Abstract
Hemerocallis citrina is a perennial herbaceous plant that is dedicated to mothers in Chinese culture and is widely distributed across the country. As a popular species with a long history of cultivation and utilization, it is renowned for its remarkable edible and medicinal value. In this study, we integrated Illumina short-read and Oxford Nanopore long-read sequencing to generate a complete mitochondrial genome (mitogenome) assembly of H. citrina. The H. citrina mitogenome has a multiple chromosomal structure consisting of three circular molecules that are 45,607 bp, 239,991 bp, and 182,864 bp long. We correspondingly annotated 66 genes, comprising 45 protein-coding genes (PCGs), 17 tRNA genes, and 4 rRNA genes. Comparative analysis of gene organization indicated that six syntenic gene clusters were conserved in the mitogenomes of the compared plants. The investigation of repeat content revealed repeat-rich nature of the H. citrina mitogenome, for which plentiful dispersed repeats were characterized to correlate with the size of the mitogenome. The codon usage behavior disclosed that Leucine (Leu) and Serine (Ser) were the most preferred amino acids in H. citrina, and nearly all of the codons with relative synonymous codon usage (RSCU) values greater than 1 showed the preference of A or T ending. Moreover, we inferred a total of 679 RNA editing sites in all mitochondrial PCGs, which presented perfect C-to-U types and tended to lead to the alteration of internal codons. Subsequent selective pressure analysis showed that the majority of the PCGs had undergone evolutionary negative selections, with atp9 in particular undergoing strong stabilizing selection, reflecting its indispensable function in mitogenomes. According to the phylogenetic analysis, H. citrina is close to the species Allium cepa (Amaryllidaceae) and Asparagus officinalis (Asparagaceae) in evolutionary terms. Overall, this project presents the first complete mitogenome of H. citrina, which could provide a reference genome for the comprehensive exploration of the Asphodelaceae family and can facilitate further genomic breeding and evolutionary research on this medicine–food homologous plant.
Introduction
Mitochondria (mt) are critical organelles that are ubiquitous in eukaryotic cells and drive the energy metabolism for biological growth, development, and reproductive processes (Liberatore et al., 2016). They retain their own genomes that are inherited independently from nuclear genomes through ancient endosymbiotic events (Timmis et al., 2004). With the ongoing advancements in genome sequencing and assembly technologies, there has been rapid progress in deciphering plant mt genomes (mitogenomes). In contrast to conserved plastid genomes, plant mitogenomes exhibit more complex genome structures, broad genome size variations, numerous RNA editing modifications, abundant repeated sequences, sparse gene distribution, and continual gene relocation during evolution (Handa, 2003; Kubo and Mikami, 2007; Alverson et al., 2010; Sloan et al., 2012; Zhang et al., 2019; Kan et al., 2020). These unique features complicate the sequencing and annotation of plant mitogenomes. Although more than 350 complete mitogenomes of land plants are available in the NCBI (National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/genome/browse#!/organelles/., 18/07/2022), this number is miniscule compared to the complete genome data for thousands of plant chloroplasts (cp) and animal mt.
The evolution of plant mitogenomes has involved complex genomic rearrangements, contributing to tremendous variations in the size and structure of mitogenomes across different species (Wu et al., 2020). Current known sizes range from the standard dozens and hundreds of kilobases to a few notable exceptions that are larger than one megabase (Sloan et al., 2012; Skippington et al., 2015; Jackman et al., 2020; Li et al., 2021a). As already researched and documented, mitogenome size can fluctuate greatly, even among species that are closely related in evolutionary terms (Alverson et al., 2010). The divergence of mitogenome size is principally driven by the accumulation of noncoding regions, consisting of dispersed repeats, introns, intergenic segments, and the integration of foreign DNA, rather than by differences in gene content (Allen et al., 2007; Alverson et al., 2011; Goremykin et al., 2012; Rice et al., 2013). Thus, plant mitogenomes present comparative conservation in functional genes, and in particular, genes associated with mt respiration and energy synthesis are deemed to be highly conserved in most plant mitogenomes. However, there is also evidence that Viscum scurruloideum has lost all nad genes contributing to respiratory Complex I (NADH dehydrogenase) (Skippington et al., 2015), resulting in diverse and complex plant mitogenomes in terms of the gene content. In addition to their size and gene category, plant mitogenomes have also been found to possess multiple structures. The majority of plant mitogenomes established so far can be portrayed as single circular chromosomes, which were once assumed to be the dominant form of the mt structure. However, subsequent studies confirmed that the actual architecture of the mitogenome appears to occur in circular, linear, and complex branched arrangements (Backert and Börner, 2000; Smith and Keeling, 2015). These variable features render the plant mitogenome an attractive genetic system for the study of issues relevant to evolutionary biology, as well as laborious to assemble and annotate. Nevertheless, plant mitogenomes remain somewhat mysterious because of limited sampling, which hinders an overall understanding of their diversity and evolution.
Hemerocallis citrina Borani, a popular perennial herbaceous plant mainly distributed in China, belongs to the family Asphodelaceae. H. citrina, also known as “Daylily” or “Huang hua cai”, has been respected as the “mother flower” in traditional Chinese culture for thousands of years (Ma et al., 2018; Liu et al., 2020). The immature flower buds are commonly processed into a dried vegetable and consumed as a nutraceutical food in Asia. Due to its profusely high flavonoid and polyphenol contents (Xu et al., 2016), H. citrina has the concomitant function of both a medicine and a foodstuff. H. citrina has been proposed as a Chinese herbal medicine in antioxidant, anti-inflammatory, and antidepressant treatments for a long time. Research on H. citrina has mainly focused on morphological assessment (Li et al., 2021b), pharmaceutical properties (Wang et al., 2018), tissue culture (Matand et al., 2020), and circadian flowering rhythm (Ren et al., 2019). In recent years, sequencing information of the cp (Ou et al., 2020) and nuclear (Qing et al., 2021) genomes in H. citrina has been successively obtained and utilized. However, further knowledge of the mt genome is urgently needed for the evolutionary understanding and genetic improvement of H. citrina.
In this study, we sequenced and completed the first H. citrina mitogenome from Asphodelaceae utilizing the Illumina short-read and Nanopore long-read integrated pipeline. The assembled H. citrina genome was characterized for gene annotation, sequence variation, selection pressure, RNA editing events, and phylogenetic position by comparing its counterparts with published plant mitogenomes. This information on the H. citrina mitogenome will fill the mitogenomic evolution knowledge gap for the Hemerocallis genus and will further facilitate genetic and evolutionary studies of this extremely valuable plant.
Materials and methods
Plant materials and genome sequencing
For DNA extraction, fresh leaves of H. citrina (cultivar Datong hua) were collected from Datong, Shanxi Province, China. High-quality genomic DNA was isolated employing a modified CTAB protocol (Porebski et al., 1997). The purity, concentration, and integrity of the extracted DNA were examined using a NanoDrop (Thermo Scientific, USA), a Qubit fluorometer (Thermo Scientific, USA), and 0.35% agarose gel electrophoresis, respectively. Both short-read (Illumina) and long-read (Oxford Nanopore) sequencing technologies were implemented to obtain full-length mitogenome sequences. To collect large DNA sequences, optional fragmentation was performed using the BluePippin system (Sage Science, USA). Subsequently, the fragmented DNA was treated using damage repair, end preparation, A-tailing, and adapter ligation and then cleaned with magnetic beads. A library > 8 kb was constructed following the SQK-LSK109 (Oxford Nanopore Technologies, ONT, UK) sequencing kit protocol and loaded into a Nanopore GridION Sequencer (ONT, UK).
Genome assembly and annotation
This whole genome sequencing produced 12.6 Gb in 1.3 million long-reads (SRA accession SRR15370204). The raw long-reads were filtered and re-edited using NanoFilt and NanoPlot in Nanopack (De Coster et al., 2018), then a total of 11.6 Gb clean data were generated. Meanwhile, libraries with an average fragment length of 350 bp were prepared utilizing the NEBNext® Ultra™ DNA Library Prep Kit (New England Biolabs, England) and then sequenced on the Illumina Novaseq 6000 platform (Illumina, USA) with 150 bp paired-end reads. The Illumina short-read sequencing yielded 10.2 Gb in 34.1 million reads (SRA accession SRR15370205). The quality of the raw short-reads was assessed using the NGS QC Tool Kit v2.3.3 (Patel and Jain, 2012). A total of 10.1 Gb clean short-reads were obtained by removing adapter sequences and low-quality reads using Cutadapt (v1.9.1). First, we obtained a rough but computationally efficient assembly via Miniasm (Li, 2016) after trimming adapter sequences with Porechop v0.2.4 (Wick et al., 2017) and polishing the resulting assembly with Racon v1.4.7 (Vaser et al., 2017). Subsequently, potential mt contigs with homology to the Asparagus officinalis mitogenome (NCBI Reference Sequence: MT483944.1) were obtained using Bandage (Wick et al., 2015). We then proceeded to align the Nanopore reads to our draft H. citrina assembly using minimap2 (Li, 2018) and segregated aligned reads and reassembled them, first using Flye v2.6 (Kolmogorov et al., 2019), and then using Canu v2.1.1 (Koren et al., 2017). The final genome sequence was obtained by polishing with Pilon (Walker et al., 2014) using Illumina Novaseq sequencing reads. Based on the above assembly procedures, we obtained a three-ring structure of the H. citrina mitogenome.
The complete mitogenome of H. citrina was annotated using MITOFY (Alverson et al., 2010) and MFANNOT (Gautheret and Lambert, 2001), utilizing previous angiosperm mt genes to query sequences in the NCBI database (https://www.blast.ncbi.nlm.nih.gov). The tRNA genes were detected using tRNAscan-SE 2.0 (Schattner et al., 2005). The chloroplasts (cp) genome of H. citrina (MN872235.1) was used to determine the tRNA genes transferred from cp to mt. The origins of tRNA genes were identified using BLASTn on tRNAscan-SE 2.0 with the matching rate set to ≥ 70%, length ≥ 30, and E-value ≤ 1e-3. The relative synonymous codon usage (RSCU) values and amino acid composition of PCGs were calculated using CodonW (http://codonw.sourceforge.net/). The circular and syntenic gene cluster maps of the H. citrina mitogenome were visualized using the OGDRAW program (Greiner et al., 2019). The assembled complete mitogenome sequence of H. citrina has been submitted to GenBank (NCBI) and is openly available under accession numbers: MZ726801.1, MZ726802.1, and MZ726803.1.
Genome analysis
To identify mitochondrial synteny blocks of the H. citrina compared with representative species, 7 pairs of monocotyledonous mitogenomes were aligned using Mauve v2.4.0 with a LCB cutoff of 42 (Darling et al., 2010). The amount of shared mtDNA was determined using BLASTn with the parameters word_size 7 and e-value 1×10−6. Three types of repeat sequences were identified in the H. citrina mitogenome. Simple sequence repeats (SSRs) were searched using MISA-web (https://webblast.ipk-gatersleben.de/misa/) (Beier et al., 2017) with preset thresholds of eight, four, four, three, three, and three corresponding to the motif size of one to six nucleotides, respectively. The Finder 4.09 program (http://tandem.bu.edu/trf/trf.html) was utilized to confirm and locate tandem repeats using default parameters (Benson, 1999). Dispersed repeats were identified using the public tool REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer) (Kurtz et al., 2001) with the repeat size set to > 30 bp. To infer the selection pressure of PCGs during the evolution of H. citrina, the nonsynonymous (Ka) and synonymous (Ks) substitution rates of each PCG were calculated for H. citrina and five other species (Allium cepa, A. officinalis, Arabidopsis thaliana, Nelumbo nucifera, and Oryza sativa). ParaAT2.0 was used with default settings to align and format the PCGs (Zhang et al., 2012). The calculations of the Ka, Ks, and Ka/Ks values were performed using KaKs_Calculator v.2.0 following the YN method (Wang et al., 2010). Putative RNA editing sites in the PCGs of H. citrina and the five other species were projected using PREP-Mt (http://prep.unl.edu/). To detect more true edits, the cutoff parameter was set to 0.2 (Mower, 2009).
Phylogenetic tree construction
Nine conserved mt PCGs (atp1, ccmB, ccmC, cob, nad3, nad4, nad4L, nad7, and nad9) were lined up to construct a contiguous sequence and were then individually aligned using MAFFT 7.017 (Katoh and Standley, 2013) among the 29 species analyzed. The NCBI accession numbers of all these observed mitogenomes are listed in Supplementary Table 1. After ModelFinder was used to select the most appropriate amino acid substitution pattern, the maximum likelihood (ML) phylogenetic tree was established based on a GTR+F+R3 model using IQTREE v1.6 (Trifinopoulos et al., 2016) with 1000 bootstraps and Ginkgo biloba as an outgroup.
Results
Mitogenome assembly and genomic features
As visualized in Figure 1A, we obtained six segments to construct the completed draft mt graph for assembling the H. citrina mitogenome. All segment adjacencies are supported by the long reads, suggesting that several combinations exist in the mitogenome of H. citrina. Since the three loops are the smallest indivisible units, the H. citrina mitogenome was assembled into three complete circular molecules that were 45,607 bp (molecule 1), 239,991 bp (molecule 2), and 182,864 bp (molecule 3) long (Figures 1B-D, Supplementary Table 2). The three molecules of the mitogenomes had an overall GC content of 44.4%, 45.1%, and 45.7%, respectively (Table 1). Notably, the lengths of the coding regions, without exception, occupied only a small part of the three mitogenomes.
Figure 1
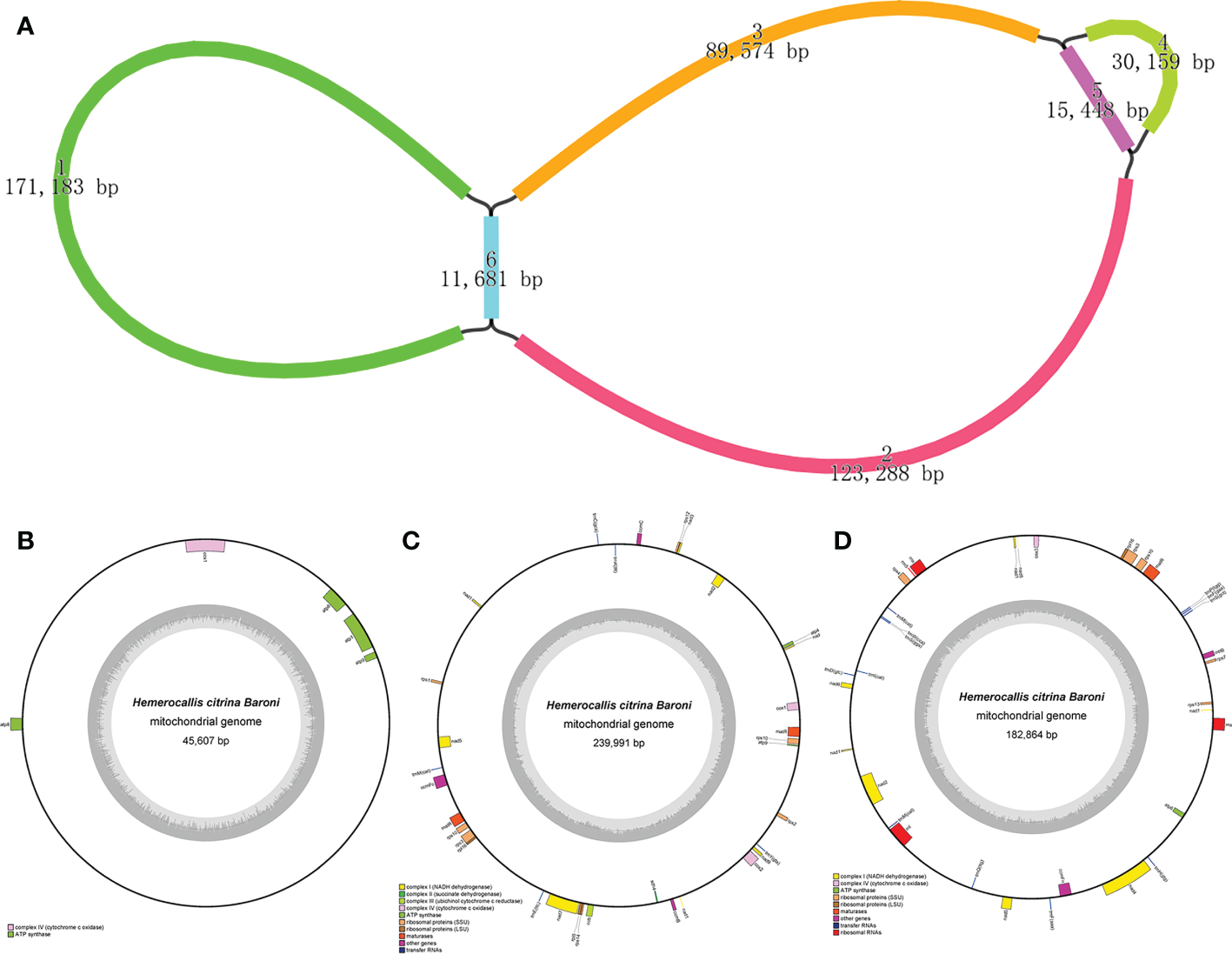
Assembly results of the H. citrina mitogenome. (A) The assembly graph of the H. citrina mitogenome. Each colored segment is labeled with its size and named 1-6 by rank of size. (B-D) Circular map of the H. citrina mitogenome. Genes presented on the outer circle are transcribed, whereas genes on the inside are counterclockwise. The inner circle reveals the GC content as a dark gray plot with the gray line in the middle as the 50% threshold line. (B) Schematic diagram of molecule 1 with a length of 45,607 bp and five annotated genes; (C, D) Similar maps for molecule 2 and molecule 3 with lengths of 239,991 bp and 182,864 bp, respectively. Because the three genes (nad1, nad2, and nad5) encoding the subunits of Complex I (NADH dehydrogenase) are fragmented in molecules 2 and 3, the two mt circular molecules encode a total of 61 genes.
Table 1
| Genome | Feature | A % | C % | G % | T % | GC % | Size (bp) | Proportion in Genome (%) |
|---|---|---|---|---|---|---|---|---|
| Molecule 1 | Whole genome | 27.9 | 22.4 | 22.0 | 27.7 | 44.4 | 45,607 | 100 |
| Protein-coding genesa | 26.5 | 21.3 | 21.9 | 30.3 | 43.2 | 4,683 | 10.3 | |
| cis-spliced intronsa | – | – | – | – | – | – | – | |
| tRNA genesa | – | – | – | – | – | – | – | |
| rRNA genesa | – | – | – | – | – | – | – | |
| Non-coding regions | 27.7 | 22.4 | 22.1 | 27.8 | 44.5 | 40,924 | 89.7 | |
| Molecule 2 | Whole genome | 27.4 | 22.5 | 22.5 | 27.6 | 45.1 | 239,991 | 100 |
| Protein-coding genesa | 26.7 | 21.9 | 22.3 | 29.1 | 44.2 | 21,604 | 9.0 | |
| cis-spliced intronsa | 23.4 | 26.1 | 27.6 | 22.9 | 53.7 | 11,094 | 4.6 | |
| tRNA genesa | 23.7 | 22.0 | 26.6 | 27.7 | 48.6 | 372 | 0.2 | |
| rRNA genesa | – | – | – | – | – | – | – | |
| Non-coding regions | 27.5 | 22.4 | 22.3 | 27.8 | 44.7 | 206,949 | 86.2 | |
| Molecule 3 | Whole genome | 27.4 | 22.7 | 23.0 | 26.9 | 45.7 | 182,864 | 100 |
| Protein-coding genesa | 26.3 | 22.3 | 21.8 | 27.5 | 44.2 | 14,495 | 7.9 | |
| cis-spliced intronsa | 25.3 | 24.1 | 26.9 | 23.7 | 51.0 | 12,046 | 6.6 | |
| tRNA genesa | 24.1 | 21.5 | 27.0 | 27.4 | 48.5 | 910 | 0.5 | |
| rRNA genesa | 25.4 | 23.2 | 30.0 | 21.3 | 53.3 | 7300 | 4.0 | |
| Non-coding regions | 27.6 | 22.5 | 22.5 | 27.4 | 45.0 | 148,142 | 81.0 |
Genomic features of the H. citrina mitogenome.
a Protein-coding genes, cis-spliced introns, tRNAs, and rRNAs belong to coding regions.
-, absent.
To investigate the genome rearrangements in H. citrina, the quantity of shared DNA was measured between the H. citrina and the other 7 monocotyledons. As shown in Figure 2, the mitogenome sequence of H. citrina exhibited a low level of synteny and shared DNA compared with the other monocotyledons. For instance, only 30% (141 kb) of the 468 kb mitogenome of H. citrina is homologous to the A. officinalis mitogenome. Lower amounts of DNA are shared with the 26% (124 kb) of the shared DNA between H. citrina and Phoenix dactylifera, 26% (120 kb) shared between H. citrina and O. sativa, and 24% (114 kb) shared between H. citrina and Cocos nucifera. In stark contrast, the H. citrina and A. cepa mitogenomes retain merely 70 kb of shared DNA.
Figure 2
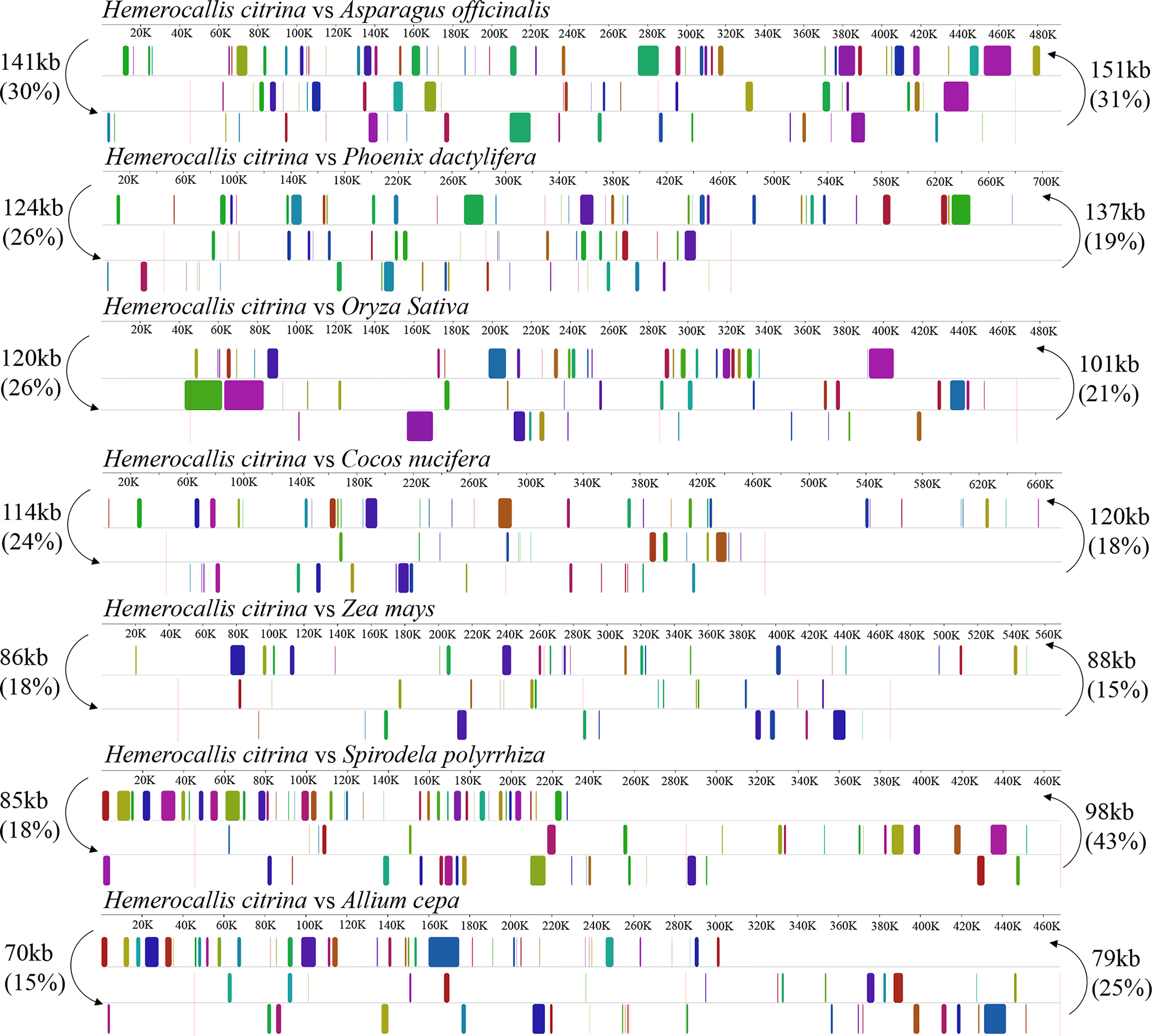
Synteny blocks and shared mtDNA of the H. citrina and seven other monocotyledons based on Mauve alignments. Left arrows indicate the amount of shared mtDNA, in kb and percentage, of the H. citrina and one other species, and right arrows present the reciprocal values.
Gene annotation and organization
The H. citrina mitogenome contains 66 annotated genes, representing 53 unique genes (Table 2). Forty-five protein-coding genes (PCGs) responsible for electron transport, oxidative phosphorylation, cytochrome c biogenesis, ATP synthesis, and ribosomal protein were identified in the mitogenome. PCGs comprised most of the coding regions, whereas their GC content was the smallest compared with other regions (Table 1). The genome includes 17 tRNA genes, of which 13 and 4 genes were of mt and cp origin, respectively. In addition, four rRNA genes were also found in H. citrina. Introns emerged in seven of the annotated genes, namely, nad1, nad2, nad5, nad7, cox2, ccmFc, and rps10. Additionally, three genes (nad1, nad2, and nad5) encoding subunits of Complex I were fragmented in molecules 2 and 3.
Table 2
| Group of Genes | Gene Name | |||
|---|---|---|---|---|
| Molecule 1 | Molecule 2 | Molecule 3 | Numbers | |
| Complex I (NADH dehydrogenase) | nad1#, nad2#, nad3, nad4L, nad5#, nad7#, nad9 | nad1#, nad2#, nad4, nad5#, nad6 | 9 | |
| Complex II (succinate dehydrogenase) | sdh4 | 1 | ||
| Complex III (ubiquinol cytochrome c reductase) | cob | 1 | ||
| Complex IV (cytochrome c oxidase) | cox1 | cox1, cox2# | cox3 | 4 |
| Complex V (ATP synthase) | atp1, atp8(2)*, atp9 | atp4, atp9 | atp6 | 7 |
| Cytochrome c biogenesis | ccmB, ccmC, ccmFc# | ccmFn | 4 | |
| Ribosomal protein | rpl5, rpl16, rps1, rps2, rps3, rps10#(2)*, rps12, rps14 | rpl16, rps3, rps4, rps7, rps10#, rps13 | 15 | |
| Ribosomal RNA | rrn5, rns(2)*, rnl | 4 | ||
| Transfer RNA | trnC, trnE, trnK, trnM-cp, trnY | trnD, trnF(2)*, trnH-cp, trnI, trnM, trnM-cp, trnP, trnQ, trnS(2)*, trnW-cp | 17 | |
| Others | matR(2)* | matR, mttB | 4 | |
| Total | 5 | 61 | 66 | |
Gene annotation of the H. citrina mitogenome.
Multicopy genes are presented with *, and bracketed numbers represent copy number of each gene. Genes with introns are denoted with #.
The mitogenome gene content and order vary during the evolution of higher plants. Frequent occurrence of PCG loss that is extensively found in the course of evolution creates diversity in plant mitogenomes. To investigate gene loss in the H. citrina mitogenome, we compared its gene content with that of the other 29 species. As exhibited in Figure 3, the genes in the categories of Complex I, Complex III, Complex V, cytochrome c biogenesis, and maturases are conserved among these mitogenomes. The conserved genes relevant to mt respiration and energy synthesis, excluding the sdh3 and sdh4 encode subunits of mt Complex II, are essential to mt function. Consequently, variation of ribosomal protein genes appears to be the dominant factor in the high diversity of the gene content among plant mitogenomes.
Figure 3
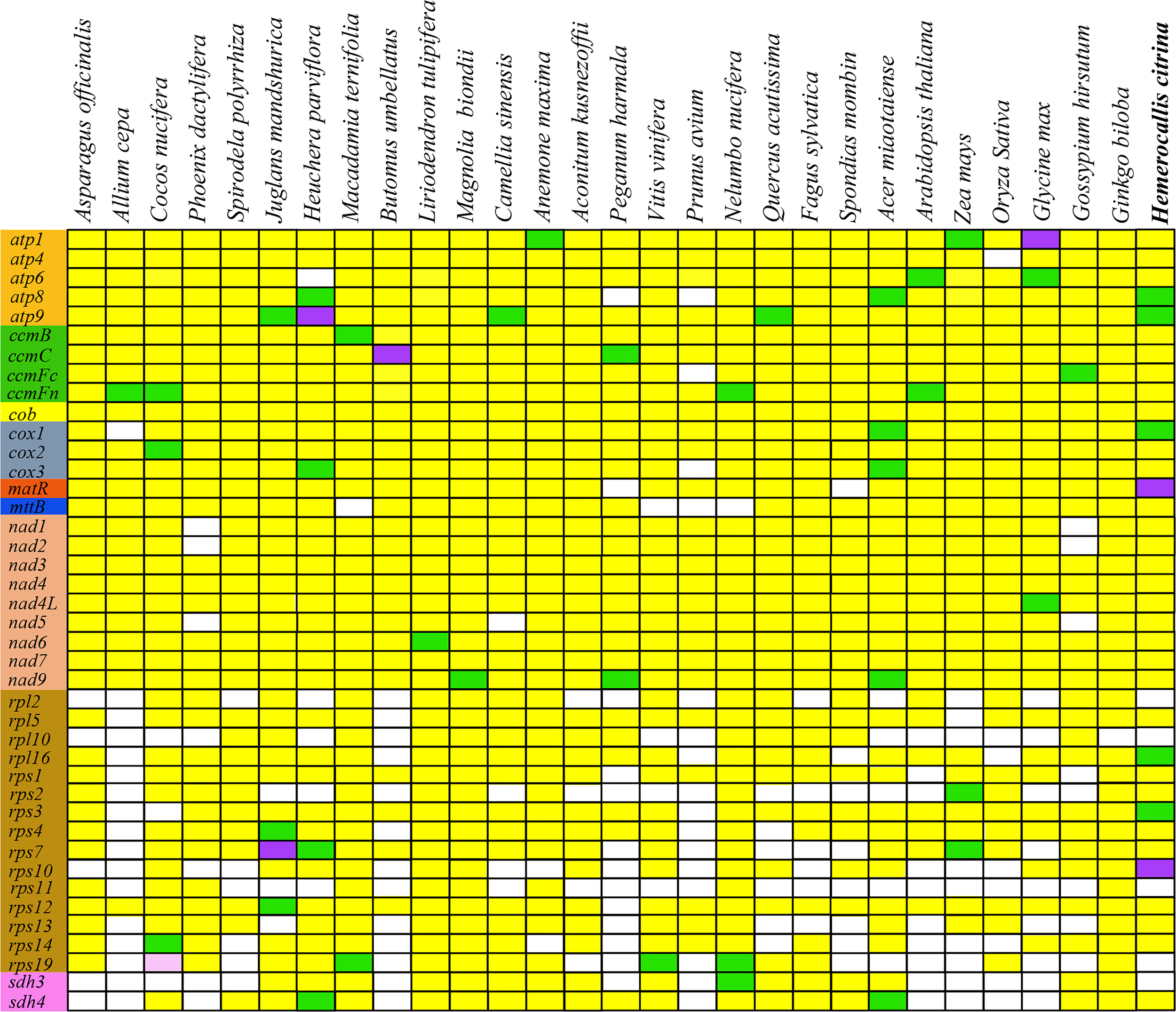
Distribution of PCGs in plant mitogenomes. White boxes indicate that the gene is absent in the mitogenome. The remaining colors of boxes indicate that one (yellow), two (green), three (purple), or five (pink) copies exist in the genomes.
In order to investigate the gene organization in H. citrina, the selected 28 species were used for mitogenome comparison and calculation of syntenic gene clusters. A total of six conserved gene clusters (i.e., rps3-rpl16, atp4-nad4L, rrn18-rrn5, rps12-nad3, cob-rpl5, and atp1-atp9) were identified in the H. citrina mitogenomes (Supplementary Figure 1, Supplementary Table 3). Furthermore, the cluster cox3-sdh4 was primarily present in dicotyledons, but rarely occurred in monocotyledons. Interestingly, the conserved cluster nad1-matR existed extensively in most species except H. citrina. It is remarkable, however, that H. citrina was one of the few species containing the atp1-atp9 gene cluster, similar to the other three monocotyledons (i.e., A. officinalis, P. dactylifera, and Butomus umbellatus). Among the dicotyledons, only Aconitum kusnezoffii had the cluster atp1-atp9.
Identification of repeat sequences
Repeat sequences are characterized as a high degree of polymorphism and extensive presence in plant mitogenomes, which are important factors for genomic structural variances. The specific forms of repeat sequences are characterized primarily as simple sequence repeats (SSRs), tandem repeats, and dispersed repeats. For the investigation of H. citrina, we identified 530 repeat sequences with 379, 24, and 127 distributed among SSRs, tandem repeats, and dispersed repeats, respectively. The SSRs in the H. citrina mitogenome were classified as 40.6% monomers, 37.2% dimers, 6.9% trimers, 14.0% tetramers, 1.0% pentamers, and 0.3% hexamers (Supplementary Table 4). The mononucleotide repeats of A/T were determined to be the most extensive repeat type with 133 repeats identified, followed by the dinucleotide repeats of AG/CT with 83 repeats. Polynucleotide repeats accounted for a small proportion and were only found in the intronic or intergenic regions, apart from a single tetranucleotide repeat of AATG/ATTC identified in the coding region (rps3). As illustrated in Supplementary Table 5, the 24 tandem repeats observed in the H. citrina mitogenome varied between 25 and 86 bp in length, and most of them contained two copies of the sequence. These tandem repeats, without exception, were localized in the intergenic spacers.
In addition to the above two types of repeat, dispersed repeats (repeat unit >30) were analyzed further, resulting in 35,337 bp occupying 7.5% of the genome. The 127 dispersed repeats that were identified consisted of 59 forward repeats and 68 palindromic repeats, and a majority of them were 30~49 bp in length (Supplementary Table 6). There were nine such repeats greater than 100 bp, of which only three were greater than 1 kb (Supplementary Table 7). Simultaneously, we compared H. citrina with the other five monocotyledons to further explore the position of repeats in the mitogenome. The results suggested that the number of repeats counted in the six comparison species ranged from 38 in Spirodela polyrrhiza to 232 in Zea mays (Supplementary Figure 2, Supplementary Table 8). Interestingly, the total length of the repeats generally correlates with their mitogenome size. C. nucifera has the largest mitogenome (678,653 bp) among these species, and its total repeats cover 81,999 bp, occupying 12.1% of the total size of the mitogenome. In contrast, the mitogenome of S. polyrrhiza is only one-third the size of that of C. nucifera, and its repeats are the minimum among the comparative species, at 3,129 bp long and making up 1.4% of the genome size. Analyses of large repeats (>1 kb) implied that species with large mitogenomes were more abundant in large repeats. Eleven large repeats covering 68,972 bp were detected in C. nucifera, followed by the four large repeats of Z. mays (48,168 bp). However, no large repeats were found in the S. polyrrhiza mitogenome. The moderately sized H. citrina genome has repeats with a longer length than those found in the similar genome size of A. officinalis, mainly resulting from the difference in the number of large repeats.
Determination of the codon usage pattern
In H. citrina, the majority of PCGs use NTG as their start codon, in which 28 unique genes have the standard ATG start codon, whereas the other two genes harbor the initiation codons GTG for rpl16 and CTG for sdh4. Other than that, the remaining three genes (nad1, nad4L, and rps10) initiate with ACG (Supplementary Table 9). The complete PCGs in this mitogenome consist of three termination codon types, of which 17, 12, and 7 unique genes utilize the termination codons TAA, TGA, and TAG, respectively. The codon usage behavior revealed that Leucine (Leu) and Serine (Ser) were the most frequently utilized amino acids in H. citrina, whereas, as in other plant mitogenomes, Tryptophane (Trp) and Cysteine (Cys) were the least popular residues (Supplementary Figure 3). Furthermore, the vast majority of amino acid residues were highly conserved in dicotyledonous (N. nucifera and Vitis vinifera) and monocotyledonous (H. citrina, A. officinalis, A. cepa, C. nucifera, S. polyrrhiza, and Z. mays) plants based on interspecies alignment. Interestingly, codon usage showed a significant bias towards five amino acid residues (i.e., Phenylalanine (Phe), Leu, Proline (Pro), Ser, and Trp) in angiosperms and gymnosperms (Cycas taitungensis and G. biloba). The investigation of relative synonymous codon usage (RSCU) indicated that all possible codons were exhibited in the H. citrina PCGs (Supplementary Figure 4). Excluding the stop codons, the H. citrina mitogenome contains a total of 13,549 codons. The most preferentially used codons in this genome are A-ended or U-ended codons, which have RSCU values greater than 1, with the exception of Ser (AGU). This AT bias might influence codon usage in proteins, a phenomenon found in previously researched mitogenomes.
Selection pressure analysis
The ratio of nonsynonymous substitutions (Ka) to synonymous substitutions (Ks) is a suitable test of the neutral evolutionary model to infer the magnitude and direction of natural selection working on homologous PCGs among diverse species. In our research, the Ka/Ks values were inspected for all 36 unique PCGs after contrasting the H. citrina mitogenome with those of A. cepa, A. officinalis, A. thaliana, N. nucifera, and O. sativa. As shown in Figure 4, most of the Ka/Ks ratios were less than 1, implying that the majority of PCGs had undergone purifying selection during the evolutionary process. Nearly all of the Complex I-V genes showed stabilizing selection, suggesting that these genes exhibited high conservation in H. citrina and the selected species during evolution. Three genes (atp4, nad3, and sdh4) encoding Complex subunit proteins were subject to positive selection with Ka/Ks values >1. Additionally, high Ka/Ks ratios were determined in some other mt genes, involving ccmB, ccmFn, matR, mttB, rps1, rps7, and rps12. As many as four positive selection genes were observed in the comparison of H. citrina with A. officinalis and N. nucifera, suggesting that those mt genes may have suffered from diverse selection pressures after diverging from their last common ancestor. Interestingly, none of the PCGs had Ka/Ks values greater than 1, which only existed between H. citrina and A. cepa.
Figure 4
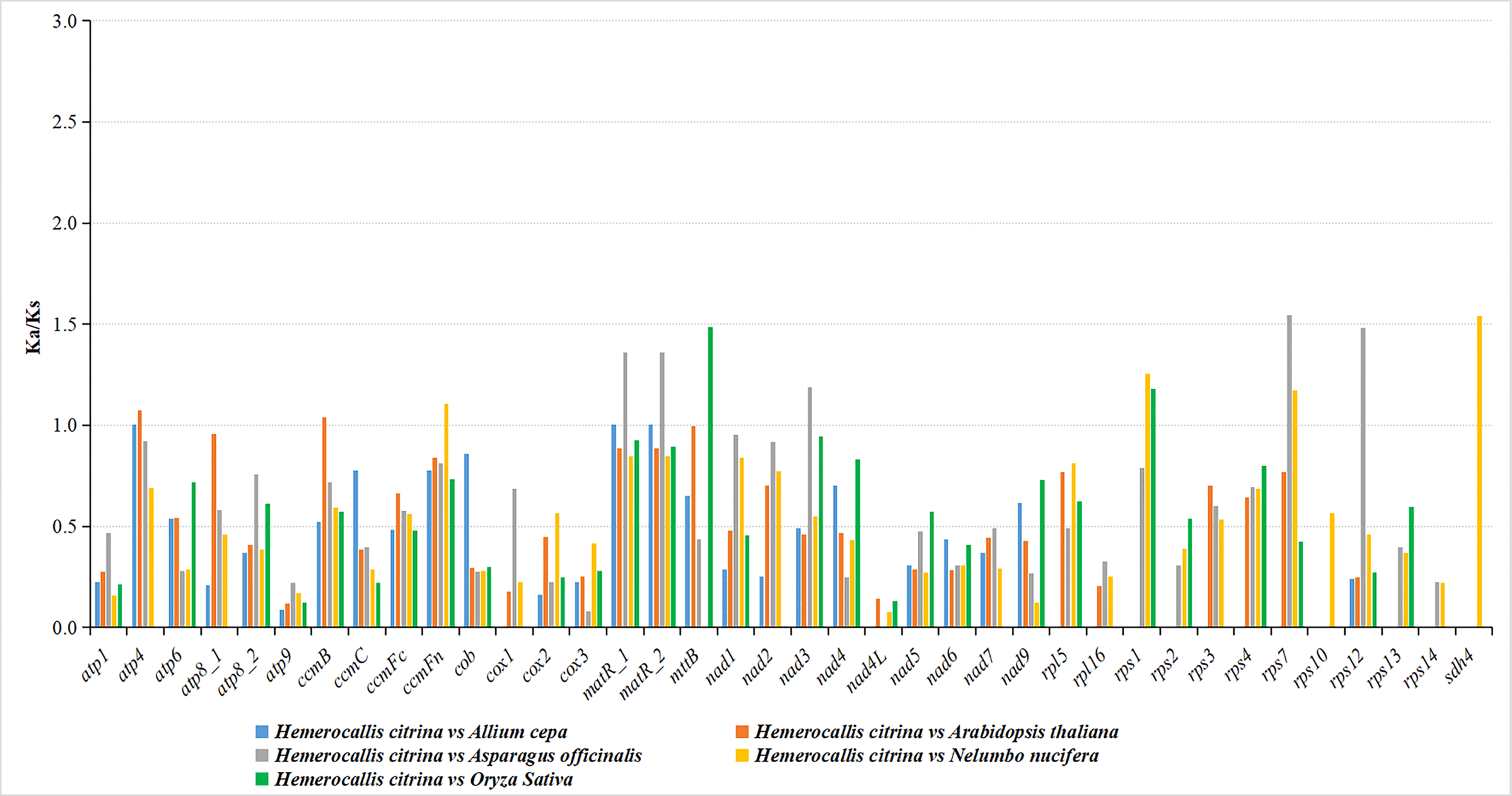
Comparison of the Ka/Ks ratios for PCGs in H. citrina and four other species.
Prediction of RNA editing sites
For the identification of H. citrina, all 45 PCGs were subject to RNA editing, and a total of 679 sites were predicted. Among the predicted editing events, the conversions carried out were uniformly post-transcriptional cytidine (C) to uridine (U) editing. This identified 679 sites where the RNA editing bases were substituted at the first 2-base positions of the codon. The frequency of the editing events in PCGs varied greatly, ranging from two editing sites in rps7 to 56 editing sites in matR. As shown in Figure 5, RNA editing was also compared across six species and ranged from 491 sites in O. sativa to 1,405 sites in G. biloba. The RNA editing site was relatively conserved among angiospermous mitogenomes, and that of H. citrina showed high similarity with that of A. officinalis, indicating that they share extremely conserved PCGs. The occurrence of RNA editing is obviously influenced by gene type, and the cytochrome c biogenesis and Complex I genes are more prone to RNA editing. Furthermore, there were ten cases involving the genes ccmB, ccmC, ccmFc, ccmFn, cox2, cox3, mttB, nad4L, nad9, and rps12, in which the number of edits was highly conserved in the provided angiosperms. Notably, the number of predicted sites of almost all genes in the gymnosperm (G. biloba) was markedly higher than that in angiosperms. However, matR was one of the exceptions, and the highest frequency of RNA editing within this gene renders H. citrina stand out in comparison to the other species.
Figure 5
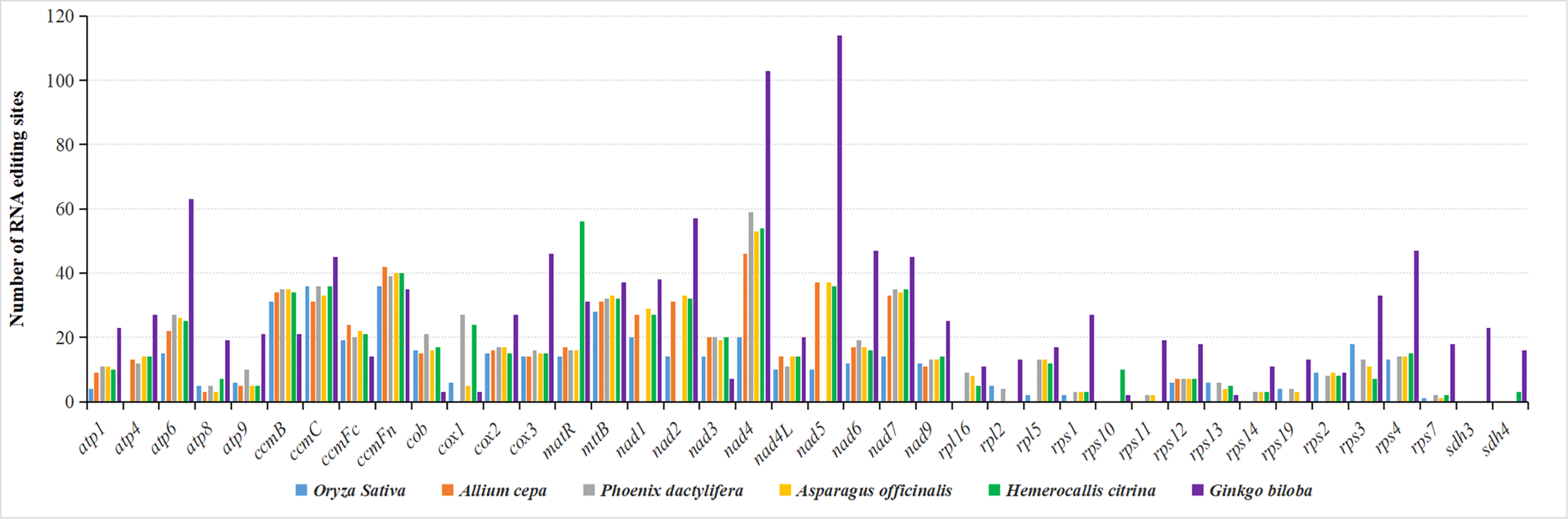
Prediction of RNA editing sites in the H. citrina mitogenome and five other species.
Phylogenetic analysis
A phylogenetic analysis of 29 complete mitogenomes was proceeded to explore the phylogenetic position of H. citrina. The results suggested that the majority of nodes had high bootstrap values for the maximum likelihood (ML) tree. Importantly, H. citrina is evolutionarily close to the species A. cepa (Amaryllidaceae) and A. officinalis (Asparagaceae) based on 100% bootstrap values (Figure 6). Our phylogenetic analysis also strongly indicated the divergence of dicotyledons and monocotyledons, as well as the divergence of angiosperms and gymnosperms.
Figure 6

Maximum likelihood phylogenetic tree for H. citrina according to 29 complete mitogenomic sequences, with Ginkgo biloba as an outgroup. The bootstrap index value in which the associated taxa clustered together is indicated next to the branches, and the NCBI accession numbers are listed beside the abbreviations of the species.
Discussion
Since the initial endosymbiosis event, mitogenomes have undergone significant and divergent changes in genome structure (Smith and Keeling, 2015). With the worldwide sequencing and assembly of mitogenomes in land plants, the true physical structure of the mitogenome is gradually emerging in multifarious forms, such as single circular molecule, multiple circular, linear, branched or composite structures (Unseld et al., 1997; Notsu et al., 2002; Alverson et al., 2011; Sloan et al., 2012; Jackman et al., 2020; Li et al., 2021a). To explore these challenging mitogenome structures in our study, we adopted a mixed assembly strategy, integrating Illumina short-read and Oxford Nanopore long-read sequencing to complete the mitogenome of H. citrina. The genome assembly presents a multiple chromosomal structure, composed of three distinct circular molecules with lengths of 45,607 bp, 239,991 bp, and 182,864 bp (Figures 1B-D). The newly sequenced H. citrina mitogenome is close to the average genome size (394.9 kb) and GC content (43.95%) of published land plants (Wu et al., 2020). Similarly, the genomes of Sitka spruce (Jackman et al., 2020), Leucaena trichandra (Kovar et al., 2018), and Lactuca sativa (Kozik et al., 2019) were successfully sequenced and precisely assembled using the combined approach. The resulting research further confirmed that second- and third-generation sequencing combined assemblies promise to obtain better and more reliable de novo assembly results for plant mitogenomes.
Plant mitogenomes are fascinating attributing to their highly conserved gene content and sequences, which are noted as having the slowest evolutionary rate of the three sets of plant genomes (nuclear, cp, and mt genomes). Despite differences in the number of mt genes across different plants, the genes required to maintain the major mt functions of respiration and energy synthesis are highly conserved. In general, the Complex genes responsible for mt respiration and metabolism are quite stable, with the exception of the succinate dehydrogenase (sdh) genes that encode subunits of mt Complex II. In comparing the gene content of different species, we found that sdh3 was lost in monocotyledons during evolution (Figure 3). In addition, sdh4 was also scarcer in the mitogenomes of most monocot plants. Our findings revealed that only sdh4 was present in the H. citrina mitogenome, which is consistent with results previously reported for two other monocot plants, C. nucifera (Aljohi et al., 2016) and S. polyrrhiza (GenBank accession: JQ804980.1). In contrast, the absence of sdh genes was observed only in a few dicotyledons. Consequently, the frequent loss of sdh3 and sdh4 tends to occur in monocotyledons. Furthermore, the plant mitogenome also harbors many ribosomal protein genes, which present substantial variation in sequence conservation and gene content across species (Liao et al., 2018). In our research of H. citrina, we annotated up to 11 unique ribosomal protein genes excluding the four genes rpl2, rpl10, rps11, and rps19. Surprisingly, in the mitogenome of A. cepa (Kim et al., 2016), there was only one ribosomal protein gene (rps12), which is far fewer than in most spermatophytes. The difference in the PCG content between H. citrina and A. cepa appears to be driven by the high diversity of ribosomal protein genes.
Although mitogenomic structures and sizes are highly fluid among spermatophytes, the generation of similar gene clusters by multiple recombination events often leads to conserved gene organizations across large phylogenetic scales (Overbeek et al., 1999; Gui et al., 2016). In the comparison of gene clusters in H. citrina and 28 other plants, six syntenic gene clusters were explored in H. citrina (Supplementary Figure 1, Supplementary Table 3). Five of those conserved clusters (rps3-rpl16, atp4-nad4L, rrn18-rrn5, rps12-nad3, and cob-rpl5) were identified in almost all the compared species and might date back to their original mitogenomes. The cox3-sdh4 gene cluster seemed to show a preference for distribution in dicotyledons, indicating its existence in the angiosperm ancestor followed by rapid degeneration due to substantial genome rearrangements. Notably, the cluster nad1-matR emerged extensively in most species but was absent from H. citrina, suggesting that recombination events occurred frequently during the evolution of the H. citrina mitogenome. Generally speaking, gene orders are more similar in species with close evolutionary relationships. The sharing of the rare atp1-atp9 cluster by H. citrina and three other monocotyledons (A. officinalis, P. dactylifera, and B. umbellatus), appears to support their close relationship during the evolution of the plant mitogenomes.
Plant mitogenomes contain sizeable fractions of repeat sequences that are assumed to be a vital factor in genome structure and size variations. Previous researches have documented that repeats in mt present high levels of polymorphism and serve as sites of intra- or intergenomic recombination to produce multiple alternative arrangements (Fujii et al., 2010; Sloan, 2013; Gualberto et al., 2014). In H. citrina, the repeat sequences had a size of 39,294 kb and accounted for 8.4% of the mitogenome. However, more than 76% of the repeats were primarily comprised of SSRs and tandem repeats and only occupied 0.9% of the genome (Supplementary Tables 4, 5). In contrast, the 127 dispersed repeats identified amounted to 35,337 kb and occupied 7.5% of the H. citrina mitogenome (Supplementary Table 8). From the comparison of H. citrina with other monocots (Supplementary Figure 2, Supplementary Table 8), we were encouraged to acquire evidence that the scale of dispersed repeats largely correlates with the size of their mitogenome despite their poor conservation across species. Although only three large repeats (>1 kb) were detected in H. citrina, the presence of large segmental duplications possibly led to mitogenome expansion. In conclusion, the abundant repetitive sequences may accelerate the rearrangement of the H. citrina mitogenome.
As a post-transcriptional processing event, RNA editing is pervasive among higher plants, contributing to maintaining the amino acid sequence conservation of important functional proteins in mt. The occurrence of RNA editing generally converts cytidine (C) to uridine (U), sometimes causing the alteration of initiation and termination codons, but more often creating internal codons with powerful functional correlations that alter coding messages (Handa, 2003). For the H. citrina investigation, 679 RNA editing sites within all 45 PCGs were inferred throughout the mitogenome. Unsurprisingly, 100% of the potential editing sites were the expected C-to-U type, which also correspond to certain activities in most spermatophytes. The majority of editing sites led to the alteration of internal codons, and in particular the amino acids exhibited a significant preference in 43.0% (292 sites) of the edits and were transformed to Leucine after RNA editing. This particular phenomenon has also been documented in a previous report (Cheng et al., 2021). Serine and Proline were more susceptible to editing in H. citrina, occupying 37.6% (255 sites) and 34.0% (231 sites), respectively, of the identified edits, which is consistent with the conclusions of a previous study (Sloan and Taylor, 2010). Furthermore, the regulation process created initiation codons by altering ACG to AUG for the transcripts of nad1, nad4L, and rps10. In a similar pattern, atp6 and ccmFc used alternative termination codons by editing CAA to UAA or CGA to UGA. The prediction of RNA editing events could provide possibilities for deciphering gene functions with novel codons. In closely related lineages, RNA editing sites are universally thought to be conserved, such as in Cruciferae (Handa, 2003) and Cucurbitaceae (Alverson et al., 2010). In a comparison of multiple species, the type and frequency of RNA editing in H. citrina were very similar to those of A. officinalis (Figure 5), which provided further evidence of their close evolutionary relationship.
When discussing the evolutionary dynamics of PCG sequences for genetically close species, it is essential to analyze the nucleotide substitution rates to comprehensively understand the evolution of plant mt genes. The Ka/Ks ratio is typically applied to estimate the selective pressure to which specific mitogenome PCGs are subjected during evolution (Zhang et al., 2006). In general, most mt genes have undergone negative or neutral selection considering their high level of conservation, resulting in Ka/Ks values that do not exceed 1 (Bi et al., 2020; Cheng et al., 2021; Yu et al., 2022). From Ka/Ks analysis of all 36 unique PCGs from the H. citrina mitogenome with five other species, we detected that the majority of PCGs had undergone purifying selection during the evolutionary process (Figure 4). However, the Ka/Ks values of atp4, nad3, and sdh4 were greater than 1, implying positive selections of these genes in H. citrina. Since the three genes encode the vital components of the mt Complex subunit, we conjecture that the adaptive evolution of H. citrina has been to improve respiratory electron transport and oxidative phosphorylation. An additional seven genes (ccmB, ccmFn, matR, mttB, rps1, rps7, and rps12) were identified as having been subjected to positive selection pressure in H. citrina. Genes ccmFn and ccmB are believed to participate in the cytochrome c maturation process of mt by affecting the export and metabolism of heme (Giegé et al., 2008; Verrier et al., 2008). The matR gene exists within nad1 intron 4 as the only maturase retained in the angiosperm mt (Sultan et al., 2016), but the biological function of matR remains elusive. The product of mttB is a membrane transport protein that participates in proton transmembrane transfer activity (Gaudet et al., 2011). Additionally, genes rps1, rps7, and rps12 encode the small mitoribosomal subunit proteins in various biological pathways. Altogether, these genes that have undergone positive selection might have developed novel functions and played a crucial role in the evolution of H. citrina. Interestingly, the Ka/Ks values observed for atp9 were notably lower than those for all other PCGs in the H. citrina investigation, implying that strong negative selection was acting on atp9 to maintain its indispensable function in these mitogenomes. The atp9 gene is considered to be involved in proton transport (Gaudet et al., 2011), and, more importantly, it is known to be intimately associated with cytoplasmic male sterility (Reddemann and Horn, 2018). We were surprised to find that the Ka/Ks values did not exceed 1 for all PCGs between H. citrina and A. cepa, which hints strongly at their close evolutionary relationship.
Conclusions
Currently, we assembled and depicted the complete mitogenome of H. citrina based on a strategy using Oxford Nanopore long-read and Illumina short-read sequencing. The multiple chromosomal structure of H. citrina broadens our understanding of structural complexity in plant mitogenomes. Extensive analyses were performed by comparing the genomic characteristics of the H. citrina mt with those of multiple plant mitogenomes, as illustrated by the conservation of the gene content, syntenic gene clusters, repeat sequences, codon usage preferences, selection pressure, prediction of RNA edits, and phylogenetic tree construction. The enormous genomic variability discovered sheds new light on the evolutionary process of higher plant mitogenomes. In summary, the exhaustive nature of the H. citrina mitogenome should offer deeper insights into the evolution of the genus Hemerocallis and should serve as a fundamental reference for further molecular breeding.
Funding
This research was funded by Shanxi Province Science Foundation for Youths (Grant No. 201901D211426), Datong Science and Technology Bureau Applied Basic Research Project (Grant No. 2020137), Youth Science and Technology Innovation Project of Tianjin Academy of Agricultural Sciences (Grant No. 2022014).
Acknowledgments
The authors thank Shenzhen Huitong Biotechnology Co., Ltd., China for sequencing and MDPI for English language editing.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/nuccore/MZ726801.1/, MZ726801.1, https://www.ncbi.nlm.nih.gov/nuccore/MZ726802.1/, MZ726802.1, https://www.ncbi.nlm.nih.gov/nuccore/MZ726803.1/, MZ726803.1, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA752889, PRJNA752889, https://www.ncbi.nlm.nih.gov/sra/SRR15370204, SRR15370204, https://www.ncbi.nlm.nih.gov/sra/SRR15370205, SRR15370205, https://www.ncbi.nlm.nih.gov/biosample/SAMN20667040/, SAMN20667040.
Author contributions
KZ and YW conceived the idea. KZ and XS performed data analysis and wrote the manuscript. YW, XZ, and ZH supervised the research and revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1051221/full#supplementary-material
References
1
Aljohi H. A. Liu W. F. Lin Q. Zhao Y. H. Zeng J. Y. Alamer A. et al . (2016). Complete sequence and analysis of coconut palm (Cocos nucifera) mitochondrial genome. PloS One11, e0163990. doi: 10.1371/journal.pone.0163990
2
Allen J. O. Fauron C. M. Minx P. Roark L. Oddiraju S. Lin G. N. et al . (2007). Comparisons among two fertile and three male-sterile mitochondrial genomes of maize. Genetics177, 1173–1192. doi: 10.1534/genetics.107.073312
3
Alverson A. J. Rice D. W. Dickinson S. Barry K. Palmer J. D. (2011). Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell23, 2499–2513. doi: 10.1105/tpc.111.087189
4
Alverson A. J. Wei X. X. Rice D. W. Stern D. B. Barry K. Palmer J. D. (2010). Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol.27, 1436–1448. doi: 10.1093/molbev/msq029
5
Backert S. Börner T. (2000). Phage T4-like intermediates of DNA replication and recombination in the mitochondria of the higher plant Chenopodium album (L.). Curr. Genet.37, 304–314. doi: 10.1007/s002940050532
6
Beier S. Thiel T. Münch T. Scholz U. Mascher M. (2017). MISA-web: A web server for microsatellite prediction. Bioinformatics33, 2583–2585. doi: 10.1093/bioinformatics/btx198
7
Benson G. (1999). Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res.27, 573–580. doi: 10.1093/nar/27.2.573
8
Bi C. W. Lu N. Xu Y. Q. He C. P. Lu Z. H. (2020). Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. Int. J. Mol. Sci.21, 3778–3797. doi: 10.3390/ijms21113778
9
Cheng Y. He X. X. Priyadarshani S. V. G. N. Wang Y. Ye L. Shi C. et al . (2021). Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genomics22, 167–181. doi: 10.1186/s12864-021-07490-9
10
Darling A. E. Mau B. Perna N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PloS One5, e11147. doi: 10.1371/journal.pone.0011147
11
De Coster W. D’Hert S. Schultz D. T. Cruts M. Van Broeckhoven C. (2018). NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics34, 2666–2669. doi: 10.1093/bioinformatics/bty149
12
Fujii S. Kazama T. Yamada M. Toriyama K. (2010). Discovery of global genomic re-organization based on comparison of two newly sequenced rice mitochondrial genomes with cytoplasmic male sterility-related genes. BMC Genomics11, 209–223. doi: 10.1186/1471-2164-11-209
13
Gaudet P. Livstone M. S. Lewis S. E. Thomas P. D. (2011). Phylogenetic-based propagation of functional annotations within the gene ontology consortium. Brief. Bioinform.12, 449–462. doi: 10.1093/bib/bbr042
14
Gautheret D. Lambert A. (2001). Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J. Mol. Biol.313, 1003–1011. doi: 10.1006/jmbi.2001.5102
15
Giegé P. Grienenberger J. M. Bonnard G. (2008). Cytochrome c biogenesis in mitochondria. Mitochondrion8, 61–73. doi: 10.1016/j.mito.2007.10.001
16
Goremykin V. V. Lockhart P. J. Viola R. Velasco R. (2012). The mitochondrial genome of malus domestica and the import-driven hypothesis of mitochondrial genome expansion in seed plants. Plant J.71, 615–626. doi: 10.1111/j.1365-313X.2012.05014.x
17
Greiner S. Lehwark P. Bock R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res.47, W59–W64. doi: 10.1093/nar/gkz238
18
Gualberto J. M. Mileshina D. Wallet C. Niazi A. K. Weber-Lotfi F. Dietrich A. (2014). The plant mitochondrial genome: dynamics and maintenance. Biochimie100, 107–120. doi: 10.1016/j.biochi.2013.09.016
19
Gui S. T. Wu Z. H. Zhang H. Y. Zheng Y. Z. Zhu Z. X. Liang D. Q. et al . (2016). The mitochondrial genome map of Nelumbo nucifera reveals ancient evolutionary features. Sci. Rep.6, 30158. doi: 10.1038/srep30158
20
Handa H. (2003). The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus l.): Comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res.31, 5907–5916. doi: 10.1093/nar/gkg795
21
Jackman S. D. Coombe L. Warren R. L. Kirk H. Trinh E. MacLeod T. et al . (2020). Complete mitochondrial genome of a gymnosperm, sitka spruce (Picea sitchensis), indicates a complex physical structure. Genome Biol. Evol.12, 1174–1179. doi: 10.1093/gbe/evaa108
22
Kan S. L. Shen T. T. Gong P. Ran J. H. Wang X. Q. (2020). The complete mitochondrial genome of Taxus cuspidata (Taxaceae): eight protein-coding genes have transferred to the nuclear genome. BMC Evol. Biol.20, 10–26. doi: 10.1186/s12862-020-1582-1
23
Katoh K. Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol.30, 772–780. doi: 10.1093/molbev/mst010
24
Kim B. Kim K. Yang T. J. Kim S. (2016). Completion of the mitochondrial genome sequence of onion (Allium cepa l.) containing the CMS-s male-sterile cytoplasm and identification of an independent event of the ccmFN gene split. Curr. Genet.62, 873–885. doi: 10.1007/s00294-016-0595-1
25
Kolmogorov M. Yuan J. Lin Y. Pevzner P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol.37, 540–546. doi: 10.1038/s41587-019-0072-8
26
Koren S. Walenz B. P. Berlin K. Miller J. R. Bergman N. H. Phillippy A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res.27, 722–736. doi: 10.1101/gr.215087.116
27
Kovar L. Nageswara-Rao M. Ortega-Rodriguez S. Dugas D. V. Straub S. Cronn R. et al . (2018). PacBio-based mitochondrial genome assembly of Leucaena trichandra (Leguminosae) and an intrageneric assessment of mitochondrial RNA editing. Genome Biol. Evol.10, 2501–2517. doi: 10.1093/gbe/evy179
28
Kozik A. Rowan B. A. Lavelle D. Berke L. Schranz M. E. Michelmore R. W. et al . (2019). The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PloS Genet.15, e1008373. doi: 10.1371/journal.pgen.1008373
29
Kubo T. Mikami T. (2007). Organization and variation of angiosperm mitochondrial genome. Physiol. Plantarum129, 6–13. doi: 10.1111/j.1399-3054.2006.00768.x
30
Kurtz S. Choudhuri J. V. Ohlebusch E. Schleiermacher C. Stoye J. Giegerich R. (2001). Reputer: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res.29, 4633–4642. doi: 10.1093/nar/29.22.4633
31
Li H. (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics32, 2103–2110. doi: 10.1093/bioinformatics/btw152
32
Li H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics34, 3094–3100. doi: 10.1093/bioinformatics/bty191
33
Liao X. F. Zhao Y. H. Kong X. J. Khan A. Zhou B. J. Liu D. M. et al . (2018). Complete sequence of kenaf (Hibiscus cannabinus) mitochondrial genome and comparative analysis with the mitochondrial genomes of other plants. Sci. Rep.8, 12714. doi: 10.1038/s41598-018-30297-w
34
Liberatore K. L. Dukowic-Schulze S. Miller M. E. Chen C. Kianian S. F. (2016). The role of mitochondria in plant development and stress tolerance. Free Radical Biol. Med.100, 238–256. doi: 10.1016/j.freeradbiomed.2016.03.033
35
Li S. Ji F. F. Hou F. F. Shi Q. Q. Xing G. M. Chen H. et al . (2021b). Morphological, palynological and molecular assessment of Hemerocallis core collection. Sci. Hortic.285, 110181. doi: 10.1016/j.scienta.2021.110181
36
Liu J. H. Zhong X. H. Jiang Y. Y. Yu L. Y. Huang X. Q. Dong Z. et al . (2020). Systematic identification metabolites of Hemerocallis citrina borani by high-performance liquid chromatography/quadrupole-time-of-flight mass spectrometry combined with a screening method. J. Pharm. Biomed. Anal.186, 113314. doi: 10.1016/j.jpba.2020.113314
37
Li J. L. Xu Y. C. Shan Y. Y. Pei X. Y. Yong S. Y. Liu C. et al . (2021a). Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta254, 36–51. doi: 10.1007/s00425-021-03684-3
38
Ma G. Y. Shi X. H. Zou Q. C. Tian D. Q. (2018). iTRAQ-based quantitative proteomic analysis reveals dynamic changes during daylily flower senescence. Planta248, 859–873. doi: 10.1007/s00425-018-2943-5
39
Matand K. Shoemake M. Li C. X. (2020). High frequency in vitro regeneration of adventitious shoots in daylilies (Hemerocallis sp) stem tissue using thidiazuron. BMC Plant Biol.20, 31–40. doi: 10.1186/s12870-020-2243-7
40
Mower J. P. (2009). The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res.37, W253–W259. doi: 10.1093/nar/gkp337
41
Notsu Y. Masood S. Nishikawa T. Kubo N. Akiduki G. Nakazono M. et al . (2002). The complete sequence of the rice (Oryza sativa l.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genomics268, 434–445. doi: 10.1007/s00438-002-0767-1
42
Ou X. B. Liu G. Wu L. H. (2020). The complete chloroplast genome of Hemerocallis citrina (Asphodelaceae), an ornamental and medicinal plant. Mitochondrial DNA B Resour.5, 1109–1110. doi: 10.1080/23802359.2020.1726227
43
Overbeek R. Fonstein M. D’Souza M. Pusch G. D. Maltsev N. (1999). The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. U.S.A.96, 2896–2901. doi: 10.1073/pnas.96.6.2896
44
Patel R. K. Jain M. (2012). NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PloS One7, e30619. doi: 10.1371/journal.pone.0030619
45
Porebski S. Bailey L. G. Baum B. R. (1997). Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep.15, 8–15. doi: 10.1007/BF02772108
46
Qing Z. X. Liu J. H. Yi X. X. Liu X. B. Hu G. A. Lao J. et al . (2021). The chromosome-level Hemerocallis citrina borani genome provides new insights into the rutin biosynthesis and the lack of colchicine. Hortic. Res.8, 89–98. doi: 10.1038/s41438-021-00539-6
47
Reddemann A. Horn R. (2018). Recombination events involving the atp9 gene are associated with male sterility of CMS PET2 in sunflower. Int. J. Mol. Sci.19, 806–822. doi: 10.3390/ijms19030806
48
Ren Y. Gao Y. K. Gao S. Y. Yuan L. Wang X. Q. Zhang Q. X. (2019). Genetic characteristics of circadian flowering rhythm in. Hemerocallis Sci. Hortic.250, 19–26. doi: 10.1016/j.scienta.2019.01.052
49
Rice D. W. Alverson A. J. Richardson A. O. Young G. J. Sanchezpuerta M. V. Munzinger J. et al . (2013). Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm amborella. Science342, 1468–1473. doi: 10.1126/science.1246275
50
Schattner P. Brooks A. N. Lowe T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res.33, W686–W689. doi: 10.1093/nar/gki366
51
Skippington E. Barkman T. J. Rice D. W. Palmer J. D. (2015). Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. U.S.A.112, E3515–E3524. doi: 10.1073/pnas.1504491112
52
Sloan D. B. (2013). One ring to rule them all? genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol.200, 978–985. doi: 10.1111/nph.12395
53
Sloan D. B. Alverson A. J. Chuckalovcak J. P. Wu M. McCauley D. E. Palmer J. D. et al . (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PloS Biol.10, e1001241. doi: 10.1371/journal.pbio.1001241
54
Sloan D. B. Taylor D. R. (2010). Testing for selection on synonymous sites in plant mitochondrial DNA: the role of codon bias and RNA editing. J. Mol. Evol.70, 479–491. doi: 10.1007/s00239-010-9346-y
55
Smith D. R. Keeling P. J. (2015). Mitochondrial and plastid genome architecture: Reoccurring themes, but significant differences at the extremes. Proc. Natl. Acad. Sci. U.S.A.112, 10177–10184. doi: 10.1073/pnas.1422049112
56
Sultan L. D. Mileshina D. Grewe F. Rolle K. Abudraham S. Głodowicz P. et al . (2016). The reverse transcriptase/RNA maturase protein MatR is required for the splicing of various group II introns in brassicaceae mitochondria. Plant Cell28, 2805–2829. doi: 10.1105/tpc.16.00398
57
Timmis J. N. Ayliffffe M. A. Huang C. Y. Martin W. (2004). Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet.5, 123–135. doi: 10.1038/nrg1271
58
Trifinopoulos J. Nguyen L. T. von Haeseler A. Minh B. Q. (2016). W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res.44, W232–W235. doi: 10.1093/nar/gkw256
59
Unseld M. Marienfeld J. R. Brandt P. Brennicke A. (1997). The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet.15, 57–61. doi: 10.1038/ng0197-57
60
Vaser R. Sović I. Nagarajan N. Šikicí M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res.27, 737–746. doi: 10.1101/gr.214270.116
61
Verrier P. J. Bird D. Burla B. Dassa E. Forestier C. Geisler M. et al . (2008). Plant ABC proteins–a unified nomenclature and updated inventory. Trends Plant Sci.13, 151–159. doi: 10.1016/j.tplants.2008.02.001
62
Walker B. J. Abeel T. Shea T. Priest M. Abouelliel A. Sakthikumar S. et al . (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One9, e112963. doi: 10.1371/journal.pone.0112963
63
Wang J. Hu D. M. Hou J. Li S. S. Wang W. P. Li J. et al . (2018). Ethyl acetate fraction of Hemerocallis citrina baroni decreases tert-butyl hydroperoxide-induced oxidative stress damage in BRL-3A cells. Oxid. Med. Cell. Longev.1526125, 1–13. doi: 10.1155/2018/1526125
64
Wang D. Zhang Y. Zhang Z. Zhu J. Yu J. (2010). KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinf.8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
65
Wick R. R. Judd L. M. Gorrie C. L. Holt K. E. (2017). Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom.3, e000132. doi: 10.1099/mgen.0.000132
66
Wick R. R. Schultz M. B. Zobel J. Holt K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics31, 3350–3352. doi: 10.1093/bioinformatics/btv383
67
Wu Z. Q. Liao X. Z. Zhang X. N. Tembrock L. R. Broz A. (2020). Genomic architectural variation of plant mitochondria-a review of multichromosomal structuring. J. Syst. Evol.60, 160–168. doi: 10.1111/jse.12655
68
Xu P. Wang K. Z. Lu C. Dong L. M. Zhai J. L. Liao Y. H. et al . (2016). Antidepressant-like effects and cognitive enhancement of the total phenols extract of Hemerocallis citrina baroni in chronic unpredictable mild stress rats and its related mechanism. J. Ethnopharmacol.194, 819–826. doi: 10.1016/j.jep.2016.09.023
69
Yu X. L. Duan Z. G. Wang Y. J. Zhang Q. X. Li W. (2022). Sequence analysis of the complete mitochondrial genome of a medicinal plant, Vitex rotundifolia Linnaeus f. (Lamiales: Lamiaceae). Genes (Basel)13, 839–848. doi: 10.3390/genes13050839
70
Zhang F. Li W. Gao C. W. Zhang D. Gao L. Z. (2019). Deciphering tea tree chloroplast and mitochondrial genomes of Camellia sinensis var. assamica. Sci. Data6, 209–218. doi: 10.1038/s41597-019-0201-8
71
Zhang Z. Li J. Zhao X. Q. Wang J. Wong G. K. Yu J. (2006). KaKs_Calculator: calculating ka and ks through model selection and model averaging. Genom. Proteom. Bioinf.4, 259–263. doi: 10.1016/S1672-0229(07)60007-2
72
Zhang Z. Xiao J. Wu J. Zhang H. Liu G. Wang X. et al . (2012). ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun.419, 779–781. doi: 10.1016/j.bbrc.2012.02.101
Summary
Keywords
Hemerocallis citrina , Asphodelaceae, mitochondrial genome, combined sequencing, comparative genomics
Citation
Zhang K, Wang Y, Zhang X, Han Z and Shan X (2022) Deciphering the mitochondrial genome of Hemerocallis citrina (Asphodelaceae) using a combined assembly and comparative genomic strategy. Front. Plant Sci. 13:1051221. doi: 10.3389/fpls.2022.1051221
Received
22 September 2022
Accepted
04 November 2022
Published
18 November 2022
Volume
13 - 2022
Edited by
Zhenyu Gao, China National Rice Research Institute (CAAS), China
Reviewed by
Chuyu Ye, Zhejiang University, China; Lianguang Shang, Agricultural Genomics Institute at Shenzhen (CAAS), China; Jian Sun, Shenyang Agricultural University, China
Updates
Copyright
© 2022 Zhang, Wang, Zhang, Han and Shan.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun Zhang, 876828320@qq.com
†These authors have contributed equally to this work and share first authorship
This article was submitted to Functional and Applied Plant Genomics, a section of the journal Frontiers in Plant Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.