 Mengjie Cui1,2,3,4,5
Mengjie Cui1,2,3,4,5 Suoyi Han1,2,3,4,5
Suoyi Han1,2,3,4,5 Du Wang6
Du Wang6 Muhammad Salman Haider7
Muhammad Salman Haider7 Junjia Guo2,3,4,5Qi Zhao2,3,4
Junjia Guo2,3,4,5Qi Zhao2,3,4 Pei Du1,2,3,4,5Ziqi Sun2,3,4,5
Pei Du1,2,3,4,5Ziqi Sun2,3,4,5 Feiyan Qi2,3,4,5
Feiyan Qi2,3,4,5 Zheng Zheng2,3,4,5Bingyan Huang2,3,4,5Wenzhao Dong2,3,4,5
Zheng Zheng2,3,4,5Bingyan Huang2,3,4,5Wenzhao Dong2,3,4,5 Peiwu Li6*Xinyou Zhang1,2,3,4,5*
Peiwu Li6*Xinyou Zhang1,2,3,4,5*- 1College of Agriculture, Nanjing Agricultural University, Nanjing, China
- 2The Shennong Laboratory, Henan Academy of Crops Molecular Breeding, Henan Academy of Agricultural Science, Zhengzhou, China
- 3Key Laboratory of Oil Crops in Huang-Huai-Hai Plains, Ministry of Agriculture, Zhengzhou, China
- 4Henan Provincial Key Laboratory for Oil Crop Improvement, Zhengzhou, China
- 5National Centre for Plant Breeding, Xinxiang, China
- 6Key Laboratory of Detection for Mycotoxins, Oil Crops Research Institute of the Chinese Academy of Agricultural Sciences, Ministry of Agriculture and Rural Affairs, Wuhan, China
- 7Department of Horticulture, Ghazi University, Dera Ghazi Khan, Pakistan
Cultivated peanut (Arachis hypogaea L.), a cosmopolitan oil crop, is susceptible to a variety of pathogens, especially Aspergillus flavus L., which not only vastly reduce the quality of peanut products but also seriously threaten food safety for the contamination of aflatoxin. However, the key genes related to resistance to Aspergillus flavus L. in peanuts remain unclear. This study identifies hub genes positively associated with resistance to A. flavus in two genotypes by comparative transcriptome and weighted gene co-expression network analysis (WGCNA) method. Compared with susceptible genotype (Zhonghua 12, S), the rapid response to A. flavus and quick preparation for the translation of resistance-related genes in the resistant genotype (J-11, R) may be the drivers of its high resistance. WGCNA analysis revealed that 18 genes encoding pathogenesis-related proteins (PR10), 1-aminocyclopropane-1-carboxylate oxidase (ACO1), MAPK kinase, serine/threonine kinase (STK), pattern recognition receptors (PRRs), cytochrome P450, SNARE protein SYP121, pectinesterase, phosphatidylinositol transfer protein, and pentatricopeptide repeat (PPR) protein play major and active roles in peanut resistance to A. flavus. Collectively, this study provides new insight into resistance to A. flavus by employing WGCNA, and the identification of hub resistance-responsive genes may contribute to the development of resistant cultivars by molecular-assisted breeding.
Introduction
As an important oilseed crop and a major source of vegetable oil and protein worldwide, peanut can be easily infected by Aspergillus flavus L. during drying, storage, and transportation processes (Nigam et al., 2009; Pandey et al., 2019; Soni et al., 2020), resulting in kernel rot and subsequent contamination of aflatoxins, which seriously threaten the safety of peanut products (Liang et al., 2006; Guo et al., 2008b; Passone et al., 2009; Ding et al., 2012). To date, breeding of cultivars with resistance to A. flavus has been widely accepted as the most cost-effective way to mitigate aflatoxin contamination, and the identification of hub resistance genes is recognized as a fundamental premise for breeding of resistance cultivars.
In general, pre- and post-harvest contamination are two major types caused by A. flavus (Guo et al., 2008a,b, 2011; Liao et al., 2009; Wang et al., 2013; Clevenger et al., 2016; Zhao et al., 2019; Soni et al., 2021). Two types of mechanism for host resistance to A. flavus, in vitro seed colonization and aflatoxin production, have been documented, which were further proved to be independently inherited (Liao et al., 2009; Nigam et al., 2009; Pandey et al., 2019; Soni et al., 2020), thus making it a great challenge for the researchers to elucidate resistance mechanism, breed resistant lines, and eventually control the disease in peanuts. Over the past two decades, studies have been conducted to characterize differentially expressed genes (DEGs) and main signaling pathways that were involved in resistance of A. flavus, but relatively limited information is available for hub genes associated with resistance to A. flavus stress, thus restricting the elucidation of A. flavus-resistance mechanism (Wang et al., 2016; Nayak et al., 2017; Walid et al., 2017; Korani et al., 2018). Recently, quantitative trait locus (QTLs) and single nucleotide polymorphisms (SNPs) related to peanut resistance to A. flavus have been reported (Liang et al., 2009; Pandey et al., 2014; Yu et al., 2019, 2020; Khan et al., 2020). However, no gene that responds to A. flavus in peanuts has been cloned by forward genetics. Meanwhile, studies on reverse genetics have demonstrated that pathogenesis-related proteins (PRs), such as PR10 (Luo et al., 2005; Guo et al., 2008a,b; Xie et al., 2013) and chitinase (Prasad et al., 2013), acted in the resistance to Aspergillus flavus L. infection. Although specific genes have been linked to peanut seed resistance, mining of hub resistance associated genes deserves further investigation.
Weighted gene co-expression network analysis (WGCNA), an approach that can be used to identify certain traits-related modules (Langfelder and Horvath, 2008; Menon, 2018), has been widely used in identification of hub resistance associated genes and clarification of molecular mechanisms of stresses in various plants (Hopper et al., 2016; Tan et al., 2017; Lin et al., 2019; Li et al., 2020; Liu et al., 2021; Yan et al., 2021). In this study, we aimed to systematically and comprehensively illustrate the resistance mechanism of two peanut genotypes that differ in their resistance to A. flavus, as well as identify hub genes positively associated with A. flavus resistance using WGCNA methods within RNA-seq analysis.
Experimental Procedures
Phenotypic Evaluation on the Resistance to A. flavus of Cultivated Peanut Genotypes
Experiments were conducted at the Henan Provincial Key Laboratory for Oil Crops Improvement, Henan Institute of Crop Molecular Breeding, Zhengzhou city, Henan, China. Highly toxigenic strain A. favus 3.4408 was cultured on dichloranglycerol-18 (DG-18) agar plates. After incubation for 7 days at 30°C, conidia were collected and suspended in sterile water containing 0.05% tween-80 with a concentration of 2 × 106 CFU (spores/ml).
Approximately 200 healthy and mature kernels of each R (J-11) and S (Zhonghua 12) genotypes were collected for the experiment. Samples were collected at 0 (T0), 24 (T1), 48 (T2), 72 (T3), 120 (T5), and 168 h (T7) after inoculation from the infected samples of R and S genotypes. In total, 36 samples (2 genotypes × 6 stages × 3 replicates) were analyzed. At each time interval (T0, T1, T2, T3, T5, T7), 10–12 seeds were frozen in liquid nitrogen for RNA isolation and subsequently RNA-seq. Three seeds were immediately fixed by electron microscopy fixative and scanned in Wuhan Sevicebio Biological Technology, Wuhan city, Hubei, China. Seven days after inoculation, the infection index was scored according to the previously described method (Khan et al., 2020). Seeds were then autoclaved at 121°C for 30 min, and dried at 110°C for 3 h for aflatoxin B1 quantification by high-performance liquid chromatography (HPLC) method (Ma et al., 2013).
RNA-Sequencing and Data Analysis
A total of 36 cDNA libraries were constructed using NEBNext Ultra RNA Library Prep Kit (NEB, USA) following the instructions of the manufacturer and deep sequenced by GENE DENOVO (Guangzhou, China), using Illumina sequencing platform. Three independent biological replications were used, and each biological replication contained five samples.
Reads were aligned to the reference Arachis hypogaea L. genome (GCA_003086295.2, https://www.ncbi.nlm.nih.gov/assembly) using HISAT 2. 2.4 with “-RNA-strandness RF” and other parameters set as a default (Kim et al., 2015). RSEM software was used to calculate the abundance values of the transcript based on the fragments per kilobase of exon per million mapped reads (FPKM) (Dewey and Li, 2011). DEGs were analyzed using DESeq2 software (Love et al., 2014), with the estimated absolute log2 fold change (FC) > 2 and false discovery rate (FDR) < 0.01. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of DEGs were also carried out to identify A. flavus stress-related genes involved in the key biological processes and metabolic pathways associated with A. flavus response. Mapping of the top 30 significant enriched ones (p < 0.05) was generated on the online website (https://www.omicshare.com/tools/) by GENE DENOVO (Guangzhou, China), as described in the study by Li et al. (2021).
Data Integration and Network Construction
Co-expression networks were constructed by the R package WGCNA (Langfelder and Horvath, 2008). Co-expression transcript was clustered into 16 modules after filtering the genes of which FPKM < 1 in more than half of the samples. CYTOSCAPE (v3.7.1) was then used to visualize the networks of genes within module and to present biological interaction of core genes (Shannon et al., 2003).
RT-qPCR Analysis
Twelve genes were randomly selected for validating the repeatability and authenticity of gene expression patterns by RT-qPCR, as described previously (Guimaraes et al., 2012; Yin et al., 2013; Chen et al., 2014; Li et al., 2014; Cui et al., 2021). The alcohol dehydrogenase class III (AhADH3, Arahy. VYWU26.2) was selected as the internal reference (Supplementary Table S1) (Brand and Hovav, 2010).
Results
Cultivated Peanut Cultivars Exhibiting Higher Resistance (R) and Susceptibility (S) to A. flavus
Evaluation experiment of peanut resistance to A. flavus was performed by genotypes materials, which were grown in Yuanyang, China (N35°18′, E113°55′) (2020). Peanut accessions with higher resistance (J11, R cultivar) and susceptibility (Zhonghua 12, S cultivar) to A. flavus (Figure 1) as a comparative experimental material were selected. As seed coat of peanut, which is the outermost layer, may act as a physical barrier, the A. flavus infection process was observed by scanning electron microscopy (Figure 1A). Obviously, mycelial on the seeds of both of R and S penetrated the seed coat on the second day of infection (T2) and reached the cotyledons, where they acquired nutrients and produced aflatoxin (Figure 1A), and very little mycelial was observed in the R seed coat compared with S (Figure 1A). Furthermore, profuse mycelial growth and sporulation in genotype S was compared to genotype R on the third day after inoculation (T3) (Figure 1A). At T7, kernels of S were almost covered by green sporulation, and 115,391 (μg/kg) aflatoxin B1 was detected (Figures 1B,C). These results suggest that seed coat may be not the main reason for the phenotypic differences between R and S, but were more mechanism-based. The two genotypes are ideal candidates for studying the resistance mechanism of A. flavus stress on peanut seeds.
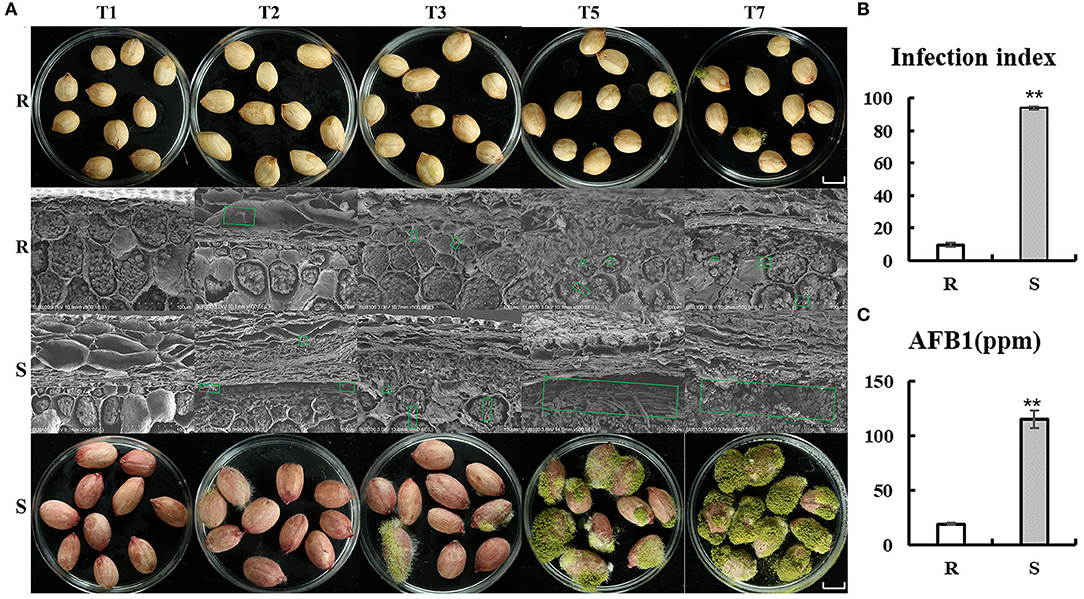
Figure 1. Comparison of R and S on the phenotypes of peanut seeds responding to A. flavus. (A) Mycelia growth on peanut seeds at different inoculation processes (T1, T2, T3, T5, and T7). Bars = 1 cm. (B) Infection index and aflatoxin B1 (AFB1) content (C) of the R and S genotypes after inoculation of A. flavus for 7 days. Green box shows mycelial of Aspergillus flavus L. Error bars indicated ± standard errors (SEs) of three independent biological replicates (n = 3). **P < 0.01 (t-test) compared with R.
Transcriptome Profiles of 36 RNA Libraries From Peanut Seeds Infected With A. flavus at Different Time Points
A total of 380,590,871,700 raw reads and 377,441,077,140 clean reads (clean ratio > 99.15 %) were obtained after filtering reads with low-quality. On average, 91.77% of reads could be mapped to the reference genome of peanut, except for S-T5 and S-T7, which contained more mycelium and spores in kernels, the average mapping genome ratio of which was only 23.12 and 17.27%, respectively (Supplementary Table S2). As the amount of sequencing (reads) increases, the number of genes detected for S-T5 and S-T7 also increased and eventually tended to be saturated, implying the high accuracy of transcriptome sequencing results (Supplementary Figure S1). Pearson correlation coefficients among samples (Supplementary Figure S2) and principal component analyses (PCA) (Supplementary Figure S3) manifested a tremendous difference between samples at T0 and other time points (T1, T2, T3, T5, and T7), indicating the infection of A. flavus L. induced large transcription level perturbation in the peanut. The raw transcriptome reads were submitted to the NCBI Sequence Read Archive (SRA) database under accession: PRJNA825125.
Identification of Differentially Expressed Genes
According to the criteria of |log2fold change| > 2 and FDR < 0.01, a total of 3,670, 7,888, 4,708, 6,545, and 7,533, respectively, upregulated DEGs and 2,392, 3,872, 2,910, 3,138, and 3,273 downregulated DEGs in R were screened, while 6,601, 5,870, 6,838, 6,425, and 6,732 DEGs were significantly upregulated and 3,649, 3,389, 3,731, 4,444, and 5,057 DEGs downregulated in S at T1, T2, T3, T5, and T7 after infection, respectively (Figure 2). It followed that DEGs (6,062–10,806) in R were fewer than those of S (10,250–11,789) at the stage of T1, T3, T5, and T7. Whereas the number of DEGs (11,760) in R was significantly higher than that in S (9,259) at T2, which was the time when mycelial penetrated seed coat, implying that R developed a very strong and quick resistant response to A. flavus at T2, thus preventing the seed from being contaminated by aflatoxin (Figure 2).
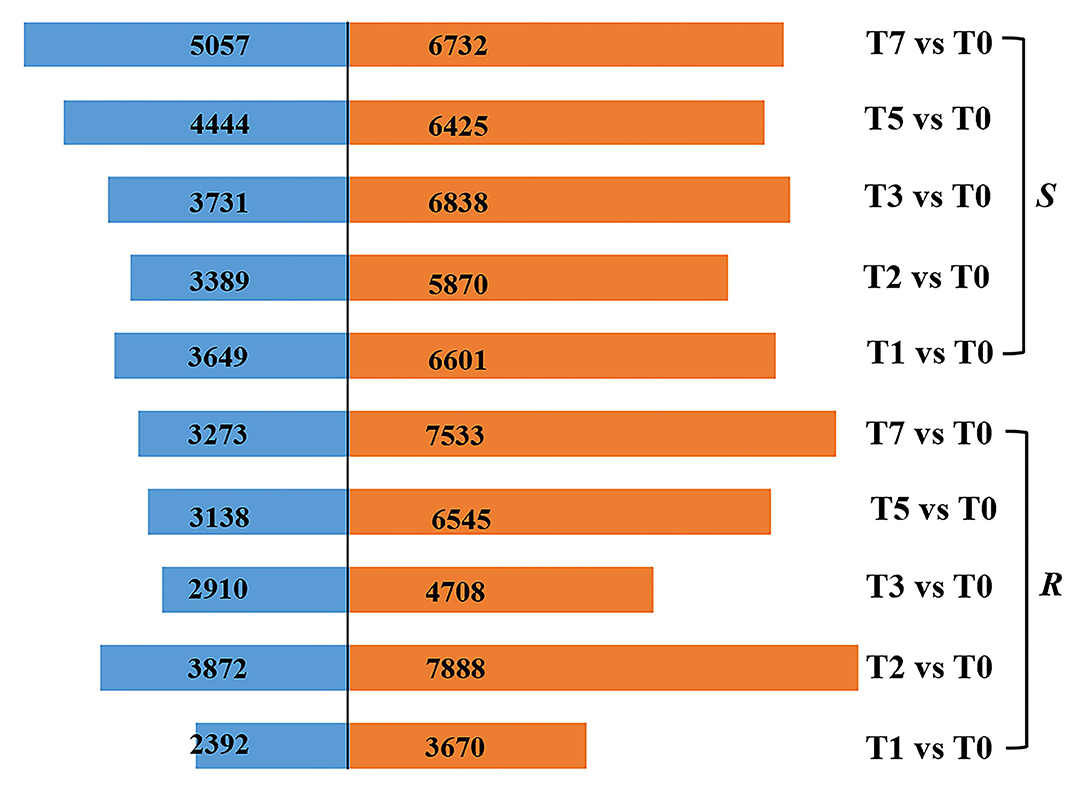
Figure 2. DEGs in R and S at five time points post-inoculation (T1, T2, T3, T5, and T7) compared with T0.
DEGs Upregulated Uniquely in R Compared With S
As the upregulated genes may be positively responsible for resistance (Yan et al., 2021), we further conducted the overlap analysis at five-time points of R and S. The genes 128, 2,219, 196, 118, and 1,641 upregulated specifically DEGs in R, while 1,769, 274, 356, 1,242, and 1,269 in S were found at each time point (Figure 3A). For better identifying the critical time points of resistance in R and dissecting the resistance process, DEGs upregulated uniquely in R compared with S were explored further (Figure 3B). At T1, 111 DEGs were upregulated uniquely in R, while 1,752 DEGs were upregulated in S. GO analysis showed that the upregulated DEGs were enriched in “ribosome synthesis related process” and “response to endogenous stimulus” (Supplementary Figure S4). KEGG analysis indicated that these DEGs were enriched in “Brassinosteroid biosynthesis,” “MAPK signaling pathway—plant,” “Plant-pathogen interaction,” “Phenylpropanoid biosynthesis,” and “Plant hormone signal transduction” (Figure 4 and Supplementary Figure S5), which were reported to be main pathways of plants resistance to biotic stresses (Dixon and Paiva, 1995; Zhang and Klessig, 2001; Yan et al., 2018; Polturak and Osbourn, 2021). The same KEGG enrichment analysis revealed that the 1,752 DEGs, upregulated uniquely in S, were enriched in “Ribosome biogenesis in eukaryotes,” “Proteasome,” “RNA transport,” and “ABC transporters” (Figure 4 and Supplementary Figures S6, S7), but no resistance-associated pathways were significantly enriched uniquely in S at T1 (Supplementary Figures S6, S7), illustrating that R may respond more quickly than S and is prepared for resistance of infection at the level of transcription.
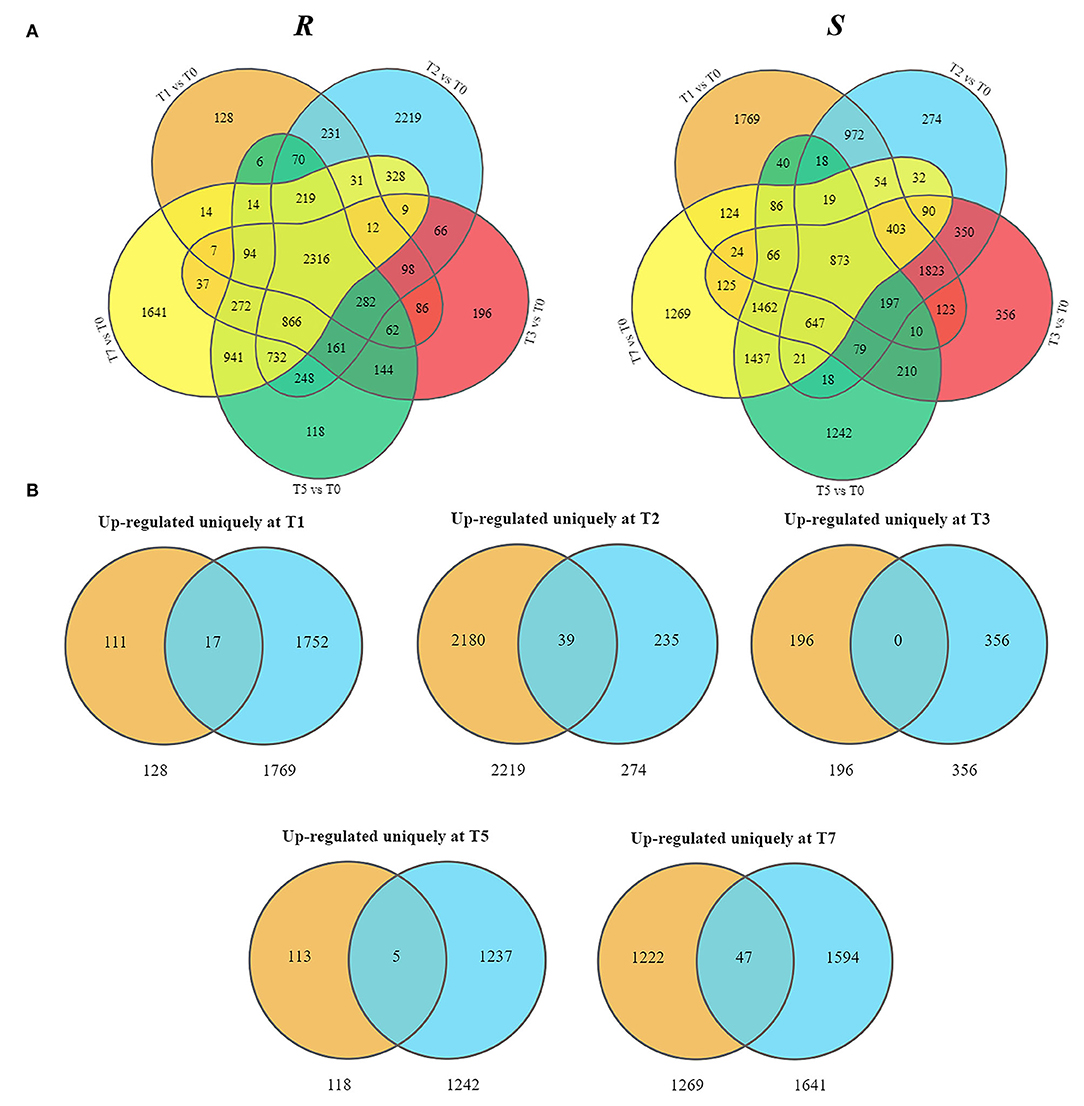
Figure 3. Presentation of upregulated DEGs in R and S at five time points post-inoculation (T1, T2, T3, T5, and T7). (A) Overlap analysis of upregulated DEGs. (B) Uniquely upregulated DEGs in R compared with S.
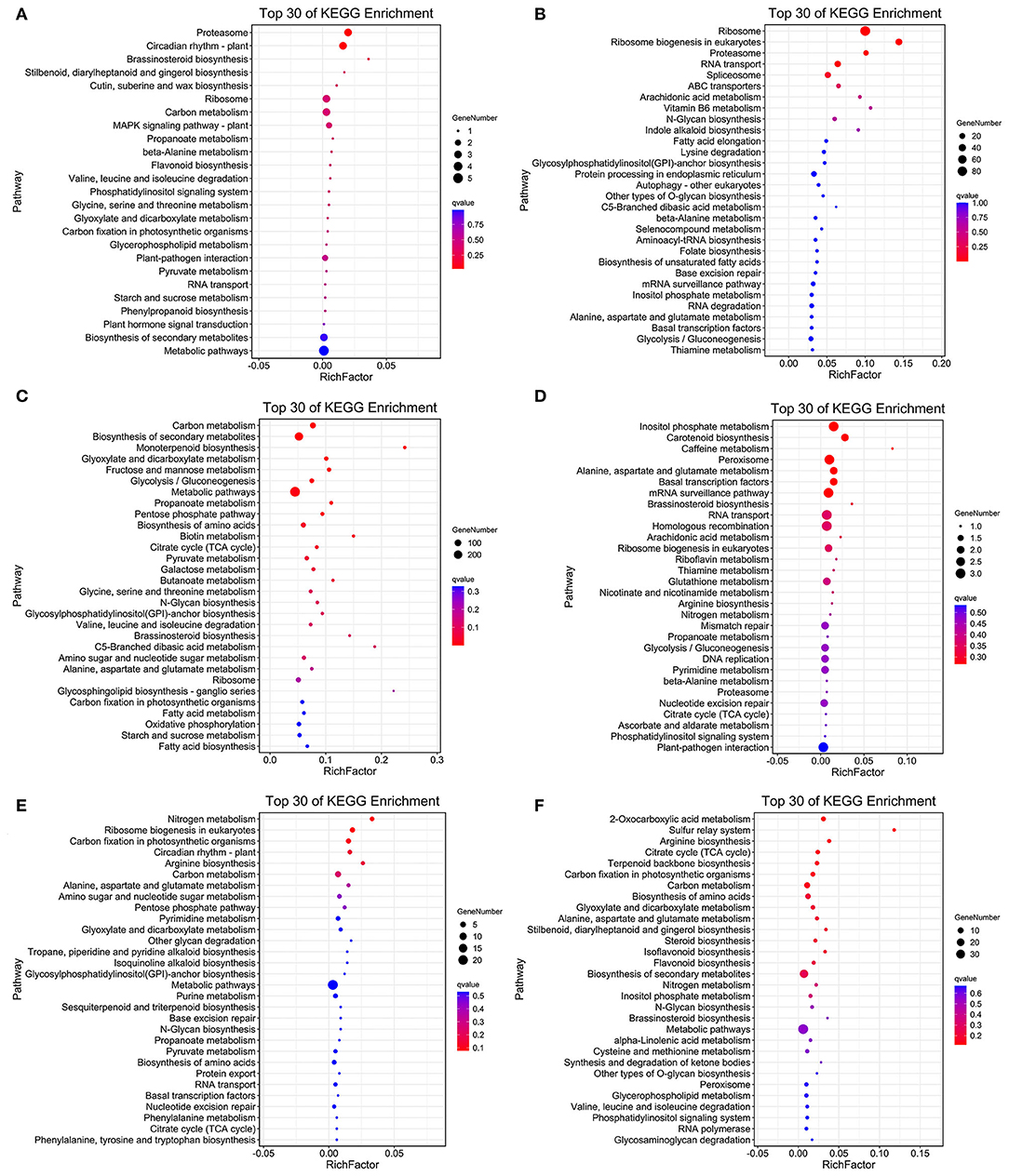
Figure 4. Top 30 of KEGG enrichment genes of uniquely upregulated DEGs in R and S at T1 (A,B), T2 (C,D), and T3 (E,F).
At T2, the unique upregulated DEGs (2,180) in genotype R were enriched in KEGG pathways such as “biosynthesis of secondary metabolites,” “Monoterpenoid biosynthesis,” “Fructose and mannose metabolism,” “Brassinosteroid biosynthesis,” and “Oxidative phosphorylation” (Figure 4 and Supplementary Figure S5), suggesting that genotype R may prepare for the translation and transportation of tolerance-related secondary metabolites at T2. Whereas in S, few pathways related to biotic resistance were enriched at T2 (Figure 4). Meanwhile, KEGG analysis of upregulated DEGs uniquely at T3, T5, and T7 DEGs in R was also enriched in disease resistance related pathways like “Plant-pathogen interaction,” “Autophagy—other eukaryotes,” “Phenylpropanoid biosynthesis,” “MAPK signaling pathway—plant,” “Ubiquitin mediated proteolysis,” and “SNARE interactions in vesicular transport” (Figure 4 and Supplementary Figure S5). It was observed that the pathways related to biotic resistance were significantly enriched in S at T3 (Figure 4 and Supplementary Figure S7), when profuse mycelial growth and sporulation started to appear (Figure 1). Taken together, it seems that the rapid responses of A. flavus in genotype R and activation of specific disease-related signaling pathways at T1 and T2 might lead to the high resistance.
Co-Expression Network Analysis Identified Key Modules Correlated With Resistance to A. flavus
To identify specific genes that were highly associated with resistance to A. flavus, WGCNA of 28,579 DEGs was carried out. We chose a power of β=12 based on the scale-free topology criterion to generate a hierarchical tree. All genes were assigned into 16 distinct modules (mergeCutHeight = 0.25) based on the similarity of their expression patterns (Figure 5A). The numbers of the genes in each module varied greatly ranging from 107 to 14,491 (Supplementary Table S3).
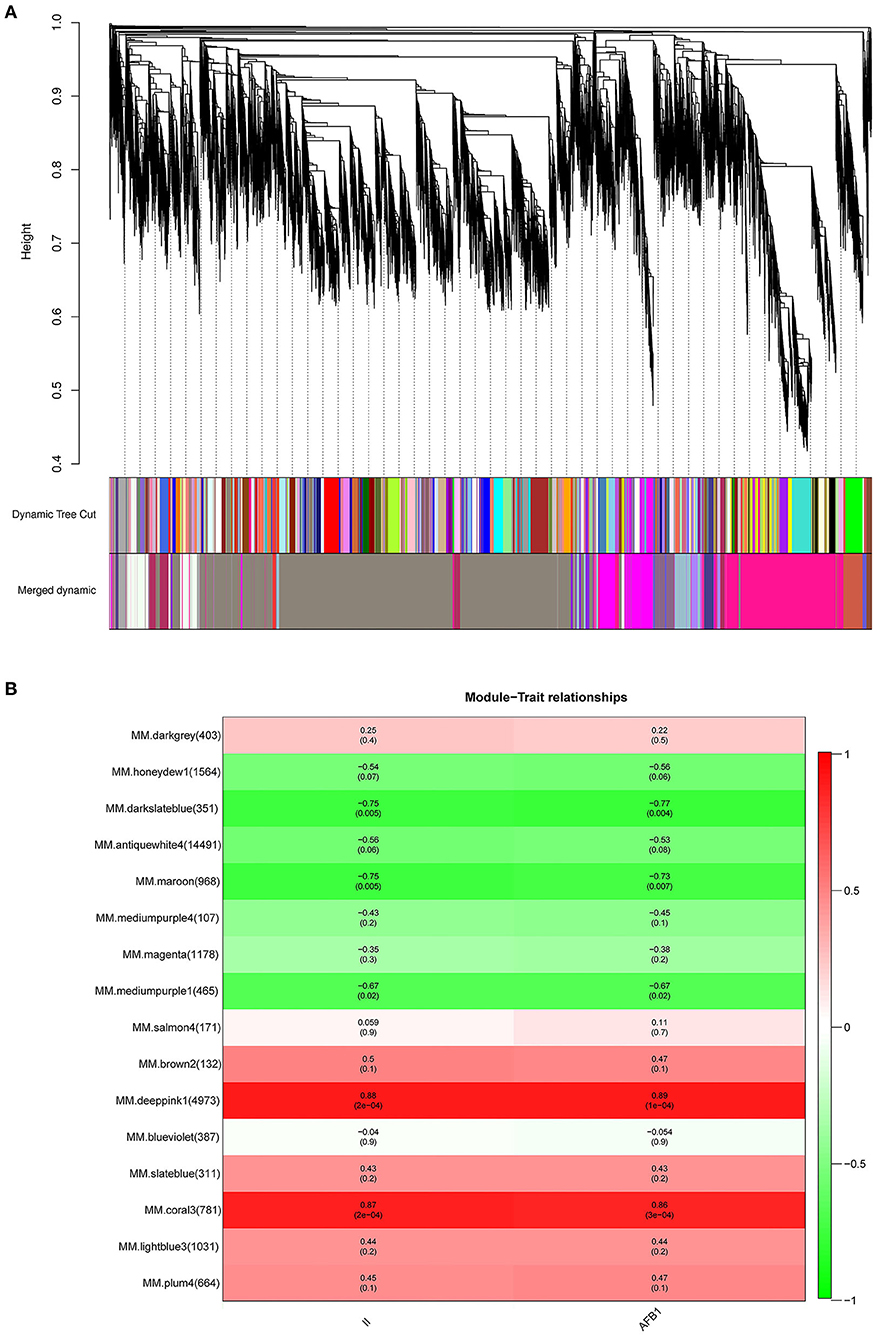
Figure 5. Co-expression network analysis of DEGs and module-trait relationship (MTRs) in response to A. flavus. (A) Cluster dendrogram of different genes in co-expression modules. (B) Relationships between modules (left) and traits (bottom). The numbers in brackets on the left show the number of genes in each module. Red and green represent positive and negative correlations, respectively. The darker colors indicate higher correlation coefficients. Numbers represent Pearson's correlation coefficients r2-values and the P-value for the correlation (in brackets).
To characterize the key modules associated with A. flavus infection and aflatoxin production in peanut seeds, the module-trait relationships (MTRs) were analyzed subsequently (Figure 5B). Modules with MTR > 0.7 were selected as the key ones that were significantly associated with the growth and reproduction of A. flavus in seeds. Obviously, deeppink1 and coral3 were positively correlated with the infection index (II) and AFB1 content, whereas darkslateblue (r2 = −0.75/−0.77) and maroon (r2 = −0.75/−0.73) were negatively correlated with the corresponding traits, implying that DEGs in darkslateblue and maroon may act positively in inhibiting the growth of A. flavus and aflatoxin production in seeds. Subsequently, sample expression patterns were clustered and visualized by heatmap to clearly understand the expression of genes in modules at different time points after inoculation by A. flavus. As shown in Figure 6, expression level of genes in maroon and Salmon4 was increasing in R but with opposite trends in S with the extension of time after inoculation of A. flavus, and was higher in R than in S from T1 to T7, implying their positive roles in A. flavus resistance in R. Whereas the genes in darkslateblue expressed more strongly in S than in R from T1 to T3, suggesting its relatively low correlation with A. flavus defense. Heatmap of module-module relationship showed that maroon was significantly negatively correlated with deeppink1 (Supplementary Figure S8). In addition, plum 4 showed a significantly positive correlation with light-blue3 and coral3 but a negative correlation with magenta. And light-blue3 was significantly correlated with magenta and medium-purple1. Whereas there were no significant positive or negative correlation modules found with salmon4, implying the specificity of the module. All in all, it appears that genes in maroon and salmon4 may be the ones closely associated with resistance to A. flavus stress.
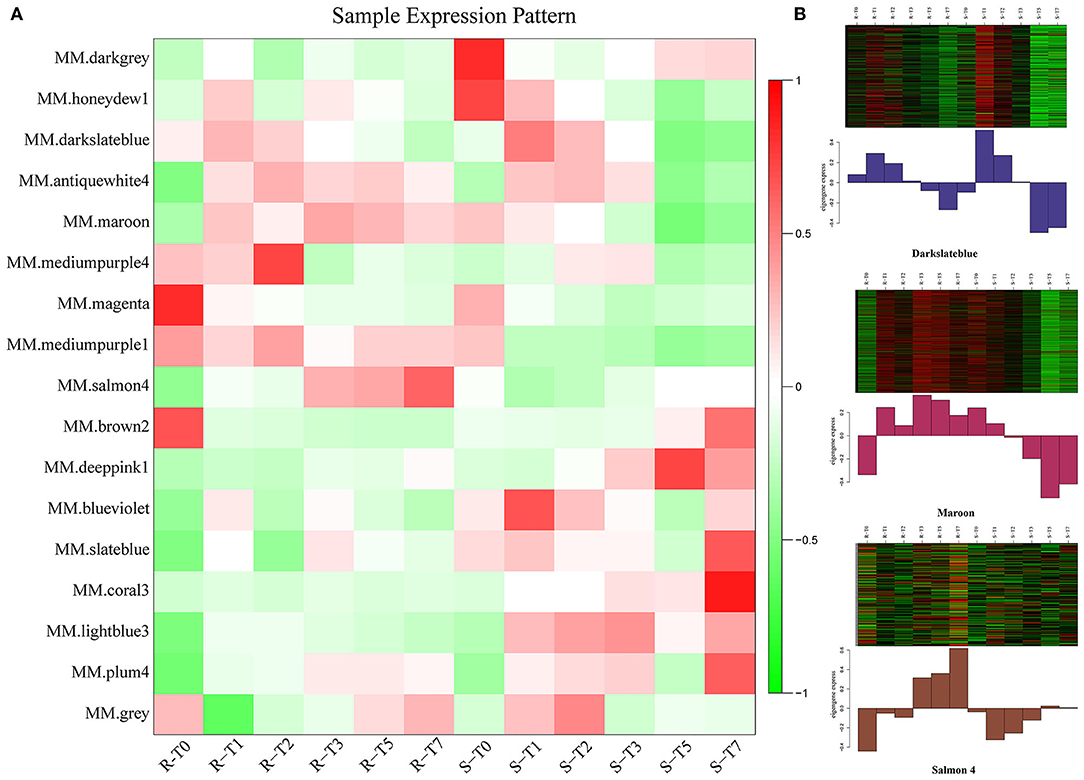
Figure 6. Expression patterns of genes in modules. (A) Heatmap of sample expression pattern. (B) Expression patterns in key modules at different time-points post-inoculation.
GO and KEGG Enrichment Analysis of the Key Modules
GO and KEGG analysis of DEGs from the two key modules, salmon4 and maroon, was performed to clarify the specific functions of each module. GO analysis indicated that “response to biotic stimulus,” “defense response,” “response to stress,” “oxidoreductase activity,” and “protein kinase activity” were the most significantly enriched terms in Salman 4 (Supplementary Figure S9A). KEGG analysis revealed that “MAPK signaling pathway—plant,” “Plant-pathogen interaction,” “Phenylalanine, tyrosine, and tryptophan biosynthesis,” “Oxidative phosphorylation,” and “Plant hormone signal transduction” as the most significantly enriched metabolic pathways, indicating that genes in Salman 4 conferred the resistance to A. flavus by regulating “MAPK signaling pathway,” “Plant-pathogen interaction,” “Phenylalanine,” and “Oxidative phosphorylation,” and “Plant hormone signal transduction.” In Maroon, GO analysis identified “zinc ion binding,” “pyrophosphatase activity,” “hydrolase activity,” “cellular response to DNA damage stimulus,” “intracellular transport” as the most significantly enriched categories, and KEGG analysis identified “mRNA surveillance pathway,” “Basal transcription factors,” “Base excision repair,” “Glycine, serine and threonine metabolism,” and “Ubiquitin mediated proteolysis” as the most significantly enriched metabolic pathways, which showed that genes in Maroon module contribute to resistance of A. flavus by regulating mRNA surveillance, intracellular transport, and ubiquitin-mediated proteolysis (Supplementary Figure S9B).
Hub Genes Involved in Resistance to A. flavus Screened via WGCNA
To identify the key genes associated with resistance to A. flavus in salmon4 and maroon, gene network analysis was conducted by CYTOSCAPE software (the first 2,000 edges) (Supplementary Tables S4, S5). After removing the unknown genes, the top 20 genes with the largest hubness with others were regarded as “hub genes” and shown as red nodes (Figure 7). Further information of the other genes in salmon4 and maroon is provided in Supplementary Tables S4, S5.
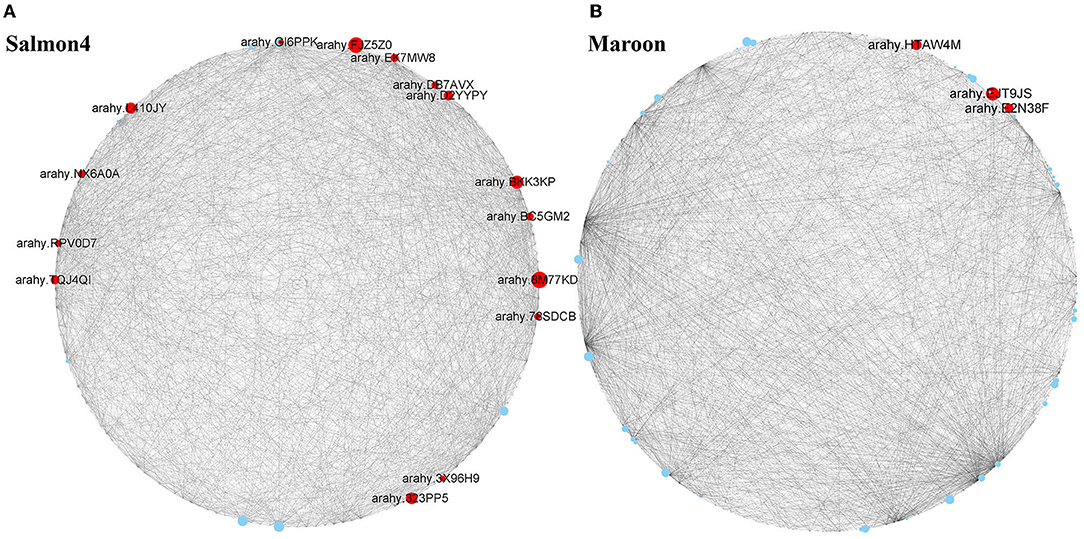
Figure 7. Hub genes identified in maroon and salmon4. Co-expressed network analysis of salmon4 module (A) and maroon module (B). The size of the node circle is positively correlated with the number of genes it interacts with. Hub genes are shown as red nodes.
According to the function annotated in the reference genome, and annotation related to pathogen resistance (Ruperti et al., 2001; Assaad, 2004; Hollenstein et al., 2007; Jérme et al., 2007; Vierstra, 2009; Jayaprakash et al., 2019; Pandey et al., 2019; Zhao et al., 2019; Soni et al., 2020, 2021), 18 hub genes in the two modules were selected (Figure 7 and Table 1). In salmon4, six genes encoding pathogenesis-related 10 protein (PR10, Arahy. 8M77KD, arahy. FJZ5Z0, arahy. BKK3KP, arahy. TQJ4QI, arahy. EK7MW8 and arahy. 3X96H9) was characterized as members of Bet V 1 family protein, which were reported to function in degrading microbial nucleic acid for its ribonuclease activity (Bufe et al., 1996; Agarwal et al., 2013). Arahy.78SDCB and Arahy.GI6PPK are the ones encoding 1-aminocyclopropane-1-carboxylate oxidase (ACO1), which catalyze the formation of ethylene from 1-aminocyclopropane-1-carboxylic acid (ACC) and play key roles in ethylene signaling pathway (Johnson and Ecker, 1998; Bleecker and Kende, 2000; Lin et al., 2009). Arahy. L410JY and arahy. BC5GM2 encode mitogen-activated protein kinase (MAPK protein) in MAPK cascades, which could sense the extracellular stimuli and conduct signaling transduction process (Zhang and Klessig, 2001). arahy. D2YYPY encodes serine/threonine kinase, which is one of the major types of disease resistance proteins (R) (Heierhorst et al., 2000). In addition, gene encoding pattern-recognition receptors (PRRs, arahy. RPV0D7), cytochrome P450 (arahy. DB7AVX), syntaxin of plants 121 (SYP121, Arahy. NX6A0A), and pectinesterase (Arahy. 323PP5) were also identified as hub genes with resistance to A. flavus. Three genes were identified in maroon. Arahy. HTAW4M and arahy. E2N38F encode phosphatidylinositol transfer family protein (PITPs), which is a class of proteins ubiquitous in eukaryotes that can promote the transfer of lipid molecules between intracellular membrane components, and participate in the signal transduction process of plant stresses (Phillips et al., 2006; Thole and Nielsen, 2008; Ghosh and Bankaitis, 2011). Arahy. EJT9JS is the one encodes pentatricopeptide repeat (PPR) superfamily protein that mainly regulates the expression of genes related to plant stress resistance through post-transcriptional modification of RNA (Small and Peeters, 2000; Ichinose and Sugita, 2017). All in all, 18 hub genes were identified as hub (key) genes that acted positively in resistance to A. flavus in peanuts and were classified into 10 categories (Table 1).
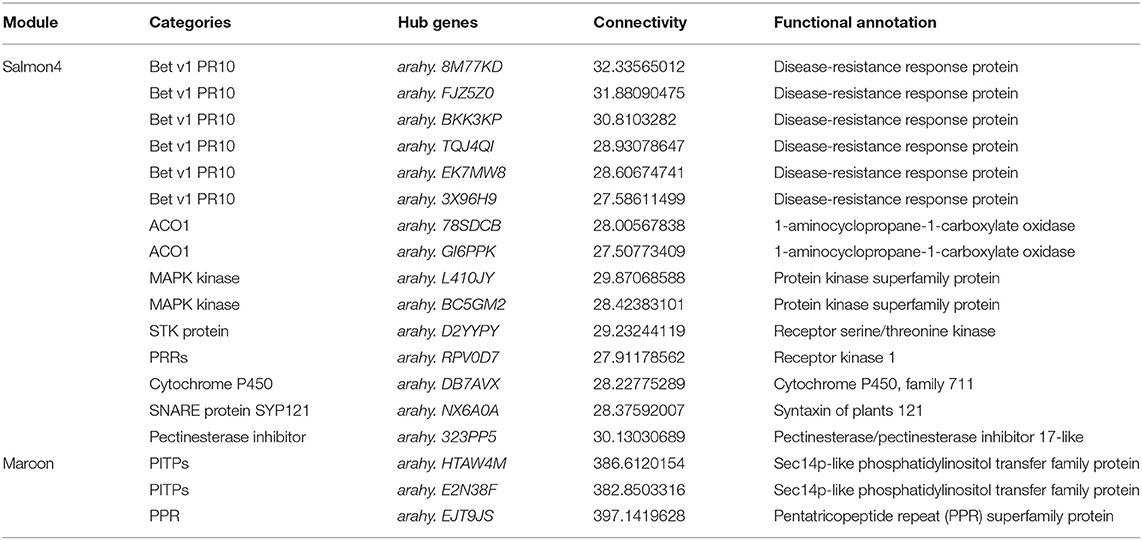
Table 1. Hub genes involved in resistance to A. flavus.
Quantitative Real-Time PCR Validation of RNA-Seq Data
To validate the reliability of the RNA-seq data and differential expression level data, 12 genes were selected randomly from the DEGs to perform quantitative RT-qPCR (Figure 8). As shown in Figure 8, RT-qPCR detected the same expression tendency with the RNA-seq analysis. The validation experiments demonstrated that RNA-seq used in this study was highly reliable.
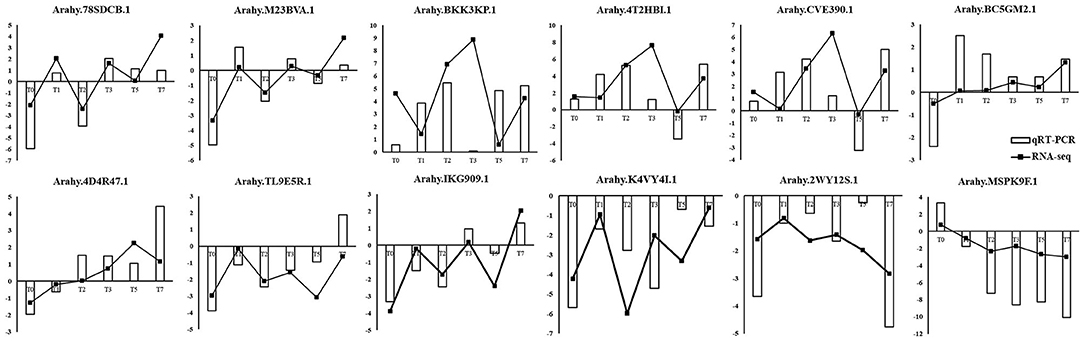
Figure 8. Validation of RNA-seq data by RT-qPCR. Y-axis showed the log2(R/S) between R and S. Positive value indicated upregulated in R and negative value indicated downregulated in R.
Discussion
Co-Expression Networks Were Constructed From Two Cultivated Peanuts With Differing Resistance to Aflatoxin Contamination Using the WGCNA Method
Plants are often subjected to a variety of biotic and abiotic stresses, of which A. flavus and subsequent aflatoxin contamination is an important example of biotic ones affecting food safety and peanut industry (Wild and Gong, 2010; Bryden, 2012; Sarma et al., 2017; Soni et al., 2020). Whereas the mechanism of resistance to A. flavus has not been elucidated and no hub resistance associated gene have been mined because of the lack of systematic screening method. In recent years, WGCNA have been recognized as an efficient method exploring hub genes related to certain traits in various plants (Amrine et al., 2015; Hopper et al., 2016; Kost et al., 2017; Tan et al., 2017; Lin et al., 2019; Wu et al., 2020).
In this study, we confirmed that R and S exhibit different responses to A. flavus stress based on RNA-seq data and morphological studies. The R and S cultivars exhibited reverse response phenotypes during the process of A. flavus puncturing kernel (Figure 1). Enrichment analysis of uniquely upregulated DEGs in R compared with S implied that high reactivity in R in T1 and T2 compared with other time points of inoculation. R genotype may respond more quickly than S genotype and prepare for the transcription and translation of resistance-related genes during the initial stage of infection. In addition, S had more DEGs than R regardless of the post-inoculation time except for T2, indicating that S needs to trigger more metabolic responsive processes and more genes to cope with the stress, which may owe to the lack of the coordination mechanism in S to adapt to stress. The responses shown by R and S during A. flavus infection process may elaborate differences for their resistance.
Moreover, modules closely associated with resistance to A. flavus were identified by WGCNA for the first time, and genes with the highest correlation with others were found and characterized as hub genes (Figure 7). Differential expression patterns of hub genes may lead to the obvious differences in phenotypes.
Aspergillus flavus L. Perception and Recognition by PRRs in Peanut
Pattern recognition receptors (PRRs) play an essential role in pattern-triggered immunity (PTI) response (Shiu and Bleecker, 2001; Couto et al., 2016; Boutrot and Zipfel, 2017) for the detection of pathogen-associated molecular patterns (PAMPs) and initiation of immune signaling transduction (Jones and Dangl, 2006; Cao, 2016) by binding with various co-receptors, receptor-like kinases (RLK), and receptor-like cytoplasmic kinases (RLCK). And some studies also showed that PRRs, brassinosteroid insensitive 1-associated kinase 1 (BAK1) (Li et al., 2012; Zhang et al., 2020), fagellin sensitive 2 (FLS2) (Zhang et al., 2017), EF-Tu receptor (EFR) (Kim et al., 2011), chitin elicitor receptor kinase (CERK) (Petutschnig et al., 2010), and chitin elicitor-binding protein (CEBiP) (Dodds and Rathjen, 2010) were upregulated in the resistant lines of crops, thus acting positively in resistance to pathogens. In this study, one PRR gene, arahy. RPV0D7, was identified as hub gene that acted positively in the defense of A. flavus. It is inferred that peanuts can identify PAMPs of A. flavus, and induce PTI quickly when infected by A. flavus, and the rapid and active response of PRRs in R may be the major reason for the difference in the phenotypes between R and S.
Serine/Threonine Kinase Disease Resistant (R) Genes
Serine/threonine kinase (STK) is one of the major types of disease resistance proteins (R), which are sorted into other four categories, namely, detoxifying enzymes, NB-LRR proteins, transmembrane receptor protein with leucine-rich repeat structure, and protein kinase with leucine-rich repeat structure (Pamela, 1997; Heierhorst et al., 2000). In previous research, Xa21 (Song et al., 1995), Pto (Martin et al., 1993), and Lr10 (Feuillet et al., 1997) were reported to belong to the STK group and were considered as candidate genes to induce resistance against diseases. Arahy.D2YYPY.1 from salmon 4 module was identified as R gene by WGCNA, suggesting that R genotype triggered stronger ETI responses than S at the initial stages of stress and conferred resistance against A. flavus.
Pathogenesis-Related Proteins
Previous studies have shown that PR family genes, especially PR10 genes, can enhance the resistance against both biotic and abiotic stresses in plants (Wan et al., 2008; Gupta et al., 2013), such as sugarcane (Peng et al., 2017), plum (El-Kereamy et al., 2008), and maize (Xie et al., 2010). In this study, six PR10 genes were identified as hub genes by WGCNA analysis from Salmon 4 and showed continuous upregulation from T1 to T7 in R vs. S genotype, signifying that PR10 members were closely related to peanut seed resistance to A. flavus stress (Figure 7). Based on our data, it is speculated that the upregulation of PR10 in R genotype from T1 to T7 may contribute to inhibiting the growth of A. flavus in the seed.
The Other Key Genes Involved in Resistance to A. flavus
It is reported that phosphorylation cascades, containing calcium-dependent protein kinases (CDPKs) and mitogen-activated protein kinase (MAPK), play a major role in PRR-derived signals transmission (Young et al., 2010; Saijo et al., 2018). Correspondingly, in the process of peanut resistance to A. flavus, there were two hub MAPK genes (arahy. L410JY, arahy. BC5GM2) detected, which were upregulated in R compared with S (Table 1). Another important hub resistance-associated gene set identified was cytochrome P450 (arahy. DB7AVX). Gunupuru et al. (2018) reported that TaCYP72A, one of the members of cytochrome P450 genes in wheat, indirectly improved the early resistance of wheat to F. graminearum. Likewise, GbCYP86A1-1 gene in Gossypium barbadense was conformed to confer resistance against Verticillium dahlia by significantly increasing the expression of disease-associated genes (Wang et al., 2020). In addition, as a key enzyme in ethylene biosynthetic pathway, 1-aminocyclopropane-1-carboxylic acid (ACC) is widely used as a proxy for ethylene, given that nearly all plant tissues readily convert it into ethylene (Yu et al., 1979). Helliwell et al. (2013) reported that overexpression of the ACC synthase gene (OsACS2) significantly increases the endogenous ethylene content of rice and enhances rice sheath blight resistance. Two ACC genes (Arahy.78SDCB.1 and Arahy.GI6PPK.1) were characterized as hub genes associated with A. flavus stress in peanut, suggesting that ethylene-mediated resistance responses may play an unignored role in resistance to A. flavus. Another hub gene identified in salmon4 was arahy. 323PP5 encoding pectinesterase or pectinesterase inhibitor. It is reported that pectin is the main component of plant cell wall, exists in the intercellular layer and the middle layer, and affects the rheology and adhesion properties of cells. Overexpression of AtPMEI2 and AtPMEI3 enhanced the resistance to Botrytis cinerea (Lionetti et al., 2017). Yang et al. (2019) reported that the overexpression of poplar PtoPME35 in Arabidopsis thaliana controls stomatal opening and closing of leaf under mannitol stress, thereby regulating plant stress resistance (Yang et al., 2019). In Arabidopsis, SYP121, also known as SYNTAXIN RELATED PROTEIN1/PENETRATION1 (PEN1), is one of the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins and encodes syntaxin that has been shown to reside on the plasma membrane (Collins et al., 2003) and is more specifically involved in the polarized secretion events that give rise to papilla formation during fungi attack in Arabidopsis (Assaad, 2004). One hub gene encoding SYP121 (Arahy.NX6A0A.1) identified from Salmon 4 module by WGCNA analysis implied that a more severe papilla response exists in R compared with S genotype, thus eventually preventing the A. flavus infection in kernels.
In addition, two genes annotated as member of sec14p-like phosphatidylinositol transfer family (PITPS, arahy. HTAW4M and arahy. E2N38F) were identified by the WGCNA method from DEGs (Table 1). As one of the proteins responsive for the transferring of lipid molecules between intracellular membrane components, PITPs were reported to be involved in the signal transduction process of plant stresses (Thole and Nielsen, 2008). Therefore, arahy. HTAW4M, arahy. E2N38F may also be a core factor in the responsive process. PPR is a type of protein containing PPRs (Small and Peeters, 2000). As a trans-acting factor, PPRs mainly regulate the expression of genes related to plant growth and development through post-transcriptional modification of RNA (Ichinose and Sugita, 2017). Studies have shown that stresses usually cause severe damage to the structure and function of plant mitochondria, and PPR protein can regulate mRNA processing by editing and splicing mitochondrial RNA, and thus playing an indispensable role in the response of plants to stress (Umbach et al., 2005; Baldwin and Dombrowski, 2006; Ma et al., 2006; Yan et al., 2006; Baxter et al., 2007). Arabidopsis AtPPR96 is involved in mediating oxidative stress responses (Liu et al., 2016), and overexpression of the Arabidopsis PPR gene SOAR1 can enhance the tolerance to salt, drought, and chilling damage (Tan et al., 2014; Jiang et al., 2015; Xing et al., 2018). Similarly, the hub gene arahy. EJT9JS encoding PRR gene was upregulated in R relative to S genotype, implying the positive regulation in resistance to A. flavus.
Overall, 18 hub genes identified from salmon4 and maroon were the candidate genes in resistance to A. flavus stress. It is inferred that peanuts can identify PAMPs of A. flavus, and induce PTI and subsequently ETI when infected by A. flavus (Figure 9). The rapid and active response of PRRs, R genes, and other genes involved in a series of signaling pathways in R may be the major reason for the difference in the phenotypes between R and S. Certain regulatory function of hub genes will be further investigated in the future study.
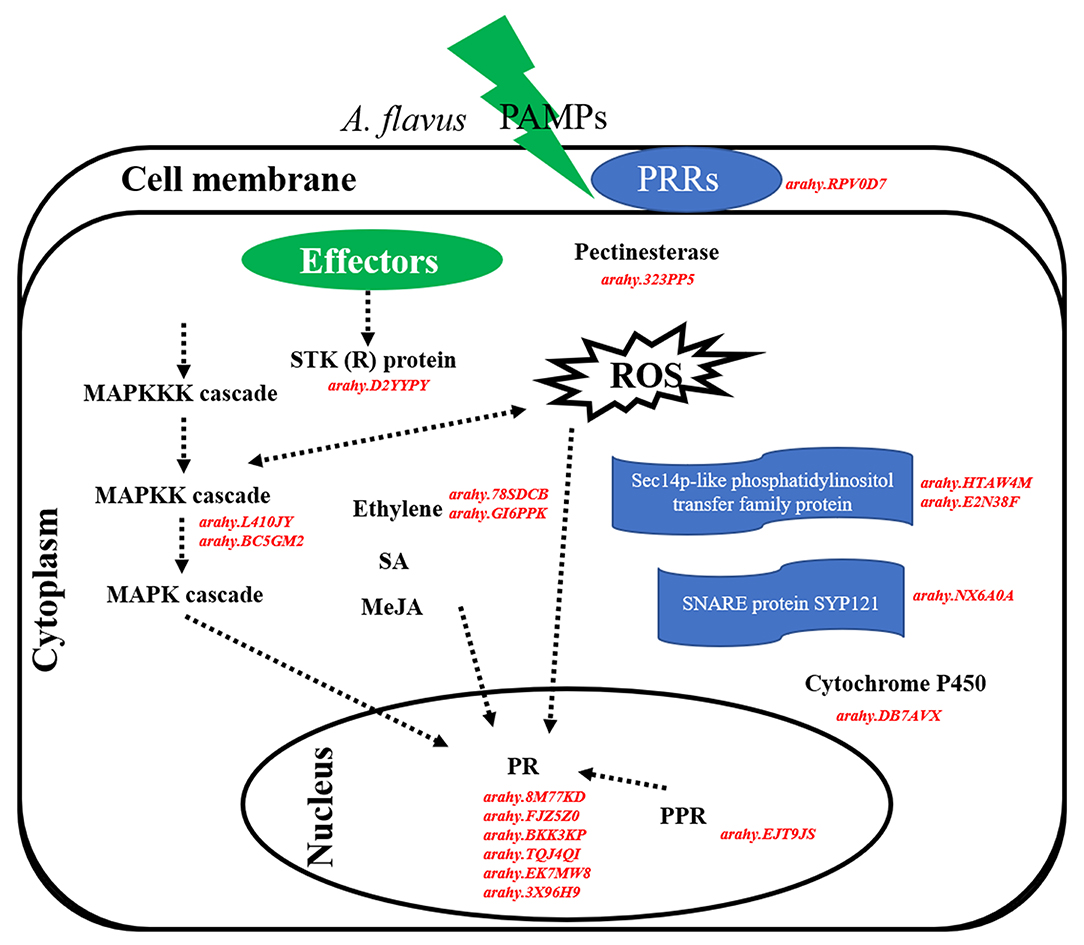
Figure 9. Hypothetical pathways and reactions present in response of peanut seeds to A. flavus at the gene expression level. The components represented in red color are hub genes resistance to A. flavus stress identified in our study. In brief, PAMPs of A. flavus combined with PRRs at the cell membrane and activate PTI response in peanut to limit the growth and reproduction of A. flavus. Subsequently, effectors of A. flavus were released into the cell and were recognized by R proteins (STK), thus triggering ETI response. In this defense responsive mechanism, PR10, pathogenesis-related proteins; ACO1, 1-aminocyclopropane-1-carboxylate oxidase; MAPK kinase, STK, serine/threonine kinase, PRRs, cytochrome P450, SNARE protein SYP121, pectinesterase, phosphatidylinositol transfer protein, and PPR protein are expressed during A. flavus infection process and play main role in the resistance mechanism.
Conclusion
A total of 18 genes were identified, which might be associated with resistance to A. flavus in peanut. The upregulation of genes encoding pathogenesis-related proteins (PR10), 1-aminocyclopropane-1-carboxylate oxidase (ACO1), MAPK kinase, STK, PRRs, cytochrome P450, SNARE protein SYP121, pectinesterase, phosphatidylinositol transfer protein, and PPR protein involved in PTI and ETI response in R compared with S from T3 to T7 may be the cause of R showing resistance to A. flavus. Our study provides a new insight into future peanut breeding and development of A. flavus resistant peanut varieties to mitigate aflatoxin contamination for food safety and peanut industry.
Data Availability Statement
The data presented in the study are deposited in the NCBI Sequence Read Archive (SRA) repository, accession number PRJNA825125.
Author Contributions
MC, SH, and XZ conceived the study and research plans. MC, JG, and DW collected plant materials and performed the experiments. MC, QZ, and MH analyzed the data. PD, ZS, and FQ participated in handling figures and tables. MC, ZZ, and BH drafted the manuscript. WD, PL, and XZ revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the China Agriculture Research System (CARS-13), the Key Scientific and Technological Project of Henan Province (201300111000), and the Henan Provincial Agriculture Research System, China (S2012-5).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.899177/full#supplementary-material
Supplementary Figure S1. Distribution of sequencing saturation of S-T5 and S-T7.
Supplementary Figure S2. Pairwise Pearson's correlation coefficients of the sequencing data of 36 samples.
Supplementary Figure S3. Principal component analysis of the sequencing data of 36 samples.
Supplementary Figure S4. Top 30 GO term enriched functional categories of DEGs upregulated uniquely in R at T1 (A), T2 (B), T3 (C), T5 (D), and T7 (E).
Supplementary Figure S5. Top 30 of KEGG enrichment of DEGs upregulated uniquely in R at T1 (A), T2 (B), T3 (C), T5 (D), and T7 (E).
Supplementary Figure S6. Top 30 GO term enriched functional categories of DEGs upregulated uniquely in S at T1 (A), T2 (B), T3 (C), T5 (D), and T7 (E).
Supplementary Figure S7. Top 30 of KEGG enrichment of DEGs up-regulated uniquely in S at T1 (A), T2 (B), T3 (C), T5 (D), and T7 (E).
Supplementary Figure S8. Heat map of module-module relationship.
Supplementary Figure S9. Top 30 GO term enriched functional categories and top 30 KEGG enrichments of DEGs in salmon4 and maroon.
Supplementary Table S1. Primers used for RT-qPCR analysis.
Supplementary Table S2. Detail information of sequencing reads from different sample groups.
Supplementary Table S3. Gene number of each module.
Supplementary Table S4. Connectivity of genes in salmon4.
Supplementary Table S5. Connectivity of genes in maroon.
Abbreviations
ACO1, Aminocyclopropane-1-carboxylate oxidase; A. flavus, Aspergillus flavus L.; AP, Aflatoxin production; BAK1, Brassinosteroid insensitive 1-associated kinase 1; CDPKs, calcium-dependent protein kinases; CEBiP, chitin elicitor-binding protein; CERK, Chitin elicitor receptor kinase; DEGs, Differentially expressed genes; EFR, EF-Tu receptor; ETI, Effector-triggered immunity; FLS2, Fagellin sensitive 2; GO, Gene Ontology; HPLC, High performance liquid chromatography; II, Infection index; IVSC, In vitro seed colonization; KEGG, Kyoto Encyclopedia of Genes and Genomes; MAPK, Mitogen-activated protein kinase; PAMPs, pathogen-associated molecular patterns; PCA, principal component analyses; PRs, Pathogenesis-related proteins; PRRs, Pattern recognition receptors; PTI, pattern-triggered immunity; QTLs, Quantitative trait locus; R, Resistant; RLCK, receptor-like cytoplasmic kinases; RLK, receptor-like kinases; RT-qPCR, Real-time quantitative polymerase chain reaction; S, Susceptible; SNPs, Single nucleotide polymorphisms; STK, Serine/threonine kinase; SYP121, Syntaxin of plants 121; TF, Transcription factor; WGCNA, Weighted gene co-expression network analysis.
References
Agarwal, P., Bhatt, V., Singh, R., Das, M., Sopory, S. K., and Chikara, J. (2013). Pathogenesis-related gene, JcPR-10a from Jatropha curcas exhibit RNase and antifungal activity. Mol. Biotech. 54, 412–425. doi: 10.1007/s12033-012-9579-7
Amrine, K. C., Blanco-Ulate, B., and Cantu, D. (2015). Discovery of core biotic stress responsive genes in Arabidopsis by weighted gene co-expression network analysis. PLoS ONE 10, e0118731. doi: 10.1371/journal.pone.0118731
Assaad, F. F. (2004). The PEN1 syntaxin defines a novel cellular compartment upon fungal attack and is required for the timely assembly of papillae. Mol. Biol. Cell 15, 5118–5129. doi: 10.1091/mbc.E04-02-0140
Baldwin, J. C., and Dombrowski, J. E. (2006). Evaluation of Lolium temulentum as a model grass species for the study of salinity stress by PCR-based subtractive suppression hybridization analysis. Plant Sci. 171, 459–469. doi: 10.1016/j.plantsci.2006.05.003
Baxter, C. J., Redestig, H., Schauer, N., Repsilber, D., Patil, K. R., Nielsen, J., et al. (2007). The metabolic response of heterotrophic Arabidopsis cells to oxidative stress. Plant Physiol. 143, 312–325. doi: 10.1104/pp.106.090431
Bleecker, A. B., and Kende, H. (2000). Ethylene: a gaseous signal molecule in plants. Annu. Rev. Cell Dev. Biol. 16, 1–18. doi: 10.1146/annurev.cellbio.16.1.1
Boutrot, F., and Zipfel, C. (2017). Function, discovery, and exploitation of plant pattern recognition receptors for broad-Spectrum disease resistance. Annu. Rev. Phytopathol. 55, 257–286. doi: 10.1146/annurev-phyto-080614-120106
Brand, Y., and Hovav, R. (2010). Identification of suitable internal control genes for quantitative real-time PCR expression analyses in peanut (Arachis hypogaea L.). Peanut Sci. 37, 12–19. doi: 10.3146/PS09-014.1
Bryden, W. L. (2012). Mycotoxin contamination of the feed supply chain: implications for animal productivity and feed security. Anim. Feed Sci. Tech. 173, 134–158. doi: 10.1016/j.anifeedsci.2011.12.014
Bufe, A., Spangfort, M. D., Kahlert, H., Schlaak, M., and Becker, W. (1996). The major birch pollen allergen, Bet v1, shows ribonuclease activity. Planta 199, 413–415. doi: 10.1007/bf00195733
Cao, X. (2016). Self-regulation and cross-regulation of pattern-recognition receptor signaling in health and disease. Nat. Rev. Immunol. 16, 35–50. doi: 10.1038/nri.2015.8
Chen, Y., Ren, X., Zhou, X., Huang, L., Yan, L., Lei, Y., et al. (2014). Dynamics in the resistant and susceptible peanut (Arachis hypogaea L.) root transcriptome on infection with the Ralstonia solanacearum. BMC Genom. 15, 1078–1093. doi: 10.1186/1471-2164-15-1078
Clevenger, J., Marasigan, K., Liakos, V., Sobolev, V., Vellidis, G., Holbrook, C., et al. (2016). RNA sequencing of contaminated seeds reveals the state of the seed permissive for pre-harvest aflatoxin contamination and points to a potential susceptibility factor. Toxins 8, 317–334. doi: 10.3390/toxins8110317
Collins, N. C., Christensen, H. T., Lipka, V.olker., Bau, S., Kombrink, E., Qiu, J. L., et al. (2003). SNARE-protein-mediated disease resistance at the plant cell wall. Nature 425, 973–977. doi: 10.1038/nature02076
Couto, D., Niebergall, R., Liang, X., Bücherl, C. A., Sklenar, J., Macho, A. P., et al. (2016). The Arabidopsis protein phosphatase PP2C38 negatively regulates the central immune kinase BIK1. PLoS Pathog. 12, e1005811. doi: 10.1371/journal.ppat.1005811
Cui, M., Muhammad, H., Chai, P., Guo, J., Du, P., Li, H., et al. (2021). Genome-wide identification and expression analysis of AP2/ERF transcription factor related to drought stress in cultivated peanut (Arachis hypogaea L.). Front. Genet. 12, 750761–750761. doi: 10.3389/fgene.2021.750761
Dewey, C. N., and Li, B. (2011). RSEM, accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323–338. doi: 10.1186/1471-2105-12-323
Ding, X., Li, P., Bai, Y., and Zhou, H. (2012). Aflatoxin B1 in post-harvest peanuts and dietary risk in China. Food Control 23, 143–148. doi: 10.1016/j.foodcont.2011.06.026
Dixon, R. A., and Paiva, N. L. (1995). Stress-induced phenylpropanoid metabolism. Plant Cell 7, 1085–1097. doi: 10.org/10.1105/tpc.7.7.1085
Dodds, P. N., and Rathjen, J. P. (2010). Plant immunity: towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 11, 539–548. doi: 10.1038/nrg2812
El-Kereamy, A., Jayasankar, S., Taheri, A., Errampalli, D., and Paliyath, G. (2008). Expression analysis of a plum pathogenesis related 10 (PR10) protein during brown rot infection. Plant Cell Rep. 28, 95–102. doi: 10.1007/s00299-008-0612-z
Feuillet, C., Schachermayr, G., and Keller, B. (1997). Molecular cloning of a new receptor-like kinase gene encoded at the Lr10 disease resistance locus of wheat. Plant J. 11, 45–52. doi: 10.1046/j.1365-313X.1997.11010045.x
Ghosh, R., and Bankaitis, V. A. (2011). Phosphatidylinositol transfer proteins: negotiating the regulatory interface between lipid metabolism and lipid signaling in diverse cellular processes. Biofactors 37, 290–308. doi: 10.1002/biof.180
Guimaraes, P. M., Brasileiro, A. C. M., Morgante, C. V., Martins, A. C., Pappas, G., Silva, O. B., et al. (2012). Global transcriptome analysis of two wild relatives of peanut under drought and fungi infection. BMC Genomics 13, 387–401. doi: 10.1186/1471-2164-13-387
Gunupuru, L. R., Arunachalam, C., Malla, K. B., Kahla, A., Perochon, A., Jia, J., et al. (2018). A wheat cytochrome P450 enhances both resistance to deoxynivalenol and grain yield. PLoS ONE 13, e0204992. doi: 10.1371/journal.pone.0204992
Guo, B., Chen, X., Dang, P., Scully, B. T., and Culbreath, A. K. (2008a). Peanut gene expression profiling in developing seeds at different reproduction stages during Aspergillus parasiticus infection. BMC dev. Biol. 8, 12–17. doi: 10.1186/1471-213X-8-12
Guo, B., Chen, Z. Y., Lee, R. D., and Scully, B. T. (2008b). Drought stress and preharvest aflatoxin contamination in agricultural commodity: genetics, genomics and proteomics. J. Integr. Plant Biol. 50, 1281–1291. doi: 10.1111/j.1744-7909.2008.00739.x
Guo, B., Fedorova, N. D., Chen, X., Wan, C. H., Wang, W., Nierman, W. C., et al. (2011). Gene expression profiling and identification of resistance genes to Aspergillus flavus infection in peanut through EST and microarray strategies. Toxins 3, 737–753. doi: 10.3390/toxins3070737
Gupta, P., Ravi, I., and Sharma, V. (2013). Induction of b-1,3-glucanase and chitinase activity in the defense response of Eruca sativa plants against the fungal pathogen Alternaria brassicicola. J. Plant Interact. 8, 155–161. doi: 10.1080/17429145.2012.679705
Heierhorst, J., Pearson, R., Horne, J., Bozinovski, S., and Kemp, B. E. (2000). Protein serine/threonine kinases. Annu. Rev. Biochem. 56, 567–613.
Helliwell, E. E., Wang, Q., and Yang, Y. (2013). Transgenic rice with inducible ethylene production exhibits broad-spectrumdisease resistance to the fungal pathogens Magnaporthe oryzae and rhizoctonia solani. Plant Biotechnol. J. 11, 33–42. doi: 10.1111/pbi.12004
Hollenstein, K., Frei, D. C., and Locher, K. P. (2007). Structure of an ABC transporter in complex with its binding protein. Nature 446, 213–216. doi: 10.1038/nature05626
Hopper, D. W., Ghan, R., Schlauch, K. A., and Cramer, G. R. (2016). Transcriptomic network analyses of leaf dehydration responses identify highly connected ABA and ethylene signaling hubs in three grapevine species differing in drought tolerance. BMC Plant. Biol. 16, 118–137. doi: 10.1186/s12870-016-0804-6
Ichinose, M., and Sugita, M. (2017). RNA editing and its molecular mechanism in plant organelles. Genes 8, 5–19. doi: 10.3390/genes8010005
Jayaprakash, A., Thanmalagan, R. R., Roy, A., Arunachalam, A., and Ptv, L. (2019). Strategies to understand Aspergillus flavus resistance mechanism in Arachis hypogaea L. Curr. Plant Biol. 20, 100123–100159. doi: 10.1016/j.cpb.2019.100123
Jérme, P., Rustérucci, C., and Mellerowicz, E. J. (2007). New insight into pectin methylesterase structure and function. Trends Plant Sci. 12, 267–277. doi: 10.1016/j.tplants.2007.04.001
Jiang, S. C., Mei, C., Liang, S., Yu, Y. T., Lu, K., Wu, Z., et al. (2015). Crucial roles of the pentatricopeptide repeat protein soar1 in Arabidopsis response to drought, salt and cold stresses. Plant Mol. Biol. 88, 369–385. doi: 10.1007/s11103-015-0327-9
Johnson, P. R., and Ecker, J. R. (1998). The ethylene gas signal transduction pathway: a molecular perspective. Annu. Rev. Genet. 32, 227–254. doi: 10.1146/annurev.genet.32.1.227
Jones, J., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Khan, S. A., Zhang, C., Ali, N., and Gandeka, M. (2020). Highdensity SNP map facilitates fine mapping of QTLs and candidate genes discovery for Aspergillus favus resistance in peanut (Arachis hypogaea L.). Theor. Appl. Genet. 133, 2239–2257. doi: 10.1007/s00122-020-03594-0
Kim, D., Ben, L., Steven, L., and Salzberg. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kim, K. H., Yang, J. K., Dong, H. K., Min, Y. Y., Moon, J. K., Kim, M. Y., et al. (2011). RNA-seq analysis of a soybean near-isogenic line carrying bacterial leaf pustule-resistant and-susceptible alleles. DNA Res. 18, 483–497. doi: 10.1093/dnares/dsr033
Korani, W., Chu, Y., Holbrook, C. C., and Ozias-Akins, P. (2018). Insight into genes regulating postharvest aflatoxin contamination of tetraploid peanut from transcriptional profiling. Genet 209, 143–156. doi: 10.1534/genetics.118.300478
Kost, M. A., Perales, H. R., Wijeratne, S., Wijeratne, A. J., Stockinger, E., and Mercer, K. L. (2017). Differentiated transcriptional signatures in the maize landraces of Chiapas, Mexico. BMC Genom. 18, 707–720. doi: 10.1186/s12864-017-4005-y
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559–571. doi: 10.1186/1471-2105-9-559
Li, C., Li, K., Liu, X., Hui, R., and Yang, S. (2021). Transcription factor GmWRKY46 enhanced phosphate starvation tolerance and root development in transgenic plants. Front. Plant Sci. 12, 700651. doi: 10.3389/fpls.2021.700651
Li, C. Y., Deng, G. M., Yang, J., Viljoen, A., Jin, Y., Kuang, R. B., et al. (2012). Transcriptome profiling of resistant and susceptible Cavendish banana roots following inoculation with Fusariumo xysporum f. sp. cubense tropical race 4. BMC Genom. 13, 374-384. doi: 10.1186/1471-2164-13-374
Li, H., Wu, C., Lv, Q., Shi, T., and Chen, Q. (2020). Comparative cellular, physiological and transcriptome analyses reveal the potential easy dehulling mechanism of rice tartary buckwheat (Fagopyrum Tararicum). BMC Plant Biol. 20, 505–535. doi: 10.21203/rs.3.rs-58157/v1
Li, X., Lu, J., Liu, S., Liu, X., Lin, Y., and Li, L. (2014). Identification of rapidly induced genes in the response of peanut (Arachis hypogaea L.) to water deficit and abscisic acid. BMC Biotechnol. 14, 58–66. doi: 10.1186/1472-6750-14-58
Liang, X., Luo, M., and Guo, B. (2006). Resistance mechanisms to Aspergillus flavus infection and aflatoxin contamination in peanut (Arachis hypogaea L.). Plant Pathol J. 5, 115–124. doi: 10.3923/ppj.2006.115.124
Liang, X., Zhou, G., Hong, Y., Chen, X., Liu, H., and Li, S. (2009). Overview of research progress on peanut (Arachis hypogaea L.) host resistance to aflatoxin contamination and genomics at the Guangdong Academy of Agricultural Sciences. Peanut Sci. 36, 29–34. doi: 10.3146/AT07-003.1
Liao, B., Zhuang, W., Tang, R., Zhang, X., Shan, S., Jiang, H., et al. (2009). Peanut aflatoxin and genomics research in China: progress and perspectives. Peanut Sci. 36, 21–28. doi: 10.3146/at07-004.1
Lin, C. W., Fu, S. F., Liu, Y. J., Chen, C. C., Chang, C. H., Yang, Y. W., et al. (2019). Analysis of ambient temperature-responsive transcriptome in shoot apical meristem of heat-tolerant and heat-sensitive broccoli inbred lines during floral head formation. BMC Plant Biol. 19, 3–18. doi: 10.1186/s12870-018-1613-x
Lin, Z. F., Zhong, S. L., and Grierson, D. (2009). Recent advances in ethylene research. J. Exp. Bot. 60, 3311–3336. doi: 10.1093/jxb/erp204
Lionetti, V., Fa, B. E., Caroli, M. D., Hansen, A. R., Willats, W. G. T., Piro., G., et al. (2017). Three Pectin methylesterase inhibitors protect cell wall integrity for Arabidopsis immunity to botrytis. Plant Physiol. 173, 1844–1863. doi: 10.1104/pp.16.01185
Liu, H., Wu, H., Wang, Y., Wang, H., and Yin, Z. (2021). Comparative transcriptome profiling and co-expression network analysis uncover the key genes associated with early-stage resistance to Aspergillus flavus in maize. BMC Plant Biol. 21, 216–233. doi: 10.1186/s12870-021-02983-x
Liu, J. M., Zhao, J. Y., Lu, P. P., Chen, M., Guo, C., Xu, Z., et al. (2016). The E-subgroup pentatricopeptide repeat protein family in Arabidopsis thaliana and confirmation of the responsiveness PPR96 to abiotic stresses. Front. Plant Sci. 7, 1825. doi: 10.3389/fpls.2016.01825
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–570. doi: 10.1186/s13059-014-0550-8
Luo, M., Dang, P., Guo, B. Z., He, G., Holbrook, C. C., Bausher, M. G., et al. (2005). Generation of expressed sequence tags (ESTs) for gene discovery and marker development in cultivated peanut. Crop Sci. 45, 346–353. doi: 10.2135/cropsci2005.0346
Ma, F., Chen, R., Li, P., Zhang, Q., Zhang, W., and Hu, X. (2013). Preparation of an immunoaffinity column with amino-silica gel microparticles and its application in sample cleanup for aflatoxin detection in agri-products. Molecules 18, 2222–2235. doi: 10.3390/molecules18022222
Ma, S., Gong, Q., and Bohnert, H. J. (2006). Dissecting salt stress pathways. J. Exp. Bot. 57, 1097–1107. doi: 10.1093/jxb/erj098
Martin, G., Brommonschenkel, S., Chunwongse, J., Frary, A., Ganal, M., Spivey, R., et al. (1993). Map based cloning of a protein kinase gene conferring disease resistance in tomato. Science 262, 1432–1436. doi: 10.1126/science.7902614
Menon, V. (2018). Clustering single cells: a review of approaches on high and low-depth single-cell RNA-seq data. Brief Funct. Genomics 17, 240–245. doi: 10.1093/bfgp/elx044
Nayak, S. N., Agarwal, G., Pandey, M. K., Sudini, H. K., Jayale, A. S., Purohit, S., et al. (2017). Aspergillus flavus infection triggered immune responses and host-pathogen cross-talks in groundnut during in-vitro seed colonization. Sci. Rep. 7, 9659–9673. doi: 10.1038/s41598-017-09260-8
Nigam, S. N., Waliyar, F., Aruna, R., Reddy, S. V., Kumar, P. L., Craufurd, P. Q., et al. (2009). Breeding peanut for resistance to aflatoxin contamination at ICRISAT. Peanut Sci. 36, 42–49. doi: 10.3146/AT07-008.1
Pamela, R. (1997). The molecular basis of disease resistance in rice. Plant Mol. Biol. 35, 179–186. doi: 10.1023/A:1005750811983
Pandey, M. K., Kumar, R., Pandey, A. K., Soni, P., Gangurde, S. S., Sudini, H. K., et al. (2019). Mitigating aflatoxin contamination in groundnut through a combination of genetic resistance and post-harvest management practices. Toxins 11, 1–21. doi: 10.3390/toxins11060315
Pandey, M. K., Upadhyaya, H. D., Rathore, A., Vadez, V., Sheshshayee, M. S., Sriswathi, M., et al. (2014). Genome wide association studies for 50 agronomic traits in peanut using the “reference set” comprising 300 genotypes from 48 countries of the semi-arid tropics of the world. PLoS ONE 9, e105228. doi: 10.1371/journal.pone.0113326
Passone, M. A., Ruffino, M., Ponzio, V., Resnik, S., and Etcheverry, M. G. (2009). Postharvest control of peanut Aspergillus section Flavi populations by a formulation of food-grade antioxidants. Int. J. Food Microbiol. 131, 211–217. doi: 10.1016/j.ijfoodmicro.2009.02.023
Peng, Q., Su, Y., Ling, H., Ahmad, W., Gao, S., Guo, J., et al. (2017). A sugarcane pathogenesis-related protein, ScPR10, plays a positive role in defense responses under Sporisorium scitamineum, SrMV, SA, and MeJA stresses. Plant Cell Rep. 36, 1427–1440. doi: 10.1007/s00299-017-2166-4
Petutschnig, E. K., Jones, A. M., Serazetdinova, L., Lipka, U., and Lipka, V. (2010). The lysin motif receptor-like kinase (LysM-RLK) CERK1 is a major chitin-binding protein in Arabidopsis thaliana and subject to chitin-induced phosphorylation. J. Biol. Chem. 285, 28902–28911. doi: 10.1074/jbc.M110.116657
Phillips, S. E., Vincent, P., Rizzieri, K. E., Schaaf, G., Bankaitis, V. A., and Gaucher, E. A. (2006). The diverse biological functions of phosphatidylinositol transfer proteins in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 41, 21–49. doi: 10.1080/10409230500519573
Polturak, G., and Osbourn, A. (2021). Defense-related phenylpropanoid biosynthetic gene clusters in rice. Sci. Bull. 67, 13–16. doi: 10.1016/j.scib.2021.09.021
Prasad, K., Bhatnagar-Mathur, P., Waliyar, F., and Sharma, K. K. (2013). Overexpression of a chitinase gene in transgenic peanut confers enhanced resistance to major soil borne and foliar fungal pathogens. J. Plant Biochem. Biotechnol. 22, 222–233. doi: 10.1007/s13562-012-0155-9
Ruperti, B., Bonghi, C., Rasori, A., Ramina, A., and Tonutti, P. (2001). Characterization and expression of two members of the peach 1-aminocyclopropane-1-carboxylate oxidase gene family. Physiol. Plant 111, 336–344. doi: 10.1034/j.1399-3054.2001.1110311.x
Saijo, Y., Loo, E. P., and Yasuda, S. (2018). Pattern recognition receptors and signaling in plant-microbe interactions. Plant J. 93, 592–613. doi: 10.1111/tpj.13808
Sarma, U. P., Bhetaria, P. J., Devi, P., and Varma, A. (2017). Aflatoxins: implications on health. Indian J. Clin. Biochem. 32, 124–133. doi: 10.1007/s12291-017-0649-2
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shiu, S. H., and Bleecker, A. B. (2001). Receptor-like kinase a from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc. Natl. Acad. Sci. U.S.A. 98, 10763–10768. doi: 10.1073/pnas.181141598
Small, I. D., and Peeters, N. (2000). The PPR motif: A TPR-related motif prevalent in plant organellar proteins. Trends Biochem. Sci. 25, 46–47. doi: 10.1016/S0968-0004(99)01520-0
Song, W. Y., Wang, G. L., and Chen, L. L. (1995). A receptor kinase- like protein encoded by the rice disease resistance gene, Xa21. Science 270, 1804–1806.
Soni, P., Gangurde, S. S., Ortega-Beltran, A., Kumar, R., Parmar, S., Sudini, H. K., et al. (2020). Functional biology and molecular mechanisms of host-pathogen interactions for aflatoxin contamination in groundnut (Arachis hypogaea L.) and maize (Zea mays L.). Front. Microbiol. 11, 227. doi: 10.3389/fmicb.2020.00227
Soni, P., Pandey, A. K., Nayak, S. N., Pandey, M. K., Tolani, P., Pandey, S., et al. (2021). Global transcriptome profiling identified transcription factors, biological process, and associated pathways for pre-harvest aflatoxin contamination in groundnut. J. Fungi 7, 413–430. doi: 10.3390/jof7060413
Tan, J. J., Tan, Z. H., Wu, F. Q., Sheng, P., Heng, Y., Wang, X., et al. (2014). A novel chloroplast-localized pentatricopeptide repeat protein involved in splicing affects chloroplast development and abiotic stress response in rice. Mol. Plant 7, 1329–1349. doi: 10.1093/mp/ssu054
Tan, M., Cheng, D., Yang, Y., Zhang, G., Qin, M., Chen, J., et al. (2017). Co-expression network analysis of the transcriptomes of rice roots exposed to various cadmium stresses reveals universal cadmium-responsive genes. BMC Plant Biol. 17, 194–211. doi: 10.1186/s12870-017-1143-y
Thole, J. M., and Nielsen, E. (2008). Phosphoinositides in plants: novel functions in membrane trafficking. Curr. Opin. Plant Biol. 11, 620–631. doi: 10.1016/j.pbi.2008.10.010
Umbach, A. L., Fiorani, F., and Siedow, J. N. (2005). Characterization of transformed Arabidopsis with altered alternative oxidase levels and analysis of effects on reactive oxygen species in tissue. Plant Physiol. 139, 1806–1820. doi: 10.1104/pp.105.070763
Vierstra, R. D. (2009). The ubiquitin-26S proteasome system at the nexus of plant biology. Nat. Rev. Mol. Cell Biol. 10, 385–397. doi: 10.1038/nrm2688
Walid, K., Chu, Y., Holbrook, C., Clevenger, J., Ozias-Akins, P., and Corley, H. (2017). Genotypic regulation of aflatoxin accumulation but not aspergillus fungal growth upon post-harvest infection of peanut (Arachis hypogaea L.) seeds. Toxins 9, 218–229. doi: 10.3390/toxins9070218
Wan, J., Zhang, X. C., Neece, D., Ramonell, K. M., Clough, S., Kim, S. Y., et al. (2008). A LysM receptor-like kinase plays a critical role in chitin signaling and fungal resistance in Arabidopsis. Plant Cell 20, 471–481. doi: 10.1105/tpc.107.056754
Wang, G., Xu, J., Li, L., Guo, Z., Si, Q., Zhu, G., et al. (2020). GbCYP86A1-1 from Gossypium barbadense positively regulates defense against Verticillium dahliae by cell wall modification and activation of immune pathways. Plant Biotechnol J. 18, 222–238. doi: 10.1111/pbi.13190
Wang, H. M., Lei, Y., Wan, L. Y., Yan, L., Lv, J., Dai, X., et al. (2016). Comparative transcript profiling of resistant and susceptible peanut post-harvest seeds in response to aflatoxin production by Aspergillus flavus. BMC Plant Biol. 2016, 16, 54–69. doi: 10.1186/s12870-016-0738-z
Wang, T., Chen, X. P., Li, H. F., Liu, H., Hong, Y., and Li, L. (2013). Transcriptome identification of the resistance-associated genes (RAGs) to Aspergillus flavus infection in pre-harvested peanut (Arachis hypogaea). Funct. Plant Biol. 40, 292–303. doi: 10.1071/FP12143
Wild, C. P., and Gong, Y. Y. (2010). Mycotoxins and human disease: a largely ignored global health issue. Carcinogenesis 31, 71–82. doi: 10.1093/carcin/bgp264
Wu, Y., Xu, J., Han, X., Qiao, G., Yang, K., Wen, Z., et al. (2020). Comparative transcriptome analysis combining SMRT- and Illumina-based RNA-Seq identifies potential candidate genes involved in betalain biosynthesis in Pitaya fruit. Int. J. Mol Sci. 21, 3288–3304. doi: 10.3390/ijms21093288
Xie, C. Z., Wen, S. J., Liu, H., Chen, X., and Li, H. (2013). Overexpression of ARAhPR10, a member of the PR10 family, decreases levels of Aspergillus flavus infection in peanut seeds. Am. J. Plant Sci. 4, 602–607. doi: 10.4236/ajps.2013.43079
Xie, Y. R., Chen, Z. Y., Brown, R. L., and Bhatnagar, D. (2010). Expression and functional characterization of two pathogenesis-related protein 10 genes from Zea mays. J. Plant Physiol. 167, 121–130. doi: 10.1016/j.jplph.2009.07.004
Xing, H. T., Fu, X. K., Yang, C., Tang, X., Guo, L., Li, C., et al. (2018). Genome-wide investigation of pentatricopeptide repeat gene family in poplar and their expression analysis in response to biotic and abiotic stresses. Sci. Rep. 8, 2817–2826. doi: 10.1038/s41598-018-21269-1
Yan, H. J., Zhao, Y. F., Wang, Y. C., and Tang, D. Z. (2018). BRASSINOSTEROID-SIGNALING KINASE1 phosphorylates MAPKKK5 to regulate immunity in arabidopsis. Plant Physiol. 176, 2991–3002. doi: 10.1104/pp.17.01757
Yan, S. P., Zhang, Q. Y., Tang, Z. C., Su, W. A., and Sun, W. N. (2006). Comparative proteomic analysis provides new insights into chilling stress responses in rice. Mol. Cell. Proteomics 5, 484–496. doi: 10.1074/mcp.M500251-MCP200
Yan, W., Ni, Y., Liu, X., Zhao, H., and Tian, B. (2021). The mechanism of sesame resistance against Macrophomina phaseolina was revealed via a comparison of transcriptomes of resistant and susceptible sesame genotypes. BMC Plant Biol. 21, 159–179. doi: 10.1186/s12870-021-02927-5
Yang, W., Mei, R., Xiang, M., Deng, A., and Xiao, C. (2019). Overexpression of a pectin methylesterase gene PtoPME35 from Populus tomentosa influences stomatal function and drought tolerance in Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 523, 416–422. doi: 10.1016/j.bbrc.2019.12.073
Yin, D., Wang, Y., Zhang, X., Li, H., Lu, X., Zhang, J., et al. (2013). De novo assembly of the peanut (Arachis hypogaea L.) seed transcriptome revealed candidate unigenes for oil accumulation pathways. PLoS ONE 8, e73767. doi: 10.1371/journal.pone.0073767
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14–R25. doi: 10.1186/gb-2010-11-2-r14
Yu, B., Huai, D., Huang, L., Kang, Y., Ren, X., et al. (2019). Identification of genomic regions and diagnostic markers for resistance to aflatoxin contamination in peanut (Arachis hypogaea L.). BMC Genet. 20, 32–44. doi: 10.1186/s12863-019-0734-z
Yu, B., Jiang, H., Pandey, M. K., Huang, L., Huai, D., Zhou, X., et al. (2020). Identification of two novel peanut genotypes resistant to aflatoxin production and their SNP markers associated with resistance. Toxins 12, 156–171. doi: 10.3390/toxins12030156
Yu, Y. B., Adams, D. O., and Shang, F. Y. (1979). 1-Aminocyclopropanecarboxylate synthase, a key enzyme in ethylene biosynthesis. Arch. Biochemi. Biophys. 198, 280–286. doi: 10.1016/0003-9861(79)90420-x
Zhang, B., Shao, L., Wang, J., Zhang, Y., Guo, X., Peng, Y., et al. (2020). Phosphorylation of ATG18a by BAK1 suppresses autophagy and attenuates plant resistance against necrotrophic pathogens. Autophagy 26, 1–18. doi: 10.1080/15548627.2020.1810426
Zhang, S., and Klessig, D. F. (2001). MAPK cascades in plant defense signaling. Trends Plant Sci. 6, 0–527. doi: 10.1016/s1360-1385(01)02103-3
Zhang, X., Valdés-López, O., Arellano, C., Stacey, G., and Balint-Kurti, P. (2017). Genetic dissection of the maize (Zea mays L.) MAMP response. Theor. Appl. Genet. 130, 1155–1168. doi: 10.1007/s00122-017-2876-6
Keywords: peanut, Aspergillus flavus L., resistance, transcriptome analysis, weighted gene co-expression network analysis (WGCNA)
Citation: Cui M, Han S, Wang D, Haider MS, Guo J, Zhao Q, Du P, Sun Z, Qi F, Zheng Z, Huang B, Dong W, Li P and Zhang X (2022) Gene Co-expression Network Analysis of the Comparative Transcriptome Identifies Hub Genes Associated With Resistance to Aspergillus flavus L. in Cultivated Peanut (Arachis hypogaea L.). Front. Plant Sci. 13:899177. doi: 10.3389/fpls.2022.899177
Received: 18 March 2022; Accepted: 06 May 2022;
Published: 15 June 2022.
Edited by:
Guowei Li, Shandong Academy of Agricultural Sciences, ChinaReviewed by:
Kai-Hua Jia, Shandong Academy of Agricultural Sciences, ChinaPeijian Cao, Zhengzhou Tobacco Research Institute of CNTC, China
Copyright © 2022 Cui, Han, Wang, Haider, Guo, Zhao, Du, Sun, Qi, Zheng, Huang, Dong, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peiwu Li, cGVpd3VsaUBvaWxjcm9wcy5jbg==; Xinyou Zhang, aGFhc3pAMTI2LmNvbQ==