 Paul Vollrath1
Paul Vollrath1 Harmeet S. Chawla2
Harmeet S. Chawla2 Dima Alnajar3
Dima Alnajar3 Iulian Gabur1,4
Iulian Gabur1,4 HueyTyng Lee1
HueyTyng Lee1 Sven Weber1
Sven Weber1 Lennard Ehrig1
Lennard Ehrig1 Birger Koopmann3
Birger Koopmann3 Rod J. Snowdon1
Rod J. Snowdon1 Christian Obermeier1*
Christian Obermeier1*- 1Department of Plant Breeding, IFZ Research Centre for Biosystems, Land Use and Nutrition, Justus Liebig University Giessen, Giessen, Germany
- 2Department of Plant Sciences, Crop Development Centre, University of Saskatchewan, Saskatoon, SK, Canada
- 3Plant Pathology and Crop Protection Division, Department of Crop Sciences, Georg August University of Göttingen, Göttingen, Germany
- 4Department of Plant Sciences, Faculty of Agriculture, Iasi University of Life Sciences, Iaşi, Romania
Blackleg is one of the major fungal diseases in oilseed rape/canola worldwide. Most commercial cultivars carry R gene-mediated qualitative resistances that confer a high level of race-specific protection against Leptosphaeria maculans, the causal fungus of blackleg disease. However, monogenic resistances of this kind can potentially be rapidly overcome by mutations in the pathogen’s avirulence genes. To counteract pathogen adaptation in this evolutionary arms race, there is a tremendous demand for quantitative background resistance to enhance durability and efficacy of blackleg resistance in oilseed rape. In this study, we characterized genomic regions contributing to quantitative L. maculans resistance by genome-wide association studies in a multiparental mapping population derived from six parental elite varieties exhibiting quantitative resistance, which were all crossed to one common susceptible parental elite variety. Resistance was screened using a fungal isolate with no corresponding avirulence (AvrLm) to major R genes present in the parents of the mapping population. Genome-wide association studies revealed eight significantly associated quantitative trait loci (QTL) on chromosomes A07 and A09, with small effects explaining 3–6% of the phenotypic variance. Unexpectedly, the qualitative blackleg resistance gene Rlm9 was found to be located within a resistance-associated haploblock on chromosome A07. Furthermore, long-range sequence data spanning this haploblock revealed high levels of single-nucleotide and structural variants within the Rlm9 coding sequence among the parents of the mapping population. The results suggest that novel variants of Rlm9 could play a previously unknown role in expression of quantitative disease resistance in oilseed rape.
Introduction
Oilseed rape/canola (Brassica napus L.) is one of the most important vegetable oil crops. As a recent allotetraploid crop, originating from an interspecific hybridization event between its two diploid ancestors Brassica rapa (2n = 2x = 20, AA) and Brassica oleracea (2n = 2x = 18, CC) (Nagaharu, 1935), B. napus (2n = 4x = 38, AACC) carries a highly complex and dynamic genome which is affected by many small-scale and large-scale structural variations (Parkin et al., 1995; Chalhoub et al., 2014; Stein et al., 2017; Hurgobin et al., 2018; Chawla et al., 2020). Many studies revealed high frequencies of homoeologous exchanges between the highly similar A and C subgenomes (Szadkowski et al., 2010; Chalhoub et al., 2014; Samans et al., 2017).
The hemibiotrophic fungal pathogen Leptosphaeria maculans (Desm.) Ces. & de Not. [anamorph: Phoma lingam (Tode ex. Fr.) Desm.] causes stem canker in B. napus. This disease, also known as blackleg, is a major problem in almost all oilseed rape and canola-growing regions around the globe. Substantial yield losses have been reported in Australia, North America and several European countries (Fitt et al., 2006). The primary infection of winter oilseed rape takes place in autumn via airborne ascospores. Additionally, secondary infections are likely through pycnidiospores formed within the asexual pycnidia. The spores penetrate the host tissue via stomata or wounds and colonize intercellular spaces of the mesophyll. From there, the fungus starts its symptomless, biotrophic growth systematically through the petiole into the stem. Here the pathogen kills the cells, leading to girdling and rotting of the stem base. As a result, the plant tends to ripen prematurely and severe infections can lead to serious lodging and death (West et al., 2001). Resistance breeding is the most sustainable and effective method to counteract L. maculans. Resistance of B. napus against L. maculans is often divided into two classes of resistance mechanisms. A distinction is made between race-specific, qualitative resistance determined by major genes, and non-race-specific quantitative resistance provided by numerous minor effect genes. Qualitative resistance against L. maculans, considered as complete resistance, has been investigated in considerable detail and used extensively in commercial breeding programs of B. napus, due to the high efficacy and convenient assessment at the cotyledon stage (Rimmer and van den Berg, 1992; Balesdent et al., 2005; Delourme et al., 2006; Elliott et al., 2016). However, it has been observed that rapid adaptation of the pathogen populations can overcome R gene-mediated resistance in the field within a few seasons (Rouxel et al., 2003; Sprague et al., 2006; Brun et al., 2010; Zhang et al., 2016; van de Wouw et al., 2017). Thus, commercial breeders place special focus on quantitative disease resistance. Quantitative resistance is influenced by multiple genes, and the incomplete nature of the resistance decreases the selection pressure on the pathogen population and consequently increases the durability of the resistance (St Clair, 2010; Delourme et al., 2014). In contrast to qualitative resistance, the assessment of quantitative resistance is more challenging as it is mainly expressed at adult plant stages and highly influenced by environmental conditions (Fitt et al., 2006; Huang et al., 2009). A main target of breeding is therefore the combination of highly effective R gene-mediated resistance with a broad and durable quantitative resistance (Brun et al., 2010; Pilet-Nayel et al., 2017). However, this clear distinction between the two types of resistance has recently been questioned in several studies (Thomma et al., 2011; Delplace et al., 2020). Also, in some cases the expression of partial resistance in adult plants imparted by major resistance genes has been demonstrated (Chantret et al., 1999; Raman et al., 2018). The complexity of the B. napus–L. maculans pathosystem and possible corresponding genes often makes the assessment of quantitative resistance difficult. Therefore, the objective of the present study was to identify genomic regions involved in quantitative blackleg resistance by excluding the possibility of effective R gene-Avr gene interactions. A multiparental mapping population was tested under controlled greenhouse conditions with a selected highly virulent fungal isolate. In addition, whole-genome long-read re-sequencing using Oxford Nanopore Technology (ONT) was conducted to reveal the implications of single nucleotide variants (SNV) and structural genome variations (SV) on an agronomical highly important trait within the narrow genepool of European elite winter oilseed rape. Recently, Chawla et al. (2020) demonstrated a previously unknown extent of genome-wide, small to medium-sized SV events within B. napus genes using ONT long-read sequencing technology. In contrast, SNV have to date been largely ignored in quantitative trait analysis of important crop traits, due to the difficulty of assaying SV on a genome-wide scale in complex crop genomes. In the past few years, however, rapidly decreasing costs and increasing accuracy of long-read sequencing from the ONT or Pacific Biosciences platforms has opened the way to include genome-wide SV data from long-read sequences in QTL analysis and interpretation. Here we successfully called SNV in ONT data from the seven elite winter oilseed rape parents of the multiparental mapping population, enabling us to associate genome-wide SV with single nucleotide polymorphism (SNP) haplotypes carrying blackleg resistance QTL.
Materials and Methods
Plant Material
A B. napus multiparental population comprising 354 double haploid (DH) lines derived from seven European elite winter oilseed rape varieties was tested for quantitative blackleg resistance in multiple greenhouse screenings. The mapping population consisted of six subfamilies derived from crosses of the elite parent “Lorenz” to six elite founder lines (“Adriana,” “Alpaga,” “DK Cabernet,” “Galileo,” “King 10,” and the DH line “JN”). Each of the subfamilies comprised 60 DH lines except for the cross Lorenz × Galileo, which comprised 54 DH lines. The common parent Lorenz was previously classified as highly susceptible to blackleg disease, whereas the other six founder lines were all known to carry quantitative blackleg resistance (unpublished data, breeding companies). The German breeding companies NPZ Innovation GmbH (Holtsee, Germany), Syngenta Seeds GmbH (Bad Salzuflen, Germany), and KWS SAAT SE & Co. KGaA (Einbeck, Germany) produced and provided the DH families. In parallel, another panel of 256 diverse winter oilseed rape inbred lines was tested for blackleg resistance in a 2-year field trial with one plot per field. These accessions were part of the ERANET-ASSYST B. napus diversity set, previously described by Bus et al. (2011).
Resistance Screenings and Data Analysis
The multiparental mapping population was screened for adult-plant blackleg resistance under controlled conditions in the greenhouse of Georg August University of Göttingen in 11 independent screening experiments, each individual screening included all 354 genotypes with two plant individuals (replicates) per genotype in a completely randomized design. In total 22 plant individuals were tested per genotype across 11 screening experiments (22 replicates per genotype). Manual infection was carried out at developmental stage BBCH 13–14 (3–4 true leaves, leaf pairs, or whorls unfolded) (Lancashire et al., 1991). L. maculans was propagated on oatmeal agar medium 2 weeks before infection. A mycelial agar plug was then placed at the stem base, slightly above the axil of the first true leaf, after wounding of the infection site using a needle. Subsequently, plants were grown under foil tunnels for 72 h to ensure appropriate humidity and temperature for a successful infection. At 49 days post infection (dpi), a cross section was cut at the stem base to estimate the length (L), girdling (G), and penetration depth (P) of the blackleg lesions. L was measured in mm whereas G and P were visually scored as percentage of the total circumference and diameter, respectively. Next, these scores were converted into individual 0–9 scales for each score. Using L, G, and P scoring values at 49 dpi, the Volume of Diseased Tissue (VDT) value was calculated using a formula modified from Kutcher et al. (1993):
Also, single screening means of the two replicates per screening, adjusted means across all the eleven screenings, were calculated using the R packages lmerTest version 3.1-2 (Kuznetsova et al., 2017) and lsmeans version 2.30-0 (Lenth, 2016). This approach allowed the assessment of QTL stability across different screenings.
The diversity set was grown in 2016/2017 and 2017/2018 in field trials in Rauischholzhausen, Germany. It was grown under normal farming practices with no use of fungicides. The fields were chosen based on close crop rotation and known high natural blackleg infection pressure. Single plots per genotype were sown and analyzed in a randomized complete block design, with plot sizes of 12.5 m2 (10 m × 1.25 m). At developmental stage BBCH 83–85 (30–50% of pods ripe, seeds black, and hard), 20 plants from the middle row of each plot were uprooted and cut at the stem base. Visual scores from 1 to 6 for blackleg infestation at the resulting cross section were used to calculate the G2 index for blackleg adult plant stem infection (Pilet et al., 1998; Aubertot et al., 2004) using the formula:
where N1, N2, N3, N4, N5, and N6 are the number of stems with scores 1, 2, 3, 4, 5, and 6, respectively, and Nt is the total number of stems assessed.
Characterization of Fungal Isolates
Leaf samples with characteristic lesions of L. maculans were collected in the field trials and dried. Leave segments were incubated in humid chambers to allow spore release from pycnidia. Single pycnidium isolates were prepared by plating spores on SNA medium amended with 200 ppm streptomycin for 6 days. Petri dishes were incubated under UV light at 20°C. Subsequently, a mycelial plug was transferred to V8 medium supplemented with 200 ppm streptomycin and incubated for 2 weeks under the same conditions. Spore suspensions were prepared by adjustment to 1 × 107 spores/ml using a hemocytometer. To characterize L. maculans isolates, cotyledon tests were applied using a differential set of B. napus harboring major resistance genes (Supplementary Table 3; Balesdent et al., 2002, 2006; Marcroft et al., 2012). Shortly, 10 μl spore suspension was applied on each lobe of cotyledon after injuring it with a needle. Eight replications were used. Symptoms were evaluated 14 days post inoculation according to the IMASCORE rating scale, where class 1 shows typical hypersensitive reactions and class 6 reflects tissue collapse with sporulation. Classes 1–3 were considered as resistance reactions, while classes 4–6 were noted as susceptible ones (Balesdent et al., 2001).
Single Nucleotide Polymorphism Genotyping and Analysis of Linkage Disequilibrium
All the investigated B. napus accessions were genotyped using the Brassica 60k Illumina Infinium™ SNP array. The B. napus reference genome assembly Darmor-bzh version 10 (Rousseau-Gueutin et al., 2020) was used to anchor 34,079 markers uniquely to a single position of the genome (Supplementary Table 1). Markers mapping unspecifically to multiple positions were excluded from further analyses. Finally, single hits were filtered for a cut-off e-value of 1e––15. Heterozygote SNP calls were considered as missing data since we should not expect heterozygote calls for DH or inbred lines, hence it can be assumed that these calls are mostly due to technical artifacts. Genome-wide linkage disequilibrium (LD) was calculated using the R package SelectionTools version 19.41. Prior to LD analysis, markers were filtered for minor allele frequency (MAF) ≥0.05 and a maximum of 10% missing data per marker and DH line. A tolerance threshold of r2 > 0.4 was set to assign markers in strong LD to respective LD blocks.
Genome-Wide Association Studies
Genome-wide association studies were conducted using the R package GenABEL version 1.8-0 (Aulchenko et al., 2007). Markers for the multiparental population were filtered as described above for LD analysis. This approach led to a set of 17,869 polymorphic and unique markers, including 16,400 SNP markers and 1,469 Single Nucleotide absence Polymorphism (SNaP) markers called as described by Gabur et al. (2018). The linear mixed model was adjusted for population structure by consideration of identity-by-state estimates and the first two principal components (PC) as covariates. To reduce false positive rates, LD blocks were determined as suggestive QTL when a minimum of two markers per block showed trait associations in at least two individual greenhouse screening rounds. A LOD score of −log10 (p-value) ≥3.0 was applied as threshold for suggestive marker-trait associations. Finally, QTL were determined after correction for false discovery rate (FDR ≤ 0.1) when performing GWAS with the adjusted means across all greenhouse screening rounds.
In order to validate QTL discovered in the multiparental population using the greenhouse data, we also performed an independent GWAS in the diversity panel using phenotypic data from the field trials. Similar filtering steps led to 23,603 polymorphic SNP markers. Here, LD-based QTL were defined by considering kinship and PC and applying a LOD score of −log10 (p-value) ≥3.0 as an arbitrary threshold for putative marker-trait associations.
Functional Annotation of Darmor-bzh Genes
Functional annotation data for Darmor-bzh v4.1 produced by Gabur et al. (2020) were used together with genome-wide functional annotation of Darmor-bzh v10 performed using the ‘‘Automatic assignment of Human Readable Descriptions’’ (AHRD)2 package (Supplementary Table 2). AHRD obtains the functional annotations for gene models by blasting them to various publicly available protein databases such as Swiss-Prot, TAIR or trEMBL. Two hundred best scoring blast results (based on e-value) were chosen from each of the above-mentioned databases. Description for all the resulting blast hits was then assigned a score using a multi-step approach. In the first step every description line was subjected to a couple of regular expression filters, removing descriptions such as “Whole genome shotgun sequence” and other vague terms like “OS = Arabidopsis thaliana.” In the subsequent step the description lines were broken down into single tokens. These tokens were then pushed through a blacklist filter, thereby discarding all the tokens present in the blacklist. Every token was then assigned an overlap score based on the bit score, the database score, and the overlap score of the blast match. In the last step the token score was divided by a correction factor to remove any bias toward longer or shorter description lines. For the exact database and software versions please refer to the “Darmor10_input_go_prediction.yaml” file in the Supplementary Material.
Whole Genome Long-Read Resequencing and Variant Calling
Long-read sequencing was performed for all the seven parental lines of the mapping population using Oxford Nanopore Technologies (ONT). DNA extraction was conducted as described in Chawla et al. (2020). In addition, DNA size selection was performed using the Circulomics Short Read Eliminator Kit (Circulomics Inc.). The recommended kit LSK-109 from ONT was used for DNA library preparation. Long-read sequencing was performed using the MinION device from ONT. Subsequently, basecalling was executed with Guppy version 4.0.14 and reads were aligned to the B. napus reference assembly Darmor-bzh v10 using the long-read mapper NGMLR version 0.2.7 (Sedlazeck et al., 2018). BAM files were created using samtools version 1.9 (Li et al., 2009). For genome-wide SV detection the variant caller Sniffles version 1.0.12 was used with default settings (Sedlazeck et al., 2018). Deletions and insertions with a minimum size of 30 bp were classified as SV. In addition, SNV calling was conducted using the deep neural network based variant caller Clair version 2 (Luo et al., 2020). The Clair module callVarBam, with a model for ONT data, was used to call SNV from BAM files. To reduce false positive calls, SNV calls were filtered for a minimum quality using a shell script kindly provided by Fritz Sedlazeck and Medhat Mahmoud, Baylor College of Medicine, Human Genome Sequencing Center, Houston, TX, United States. The script calculates the most appropriate cut-off value for SNV quality filtering according to the recommendations of the authors of Clair. Luo et al. (2020) observed that quality scores of variants derived from ONT data are usually bimodally distributed, so that high-quality base calls can be extracted by setting a quality cut-off at a value corresponding to the bottom of the valley between the two peaks plus 50. In addition to quality filtering, a stringent filtering for only homozygous calls and a minimum allele frequency (AF) of 0.5 for the variant were applied. Then, the files of the single genotypes were merged using bcftools version 1.10.2 and were subsequently used as an input to invoke the force-calling parameter of Clair. Next, all samples were run again to force-call the SNV provided within the merged file. This again was followed by filtering for quality, homozygous calls and a minimum AF of 0.1. A lower threshold for AF was chosen after force-calling, since previous SNV calling already indicates the presence of these variants. Subsequently, CLC Genomics Workbench (v9.0, QIAGEN Digital Insights, Aarhus, Denmark) was used to align the sequences of the parental lines and to predict the impacts of amino acid changes caused by SNV.
Single Nucleotide Variants Validation
Due to the high number of SNV, the sequence of the gene A07p27010.1_BnaDAR was selected to validate the SNV calling method. For this purpose, sets of primers were designed to amplify the selected region and Sanger Sequencing was performed in the mapping parents. Primers were designed using the online tool Primer3Plus (Untergasser et al., 2007).
Results
Identification of a L. maculans Isolate Without R Gene Interaction for Use in Quantitative Resistance Screening
From the 644 L. maculans isolates collected in Northern Germany and characterized by Winter and Koopmann (2016), one isolate was selected (isolate 1.4.1.15). This isolate has been shown in cotyledon test with a B. napus differential set to harbor virulence alleles against R genes Rlm1, Rlm2, Rlm4, Rlm7, Rlm9, LepR2, and LepR3. The isolate was tested on the cotyledons of the seven elite oilseed rape parents of the multiparental mapping population. It showed a susceptible interaction in all cotyledon tests indicating that none of the parents exhibit any qualitative major resistance against this fungal isolate based on plant R gene and fungal avirulence gene interaction (Supplementary Table 3). Thus, this virulent isolate 1.4.1.15 recovered from a field in Peine (Germany) in 2013 was used subsequently for the greenhouse screenings for quantitative resistance, in order to avoid interaction of major monogenic resistance genes with fungal avirulence genes that could putatively mask the effects of minor quantitative resistance loci in the mapping population.
European Elite B. napus Accessions Show Genetic Variation for Quantitative Blackleg Resistance
Mixed linear models (MLM) demonstrated significant genotypic variation within the multiparental population (p < 0.001). VDT values from greenhouse screenings showed a normal distribution within the six subfamilies as well as in the entire mapping population, confirming the quantitative inheritance of blackleg resistance in this population (Supplementary Figure 1). LD analysis resulted in an average number of 33 LD blocks per chromosome. Single greenhouse screening rounds using mean VDT values from 2 replicates per genotype revealed together up to 326 marker-trait associations in GWAS. GWAS using mean VDT values composed of 22 replicates for each genotype merged from a total number of 11 screening rounds identified 84 significant marker-trait associations (Supplementary Table 4). After FDR correction a total of eight QTL regions were identified (Table 1), seven on chromosome A09 and one on chromosome A07. No QTL were detected on C-subgenome chromosomes. As expected for quantitative resistance, phenotypic variation explained by individual SNP markers was low and was ranging from 3.2 to 5.6% (Table 1). QTL stacking in the multiparental population revealed that the allele combination present in the common parent Lorenz led to the highest susceptibility compared to alleles from the other parental lines. This confirmed the initial assumption and the choice of Lorenz as the susceptible common parent. Almost all resistance alleles were derived from the six founder lines, whereas only one resistance allele was derived from the common parent Lorenz. The ideal allele combination of the eight QTL resulted in an estimated effect on resistance of 35.5% (−2.06 on the VDT scale). This beneficial combination was already present in the two founder lines DK Cabernet and JN (Supplementary Table 5).
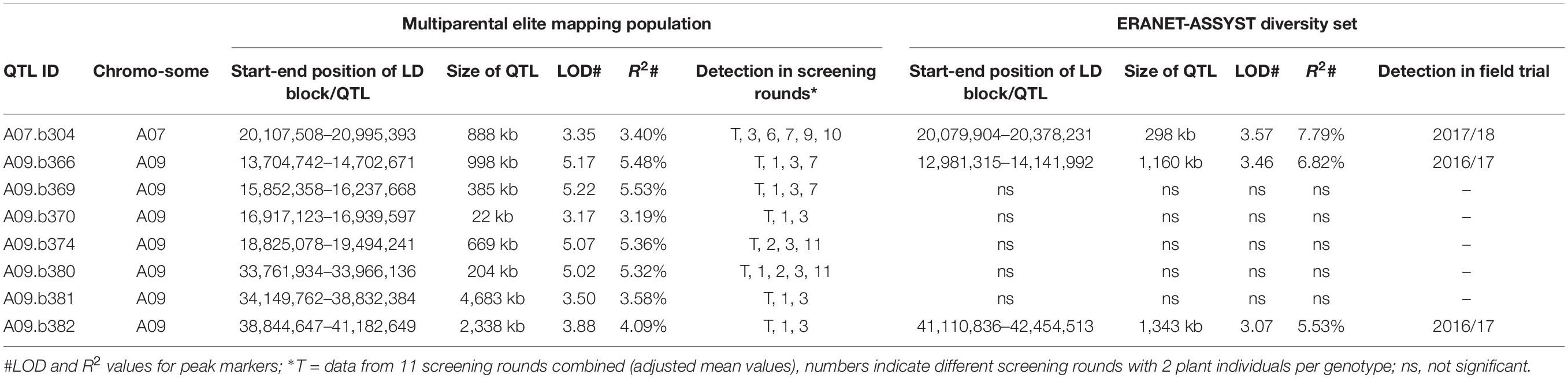
Table 1. Blackleg resistance QTL (VDT trait) identified in a B. napus multiparental elite mapping population in 11 independent greenhouse screening rounds (n = 354) and overlapping resistance QTL (G2 or stem lesion) in the ERANET-ASSYST diversity set in two field trials (n = 256).
Candidate Gene Analysis for Genes Involved in Quantitative Resistance
Based on two approaches together we identified 128 genes across the eight QTL regions that are associated to defense response and/or resistance in at least one reference genome (Supplementary Table 6). The QTL on chromosome A07 (haploblock A07.b304) was detected in the highest number of individual screening rounds (Table 1). The haploblock had a size of 888 kb in the investigated multiparental mapping population and contained 53 SNP markers and 193 genes in Darmor-bzh v10 (Supplementary Table 7). However, some genes in the interval revealed no annotation in Darmor-bzh v4.1 because no homologous genes exist between Darmor-bzh v4.1 and v10 or because Blast2GO revealed no annotation for the Darmor-bzh v4.1 genes (60 of 193). Thus, the protein sequences of the 193 B. napus Darmor-bzh v10 genes from the QTL interval were additionally aligned to the Arabidopsis reference genome Araport11 (Cheng et al., 2017) and literature links were evaluated exhibiting some additional, more detailed functional annotations. Out of 193 genes, 17 were associated to defense response and/or resistance (Supplementary Table 7). In particular, we found substantiated evidence in the literature that three of these 17 genes have a function related to fungal plant resistance in A. thaliana and B. napus in interaction with common B. napus fungal pathogens (A07p26890.1_BnaDAR, A07p28430.1_BnaDAR, A07p27010.1_BnaDAR). Most interestingly, the gene A07p27010.1_BnaDAR has been shown to be the major resistance gene Rlm9, which imparts qualitative resistance against L. maculans in B. napus (Larkan et al., 2020). To evaluate for the presence of resistance gene analogs (RGAs) in the QTL region of A07.b304 and their polymorphisms between the parents of the multiparental mapping population, we also located the QTL interval for Darmor-bzh versions 4.1 (Chalhoub et al., 2014) and 8.1 (Bayer et al., 2017) based on the flanking SNP markers (Supplementary Table 8). However, two out of five putative RGAs identified in this interval showed no polymorphism and none of them had been annotated to be involved in plant resistance.
Identification of Major Resistance Gene Rlm9 as Candidate Gene for Quantitative Blackleg Resistance
The detected quantitative resistance region has been mapped before for the qualitative resistance gene Rlm9 in young seedlings (Larkan et al., 2016b; Raman H. et al., 2020). However, testing of virulence complexity proved that the isolate used for greenhouse screening for quantitative resistance in our experiments shows no gene-for-gene interaction with Rlm1, Rlm2, Rlm4, Rlm7, Rlm9, LepR2, and LepR3 (see above). On the other hand, variant calling using ONT long-read sequencing data revealed that the sequence diversity in the gene A07p27010.1_BnaDAR (Rlm9) was considerably higher than in all other genes within the QTL interval and all putative resistance genes in any of the other identified QTL (Supplementary Table 7). Within the QTL interval, the peak SNP marker lies just 35 kb away from the Rlm9 gene. Within the sequence of Rlm9, we found 142 polymorphic SNV calls and a 6 kb insertion in four of the seven parental lines (Adriana, Alpaga, Galileo, and King 10). Parental lines Lorenz (susceptible), DK Cabernet and JN displayed an identical haplotype, comprising 24 SNPs in LD to the peak SNP marker, whereas the resistant genotypes harboring the insertion within the sequence of Rlm9 (Adriana, Alpaga, Galileo, King 10) showed a deviating haplotype (Figure 1). PCR and Sanger sequencing proved both, the authenticity of the 6 kb insertion and the correctness of the 142 intragenic SNV calls in A07p27010.1_BnaDAR. Each of the 142 SNV calls from ONT data was confirmed to be correct, whereby the Sanger sequencing also revealed a further 60 SNV within this particular gene. These 60 false negatives can be explained by the strict filtering process applied to eliminate false positive calls in the SNV calling approach. Nevertheless, the results indicate that SNV calling from “noisy” long reads produced with ONT provides reliable variant calls for genetic analysis.

Figure 1. LD based haploblock A07.b304 (r2 = 0.4) containing 54 single nucleotide polymorphism (SNP) markers and Rlm9 (A07p27010.1_BnaDAR). Green color indicates the two haplotypes with 24 identical SNP markers between the three parental lines Lorenz, DK Cabernet and JN. Haplotypes 2, 3, and 5 are harboring the 6 kb insertion in the sequence of Rlm9 (orange). Purple bar highlights the overlap with the LD based QTL identified in the diversity set in field trials. Gray highlight indicates Rlm9 gene, yellow highlight indicates SNP markers identified in one individual greenhouse screening round, red color indicates SNP markers identified in at least two individual greenhouse screening rounds.
Rlm9 codes for a wall-associated kinase like (WAKL) protein. A search for motifs using the Conserved Domain Search tool implemented in NCBI along with the Pfam database revealed three conserved domains, two within exon 1 and one within exon 3. These comprise an extracellular galacturonan-binding domain (GUB_WAK), a C-terminal wall-associated kinase (WAK) and an intracellular Serine/Threonine protein kinase domain (Ser/Thr_kinase). In addition, an EGF-like domain is located in exon 2 (Larkan et al., 2020). Based on the Sanger sequencing we found that three of the seven parental genotypes (Lorenz, DK Cabernet, JN) carry an identical Rlm9 allele to Darmor-bzh, whereas the other four harbor a 6 kb insertion within the second exon along with 202 SNV throughout the entire gene (Adriana, Alpaga, Galileo, King 10). These SNV caused in total 92 non-synonymous amino acid changes. We observed 19 amino acid changes within the GUB_WAK domain (84.2% identity to Darmor-bzh), 15 amino acid changes within the WAK domain (86.7% identity to Darmor-bzh) and 23 amino acid changes as well as a stop codon within the Ser/Thr_kinase domain (91.3% identity to Darmor-bzh). The EGF-like domain in exon 2 was disrupted by the 6 kb insertion in genotypes Adriana, Alpaga, Galileo, and King 10 (Figure 2).
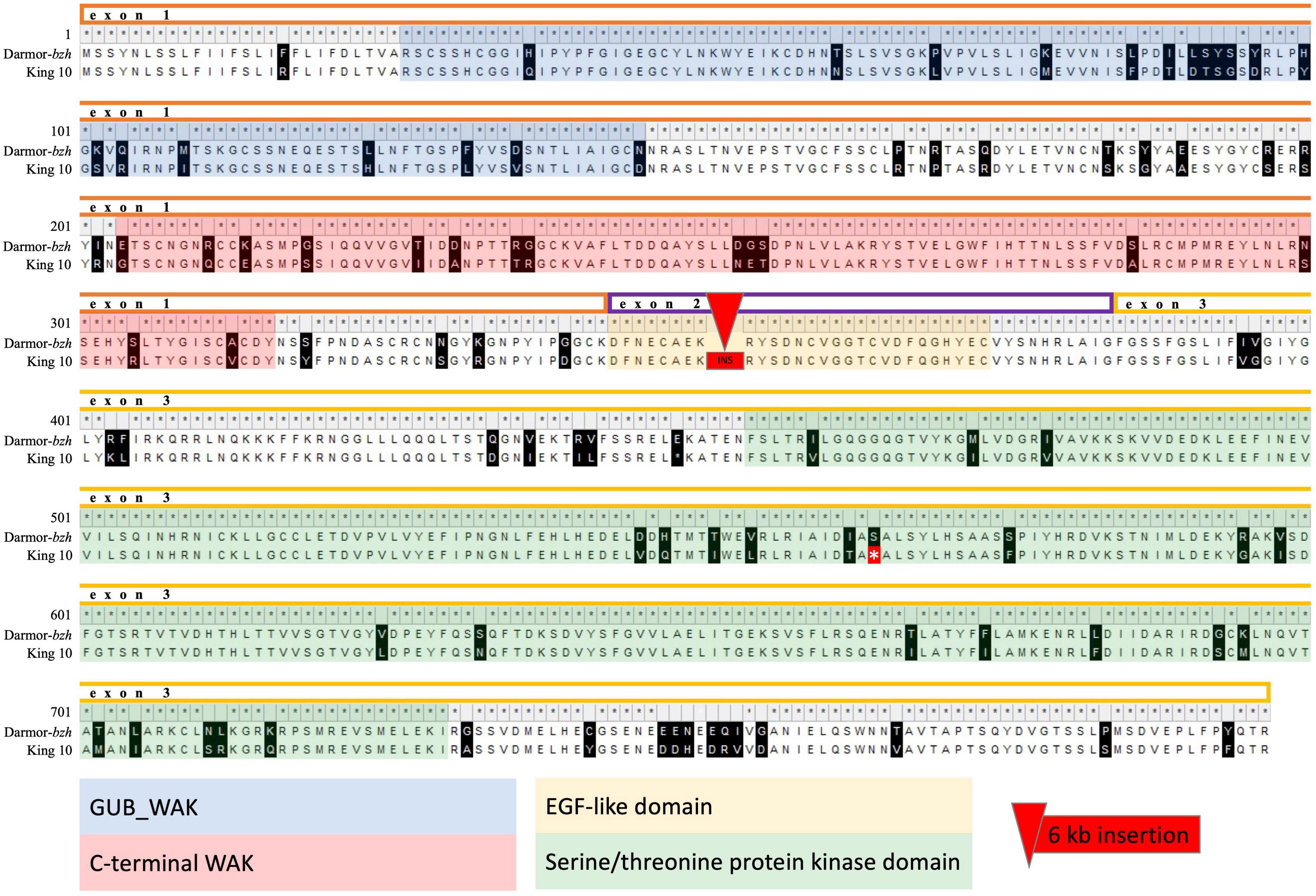
Figure 2. Alignment of the protein sequence of King 10 against the reference Darmor-bzh v10. King 10 is representative for the four parental lines harboring a 6 kb insertion and 202 SNV leading to 92 non-synonymous amino acid changes. Different colors indicate exons and conserved domains. The asterisk indicates a stop codon.
Candidate Genes in Co-localizing Quantitative Trait Loci From Greenhouse and Field Trials
The QTL regions and candidate genes where initially identified in the elite population under controlled conditions excluding R gene interaction. To putatively support these candidate genes, we also identified QTL for adult plant resistance under field conditions in a diversity set. The field trials using the ERANET-ASSYST diversity set relied on natural infection. Characterization of field isolates sampled at the field site in Rauischholzhausen (Germany) revealed fungal isolates harboring the avirulence alleles AvrLm3 or AvrLm7 in combination with virulence alleles avrLm1, avrLm2, avrLm4, and avrLm9. This situation represents the typical German field situation as reported previously (Winter and Koopmann, 2016). This implies that monogenic Rlm3 and Rlm7, also located on chromosome A07 (Larkan et al., 2016b; Raman R. et al., 2020) and potentially harbored by some of the genotypes of the diversity set, could mask quantitative resistance associated with the investigated genomic region on chromosome A07. However, monogenic Rlm9 effects should not mask quantitative resistance effects associated with Rlm9 in this field situation. Three of the eight QTL found in the analysis with the multiparental mapping population were also found in field trials for blackleg stem lesions in the ERANET-ASSYST diversity set (QTL A07.b304, A09.b366 and A09.b382, Table 1 and Supplementary Table 9). The LD blocks for these three QTL partly overlap in both populations. Assuming that the same genes are involved in quantitative resistance expression in both winter oilseed rape populations, the overlap in the QTL interval might be useful to narrow the search for potential candidate genes. By considering overlapping QTL regions as high-confidence QTL intervals, we reduced the areas of interest from 888 kb to 271 kb, 998 kb to 473 kb, and 2,338 kb to 72 kb, respectively. In total, these three intervals contained 133 genes (68, 54, and 11). The number of candidate genes was reduced from 68 to 5, 54 to 2, and 11 to 1 for the three co-localizing QTL intervals for QTL A07.b304, A09.b366, and A09.b382 by filtering for GO terms associated to defense response and/or resistance (Table 2 and Supplementary Table 10). Rlm9 is localized in the overlapping QTL region of both populations, the multiparental mapping population and the diversity set tested under controlled conditions in the greenhouse and in the field, respectively.
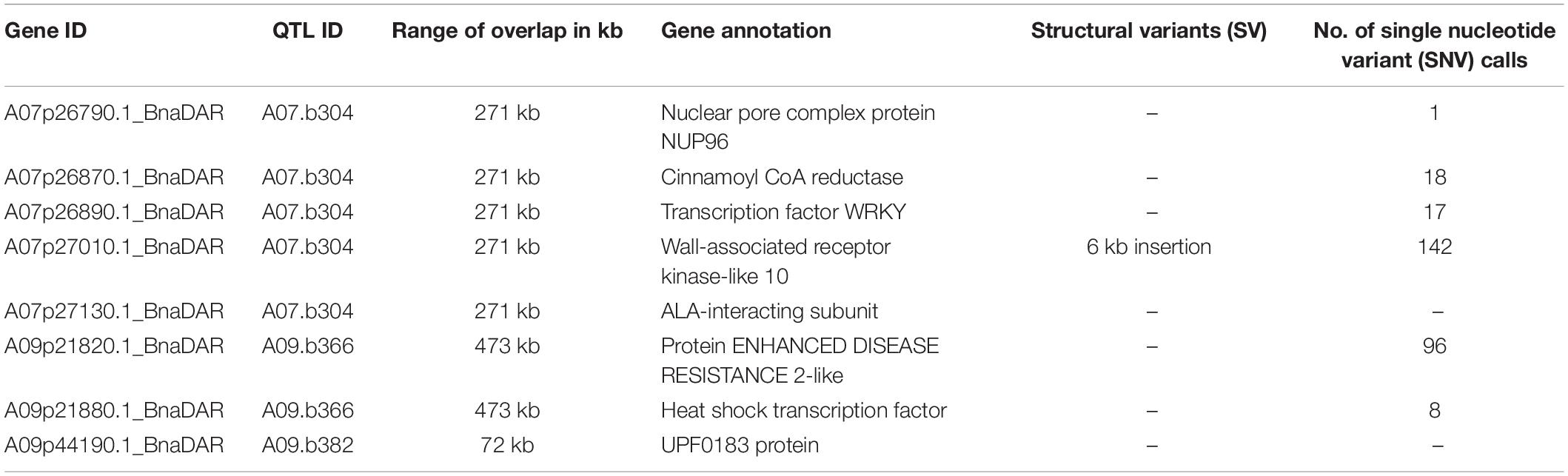
Table 2. Genes within the L. maculans resistance QTL of the multiparental population overlapping with QTL of the diversity set based on GO terms that can be associated with plant resistance and numbers of genomic variants in the multiparental population.
Discussion
Quantitative resistance against L. maculans exhibiting minor effects in B. napus is difficult to detect as it is frequently masked by qualitative resistances exhibiting major effects controlled by race-specific R genes in the host and pathogen. Pathogen-associated molecular patterns (PAMPs) are considered to initiate a broad-spectrum resistance against a pathogen species, termed PAMP-triggered immunity (PTI), whereas race-specific pathogen effectors contribute to pathogen virulence and can induce effector-triggered immunity (ETI) in the plant host on a gene-for-gene interaction model, also called qualitative resistance (Jones and Dangl, 2006). However, in the last decade it has become clear that this strict distinction between PTI and ETI might need reconsideration (Thomma et al., 2011). In addition, the extent to which PTI is associated with quantitative resistance expression also remains to be clarified (Delplace et al., 2020). ETI and PTI are both mainly involved in pathogen perception and signaling, whereas quantitative resistance goes beyond that and is controlled by numerous genes with diversified functions (Corwin and Kliebenstein, 2017). Qualitative and quantitative resistance cannot normally be clearly distinguished in field studies. The complexity of the fungal population in the field, harboring different avirulence genes, and the genetic composition of the plant population, harboring unknown minor and major resistance allele combinations, generally cause difficulties in association studies of quantitative resistance under field conditions. Thus, our strategy in this study was to identify and use a fungal L. maculans isolate that has no qualitative resistance R gene interaction with the parental oilseed rape cultivars of our multiparental population, and to use this isolate to map quantitative minor effect QTL by GWAS under controlled conditions.
Using this strategy, we identified a normal distribution of disease, indicating the exclusively quantitative inheritance of blackleg resistance in the mapping population. The absence of effective gene-for-gene interactions that can usually be observed in the B. napus–L. maculans pathosystem allowed us to identify genomic regions explaining only a small portion of phenotypic variation (<6%). In accordance with a previous study, we also observed high QTL-by-environment interactions, which is common for quantitatively inherited disease resistance even under controlled greenhouse conditions (Obermeier et al., 2013) and is even more common due to varying disease pressure throughout field trials (Huang et al., 2016; Larkan et al., 2016a; Kumar et al., 2018; Raman et al., 2018).
In most studies mapping quantitative blackleg resistance, different methods for disease scoring of cross sections at the crown or scoring of survival rate were used to estimate the disease severity (Huang et al., 2016; Larkan et al., 2016a; Gabur et al., 2018; Kumar et al., 2018; Raman et al., 2018; Raman R. et al., 2020). In the field trials of the present study, G2 index scoring of cross sections at the crown was applied due to the immense workload that needs to be achieved within a short period of time. However, the G2 index depends strongly on precise cuts to accurately assess the cross sections. This phenotyping method is time-saving compared to VDT scoring and provides useful and informative data from field evaluations of blackleg disease. In addition, to gain more detailed insights into the growth of the fungus, the VDT scoring method of Kutcher et al. (1993) was used for the assessments in the greenhouse trials. The high overlap of QTL results with previous studies underpins the relevance of the applied phenotyping method and further reveals that some of the identified QTL regions are crucial within international germplasm (Jestin et al., 2015; Raman et al., 2016, 2018; Kumar et al., 2018; Fikere et al., 2020; Raman H. et al., 2020; Raman R. et al., 2020; Supplementary Table 11).
We mapped quantitative resistance on chromosomes A09 and A07 in the multiparental population in the greenhouse. All QTL explained less than 5% of the phenotypic variation suggesting that genomic selection approaches are more suitable than marker-assisted selection for breeding toward quantitative L. maculans resistance in oilseed rape. Some of the QTL regions identified in the present study on chromosome A09 and A07 overlapped with previously described QTL (Supplementary Table 11). However, chromosome A07, in contrast to A09, is mainly known to harbor qualitative resistance genes providing race-specific resistances (Delourme et al., 2006). Although we ensured the specific assessment of quantitative resistance by selecting a fungal isolate with no gene-for-gene interaction with the mapping population in our greenhouse trials, to our surprise a QTL explaining less than 5% of the phenotypic variance was identified in a genome region on chromosome A07 known to harbor a cluster of R genes (Rlm3, Rlm4, Rlm7, and Rlm9) involved in qualitative resistance expression. This suggests that this genome region is either a genomic hot spot where genes involved in qualitative as well as quantitative resistance are tightly clustered (linkage) or that some genes known to be involved in major qualitative resistance can also impart quantitative effects on resistance. Although it is possible that linkage exists between R gene clusters which are non-functional at the cotyledon stage in our population and genes involved explicitly in quantitative resistance expression in the LD block on chromosome A07, no clear candidate genes could be identified by GO analysis or polymorphism detection between the parental genotypes. The most striking polymorphism was detected in the well described major resistance gene Rlm9, which could suggest that this gene is also involved in adult plant resistance expression in the investigated multiparental population. In addition, Rlm9 co-localized with an overlapping QTL region in a diversity set for field resistance in adult plants subjected to infection by an L. maculans population with an avirulent AvrRlm9 gene composition. This observation supports the involvement of this candidate gene in quantitative resistance expression in these environments and populations.
This result is in accordance with previous reports that some major Rlm/LepR genes also have quantitative effects on adult plant resistance. For example, the fungal AvrLmS-Lep2 gene-for-gene interaction with a B. napus R gene, LepR2, shows a qualitative intermediate resistance response at the cotyledon stage and partial resistance at the adult plant stage (Long et al., 2011; Dandena et al., 2019; Neik et al., 2020, preprint). The hypothesis that Rlm9 is involved in quantitative resistance is also supported by Raman et al. (2018), who reported a quantitative resistance effect of the location harboring Rlm9 in the greenhouse on adult plants, using a plant population segregating for Rlm9 and an L. maculans isolate carrying a corresponding functional avirulence gene AvrLm5-9 allele. That result also suggested that Rlm9-mediated resistance was expressed at the adult plant stage. The concept of R genes mediating quantitative resistance has been previously suggested and discussed in other studies that made similar observations (Chantret et al., 1999; Raman et al., 2018). A possible weak, constitutive expression of R genes at adult plant stage may lead to partial resistance with only minor effects. Our results suggest that further studies investigating R gene expression at adult plant stage or even accompanying an entire growth period could give deeper insights into these repeatedly observed findings.
Our detailed analysis of the molecular polymorphism for Rlm9 in the parents of the mapping population also supports this hypothesis. In particular, non-synonymous amino acid changes, an inserted stop codon and especially the large insertion in Rlm9 most likely alter the transcript or even interrupt the transcription of the gene in four of the seven parents of the mapping population. Usually, R genes encode nucleotide-binding site leucine-rich repeat (NBS-LRR) proteins (McHale et al., 2006). Wall-associated kinase (WAK) and wall-associated kinase-like (WAKL) genes are a newly discovered class of race-specific plant receptor-like kinase resistance genes involved in qualitative resistance (Larkan et al., 2020). However, although some WAKL genes have been shown to be involved in race-specific gene-for-gene interactions, for example Rlm9 in oilseed rape and Stb9 in wheat (Keller and Krattinger, 2018), other WAKL genes like RFO1 (RESISTANCE TO FUSARIUM OXYSPORUM 1)/WAKL22 in Arabidopsis have been found to be involved in broad-spectrum resistance against F. oxysporum f. sp. matthioli and other races (Diener and Ausubel, 2005) and against Verticillium longisporum (Johansson et al., 2006) in Arabidopsis, while ZmWAK1 confers quantitative resistance to northern corn leaf blight in maize (Hurni et al., 2015). The protein structure of the WAKs and some WAKLs can be divided into an extracellular and an intracellular compartment, which are connected by a transmembrane domain. These proteins are characterized by a cytoplasmic Ser/Thr kinase domain in the cell interior and an extracellular domain that is similar to epidermal growth factor domains in vertebrates (epidermal growth factor; EGF-like domain). This occurs as a calcium-binding EGF domain (EGF-Ca2+) and/or as an EGF2-like domain, although in some cases it is slightly degenerate (Verica and He, 2002). The exact function of these EGF-like domains is still largely unclear. However, previous studies demonstrated that EGF-like domains are involved in protein-protein interactions (Kuroda and Tanizawa, 1999). This extracellular domain is disrupted in four of the seven parents in our study by a 6 kb insertion, which might thus be expected to potentially interrupt (unknown) protein-protein-interactions and could consequently impart a quantitative impact on resistance activity. Other parts of the extracellular domain are bound to the pectin of the cell wall (Wagner and Kohorn, 2001), but can also serve as receptors for oligogalacturonides (OGs), which, among other things, arise from mechanical destruction of the pectin and act as DAMPs (damage-associated molecular patterns) to activate the plant immune system (Brutus et al., 2010). This part contains a conserved GUB_WAK domain (galacturonan-binding wall-associated receptor kinase). This suggests that WAKL genes might be involved in more broad-spectrum resistance in some pathogen-host interactions by sensing DAMPs. Hence, our results in oilseed rape suggest a possible dual function of Rlm9. On the one hand, Rlm9 is expected to be involved in race-specific PTI in oilseed rape when challenged with L. maculans isolates carrying AvrLm5-9, which trigger a strong qualitative gene-for-gene resistance effect in plant genotypes with a functional interacting Rlm9 gene as in Raman et al. (2018). However, in contrast to Raman et al. (2018), the results in our study suggest that the gene-for-gene interaction for Rlm9 is not only expressed at the cotyledon stage, but also to a lesser extent in adult plants. In contrast to Raman et al. (2018), we used a L. maculans isolate which did not harbor a corresponding avirulence gene (avrLm5-9). Thus, even though no gene-for-gene interaction of Rlm9 with its corresponding avirulence gene is expected in our experiment, still we found a weak expression of quantitative resistance of 5%. This indicates that Rlm9 may have other additional features triggering quantitative resistance, for example by sensing DAMPs. Only if some functional domains are disrupted, like the EGF-like domain in four of the seven genotypes in our study, might this weak quantitative resistance effect also be lost. All in all, the results of the present study confer with previous studies that observed R gene mediated resistance under controlled and field conditions at adult plant stages in B. napus (Delourme et al., 2004; Raman et al., 2012). Based on these collective observations, we hypothesize that the role of Rlm9 in this interaction deserves more detailed functional analysis in future.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA751459).
Author Contributions
CO and RS conceived the idea and sourced the funding. PV, HC, and CO developed the methodology. PV generated the genetic and field data. DA and BK generated the greenhouse data. PV and CO performed data curation. PV, HC, and HL analyzed the sequence and SV data. PV, SW, LE, and IG performed the quantitative genetic analysis. PV, RS, and CO drafted and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
PV and CO received funding from the German Federal Ministry of Education and Research (BMBF) within the project PhomaDur (Grant Number BLE: 2818205115). This work was also supported by the BMBF-funded de.NBI Cloud within the German Network for Bioinformatics Infrastructure (de.NBI) (031A537B, 031A533A, 031A538A, 031A533B, 031A535A, 031A537C, 031A534A, and 031A532B).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Regina Illgner, Jutta Schaper, and Anita Popovska for excellent technical help. We also thank all German breeding companies organized in the GFPi (Gemeinschaft zur Förderung von Pflanzeninnovation e. V.) for production of the elite NAM population, for performing field trials, and for providing phenotyping data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.749491/full#supplementary-material
Supplementary Figure 1 | Phenotypic distribution of VDT values from greenhouse screenings for the entire mapping population (A) and the individual subfamilies: Adriana × Lorenz (B), Lorenz × Alpaga (C), Lorenz × DK Cabernet (D), Lorenz × Galileo (E), JN × Lorenz (F), and King 10 × Lorenz (G).
Footnotes
References
Aubertot, J.-N., Schott, J.-J., Penaud, A., Brun, H., and Doré, T. (2004). Methods for sampling and assessment in relation to the spatial pattern of phoma stem canker (Leptosphaeria maculans) in oilseed rape. Eur. J. Plant Pathol. 110, 183–192. doi: 10.1023/B:EJPP.0000015359.61910.3b
Aulchenko, Y. S., Koning, D.-J. D., and Haley, C. (2007). Genomewide Rapid Association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait Loci Association analysis. Genetics 177, 577–585. doi: 10.1534/genetics.107.075614
Balesdent, M. H., Attard, A., Ansan-Melayah, D., Delourme, R., Renard, M., and Rouxel, T. (2001). Genetic Control and host range of avirulence toward Brassica napus cultivars quinta and jet neuf in Leptosphaeria maculans. Phytopathology 91, 70–76. doi: 10.1094/PHYTO.2001.91.1.70
Balesdent, M. H., Attard, A., Kühn, M. L., and Rouxel, T. (2002). New avirulence genes in the phytopathogenic fungus Leptosphaeria maculans. Phytopathology 92, 1122–1133. doi: 10.1094/PHYTO.2002.92.10.1122
Balesdent, M. H., Barbetti, M. J., Li, H., Sivasithamparam, K., Gout, L., and Rouxel, T. (2005). Analysis of Leptosphaeria maculans race structure in a worldwide collection of isolates. Phytopathology 95, 1061–1071. doi: 10.1094/PHYTO-95-1061
Balesdent, M.-H., Louvard, K., Pinochet, X., and Rouxel, T. (2006). A large-scale survey of races of Leptosphaeria maculans occurring on oilseed rape in France. Eur. J. Plant Pathol. 114, 53–65. doi: 10.1007/s10658-005-2104-0
Bayer, P. E., Hurgobin, B., Golicz, A. A., Chan, C.-K. K., Yuan, Y., Lee, H., et al. (2017). Assembly and comparison of two closely related Brassica napus genomes. Plant Biotechnol. J. 15, 1602–1610. doi: 10.1111/pbi.12742
Brun, H., Chèvre, A.-M., Fitt, B. D. L., Powers, S., Besnard, A.-L., Ermel, M., et al. (2010). Quantitative resistance increases the durability of qualitative resistance to Leptosphaeria maculans in Brassica napus. New Phytol. 185, 285–299. doi: 10.1111/j.1469-8137.2009.03049.x
Brutus, A., Sicilia, F., Macone, A., Cervone, F., and de Lorenzo, G. (2010). A domain swap approach reveals a role of the plant wall-associated kinase 1 (WAK1) as a receptor of oligogalacturonides. Proc. Natl. Acad. Sci. U.S.A. 107, 9452–9457. doi: 10.1073/pnas.1000675107
Bus, A., Körber, N., Snowdon, R. J., and Stich, B. (2011). Patterns of molecular variation in a species-wide germplasm set of Brassica napus. Theor. Appl. Genet. 123, 1413–1423. doi: 10.1007/s00122-011-1676-7
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A. P., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Chantret, N., Pavoine, M. T., and Doussinault, G. (1999). The race-specific resistance gene to powdery mildew, MlRE, has a residual effect on adult plant resistance of winter wheat line RE714. Phytopathology 89, 533–539. doi: 10.1094/PHYTO.1999.89.7.533
Chawla, H. S., Lee, H., Gabur, I., Vollrath, P., Tamilselvan-Nattar-Amutha, S., Obermeier, C., et al. (2020). Long-read sequencing reveals widespread intragenic structural variants in a recent allopolyploid crop plant. Plant Biotechnol. J. 19, 240–250. doi: 10.1111/pbi.13456
Cheng, C. Y., Krishnakumar, V., Chan, A. P., Thibaud-Nissen, F., Schobel, S., and Town, C. D. (2017). Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 89, 789–804. doi: 10.1111/tpj.13415
Corwin, J. A., and Kliebenstein, D. J. (2017). Quantitative resistance: more than just perception of a pathogen. Plant Cell 29, 655–665. doi: 10.1105/tpc.16.00915
Dandena, H. B., Zhang, Q., Zhou, T., Hirani, A. H., Liu, Z., Fernando, D. W. G., et al. (2019). Analysis of quantitative adult plant resistance to blackleg in Brassica napus. Mol. Breed. 39:124. doi: 10.1007/s11032-019-1035-y
Delourme, R., Bousset, L., Ermel, M., Duffé, P., Besnard, A. L., Marquer, B., et al. (2014). Quantitative resistance affects the speed of frequency increase but not the diversity of the virulence alleles overcoming a major resistance gene to Leptosphaeria maculans in oilseed rape. Infect. Genet. Evol. 27, 490–499. doi: 10.1016/j.meegid.2013.12.019
Delourme, R., Chèvre, A. M., Brun, H., Rouxel, T., Balesdent, M. H., Dias, J. S., et al. (2006). Major gene and polygenic resistance to Leptosphaeria maculans in oilseed rape (Brassica napus). Eur. J. Plant Pathol. 114, 41–52. doi: 10.1007/s10658-005-2108-9
Delourme, R., Pilet-Nayel, M.-L., Archipiano, M., Horvais, R., Tanguy, X., Rouxel, T., et al. (2004). A cluster of major specific resistance genes to Leptosphaeria maculans in Brassica napus. Phytopathology 94, 578–583. doi: 10.1094/phyto.2004.94.6.578
Delplace, F., Huard-Chauveau, C., Dubiella, U., Khafif, M., Alvarez, E., Langin, G., et al. (2020). Robustness of plant quantitative disease resistance is provided by a decentralized immune network. Proc. Natl. Acad. Sci. U.S.A. 117, 18099–18109. doi: 10.1073/pnas.2000078117
Diener, A. C., and Ausubel, F. M. (2005). RESISTANCE TO FUSARIUM OXYSPORUM 1, a dominant Arabidopsis disease-resistance gene, is not race specific. Genetics 171, 305–321. doi: 10.1534/genetics.105.042218
Elliott, V. L., Marcroft, S. J., Howlett, B. J., and van de Wouw, A. P. (2016). Gene-for-gene resistance is expressed in cotyledons, leaves and pods, but not during late stages of stem colonization in the Leptosphaeria maculans-Brassica napus pathosystem. Plant Breed. 135, 200–207. doi: 10.1111/pbr.12343
Fikere, M., Barbulescu, D. M., Malmberg, M. M., Spangenberg, G. C., Cogan, N. O. I., and Daetwyler, H. D. (2020). Meta-analysis of GWAS in canola blackleg (Leptosphaeria maculans) disease traits demonstrates increased power from imputed whole-genome sequence. Sci. Rep. 10:14300. doi: 10.1038/s41598-020-71274-6
Fitt, B. D. L., Brun, H., Barbetti, M. J., and Rimmer, S. R. (2006). World-wide importance of phoma stem canker (Leptosphaeria maculans and L. biglobosa) on oilseed rape (Brassica napus). Eur. J. Plant Pathol. 114, 3–15. doi: 10.1007/s10658-005-2233-5
Gabur, I., Chawla, H. S., Liu, X., Kumar, V., Faure, S., Tiedemann, A. V., et al. (2018). Finding invisible quantitative trait loci with missing data. Plant Biotechnol. J. 16, 2102–2112. doi: 10.1111/pbi.12942
Gabur, I., Chawla, H. S., Lopisso, D. T., Tiedemann, A. V., Snowdon, R. J., and Obermeier, C. (2020). Gene presence-absence variation associates with quantitative Verticillium longisporum disease resistance in Brassica napus. Sci. Rep. 10:4131. doi: 10.1038/s41598-020-61228-3
Huang, Y. J., Jestin, C., Welham, S. J., King, G. J., Manzanares-Dauleux, M. J., Fitt, B. D. L., et al. (2016). Identification of environmentally stable QTL for resistance against Leptosphaeria maculans in oilseed rape (Brassica napus). Theor. Appl. Genet. 129, 169–180. doi: 10.1007/s00122-015-2620-z
Huang, Y. J., Pirie, E. J., Evans, N., Delourme, R., King, G. J., and Fitt, B. D. L. (2009). Quantitative resistance to symptomless growth of Leptosphaeria maculans (phoma stem canker) in Brassica napus (oilseed rape). Plant Pathol. 58, 314–323. doi: 10.1111/j.1365-3059.2008.01957.x
Hurgobin, B., Golicz, A. A., Bayer, P. E., Chan, C.-K. K., Tirnaz, S., Dolatabadian, A., et al. (2018). Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 16, 1265–1274. doi: 10.1111/pbi.12867
Hurni, S., Scheuermann, D., Krattinger, S. G., Kessel, B., Wicker, T., Herren, G., et al. (2015). The maize disease resistance gene Htn1 against northern corn leaf blight encodes a wall-associated receptor-like kinase. Proc. Natl. Acad. Sci. U.S.A. 112, 8780–8785. doi: 10.1073/pnas.1502522112
Jestin, C., Bardol, N., Lodé, M., Duffé, P., Domin, C., Vallée, P., et al. (2015). Connected populations for detecting quantitative resistance factors to phoma stem canker in oilseed rape (Brassica napus L.). Mol. Breed. 35:167. doi: 10.1007/s11032-015-0356-8
Johansson, A., Staal, J., and Dixelius, C. (2006). Early responses in the Arabidopsis-Verticillium longisporum pathosystem are dependent on NDR1, JA- and ET-associated signals via cytosolic NPR1 and RFO1. Mol. Plant Microbe Interact. 19, 958–969. doi: 10.1094/MPMI-19-0958
Jones, J. D. G., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Keller, B., and Krattinger, S. G. (2018). A new player in race-specific resistance. Nat. Plants 4, 197–198. doi: 10.1038/s41477-018-0126-9
Kumar, V., Paillard, S., Fopa Fomeju, B., Falentin, C., Deniot, G., Baron, C., et al. (2018). Multi-year linkage and association mapping confirm the high number of genomic regions involved in oilseed rape quantitative resistance to blackleg. Theor. Appl. Genet. 131, 1627–1643. doi: 10.1007/s00122-018-3103-9
Kuroda, S., and Tanizawa, K. (1999). Involvement of epidermal growth factor-like domain of NELL proteins in the novel protein-protein interaction with protein kinase C. Biochem. Biophys. Res. Commun. 265, 752–757. doi: 10.1006/bbrc.1999.1753
Kutcher, H. R., van den Berg, C. G. J., and Rimmer, S. R. (1993). Variation in pathogenicity of Leptosphaeria maculans on Brassica spp. based on cotyledon and stem reactions. Can. J. Plant Pathol. 15, 253–258. doi: 10.1080/07060669309501920
Kuznetsova, A., Brockhoff, P. B., and Christensen, R. H. B. (2017). lmertest package: tests in linear mixed effects models. J. Stat. Softw. 82, 1–26. doi: 10.18637/jss.v082.i13
Lancashire, P. D., Bleiholder, H., van den Boom, T., Langelüddeke, P., Stauss, R., Elfriede, W., et al. (1991). A uniform decimal code for growth stages of crops and weeds. Ann. Appl. Biol. 1991, 561–601. doi: 10.1111/j.1744-7348.1991.tb04895.x
Larkan, N. J., Ma, L., Haddadi, P., Buchwaldt, M., Parkin, I. A. P., Djavaheri, M., et al. (2020). The Brassica napus wall-associated kinase-like (WAKL) gene Rlm9 provides race-specific blackleg resistance. Plant J. 104, 892–900. doi: 10.1111/tpj.14966
Larkan, N. J., Raman, H., Lydiate, D. J., Robinson, S. J., Yu, F., Barbulescu, D. M., et al. (2016a). Multi-environment QTL studies suggest a role for cysteine-rich protein kinase genes in quantitative resistance to blackleg disease in Brassica napus. BMC Plant Biol. 16:183. doi: 10.1186/s12870-016-0877-2
Larkan, N. J., Yu, F., Lydiate, D. J., Rimmer, S. R., and Borhan, M. H. (2016b). Single R gene introgression lines for accurate dissection of the Brassica–Leptosphaeria pathosystem. Front. Plant Sci. 7:1771. doi: 10.3389/fpls.2016.01771
Lenth, R. V. (2016). Least-squares means: the R package lsmeans. J. Stat. Softw. 69, 1–33. doi: 10.18637/jss.v069.i01
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Long, Y., Wang, Z., Sun, Z., Fernando, D. W. G., McVetty, P. B. E., and Li, G. (2011). Identification of two blackleg resistance genes and fine mapping of one of these two genes in a Brassica napus canola cultivar ‘Surpass 400’. Theor. Appl. Genet. 122, 1223–1231. doi: 10.1007/s00122-010-1526-z
Luo, R., Wong, C.-L., Wong, Y.-S., Tang, C.-I., Liu, C.-M., Leung, C.-M., et al. (2020). Exploring the limit of using a deep neural network on pileup data for germline variant calling. Nat. Mach. Intell. 2, 220–227. doi: 10.1038/s42256-020-0167-4
Marcroft, S. J., Elliott, V. L., Cozijnsen, A. J., Salisbury, P. A., Howlett, B. J., and van de Wouw, A. P. (2012). Identifying resistance genes to Leptosphaeria maculans in Australian Brassica napus cultivars based on reactions to isolates with known avirulence genotypes. Crop Pasture Sci. 63, 338–350. doi: 10.1071/CP11341
McHale, L., Tan, X., Koehl, P., and Michelmore, R. W. (2006). Plant NBS-LRR proteins: adaptable guards. Genome Biol. 7:212. doi: 10.1186/gb-2006-7-4-212
Neik, T. X., Ghanbarnia, K., Ollivier, B., Scheben, A., Severn-Ellis, A., Larkan, N. J., et al. (2020). Two independent approaches converge to the cloning of a new Leptosphaeria maculans avirulence effector gene, AvrLmS-Lep2. bioRxiv [Preprint]. doi: 10.1101/2020.10.02.322479
Obermeier, C., Hossain, M. A., Snowdon, R., Knüfer, J., von Tiedemann, A., and Friedt, W. (2013). Genetic analysis of phenylpropanoid metabolites associated with resistance against Verticillium longisporum in Brassica napus. Mol. Breed. 31, 347–361. doi: 10.1007/s11032-012-9794-8
Parkin, I. A., Sharpe, A. G., Keith, D. J., and Lydiate, D. J. (1995). Identification of the A and C genomes of amphidiploid Brassica napus (oilseed rape). Genome 38, 1122–1131. doi: 10.1139/g95-149
Pilet, M. L., Delourme, R., Foisset, N., and Renard, M. (1998). Identification of QTL involved in field resistance to light leaf spot (Pyrenopeziza brassicae) and blackleg resistance (Leptosphaeria maculans) in winter rapeseed (Brassica napus L.). Theor. Appl. Genet. 97, 398–406. doi: 10.1007/s001220050909
Pilet-Nayel, M.-L., Moury, B., Caffier, V., Montarry, J., Kerlan, M.-C., Fournet, S., et al. (2017). Quantitative resistance to plant pathogens in pyramiding strategies for durable crop protection. Front. Plant Sci. 8:1838. doi: 10.3389/fpls.2017.01838
Raman, H., McVittie, B., Pirathiban, R., Raman, R., Zhang, Y., Barbulescu, D. M., et al. (2020). Genome-Wide Association mapping identifies novel Loci for quantitative resistance to blackleg disease in canola. Front. Plant Sci. 11:1184. doi: 10.3389/fpls.2020.01184
Raman, H., Raman, R., Coombes, N., Song, J., Diffey, S., Kilian, A., et al. (2016). Genome-wide Association study identifies new loci for resistance to Leptosphaeria maculans in canola. Front. Plant Sci. 7:1513. doi: 10.3389/fpls.2016.01513
Raman, H., Raman, R., Diffey, S., Qiu, Y., McVittie, B., Barbulescu, D. M., et al. (2018). Stable quantitative resistance loci to blackleg disease in canola (Brassica napus L.) over continents. Front. Plant Sci. 9:1622. doi: 10.3389/fpls.2018.01622
Raman, R., Diffey, S., Barbulescu, D. M., Coombes, N., Luckett, D., Salisbury, P., et al. (2020). Genetic and physical mapping of loci for resistance to blackleg disease in canola (Brassica napus L.). Sci. Rep. 10:4416. doi: 10.1038/s41598-020-61211-y
Raman, R., Taylor, B., Marcroft, S., Stiller, J., Eckermann, P., Coombes, N., et al. (2012). Molecular mapping of qualitative and quantitative loci for resistance to Leptosphaeria maculans causing blackleg disease in canola (Brassica napus L.). Theor. Appl. Genet. 125, 405–418. doi: 10.1007/s00122-012-1842-6
Rimmer, S. R., and van den Berg, C. G. J. (1992). Resistance of oilseed Brassica spp. to blackleg caused by Leptosphaeria maculans. Can. J. Plant Pathol. 14, 56–66. doi: 10.1080/07060669209500906
Rousseau-Gueutin, M., Belser, C., Da Silva, C., Richard, G., Istace, B., Cruaud, C., et al. (2020). Long-read assembly of the Brassica napus reference genome Darmor-bzh. Gigascience 9, 1–16. doi: 10.1093/gigascience/giaa137
Rouxel, T., Penaud, A., Pinochet, X., Brun, H., Gout, L., Delourme, R., et al. (2003). A 10-year survey of populations of Leptosphaeria maculans in France indicates a rapid adaptation towards the Rlm1 resistance gene of oilseed rape. Eur. J. Plant Pathol. 109, 871–881. doi: 10.1023/A:1026189225466
Samans, B., Chalhoub, B., and Snowdon, R. J. (2017). Surviving a genome collision: genomic signatures of allopolyploidization in the recent crop species Brassica napus. Plant Genome 10, 1–15. doi: 10.3835/plantgenome2017.02.0013
Sedlazeck, F. J., Rescheneder, P., Smolka, M., Fang, H., Nattestad, M., Haeseler, A. V., et al. (2018). Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468. doi: 10.1038/s41592-018-0001-7
Sprague, S. J., Balesdent, M.-H., Brun, H., Hayden, H. L., Marcroft, S. J., Pinochet, X., et al. (2006). Major gene resistance in Brassica napus (oilseed rape) is overcome by changes in virulence of populations of Leptosphaeria maculans in France and Australia. Eur. J. Plant Pathol. 114, 33–40. doi: 10.1007/s10658-005-3683-5
St Clair, D. A. (2010). Quantitative disease resistance and quantitative resistance loci in breeding. Annu. Rev. Phytopathol. 48, 247–268. doi: 10.1146/annurev-phyto-080508-081904
Stein, A., Coriton, O., Rousseau-Gueutin, M., Samans, B., Schiessl, S. V., Obermeier, C., et al. (2017). Mapping of homoeologous chromosome exchanges influencing quantitative trait variation in Brassica napus. Plant Biotechnol. J. 15, 1478–1489. doi: 10.1111/pbi.12732
Szadkowski, E., Eber, F., Huteau, V., Lodé, M., Huneau, C., Belcram, H., et al. (2010). The first meiosis of resynthesized Brassica napus, a genome blender. New Phytol. 186, 102–112. doi: 10.1111/j.1469-8137.2010.03182.x
Thomma, B., Nürnberger, T., and Joosten, M. H. A. J. (2011). Of PAMPs and effectors: the blurred PTI-ETI dichotomy. Plant Cell 23, 4–15. doi: 10.1105/tpc.110.082602
Nagaharu, U. (1935). Genome analysis in Brassica with special reference to the experimental formation of Brassica napus and peculiar mode of fertilization. Jpn. J. Bot. 7, 389–453.
Untergasser, A., Nijveen, H., Rao, X., Bisseling, T., Geurts, R., and Leunissen, J. A. M. (2007). Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 35, W71–W74. doi: 10.1093/nar/gkm306
van de Wouw, A. P., Howlett, B. J., and Idnurm, A. (2017). Changes in allele frequencies of avirulence genes in the blackleg fungus, Leptosphaeria maculans, over two decades in Australia. Crop Pasture Sci. 69, 20. doi: 10.1071/CP16411
Verica, J. A., and He, Z. (2002). The cell Wall-Associated Kinase (WAK) and WAK-like kinase gene family. Plant Physiol. 129, 455–459.
Wagner, T. A., and Kohorn, B. D. (2001). Wall-Associated Ainases are expressed throughout plant development and are required for cell expansion. Plant Cell 13, 303–318. doi: 10.1105/tpc.13.2.303
West, J. S., Kharbanda, P. D., Barbetti, M. J., and Fitt, B. D. L. (2001). Epidemiology and management of Leptosphaeria maculans (phoma stem canker) on oilseed rape in Australia, Canada and Europe. Plant Pathol. 50, 10–27. doi: 10.1046/j.1365-3059.2001.00546.x
Winter, M., and Koopmann, B. (2016). Race spectra of Leptosphaeria maculans, the causal agent of blackleg disease of oilseed rape, in different geographic regions in northern Germany. Eur. J. Plant Pathol. 145, 629–641. doi: 10.1007/s10658-016-0932-8
Keywords: ONT, structural variation, blackleg, Brassica napus, long-read sequencing, Rlm9
Citation: Vollrath P, Chawla HS, Alnajar D, Gabur I, Lee H, Weber S, Ehrig L, Koopmann B, Snowdon RJ and Obermeier C (2021) Dissection of Quantitative Blackleg Resistance Reveals Novel Variants of Resistance Gene Rlm9 in Elite Brassica napus. Front. Plant Sci. 12:749491. doi: 10.3389/fpls.2021.749491
Received: 29 July 2021; Accepted: 29 October 2021;
Published: 18 November 2021.
Edited by:
Jacqueline Batley, University of Western Australia, AustraliaReviewed by:
Angela Van De Wouw, The University of Melbourne, AustraliaFrancis Chuks Ogbonnaya, Grains Research and Development Corporation, Australia
Copyright © 2021 Vollrath, Chawla, Alnajar, Gabur, Lee, Weber, Ehrig, Koopmann, Snowdon and Obermeier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Obermeier, Y2hyaXN0aWFuLm9iZXJtZWllckBhZ3Jhci51bmktZ2llc3Nlbi5kZQ==