 Stefania Savoi
Stefania Savoi Antonio Santiago
Antonio Santiago Luis Orduña
Luis Orduña José Tomás Matus
José Tomás Matus- 1Department of Agricultural, Forest and Food Sciences, University of Turin, Grugliasco, Italy
- 2Institute for Integrative Systems Biology (I2SysBio), Universitat de València-CSIC, Paterna, Spain
Transcriptomics and metabolomics are methodologies being increasingly chosen to perform molecular studies in grapevine (Vitis vinifera L.), focusing either on plant and fruit development or on interaction with abiotic or biotic factors. Currently, the integration of these approaches has become of utmost relevance when studying key plant physiological and metabolic processes. The results from these analyses can undoubtedly be incorporated in breeding programs whereby genes associated with better fruit quality (e.g., those enhancing the accumulation of health-promoting compounds) or with stress resistance (e.g., those regulating beneficial responses to environmental transition) can be used as selection markers in crop improvement programs. Despite the vast amount of data being generated, integrative transcriptome/metabolome meta-analyses (i.e., the joint analysis of several studies) have not yet been fully accomplished in this species, mainly due to particular specificities of metabolomic studies, such as differences in data acquisition (i.e., different compounds being investigated), unappropriated and unstandardized metadata, or simply no deposition of data in public repositories. These meta-analyses require a high computational capacity for data mining a priori, but they also need appropriate tools to explore and visualize the integrated results. This perspective article explores the universe of omics studies conducted in V. vinifera, focusing on fruit-transcriptome and metabolome analyses as leading approaches to understand berry physiology, secondary metabolism, and quality. Moreover, we show how omics data can be integrated in a simple format and offered to the research community as a web resource, giving the chance to inspect potential gene-to-gene and gene-to-metabolite relationships that can later be tested in hypothesis-driven research. In the frame of the activities promoted by the COST Action CA17111 INTEGRAPE, we present the first grapevine transcriptomic and metabolomic integrated database (TransMetaDb) developed within the Vitis Visualization (VitViz) platform (https://tomsbiolab.com/vitviz). This tool also enables the user to conduct and explore meta-analyses utilizing different experiments, therefore hopefully motivating the community to generate Findable, Accessible, Interoperable and Reusable (F.A.I.R.) data to be included in the future.
Introduction
There has been more than 15 years of omics studies accumulated in grapevine (Vitis vinifera L.) to understand processes related to its development and interaction with the environment. The vast amount of data generated has allowed us to understand this species singularity in terms of its adaptive traits, but has also permitted us to explore its diversity, especially related to plant performance (e.g., stress resistance, vigor, yield, etc.) and fruit quality. This data includes genome assemblies of many different cultivars and clones of V. vinifera and their wild American or Asian-related species and, in decreasing order of the number of published studies, transcriptomic, metabolomic, proteomic, and ionomic data (Figure 1 and Supplementary Table 1).
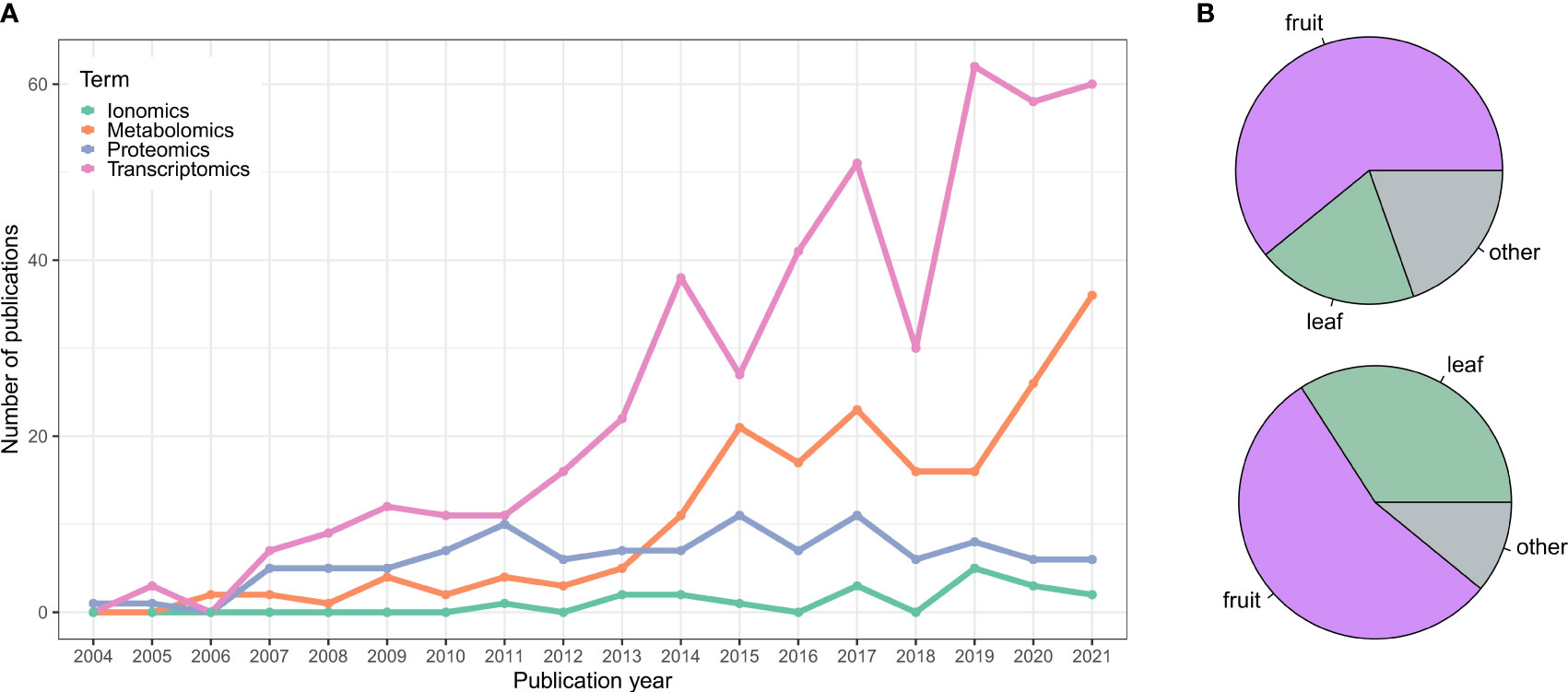
Figure 1 (A) Publication trend of grapevine omics studies. Data was collected from NCBI using different queries and keywords for each category to be as comprehensive as possible and retrieve most of the works. Transcriptomics query: 1) RNA-Seq (excluding microarray data): (transcriptom*[Title/Abstract] OR transcript profiling[Title/Abstract] OR mRNA expression[Title/Abstract] OR RNA-Seq[Title/Abstract] OR RNASeq[Title/Abstract] OR RNA Sequencing[Title/Abstract] OR RNA-Sequencing[Title/Abstract]) AND (grapevine[Title/Abstract] OR grape[Title/Abstract] OR Vitis[Title/Abstract] OR V. vinifera[Title/Abstract]). 2) Microarray: (microarray[Title/Abstract] OR Affymetrix[Title/Abstract] OR CombiMatrix[Title/Abstract] OR NimbleGen[Title/Abstract]) AND (grapevine[Title/Abstract] OR grape[Title/Abstract] OR Vitis[Title/Abstract] OR V. vinifera[Title/Abstract]). Proteomics query: (proteom*[Title/Abstract] OR protein profiling[Title/Abstract]) AND (grapevine[Title/Abstract] OR grape[Title/Abstract] OR Vitis[Title/Abstract] OR V. vinifera[Title/Abstract]). Metabolomics query: (metabolom*[Title/Abstract] OR metabolite profiling[Title/Abstract] OR metabolite analyses[Title/Abstract] OR metabolic response[Title/Abstract]) AND (grapevine[Title/Abstract] OR grape[Title/Abstract] OR Vitis[Title/Abstract] OR V. vinifera[Title/Abstract]). Ionomics query: (ionom*[Title/Abstract] OR elemental profiling[Title/Abstract] OR mineral elements[Title/Abstract] OR cations[Title/Abstract]) AND (grapevine[Title/Abstract] OR grape[Title/Abstract] OR Vitis[Title/Abstract] OR V. vinifera[Title/Abstract]). Only the studies published until 12/2021 were considered. Data were manually curated to remove outliers. (B) Pie chart showing the percentage of transcriptomics (top panel) and metabolomics (bottom panel) studies focusing on fruit tissues, leaves or other grapevine organs.
A significant achievement in the grapevine omics era was the release of two independent genome sequences in 2007, with one being accomplished by a French-Italian Public Consortium (Jaillon et al., 2007), and the second through an Italian-American Collaboration (Velasco et al., 2007). Grapevine then became the first fruit genome to be sequenced and the fourth among plants after Arabidopsis thaliana (Arabidopsis Genome Initiative, 2000), rice [Oryza sativa subsp. indica and subsp. japonica (Goff et al., 2002; Yu et al., 2002)] and poplar (Populus trichocarpa) (Tuskan et al., 2006). Since then, the grapevine community has witnessed an increase of high-throughput next-generation sequencing techniques (e.g., long-read sequencing of RNA and DNA) and the availability of updated grapevine genome assemblies of the reference PN40024 (Canaguier et al., 2017; unpublished data), with updated gene functional annotations incorporated in the recently developed Grape Gene Reference Catalogue (Navarro-Payá et al., 2022). We have observed a cascade of genome sequences of V. vinifera top-quality wine-making cultivars, and genomes of Vitis species important for breeding purposes (da Silva et al., 2013; Venturini et al., 2013; Chin et al., 2016; Minio et al., 2019a; Minio et al., 2019b Vondras et al., 2019; Zhou et al., 2019; Massonnet et al., 2020; Magris et al., 2021). In parallel with genomic/transcriptomic advances, the technological improvement of analytical techniques such as high-resolution liquid and gas chromatography coupled to mass spectrometry, in terms of sensitivity, accuracy, and resolution, has led to a massive amount of metabolomic data from heterogeneous experimental designs, many of which are not public to date.
Comprehensive studies pointing to the expression of the transcriptome or the abundance of the so-far known grape metabolites have boosted the understanding of grapevine physiology in the context of crop and fruit improvement. However, data/metadata interpretation generally encounters difficulties of reusability, for example in meta-analysis studies, either due to high variability and heterogeneity of the associated data or to the presence of partial, misleading, or incomplete experimental descriptions. These descriptors should typically include detailed information on the experimental set-up, report the plant materials used and adopt standardized cultivar names, organs, and developmental stages. Although this comprehensive praxis is highly recommended, in most cases, we notice that even raw data is not entirely available in public databases. Different guidelines have been generated to fill this gap, focusing on harmonizing plant and experiment descriptors. These include standard ontologies, lists of tools, and systematic information for describing omics analyses and tutorials for data submission in public repositories. These guidelines have been recently adopted by the viti-oenology community, sponsored by the COST Action CA17111 INTEGRAPE, in order to adhere to the findable, accessible, interoperable and reusable (F.A.I.R) principles (Wilkinson et al., 2016). A section specifically related to grape and wine metabolomics has been recently published (Savoi et al., 2021a) to encourage the grapevine community to follow these guidelines and share metabolomic data in open repositories such as MetaboLights (Haug et al., 2020). These efforts were made because, contrary to the wave of transcriptomic data available online, unfortunately, only very few metabolomic studies are so far accessible to the public.
History of omics studies in grapevine
Transcriptomics
The first records of high-throughput grapevine transcriptomic studies date back to 2005, exploring the changes in gene expression during berry development. Terrier et al. (2005) generated 50-mers oligoarrays bearing a set of approximately 3,200 unigenes from Vitis vinifera from nine berry developmental stages to provide the first global picture of the genetic program of grape berry development. The authors were able to discriminate differences in gene expression between hard green and soft green berries at the onset of ripening (veraison), pointing out that remarkable changes may occur within a short period and that 25% of the transcripts were significantly activated or repressed between the green and the ripening phases. In a second study (da Silva et al., 2005), the authors retrieved all the available Vitis sequences deposited in GenBank representing numerous Vitis species, cultivars, organs, plant developmental stages, and stress treatments. The analysis concluded that each stage of development was characterized by distinct gene expression patterns, including numerous stage-specific transcripts. Interestingly, they identified a MADS-box gene as a putative regulator of grape berry development. A third study (Waters et al., 2005) used the same technology to explore gene expression patterns throughout grape berry development revealing sets of genes with distinctive or similar expression profiles over the course of berry development. Finally, another pioneering study (Grimplet et al., 2007) used a brand new Affymetrix GeneChip® Vitis vinifera oligonucleotide microarray to study tissue-specific mRNA expression in berry skin, flesh, and seeds in well-watered and water deficit plants at fruit maturity, listing a repertoire of expressed genes, highlighting those modulated by drought stress. In particular, stress modulated around 13% of the genes, mainly in the pulp and skin. In synthesis, these groundbreaking studies paved the way for future grapevine transcriptomic works developing compendiums of gene expression, studying gene profiles during grape berry development and setting the first milestone in understanding grapevine physiology.
The first grapevine study that specifically employed RNA sequencing using the Illumina platform corresponds to that of Zenoni et al. (2010). The authors focused on three stages of berry development indicated as post-setting, veraison, and ripening, to gain insight into the wide range of transcriptional responses associated with the development of fruits. They detected the expression of 17,324 genes during berry development, of which 6,695 were expressed in a stage-specific manner. Moreover, they identified alternative splicing events for 385 genes suggesting a considerable transcript complexity in developing berries. The grapevine expression atlas of the cultivar (cv.) ‘Corvina’ (Fasoli et al., 2012) was later released based on the Nimblegen microarray platform (29,000 genes spotted) representing the first wide study of global gene expression in a complete repertoire of grape organs, including 54 reproductive and vegetative tissues at different developmental stages. This work highlighted a fundamental transcriptome reprogramming during maturation with the activation of a mature/woody developmental program, which was mainly inactive during the vegetative/green stage. Subsequent studies also focused on berry development, ripening, and post-harvest of different cultivars (Pilati et al., 2007; Lijavetzky et al., 2012; Sweetman et al., 2012; Cramer et al., 2014; Gouthu et al., 2014; Pilati et al., 2014; Guo et al., 2016; Zenoni et al., 2016; Ghan et al., 2017; Massonnet et al., 2017; Shangguan et al., 2017; Balic et al., 2018; Fasoli et al., 2018; Savoi et al., 2019; Cramer et al., 2020; Guo et al., 2020; Savoi et al., 2021b; Theine et al., 2021). At the same time, other studies considered the development of tendrils and inflorescences (Díaz-Riquelme et al., 2014), buds (Díaz-Riquelme et al., 2012; Pucker et al., 2020; Shangguan et al., 2020), flower (Sreekantan et al., 2010; Grimplet et al., 2017; Vannozzi et al., 2021), leaf (Pervaiz et al., 2016), fruits of seeded/seedless cultivars (Nwafor et al., 2014; Royo et al., 2016) and roots from V. vinifera and Vitis rootstocks (Cookson et al., 2013; Corso et al., 2015; Cochetel et al., 2017; Livigni et al., 2019). Further studies investigated the plant responses to ozonated water applications (Campayo et al., 2021), the circadian cycle (Carbonell-Bejerano et al., 2014b; Rienth et al., 2014a), the interaction with abiotic stresses such as temperature (Liu et al., 2012; Carbonell-Bejerano et al., 2013; Xin et al., 2013; Rienth et al., 2014b; Rienth et al., 2016), light (Pontin et al., 2010; Carbonell-Bejerano et al., 2014a) and water availability (Perrone et al., 2012; Dal Santo et al., 2016b).
Regarding pathogens, grapevine is highly susceptible to a range of fungal diseases such as downy mildew (Plasmopara viticola) and powdery mildew (Erysiphe necator), and the bunch or noble rot (Botrytis cinerea). Therefore, several efforts have been made to study these interactions by performing transcriptomic analyses, although most of these have been performed on leaves (Fung et al., 2008; Polesani et al., 2010; Wu et al., 2010; Weng et al., 2014; Amrine et al., 2015; Li et al., 2015).
A few studies have used transcriptomics to better understand the impact of agronomic practices such as defoliation (Pastore et al., 2013; Zenoni et al., 2017) or cluster thinning (Pastore et al., 2011) on fruit quality, showing that these techniques, when applied at the appropriate phenological stage, can improve the quality of ripening fruits, in term of sugars and colors. Moreover, an increasing interest in the transcriptome profiles of other Vitis species has been asserted. A complete list of all transcriptomic grapevine studies in fruits is available in Supplementary Table 1A.
Thanks to the large number of public experiments, several transcriptome-related tools have been developed to explore this type of data. The Vitis Co-expression database ‘VTCdb’ (Wong et al., 2013) offered an online platform for exploring potential regulatory networks by addressing gene co-expression. The VTCdb was replaced by ‘VTC-Agg’ in Wong, 2020, by gene rank of correlations and aggregate networks, being constructed from 1,359 microarray samples (33 experiments). More recent applications include ‘AggGCN’, present in the VitViz platform (unpublished), which offers condition-dependent and independent aggregated networks from all SRA-located RNA-seq experiments, and ‘VESPUCCI’ (Moretto et al., 2016; Moretto et al., 2022), a cross-platform expression compendium that was carefully constructed by collecting, homogenizing, and re-annotating the metadata of publicly available microarray and RNA-Seq data (271 experiments). The GRape Expression Atlas ‘GREAT’ (unpublished) allows to query, visualize and analyze genes of interest with interactive heatmaps or expression tables by inferring public RNA-seq data (about 2,000 samples) from Vitis vinifera. Finally, ‘OneGenE’ (One Gene network Expansion) (Pilati et al., 2021) is a transcriptomic data mining tool that finds direct correlations between genes, thus producing association networks by using a causality-based method.
Metabolomics
The use of plant metabolite profiling as a new tool for gene functional analyses and plant phenotyping has been incorporated in the last two decades (Fiehn et al., 2000). However, given the vast diversity of plant metabolites, metabolomic approaches are often based on separately analyzing different classes of metabolites having similar physical or chemical properties or functional groups. Hence metabolome studies are based on applying different analytical methods, including variable extraction protocols and instruments. A complete list of metabolomic studies in grapevine is available in Supplementary Table 1B.
The first metabolomic profiling study comprehensively reporting important grape secondary metabolites was achieved by Mattivi et al. (2006), where 91 grape cultivars were characterized for different polyphenols (focusing on the type and amount of flavonols and anthocyanins) at ripening on the berry skins. In particular and on average, the main flavonols found in red and white grapes differed in abundance order, and interestingly, the delphinidin-like flavonols were missing in all white cultivars, suggesting that the genes coding for flavonoid 3′,5′-hydroxylases were not expressed in these cases. A further study identified a hundred grape polyphenols by UHPLC/QTOF, providing a compendium of grape flavonols, anthocyanins, and stilbenes (Flamini et al., 2015). Other studies examined anthocyanin profiles in berry skins during the progress of ripening (Castellarin et al., 2006), the polyphenolic composition of PIWI grape cultivars (from the German fungus-resistant cv. ‘Pilzwiderstandsfähige’) (Ehrhardt et al., 2014; Gratl et al., 2021), or of wild American genotypes (Narduzzi et al., 2015; Ruocco et al., 2017). Cultivar-specific compositions of polyphenols have also been assessed, for instance, in ‘Muscat’ cultivars with anthocyanin profiles driving the main divergence between red and white cultivars, followed by flavonols and flavanols discriminating among the white accessions (Degu et al., 2015). Additionally, the responses to several stresses (Degu et al., 2016), the changes in the flavonoid profiles by early mechanical leaf removal (VanderWeide et al., 2018), or the stimulatory effect of kaolin application in leaves (Conde et al., 2016) have also been accomplished. Metabolomic analyses have also been performed to compare the primary metabolites of fruits (Dai et al., 2013; Cuadros-Inostroza et al., 2016; Zhao et al., 2016) and leaves (Harb et al., 2015; Conde et al., 2018). Other studies have focused on the analysis of the volatile compounds in roots (Lawo et al., 2011), leaves (Weingart et al., 2012), and berries (Vrhovsek et al., 2014).
Research has also been carried out to study the berry late-ripening program in skin, flesh, and seed (Vondras et al., 2017) or to understand the role of drought conditions affecting several metabolic pathways (Hochberg et al., 2013; Griesser et al., 2015; Hochberg et al., 2015b; Herrera et al., 2017; Pinasseau et al., 2017). In addition, the effect of temperature (Hochberg et al., 2015a) and light (Reshef et al., 2017; Reshef et al., 2018) have also been investigated.
Like for transcriptomics, metabolomic approaches have also been deployed to understand plant-pathogen interactions. For example, the identification of biomarkers of defense response to Plasmopara viticola has been a trending topic (Adrian et al., 2017; Chitarrini et al., 2017; Negrel et al., 2018; Ciubotaru et al., 2021), as well as the action of Botrytis cinerea on the fruit metabolism (Hong et al., 2012; Negri et al., 2017).
In recent years, metabolomics in grapevine has been coupled to transcriptome analyses to understand in detail the physiological mechanisms of berry ripening and the interaction with biotic or abiotic stresses. This topic will be further discussed later on.
Proteomics
Approximately one-hundred grapevine proteomic studies can be found in the literature (a complete list of proteomic studies in grapevine is available in Supplementary Table 1C). The first extensive study on grapevine proteomics analyzed the mesocarp (flesh)-allocated proteins of ripe berries in six different cultivars using two-dimensional electrophoresis followed by MALDI-TOF peptide mass fingerprinting (Sarry et al., 2004). The authors determined the composition of 67 major proteins in ripe fruits and provided new evidence on the metabolism of sugar and organic acids in fruits. A second extensive work studied the proteomic dynamic changes during berry development and ripening in the mesocarp of cv. ‘Nebbiolo’ by profiling 101 proteins over seven time points (Giribaldi et al., 2007). The authors found that the majority of proteins were linked to metabolism, energy and protein synthesis and fate. In the same year, another study investigated the impact of two major abiotic stresses, water deficit and salt stress, on the shoot proteome of two cultivars, cv. ‘Chardonnay’ and cv. ‘Cabernet Sauvignon’, showing that the protein concentration varied mainly in response to the cultivars, then with time, and lastly with the abiotic stress (Vincent et al., 2007).
The knowledge of the grapevine berry proteome has been improved over the years by different studies focusing on skin proteome dynamics throughout berry ripening (Deytieux et al., 2007; Negri et al., 2008a; Martínez-Esteso et al., 2011a), on the identification of the different proteins present in skin, flesh or seed (Tian et al., 2015), on plasma membrane protein expression either during berry ripening (Zhang et al., 2008) or at maturity (Negri et al., 2008b), on the changes of the vacuolar proteome during ripening (Kuang et al., 2019), on single-berry proteome during development and ripening (Martínez-Esteso et al., 2011b; Martínez-Esteso et al., 2013) or in the protein expression profiles of grape berry during postharvest withering process (Di Carli et al., 2011).
Proteomics has also been employed to identify proteins associated with flavor volatile compounds found in fruits, such as proteins involved in the phenylpropanoid pathway, terpene synthesis, fatty acid derived volatiles and esters (Kambiranda et al., 2016; Kambiranda et al., 2018), or with the effects of ABA treatments on ripening Vitis vinifera berries (Giribaldi et al., 2010), sunlight exposure (Niu et al., 2013) and water deficit (Cramer et al., 2013). In addition, many other studies have tested proteome changes in plant-pathogen responses. For example, several studies investigated the leaf response to biotic stress, such as Plasmopara viticola (Milli et al., 2012; Figueiredo et al., 2017; Nascimento-Gavioli et al., 2017; Santos et al., 2020; Liu et al., 2021), while another study identified potential protein markers in berries affected by noble rot (Lorenzini et al., 2015).
Ionomics
So far, only a few ionomic studies have been performed in grapevine. In 2011 the first ionomic work was published, describing the accumulation pattern of 42 mineral elements in cv. ‘Chardonnay’ berries during development and ripening (Bertoldi et al., 2011). The authors described that seven elements accumulate prior to veraison, other eighteen accumulate mainly prior to veraison but also during ripening, and seventeen progressively during growth and ripening. With regard to distribution, eight, sixteen and eighteen elements specifically accumulated in seeds, skin and flesh, respectively. In another study, the concentration of 34 mineral elements in grapevine berries was determined by ICP-MS in cv. ‘Corvina’ berries, harvested from eleven vineyards, to trace their geographical origin (Pii et al., 2017). Moreover, the analysis of cations such as K+, Mg2+, Ca2+, in berries at different developmental stages showed the diversity of their concentration in several cultivars, providing additional information for the selection of genotypes able to cope with the adverse effects of climate change on fruit quality (Bigard et al., 2020). Finally, the ionomic signature was also studied in leaves identifying a mineral element-based response to Xylella fastidiosa (De La Fuente et al., 2013) and the changes in mineral distribution after Plasmopara viticola infection (Cesco et al., 2020). A complete list of ionomic grapevine studies is available in Supplementary Table 1D.
Multi-omics integration of transcriptomics and metabolomics
In addition to single-omic studies, integrative omics analyses can be applied to provide a deeper understanding of the regulatory processes controlling different molecular phenotypes. To date, around sixty multi-omic studies combine transcriptomics and metabolomics, to study either the early and late responses to abiotic stresses, such as water and salinity stress (Cramer et al., 2007), the molecular mechanisms involved in berry development (Deluc et al., 2007; Fortes et al., 2011; Degu et al., 2014; Fasoli et al., 2018; Griesser et al., 2020) and over-ripening (i.e., post-harvest withering; Zenoni et al., 2016; Zenoni et al., 2020), or the changes in polyphenol and aromatic compound content in ripening berries (Costantini et al., 2015; Malacarne et al., 2015; Costantini et al., 2017). Other integrated approaches have studied berry responses during ripening to water deficit (Deluc et al., 2009; Savoi et al., 2016; Savoi et al., 2017; Yang et al., 2020), light (Suzuki et al., 2015; Sun et al., 2019; He et al., 2020; Zhang K et al., 2021) or temperature (Lecourieux et al., 2017). A few additional studies have also studied the ‘genotype by environment’ (GxE) interaction (i.e., the effect of terroir) on the plasticity of red and white grapes (Dal Santo et al., 2013; Anesi et al., 2015; Dal Santo et al., 2016a; Dal Santo et al., 2018).
The integration of transcript and metabolite data has contributed to the biological understanding of grape cultivar differences for fruit composition. For example, Degu et al. (2014) identified a cultivar-dependent regulation of specialized metabolism towards fruit maturation when comparing the cv. ‘Shiraz’ and ‘Cabernet Sauvignon’, the former displaying a higher upregulation of the entire polyphenol pathway, favoring the accumulation of piceid and p-coumaroylated anthocyanins. Moreover, in a post-harvest withering study, the expression of stilbene biosynthesis genes increased after harvest in a genotype-dependent manner, matching the varietal accumulation differences (Zenoni et al., 2016).
The integration of multi-omic datasets has been helpful in the identification of putative candidate genes regulating the accumulation of secondary metabolites in fruits. In this regard, a nudix hydrolase was identified as a component of a terpene synthase-independent pathway, enhancing monoterpene biosynthesis together with other genes potentially involved in terpenoid metabolism, such as cytochrome P450 hydroxylases, epoxide hydrolases, and glucosyltransferases (Costantini et al., 2015). Transcript and metabolite analyses determined that the biosynthesis of anthocyanins was a consistent hallmark of noble rot in white-skinned cv. ‘Sémillon’ (Blanco-Ulate et al., 2015), an unexpected response due to the absence of functional alleles of the MYBA1-A2 anthocyanin-regulators. This phenomenon led to the hypothesis of novel regulators controlling berry skin pigment production, which were later found by Matus et al. (2017) and D’Incà et al. (2021). The use of metabolomics (stilbenoid-oriented) and transcriptomics, coupled to the genome-wide exploration of transcription factor binding sites through DAP-seq, allowed Orduña et al. (2022) to identify new candidate genes coding for resveratrol modifying enzymes including laccasses, glycosyltransferases and O-methyl-transferases, potentially producing viniferin, piceid and pterostilbene, respectively. Finally, Savoi et al. (2016) associated the up-regulation of the MYB24 transcription factor with the observed increased biosynthesis of three key monoterpenes under water deficit in white grapes, leading to the hypothesis of its capacity to regulate terpene synthase (TPS) gene expression. Indeed, this MYB24-TPS regulatory relationship was recently confirmed by Zhang C et al. (2021), who also integrated several omics including transcriptomics, metabolomics, and DAP-seq.
Numerous works have applied integrative methods to address responses to pathogen infection, most being conducted in fruits or leaves. For example, the leaf analysis of ‘Regent’ and ‘Trincadeira’ cultivars, respectively resistant and susceptible to mildew, has provided information on the different metabolic pathways related to the defense process (Figueiredo et al., 2008). The work of Malacarne et al. (2011) combined metabolic and transcriptional profiles in a segregating population for resistance to Plasmopara viticola, while Maia et al. (2020) suggested several gene/metabolite biomarkers of fungal/oomycetes-associated disease susceptibility in eleven Vitis genotypes. In addition, combined approaches have been employed to study berries infected with the fungus Botrytis cinerea (Agudelo-Romero et al., 2015; Blanco-Ulate et al., 2015), and also to determine how the interaction with this same pathogen at flowering influences quiescence and egressed infection (Haile et al., 2017; Haile et al., 2020). Recently, a few studies have examined the defense responses in susceptible and resistant grape cultivars (Eisenmann et al., 2019; Chitarrini et al., 2020), while others have examined the transcriptional, hormonal, and metabolic changes under powdery mildew infection caused by Erysiphe necator in leaves and berries (Pagliarani et al., 2020; Pimentel et al., 2021).
To date, only three publications integrate transcriptomics, proteomics, and metabolomics in a single study. By overlapping genes, proteins, and metabolites, Zamboni et al. (2010) identified stage-specific biomarkers for berry development and withering. Moreover, multi-omic datasets were integrated to test the uniqueness of three red-skinned and two white-skinned cultivars at berry maturity, providing a detailed indication of genotype differences (Ghan et al., 2015). Finally, multi-omic analyses have been employed in the study of leaf responses to copper stress, providing agronomic knowledge to improve vineyard management and favor the breeding of copper-resistant grape cultivars (Chen et al., 2021).
An illustration of data integration and meta-analysis using the TransMetaDb app
Moving forward from single-omic studies and gene-to-gene network exploring tools, the construction of gene-to-metabolite networks via integrative approaches represents a promising strategy for identifying novel gene functions and unprecedent links between genes and metabolites (Hirai et al., 2005; Saito et al., 2008). This concept has been reviewed in grapevine (Wong and Matus, 2017; Matus et al., 2019), stating the need for appropriate tools to visualize the integration of multi-omic datasets. The first tool developed for data integration in grapevine was ‘VitisNet’ (Grimplet et al., 2009; Grimplet et al., 2012), which allowed the multiple combinations of omics data by overlapping user data to 247 curated grapevine molecular networks, reporting more than 16,000 genes; unfortunately, this tool is no longer accessible. A second developed tool was ‘VitisCyc’ (Naithani et al., 2014), a grape-specific database for browsing and visualizing metabolic pathways end enzymatic reactions, compounds, genes and proteins, and for comparing metabolic networks with other publicly-available resources from other plant species (this tool is now hosted in the Gramene database; Tello-Ruiz et al., 2021).
To date, there is no visualization tool in grape to explore the correlation of transcriptomics and metabolomics data, and even less to combine them in meta-analyses. Within the COST Action CA17111 INTEGRAPE, we have developed TransMetaDb (Figure 2), an application available in the Vitis Visualization (VitViz) platform (https://tomsbiolab.com/vitviz), to freely explore the correlation between metabolites and genes in multiple transcriptomics/metabolomics integrated datasets. The Integrated Transcriptomics and Metabolomics Database Application (TransMetaDb) was developed in the Shiny R environment and offers a user-friendly interface to explore transcriptomic data (i.e., differentially expressed genes and co-expression modules) and its correlation to metabolites quantified in the same conditions. The App takes an Excel or comma/tab-separated file as an input, with only one column containing the 12X.v2 VCost.v3 gene IDs of interest, and an optional second column with the gene names/symbols, whose purpose is to provide a descriptive name for the user-provided genes. The output is a downloadable heatmap or table, with all the genes of interest by row, compared with the different metabolites on each column. Each cell contains the Pearson’s correlation coefficient (PCC) value and its p-value in brackets, calculated using the student asymptotic method. The identification of gene candidates related to metabolite phenotypic traits has often been inferred from association measures, such as the commonly-used PCC method (to measure linear correlation between two variables). Although alternative measures can be employed, PCC is still the standard for initial exploration of gene-to-gene and gene-to-metabolite networks (Nikiforova et al., 2005; Mao et al., 2009). This is a first general approach to discover functions in a set of genes and metabolites of interest. On a second tab, the App allows the user to explore co-expression clustered modules, downloading a table with all gene-module categories.
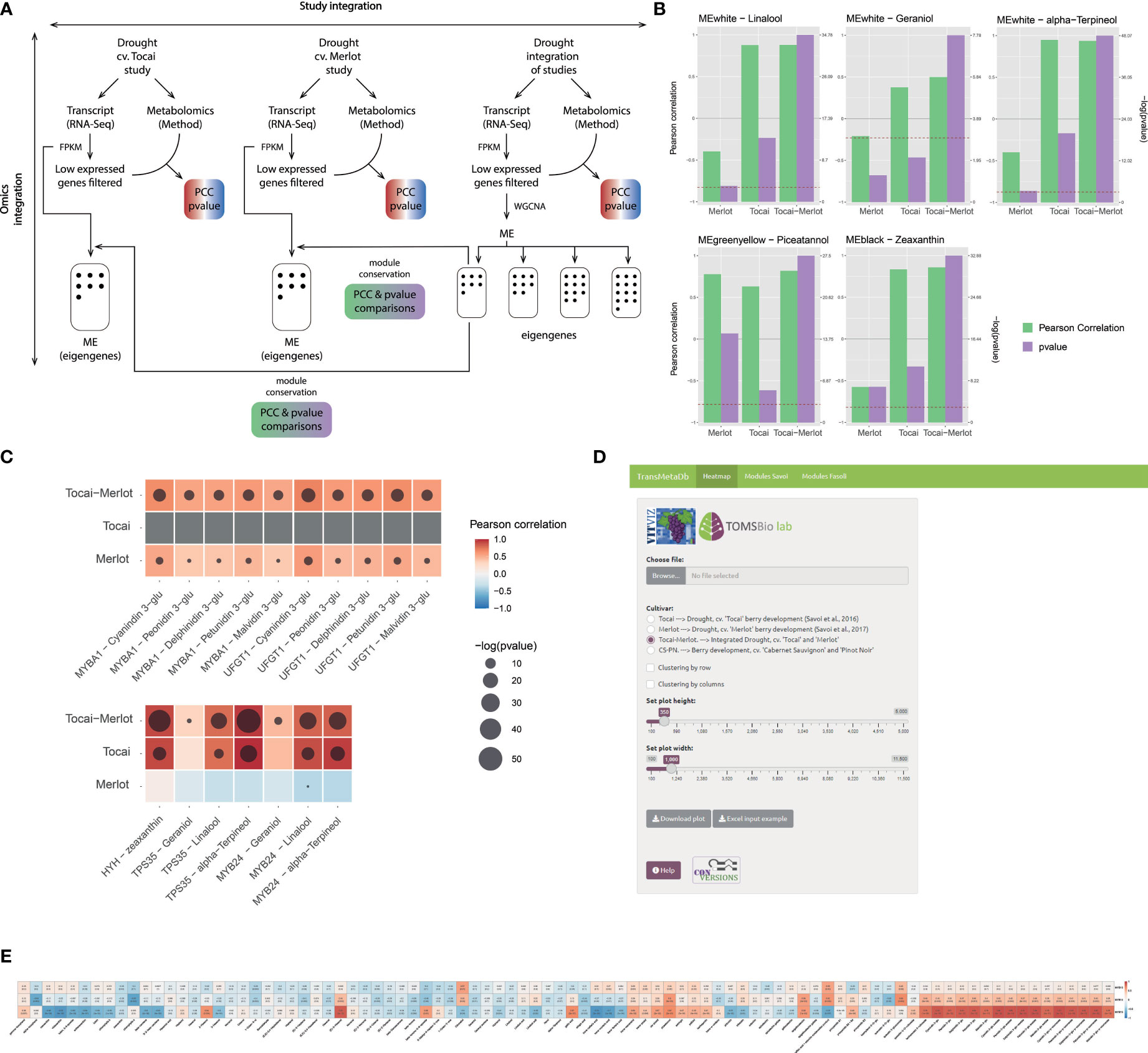
Figure 2 Omics integration in two studies that investigated the effect of a long-lasting water deficit on the metabolism of white (cv. ‘Tocai friulano’) and red (cv. ‘Merlot’) cultivars during berry development and ripening (Savoi et al., 2016; 2017). (A) Methods scheme of the analyses performed for each of the independent studies and the combination of them (i.e., study integration or meta-analysis). Genes with low expression are filtered-out before WGCNA analysis. (B, C) Results obtained from the analyses depicted in (A), where (B) Improvement of statistical significance and/or correlation between genes and metabolites for the combination of both studies. P-value representation is only shown when the value exceeds the 0.05 threshold. (C) Improvement of statistical significance and/or correlation between gene modules and metabolites for the combination of both studies. A −log10(p-value) scale is provided where a higher value represents a greater statistical significance. The red dashed line depicts −log10(p-value) corresponding to a 0.05 threshold. (D) Visual interface of the TransMetaDb App, found at the Vitis Visualization (VitViz) platform (https://tomsbiolab.com/vitviz). The resulting example heatmap is shown in (E).
So far, three studies have been uploaded to the App; Savoi et al. (2016) and Savoi et al. (2017) focused on the impact of drought on fruit secondary metabolism and have a similar experimental design, the same metabolomic method, with all data being publicly available. A third study detailed a transcriptomic and metabolomic temporal map of berry development in two cultivars for three consecutive years (Fasoli et al., 2018). This data was first reanalyzed; briefly, the transcriptomic data was re-aligned using STAR v. 2.7.3a (Dobin et al., 2013) against the PN40024 12X.v2 assembly, and a raw gene count matrix was extracted using the latest VCost.v3 annotation (Canaguier et al., 2017) and featureCounts (Liao et al., 2014). Gene expression was normalized with FPKM (Fragments per kilobase per million mapped reads) as this normalization method takes into account both sequencing depth of libraries and gene length, allowing the comparison of expression between different genes across different samples (other normalization methods will be applied in the future, such as TMM).
Clustering of gene expression and metabolite profiles can serve for mining useful information from noisy data, identifying cohorts of genes that control metabolism and understanding how metabolic pathways can be rewired in development and stress. Detection of functional gene modules by classical clustering methods, such as K-means or mean-shift, have been outperformed in transcriptomic studies by a modification of the algorithm, e.g., the fuzzy k-means applied in the fclust R package (Ferraro et al., 2019), or by correlation networks using for instance the Weighted Gene Correlation Network Analysis (WGCNA) R package (Langfelder and Horvath, 2008). In TransMetaDb, genes have been clustered in eigen modules (ME) by applying WGCNA, and metabolites have been associated to these modules in the same way as for genes. As explained in Langfelder and Horvath (2008), WGCNA works by generating a network (or adjacency matrix) from the biological data and then performing a hierarchical clustering. In order to build the adjacency matrix, an intermediate co-expression similarity matrix is defined using the biological data by computing co-expression measures between genes. Once the co-expression similarity matrix is constructed, it is transformed in the adjacency matrix using a beta power value that is chosen by the user in order to generate an adjacency matrix, and that satisfies the scale-free network topology where the distribution of node degree is adjusted to a potential law (i.e., high number of nodes with a low number of edges and few nodes with a high number of edges). For doing this analysis, the blockwiseModules function was used with a deepSplit of 4, a mergeCutHeight of 0.1 and a beta power of 30 for the integration of the ‘Tocai friulano’ and ‘Merlot’ studies and a power of 11, deepSplit of 4 and a mergeCutHeight of 0.2 for Fasoli et al. (2018). Following WGCNA authors’ recommendation, the deepSplit and mergeCutHeight parameters were adapted to obtain a number of eigen modules and a number of genes per eigen module adaptable for subsequent analysis with a minimum module size of 40 genes. Transcripts and metabolites were integrated in gene-to-metabolite and eigen module-to-metabolite matrices for each of the individual and combined studies.
While exploring the WGCNA results of the combined studies we observed that the Pearson correlation coefficients (PCC) and/or the significance (p-values) of specific eigen module (ME, Figure 2B) or gene-metabolite (Figure 2C), often increased. This is expected, because as the sample is larger the error is lower and the sample correlation converges to the population parameter, in the same way as increasing the number of replicates improves the inferences that can be made about a population. For example, in the combined eigen module “white”, PCC and p-values of linalool, geraniol, and α-terpineol were higher and more significant in the ‘Tocai friulano’+’Merlot’ set, just like the association between these same three terpenes (present in the white cultivar only) and the genes MYB24 and TPS35 (Savoi et al., 2016; Zhang C et al., 2021). Moreover, the addition of an anthocyanin-free dataset (i.e., Tocai) to the data belonging to an anthocyanin-pigmented cultivar (i.e., Merlot) also improved the transcript-metabolite correlations between different pigments and related genes as the ‘no expression’ and ‘no pigment accumulation’ is a positive correlation in itself. The five glycosylated anthocyanins (only present in the red cultivar) and the anthocyanin-related genes UFGT (Boss et al., 1996) and MYBA1 (Kobayashi et al., 2004) showed increased correlation and/or increased significance in the meta-analysis compared to individual studies (where the correlation was already high). This improvement is due to the coherent behavior of the selected proteins as they correspond to the latest enzyme of the pathway and the master regulator of anthocyanin accumulation, respectively, representing a perfect case of expected correlation. Finally, we observed the same trend for the light-induced carotenoid zeaxanthin and the light/UV signaling gene HYH (HY5 HOMOLOGUE; (Loyola et al., 2016). These results clearly imply that meta-analyses of integrated transcriptomic/metabolomic datasets improves the strength of correlation metrics in the same way as adding new samples in the construction of a gene co-expression network increases the network performance until it reaches a plateau (Wong, 2020).
We plan to increase the number of combined transcriptomic and metabolomic datasets in TransMeta Db, based on the quality of the metadata. Metabolic data will be compared in the following submitted studies to see which metabolites can be combined. If this data has been acquired by different platforms, or if it is presented in different units, one possibility would be to scale the data (e.g., Z-score) before integration. Nevertheless, this effort would certainly push the community to improve the annotation of their experiments in public repositories so their data can be fully interoperable. Once more studies have been uploaded, we also expect to showcase alternative correlation metrics using other compatible software and pipelines.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
SS and JM conceived the study and wrote the manuscript. AS and LO contributed with the reanalysis of the transcriptomic and metabolomic data set and by building the TransMetaDb app found in the VitViz platform. All authors contributed to the article and approved the submitted version.
Funding
This publication is based upon work from COST Action CA17111 INTEGRAPE, supported by COST (European Cooperation in Science and Technology) and COST grant ECOST-STSM-Request-CA17111-48997 awarded to SS. This work was also supported by Grants PGC2018-099449-A-I00, PID2021-128865NB-I00 and by the Ramón y Cajal program grant RYC-2017-23645, all awarded to JM, and to the FPI scholarship PRE2019-088044 granted to LO from the Ministerio de Ciencia, Innovación y Universidades (MCIU, Spain), Agencia Estatal de Investigación (AEI, Spain), and Fondo Europeo de Desarrollo Regional (FEDER, European Union).
Acknowledgments
We thank Anne-Marie Digby (University of Verona) for manuscript revision. We apologize to all authors whose work has not been cited owing to space constraints.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.937927/full#supplementary-material
Supplementary Table 1 | List of (A) transcriptomics, (B) metabolomics, (C) proteomics, and (D) ionomics studies retrieved from the literature. Data was collected from NCBI using different queries and keywords. Only the studies published until 12/2021 were considered. Data were manually curated to remove clear outliers.
References
Adrian, M., Lucio, M., Roullier-Gall, C., Héloir, M.-C., Trouvelot, S., Daire, X., et al. (2017). Metabolic fingerprint of PS3-induced resistance of grapevine leaves against Plasmopara viticola revealed differences in elicitor-triggered defenses. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00101
Agudelo-Romero, P., Erban, A., Rego, C., Carbonell-Bejerano, P., Nascimento, T., Sousa, L., et al. (2015). Transcriptome and metabolome reprogramming in Vitis vinifera cv. trincadeira berries upon infection with Botrytis cinerea. J. Exp. Bot. 66, 1769–1785. doi: 10.1093/jxb/eru517
Amrine, K. C. H., Blanco-Ulate, B., Riaz, S., Pap, D., Jones, L., Figueroa-Balderas, R., et al. (2015). Comparative transcriptomics of central Asian Vitis vinifera accessions reveals distinct defense strategies against powdery mildew. Hortic. Res. 2, 1–11. doi: 10.1038/hortres.2015.37
Anesi, A., Stocchero, M., Dal Santo, S., Commisso, M., Zenoni, S., Ceoldo, S., et al. (2015). Towards a scientific interpretation of the terroir concept: plasticity of the grape berry metabolome. BMC Plant Biol. 15, 191. doi: 10.1186/s12870-015-0584-4
Arabidopsis Genome Initiative (2000). Analysis of the genome sequence of the flowering plant arabidopsis thaliana. Nature 408, 796–815. doi: 10.1038/35048692
Balic, I., Vizoso, P., Nilo-Poyanco, R., Sanhueza, D., Olmedo, P., Sepúlveda, P., et al. (2018). Transcriptome analysis during ripening of table grape berry cv. Thompson seedless. PloS One 13, e0190087. doi: 10.1371/journal.pone.0190087
Bertoldi, D., Larcher, R., Bertamini, M., Otto, S., Concheri, G., Nicolini, G. (2011). Accumulation and distribution pattern of macro- and microelements and trace elements in Vitis vinifera l. cv. Chardonnay berries. J. Agric. Food Chem. 59, 7224–7236. doi: 10.1021/jf2006003
Bigard, A., Romieu, C., Sire, Y., Torregrosa, L. (2020). Vitis vinifera l. diversity for cations and acidity is suitable for breeding fruits coping with climate warming. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.01175
Blanco-Ulate, B., Amrine, K. C. H., Collins, T. S., Rivero, R. M., Vicente, A. R., Morales-Cruz, A., et al. (2015). Developmental and metabolic plasticity of white-skinned grape berries in response to Botrytis cinerea during noble rot. Plant Physiol. 169, 2422–2443. doi: 10.1104/pp.15.00852
Boss, P. K., Davies, C., Robinson, S. P. (1996). Expression of anthocyanin biosynthesis pathway genes in red and white grapes. Plant Mol. Biol. 32, 565–569. doi: 10.1007/BF00019111
Campayo, A., Savoi, S., Romieu, C., López-Jiménez, A. J., Serrano de la Hoz, K., Salinas, M. R., et al. (2021). The application of ozonated water rearranges the vitis vinifera l. leaf and berry transcriptomes eliciting defence and antioxidant responses. Sci. Rep. 11, 8114. doi: 10.1038/s41598-021-87542-y
Canaguier, A., Grimplet, J., Di Gaspero, G., Scalabrin, S., Duchêne, E., Choisne, N., et al. (2017). A new version of the grapevine reference genome assembly (12X.v2) and of its annotation (VCost.v3). Genom Data 14, 56–62. doi: 10.1016/j.gdata.2017.09.002
Carbonell-Bejerano, P., Diago, M.-P., Martínez-Abaigar, J., Martínez-Zapater, J. M., Tardáguila, J., Núñez-Olivera, E. (2014a). Solar ultraviolet radiation is necessary to enhance grapevine fruit ripening transcriptional and phenolic responses. BMC Plant Biol. 14, 183. doi: 10.1186/1471-2229-14-183
Carbonell-Bejerano, P., Rodríguez, V., Royo, C., Hernáiz, S., Moro-González, L. C., Torres-Viñals, M., et al. (2014b). Circadian oscillatory transcriptional programs in grapevine ripening fruits. BMC Plant Biol. 14, 78. doi: 10.1186/1471-2229-14-78
Carbonell-Bejerano, P., Santa María, E., Torres-Pérez, R., Royo, C., Lijavetzky, D., Bravo, G., et al. (2013). Thermotolerance responses in ripening berries of Vitis vinifera l. cv Muscat Hamburg. Plant Cell Physiol. 54, 1200–1216. doi: 10.1093/pcp/pct071
Castellarin, S. D., Di Gaspero, G., Marconi, R., Nonis, A., Peterlunger, E., Paillard, S., et al. (2006). Colour variation in red grapevines (Vitis vinifera l.): genomic organisation, expression of flavonoid 3’-hydroxylase, flavonoid 3’,5’-hydroxylase genes and related metabolite profiling of red cyanidin-/blue delphinidin-based anthocyanins in berry skin. BMC Genomics 7, 12. doi: 10.1186/1471-2164-7-12
Cesco, S., Tolotti, A., Nadalini, S., Rizzi, S., Valentinuzzi, F., Mimmo, T., et al. (2020). Plasmopara viticola infection affects mineral elements allocation and distribution in Vitis vinifera leaves. Sci. Rep. 10, 18759. doi: 10.1038/s41598-020-75990-x
Chen, M., Fang, X., Wang, Z., Shangguan, L., Liu, T., Chen, C., et al. (2021). Multi-omics analyses on the response mechanisms of ‘Shine muscat’ grapevine to low degree of excess copper stress (Low-ECS). Environ. Pollut. 286, 117278. doi: 10.1016/j.envpol.2021.117278
Chin, C.-S., Peluso, P., Sedlazeck, F. J., Nattestad, M., Concepcion, G. T., Clum, A., et al. (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054. doi: 10.1038/nmeth.4035
Chitarrini, G., Riccadonna, S., Zulini, L., Vecchione, A., Stefanini, M., Larger, S., et al. (2020). Two-omics data revealed commonalities and differences between Rpv12- and Rpv3-mediated resistance in grapevine. Sci. Rep. 10, 12193. doi: 10.1038/s41598-020-69051-6
Chitarrini, G., Soini, E., Riccadonna, S., Franceschi, P., Zulini, L., Masuero, D., et al. (2017). Identification of biomarkers for defense response to Plasmopara viticola in a resistant grape variety. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01524
Ciubotaru, R. M., Franceschi, P., Zulini, L., Stefanini, M., Škrab, D., Rossarolla, M. D., et al. (2021). Mono-locus and pyramided resistant grapevine cultivars reveal early putative biomarkers upon artificial inoculation with Plasmopara viticola. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.693887
Cochetel, N., Escudié, F., Cookson, S. J., Dai, Z., Vivin, P., Bert, P.-F., et al. (2017). Root transcriptomic responses of grafted grapevines to heterogeneous nitrogen availability depend on rootstock genotype. J. Exp. Bot. 68, 4339–4355. doi: 10.1093/jxb/erx224
Conde, A., Neves, A., Breia, R., Pimentel, D., Dinis, L.-T., Bernardo, S., et al. (2018). Kaolin particle film application stimulates photoassimilate synthesis and modifies the primary metabolome of grape leaves. J. Plant Physiol. 223, 47–56. doi: 10.1016/j.jplph.2018.02.004
Conde, A., Pimentel, D., Neves, A., Dinis, L.-T., Bernardo, S., Correia, C. M., et al. (2016). Kaolin foliar application has a stimulatory effect on phenylpropanoid and flavonoid pathways in grape berries. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01150
Cookson, S. J., Clemente Moreno, M. J., Hevin, C., Nyamba Mendome, L. Z., Delrot, S., Trossat-Magnin, C., et al. (2013). Graft union formation in grapevine induces transcriptional changes related to cell wall modification, wounding, hormone signalling, and secondary metabolism. J. Exp. Bot. 64, 2997–3008. doi: 10.1093/jxb/ert144
Corso, M., Vannozzi, A., Maza, E., Vitulo, N., Meggio, F., Pitacco, A., et al. (2015). Comprehensive transcript profiling of two grapevine rootstock genotypes contrasting in drought susceptibility links the phenylpropanoid pathway to enhanced tolerance. J. Exp. Bot. 66, 5739–5752. doi: 10.1093/jxb/erv274
Costantini, L., Kappel, C. D., Trenti, M., Battilana, J., Emanuelli, F., Sordo, M., et al. (2017). Drawing links from transcriptome to metabolites: the evolution of aroma in the ripening berry of moscato bianco (Vitis vinifera l.). Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00780
Costantini, L., Malacarne, G., Lorenzi, S., Troggio, M., Mattivi, F., Moser, C., et al. (2015). New candidate genes for the fine regulation of the colour of grapes. J. Exp. Bot. 66, 4427–40. doi: 10.1093/jxb/erv159
Cramer, G. R., Cochetel, N., Ghan, R., Destrac-Irvine, A., Delrot, S. (2020). A sense of place: transcriptomics identifies environmental signatures in Cabernet sauvignon berry skins in the late stages of ripening. BMC Plant Biol. 20, 41. doi: 10.1186/s12870-020-2251-7
Cramer, G. R., Ergül, A., Grimplet, J., Tillett, R. L., Tattersall, E. A. R., Bohlman, M. C., et al. (2007). Water and salinity stress in grapevines: early and late changes in transcript and metabolite profiles. Funct. Integr. Genomics 7, 111–34. doi: 10.1007/s10142-006-0039-y
Cramer, G. R., Ghan, R., Schlauch, K. A., Tillett, R. L., Heymann, H., Ferrarini, A., et al. (2014). Transcriptomic analysis of the late stages of grapevine (Vitis vinifera cv. Cabernet sauvignon) berry ripening reveals significant induction of ethylene signaling and flavor pathways in the skin. BMC Plant Biol. 14, 1–21. doi: 10.1186/s12870-014-0370-8
Cramer, G. R., Sluyter, S. C. V., Hopper, D. W., Pascovici, D., Keighley, T., Haynes, P. A. (2013). Proteomic analysis indicates massive changes in metabolism prior to the inhibition of growth and photosynthesis of grapevine (Vitis vinifera l.) in response to water deficit. BMC Plant Biol. 13, 1–22. doi: 10.1186/1471-2229-13-49
Cuadros-Inostroza, A., Ruíz-Lara, S., González, E., Eckardt, A., Willmitzer, L., Peña-Cortés, H. (2016). GC–MS metabolic profiling of Cabernet sauvignon and merlot cultivars during grapevine berry development and network analysis reveals a stage- and cultivar-dependent connectivity of primary metabolites. Metabolomics 12, 39. doi: 10.1007/s11306-015-0927-z
Dai, Z. W., Léon, C., Feil, R., Lunn, J. E., Delrot, S., Gomès, E. (2013). Metabolic profiling reveals coordinated switches in primary carbohydrate metabolism in grape berry (Vitis vinifera l.), a non-climacteric fleshy fruit. J. Exp. Bot. 64, 1345–1355. doi: 10.1093/jxb/ers396
Dal Santo, S., Fasoli, M., Negri, S., D’Incà, E., Vicenzi, N., Guzzo, F., et al. (2016a). Plasticity of the berry ripening program in a white grape variety. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00970
Dal Santo, S., Palliotti, A., Zenoni, S., Tornielli, G. B., Fasoli, M., Paci, P., et al. (2016b). Distinct transcriptome responses to water limitation in isohydric and anisohydric grapevine cultivars. BMC Genomics 17, 815. doi: 10.1186/s12864-016-3136-x
Dal Santo, S., Tornielli, G. B., Zenoni, S., Fasoli, M., Farina, L., Anesi, A., et al. (2013). The plasticity of the grapevine berry transcriptome. Genome Biol. 14, r54. doi: 10.1186/gb-2013-14-6-r54
Dal Santo, S., Zenoni, S., Sandri, M., Lorenzis, G. D., Magris, G., Paoli, E. D., et al. (2018). Grapevine field experiments reveal the contribution of genotype, the influence of environment and the effect of their interaction (G×E) on the berry transcriptome. Plant J. 93, 1143–1159. doi: 10.1111/tpj.13834
da Silva, F. G., Iandolino, A., Al-Kayal, F., Bohlmann, M. C., Cushman, M. A., Lim, H., et al. (2005). Characterizing the grape transcriptome. analysis of expressed sequence tags from multiple Vitis species and development of a compendium of gene expression during berry development. Plant Physiol. 139, 574–597. doi: 10.1104/pp.105.065748
da Silva, C., Zamperin, G., Ferrarini, A., Minio, A., Molin, A. D., Venturini, L., et al. (2013). The high polyphenol content of grapevine cultivar tannat berries is conferred primarily by genes that are not shared with the reference genome. Plant Cell 25, 4777–4788. doi: 10.1105/tpc.113.118810
Degu, A., Ayenew, B., Cramer, G. R., Fait, A. (2016). Polyphenolic responses of grapevine berries to light, temperature, oxidative stress, abscisic acid and jasmonic acid show specific developmental-dependent degrees of metabolic resilience to perturbation. Food Chem. 212, 828–836. doi: 10.1016/j.foodchem.2016.05.164
Degu, A., Hochberg, U., Sikron, N., Venturini, L., Buson, G., Ghan, R., et al. (2014). Metabolite and transcript profiling of berry skin during fruit development elucidates differential regulation between Cabernet sauvignon and Shiraz cultivars at branching points in the polyphenol pathway. BMC Plant Biol. 14, 188. doi: 10.1186/s12870-014-0188-4
Degu, A., Morcia, C., Tumino, G., Hochberg, U., Toubiana, D., Mattivi, F., et al. (2015). Metabolite profiling elucidates communalities and differences in the polyphenol biosynthetic pathways of red and white Muscat genotypes. Plant Physiol. Biochem. 86, 24–33. doi: 10.1016/j.plaphy.2014.11.006
De La Fuente, L., Parker, J. K., Oliver, J. E., Granger, S., Brannen, P. M., Santen, E., et al. (2013). The bacterial pathogen Xylella fastidiosa affects the leaf ionome of plant hosts during infection. PloS One 8, e62945. doi: 10.1371/journal.pone.0062945
Deluc, L. G., Grimplet, J., Wheatley, M. D., Tillett, R. L., Quilici, D. R., Osborne, C., et al. (2007). Transcriptomic and metabolite analyses of Cabernet sauvignon grape berry development. BMC Genomics 8, 1–42. doi: 10.1186/1471-2164-8-429
Deluc, L. G., Quilici, D. R., Decendit, A., Grimplet, J., Wheatley, M. D., Schlauch, K. A., et al. (2009). Water deficit alters differentially metabolic pathways affecting important flavor and quality traits in grape berries of Cabernet sauvignon and Chardonnay. BMC Genomics 10, 212. doi: 10.1186/1471-2164-10-212
Deytieux, C., Geny, L., Lapaillerie, D., Claverol, S., Bonneu, M., Donèche, B. (2007). Proteome analysis of grape skins during ripening. J. Exp. Bot. 58, 1851–1862. doi: 10.1093/jxb/erm049
Díaz-Riquelme, J., Grimplet, J., Martínez-Zapater, J. M., Carmona, M. J. (2012). Transcriptome variation along bud development in grapevine (Vitis vinifera l.). BMC Plant Biol. 12, 181. doi: 10.1186/1471-2229-12-181
Díaz-Riquelme, J., Martínez-Zapater, J. M., Carmona, M. J. (2014). Transcriptional analysis of tendril and inflorescence development in grapevine (Vitis vinifera l.). PloS One 9 (3), e92339. doi: 10.1371/journal.pone.0092339
Di Carli, M., Zamboni, A., Pè, M. E., Pezzotti, M., Lilley, K. S., Benvenuto, E., et al. (2011). Two-dimensional differential in gel electrophoresis (2D-DIGE) analysis of grape berry proteome during postharvest withering. J. Proteome Res. 10, 429–446. doi: 10.1021/pr1005313
D’Incà, E., Cazzaniga, S., Foresti, C., Vitulo, N., Bertini, E., Galli, M., et al. (2021). VviNAC33 promotes organ de-greening and represses vegetative growth during the vegetative-to-mature phase transition in grapevine. New Phytol. 231 (2), 726–746. doi: 10.1111/nph.17263
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, F., Zaleski, C., Jha, S., et al. (2013)STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29 (1), 15–21 doi: 10.1093/bioinformatics/bts635.
Ehrhardt, C., Arapitsas, P., Stefanini, M., Flick, G., Mattivi, F. (2014). Analysis of the phenolic composition of fungus-resistant grape varieties cultivated in Italy and Germany using UHPLC-MS/MS. J. Mass Spectrometry 49, 860–869. doi: 10.1002/jms.3440
Eisenmann, B., Czemmel, S., Ziegler, T., Buchholz, G., Kortekamp, A., Trapp, O., et al. (2019). Rpv3–1 mediated resistance to grapevine downy mildew is associated with specific host transcriptional responses and the accumulation of stilbenes. BMC Plant Biol. 19, 343. doi: 10.1186/s12870-019-1935-3
Fasoli, M., Richter, C. L., Zenoni, S., Bertini, E., Vitulo, N., Santo, S. D., et al. (2018). Timing and order of the molecular events marking the onset of berry ripening in grapevine. Plant Physiol. 178, 1187–1206. doi: 10.1104/pp.18.00559
Fasoli, M., Santo, S. D., Zenoni, S., Tornielli, G. B., Farina, L., Zamboni, A., et al. (2012). The grapevine expression atlas reveals a deep transcriptome shift driving the entire plant into a maturation program. Plant Cell 24, 3489–3505. doi: 10.1105/tpc.112.100230
Ferraro, M. ,. B., Giordani, P., Serafini, A. (2019). Fclust: An r package for fuzzy clustering. R J. 11 (1), 198. doi: 10.32614/RJ-2019-017
Fiehn, O., Kopka, J., Dörmann, P., Altmann, T., Trethewey, R. N., Willmitzer, L. (2000). Metabolite profiling for plant functional genomics. Nat. Biotechnol. 18, 1157–1161. doi: 10.1038/81137
Figueiredo, A., Fortes, A. M., Ferreira, S., Sebastiana, M., Choi, Y. H., Sousa, L., et al. (2008). Transcriptional and metabolic profiling of grape (Vitis vinifera l.) leaves unravel possible innate resistance against pathogenic fungi. J. Exp. Bot. 59, 3371–3381. doi: 10.1093/jxb/ern187
Figueiredo, A., Martins, J., Sebastiana, M., Guerreiro, A., Silva, A., Matos, A. R., et al. (2017). Specific adjustments in grapevine leaf proteome discriminating resistant and susceptible grapevine genotypes to Plasmopara viticola. J. Proteomics 152, 48–57. doi: 10.1016/j.jprot.2016.10.012
Flamini, R., De Rosso, M., Bavaresco, L. (2015). Study of grape polyphenols by liquid chromatography-high-resolution mass spectrometry (UHPLC/QTOF) and suspect screening analysis. J. Analytical Methods Chem. 2015, e350259. doi: 10.1155/2015/350259
Fortes, A. M., Agudelo-Romero, P., Silva, M. S., Ali, K., Sousa, L., Maltese, F., et al. (2011). Transcript and metabolite analysis in trincadeira cultivar reveals novel information regarding the dynamics of grape ripening. BMC Plant Biol. 11, 149. doi: 10.1186/1471-2229-11-149
Fung, R. W. M., Gonzalo, M., Fekete, C., Kovacs, L. G., He, Y., Marsh, E., et al. (2008). Powdery mildew induces defense-oriented reprogramming of the transcriptome in a susceptible but not in a resistant grapevine. Plant Physiol. 146, 236–249. doi: 10.1104/pp.107.108712
Ghan, R., Petereit, J., Tillett, R. L., Schlauch, K. A., Toubiana, D., Fait, A., et al. (2017). The common transcriptional subnetworks of the grape berry skin in the late stages of ripening. BMC Plant Biol. 17, 94. doi: 10.1186/s12870-017-1043-1
Ghan, R., Van Sluyter, S. C., Hochberg, U., Degu, A., Hopper, D. W., Tillet, R. L., et al. (2015). Five omic technologies are concordant in differentiating the biochemical characteristics of the berries of five grapevine (Vitis vinifera l.) cultivars. BMC Genomics 16, 946. doi: 10.1186/s12864-015-2115-y
Giribaldi, M., Gény, L., Delrot, S., Schubert, A. (2010). Proteomic analysis of the effects of ABA treatments on ripening Vitis vinifera berries. J. Exp. Bot. 61, 2447–2458. doi: 10.1093/jxb/erq079
Giribaldi, M., Perugini, I., Sauvage, F.-X., Schubert, A. (2007). Analysis of protein changes during grape berry ripening by 2-DE and MALDI-TOF. PROTEOMICS 7, 3154–70. doi: 10.1002/pmic.200600974
Goff, S. A., Ricke, D., Lan, T.-H., Presting, G., Wang, R., Dunn, M., et al. (2002). A draft sequence of the rice genome (Oryza sativa l. ssp. japonica). Science 296, 92–100. doi: 10.1126/science.1068275
Gouthu, S., O’Neil, S. T., Di, Y., Ansarolia, M., Megraw, M., Deluc, L. G. (2014). A comparative study of ripening among berries of the grape cluster reveals an altered transcriptional programme and enhanced ripening rate in delayed berries. J. Exp. Bot. 65, 5889–5902. doi: 10.1093/jxb/eru329
Gratl, V., Sturm, S., Zini, E., Letschka, T., Stefanini, M., Vezzulli, S., et al. (2021). Comprehensive polyphenolic profiling in promising resistant grapevine hybrids including 17 novel breeds in northern Italy. J. Sci. Food Agric. 101, 2380–2388. doi: 10.1002/jsfa.10861
Griesser, M., Savoi, S., Supapvanich, S., Dobrev, P., Vankova, R., Forneck, A. (2020). Phytohormone profiles are strongly altered during induction and symptom development of the physiological ripening disorder berry shrivel in grapevine. Plant Mol. Biol 103, 141–157. doi: 10.1007/s11103-020-00980-6
Griesser, M., Weingart, G., Schoedl-Hummel, K., Neumann, N., Becker, M., Varmuza, K., et al. (2015). Severe drought stress is affecting selected primary metabolites, polyphenols, and volatile metabolites in grapevine leaves (Vitis vinifera cv. pinot noir). Plant Physiol. Biochem. 88, 17–26. doi: 10.1016/j.plaphy.2015.01.004
Grimplet, J., Cramer, G. R., Dickerson, J. A., Mathiason, K., Hemert, J. V., Fennell, A. Y. (2009). VitisNet: “Omics“ integration through grapevine molecular networks. PloS One 4, e8365. doi: 10.1371/journal.pone.0008365
Grimplet, J., Deluc, L. G., Tillett, R. L., Wheatley, M. D., Schlauch, K. A., Cramer, G. R., et al. (2007). Tissue-specific mRNA expression profiling in grape berry tissues. BMC Genomics 8, 187. doi: 10.1186/1471-2164-8-187
Grimplet, J., Hemert, J. V., Carbonell-Bejerano, P., Díaz-Riquelme, J., Dickerson, J., Fennell, A., et al. (2012). Comparative analysis of grapevine whole-genome gene predictions, functional annotation, categorization and integration of the predicted gene sequences. BMC Res. Notes 5, 213. doi: 10.1186/1756-0500-5-213
Grimplet, J., Tello, J., Laguna, N., Ibáñez, J. (2017). Differences in flower transcriptome between grapevine clones are related to their cluster compactness, fruitfulness, and berry size. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00632
Guo, D. L., Wang, Z., Pei, M.-S., Guo, L.-L., Yu, Y.-H. (2020). Transcriptome analysis reveals mechanism of early ripening in kyoho grape with hydrogen peroxide treatment. BMC Genomics 21, 784. doi: 10.1186/s12864-020-07180-y
Guo, D.-L., Xi, F.-F., Yu, Y.-H., Zhang, X.-Y., Zhang, G.-H., Zhong, G.-Y. (2016). Comparative RNA-seq profiling of berry development between table grape ‘Kyoho’ and its early-ripening mutant ’Fengzao’. BMC Genomics 17, 795. doi: 10.1186/s12864-016-3051-1
Haile, Z. M., Malacarne, G., Pilati, S., Sonego, P., Moretto, M., Masuero, D., et al. (2020). Dual transcriptome and metabolic analysis of Vitis vinifera cv. pinot noir berry and Botrytis cinerea during quiescence and egressed infection. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01704
Haile, Z. M., Pilati, S., Sonego, P., Malacarne, G., Vrhovsek, U., Engelen, K., et al. (2017). Molecular analysis of the early interaction between the grapevine flower and Botrytis cinerea reveals that prompt activation of specific host pathways leads to fungus quiescence. Plant Cell Environ. 40, 1409–28. doi: 10.1111/pce.12937
Harb, J., Alseekh, S., Tohge, T., Fernie, A. R. (2015). Profiling of primary metabolites and flavonols in leaves of two table grape varieties collected from semiarid and temperate regions. Phytochemistry 117, 444–55. doi: 10.1016/j.phytochem.2015.07.013
Haug, K., Cochrane, K., Nainala, V. C., Williams, M., Chang, J., Jayaseelan, K. V., et al. (2020). MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 48, D440–D444. doi: 10.1093/nar/gkz1019
Herrera, J. C., Hochberg, U., Degu, A., Sabbatini, P., Lazarovitch, N., Castellarin, S. D., et al. (2017). Grape metabolic response to postveraison water deficit is affected by interseason weather variability. J. Agric. Food Chem. 65, 5868–5878. doi: 10.1021/acs.jafc.7b01466
He, L., Xu, X.-Q., Wang, Y., Chen, W.-K., Sun, R.-Z., Cheng, G., et al. (2020). Modulation of volatile compound metabolome and transcriptome in grape berries exposed to sunlight under dry-hot climate. BMC Plant Biol. 20, 59. doi: 10.1186/s12870-020-2268-y
Hirai, M. Y., Klein, M., Fujikawa, Y., Yano, M., Goodenowe, D. B., Yamazaki, Y., et al. (2005). Elucidation of gene-to-gene and metabolite-to-gene networks in arabidopsis by integration of metabolomics and transcriptomics. J. Biol. Chem. 280, 25590–25595. doi: 10.1074/jbc.M502332200
Hochberg, U., Batushansky, A., Degu, A., Rachmilevitch, S., Fait, A. (2015a). Metabolic and physiological responses of Shiraz and Cabernet sauvignon (Vitis vinifera l.) to near optimal temperatures of 25 and 35 °C. Int. J. Mol. Sci. 16, 24276–24294. doi: 10.3390/ijms161024276
Hochberg, U., Degu, A., Cramer, G. R., Rachmilevitch, S., Fait, A. (2015b). Cultivar specific metabolic changes in grapevines berry skins in relation to deficit irrigation and hydraulic behavior. Plant Physiol. Biochem. 88, 42–52. doi: 10.1016/j.plaphy.2015.01.006
Hochberg, U., Degu, A., Toubiana, D., Gendler, T., Nikoloski, Z., Rachmilevitch, S., et al. (2013). Metabolite profiling and network analysis reveal coordinated changes in grapevine water stress response. BMC Plant Biol. 13, 184. doi: 10.1186/1471-2229-13-184
Hong, Y.-S., Martinez, A., Liger-Belair, G., Jeandet, P., Nuzillard, J.-M., Cilindre, C. (2012). Metabolomics reveals simultaneous influences of plant defence system and fungal growth in Botrytis cinerea-infected Vitis vinifera cv. Chardonnay berries. J. Exp. Bot. 63, 5773–5785. doi: 10.1093/jxb/ers228
Jaillon, O., Aury, J. M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467. doi: 10.1038/nature06148
Kambiranda, D., Basha, S. M., Singh, R. K., He, H., Calvin, K., Mercer, R. (2016). In depth proteome analysis of ripening muscadine grape berry cv. carlos reveals proteins associated with flavor and aroma compounds. J. Proteome Res. 15, 2910–2923. doi: 10.1021/acs.jproteome.5b01064
Kambiranda, D., Basha, S. M., Singh, R., Snowden, J., Mercer, R. (2018). Proteome profile of american hybrid grape cv. blanc du Bois during ripening reveals proteins associated with flavor volatiles and ethylene production. PROTEOMICS 18, 1700305. doi: 10.1002/pmic.201700305
Kobayashi, S., Goto-Yamamoto, N., Hirochika, H. (2004). Retrotransposon-induced mutations in grape skin color. Science 304, 982–982. doi: 10.1126/science.1095011
Kuang, L., Chen, S., Guo, Y., Ma, H. (2019). Quantitative proteome analysis reveals changes in the protein landscape during grape berry development with a focus on vacuolar transport proteins. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00641
Langfelder, P., Horvath, S. (2008). WGCNA: an r package for weighted correlation network analysis. BMC Bioinf. 9, 559. doi: 10.1186/1471-2105-9-559
Lawo, N. C., Weingart, G. J. F., Schuhmacher, R., Forneck, A. (2011). The volatile metabolome of grapevine roots: First insights into the metabolic response upon phylloxera attack. Plant Physiol. Biochem. 49, 1059–1063. doi: 10.1016/j.plaphy.2011.06.008
Lecourieux, F., Kappel, C., Pieri, P., Charon, J., Pillet, J., Hilbert, G., et al. (2017). Dissecting the biochemical and transcriptomic effects of a locally applied heat treatment on developing Cabernet sauvignon grape berries. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00053
Liao, Y., Smyth, G. K., Shi, W. (2014)featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30 (7), 923–930 doi: 10.1093/bioinformatics/btt656.
Lijavetzky, D., Carbonell-Bejerano, P., Grimplet, J., Bravo, G., Flores, P., Fenoll, J., et al. (2012). Berry flesh and skin ripening features in Vitis vinifera as assessed by transcriptional profiling. PloS One 7, e39547. doi: 10.1371/journal.pone.0039547
Liu, G.-T., Wang, J.-F., Cramer, G., Dai, Z.-W., Duan, W., Xu, H.-G., et al. (2012). Transcriptomic analysis of grape (Vitis vinifera l.) leaves during and after recovery from heat stress. BMC Plant Biol. 12, 174. doi: 10.1186/1471-2229-12-174
Liu, G.-T., Wang, B.-B., Lecourieux, D., Li, M.-J., Liu, M.-B., Liu, R.-Q., et al. (2021). Proteomic analysis of early-stage incompatible and compatible interactions between grapevine and p. viticola. Hortic. Res. 8, 1–21. doi: 10.1038/s41438-021-00533-y
Livigni, S., Lucini, L., Sega, D., Navacchi, O., Pandolfini, T., Zamboni, A., et al. (2019). The different tolerance to magnesium deficiency of two grapevine rootstocks relies on the ability to cope with oxidative stress. BMC Plant Biol. 19, 148. doi: 10.1186/s12870-019-1726-x
Li, X., Wu, J., Yin, L., Zhang, Y., Qu, J., Lu, J. (2015). Comparative transcriptome analysis reveals defense-related genes and pathways against downy mildew in Vitis amurensis grapevine. Plant Physiol. Biochem. 95, 1–14. doi: 10.1016/j.plaphy.2015.06.016
Lorenzini, M., Millioni, R., Franchin, C., Zapparoli, G., Arrigoni, G., Simonato, B. (2015). Identification of potential protein markers of noble rot infected grapes. Food Chem. 179, 170–174. doi: 10.1016/j.foodchem.2015.01.112
Loyola, R., Herrera, D., Mas, A., Wong, D. C. J., Höll, J., Cavallini, E., et al. (2016). The photomorphogenic factors UV-b RECEPTOR 1, ELONGATED HYPOCOTYL 5, and HY5 HOMOLOGUE are part of the UV-b signalling pathway in grapevine and mediate flavonol accumulation in response to the environment. J. Exp. Bot. 67, 5429–5445. doi: 10.1093/jxb/erw307
Magris, G., Jurman, I., Fornasiero, A., Paparelli, E., Schwope, R., Marroni, F., et al. (2021). The genomes of 204 Vitis vinifera accessions reveal the origin of European wine grapes. Nat. Commun. 12, 7240. doi: 10.1038/s41467-021-27487-y
Maia, M., Ferreira, A. E. N., Nascimento, R., Monteiro, F., Traquete, F., Marques, A. P., et al. (2020). Integrating metabolomics and targeted gene expression to uncover potential biomarkers of fungal/oomycetes-associated disease susceptibility in grapevine. Sci. Rep. 10, 15688. doi: 10.1038/s41598-020-72781-2
Malacarne, G., Costantini, L., Coller, E., Battilana, J., Velasco, R., Vrhovsek, U., et al. (2015). Regulation of flavonol content and composition in (Syrah×Pinot noir) mature grapes: integration of transcriptional profiling and metabolic quantitative trait locus analyses. J. Exp. Bot. 66, 4441–4453. doi: 10.1093/jxb/erv243
Malacarne, G., Vrhovsek, U., Zulini, L., Cestaro, A., Stefanini, M., Mattivi, F., et al. (2011). Resistance to Plasmopara viticola in a grapevine segregating population is associated with stilbenoid accumulation and with specific host transcriptional responses. BMC Plant Biol. 11, 114. doi: 10.1186/1471-2229-11-114
Mao, L., Van Hemert, J. L., Dash, S., Dickerson, J. A. (2009). Arabidopsis gene co-expression network and its functional modules. BMC Bioinf. 10 (1), 346. doi: 10.1186/1471-2105-10-346
Martínez-Esteso, M. J., Casado-Vela, J., Sellés-Marchart, S., Elortza, F., Pedreño, M. A., Bru-Martínez, R. (2011a). iTRAQ-based profiling of grape berry exocarp proteins during ripening using a parallel mass spectrometric method. Mol. Biosyst. 7, 749–765. doi: 10.1039/C0MB00194E
Martínez-Esteso, M. J., Sellés-Marchart, S., Lijavetzky, D., Pedreño, M. A., Bru-Martínez, R. (2011b). A DIGE-based quantitative proteomic analysis of grape berry flesh development and ripening reveals key events in sugar and organic acid metabolism. J. Exp. Bot. 62, 2521–2569. doi: 10.1093/jxb/erq434
Martínez-Esteso, M. J., Vilella-Antón, M. T., Pedreño, M. Á., Valero, M. L., Bru-Martínez, R. (2013). iTRAQ-based protein profiling provides insights into the central metabolism changes driving grape berry development and ripening. BMC Plant Biol. 13, 167. doi: 10.1186/1471-2229-13-167
Massonnet, M., Cochetel, N., Minio, A., Vondras, A. M., Lin, J., Muyle, A., et al. (2020). The genetic basis of sex determination in grapes. Nat. Commun. 11, 2902. doi: 10.1038/s41467-020-16700-z
Massonnet, M., Fasoli, M., Tornielli, G. B., Altieri, M., Sandri, M., Zuccolotto, P., et al. (2017). Ripening transcriptomic program in red and white grapevine varieties correlates with berry skin anthocyanin accumulation. Plant Physiol. 174, 2376–2396. doi: 10.1104/pp.17.00311
Mattivi, F., Guzzon, R., Vrhovsek, U., Stefanini, M., Velasco, R. (2006). Metabolite profiling of grape: flavonols and anthocyanins. J. Agric. Food Chem. 54, 7692–7702. doi: 10.1021/jf061538c
Matus, J. T., Cavallini, E., Loyola, R., Höll, J., Finezzo, L., Dal Santo, S., et al. (2017). A group of grapevine MYBA transcription factors located in chromosome 14 control anthocyanin synthesis in vegetative organs with different specificities compared with the berry color locus. Plant J. 91 (2), 220–36. doi: 10.1111/tpj.13558
Matus, J. T., Ruggieri, V., Romero, F. J., Moretto, M., Wong, D. C. J. (2019). “Status and prospects of systems biology in grapevine research,” in The grape genome, compendium of plant genomes. Eds. Cantu, D., Walker, M. A. (Cham: Springer International Publishing), 137–166. doi: 10.1007/978-3-030-18601-2_8
Milli, A., Cecconi, D., Bortesi, L., Persi, A., Rinalducci, S., Zamboni, A., et al. (2012). Proteomic analysis of the compatible interaction between Vitis vinifera and Plasmopara viticola. J. Proteomics 75, 1284–1302. doi: 10.1016/j.jprot.2011.11.006
Minio, A., Massonnet, M., Figueroa-Balderas, R., Castro, A., Cantu, D. (2019a). Diploid genome assembly of the wine grape carménère. G3 Genes|Genomes|Genetics 9, 1331–1337. doi: 10.1534/g3.119.400030
Minio, A., Massonnet, M., Figueroa-Balderas, R., Vondras, A. M., Blanco-Ulate, B., Cantu, D. (2019b). Iso-seq allows genome-independent transcriptome profiling of grape berry development. G3: Genes Genomes Genet. 9, 755–767. doi: 10.1534/g3.118.201008
Moretto, M., Sonego, P., Pilati, S., Malacarne, G., Costantini, L., Grzeskowiak, L., et al. (2016). VESPUCCI: Exploring patterns of gene expression in grapevine. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00633
Moretto, M., Sonego, P., Pilati, S., Matus, J. T., Costantini, L., Malacarne, G., et al. (2022). A COMPASS for VESPUCCI: A FAIR way to explore the grapevine transcriptomic landscape. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.815443
Naithani, S., Raja, R., Waddell, E. N., Elser, J., Gouthu, S., Deluc, L. G., et al. (2014). VitisCyc: a metabolic pathway knowledgebase for grapevine (Vitis vinifera). Front. Plant Sci. 5. doi: 10.3389/fpls.2014.00644
Narduzzi, L., Stanstrup, J., Mattivi, F. (2015). Comparing wild American grapes with Vitis vinifera: a metabolomics study of grape composition. J. Agric. Food Chem. 63, 6823–6834. doi: 10.1021/acs.jafc.5b01999
Nascimento-Gavioli, M. C. A., Agapito-Tenfen, S. Z., Nodari, R. O., Welter, L. J., Sanchez Mora, F. D., Saifert, L., et al. (2017). Proteome of Plasmopara viticola-infected Vitis vinifera provides insights into grapevine Rpv1/Rpv3 pyramided resistance to downy mildew. J. Proteomics Braz. Proteomics 151, 264–274. doi: 10.1016/j.jprot.2016.05.024
Navarro-Payá, D., Santiago, A., Orduña, L., Zhang, C., Amato, A., D’Inca, E., et al. (2022). The grape gene reference catalogue as a standard resource for gene selection and genetic improvement. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.803977
Negrel, L., Halter, D., Wiedemann-Merdinoglu, S., Rustenholz, C., Merdinoglu, D., Hugueney, P., et al. (2018). Identification of lipid markers of Plasmopara viticola infection in grapevine using a non-targeted metabolomic approach. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00360
Negri, S., Lovato, A., Boscaini, F., Salvetti, E., Torriani, S., Commisso, M., et al. (2017). The induction of noble rot (Botrytis cinerea) infection during postharvest withering changes the metabolome of grapevine berries (Vitis vinifera l., cv. garganega). Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01002
Negri, A. S., Prinsi, B., Rossoni, M., Failla, O., Scienza, A., Cocucci, M., et al. (2008a). Proteome changes in the skin of the grape cultivar barbera among different stages of ripening. BMC Genomics 9, 378. doi: 10.1186/1471-2164-9-378
Negri, A. S., Prinsi, B., Scienza, A., Morgutti, S., Cocucci, M., Espen, L. (2008b). Analysis of grape berry cell wall proteome: A comparative evaluation of extraction methods. J. Plant Physiol. 165, 1379–1389. doi: 10.1016/j.jplph.2007.10.011
Nikiforova, V. J., Daub, C. O., Hesse, H., Willmitzer, L., Hoefgen, R. (2005). Integrative gene-metabolite network with implemented causality deciphers informational fluxes of sulphur stress response. J. Exp. Bot. 56 (417), 1887–1896. doi: 10.1093/jxb/eri179
Niu, N., Cao, Y., Duan, W., Wu, B., Li, S. (2013). Proteomic analysis of grape berry skin responding to sunlight exclusion. J. Plant Physiol. 170, 748–757. doi: 10.1016/j.jplph.2012.12.020
Nwafor, C. C., Gribaudo, I., Schneider, A., Wehrens, R., Grando, M. S., Costantini, L. (2014). Transcriptome analysis during berry development provides insights into co-regulated and altered gene expression between a seeded wine grape variety and its seedless somatic variant. BMC Genomics 15, 1030. doi: 10.1186/1471-2164-15-1030
Orduña, L., Li, M., Navarro-Payá, D., Zhang, C., Santiago, A., Romero, P., et al. (2022). Direct regulation of shikimate, early phenylpropanoid and stilbenoid pathways by subgroup 2 R2R3-MYBs in grapevine. Plant J 110 (2), 529–547. doi: 10.1111/tpj.15686
Pagliarani, C., Moine, A., Chitarra, W., Meloni, G. R., Abbà, S., Nerva, L., et al. (2020). The molecular priming of defense responses is differently regulated in grapevine genotypes following elicitor application against powdery mildew. Int. J. Mol. Sci. 21, 6776. doi: 10.3390/ijms21186776
Pastore, C., Zenoni, S., Fasoli, M., Pezzotti, M., Tornielli, G. B., Filippetti, I. (2013). Selective defoliation affects plant growth, fruit transcriptional ripening program and flavonoid metabolism in grapevine. BMC Plant Biol. 13, 30. doi: 10.1186/1471-2229-13-30
Pastore, C., Zenoni, S., Tornielli, G. B., Allegro, G., Dal Santo, S., Valentini, G., et al. (2011). Increasing the source/sink ratio in Vitis vinifera (cv sangiovese) induces extensive transcriptome reprogramming and modifies berry ripening. BMC Genomics 12, 631. doi: 10.1186/1471-2164-12-631
Perrone, I., Pagliarani, C., Lovisolo, C., Chitarra, W., Roman, F., Schubert, A. (2012). Recovery from water stress affects grape leaf petiole transcriptome. Planta 235, 1383–1396. doi: 10.1007/s00425-011-1581-y
Pervaiz, T., Haifeng, J., Haider, M. S., Cheng, Z., Cui, M., Wang, M., et al. (2016). Transcriptomic analysis of grapevine (cv. summer black) leaf, using the illumina platform. PloS One 11, e0147369. doi: 10.1371/journal.pone.0147369
Pii, Y., Zamboni, A., Dal Santo, S., Pezzotti, M., Varanini, Z., Pandolfini, T. (2017). Prospect on ionomic signatures for the classification of grapevine berries according to their geographical origin. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00640
Pilati, S., Brazzale, D., Guella, G., Milli, A., Ruberti, C., Biasioli, F., et al. (2014). The onset of grapevine berry ripening is characterized by ROS accumulation and lipoxygenase-mediated membrane peroxidation in the skin. BMC Plant Biol. 14, 87. doi: 10.1186/1471-2229-14-87
Pilati, S., Malacarne, G., Navarro-Payá, D., Tomè, G., Riscica, L., Cavecchia, V., et al. (2021). Vitis OneGenE: a causality-based approach to generate gene networks in vitis vinifera sheds light on the laccase and dirigent gene families. Biomolecules 11, 1744. doi: 10.3390/biom11121744
Pilati, S., Perazzolli, M., Malossini, A., Cestaro, A., Demattè, L., Fontana, P., et al. (2007). Genome-wide transcriptional analysis of grapevine berry ripening reveals a set of genes similarly modulated during three seasons and the occurrence of an oxidative burst at vèraison. BMC Genomics 8, 428. doi: 10.1186/1471-2164-8-428
Pimentel, D., Amaro, R., Erban, A., Mauri, N., Soares, F., Rego, C., et al. (2021). Transcriptional, hormonal, and metabolic changes in susceptible grape berries under powdery mildew infection. J. Exp. Bot. 72, 6544–6569. doi: 10.1093/jxb/erab258
Pinasseau, L., Vallverdú-Queralt, A., Verbaere, A., Roques, M., Meudec, E., Le Cunff, L., et al. (2017). Cultivar diversity of grape skin polyphenol composition and changes in response to drought investigated by LC-MS based metabolomics. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01826
Polesani, M., Bortesi, L., Ferrarini, A., Zamboni, A., Fasoli, M., Zadra, C., et al. (2010). General and species-specific transcriptional responses to downy mildew infection in a susceptible (Vitis vinifera) and a resistant (V. riparia) grapevine species. BMC Genomics 11, 117. doi: 10.1186/1471-2164-11-117
Pontin, M. A., Piccoli, P. N., Francisco, R., Bottini, R., Martinez-Zapater, J. M., Lijavetzky, D. (2010). Transcriptome changes in grapevine (Vitis vinifera l.) cv. Malbec leaves induced by ultraviolet-b radiation. BMC Plant Biol. 10, 224. doi: 10.1186/1471-2229-10-224
Pucker, B., Schwandner, A., Becker, S., Hausmann, L., Viehöver, P., Töpfer, R., et al. (2020). RNA-Seq time series of Vitis vinifera bud development reveals correlation of expression patterns with the local temperature profile. Plants 9, 1548. doi: 10.3390/plants9111548
Reshef, N., Agam, N., Fait, A. (2018). Grape berry acclimation to excessive solar irradiance leads to repartitioning between major flavonoid groups. J. Agric. Food Chem. 66, 3624–3636. doi: 10.1021/acs.jafc.7b04881
Reshef, N., Walbaum, N., Agam, N., Fait, A. (2017). Sunlight modulates fruit metabolic profile and shapes the spatial pattern of compound accumulation within the grape cluster. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00070
Rienth, M., Torregrosa, L., Kelly, M. T., Luchaire, N., Pellegrino, A., Grimplet, J., et al. (2014a). Is transcriptomic regulation of berry development more important at night than during the day? PloS One 9, e88844. doi: 10.1371/journal.pone.0088844
Rienth, M., Torregrosa, L., Luchaire, N., Chatbanyong, R., Lecourieux, D., Kelly, M. T., et al. (2014b). Day and night heat stress trigger different transcriptomic responses in green and ripening grapevine (Vitis vinifera) fruit. BMC Plant Biol. 14, 108. doi: 10.1186/1471-2229-14-108
Rienth, M., Torregrosa, L., Sarah, G., Ardisson, M., Brillouet, J.-M., Romieu, C. (2016). Temperature desynchronizes sugar and organic acid metabolism in ripening grapevine fruits and remodels their transcriptome. BMC Plant Biol. 16, 164. doi: 10.1186/s12870-016-0850-0
Royo, C., Carbonell-Bejerano, P., Torres-Pérez, R., Nebish, A., Martínez, Ó., Rey, M., et al. (2016). Developmental, transcriptome, and genetic alterations associated with parthenocarpy in the grapevine seedless somatic variant corinto bianco. J. Exp. Bot. 67, 259–273. doi: 10.1093/jxb/erv452
Ruocco, S., Stefanini, M., Stanstrup, J., Perenzoni, D., Mattivi, F., Vrhovsek, U. (2017). The metabolomic profile of red non-v. vinifera genotypes. Food Research International 98, 10–19. doi: 10.1016/j.foodres.2017.01.024
Saito, K., Hirai, M. Y., Yonekura-Sakakibara, K. (2008). Decoding genes with coexpression networks and metabolomics – ‘majority report by precogs. Trends Plant Sci. 13, 36–43. doi: 10.1016/j.tplants.2007.10.006
Santos, R. B., Nascimento, R., V. Coelho, A., Figueiredo, A. (2020). Grapevine–downy mildew rendezvous: proteome analysis of the first hours of an incompatible interaction. Plants 9, 1498. doi: 10.3390/plants9111498
Sarry, J.-E., Sommerer, N., Sauvage, F.-X., Bergoin, A., Rossignol, M., Albagnac, G., et al. (2004). Grape berry biochemistry revisited upon proteomic analysis of the mesocarp. Proteomics 4, 201–15. doi: 10.1002/pmic.200300499
Savoi, S., Arapitsas, P., Duchêne, É., Nikolantonaki, M., Ontañón, I., Carlin, S., et al. (2021a). Grapevine and wine metabolomics-based guidelines for FAIR data and metadata management. Metabolites 11, 757. doi: 10.3390/metabo11110757
Savoi, S., Herrera, J. C., Forneck, A., Griesser, M. (2019). Transcriptomics of the grape berry shrivel ripening disorder. Plant Mol. Biol. 100, 285–301. doi: 10.1007/s11103-019-00859-1
Savoi, S., Torregrosa, L., Romieu, C. (2021b). Transcripts switched off at the stop of phloem unloading highlight the energy efficiency of sugar import in the ripening v. vinifera fruit. Hortic. Res. 8, 1–15. doi: 10.1038/s41438-021-00628-6
Savoi, S., Wong, D. C. J., Arapitsas, P., Miculan, M., Bucchetti, B., Peterlunger, E., et al. (2016). Transcriptome and metabolite profiling reveals that prolonged drought modulates the phenylpropanoid and terpenoid pathway in white grapes (Vitis vinifera l.). BMC Plant Biol. 16, 67. doi: 10.1186/s12870-016-0760-1
Savoi, S., Wong, D. C. J., Degu, A., Herrera, J. C., Bucchetti, B., Peterlunger, E., et al. (2017). Multi-omics and integrated network analyses reveal new insights into the systems relationships between metabolites, structural genes, and transcriptional regulators in developing grape berries (Vitis vinifera l.) exposed to water deficit. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01124
Shangguan, L., Chen, M., Fang, X., Xie, Z., Gong, P., Huang, Y., et al. (2020). Comparative transcriptome analysis provides insight into regulation pathways and temporal and spatial expression characteristics of grapevine (Vitis vinifera) dormant buds in different nodes. BMC Plant Biol. 20, 390. doi: 10.1186/s12870-020-02583-1
Shangguan, L., Mu, Q., Fang, X., Zhang, K., Jia, H., Li, X., et al. (2017). RNA-Sequencing reveals biological networks during table grapevine (‘Fujiminori’) fruit development. PloS One 12, e0170571. doi: 10.1371/journal.pone.0170571
Sreekantan, L., Mathiason, K., Grimplet, J., Schlauch, K., Dickerson, J. A., Fennell, A. Y. (2010). Differential floral development and gene expression in grapevines during long and short photoperiods suggests a role for floral genes in dormancy transitioning. Plant Mol. Biol. 73, 191–205. doi: 10.1007/s11103-010-9611-x
Sun, R.-Z., Cheng, G., Li, Q., Zhu, Y.-R., Zhang, X., Wang, Y., et al. (2019). Comparative physiological, metabolomic, and transcriptomic analyses reveal developmental stage-dependent effects of cluster bagging on phenolic metabolism in Cabernet sauvignon grape berries. BMC Plant Biol. 19, 583. doi: 10.1186/s12870-019-2186-z
Suzuki, M., Nakabayashi, R., Ogata, Y., Sakurai, N., Tokimatsu, T., Goto, S., et al. (2015). Multiomics in grape berry skin revealed specific induction of the stilbene synthetic pathway by ultraviolet-c irradiation. Plant Physiol. 168, 47–59. doi: 10.1104/pp.114.254375
Sweetman, C., Wong, D. C., Ford, C. M., Drew, D. P. (2012). Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genomics 13, 691. doi: 10.1186/1471-2164-13-691
Tello-Ruiz, M. K., Naithani, S., Gupta, P., Olson, A., Wei, S., Preece, J., et al. (2021). Gramene 2021: harnessing the power of comparative genomics and pathways for plant research. Nucleic Acids Res. 49, D1452–D1463. doi: 10.1093/nar/gkaa979
Terrier, N., Glissant, D., Grimplet, J., Barrieu, F., Abbal, P., Couture, C., et al. (2005). Isogene specific oligo arrays reveal multifaceted changes in gene expression during grape berry (Vitis vinifera l.) development. Planta 222, 832–847. doi: 10.1007/s00425-005-0017-y
Theine, J., Holtgräwe, D., Herzog, K., Schwander, F., Kicherer, A., Hausmann, L., et al. (2021). Transcriptomic analysis of temporal shifts in berry development between two grapevine cultivars of the pinot family reveals potential genes controlling ripening time. BMC Plant Biol. 21, 327. doi: 10.1186/s12870-021-03110-6
Tian, B., Harrison, R., Morton, J., Deb-Choudhury, S. (2015). Proteomic analysis of sauvignon blanc grape skin, pulp and seed and relative quantification of pathogenesis-related proteins. PloS One 10, e0130132. doi: 10.1371/journal.pone.0130132
Tuskan, G. A., DiFazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604. doi: 10.1126/science.1128691
VanderWeide, J., Medina-Meza, I. G., Frioni, T., Sivilotti, P., Falchi, R., Sabbatini, P. (2018). Enhancement of fruit technological maturity and alteration of the flavonoid metabolomic profile in merlot (Vitis vinifera l.) by early mechanical leaf removal. J. Agric. Food Chem. 66, 9839–9849. doi: 10.1021/acs.jafc.8b02709
Vannozzi, A., Palumbo, F., Magon, G., Lucchin, M., Barcaccia, G. (2021). The grapevine (Vitis vinifera l.) floral transcriptome in pinot noir variety: identification of tissue-related gene networks and whorl-specific markers in pre- and post-anthesis phases. Hortic. Res. 8, 1–20. doi: 10.1038/s41438-021-00635-7
Velasco, R., Zharkikh, A., Troggio, M., Cartwright, D. A., Cestaro, A., Pruss, D., et al. (2007). A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PloS One 2, e1326. doi: 10.1371/journal.pone.0001326
Venturini, L., Ferrarini, A., Zenoni, S., Tornielli, G. B., Fasoli, M., Santo, S. D., et al. (2013). De novo transcriptome characterization of Vitis vinifera cv. corvina unveils varietal diversity. BMC Genomics 14, 41. doi: 10.1186/1471-2164-14-41
Vincent, D., Ergül, A., Bohlman, M. C., Tattersall, E. A. R., Tillett, R. L., Wheatley, M. D., et al. (2007). Proteomic analysis reveals differences between Vitis vinifera l. cv. Chardonnay and cv. Cabernet sauvignon and their responses to water deficit and salinity. J. Exp. Bot. 58, 1873–1892. doi: 10.1093/jxb/erm012
Vondras, A. M., Commisso, M., Guzzo, F., Deluc, L. G. (2017). Metabolite profiling reveals developmental inequalities in pinot noir berry tissues late in ripening. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01108
Vondras, A. M., Minio, A., Blanco-Ulate, B., Figueroa-Balderas, R., Penn, M. A., Zhou, Y., et al. (2019). The genomic diversification of grapevine clones. BMC Genomics 20, 972. doi: 10.1186/s12864-019-6211-2
Vrhovsek, U., Lotti, C., Masuero, D., Carlin, S., Weingart, G., Mattivi, F. (2014). Quantitative metabolic profiling of grape, apple and raspberry volatile compounds (VOCs) using a GC/MS/MS method. J. Chromatogr. B Metabolomics II 966, 132–9. doi: 10.1016/j.jchromb.2014.01.009
Waters, D. L. E., Holton, T. A., Ablett, E. M., Lee, L. S., Henry, R. J. (2005). cDNA microarray analysis of developing grape (Vitis vinifera cv. Shiraz) berry skin. Funct. Integr. Genomics 5, 40–58. doi: 10.1007/s10142-004-0124-z
Weingart, G., Kluger, B., Forneck, A., Krska, R., Schuhmacher, R. (2012). Establishment and application of a metabolomics workflow for identification and profiling of volatiles from leaves of Vitis vinifera by HS-SPME-GC-MS. Phytochemical Anal. 23, 345–58. doi: 10.1002/pca.1364
Weng, K., Li, Z.-Q., Liu, R.-Q., Wang, L., Wang, Y.-J., Xu, Y. (2014). Transcriptome of Erysiphe necator-infected Vitis pseudoreticulata leaves provides insight into grapevine resistance to powdery mildew. Hortic. Res. 1, 1–12. doi: 10.1038/hortres.2014.49
Wilkinson, M. D., Dumontier, M., Aalbersberg, I., Appleton, G., Axton, M., Baak, A., et al. (2016). The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 160018. doi: 10.1038/sdata.2016.18
Wong, D. C. J. (2020). Network aggregation improves gene function prediction of grapevine gene co-expression networks. Plant Mol. Biol. 103, 425–41. doi: 10.1007/s11103-020-01001-2
Wong, D. C. J., Matus, J. T. (2017). Constructing integrated networks for identifying new secondary metabolic pathway regulators in grapevine: Recent applications and future opportunities. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00505
Wong, D. C., Sweetman, C., Drew, D. P., Ford, C. M. (2013). VTCdb: a gene co-expression database for the crop species vitis vinifera (grapevine). BMC Genomics 14, 882. doi: 10.1186/1471-2164-14-882
Wu, J., Zhang, Y., Zhang, H., Huang, H., Folta, K. M., Lu, J. (2010). Whole genome wide expression profiles of Vitis amurensis grape responding to downy mildew by using solexa sequencing technology. BMC Plant Biol. 10, 234. doi: 10.1186/1471-2229-10-234
Xin, H., Zhu, W., Wang, L., Xiang, Y., Fang, L., Li, J., et al. (2013). Genome wide transcriptional profile analysis of Vitis amurensis and Vitis vinifera in response to cold stress. PloS One 8, e58740. doi: 10.1371/journal.pone.0058740
Yang, B., He, S., Liu, Y., Liu, B., Ju, Y., Kang, D., et al. (2020). Transcriptomics integrated with metabolomics reveals the effect of regulated deficit irrigation on anthocyanin biosynthesis in Cabernet sauvignon grape berries. Food Chem. 314, 126170. doi: 10.1016/j.foodchem.2020.126170
Yu, J., Hu, S., Wang, J., Wong, G. K.-S., Li, S., Liu, B., et al. (2002). A draft sequence of the rice genome (Oryza sativa l. ssp. indica). Sci. 296, 79–92. doi: 10.1126/science.1068037
Zamboni, A., Carli, M. D., Guzzo, F., Stocchero, M., Zenoni, S., Ferrarini, A., et al. (2010). Identification of putative stage-specific grapevine berry biomarkers and omics data integration into networks. Plant Physiol. 154, 1439–1459. doi: 10.1104/pp.110.160275
Zenoni, S., Amato, A., D’Incà, E., Guzzo, F., Tornielli, G. B. (2020). Rapid dehydration of grape berries dampens the post-ripening transcriptomic program and the metabolite profile evolution. Horticulture Res. 7, 1–15. doi: 10.1038/s41438-020-00362-5
Zenoni, S., Dal Santo, S., Tornielli, G. B., D’Incà, E., Filippetti, I., Pastore, C., et al. (2017). Transcriptional responses to pre-flowering leaf defoliation in grapevine berry from different growing sites, years, and genotypes. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00630
Zenoni, S., Fasoli, M., Guzzo, F., Santo, S. D., Amato, A., Anesi, A., et al. (2016). Disclosing the molecular basis of the postharvest life of berry in different grapevine genotypes. Plant Physiol. 172, 1821–1843. doi: 10.1104/pp.16.00865
Zenoni, S., Ferrarini, A., Giacomelli, E., Xumerle, L., Fasoli, M., Malerba, G., et al. (2010). Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-seq. Plant Physiol. 152, 1787–95. doi: 10.1104/pp.109.149716
Zhang, C., Dai, Z., Ferrier, T., Orduña, L., Santiago, A., Peris, A., et al. (2021). The grape MYB24 mediates the coordination of light-induced terpene and flavonolaccumulation in response to berry anthocyanin sunscreen depletio. BioRxiv. doi: 10.1101/2021.12.16.472692
Zhang, K., Li, W., Ju, Y., Wang, X., Sun, X., Fang, Y., et al. (2021). Transcriptomic and metabolomic basis of short- and long-term post-harvest UV-c application in regulating grape berry quality development. Foods 10, 625. doi: 10.3390/foods10030625
Zhang, J., Ma, H., Feng, J., Zeng, L., Wang, Z., Chen, S. (2008). Grape berry plasma membrane proteome analysis and its differential expression during ripening. J. Exp. Bot. 59, 2979–90. doi: 10.1093/jxb/ern156
Zhao, L., Chanon, A. M., Chattopadhyay, N., Dami, I. E., Blakeslee, J. J. (2016). Quantification of carbohydrates in grape tissues using capillary zone electrophoresis. Front. Plant Sci. 0. doi: 10.3389/fpls.2016.00818
Keywords: omics integration, Vitviz, database, integrape, Vitis vinifera
Citation: Savoi S, Santiago A, Orduña L and Matus JT (2022) Transcriptomic and metabolomic integration as a resource in grapevine to study fruit metabolite quality traits. Front. Plant Sci. 13:937927. doi: 10.3389/fpls.2022.937927
Received: 06 May 2022; Accepted: 09 September 2022;
Published: 20 October 2022.
Edited by:
Itay Maoz, Agricultural Research Organization (ARO), IsraelCopyright © 2022 Savoi, Santiago, Orduña and Matus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefania Savoi, c3RlZmFuaWEuc2F2b2lAdW5pdG8uaXQ=; José Tomás Matus, dG9tYXMubWF0dXNAdXYuZXM=