenous cells displayed loose and irregular arrangements in sterile anthers, unlike the regular three layers of cells in fertile anthers. The cells in the exothecium, endothecium, and middle layer of the sterile anther walls began to shrink and degenerate. The tapetum cells started to detach from the anther walls and gradually degrade (Figures 2A2,B2). At the microspore mother cell and tetrad stages, microspore mother cells developed normally and then formed numerous tetrads via meiosis in the fertile anthers, and the tapetum cells were regularly arranged. In contrast, the microspore mother cells of the sterile anthers were more scattered, the tetrads were not formed, and the tapetum cells were increasingly abnormal (Figures 2A3,A4,B3,B4).
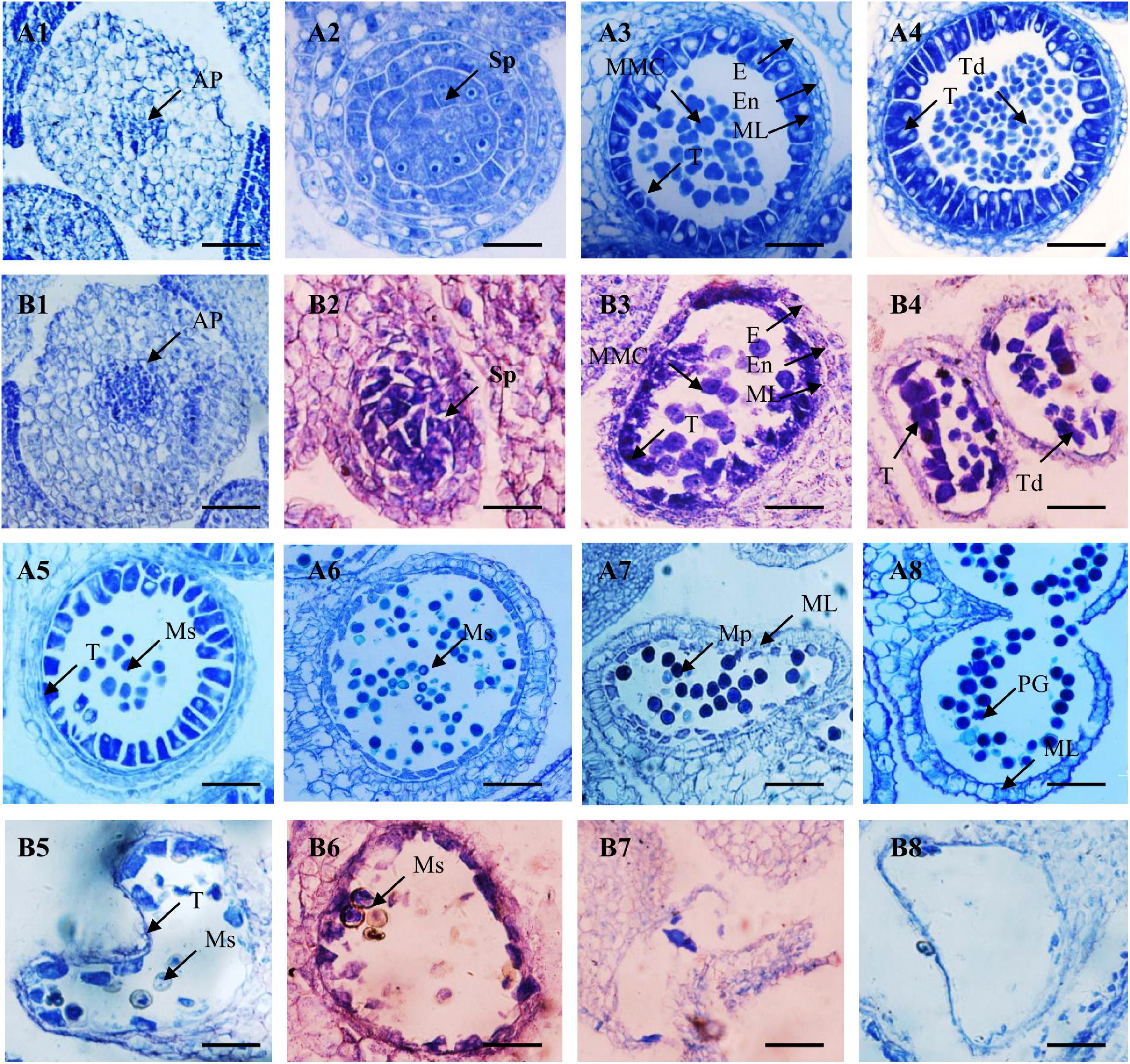
Figure 2. Microstructure of fertile (A1–A8) and sterile (B1–B8) anthers from F2 plants. (A1,B1) Anther primordium stage; (A2,B2) sporoblast stage; (A3,B3) microspore mother cell stage; (A4,B4) tetrad stage; (A5,B5) early mononuclear stage; (A6,B6) mononuclear stage; (A7,B7) dinuclear stage; (A8) mature microspores; and (B8) empty pollen sac. AP, anther primordium; Sp., sporogenous cell; E, epidermis; En, endothecium; ML, middle layer; T, tapetum; MMC, microspore mother cells; Ms, microspore; MP, mature pollen; PG, pollen grain. Bars = 100 μm.
In the remaining developmental stages (early mononuclear, mononuclear, dinuclear, and mature microspore stages) of the fertile anthers, microspores were released from the tetrads and then many mature pollen grains were found in the anther chambers. The tapetums disintegrated and gradually disappeared, and only the epidermis and fiber layer remained in the pollen sac wall (Figures 2A5–A8). However, in the sterile anthers, the tapetum cells degraded prematurely. Numerous pollen grains became vacuolated and eventually completely degraded, emptying the anther chamber (Figures 2B5–B8).
These results indicate that the sporogenous cell stage is critical for anther development, based on the abnormal WNJ01A CMS plants. Equally, this stage is an important stage for Rfw to begin to prevent pollen abortion. Furthermore, the anther sterility characteristics of WNJ01A CMS were obviously different from the previously studied pol, ogu, Nsa, hau, and inap CMS Brassica systems (Wan et al., 2008; An et al., 2014; Kang et al., 2017; Ding et al., 2018).
Mapping of Rfw Gene
To identify the Rfw region, F- and S-pools were constructed by subjecting extremely fertile and sterile F2 plants to high-throughput sequencing. A total of 229,832,439 and 167,518,595 reads were obtained from the F- and S-pool, respectively, with a sequencing coverage of 92.14% regarding the B. napus cv. Darmor-bzh reference genome, and a GC content of 37%. After trimming the raw reads, 82.21 and 60.14 Gb of clean data, respectively, were generated.
Based on comparison with the reference genome, there were 58,610 SNPs and 120,651 InDels in the F- and S-pools. The low-quality SNPs/InDels (SNP/InDel index < 0.3) in both pools were deleted. The indices related to the remaining SNPs/InDels calculated for each pool were plotted onto the 19 B. napus chromosomes. Additionally, Δ(SNPs/InDel-index) values (calculated using the sliding window method) were plotted on the 19 B. napus chromosomes. The phenotypic difference between the fertile and sterile plants resulted in Δ(SNPs/InDel-index) ≥ 0.5 in a specific genomic locus, which indicates that this region contains the locus underlying the phenotypic difference (i.e., the Rfw locus). The significant signal region was only observed on chromosome A09 (not on the other 18 chromosomes) (Figures 3A,B). More specifically, the Δ(SNP/InDel-index) analysis preliminarily mapped Rfw to a 27.62–33.87 Mb region (6.25 Mb) on chromosome A09 (Figures 3C,D).
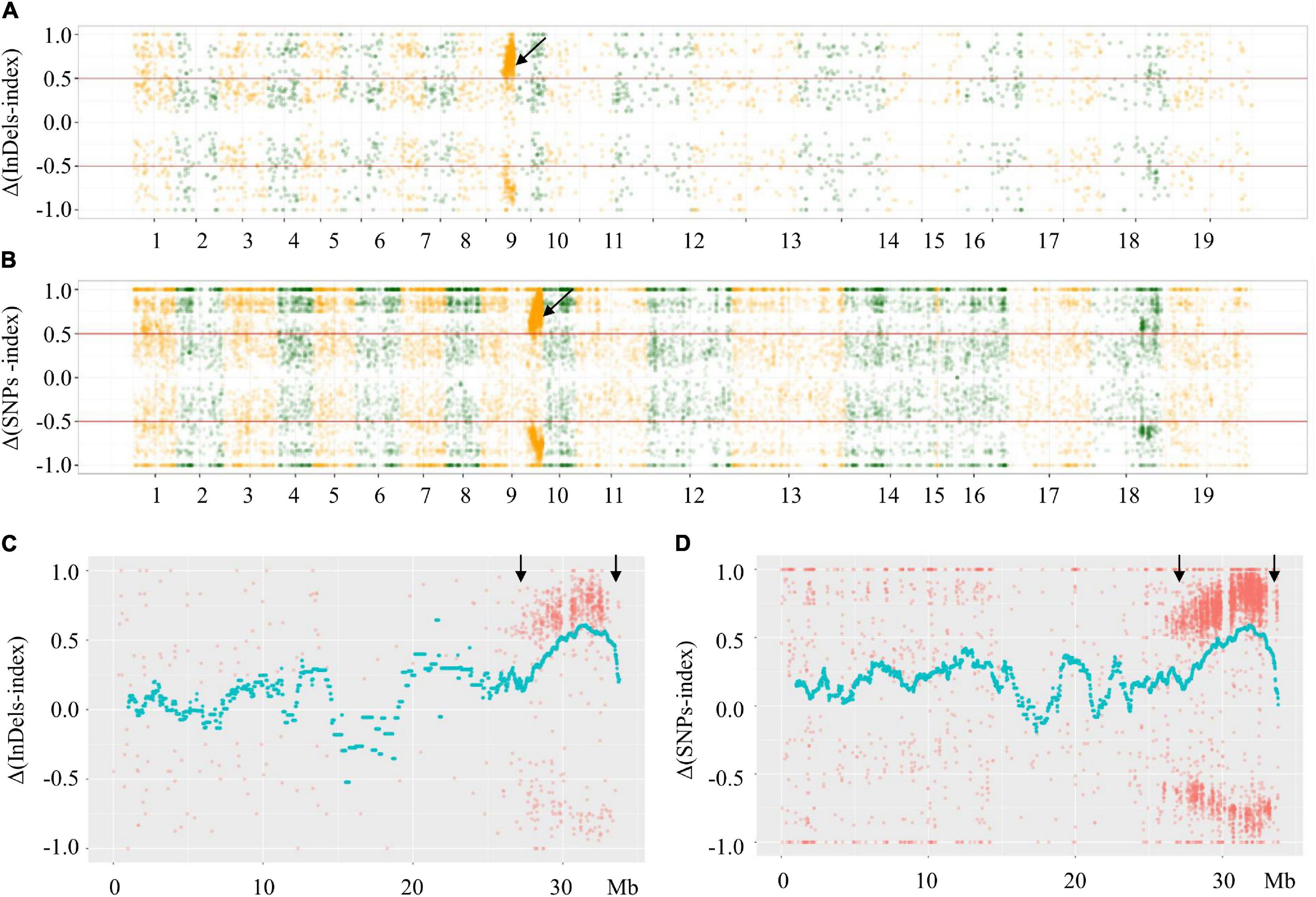
Figure 3. Distributions of Δ(SNPs/InDels-index) across B. napus chromosomes. (A) Δ(InDels-index) plot obtained by subtraction of F-pool InDel-index from S-pool InDel-index for F2 population; (B) Δ(SNPs-index) plot obtained by subtraction of F-pool SNP-index from S-pool SNP-index for F2 population; (C) Δ(InDels-index) on chromosome A09; (D) Δ(SNPs-index) on chromosome A09. The x-axes show the B. napus genome position. The y-axes show the index values; red lines in (A,B) represent index = | 0.5|. Each spot corresponds to an index value. The fitted curves indicate the average values of Δ(SNPs-index) or Δ(InDels-index) based on sliding window analysis. The black arrows indicate the candidate regions.
To verify and narrow down the Rfw region, 555 sterile F2 plants were used for further mapping. We selected 203 InDel markers in the candidate interval, and 13 of them were found to exhibit polymorphisms between reconstructed F- and S-pools from other fertile and sterile plants, respectively. These 13 markers (Figure 4) were used to assay the 555 sterile plants. We identified 4 and 2 recombinant plants using the markers ID-551A and IDC-705A, respectively, which were the closest markers flanking Rfw. Thus, Rfw was further mapped to a 30.88–31.98 Mb region on chromosome A09 (Figure 4).
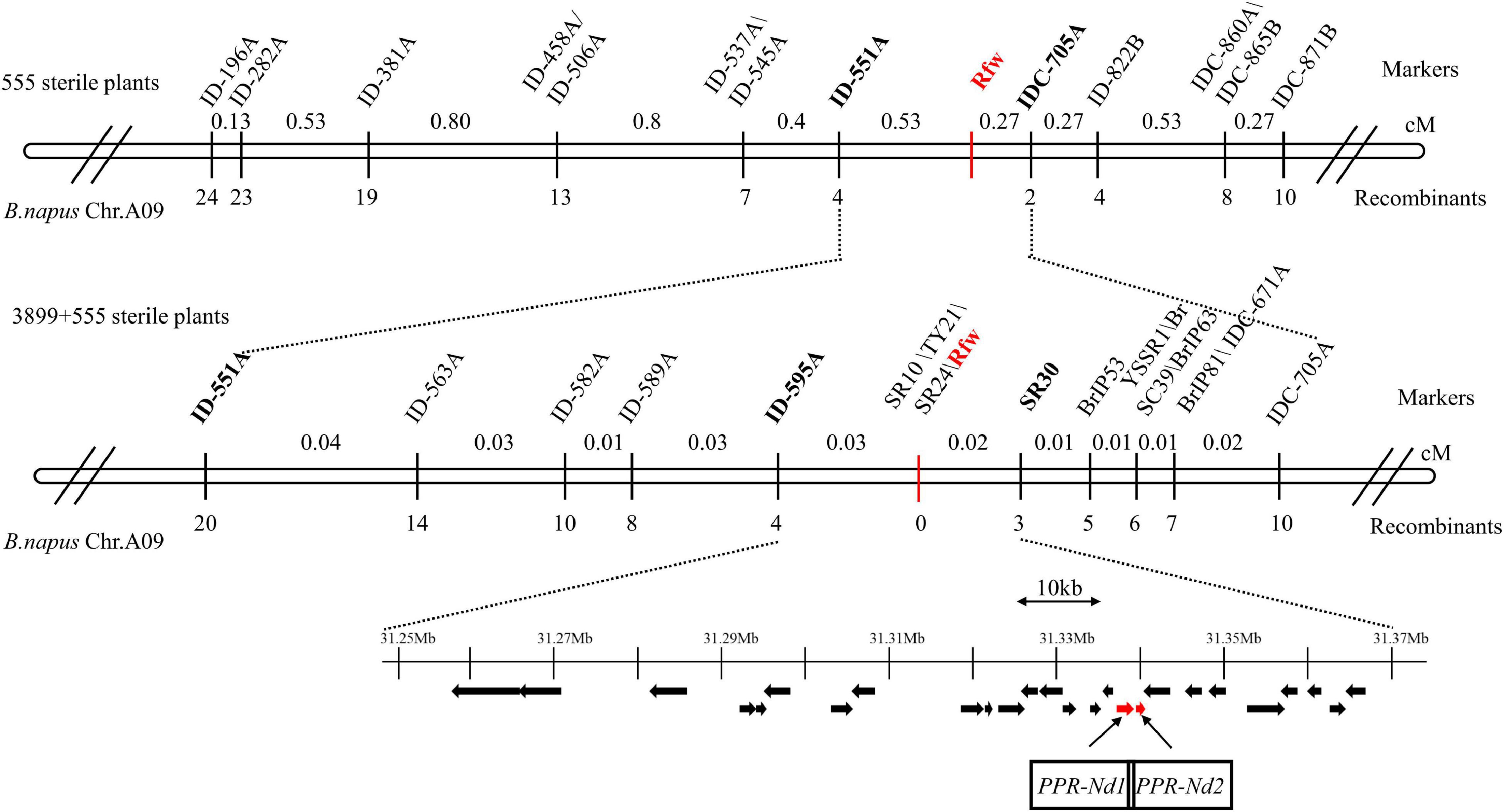
Figure 4. Fine mapping of the Rfw locus in Brassica napus. The number below each marker indicates the number of recombinant plants (with respect to each given marker and the Rfw locus). Black arrows represent predicted genes in the 115.62 kb target region on chromosome A09 of B. napus. Red arrows indicate candidate Rfw genes.
To finely map Rfw, 3899 sterile F2 plants were further analyzed using the ID-551A and IDC-705A markers, and another 16 and 8 recombinant plants were identified, respectively. Next, we selected 130 markers (81 InDels, 24 SSRs, and 25 publicly available molecular markers) in the 1.1 Mb candidate region, and 16 dominant markers (Figure 4) were found to exhibit polymorphism between reconstructed F- and S-pools. We identified 4 and 3 recombinant plants using ID-595A and SR30, respectively, which were the closest markers flanking Rfw. Thus, Rfw was located within a genetic distance of 0.05 cM in B. napus. Additionally, 3 markers (SR10, TY21, and SR24) co-segregated with the Rfw locus in the assayed plants (no recombinant plants) (Figure 4).
Candidate Rfw Genes
To isolate the candidate Rfw gene, all the genes (i.e., 26 genes, BnaA09g45870D–BnaA09g46120D) in the candidate region of the B. napus genome (Darmor-bzh and Zhongshuang11) were analyzed. One of these genes is a PPR gene (BnaA09g46030D). As the restorer line Hui01 originated from crossing WNJ01A CMS and B. rapa, and WNJ01A CMS originated from crossing B. juncea and B. napus, we also analyzed the candidate interval of chromosome A09 in B. rapa and B. juncea. There were 28 (Bra026863–Bra026890) and 26 (BjuA044103–BjuA044078) genes, including two PPR genes (Bra026882/Bra026884 and BjuA044085/BjuA044087) in the A genome of B. rapa cv. chiifu-401 and the AB genome of B. juncea var. tumida, respectively.
To determine the candidate region sequence, we constructed a BAC library for Hui01. Of the nine positive pools, three were randomly selected, and four positive clones were then identified in these pools. The previously identified closest flanking markers (ID-595A and SR30) were used to further confirm the physical location of the inserted fragment in these four positive clones. One clone (M36A7E6) contained the ID-595A locus, and three clones (M16A6E7, M8A5D1, and M8A1H10) contained the SR30 locus (Supplementary Figure 1). Among them, the M36A7E6 and M16A6E7 clones were selected for Illumina HiSeq and PacBio sequencing to obtain the candidate region sequence in Hui01.
The DNA insert size of M16A6E7 and M36A7E6 were 99,119 and 101,041 bp and the GC content were 35 and 37%, respectively. The interval between the closest flanking markers ID-595A and SR30 was 99.75 kb, with a 15.87 kb difference between it and the corresponding region (115.62 kb) in the reference genome of Darmor-bzh. Among the 28 predicted genes in this candidate region, there were two PPR genes [PPR-Nd1:(ORF19) and PPR-Nd2:(ORF20)], which were both only identified in Hui01 (not in WNJ01A CMS). PPR-Nd1 corresponded to BnaA09g46030D, Bra026882 and BjuA044087 while PPR-Nd2 corresponded to Bra026884 and BjuA044085 in the reference genomes. Because the majority of the cloned restorer genes belong to PPR family and the other genes except for PPR-Nd1 and PPR-Nd2 in this candidate interval have not been verified to be responsible for restoring fertility previously (Supplementary Table 3), the two PPR genes were further analyzed.
The CDS length of PPR-Nd1 in Hui01 is 1923 bp (with no intron). In comparison, the CDS length is 1794 bp (with a 110-bp intron) in the homolog BnaA09g46030D of Darmor-bzh, 1887 bp (with a 29-bp intron) in BjuA044087 of B. juncea var. tumida, and 1815 bp (with a 100-bp intron) in Bra026882 of B. rapa cv. chiifu-40. In the 5′ non-PPR domain region of PPR-Nd1, there was one InDel (–3 bp) and 24 SNPs compared to its Darmor-bzh homolog BnaA09g46030D, along with many InDels and SNPs in the PPR domain region (Supplementary Figure 2). Amino acid sequence alignment showed that there was an amino acid deletion and 10 amino acid substitutions in the 5′ non-PPR domain region that were specific to PPR-Nd1. Additionally, there were differences in motifs I and II of the PPR domain. The motif I sequence of PPR-Nd1 was identical to that of BjuA044087, but BnaA09g46030D and Bra026882 had long-fragment deletions and substitutions. The motif II sequence of PPR-Nd1 was identical to that of BnaA09g46030D and almost identical to that of Bra026882, while BjuA044087 had two deletions and one long-fragment substitution (Supplementary Figure 3).
PPR-Nd2 corresponds to homologs Bra026884 of B. rapa and BjuA044085 of B. juncea (none of them have introns). There were a variety of InDels/SNPs in the 5′ region (Supplementary Figure 4) and multiple length reductions and SNPs in the 5′ terminal amino acid sequence (Supplementary Figure 5). As PPR-Nd2 is not present in the Darmor-bzh genome, PCR amplification from 18 varieties of B. napus was carried out, and the gene was found to be present in some of them, such as Yangguang2009 (YG2009). The gene sequence in YG2009 was identical to that in Hui01. However, YG2009 appears not to be capable of restoring the fertility of WNJ01A CMS (data not shown), which means that this gene is not a candidate Rfw gene.
Analysis of the Expression Levels of Mitochondrial Gene Transcripts
Cytoplasmic male sterility in plants is generally caused by the formation of chimeric ORFs via mitochondrial genome rearrangement, which are usually co-transcribed with known genes or other ORFs (Chen and Liu, 2014). During the fertility restoration of CMS lines, the restorer genes regulate mitochondrial transcription, especially regarding the sterility genes (Li et al., 2018). To identify the mitochondrial genes regulated by Rfw, we selected 32 known protein-coding genes (Supplementary Table 2) based on two mitochondrial genomes (NCBI accession no. KJ461445 and JF920288) of B. juncea var. tumida and jiangpu, respectively (Chang S. et al., 2011; Zhao et al., 2016) and assessed their transcriptional levels in fertile and sterile F2 buds by RT-qPCR.
Most of the 32 known mitochondrial genes showed significant differences in transcription, except for eight genes (Figure 5). Regarding mtETC complexes I, III, IV, and V, nine nad genes, three cox genes, and four atp genes, ccmC, and ccmFN2 were significantly upregulated in fertile buds (and atp4, ccmB, ccmFC, and ccmFN1 were non-significantly upregulated while cob was non-significantly downregulated). Regarding the ribosome small subunit family, rps4, rps14, and rps3 were significantly upregulated (and rps12 was non-significantly upregulated). Regarding the ribosome large subunit family, only rpl16 was significantly upregulated (and rpl2 and rpl5 were non-significantly upregulated). Both tatC and matR were also significantly upregulated. These differentially expressed genes may participate in fertility restoration after regulation by Rfw.
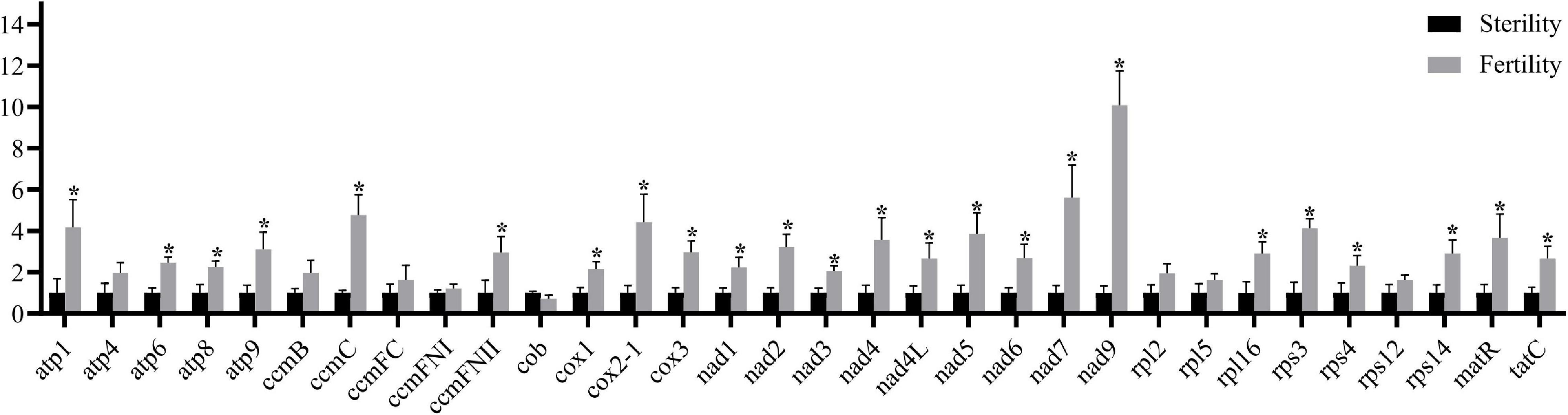
Figure 5. Mitochondrial gene transcriptional levels in fertile and sterile buds (0–2 mm) based on RT-qPCR. The x-axis shows the 32 known protein-coding genes from B. juncea mitochondrial genomes (NCBI accession nos. KJ461445 and JF920288). Bna.Actin7 was used as the internal control. Error bars indicate standard deviation (n = 3). *P < 0.05.
DEGs Analysis
To explore the molecular mechanism of Rfw regulation of fertility restoration, the transcriptomes of three fertile (Rfw1, Rfw2, and Rfw3) and three sterile (rfw1, rfw2, and rfw3) replicates from the F2 population were sequenced. After trimming the raw reads, the number of clean reads in the six samples ranged from 39,052,902 to 61,894,692. A total of 86.43 Gb clean bases was obtained, with each sample having > 11.66 Gb. The GC content was >47% and the Q30 percentage was >90%. Pearson’s correlation coefficients regarding the transcriptional levels among the three sterile bud replicates were ≥0.935 and those among the three fertile bud replicates were ≥0.889 (Supplementary Figure 6). Therefore, the RNA-seq data were suitable for further analysis.
Next, >74% of the clean reads (>70% unique mapped reads and 3.84–4.90% multiple mapped reads) were mapped to the B. napus cv. Darmor-bzh reference genome (Table 1).
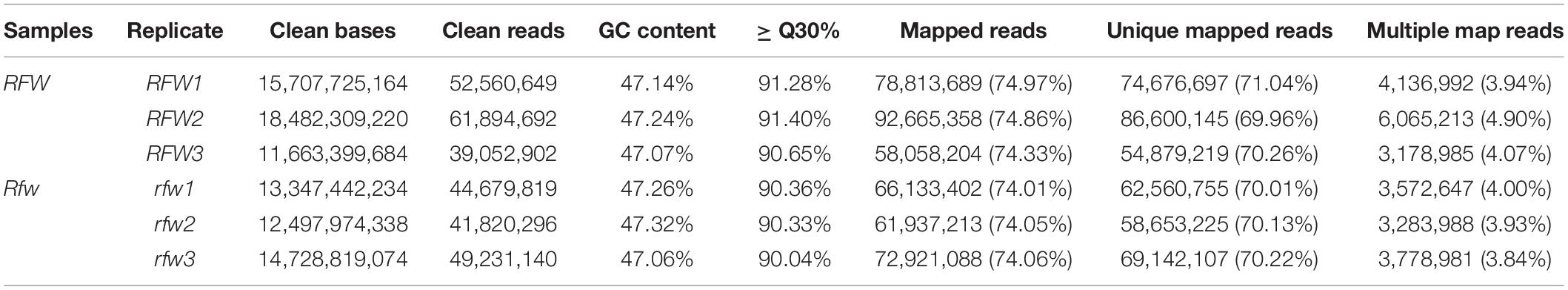
Table 1. Sequencing and mapped reads data.
We detected 104,348 genes in the RNA-seq data from the six replicates (3229 genes were novel, based on comparison to the reference genome). Subjecting the genes to BLASTx searches against COG, GO, KEGG, Swiss-Prot, eggNOG, and NR databases resulted in 102,843 annotated genes, of which 29253 (28.44%), 78382 (76.22%), 32214 (31.32%), 65560 (63.75%), 2305 (2.24%), and 102796 (99.95%) genes were aligned to the proteins in the six databases, respectively.
There were 1679 DEGs (1023 upregulated and 656 downregulated) between the fertile and sterile buds. The number of upregulated DEGs in the fertile buds was much higher than the number of downregulated DEGs.
GO and KEGG Analyses of DEGs
Of the 1518 GO annotated DEGs (905 upregulated and 613 downregulated DEGs) in the fertile buds, there were 130, 10, and 69 significantly enriched DEGs (Kolmogorov–Smirnov P-value < 0.05) in the GO biological process (BP), cellular component (CC), and molecular function (MF) GO categories, respectively (Supplementary Table 4), comprising 47 functional groups (Figure 6).
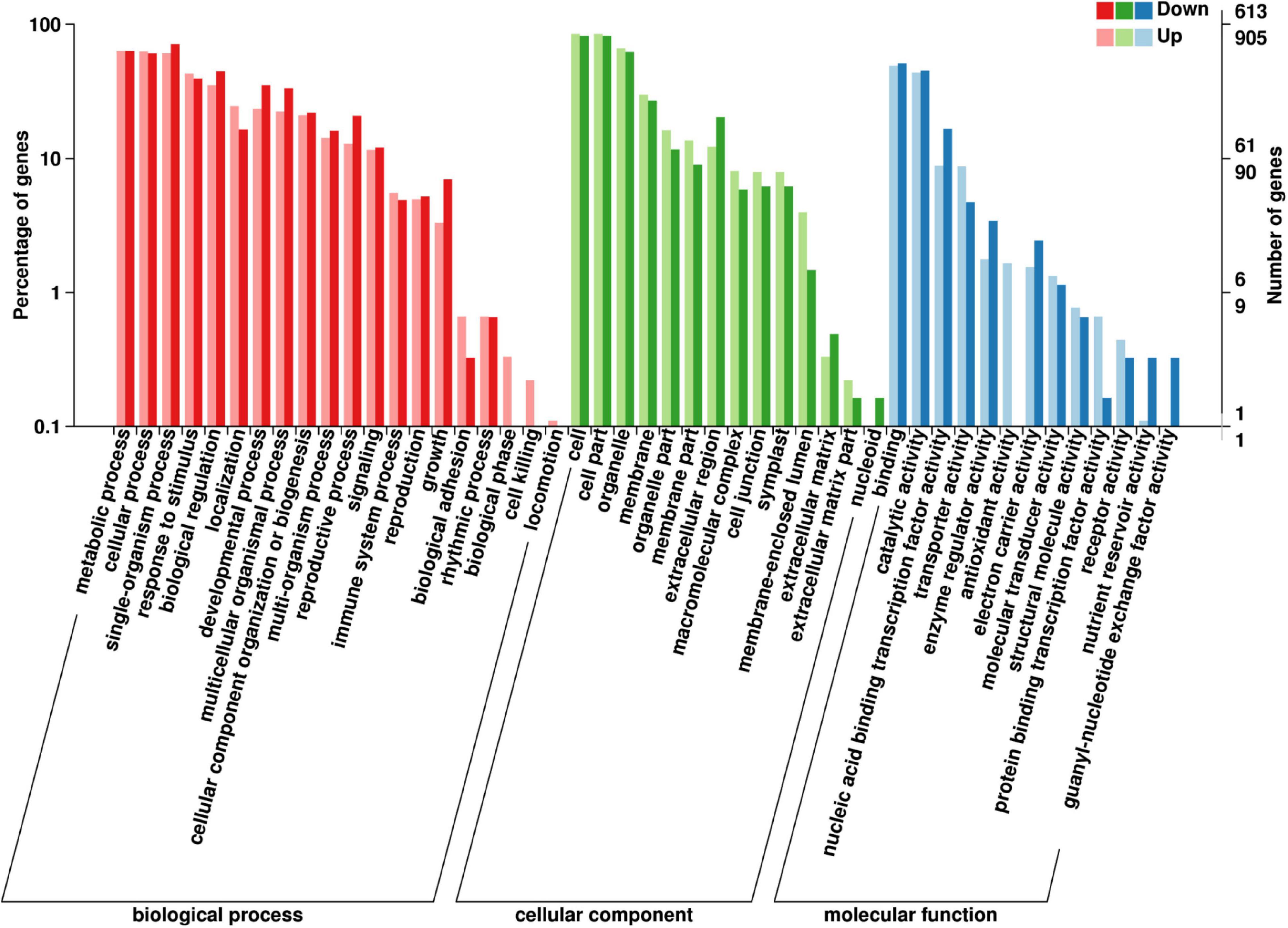
Figure 6. GO annotation of differentially expressed genes (DEGs) between fertile and sterile buds. The x-axis shows the terms in the three GO categories, the left y-axis shows the percentage of DEGs enriched in a specific term (out of all the DEGs in each category), and the right y-axis shows the number of DEGs enriched in a specific term.
In the BP category, the dominant groups were metabolic process (63.04%), cellular process (61.73%), and single-organism process (65.02%). Regarding the most significant term, specification of organ position (GO:0010159), five downregulated DEGs belong to the plant-specific TF YABBY family (Supplementary Figure 7A), which is involved in the feedback regulation of gibberellin biosynthesis in rice (Dai et al., 2007). In the CC category, the dominant groups were cell (83.33%) and cell part (83.33%). Regarding the plant-type cell wall term (GO:0009505), seven aspartic proteinase (AP) and nine pectin methyl-esterase (PME) DEGs were enriched in this term (Supplementary Figure 7A). Aspartic protease participates in stamen development and cell death in rice (Niu et al., 2013; Ko et al., 2014) and PME genes are involved in pollen development and pollen tube growth in B. campestris (Xiong et al., 2019). In the MF category, the dominant groups were binding (49%) and catalytic activity (44%). Regarding the sugar transmembrane transporter activity term (GO:0051119), six upregulated SWEET DEGs in fertile buds were enriched in this term (Supplementary Figure 7A). SWEET1/8, which are highly expressed in Arabidopsis stamens, play important roles in glucose transport for pollen tube growth and starch accumulation in pollen (Chen et al., 2010).
The 581 KEGG-annotated DEGs (382 upregulated and 199 downregulated) were assigned to 107 KEGG pathways. The five most enriched categories were cellular processes (2.6%), environmental information processing (5.3%), genetic information processing (14.5%), metabolism (76.1%), and organismal systems (1.5%) (Supplementary Table 5). Pollen abortion and fertility restoration were shown to involve transport and catabolism, signal transduction, amino acid metabolism, carbohydrate metabolism, energy metabolism, and metabolism of terpenoids and polyketides, etc. (Supplementary Table 5). More specifically, the upregulated DEGs were significantly enriched in 12 pathways, like lysine degradation (ko00310) and phenylalanine metabolism (ko00360) (Figure 7A). The downregulated DEGs were significantly enriched in 18 pathways, including cutin, suberine, and wax biosynthesis (ko00073), pentose and glucuronate interconversions (ko00040), and fatty acid metabolism (ko01212) (Figure 7B).
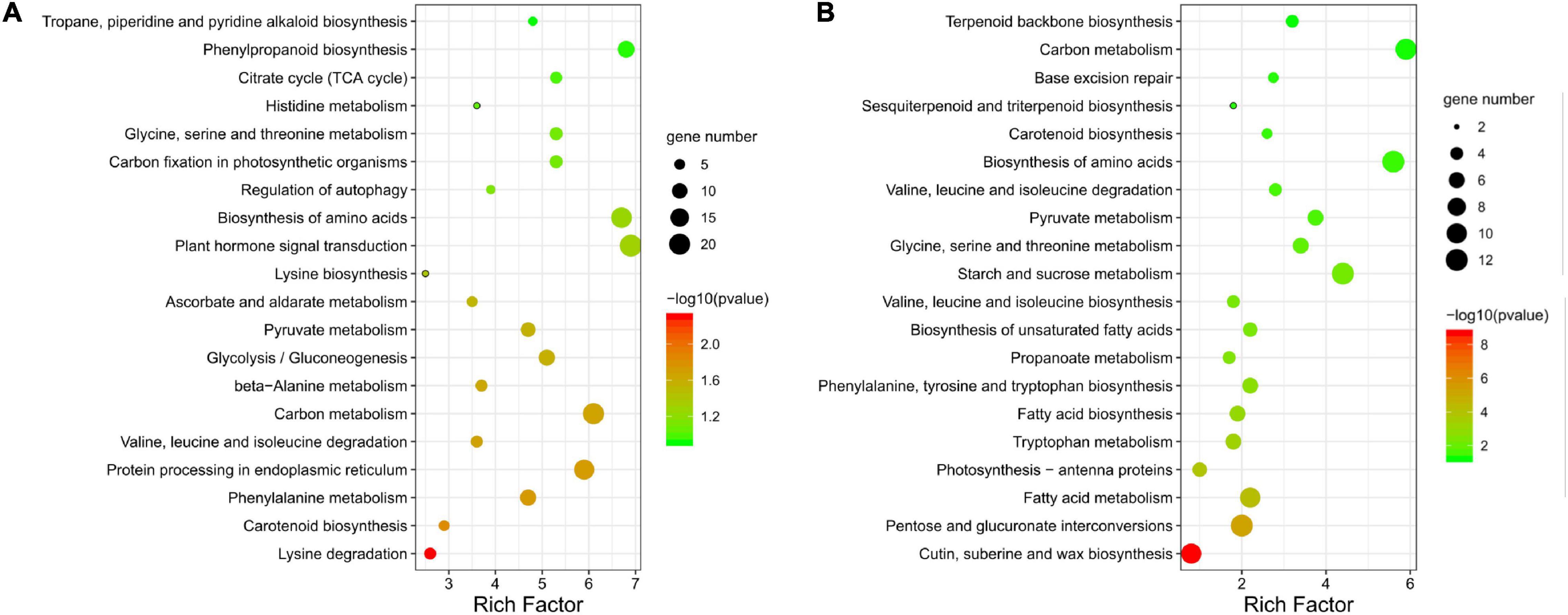
Figure 7. KEGG pathway annotation of differentially expressed genes (DEGs). Top 20 enriched KEGG pathways for (A) upregulated and (B) downregulated DEGs between fertile and sterile buds. The rich factor is the ratio of the number of DEGs to the total number of genes in each pathway. The color and size of the dots indicate the P-value and the number of DEGs mapped to the pathway, respectively.
DEGs Specific to Fertile and Sterile Buds
Differentially expressed genes specific to fertile buds may be involved in fertility restoration, while DEGs specific to sterile buds may be associated with CMS. Among the DEGs, 261 specifically expressed genes were identified (9 and 252 in sterile and fertile buds, respectively). GO analysis of the fertile bud-specific DEGs showed that the enriched BP terms included pollen exine formation (GO:0010584), carbohydrate metabolic process (GO:0005975), and SKP1-CUL-F-box (SCF) complex assembly (GO:0010265). The enriched CC terms included integral component of plasma membrane (GO:0005887), cullin-RING ubiquitin ligase complex (GO:0031461), and condensed nuclear chromosome (GO:0000794). Lastly, the enriched MF terms included sugar transmembrane transporter activity (GO:0051119), protein-lysine N-methyltransferase activity (GO:0016279), glucan endo-1,3-beta-D-glucosidase activity (GO:0042973), cation binding (GO:0043169), and oxidoreductase activity (acting on the aldehyde or oxo group of donors, NAD or NADP as acceptor, GO:0016620) (Figure 8A).
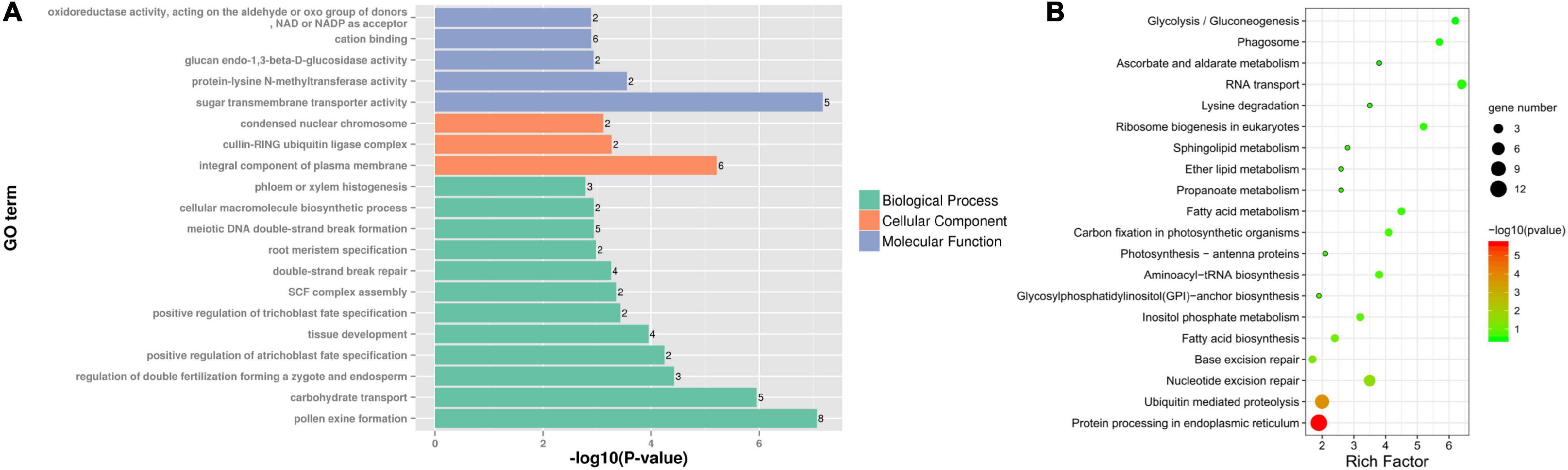
Figure 8. GO and KEGG analyses of differentially expressed genes (DEGs) specific to fertile buds. Enriched (A) GO terms and (B) KEGG pathways of DEGs specific to fertile buds.
Additionally, 55 fertile bud-specific DEGs were annotated with KEGG pathways. The four most significant enriched pathways were protein processing in endoplasmic reticulum (ko04141), ubiquitin-mediated proteolysis (ko04120), nucleotide excision repair (ko03420), and base excision repair (ko03410) (Figure 8B).
DEGs Related to Hormones and Anther/Pollen Development
Stamen development is often modulated by hormone biosynthesis and signaling pathways, and certain hormone-related mutants often display male sterility (Song et al., 2013). We identified 31 hormone-related DEGs. Of these, 3, 9, 7, 4, 1, 2, and 5 were related to gibberellin, auxin, abscisic acid (ABA), cytokinin, ethylene, jasmonic acid (JA) and salicylic acid, respectively (Supplementary Figure 7B). All DEGs related to JA and salicylic acid were upregulated in the fertile compared to the sterile buds. Additionally, BnaA09g48160D, which encodes the homologous protein Arabidopsis response regulator 4 (ARR4), was specifically expressed in fertile buds. ARR4 modulates photomorphogenesis by interacting with the red/far-red light photoreceptor phytochrome B and regulates the cytokinin response pathway (Mira-Rodado et al., 2007).
After using The Arabidopsis Information Resource (TAIR) database to annotate the DEGs, we discovered that 44 DEGs (35 upregulated and 9 downregulated in fertile buds) were related to anther and pollen development (Supplementary Table 6). The upregulated genes included ALA6, UGE3, DL1C, COX11, PSS1, ARID1, P5CS1, MAZ1, ABCG6, INP1, SPS2F, KOM, UPEX1, BHLH010, BHLH089, MYB108, CALS5, PRX9, PRX40, TAP35/TAP44, AMS, SWEET8, and PIRL1. Among them, MAZ1, ABCG6, SPS2F, KOM, and UPEX1 are involved in pollen wall (including pollen exine) formation. Four INP1 genes play important roles in pollen surface aperture formation. Two CALS5 genes (which encode callose synthase 5) are responsible for the synthesis of callose that is deposited at the primary cell wall of meiocytes, tetrads, and microspores. TAP35/TAP44 and AMS participate in tapetal cell development. SWEET8 and PIRL1 function in microspore development. Lastly, five BHLH and MYB TF DEGs (four of which were upregulated) are involved in anther development and dehiscence.
The downregulated genes included POE1, IPE1, and BHLH010. Among them, all seven DEGs of the POE1 family (which encode Pollen Ole e 1 allergen and extensin family proteins) were downregulated in fertile buds. They can be epigenetically controlled via histone H3 lysine 27 trimethylation (H3K27me3) (Hu et al., 2014).
PPI Network
The PPI network contained 6077 interactions among 658 proteins, encoded by 423 upregulated, and 235 downregulated DEGs. There were ten modules, which were composed of 610, 18, 9, 5, 3, 3, 3, 3, 2, and 2 DEGs, respectively (Supplementary Figures 8A–J). The 10 hub genes with the top degree scores, were BnaA09g56400D, BnaA10g18210D, BnaA10g18220D, BnaC09g41740D, BnaC04g03080D, BnaA05g25330D, BnaC03g51080D, BnaC09g01600D, BnaA01g10430D, and BnaCnng18680D. The first 4 of these had the highest degree scores and were all upregulated in fertile buds (Supplementary Figure 8K). They all encode RAD23d (ubiquitin receptor radiation sensitive23d), which plays essential roles in the cell cycle, morphology, and fertility of higher plants by delivering ubiquitinated substrates to the 26S proteasome (Farmer et al., 2010). Rfw may upregulate these RAD23d hub genes so that they could modulate the expressions of their interacting genes, thereby mediating fertility restoration. The remaining six hub genes were annotated as MPK6, HAOX2, bt1, and GSO1 (three times), respectively.
The DEGs that interacted with the hub genes were subjected to GO and KEGG analyses. Pathways related to carbohydrate metabolism (such as N-glycan biosynthesis, glycolysis/gluconeogenesis, the TCA cycle, and starch and sucrose metabolism) tended to be upregulated. The JA response pathway was also upregulated. Other pathways potentially related to male fertility were also dramatically upregulated, such as ubiquitin-mediated proteolysis, purine metabolism, meiotic DNA double-strand break formation, protein processing in endoplasmic reticulum, and endocytosis. In addition, RNA-related pathways (spliceosome, RNA transport, and aminoacyl-tRNA biosynthesis) were upregulated. In contrast, pentose and glucuronate interconversions, fatty acid metabolism, and cutin, suberine, and wax biosynthesis pathways were highly downregulated (Figure 9).
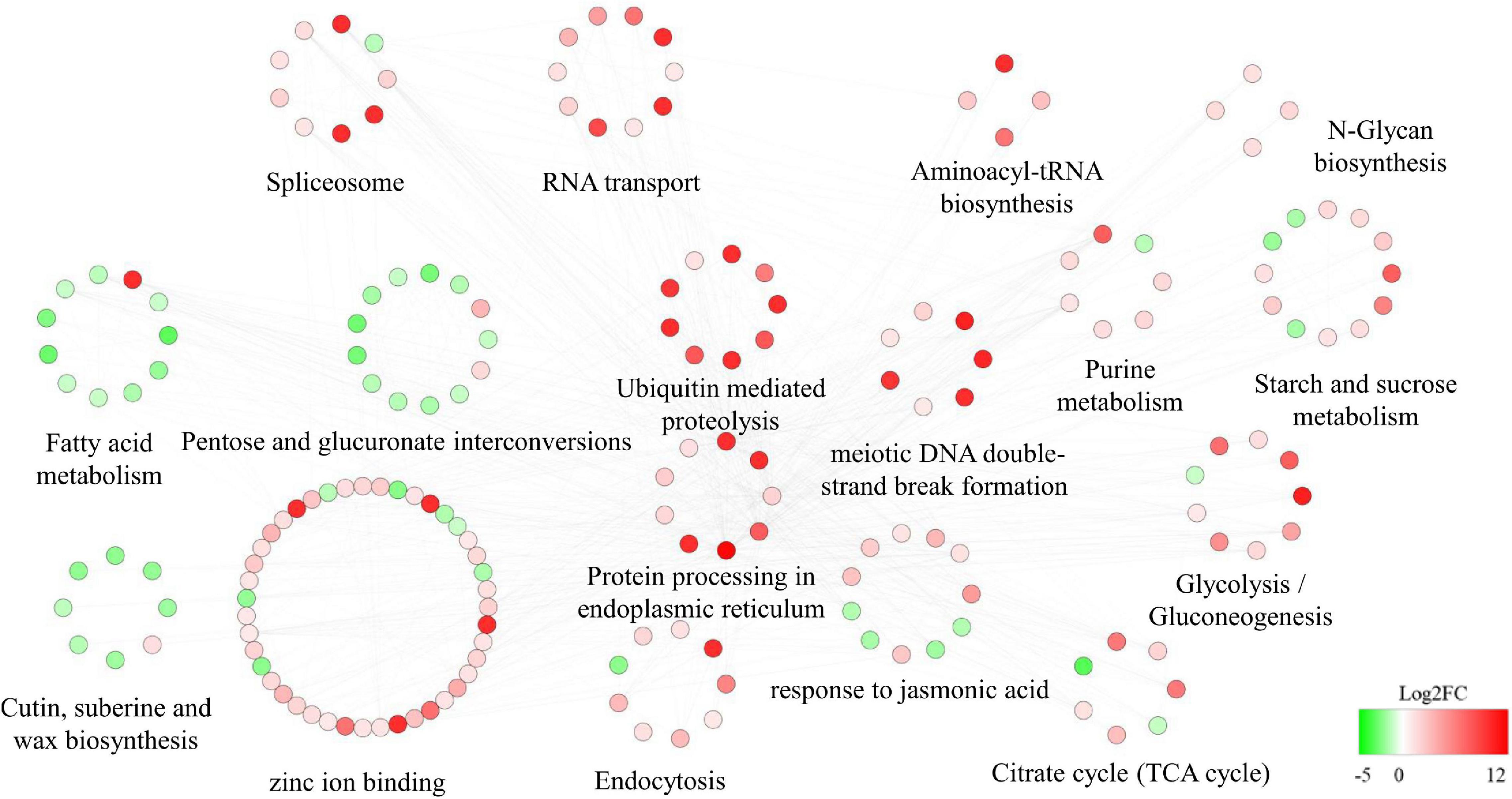
Figure 9. Protein–protein interaction (PPI) network related to the regulator Rfw. Red and green indicate upregulated and downregulated differentially expressed genes [DEGs; the shade reflects log2(fold change)].
RT-qPCR Validation
To validate the RNA-seq results, 14 DEGs in enriched pathways and related to fertility restoration were subjected to RT-qPCR (Supplementary Table 7). Of these DEGs, 10 were upregulated in fertile buds, comprising LKR, ALDH2B4, PRX40, PRX9, SWEET8, CALS5, CUL1, SKP1, RAD23A, and RAD23D. The remaining four DEGs were downregulated in fertile buds, comprising CYP86A4, CER1, HTH, and POE1. The RT-qPCR results showed that their expression tendencies were consistent with those from RNA-seq analysis, indicating the reliability and accuracy of the RNA-seq and the pathway enrichment results (Figure 10).
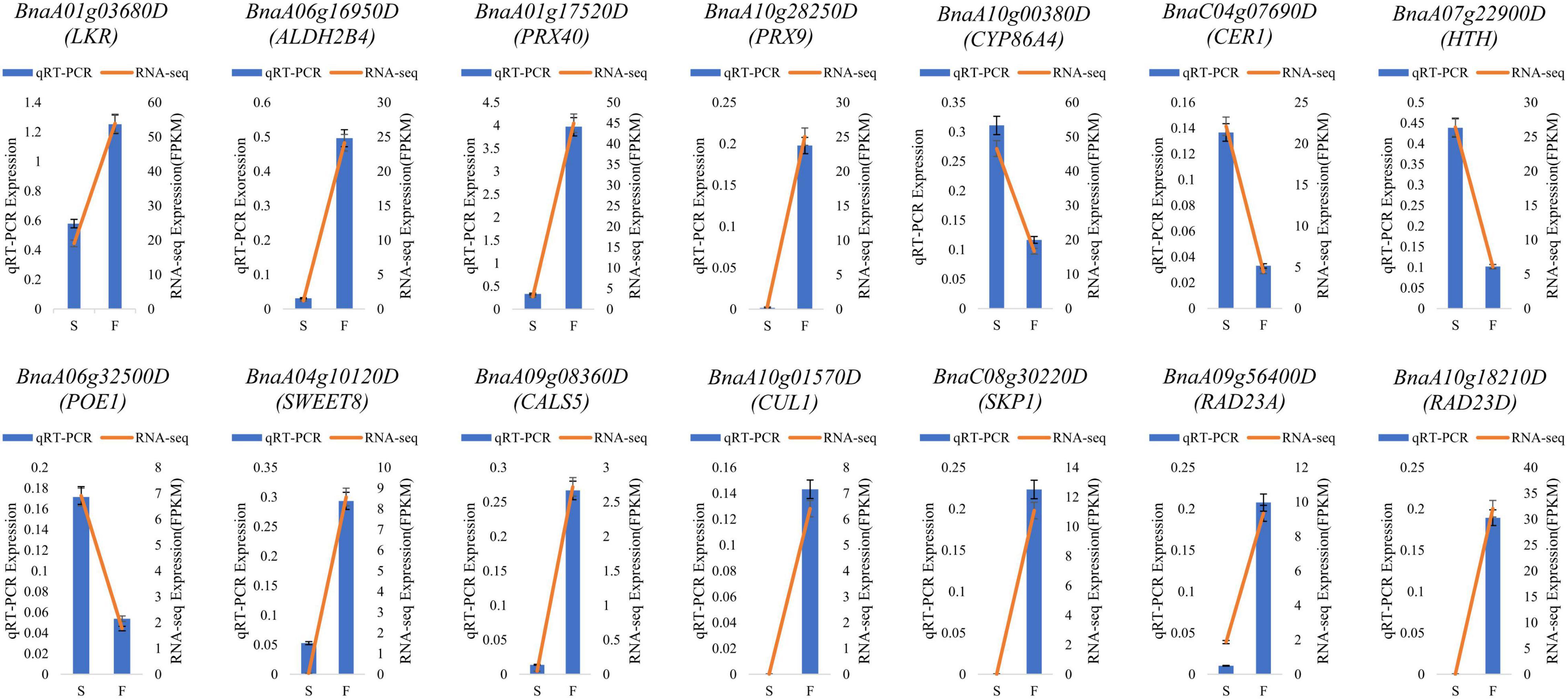
Figure 10. RT-qPCR validation. S, sterile sample; F, fertile sample.
Discussion
Cytological Characteristics and Mitochondrial Gene Transcription in Sterile and Fertile Buds
The WNJ01A CMS line displayed a complete pollen abortion phenotype, and its fertility could be restored by the restorer line Hui01. By observing paraffin-embedded sections of sterile and fertile buds, we found that the sporogenous cell stage was the critical stage regarding pollen degeneration, with major sporogenous cells exhibiting loose and irregular arrangements in sterile anthers. The pollen abortion stage of WNJ01A CMS is earlier than in pol CMS and ogu CMS. Regarding pol CMS, the pollen abortion stage is the polarization stage of the archespore, in which sporogenous cells do not differentiate and normal tetrads are not produced in the sterile anthers (An et al., 2014). Regarding ogu CMS, the pollen abortion stage involves the transition from the tetrad to single-nucleus pollen. The pollen development is impaired due to premature death of tapetal cells at the vacuolate microspore stage (Gonzalez-Melendi et al., 2008). Additionally, the pollen-abortion stage of WNJ01A CMS is later than that of hau CMS; hau CMS stamen primordia lack normal polarization and form petal primordia at the differentiation stage of stamen primordia (Wan et al., 2008).
Our RT-qPCR results regarding 32 known protein-coding mitochondrial genes indicate that most of these genes were significantly upregulated in fertile buds. For instance, genes related to mtETC complexes I, III, IV, and V were markedly upregulated in fertile buds. This implies that the mtETC was disrupted (with downregulated expression) in sterile buds. In contrast, in fertile plants, this disruption was prevented, restoring normal pollen development. Certain CMS types also display the male-sterile phenotype due to mtETC disruption (Fujii et al., 2010), such as sugar beet CMS-G (Ducos et al., 2001), pepper CMS-Peterson (Ji et al., 2013), rice CMS-HL (Peng et al., 2010; Wang et al., 2013), sunflower CMS-PET1 (Sabar et al., 2003), and pearl millet CMS plants (Kale and Munjal, 2005). Only one mtETC-related gene (cob) was downregulated (non-significantly) in fertile buds.
Mapping of Rfw Gene
Rfw was preliminarily mapped to 27.62–33.87 Mb on chromosome 9 by BSA-seq. Interestingly, there are many PPR genes in this region, including two restorer genes, Rfp (identified in Polima CMS; locus: 31.64–31.67 Mb) (Liu et al., 2012) and Rfn (identified in nap CMS; locus: 31.79–31.81 Mb) (Liu et al., 2017). Recent research has revealed that a highly dense cluster of restorer-of-fertility-like (RFL) genes exists in this region on chromosome A09 and on a collinear region on chromosome C8 in B. napus (Ning et al., 2020). A similar PPR cluster is present in the collinear region of Arabidopsis chromosome A1. The main driving force for evolution of this region may relate to the segmental duplication and retrotransposition processes of An subgenomes (Gaborieau and Brown, 2016). PPR genes are probably derived from a few common ancestors and have conserved regulatory roles regarding mitochondrial genes. During evolution, the number of PPR genes may have gradually increased and then they may have developed functional specificity (due to sequence variation) related to fertility restoration in relation to various CMS systems.
The PPR gene PPR-Nd1 in the Hui01 line (which is a homolog of BnaA09g46030D in Darmor-bzh) is a candidate Rfw gene in the finely mapped Rfw interval. BnaA09g46030D is known to contain mitochondrial localization signals and is highly upregulated in buds, indicating that it can be regulated and functions in mitochondria (Wang H. et al., 2021). However, comparing the CDS sequences of PPR-Nd1 and Rfh (BnaA09g46030D, which restores the sterile phenotype of hau CMS, Wang H. et al., 2021), there are multiple SNPs and one InDel (–3 bp) difference, and there are also differences in the amino acid sequences. Critically, whether PPR-Nd1 can restore the fertility of WNJ01A CMS, and the degree of pollen fertility after restoration, remain unknown. In addition, the maintaining and restoring relationship between WNJ01A CMS and Hau CMS is required to be further analyzed.
KEGG Analysis of DEGs
In plants, high lysine levels can be toxic to cells (Arruda et al., 2000). For instance, Arabidopsis seeds with superabundant lysine exhibited delayed germination and seedling establishment (Zhu and Galili, 2003). Our KEGG analysis of the upregulated DEGs showed that the lysine degradation pathway (ko00310) was enriched, which involved DEGs in the lysine-ketoglutarate reductase/saccharopine dehydrogenase (LKR/SDH), aldehyde dehydrogenase (ALDH), and histone-lysine N-methyltransferase/SET domain group (ASH/SDG) families. These families balance Lys levels in plants (Tang et al., 2000; Stepansky and Galili, 2003).
LKR/SDH encode the first two key enzymes in the lysine degradation pathway (Stepansky et al., 2006). They are upregulated in flowers and strongly upregulated by ABA and JA treatment in Arabidopsis. ALDHs catalyze oxidation of α-aminoadipic semialdehyde during lysine degradation. Additionally, they are upregulated in Arabidopsis anthers and can eliminate lesions induced by toxic aldehydes and reactive oxygen species (Shin et al., 2009; Brocker et al., 2010; Shen et al., 2012; Hou and Bartels, 2015; Zhao et al., 2018). In maize T-CMS, the restorer gene RF2 encodes an ALDH that can complement the lack of ALDH activity in its mitochondria to reverse the male sterility caused by the CMS protein URF13 (Liu et al., 2001). ASH/SDG genes are negative regulators of H3K27me3 (which is known to downregulate genes involved in cellular fate maintenance during development in plants and animals) (Cartagena et al., 2008; Lee et al., 2015; Crevillen, 2020; Cai et al., 2021). ASH/SDG is located upstream of ALDH in the lysine degradation pathway (ko00310). Hence, ASH/SDG probably mediate fertility restoration by affecting the H3K27me3 of the related genes. Our results indicate that upregulation of the lysine degradation pathway is very important to maintain the normal lysine level for anther development in plants with restored fertility.
Another significantly enriched pathway, phenylalanine metabolism (ko00360) involves the peroxidase (PRX) and copper amine oxidase (CuAO) families. PRX family members, such as PRX9 and PRX40, are essential extensin peroxidases that maintain the integrity of the tapetum and microspore cell walls during anther development in Arabidopsis (Jacobowitz et al., 2019). CuAOs mediate plant cell wall formation, maturation, and programmed cell death (Planas-Portell et al., 2013; Tavladoraki et al., 2016). Our results indicate that the phenylalanine metabolic pathway mediates pollen cell wall formation during fertility restoration of WNJ01A CMS.
Our results also showed that the downregulated DEGs were enriched in cutin, suberine, and wax biosynthesis (ko00073) and this pathway is involved in normal mature pollen development in B. napus (Shi et al., 2020). The DEGs included genes in the CYP86A, CER1-like, and glucose-methanol-choline (GMC) families. CYP86A are highly upregulated in mature Arabidopsis flowers, and they function as fatty acid ω-hydroxylases (Duan and Schuler, 2005) and may play indispensable roles in pollen tube growth (Koiwai and Matsuzaki, 1988; Wolters-Arts et al., 1998). Cuticular wax is composed of very-long-chain fatty acids (VLCFAs) and VLC alkane biosynthesis is regulated by CER1-like1 specific cofactors (Preuss et al., 1993; Lee and Suh, 2013; Wu et al., 2019). The GMC protein HTH regulates cutin biosynthesis and postgenital organ fusion during flower development (Krolikowski et al., 2003; Xu et al., 2017).
DEGs Related to Pollen and Anther Development
During the fertility restoration of WNJ01A by restorer line Hui01, 44 DEGs were confirmed to be involved in pollen and anther development. In fertile buds, multiple genes that play predominant roles in tapetum, pollen wall, and callose development were upregulated. These DEGs included two bHLH TFs (BHLH089 and BHLH010), two extensin peroxidases (PRX9 and PRX40), tapetum-specific TAP35/TAP44, and two CALS5 genes. During pollen formation, tapetum cells play central roles in callose degradation, pollen exine formation, and the provision of various nutrients for pollen development (Han et al., 2021). Our cytological characterization of the abortive anthers showed that the tapetum cells (which surround the microspores and provide crucial enzymes and nutrients for microsporogenesis and pollen wall development, Ferguson et al., 2017) were degraded. This likely explains why the microspores were degraded in the sterile buds. Previous studies have highlighted that, during the microspore stage, two AMS TFs (Xu et al., 2014), two SWEET8 (also called RPG1) genes (Guan et al., 2008; Chen et al., 2010), PSS1 (Forsthoefel et al., 2010; Yamaoka et al., 2011), and P5CS1 (Mattioli et al., 2018) are essential for pollen development. These genes were all upregulated in the fertile buds. In fact, most of the DEGs related to pollen and anther development were upregulated in the fertile buds, indicating that they were modulated by Rfw to achieve normal pollen and anther development.
Among the DEGs related to pollen and anther development, three types of genes were significantly downregulated, comprising seven POE1 (Pollen Ole e 1 allergen and extensin), one IPE1 (irregular pollen exine1) gene, and one bHLH10 gene. Recent research has suggested that at least 13 AtPOE1 gene loci were modified by H3K27me3 in Arabidopsis (Hu et al., 2014). Importantly, POE1 protein may participate in pollen tube emergence and guidance (Tang et al., 2002; Alche et al., 2004; Hamman-Khalifa et al., 2008), and H3K27me3 is involved in pollen sporophyte development (Hoffmann and Palmgren, 2013). Regarding IPE1, the putative oxidative pathway of ω-hydroxy fatty acids, which depends on IPE1, plays significant roles in anther cuticle and pollen exine formation in maize (Chen et al., 2017).
DEGs Specific to Fertile Buds
Among the KEGG pathways enriched in the DEGs specific to fertile buds, the ubiquitin–proteasome pathway was identified, with three SKP1-like genes, three Cullin 1 (CUL1) genes, and three RAD23D genes being specifically detected in fertile buds. In lily (Lilium longiflorum), three LSK1–3 (SKP1-like) genes play critical roles in regulating pollen tube elongation (Chang et al., 2009). In Arabidopsis, AtCUL1 plays an important role in JA signaling (Ren et al., 2005). Furthermore, in Arabidopsis, SCF complexes regulate JA-responsive genes related to pollen development (Devoto et al., 2002; Xu et al., 2002). Thus, our results indicate that fertility restoration of WNJ01A CMS relies on the ubiquitin-proteasome system (UPS).
Role of RAD23d in PPI Network
In the PPI network, the hub genes with the highest degree scores were all RAD23d genes. The RAD23 family provides an essential connection between ubiquitylated proteins and the 26S proteasome in Arabidopsis (Lahari et al., 2017). In addition, RAD23 has been identified as a ubiquitin-like (UBL)/ubiquitin-associated (UBA) protein, which is key for ubiquitin-mediated protein degradation (Andersson et al., 2005; Zuo and Mahajan, 2005; Dantuma et al., 2009). The UPS is extremely important in plant growth and development as it regulates proteasome-dependent protein turnover (Lowe et al., 2006; Wade and Auble, 2010). During the development of anthers with abnormally active meiosis, DNA repair is essential. Rad protein is involved in recognition of and binding to damaged DNA to correct DNA lesions (Gallego et al., 2000). Rad23 can also efficiently initiate the cell cycle and nucleotide excision repair-like repair pathway (Lahari et al., 2017; Okeke et al., 2020). Interestingly, the UPS not only removes incorrectly folded proteins, but also ensures appropriate spatial and temporal protein distributions (Medina et al., 2012; Liang et al., 2014; Chen and Walters, 2016). For example, RAD23B is key in pollen development as it controls the turnover of the key cell cycle protein KIP-related protein 1 (KRP1) (Li et al., 2020). In summary, RAD23d proteins may mediate fertility restoration of WNJ01A, regulated by Rfw, by repairing damaged DNA and degrading sterility-related proteins via the UPS, along with potentially regulating the degradation of other proteins and modulating hormone signals.
Mechanistic Model
After transcriptome sequencing of sterile and fertile flower buds, the fertile/sterile bud-specific DEGs, pollen/anther development- and hormone-related DEGs, and DEGs in the PPI network were subjected to GO and KEGG analysis. Based on the results, we propose a model of the mechanism of fertility restoration regulated by the restorer gene Rfw (Figure 11).
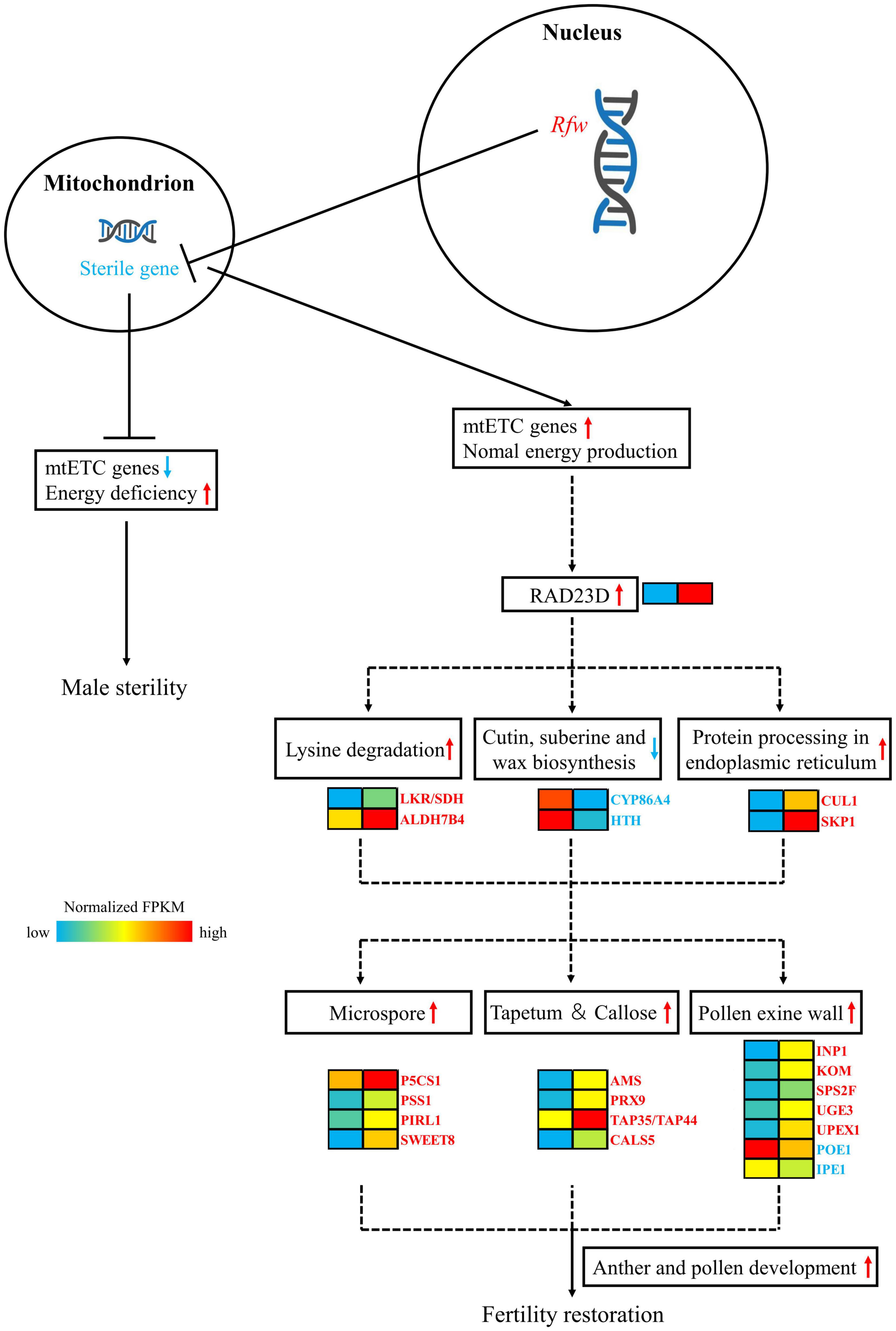
Figure 11. Mechanistic model of the role of the sterile gene of WNJ01A CMS and the role of Rfw in fertility restoration. In the heat map, the left-hand side represents sterile buds and the right-hand side represents fertile buds.
In mitochondria, the male sterility gene downregulates mtETC genes, which causes energy deficiency and results in the formation of flower buds exhibiting male sterility. The nuclear genome possesses an Rfw restorer gene, which can inhibit the expression of the male sterility gene at the level of transcription or translation or by another mechanism. The Rfw gene ensures that the function of the mtETC genes and mitochondrial energy production return to normal.
As a result of the effect of Rfw, RAD23 might be initially up-regulated in fertile buds. Then, the genes LKR/SDH, ALDH7B4, CUL1, SKP1 and their related pathways of lysine degradation, protein processing in endoplasmic reticulum are upregulated. In contrast, the genes CYP86A, HTH and their related pathways of cutin, suberine, and wax biosynthesis are downregulated. Later, the genes related to tapetum, pollen exine wall, microspore, and callose development, such as SWEET8, CALS5, TAP35/44, PRX9, PRX40, AMS, etc. are upregulated and POE1, IPE1, etc. are downregulated. Ultimately, the biological processes of anther and pollen development exhibit normal function and the flower buds restored fertility.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioProject, PRJNA739480.
Author Contributions
CL and QY conceived the experiment. FW, CZ, and JW provided advice on the experimental design. QY performed most of the experiments and analyzed the data. XN participated in paraffin-embedded sections experiments. JX participated in phenotypic investigate. FH participated in data analysis. CL and QY wrote the manuscript. All the authors reviewed and approved this submission.
Funding
This study was supported by the National Key Research and Development Program of China (grant 2016YFD0100202).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer ZL declared a shared affiliation, with no collaboration, with the authors to the handling editor at the time of the review.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank, Qinyong Yang (Huazhong Agricultural University) for his help in BSA-seq analysis, Yongtai Yin (Huazhong University of Science and Technology) for his help in PPI analysis, Qinghua Zhang (Huazhong Agricultural University) for his help in Illumina sequencing, and Yue Jia (Huazhong Agricultural University) for his help in transcriptome analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.721980/full#supplementary-material
Supplementary Figure 1 | Two sequenced BAC clones (four replicates each) verified using molecular markers. The closest flanking molecular markers (ID-595A and SR30) and a co-segregating marker (TY21) in the finely mapped candidate interval were identified by PCR amplification. PCR products were detected using 1.2% agarose gel.
Supplementary Figure 2 | Full-length CDS region alignment between PPR-Nd1, BnaA09g46030D, BjuA044087, and Bra026882. Black background: consensus sequences; red line: PPR motif region.
Supplementary Figure 3 | Full-length protein region alignment between PPR-Nd1, BnaA09g46030D, BjuA044087, and Bra026882. Black background: consensus sequences; red line: PPR motif region; purple line: motif I; blue line: motif II.
Supplementary Figure 4 | Full-length CDS region alignment between PPR-Nd2, BjuA044085, and Bra026884. Black background: consensus sequences; red line: PPR motif region.
Supplementary Figure 5 | Full-length protein region alignment between PPR-Nd2, BjuA044085 and Bra026884. Black background: consensus sequences; red line: PPR motif region.
Supplementary Figure 6 | Pearson correlation analysis of the transcript levels in three fertile plant buds (RFW1, RFW2, and RFW3) and three sterile plant buds (rfw1, rfw2, and rfw3).
Supplementary Figure 7 | Heatmap of DEGs. (A) YABBY, PEM, APs, and SWEET family differentially expressed genes (between fertile and sterile buds) that were enriched in the top ten GO terms. BP, biological process; CC, cellular component; MF, molecular function. (B) Plant hormone-related genes that were differentially expressed between fertile and sterile buds. GA, gibberellin; IAA, auxin; ABA, abscisic acid; CTK, cytokinin; ET, ethylene; JA, jasmonic acid; SA, salicylic acid.
Supplementary Figure 8 | Protein–protein interaction (PPI) network analysis of DEGs. (A–J) Ten modules in PPI network. (K) Heatmap of four hub genes.
Supplementary Table 1 | Primers used in this study.
Supplementary Table 2 | Primers for 32 known protein-coding genes.
Supplementary Table 3 | Gene annotation of 99.75 kb DNA fragment.
Supplementary Table 4 | GO annotation of DEGs.
Supplementary Table 5 | KEGG pathway annotation of DEGs.
Supplementary Table 6 | DEGs related to pollen and anther development.
Supplementary Table 7 | DEGs selected for RT-qPCR verification.
Footnotes
References
Alche, J. D., M’rani-Alaoui, M., Castro, A. J., and Rodriguez-Garcia, M. I. (2004). Ole e 1, the major allergen from olive (Olea europaea L.) pollen, increases its expression and is released to the culture medium during in vitro germination. Plant Cell Physiol. 45, 1149–1157. doi: 10.1093/pcp/pch127
An, H., Yang, Z., Yi, B., Wen, J., Shen, J., Tu, J., et al. (2014). Comparative transcript profiling of the fertile and sterile flower buds of pol CMS in B. napus. BMC Genomics 15:258. doi: 10.1186/1471-2164-15-258
Andersson, H. A., Passeri, M. F., and Barry, M. A. (2005). Rad23 as a reciprocal agent for stimulating or repressing immune responses. Hum. Gene Ther. 16, 634–641. doi: 10.1089/hum.2005.16.634
Arruda, P., Kemper, E. L., Papes, F., and Leite, A. (2000). Regulation of lysine catabolism in higher plants. Trends Plant Sci. 5, 324–330. doi: 10.1016/s1360-1385(00)01688-5
Bohra, A., Jha, U. C., Adhimoolam, P., Bisht, D., and Singh, N. P. (2016). Cytoplasmic male sterility (CMS) in hybrid breeding in field crops. Plant Cell Rep. 35, 967–993. doi: 10.1007/s00299-016-1949-3
Bonhomme, S., Budar, F., Lancelin, D., Small, I., Defrance, M. C., and Pelletier, G. (1992). Sequence and transcript analysis of the Nco2.5 Ogura-specific fragment correlated with cytoplasmic male sterility in Brassica cybrids. Mol. Gen. Genet. 235, 340–348. doi: 10.1007/BF00279379
Brocker, C., Lassen, N., Estey, T., Pappa, A., Cantore, M., Orlova, V. V., et al. (2010). Aldehyde dehydrogenase 7A1 (ALDH7A1) is a novel enzyme involved in cellular defense against hyperosmotic stress. J. Biol. Chem. 285, 18452–18463. doi: 10.1074/jbc.M109.077925
Cai, Y., Zhang, Y., Loh, Y. P., Tng, J. Q., Lim, M. C., Cao, Z., et al. (2021). H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 12:719. doi: 10.1038/s41467-021-20940-y
Cartagena, J. A., Matsunaga, S., Seki, M., Kurihara, D., Yokoyama, M., Shinozaki, K., et al. (2008). The Arabidopsis SDG4 contributes to the regulation of pollen tube growth by methylation of histone H3 lysines 4 and 36 in mature pollen. Dev. Biol. 315, 355–368. doi: 10.1016/j.ydbio.2007.12.016
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Chang, F., Wang, Y., Wang, S., and Ma, H. (2011). Molecular control of microsporogenesis in Arabidopsis. Curr. Opin. Plant Biol. 14, 66–73. doi: 10.1016/j.pbi.2010.11.001
Chang, L. C., Guo, C. L., Lin, Y. S., Fu, H., Wang, C. S., and Jauh, G. Y. (2009). Pollen-specific SKP1-like proteins are components of functional scf complexes and essential for lily pollen tube elongation. Plant Cell Physiol. 50, 1558–1572. doi: 10.1093/pcp/pcp100
Chang, S., Yang, T., Du, T., Huang, Y., Chen, J., Yan, J., et al. (2011). Mitochondrial genome sequencing helps show the evolutionary mechanism of mitochondrial genome formation in Brassica. BMC Genomics 12:497. doi: 10.1186/1471-2164-12-497
Chen, L., and Liu, Y. G. (2014). Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 65, 579–606. doi: 10.1146/annurev-arplant-050213-040119
Chen, L. Q., Hou, B. H., Lalonde, S., Takanaga, H., Hartung, M. L., Qu, X.-Q., et al. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532. doi: 10.1038/nature09606
Chen, X., and Walters, K. J. (2016). H-1, N-15, C-13 resonance assignments for Saccharomyces cerevisiae Rad23 UBL domain. Biomol. NMR Assign. 10, 291–295. doi: 10.1007/s12104-016-9686-7
Chen, X. Y., Zhang, H., Sun, H., Luo, H., Zhao, L., Dong, Z., et al. (2017). Irregular pollen exine1 is a novel factor in anther cuticle and pollen exine formation. Plant Physiol. 173, 307–325. doi: 10.1104/pp.16.00629
Crevillen, P. (2020). Histone demethylases as counterbalance to H3K27me3 silencing in plants. IScience 23:101715. doi: 10.1016/j.isci.2020.101715
Dai, M., Zhao, Y., Ma, Q., Hu, Y., Hedden, P., Zhang, Q., et al. (2007). The rice YABBY1 gene is involved in the feedback regulation of gibberellin metabolism. Plant Physiol. 144, 121–133. doi: 10.1104/pp.107.096586
Dantuma, N. P., Heinen, C., and Hoogstraten, D. (2009). The ubiquitin receptor Rad23: at the crossroads of nucleotide excision repair and proteasomal degradation. DNA Repair 8, 449–460. doi: 10.1016/j.dnarep.2009.01.005
Devoto, A., Nieto-Rostro, M., Xie, D., Ellis, C., Harmston, R., Patrick, E., et al. (2002). COI1 links jasmonate signalling and fertility to the SCF ubiquitin-ligase complex in Arabidopsis. Plant J. 32, 457–466. doi: 10.1046/j.1365-313x.2002.01432.x
Ding, B., Hao, M., Mei, D., Zaman, Q. U., Sang, S., Wang, H., et al. (2018). Transcriptome and hormone comparison of three cytoplasmic male sterile systems in Brassica napus. Int. J. Mol. Sci. 19:4022. doi: 10.3390/ijms19124022
Duan, H., and Schuler, M. A. (2005). Differential expression and evolution of the Arabidopsis CYP86A subfamily. Plant Physiol. 137, 1067–1081. doi: 10.1104/pp.104.055715
Ducos, E., Touzet, P., and Boutry, M. (2001). The male sterile G cytoplasm of wild beet displays modified mitochondrial respiratory complexes. Plant J. 26, 171–180. doi: 10.1046/j.1365-313x.2001.01017.x
Farmer, L. M., Book, A. J., Lee, K. H., Lin, Y. L., Fu, H., and Vierstra, R. D. (2010). The RAD23 family provides an essential connection between the 26S proteasome and ubiquitylated proteins in Arabidopsis. Plant Cell 22, 124–142. doi: 10.1105/tpc.109.072660
Ferguson, A. C., Pearce, S., Band, L. R., Yang, C., Ferjentsikova, I., King, J., et al. (2017). Biphasic regulation of the transcription factor ABORTED MICROSPORES (AMS) is essential for tapetum and pollen development in Arabidopsis. New Phytol. 213, 778–790. doi: 10.1111/nph.14200
Forsthoefel, N. R., Dao, T. P., and Vernon, D. M. (2010). PIRL1 and PIRL9, encoding members of a novel plant-specific family of leucine-rich repeat proteins, are essential for differentiation of microspores into pollen. Planta 232, 1101–1114. doi: 10.1007/s00425-010-1242-6
Fujii, S., and Toriyama, K. (2009). Suppressed expression of Retrograde-Regulated Male Sterility restores pollen fertility in cytoplasmic male sterile rice plants. Proc. Natl. Acad. Sci. U.S.A. 106, 9513–9518. doi: 10.1073/pnas.0901860106
Fujii, S., Yamada, M., Fujita, M., Itabashi, E., Hamada, K., Yano, K., et al. (2010). Cytoplasmic-nuclear genomic barriers in rice pollen development revealed by comparison of global gene expression profiles among five independent cytoplasmic male sterile lines. Plant Cell Physiol. 51, 610–620. doi: 10.1093/pcp/pcq026
Gaborieau, L., and Brown, G. G. (2016). Comparative genomic analysis of the compound Brassica napus Rf locus. BMC Genomics 17:834. doi: 10.1186/s12864-016-3117-0
Gaborieau, L., Brown, G. G., and Mireau, H. (2016). The propensity of pentatricopeptide repeat genes to evolve into restorers of cytoplasmic male sterility. Front. Plant Sci. 7:1816. doi: 10.3389/fpls.2016.01816
Gallego, F., Fleck, O., Li, A., Wyrzykowska, J., and Tinland, B. (2000). AtRAD1, a plant homologue of human and yeast nucleotide excision repair endonucleases, is involved in dark repair of UV damages and recombination. Plant J. 21, 507–518. doi: 10.1046/j.1365-313x.2000.00694.x
Gonzalez-Melendi, P., Uyttewaal, M., Morcillo, C. N., Hernandez, M. J., Fajardo, S., Budar, F., et al. (2008). A light and electron microscopy analysis of the events leading to male sterility in Ogu-INRA CMS of rapeseed (Brassica napus). J. Exp. Bot. 59, 827–838. doi: 10.1093/jxb/erm365
Guan, Y. F., Huang, X. Y., Zhu, J., Gao, J. F., Zhang, H. X., and Yang, Z. N. (2008). RUPTURED POLLEN GRAIN1, a member of the MtN3/saliva gene family, is crucial for exine pattern formation and cell integrity of microspores in Arabidopsis. Plant Physiol. 147, 852–863. doi: 10.1104/pp.108.118026
Hamman-Khalifa, A., Castro, A. J., Jimenez-Lopez, J. C., Rodriguez-Garcia, M. I., and Alche, J. D. (2008). Olive cultivar origin is a major cause of polymorphism for Ole e 1 pollen allergen. BMC Plant Biol. 8:10. doi: 10.1186/1471-2229-8-10
Han, Y., Zhou, S. D., Fan, J. J., Zhou, L., Shi, Q. S., Zhang, Y. F., et al. (2021). OsMS188 is a key regulator of tapetum development and sporopollenin synthesis in rice. Rice 14:4. doi: 10.1186/s12284-020-00451-y
Heng, S., Gao, J., Wei, C., Chen, F., Li, X., Wen, J., et al. (2018). Transcript levels of orf288 are associated with the hau cytoplasmic male sterility system and altered nuclear gene expression in Brassica juncea. J. Exp. Bot. 69, 455–466. doi: 10.1093/jxb/erx443
Hoffmann, R. D., and Palmgren, M. G. (2013). Epigenetic repression of male gametophyte-specific genes in the Arabidopsis sporophyte. Mol. Plant 6, 1176–1186. doi: 10.1093/mp/sst100
Hou, Q. C., and Bartels, D. (2015). Comparative study of the aldehyde dehydrogenase (ALDH) gene superfamily in the glycophyte Arabidopsis thaliana and Eutrema halophytes. Ann. Bot. 115, 465–479. doi: 10.1093/aob/mcu152
Hu, B., Liu, B., Liu, L., Liu, C., Xu, L., and Ruan, Y. (2014). Epigenetic control of Pollen Ole e 1 allergen and extensin family gene expression in Arabidopsis thaliana. Acta. Physiol. Plant 36, 2203–2209. doi: 10.1007/s11738-014-1597-6
Jacobowitz, J. R., Doyle, W. C., and Weng, J. K. (2019). PRX9 and PRX40 are extensin peroxidases essential for maintaining tapetum and microspore cell wall integrity during Arabidopsis anther development. Plant Cell 31, 848–861. doi: 10.1105/tpc.18.00907
Ji, J. J., Huang, W., Yin, C. C., and Gong, Z. H. (2013). Mitochondrial cytochrome c oxidase and F1Fo-ATPase dysfunction in peppers (Capsicum annuum L.) with cytoplasmic male sterility and its association with orf507 and psi atp6-2 genes. Int. J. Mol. Sci. 14, 1050–1068. doi: 10.3390/ijms14011050
Jing, B., Heng, S., Tong, D., Wan, Z., Fu, T., Tu, J., et al. (2012). A male sterility-associated cytotoxic protein ORF288 in Brassica juncea causes aborted pollen development. J. Exp. Bot. 63, 1285–1295. doi: 10.1093/jxb/err355
Kale, A. A., and Munjal, S. V. (2005). Mitochondrial respiration associated with cytoplasmic male sterility in pearl millet. J. Plant Biochem. Biotechnol. 14, 161–165. doi: 10.1007/BF03263245
Kang, L., Li, P., Wang, A., Ge, X., and Li, Z. (2017). A novel cytoplasmic male sterility in Brassica napus (inap CMS) with carpelloid stamens via protoplast fusion with chinese woad. Front. Plant. Sci. 8:529. doi: 10.3389/fpls.2017.00529
Kitazaki, K., Arakawa, T., Matsunaga, M., Yui-Kurino, R., Matsuhira, H., Mikami, T., et al. (2015). Post-translational mechanisms are associated with fertility restoration of cytoplasmic male sterility in sugar beet (Beta vulgaris). Plant J. 83, 290–299. doi: 10.1111/tpj.12888
Ko, S. S., Li, M. J., Sun-Ben, K. M., Ho, Y. C., Lin, Y. J., Chuang, M. H., et al. (2014). The bHLH142 transcription factor coordinates with TDR1 to modulate the expression of EAT1 and regulate pollen development in rice. Plant Cell 26, 2486–2504. doi: 10.1105/tpc.114.126292
Koiwai, A., and Matsuzaki, T. (1988). Hydroxy and normal fatty-acid distribution in stigmas of Nicotiana and other plants. Phytochemistry 27, 2827–2830. doi: 10.1016/0031-9422(88)80671-X
Krolikowski, K. A., Victor, J. L., Wagler, T. N., Lolle, S. J., and Pruitt, R. E. (2003). Isolation and characterization of the Arabidopsis organ fusion gene HOTHEAD. Plant J. 35, 501–511. doi: 10.1046/j.1365-313x.2003.01824.x
Kubo, T., Arakawa, T., Honma, Y., and Kitazaki, K. (2020). What does the molecular genetics of different types of restorer-of-fertility genes imply? Plants 9:361. doi: 10.3390/plants9030361
Lahari, T., Lazaro, J., and Schroeder, D. F. (2017). RAD4 and RAD23/HMR contribute to Arabidopsis UV tolerance. Genes 9:8. doi: 10.3390/genes9010008
Lee, J., Yun, J. Y., Zhao, W., Shen, W. H., and Amasino, R. M. (2015). A methyltransferase required for proper timing of the vernalization response in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 112, 2269–2274. doi: 10.1073/pnas.1423585112
Lee, S. B., and Suh, M. C. (2013). Recent advances in cuticular wax biosynthesis and its regulation in Arabidopsis. Mol. Plant 6, 246–249. doi: 10.1093/mp/sss159
L’Homme, Y., Stahl, R. J., Li, X. Q., Hameed, A., and Brown, G. G. (1997). Brassica nap cytoplasmic male sterility is associated with expression of a mtDNA region containing a chimeric gene similar to the pol CMS-associated orf224 gene. Curr. Genet. 31, 325–335. doi: 10.1007/s002940050212
Li, H., Li, J., Zhao, B., Wang, J., Yi, L., Liu, C., et al. (2015). Generation and characterization of tribenuron-methyl herbicide-resistant rapeseed (Brasscia napus) for hybrid seed production using chemically induced male sterility. Theor. Appl. Genet. 128, 107–118. doi: 10.1007/s00122-014-2415-7
Li, L., Li, B., Xie, C., Zhang, T., Borassi, C., Estevez, J. M., et al. (2020). Arabidopsis RAD23B regulates pollen development by mediating degradation of KRP1. J. Exp. Bot. 71, 4010–4019. doi: 10.1093/jxb/eraa167
Li, P., Kang, L., Wang, A., Cui, C., Jiang, L., Guo, S., et al. (2019). Development of a fertility restorer for inap CMS (Isatis indigotica) Brassica napus through genetic introgression of one alien addition. Front. Plant Sci. 10:257. doi: 10.3389/fpls.2019.00257
Li, S., Chen, Z., Zhao, N., Wang, Y., Nie, H., and Hua, J. (2018). The comparison of four mitochondrial genomes reveals cytoplasmic male sterility candidate genes in cotton. BMC Genom. 19:775. doi: 10.1186/s12864-018-5122-y
Liang, R. Y., Chen, L., Ko, B. T., Shen, Y. H., Li, Y. T., Chen, B. R., et al. (2014). Rad23 interaction with the proteasome is regulated by phosphorylation of its ubiquitin-like (UbL) domain. J. Mol. Biol. 426, 4049–4060. doi: 10.1016/j.jmb.2014.10.004
Lin, S., Miao, Y., Su, S., Xu, J., Jin, L., Sun, D., et al. (2019). Comprehensive analysis of Ogura cytoplasmic male sterility-related genes in turnip (Brassica rapa ssp. rapifera) using RNA sequencing analysis and bioinformatics. PLoS One 14:e218029. doi: 10.1371/journal.pone.0218029
Liu, F., Cui, X. Q., Horner, H. T., Weiner, H., and Schnable, P. S. (2001). Mitochondrial aldehyde dehydrogenase activity is required for male fertility in maize. Plant Cell 13, 1063–1078. doi: 10.1105/tpc.13.5.1063
Liu, F., and Schnable, P. S. (2002). Functional specialization of maize mitochondrial aldehyde dehydrogenases. Plant Physiol. 130, 1657–1674. doi: 10.1104/pp.012336
Liu, Z., Dong, F., Wang, X., Wang, T., Su, R., Hong, D., et al. (2017). A pentatricopeptide repeat protein restores nap cytoplasmic male sterility in Brassica napus. J. Exp. Bot. 68, 4115–4123. doi: 10.1093/jxb/erx239
Liu, Z., Liu, P., Long, F., Hong, D., He, Q., and Yang, G. (2012). Fine mapping and candidate gene analysis of the nuclear restorer gene Rfp for pol CMS in rapeseed (Brassica napus L.). Theor. Appl. Genet. 125, 773–779. doi: 10.1007/s00122-012-1870-2
Liu, Z., Yang, Z., Wang, X., Li, K., An, H., Liu, J., et al. (2016). A mitochondria-targeted PPR protein restores pol cytoplasmic male sterility by reducing orf224 transcript levels in oilseed rape. Mol. Plant 9, 1082–1084. doi: 10.1016/j.molp.2016.04.004
Lowe, E. D., Hasan, N., Trempe, J. F., Fonso, L., Noble, M. E., Endicott, J. A., et al. (2006). Structures of the Dsk2 UBL and UBA domains and their complex. Acta Crystallogr. D Biol. Crystallogr. 62, 177–188. doi: 10.1107/S0907444905037777
Mao, X. Z., Cai, T., Olyarchuk, J. G., and Wei, L. P. (2005). Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Mattioli, R., Biancucci, M., El, S. A., Mosca, L., Costantino, P., Funck, D., et al. (2018). Proline synthesis in developing microspores is required for pollen development and fertility. BMC Plant Biol. 18:356. doi: 10.1186/s12870-018-1571-3
Medina, B., Paraskevopoulos, K., Boehringer, J., Sznajder, A., Robertson, M., Endicott, J., et al. (2012). The ubiquitin-associated (UBA) 1 domain of Schizosaccharomyces pombe Rhp23 is essential for the recognition of ubiquitin-proteasome system substrates both in vitro and in vivo. J. Biol. Chem. 287, 42344–42351. doi: 10.1074/jbc.M112.419838
Mira-Rodado, V., Sweere, U., Grefen, C., Kunkel, T., Fejes, E., Nagy, F., et al. (2007). Functional cross-talk between two-component and phytochrome B signal transduction in Arabidopsis. J. Exp. Bot. 58, 2595–2607. doi: 10.1093/jxb/erm087
Morrison, M. J., Harker, K. N., Blackshaw, R. E., Holzapfel, C. J., and O’donovan, J. T. (2016). Canola yield improvement on the Canadian Prairies from 2000 to 2013. Crop Pasture Sci. 67, 245–252. doi: 10.1071/CP15348
Ning, L., Wang, H., Li, D., Li, Y., Chen, K., Zhao, W., et al. (2019). Transcriptomic and proteomic analysis of Shaan2A cytoplasmic male sterility and its maintainer line in Brassica napus. Front. Plant Sci. 10:252. doi: 10.3389/fpls.2019.00252
Ning, L., Wang, H., Li, D., Lin, Z., Li, Y., Zhao, W., et al. (2020). Genome-wide identification of the restorer-of-fertility-like (RFL) gene family in Brassica napus and expression analysis in Shaan2A cytoplasmic male sterility. BMC Genomics 21:765. doi: 10.1186/s12864-020-07163-z
Niu, N., Liang, W., Yang, X., Jin, W., Wilson, Z. A., Hu, J., et al. (2013). EAT1 promotes tapetal cell death by regulating aspartic proteases during male reproductive development in rice. Nat. Commun. 4:1445. doi: 10.1038/ncomms2396
Okeke, E., Chen, L., and Madura, K. (2020). The cellular location of Rad23, a polyubiquitin chain-binding protein, plays a key role in its interaction with substrates of the proteasome. J. Mol. Biol. 432, 2388–2404. doi: 10.1016/j.jmb.2020.03.001
Peng, X., Wang, K., Hu, C., Zhu, Y., Wang, T., Yang, J., et al. (2010). The mitochondrial gene orfH79 plays a critical role in impairing both male gametophyte development and root growth in CMS-Honglian rice. BMC Plant Biol. 10:125. doi: 10.1186/1471-2229-10-125
Planas-Portell, J., Gallart, M., Tiburcio, A. F., and Altabella, T. (2013). Copper-containing amine oxidases contribute to terminal polyamine oxidation in peroxisomes and apoplast of Arabidopsis thaliana. BMC Plant Biol. 13:109. doi: 10.1186/1471-2229-13-109
Preuss, D., Lemieux, B., Yen, G., and Davis, R. W. (1993). A conditional sterile mutation eliminates surface components from Arabidopsis pollen and disrupts cell signaling during fertilization. Genes Dev. 7, 974–985. doi: 10.1101/gad.7.6.974
Ren, C., Pan, J., Peng, W., Genschik, P., Hobbie, L., Hellmann, H., et al. (2005). Point mutations in Arabidopsis Cullin1 reveal its essential role in jasmonate response. Plant J. 42, 514–524. doi: 10.1111/j.1365-313X.2005.02394.x
Sabar, M., Gagliardi, D., Balk, J., and Leaver, C. J. (2003). ORFB is a subunit of F1FO-ATP synthase: insight into the basis of cytoplasmic male sterility in sunflower. EMBO Rep. 4, 381–386. doi: 10.1038/sj.embor.embor800
Sang, S., Cheng, H., Hao, M., Ding, B., Mei, D., Wang, H., et al. (2021). Mitochondrial localization of ORF346 causes pollen abortion in alloplasmic male sterility. Crop J. [Epub ahead of print]. doi: 10.1016/j.cj.2021.01.008
Sang, S. F., Mei, D. S., Liu, J., Zaman, Q. U., Zhang, H. Y., Hao, M. Y., et al. (2019). Organelle genome composition and candidate gene identification for Nsa cytoplasmic male sterility in Brassica napus. BMC Genomics 20:813. doi: 10.1186/s12864-019-6187-y
Schmitz-Linneweber, C., and Small, I. (2008). Pentatricopeptide repeat proteins: a socket set for organelle gene expression. Trends Plant Sci. 13, 663–670. doi: 10.1016/j.tplants.2008.10.001
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shen, Y., Zhang, Y., Yang, C., Lan, Y., Liu, L., Liu, S., et al. (2012). Mutation of OsALDH7 causes a yellow-colored endosperm associated with accumulation of oryzamutaic acid A in rice. Planta 235, 433–441. doi: 10.1007/s00425-011-1477-x
Shen, Y. F., Sun, S., Hua, S., Shen, E., Ye, C. Y., Cai, D., et al. (2017). Analysis of transcriptional and epigenetic changes in hybrid vigor of allopolyploid Brassica napus uncovers key roles for small RNAs. Plant J. 91, 874–893. doi: 10.1111/tpj.13605
Shi, H., Yu, Y., Gu, R., Feng, C., Fu, Y., Fu, Y., et al. (2020). Male sterile 305 mutation leads the misregulation of anther cuticle formation by disrupting lipid metabolism in maize. Int. J. Mol. Sci. 21:8. doi: 10.3390/ijms21072500
Shin, J. H., Kim, S. R., and An, G. (2009). Rice aldehyde dehydrogenase7 is needed for seed maturation and viability. Plant Physiol. 149, 905–915. doi: 10.1104/pp.108.130716
Singh, M., and Brown, G. G. (1993). Characterization of expression of a mitochondrial gene region associated with the Brassica “Polima” CMS: developmental influences. Curr. Genet. 24, 316–322. doi: 10.1007/BF00336783
Song, D., Tian, J., Hu, Y., Wei, Y., Lu, H., Wang, Y., et al. (2020). Identification of biomarkers associated with diagnosis and prognosis of gastroesophageal junction adenocarcinoma-a study based on integrated bioinformatics analysis in GEO and TCGA database. Medicine 99:e23605. doi: 10.1097/MD.0000000000023605
Song, S. S., Qi, T. C., Huang, H., and Xie, D. X. (2013). Regulation of stamen development by coordinated actions of jasmonate, auxin, and gibberellin in Arabidopsis. Mol. Plant 6, 1065–1073. doi: 10.1093/mp/sst054
Stepansky, A., and Galili, G. (2003). Synthesis of the Arabidopsis bifunctional lysine-ketoglutarate reductase/saccharopine dehydrogenase enzyme of lysine catabolism is concertedly regulated by metabolic and stress-associated signals. Plant Physiol. 133, 1407–1415. doi: 10.1104/pp.103.026294
Stepansky, A., Less, H., Angelovici, R., Aharon, R., Zhu, X., and Galili, G. (2006). Lysine catabolism, an effective versatile regulator of lysine level in plants. Amino Acids 30, 121–125. doi: 10.1007/s00726-005-0246-1
Su, W., Tao, R., Liu, W., Yu, C., Yue, Z., He, S., et al. (2020). Characterization of four polymorphic genes controlling red leaf colour in lettuce that have undergone disruptive selection since domestication. Plant Biotechnol. J. 18, 479–490. doi: 10.1111/pbi.13213
Takagi, H., Abe, A., Yoshida, K., Kosugi, S., Natsume, S., Mitsuoka, C., et al. (2013). QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74, 174–183. doi: 10.1111/tpj.12105
Tang, G. L., Zhu, X. H., Tang, X. H., and Galili, G. (2000). A novel composite locus of Arabidopsis encoding two polypeptides with metabolically related but distinct functions in lysine catabolism. Plant J. 23, 195–203. doi: 10.1046/j.1365-313x.2000.00770.x
Tang, H., Xie, Y., Liu, Y. G., and Chen, L. (2017). Advances in understanding the molecular mechanisms of cytoplasmic male sterility and restoration in rice. Plant Reprod. 30, 179–184. doi: 10.1007/s00497-017-0308-z
Tang, Q., Tian, M., An, G., Zhang, W., Chen, J., Yan, C., et al. (2017). Rapid identification of the purple stem (Ps) gene of Chinese kale (Brassica oleracea var. alboglabra) in a segregation distortion population by bulked segregant analysis and RNA sequencing. Mol. Breeding 37:153. doi: 10.1007/s11032-017-0752-3
Tang, W. H., Ezcurra, I., Muschietti, J., and Mccormick, S. (2002). A cysteine-rich extracellular protein, LAT52, interacts with the extracellular domain of the pollen receptor kinase LePRK2. Plant Cell 14, 2277–2287. doi: 10.1105/tpc.003103
Tavladoraki, P., Cona, A., and Angelini, R. (2016). Copper-containing amine oxidases and FAD-dependent polyamine oxidases are key players in plant tissue differentiation and organ development. Front. Plant Sci. 7:824. doi: 10.3389/fpls.2016.00824
Uyttewaal, M., Arnal, N., Quadrado, M., Martin-Canadell, A., Vrielynck, N., Hiard, S., et al. (2008). Characterization of raphanus sativus pentatricopeptide repeat proteins encoded by the fertility restorer locus for ogura cytoplasmic male sterility. Plant Cell 20, 3331–3345. doi: 10.1105/tpc.107.057208
Wade, S. L., and Auble, D. T. (2010). The Rad23 ubiquitin receptor, the proteasome and functional specificity in transcriptional control. Transcription 1, 22–26. doi: 10.4161/trns.1.1.12201
Wan, Z., Jing, B., Tu, J., Ma, C., Shen, J., Yi, B., et al. (2008). Genetic characterization of a new cytoplasmic male sterility system (hau) in Brassica juncea and its transfer to B-napus. Theor. Appl. Genet. 116, 355–362. doi: 10.1007/s00122-007-0673-3
Wang, B., Farooq, Z., Chu, L., Liu, J., Wang, H., Guo, J., et al. (2021). High-generation near-isogenic lines combined with multi-omics to study the mechanism of polima cytoplasmic male sterility. BMC Plant Biol. 21:130. doi: 10.1186/s12870-021-02852-7
Wang, H., Xiao, Q., Wei, C., Chen, H., Chen, X., Dai, C., et al. (2021). A mitochondria-localized pentatricopeptide repeat protein is required to restore hau cytoplasmic male sterility in Brassica napus. Theor. Appl. Genet. 134, 1377–1386. doi: 10.1007/s00122-021-03777-3
Wang, K., Gao, F., Ji, Y., Liu, Y., Dan, Z., Yang, P., et al. (2013). ORFH79 impairs mitochondrial function via interaction with a subunit of electron transport chain complex III in Honglian cytoplasmic male sterile rice. New Phytol. 198, 408–418. doi: 10.1111/nph.12180
Wei, W., Li, Y., Wang, L., Liu, S., Yan, X., Mei, D., et al. (2010). Development of a novel Sinapis arvensis disomic addition line in Brassica napus containing the restorer gene for Nsa CMS and improved resistance to Sclerotinia sclerotiorum and pod shattering. Theor. Appl. Genet. 120, 1089–1097. doi: 10.1007/s00122-009-1236-6
Wei, X., Lv, Y., Zhao, Y., Nath, U. K., Yuan, Y., Wang, Z., et al. (2020). Comparative transcriptome analysis in Chinese cabbage (Brassica rapa ssp. pekinesis) for DEGs of Ogura-, Polima-CMS and their shared maintainer. Physiol. Mol. Biol. Plants 26, 719–731. doi: 10.1007/s12298-020-00775-5
Wolters-Arts, M., Lush, W. M., and Mariani, C. (1998). Lipids are required for directional pollen-tube growth. Nature 392, 818–821. doi: 10.1038/33929
Wu, H., Shi, S., Lu, X., Li, T., Wang, J., Liu, T., et al. (2019). Expression analysis and functional characterization of CER1 family genes involved in very-long-chain alkanes biosynthesis in Brachypodium distachyon. Front. Plant Sci. 10:1389. doi: 10.3389/fpls.2019.01389
Xing, M., Sun, C., Li, H., Hu, S., Lei, L., and Kang, J. (2018). Integrated analysis of transcriptome and proteome changes related to the Ogura cytoplasmic male sterility in cabbage. PLoS One 13:e0193462. doi: 10.1371/journal.pone.0193462
Xiong, X., Zhou, D., Xu, L., Liu, T., Yue, X., Liu, W., et al. (2019). BcPME37c is involved in pollen intine formation in Brassica campestris. Biochem. Biophys. Res. Commun. 517, 63–68. doi: 10.1016/j.bbrc.2019.07.009
Xu, J., Ding, Z., Vizcay-Barrena, G., Shi, J., Liang, W., Yuan, Z., et al. (2014). ABORTED Microspores acts as a master regulator of pollen wall formation in Arabidopsis. Plant Cell 26, 1544–1556. doi: 10.1105/tpc.114.122986
Xu, L., Liu, F., Lechner, E., Genschik, P., Crosby, W. L., Ma, H., et al. (2002). The SCFCOl1 ubiquitin-ligase complexes are required for jasmonate response in Arabidopsis. Plant Cell 14, 1919–1935. doi: 10.1105/tpc.003368
Xu, Y., Liu, S., Liu, Y., Ling, S., Chen, C., and Yao, J. (2017). HOTHEAD-Like HTH1 is involved in anther cutin biosynthesis and is required for pollen fertility in rice. Plant Cell Physiol. 58, 1238–1248. doi: 10.1093/pcp/pcx063
Yamaoka, Y., Yu, Y., Mizoi, J., Fujiki, Y., Saito, K., and Nishijima, M. (2011). PHOSPHATIDYLSERINE SYNTHASE1 is required for microspore development in Arabidopsis thaliana. Plant J. 67, 648–661. doi: 10.1111/j.1365-313X.2011.04624.x
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11:R14. doi: 10.1186/gb-2010-11-2-r14
Zhao, J. Y., Missihoun, T. D., and Bartels, D. (2018). The ATAF1 transcription factor is a key regulator of aldehyde dehydrogenase 7B4 (ALDH7B4) gene expression in Arabidopsis thaliana. Planta 248, 1017–1027. doi: 10.1007/s00425-018-2955-1
Zhao, N., Xu, X., Wamboldt, Y., Mackenzie, S. A., Yang, X., Hu, Z., et al. (2016). MutS HOMOLOG1 silencing mediates ORF220 substoichiometric shifting and causes male sterility in Brassica juncea. J. Exp. Bot. 67, 435–444. doi: 10.1093/jxb/erv480
Zhu, X. H., and Galili, G. (2003). Increased lysine synthesis coupled with a knockout of its catabolism synergistically boosts lysine content and also transregulates the metabolism of other amino acids in Arabidopsis seeds. Plant Cell 15, 845–853. doi: 10.1105/tpc.009647
Zuo, B., Zhu, J., Xiao, F., Wang, C., Shen, Y., and Chen, X. (2020). Identification of novel biomarkers and candidate small molecule drugs in rheumatoid arthritis and osteoarthritis based on bioinformatics analysis of high-throughput data. Biosci. Rep. 40:BSR20193823. doi: 10.1042/BSR20193823
Keywords: Brassica napus, cytoplasmic male sterility, fertility restorer, map-based cloning, candidate gene, transcriptome
Citation: Yang Q, Nong X, Xu J, Huang F, Wang F, Wu J, Zhang C and Liu C (2021) Unraveling the Genetic Basis of Fertility Restoration for Cytoplasmic Male Sterile Line WNJ01A Originated From Brassica juncea in Brassica napus. Front. Plant Sci. 12:721980. doi: 10.3389/fpls.2021.721980
Received: 08 June 2021; Accepted: 05 August 2021;
Published: 31 August 2021.
Edited by:
Kun Lu, Southwest University, ChinaReviewed by:
Zhi Liu, Southwest University, ChinaJinjin Jiang, Yangzhou University, China
Shengwu Hu, Northwest A&F University, China
Xiyue Song, Northwest A&F University, China
Copyright © 2021 Yang, Nong, Xu, Huang, Wang, Wu, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chao Liu, bGl1Y2hhb0BtYWlsLmh6YXUuZWR1LmNu