 Qiang Liang1†
Qiang Liang1† Jian Ning Liu1†
Jian Ning Liu1† Hongcheng Fang1
Hongcheng Fang1 Yuhui Dong1Changxi Wang1Yan Bao1Wenrui Hou1Rui Zhou1Xinmei Ma1Shasha Gai1Lichang Wang1Shouke Li2
Yuhui Dong1Changxi Wang1Yan Bao1Wenrui Hou1Rui Zhou1Xinmei Ma1Shasha Gai1Lichang Wang1Shouke Li2 Ke Qiang Yang1,3,4*
Ke Qiang Yang1,3,4* Ya Lin Sang1,3,4*
Ya Lin Sang1,3,4*- 1College of Forestry, Shandong Agricultural University, Tai’an, Shandong, China
- 2Worth Agricultural Development Co. Ltd., Weifang, China
- 3State Forestry and Grassland Administration Key Laboratory of Silviculture in Downstream Areas of the Yellow River, Shandong Agricultural University, Tai’an, Shandong, China
- 4Shandong Taishan Forest Ecosystem Research Station, Shandong Agricultural University, Tai’an, Shandong, China
Yellowhorn (Xanthoceras sorbifolium) is an oil-bearing tree species growing naturally in poor soil. The kernel of yellowhorn contains valuable fatty acids like nervonic acid. However, the genetic basis underlying the biosynthesis of valued fatty acids and adaptation to harsh environments is mainly unexplored in yellowhorn. Here, we presented a haplotype-resolved chromosome-scale genome assembly of yellowhorn with the size of 490.44 Mb containing scaffold N50 of 34.27 Mb. Comparative genomics, in combination with transcriptome profiling analyses, showed that expansion of gene families like long-chain acyl-CoA synthetase and ankyrins contribute to yellowhorn fatty acid biosynthesis and defense against abiotic stresses, respectively. By integrating genomic and transcriptomic data of yellowhorn, we found that the transcription of 3-ketoacyl-CoA synthase gene XS04G00959 was consistent with the accumulation of nervonic and erucic acid biosynthesis, suggesting its critical regulatory roles in their biosynthesis. Collectively, these results enhance our understanding of the genetic basis underlying the biosynthesis of valuable fatty acids and adaptation to harsh environments in yellowhorn and provide foundations for its genetic improvement.
Introduction
Plant oil is the primary source of edible fatty acid in the human diet and provides a natural and renewable feedstock for diverse industrial applications (Sharma et al., 2012; Abiodun, 2017). Global plant oil production increased from approximately 161.6 million metric tons in 2012 to 213.2 million in 2022 (Statista, 2022). However, due to the decreases in agricultural land and the rapid climate changes, it has become challenging to meet the growing demands for edible plant oil (Huang et al., 2016; Balbino, 2017; Yang et al., 2018; Chen et al., 2020). To address this need, it is essential to utilize plant species with high oil production and solid environmental adaptation (Bailey-Serres et al., 2019; Ortiz et al., 2020). Among many plant species, yellowhorn (Xanthoceras sorbifolium), a solid woody oleiferous plant, is an ideal material for meeting the demand (Ruan et al., 2017).
Yellowhorn, the sole member of the Xanthoceras (Sapindaceae), is distributed widely in northern China and adapted to harsh environmental conditions in the warm temperate zone (Group, 2016; Zhao et al., 2021). This species exhibits strong resistance to drought, salt, and cold stress and has been considered a promising ecological restoration species (Ruan et al., 2017). Yellowhorn produces capsular fruits from hermaphrodite. The seed of yellowhorn is rich in oil, which has been classified as edible oil by the National Health Commission of the People’s Republic of China.1 Yellowhorn seed oil is considered one of the better among 38 plant species producing nervonic acid (Yu et al., 2017; Liu F. et al., 2021). Compared with other oleaginous woody plants producing nervonic acid, such as Malania oleifera, Ximenia caffra, Acer truncatum, yellowhorn has higher seed oil content (52.7–58.0%), lower nervonic acid (3.46–4.37%), and moderate erucic acid (8.99–9.83%) content of total fatty acid (Liang et al., 2021; Liu F. et al., 2021). Nervonic acid is crucial for nerve myelin synthesis, which is one of the fundamental components of the nerve and brain (Poulos, 1995; Merrill et al., 1997; Martínez and Mougan, 1998). It has been found that nervonic acid is essential for developing the human nervous system and can be used to prevent and treat various neurodegenerative diseases (Kageyama et al., 2021; Song et al., 2022; Terluk et al., 2022). However, long-term exposure to excessive levels of erucic acid can reduce the β-oxidation of erucic acid in animal myocardia, leading to lipid deposition and tissue damage (Bremer and Norum, 1982). According to the European Food Safety Authority, tolerable levels of daily erucic acid intake are 7 mg/kg body weight (EFSA Panel on Contaminants in the Food Chain [CONTAM], 2016). Then the European Union Commission Regulation 2019/1870 sets the maximum level for erucic acid in vegetable oils as 20.0 g/kg, except for camelina oil, mustard oil, and borage oil, in which the maximum level of erucic acid is 50.0 g/kg. Thus, it is desirable to domesticate and breed yellowhorn varieties that produce increased levels of nervonic acid and decreased levels of erucic acid.
The biosynthesis of nervonic and erucic acid consists of two spatially separated steps. The de novo fatty acid synthesis occurs in plastids and gives rise to C18:1-CoA, while very long-chain unsaturated fatty acid (VLCFA) elongation at the endoplasmic reticulum (ER) produces nervonic and erucic acid (Wang P. et al., 2022). The latter was catalyzed by 3-ketoacyl-CoA synthase (KCS), 3-ketoacyl-CoA reductase (KCR), 3-hydroxacyl-CoA dehydratase (HCD), and trans-2,3-enoyl-CoA reductase (ECR). Of the four enzymes, KCS is the rate-limiting regulator (Millar and Kunst, 1997; Joubès et al., 2008; Haslam and Kunst, 2013; Wang Y. et al., 2022). KCR, HCD, and ECR are expressed and function in all tissues with fatty acid biosynthesis (Zheng et al., 2005; Paul et al., 2006; Bach et al., 2008; Beaudoin et al., 2009; Haslam and Kunst, 2013). KCS thus determines the tissue specificity of VLCFA elongation. However, the molecular mechanism underlying nervonic acid and erucic acid biosynthesis of yellowhorn is largely unelucidated.
High-level genome assembly is essential for studying the mechanisms of economically important traits and subsequent genetic modifications. Previously, other teams and we reported the yellowhorn genome assembly (Bi et al., 2019; Liang et al., 2019; Liu H. et al., 2021). In this study, we re-assembled the yellowhorn genome at the haplotype-resolved level. Based on the new assembly, genomic and transcriptomic analyses were performed on gene families involved in fatty acid biosynthesis and stress response. The results provided new clues for understanding the molecular mechanisms of environmental adaptation and valuable fatty acid biosynthesis of yellowhorn.
Materials and methods
Plant materials
The yellowhorn cultivar ‘Shanyou 1’ (released No.: Lu S-SV-XS-021-2020, designated as ‘WF18’ previously) was employed as plant material and planted in loam soil in 2012 at the yellowhorn orchard of the Forestry Experimental Station of Shandong Agricultural University (117°8′58′′ E, 36°10′16′′N) with orchard management as previously described (Liang et al., 2019, 2021). The seed sac filled with the embryo on June 5, 2017 (50 days after flowering, DAF), and the capsules matured on July 11, 2017 (86 DAF). Therefore, we measured oil accumulation and fatty acid content every 6 days from 50 to 86 DAF. Three capsules were picked from the randomly selected infructescence at 50, 56, 62, 68, 74, 80, and 86 DAF, respectively. The kernel was stripped from each seed immediately, flash-frozen in liquid nitrogen, and then stored at −80°C. Fresh, healthy leaves were collected from each accession for DNA extraction. The high-quality genomic DNA was extracted using the NucleoSpin Plant II (MachereyeNagel, Düren, Germany) according to the recommended procedure by the Kit.
Genome assembly
The genomic DNA sequencing datasets of PacBio Sequel long reads, Illumina short reads, and Hi-C reads were used for the haplotype-resolved assembly of yellowhorn cultivar ‘Shanyou 1.’ The haplotype-resolved genome assembly was constructed with FALCON v 1.8.1 (Chin et al., 2016) and FALCON-Unzip2 using our previous datasets, which included PacBio Sequel long reads and Illumina short reads (Liang et al., 2019). Briefly, pre-assembly (error correction) was performed based on all-by-all alignments of the subreads (length > 5 kb) using the FALCON HPCdaligner module. Preassembled, error-corrected reads longer than eight kbp were selected for final assembly with the Overlap-Layout-Consensus algorithm. Next, based on the draft FALCON assembly, the FALCON-Unzip assembler was applied to separate haplotype-specific contigs, generating primary contigs (phased genome assembly) and fragmented haplotigs (alternate haplotypes). The genome assembly was then polished, firstly with the haplotype-phased reads using the FALCON-Unzip pipeline, secondly with the PacBio long reads using the Arrow algorithm in the GenomicConsensus package v2.3.3,3 and finally with the Illumina short reads using Pilon v1.23 (Walker et al., 2014). The polished primary contigs and the Hi-C reads were used to construct chromosome-length scaffolds by applying the 3D-DNA pipeline v180922 (Dudchenko et al., 2017). For this purpose, duplication-free Hi-C contact maps were generated by analyzing kilobase resolution Hi-C data with the Juicer pipeline v1.5 (Durand et al., 2016). The Hi-C contacts were put into 3D-DNA software; then the chromosome-length scaffolds were constructed using default parameters. The genome scaffolds were then manually refined using Juicebox Assembly Tools v1.8.8. The refined Hi-C contacts were selected and clustered into a chromosome-scale assembly using the 3D-DNA pipeline. Finally, the refined chromosome-length scaffolds of the assembly were elaborately optimized with gap filling using LR_Gapcloser v1.1 (Xu et al., 2019) and subsequent polished three rounds using Pilon software. The accuracy of the assembly was evaluated by identifying and calculating the percentages of homogeneous variations using GATK v3.8 (McKenna et al., 2010) based on Illumina short reads. The completeness of the assembly was assessed against the embryophyta_odb10 lineage dataset (1,614 BUSCO groups) using BUSCO v5.0.0 (Seppey et al., 2019), with the parameters “-m genome -c 20-sp arabidopsis.”
Genome annotation
An integrated de novo and sequence homology-based modeling pipeline was used to identify repetitive sequences. First, RepeatModeler v2.0.1,4 RepeatScout v1.0.5 (Price et al., 2005), LTR_Finder v1.07 (Xu and Wang, 2007), MITE-Hunter (Han and Wessler, 2010), and PILER (Edgar and Myers, 2005) were used to predict repetitive sequences. The predicted repetitive elements were classified using PASTEClassifier v2.0 (Hoede et al., 2014) and then combined with the repetitive DNA elements in Repbase v25.01 (Bao et al., 2015) to produce a repeat library. The repetitive elements in the assembly were further identified and annotated using RepeatMasker v4.1.0 (Chen, 2004) against this constructed repeat library.
Protein-coding gene identification and annotation were performed as previously described (Liang et al., 2019). The gene prediction programs GeneMark-ES/ET/EP v4.62 (Lomsadze et al., 2005), Augustus v3.0 (Stanke et al., 2008), and SNAP v2006-07-28 (Korf, 2004) were used for ab initio gene prediction based on the soft-masked genome assembly. Exonerate v2.2.0 (Slater and Birney, 2005) was used to predict genes based on protein homology by aligning the peptides of several model plant species, including Arabidopsis thaliana TAIR10 (Lamesch et al., 2012), Oryza sativa IRGSP-1.0 (Kawahara et al., 2013; Sakai et al., 2013), Populus trichocarpa v3 (Tuskan et al., 2006), Solanum lycopersicum SL3.0 (Tomato Genome Consortium, 2012), Vitis vinifera IGGP_12x (Jaillon et al., 2007), and Glycine max v2.1 (Schmutz et al., 2010) against the genome assembly. Gene structures were modeled using PASA v2.3.3 (Haas et al., 2003) based on the genome alignments of the de novo transcripts assembled by Trinity v2.8.4 (Grabherr et al., 2011). The final gene models in the assembly were generated using MAKER v 2.31.10 (Campbell et al., 2014) by integrating the ab initio and homology gene predictions. Genes were functionally annotated using the InterProScan v5.33 pipeline against InterPro database v72.0 (Mitchell et al., 2015) and the BLASTP v2.10.1+ package (Ye et al., 2006) against Swiss-Prot (Boeckmann et al., 2003), NCBI non-redundant (nr), COG (Tatusov et al., 2003), and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000). The E-value cutoff was set to 1e-5 for BLAST searches.
Comparative phylogenomic analysis
To identify homologous gene families in the yellowhorn genome, we performed a comparative genome analysis of yellowhorn and 16 other closely related angiosperm genomes (Supplementary Table 18) as previously described (Liang et al., 2019). After removing alternatively spliced transcripts and transposable elements, the most extended protein sequences (>50 amino acids) were collected. An all-vs-all comparison was performed using BLASTP with an E-value cutoff of 1e-5. The alignments were clustered into homologous groups using OrthoMCL v2.0.9 (Li et al., 2003). A maximum likelihood phylogenomic tree of the 17 species based on shared single-copy genes was constructed using RAxML v8.1.24 (Guindon et al., 2010), with parameters “-# 1000-m PROTGAMMALGX.” O. satvia was used as the outgroup. Species divergence times were estimated using the MCMCtree module in PAML v4.9 (Yang, 2007) based on three secondary calibration points (Supplementary Table 19) obtained from the TimeTree database5 (Hedges et al., 2006). The phylogenomic tree was visualized using FigTree v1.4.3 (Drummond and Rambaut, 2007).
Based on the inferred phylogenomic history, gene family expansion and contraction were detected using CAFÉ v4.2.1 (De Bie et al., 2006). The expanded and contracted gene families were annotated using HMMER v3.2.1 (Eddy, 2011) against the databases Pfam v32.0 (El-Gebali et al., 2019) and Swiss-Prot, respectively. The functional enrichment of each gene family was determined using a hypergeometric test (Cao and Zhang, 2014) under a P-value cutoff of 0.05.
Analysis of ankyrin genes in yellowhorn
The HMMs of ankyrin (ANK) repeat protein clan PF00023 (Ank), PF12796 (Ank_2), PF00023 (Ank), PF13637 (Ank_4), and PF13857 (Ank_5) were used as queries to search against protein sequences of yellowhorn using HMMER v3.2.1 with E-value cut-off of 0.01. Multiple sequence alignments of the ANK protein sequences were performed using Clustal W (Larkin et al., 2007). The phylogenetic tree was established using RAxML software with 1000 bootstraps and exhibited in the EvolView online tool6 (He et al., 2016). In addition, 27 previous published transcriptomic datasets (Wang et al., 2020a,b) were obtained from the Sequence Read Archive (SRA) database in NCBI under accession number PRJNA608707 and used to investigate expression patterns of the ANK genes.
Identification of lipid-associated genes in yellowhorn
A total of 684 lipid-associated genes of A. thaliana were obtained from the database of Arabidopsis Acyl-Lipid Metabolism (Li-Beisson et al., 2013), and used as query sequences to search against protein sequences of yellowhorn, A. truncatum, M. oleifera, O. sativa, S. lycopersicum, and V. vinifera using BLASTP v2.10.1 with parameters “E-value ≤ 1e-5, alignment identity ≥ 30%, and alignment coverage ≥ 50%.” For long-chain acyl-CoA synthetase (LACS) and KCS genes, the genes must have the same Pfam domains (PF13193 and PF13193 for LACS; PF08392 and PF08392 for KCS) as the query sequence.
Kernel oil and fatty acid composition analysis
For oil extraction, kernels at 50, 56, 62, 68, 74, 80, and 86 DAF were collected and dried at 65°C until the samples attained a uniform weight. The oil was extracted and methylated as previously described (Fu et al., 2008; Yu et al., 2017). The fatty acid methyl esters were analyzed as previously described (Liang et al., 2021) using an Agilent 7890B gas chromatograph (Agilent Technologies, Little Falls, MN, United States), connected to a flame ionization detector with a DikmaCap DM-2560 Capillary Column (inner diameter 100 m × 0.25 mm; film thickness 0.2 μm). Supelco 37 Component FAME Mix (Supelco, Bellefonte, PA, United States) was used to identify the fatty acid methyl esters. Fatty acid peaks were identified by comparison to the retention times of the standards. Each peak was quantified based on peak area using HP3398A GC Chemstation software (Hewlett Packard, Santa Clara, CA, United States) compared with the standard. The determination was run in triplicate.
Transcriptomic analysis for the biosynthesis of very long-chain unsaturated fatty acids
We analyzed the transcriptomes of kernels of the ‘Shanyou 1’ cultivar collected at 50, 56, 62, 68, and 74 DAF. Take three independent samples as biological replicates at each time point. Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, United States) following the manufacturer’s instructions. RNA purity and integrity were respectively assessed using a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, United States) and an Agilent Bioanalyzer 2100 with an RNA Nano 6000 Assay Kit (Agilent Technologies, Santa Clara, CA, United States). The RNA sequencing libraries were constructed using the TruSeq RNA Library Prep Kit v2 (Illumina, San Diego, CA, United States) following the manufacturer’s instructions and sequenced on an Illumina HiSeq 4000 platform (KeGene, Taian, China) with paired-end reads of 150 bp. Raw reads were first quality trimmed using Trimmomatic v0.39 (Bolger et al., 2014). The resulting high-quality clean reads were aligned to the yellowhorn genome assembly using HISTA2 v2.1.0 (Kim et al., 2015) and then assembled using StringTie v1.3.5 (Pertea et al., 2015). The gene expression was quantified with fragments per kilobase of exon model per million mapped fragments (FPKM). The genes that met FPKM > 1 in at least one sequencing library were retained for DEGs identification. The DEGs were analyzed using DESeq2 v1.30.0.7 Genes with P ≤ 0.05 and fold change ≥ 2.0 were considered significantly differentially expressed.
Statistical analyses
We used R v4.08 for statistical analysis. One-way analysis of variance with Tukey’s post-hoc test was used to identify statistically significant differences among groups. Data are expressed as the mean ± standard deviation (SD), and P ≤ 0.05 was considered a significant difference.
Results
Genome assembly and annotation
We assembled the whole genome sequence of the yellowhorn cultivar ‘Shanyou 1’ (WF18). However, the genome assembly of this version still contains many gaps (1,000 gaps covering 29.06 Mb) (Liang et al., 2019). To enhance the accuracy and integrity, based on a combination of PacBio, Hi-C, and Illumina reads, we reassembled the whole genome sequence using a haplotype-resolved approach (Supplementary Tables 1, 2). By executing the FALCON-Unzip assembler with PacBio long reads, the initial assembly generated 2,428 primary contigs (N50 of 408.56 kb) with a total length of 482.89 Mb, as well as 5,310 haplotigs covering 264.58 Mb, accounted for 54.79% of the genome assembly (Supplementary Table 3). The primary contigs were further scaffolded using Hi-C reads to enhance the sequence continuity. As a result, a total length of 490.44 Mb genome sequence containing 22 super-scaffolds (N50 of 34.27 Mb) was generated, of which 490.24 Mb sequences accounting for 99.96% of the whole assembly were anchored onto 15 pseudochromosomes (Figure 1A and Supplementary Table 4). The size of the reassembled genome was more extensive than that of cultivar ‘JGXP’ (470 Mb) and our last assembly (440 Mb) but smaller than that of the cultivar ‘ZS4’ (504 Mb) (Table 1). Compared with our previous version, gap filling significantly improved the integrity of the assembly and left 2,295 gaps covering only 989.98 kb. Moreover, the mapping rates of Illumina and 98.81% PacBio reads were 98.62 and 98.81%, respectively; the single-base error rates of single nucleotide polymorphisms as well as insertion and deletions were, respectively, 0.000216 and 0.000421% (Supplementary Table 5). The complete BUSCOs recovery score of the assembly was 98.7% (1,594 of 1,614 genes) (Supplementary Table 6), which was much higher than our previous version (87.4%) (Table 1).
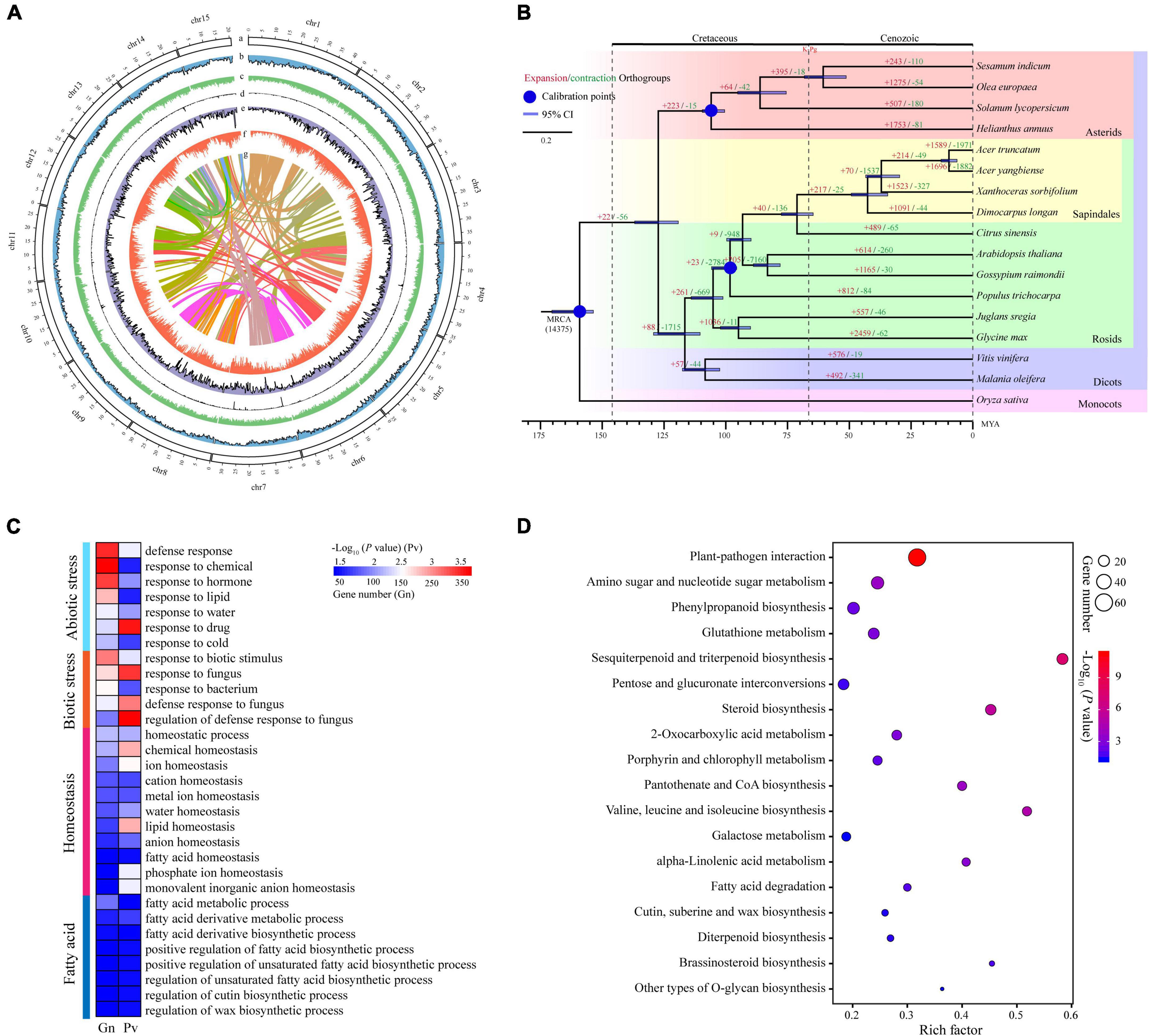
Figure 1. The characteristics of yellowhorn ‘Shanyou 1’ (WF18) genome assembly. (A) The distribution of genomic elements of yellowhorn. The items from outermost to innermost are: (a) pseudochromosomes, (b) LTR-Gypsy density, (c) LTR-Copia density, (d) transposon element density, (e) tandem repeat density, (f) gene density, and (g) intra-genome collinear blocks. Chromosome units correspond to Mbp. (B) Maximum-likelihood phylogeny of yellowhorn and 16 representative angiosperms, based on 223 shared single-copy genes. Oryza sativa was used as the outgroup. All nodes have 100% bootstrap support. Species divergence times were estimated using three secondary calibration points (blue circles) from the TimeTree database. Horizontal blue bars denote confidence intervals (CI). Values above the branches indicate the numbers of orthologous gene families that expanded (red) or contracted (green) after divergence from the most recent common ancestor (MRCA). K-Pg denotes the Cretaceous-Paleogene boundary. Abbreviation: MYA, millions of years ago. (C) The gene ontology (GO) categories are significantly enriched in the significantly expanded gene families. (D) The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enriched the significantly expanded gene families.
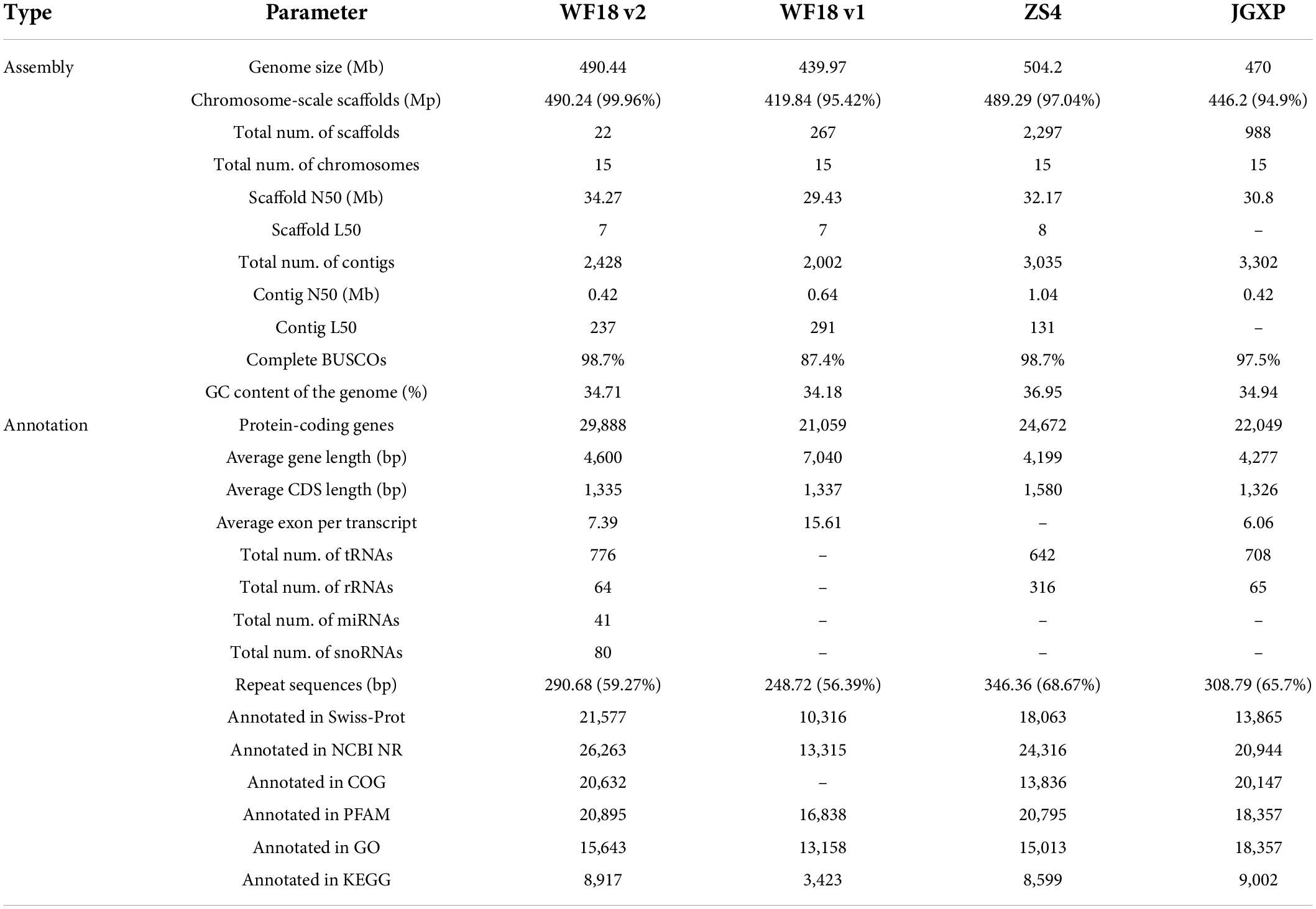
Table 1. Yellowhorn (Xanthoceras sorbifolium) genome assembly and annotation.
Genome annotation identified 290.68 Mb repetitive sequences, occupying 59.27% of the genome assembly, of which 27.79% were long terminal repeat retrotransposon elements (Figure 1A and Supplementary Table 7). A total of 29,888 protein-coding genes were predicted (Table 1); 26,331 (88.10%) of them were annotated into at least one of the databases including NCBI NR (40.03%), Swiss-Prot (42.18%), protein families (Pfam) (69.91%), InterPro (72.19%), COG (42.27%), Gene Ontology (GO) (52.34%), and KEGG (28.18%).
Gene family expansion analysis
Gene family expansion has been shown to facilitate adaptation and trait innovation in the evolution of angiosperms (Van de Peer et al., 2009; Wang et al., 2019; Ehrenreich, 2020). Therefore, to understand the genetic basis underpinning valuable fatty acids biosynthesis and environmental adaptability of yellowhorn, we analyzed gene family size using the phylogenomic tree. To this end, we first identified 223 single-copy nuclear genes conserved in 17 representative angiosperm genomes through comparative genomic analysis (Supplementary Table 8). The phylogenomic tree was then constructed using the conserved single-copy genes. The results suggested that the yellowhorn has diverged from a most recent common ancestor with Dimocarpus longan approximately 42.6 million years ago (Figure 1B and Supplementary Figure 1).
We next investigated the ancestral gene family size differences among species along the phylogenomic tree. The results showed that 1,523 gene families (e.g., LACS and ANK gene families) were significantly expanded (P < 0.01) while 327 ones were significantly contracted (Supplementary Table 9). GO functional enrichment analysis demonstrated that ten GO terms were related to fatty acid metabolic and biosynthetic processes, such as “fatty acid metabolic process,” “regulation of unsaturated fatty acid biosynthetic process,” and “regulation of cutin biosynthetic process.” Moreover, GO terms related to biotic or abiotic stress responses like “defense response,” “response to water,” “response to cold,” and “homeostatic process” were significantly enriched (P < 0.01) in the expanded gene families (Figure 1C and Supplementary Table 10). KEGG enrichment analysis also revealed that many expanded gene families were involved in fatty acid biosynthesis and stress adaptation, such as CoA biosynthesis (ko00770), plant-pathogen interaction (ko04626), and pantothenate (Figure 1D and Supplementary Table 11). Thus, these results suggested that gene family expansion was associated with fatty acid biosynthesis and stress adaptation in yellowhorn.
Analysis of lipid-associated genes
To explore genes involved in VLCFAs biosynthesis in yellowhorn, especially nervonic acid and erucic acids, we blasted against the genome assembly using genes involved in lipid synthesis and metabolism from A. thaliana as the query sequences (196 functional families containing 684 genes). As a result, the 788 homologs were identified and designated as lipid-associated genes in yellowhorn. The number was much greater than that in A. truncatum (745) and M. oleifera (566), the other two VLCFAs-producing species, suggesting an active lipid biosynthesis and metabolism in yellowhorn (Supplementary Table 12). In the yellowhorn, lipid-associated gene families encoding LACS, ATP citrate lyase A subunit (ATP-CL-alpha), and alcohol-forming fatty acyl-CoA reductase (AlcFAR), which are essential for fatty acid synthesis, contained more members than those in A. truncatum and M. oleifera. The LACS family expanded to 12 members, and the four members were organized in a tandem array located in chromosome 3 (Supplementary Figure 2A). The phylogenetic tree showed that the LACS genes were clustered into six groups, while the LACS-2 group contained four tandem genes (Figure 2A). In addition, we found that the KCS family expanded to 8 members in the yellowhorn genome. Phylogenetic analysis showed that the KCS genes were clustered into four groups (Figure 2B). Four clustered KCSs, closely related to KCS2 and KCS20 in A. thaliana, were organized in a tandem array and located in an 86 kb region of chromosome 4 (Supplementary Figure 2B). Almost all the LACS and KCS genes expressed in a set of 15 yellowhorn kernel transcriptomes with FPKM ≥ 1 (Figure 2C), demonstrating their functional activities in erucic and nervonic acids biosynthesis. Therefore, we speculate that the expanded lipid-associated gene families, especially LACS and KCS, may play crucial roles in the yellowhorn biosynthesis of erucic and nervonic acids.
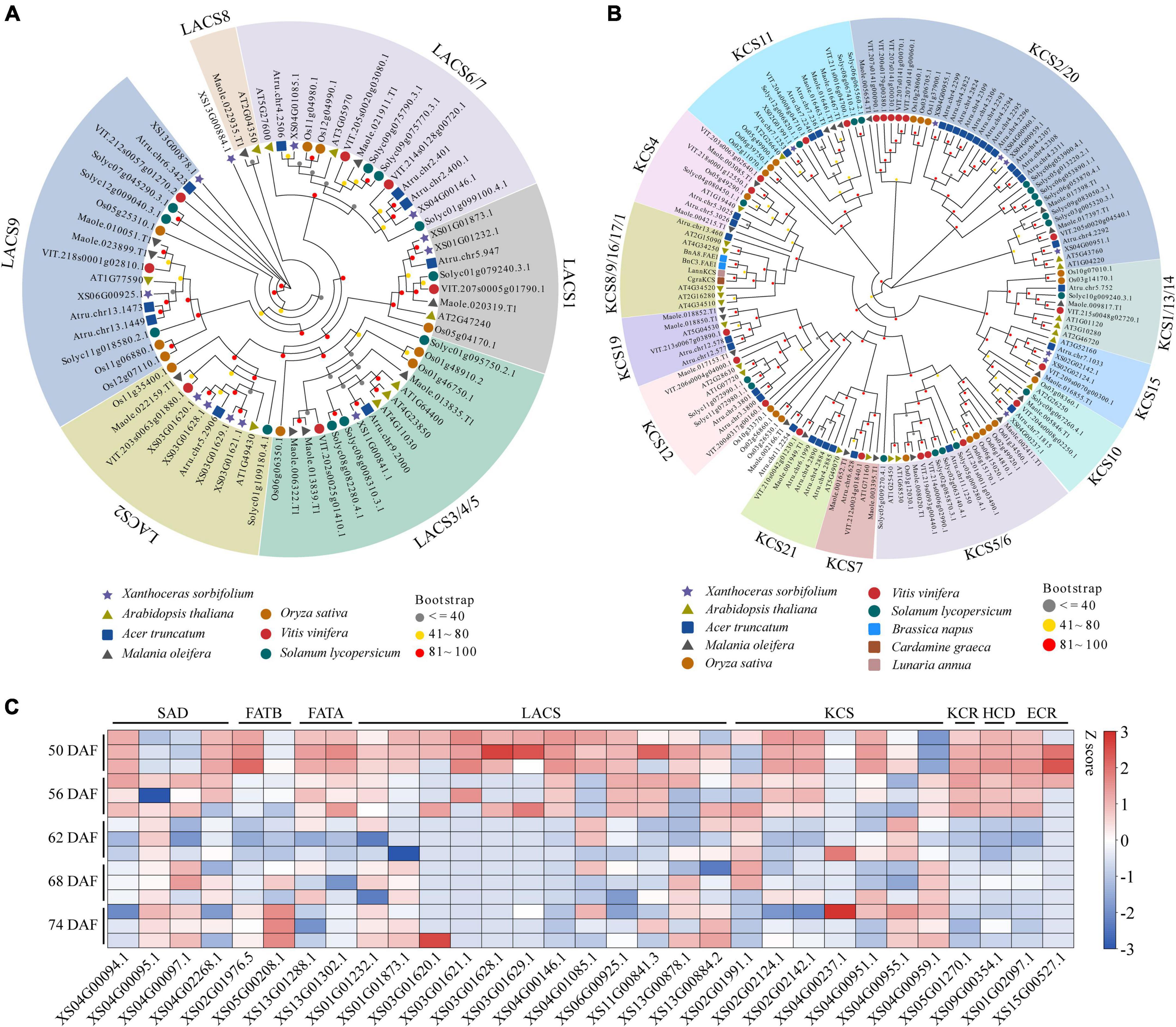
Figure 2. Gene expansion of LACS and KCS in yellowhorn. Maximum-likelihood phylogenetic tree showing expanded LACS (A) and KCS genes (B) in yellowhorn. The phylogenetic tree was constructed using RAxML software with 1000 bootstraps and exhibited in the EvolView online tool (https://www.evolgenius.info/evolview/). (C) Gene expression patterns of LACS, KCS, and other lipid-related genes among 15 kernel transcriptomes of 50, 56, 62, 68, and 74 DAF of yellowhorn.
Molecular basis of very long-chain unsaturated fatty acids biosynthesis
We performed profiles during kernel development from 50 to 86 DAF to investigate the characteristics of fatty acid composition in yellowhorn kernels. The seed oil accumulation increased rapidly from 50 to 68 DAF and reached content of 54.43% at 68 DAF (Supplementary Figure 3). Fatty acid profiling identified 12 components in kernels (Figure 3A and Supplementary Table 13). The results showed a difference in the content of different fatty acids at different time points (Figure 3A). Among these fatty acids, linoleic acid (42.24–53.48%) and oleic acid (21.73–28.08%) were the major components, whereas arachidic acid took the lowest proportion (0.20–0.34%). Nervonic and erucic acid accumulated significantly from 50 to 68 DAF and reached 4.07 and 10.41%, respectively.
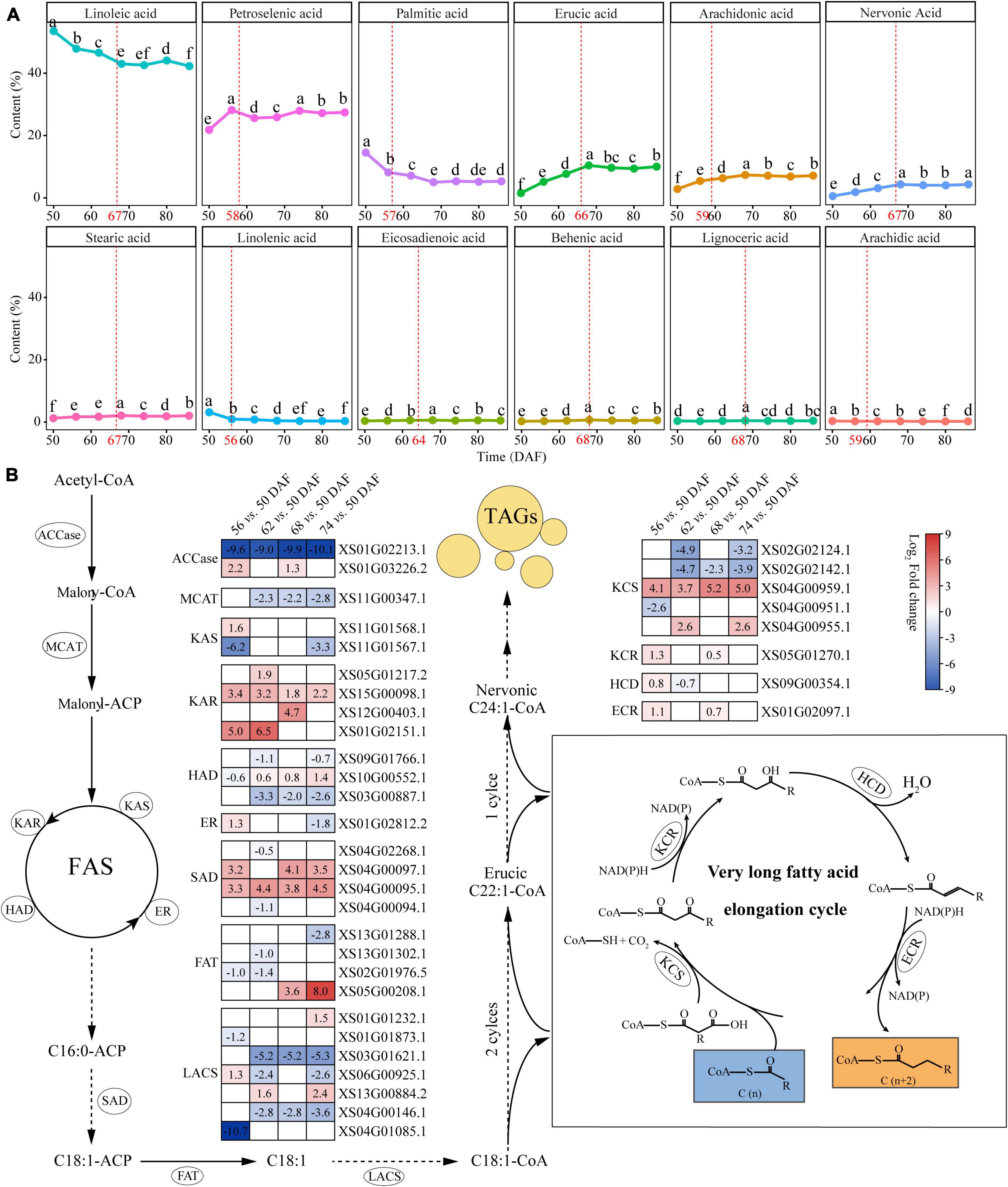
Figure 3. The biosynthetic pathways of nervonic and erucic acid in yellowhorn. (A) Fatty acid composition in yellowhorn seed oil at 50–86 DAF. Values shown are means ± SD of three biological replicates. The red dot plot represents the breakpoint analyzed using the breakpoint function of RStudio v4.0. Different letters on the dots indicate statistical significance (P ≤ 0.05). (B) The nervonic and erucic acid biosynthesis pathway, highlighting the differentially expressed with fold change ≥ 2 at 56, 62, 68, and 74 DAF compared with 50 DAF. Abbreviations: ACCase, acetyl-CoA carboxylase; MCMT, malonyl-CoA:acyl carrier protein (ACP) malonyltransferase; KAR, ketoacyl-ACP reductase; KAS, ketoacyl-ACP synthase; HAD, hydroxyacyl-ACP dehydrase; ER, enoyl-ACP reductase; SAD, stearoyl-ACP desaturase; FAT, fatty acyl thioesterase; LACS, long-chain acyl-CoA synthetase; KCS, ketoacyl-CoA synthase; KCR, ketoacyl-CoA reductase; HCD, hydroxyacyl-CoA dehydrase; ECR, enoyl-CoA reductase.
Based on the nervonic and erucic acid accumulation during the yellowhorn kernel development period, the kernel at 50, 56, 62, 68, and 74 DAF were selected for transcriptomic analysis to elucidate the genetic mechanism underlying the biosynthesis of nervonic and erucic acid. Compared with 50 DAF, 165, 229, 212, and 247 differentially expressed lipid genes (DELGs) were identified at 56, 62, 68, and 74 DAF, respectively (Supplementary Figure 4 and Supplementary Table 14). Gene functional enrichment analysis showed that the DELGs were enriched in organic acid biosynthetic and metabolic processes, fatty acid biosynthesis and metabolism, biosynthesis of unsaturated fatty acids, and fatty acid elongation (Supplementary Figure 5 and Supplementary Table 15). Fifty-three members of the DELGs were involved in VLCFAs biosynthesis (Supplementary Table 16). Nervonic acid is biosynthesized from oleyl-CoA (C18:1-CoA) by four successive enzymatic reactions at the ER catalyzed by KCS, KCR, HCD, and ECR, respectively. The DELGs contained 5 KCSs, one KCR, one HCD, and one ECR (Figure 3B and Supplementary Table 16). Four genes were significantly upregulated (P-value < 0.05) at 56, 62, 68, and 74 DAF compared with 50 DAF. Notably, the transcription level of XS04G00959, which encodes one KCS, was highly expressed at 56 (17.15-fold), 62 (13.00-fold), 68 (36.76-fold), and 74 (32.00-fold) DAF compared with 50 DAF, which was consistent with the accumulation of nervonic and erucic acid (Figure 3B and Supplementary Table 16), suggesting that XS04G00959 played essential roles in the biosynthesis of nervonic and erucic acid.
Analysis of ankyrin repeat gene family
ANK genes play crucial roles in biotic or abiotic stress responses (Becerra et al., 2004; Huang et al., 2009), the most prominent expanded gene families in yellowhorn genome assembly. To explore the potential roles of the ANK family in environmental adaptation in yellowhorn, the characteristics of ANK genes were analyzed. The results showed that the ANKs family in yellowhorn expanded to 132 members, which were further clustered into six distinct groups based on phylogenetic analysis (Figure 4A). The ANK genes were distributed across all the 15 pseudochromosomes, with 14 and 11 genes organized in tandem arrays on pseudochromosome 14 and 12 (Figure 4B). The previous published transcriptomic datasets were re-analyzed to determine the expression patterns of ANKs responding to abiotic stress. The results indicated that 53 ANK genes were significantly differentially expressed under cold, salt, or saline-alkali stresses (Figure 4C and Supplementary Table 17). Of them, six members, including XS01G01803 [accelerated cell death 6 (ACD6)], XS04G00500 [ankyrin repeat-containing protein ITN1 (ITN1)], XS14G00504, XS11G01175, XS08G00278, and XS01G03358 were significantly differentially expressed under cold stress. XS04G00933 (ACD6) was significantly upregulated after 4 and 24 h exposure to salt stress. Both cold and saline-alkali stresses significantly induced the expression of XS04G00500 (ITN1). These results suggested that ACD6 and ITN1 were related to different abiotic stresses response in yellowhorn.
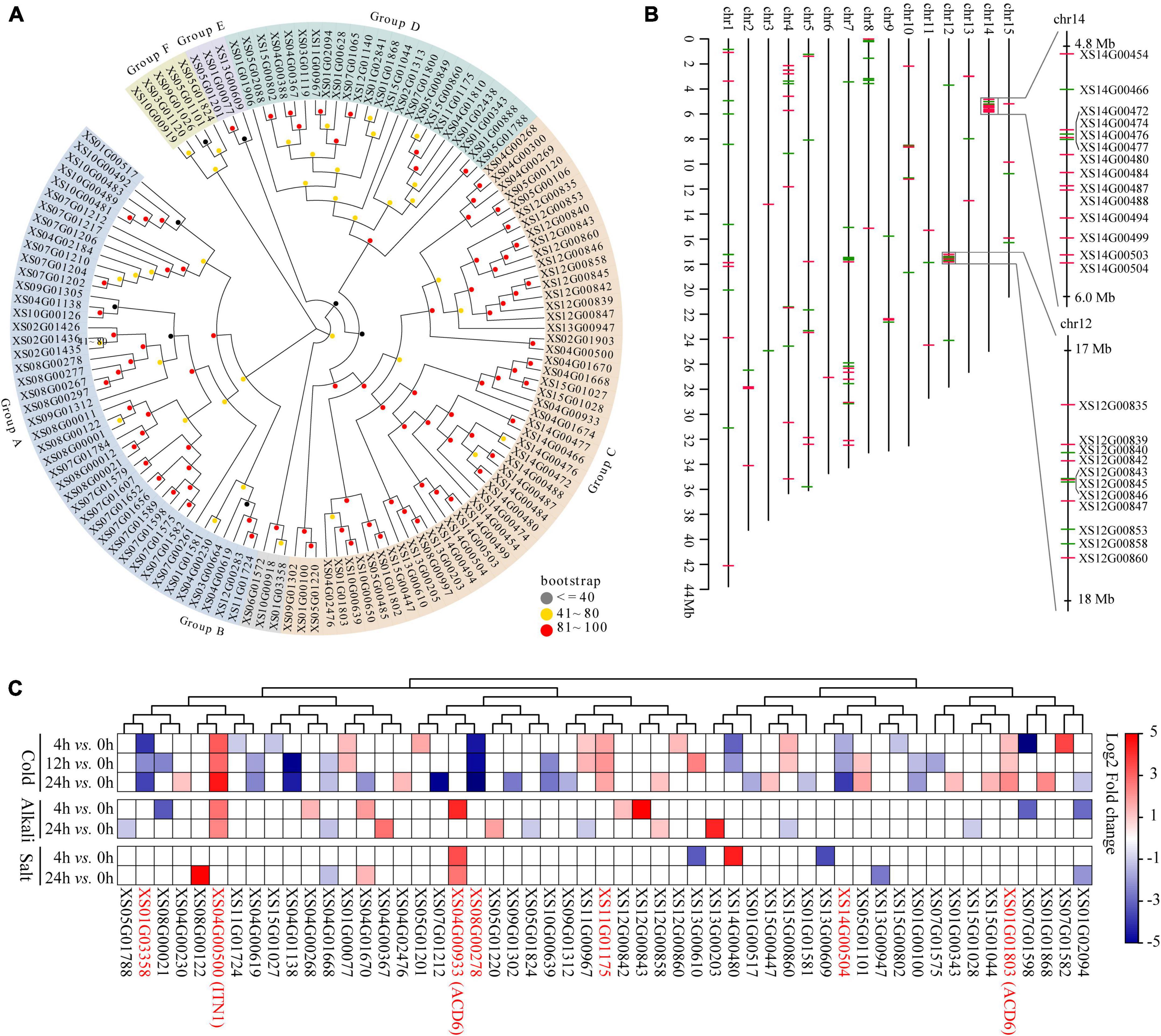
Figure 4. Gene expansion of ANK in yellowhorn. (A) Maximum-likelihood phylogenetic tree showing expanded ANK in yellowhorn. The phylogenetic tree was established using RAxML software with 1000 bootstraps and exhibited in the EvolView online tool (https://www.evolgenius.info/evolview/). (B) Distribution of the 132 ANK genes along 15 yellowhorn pseudochromosomes. The right side shows the detailed overview of ANK gene clusters on pseudochromosomes 12 (from 17 to 18 Mb) and 14 (from 4.8 to 6.0 Mb), respectively. The red and green lines represent the genes located on the senesce or antisense strand of the pseudochromosome. (C) The 53 significantly differentially expressed ANK genes were identified after cold, salt, and saline-alkali stress. The 27 transcriptomic datasets were downloaded from the NCBI Sequence Read Archive (SRA) database under accession PRJNA608707.
Discussion
High-quality genome assembly is of vital importance for biological studies and breeding applications. The yellowhorn genome assemblies have been reported previously by other teams and us (Bi et al., 2019; Liang et al., 2019; Liu H. et al., 2021). This study reconstructed the yellowhorn cultivar ‘Shanyou 1’ genome at the haplotype-resolved level based on PacBio, Hi-C, and Illumina reads (Chin et al., 2016). Compared with the previous versions, the percentage of contig anchored to the pseudochromosomes, N50 length, and complete BUSCOs were significantly increased. More than 99% of the contigs can be anchored to the pseudochromosomes, 98.7% of BUSCO genes were complete, and more than 99% of the gaps were closed. Therefore, the reassembled genome sequence is of high completeness, accuracy, and continuity and can be used as a reference for studies on genome function and genetic variation.
Our results revealed that LACS, ATP-CL-alpha, and AlcFAR families, which are responsible for fatty acid synthesis, contained more members than those in other VLCFA-producing woody plants, such as A. truncatum and M. oleifera. The LACS family genes have been shown to participate in fatty acid and glycerolipid metabolism (Lü et al., 2009; Zhao et al., 2010, 2019; Jessen et al., 2015). In Brassica napus, LACS2 plays a crucial role in seed oil production (Ding et al., 2020). Arabidopsis LACS4 and LACS9 are involved in the biosynthesis of glycolipids in plastids (Jessen et al., 2015). In the present study, we found that the LACS family expanded to 12 members in yellowhorn. Most of these genes were preferentially expressed at 56 and 62 DAF, suggesting their roles in producing fatty acyl supply flux for fatty acid synthesis (Lin et al., 2018). Moreover, the KCS family expanded to eight members, which fell into four major clades. It has been shown that ectopic expression of Lunaria and Cardamine KCSs in Brassica increased nervonic acid and reduced erucic acid accumulation in the seed oil (Guo et al., 2009; Taylor et al., 2009). Our results showed that two KCSs were significantly upregulated at 56 and 62 DAF, which is consistent with the accumulating stage of nervonic acids, suggesting their regulatory roles in this process.
Recent studies revealed that KCS also demonstrates strong substrate specificity, which defines the final chain length of the VLCFAs (Liu F. et al., 2021). Our analysis revealed that the accumulation of fatty acids and biosynthesis of both nervonic and erucic acid were mainly completed between 50 and 68 DAF. Transcriptomic analysis demonstrated that 36 genes regulating VLCFA elongation were differently expressed at 56, 62, 68, and 74 DAF compared with 50 DAF. XS04G00959 encoding KCS was significantly upregulated and exhibited an expression trend consistent with the accumulation of nervonic and erucic acid, suggesting that this gene is essential for the biosynthesis of nervonic and erucic acid.
Substantial expansions were identified in gene families associated with stress responses. In the present study, we found that the ANK family expanded significantly and contained 132 members. Several ANK genes have been shown to resist biotic or abiotic stresses (Becerra et al., 2004; Huang et al., 2009). For instance, ACD6 participates in salicylic acid signaling in Arabidopsis, and overexpressing this gene enhances pathogen resistance (Lu et al., 2003; Zhang et al., 2014). ITN1 mediates responses to salt stress by promoting the production of reactive oxygen species via the abscisic acid signaling pathway (Sakamoto et al., 2008, 2012). Based on published transcriptomic data, we found that transcription of two ACD6 copies (XS04G00933 and XS01G01803) was significantly induced by salt and cold stresses. Both cold and saline-alkali stresses significantly upregulated the ITN1 homolog (XS04G00500). These results suggested that the expansion of ANK family may contribute to the resistance against abiotic stresses in yellowhorn.
Conclusion
In summary, we reported a haplotype-resolved chromosome-scale genome assembly of yellowhorn. We identified one KCS gene, XS04G00959, which can be a vital contributor to the nervonic and erucic acid biosynthesis in yellowhorn. We revealed that ACD6 and ITN1 played crucial roles in yellowhorn against multiple abiotic stresses. Altogether, the present study provides new insights into the understanding of the economic traits such as valuable fatty acid production and environmental adaptation, thus greatly benefiting the genetic improvement of this species.
Data availability statement
The original contributions presented in this study are publicly available. This data can be found here: NCBI, PRJNA723435.
Author contributions
KY and YS designed and supervised the project and reviewed and revised the manuscript. QL and JL collected and generated the data, performed analysis, and wrote the draft manuscript. YD, RZ, and XM performed transcriptome analysis. HF, RZ, XM, and CW contributed to data analysis. CW, YB, WH, SG, LW, and SL contributed to plant material collection. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 32071802), the Improved Variety Program of Shandong Province of China (2020LZGC090402), and the Funds of Shandong “Double Tops” Program (SYL2017XTTD09).
Conflict of interest
SL was employed by the Worth Agricultural Development Co. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.991197/full#supplementary-material
Footnotes
- ^ https://slps.jdzx.net.cn/xwfb/gzcx/PassFileQuery.jsp
- ^ https://github.com/PacificBiosciences/FALCON_unzip/
- ^ https://github.com/PacificBiosciences/pbbioconda/
- ^ http://www.repeatmasker.org/RepeatModeler/
- ^ http://www.timetree.org/
- ^ https://www.evolgenius.info/evolview/
- ^ https://bioconductor.org/packages/DESeq2/
- ^ https://www.r-project.org/
References
Abiodun, O. A. (2017). “The role of oilseed crops in human diet and industrial use,” in Oilseed crops (Chichester, NY: John Wiley & Sons Ltd), 249–263. doi: 10.1002/9781119048800.ch14
Bach, L., Michaelson, L. V., Haslam, R., Bellec, Y., Gissot, L., Marion, J., et al. (2008). The very-long-chain hydroxy fatty acyl-CoA dehydratase PASTICCINO2 is essential and limiting for plant development. Proc. Natl Acad. Sci. U.S.A. 105, 14727–14731. doi: 10.1073/pnas.0805089105
Bailey-Serres, J., Parker, J. E., Ainsworth, E. A., Oldroyd, G. E. D., and Schroeder, J. I. (2019). Genetic strategies for improving crop yields. Nature 575, 109–118. doi: 10.1038/s41586-019-1679-0
Balbino, S. (2017). “Vegetable oil yield and composition influenced by environmental stress factors,” in Oilseed crops, ed. P. Ahmad (Chichester: John Wiley & Sons Ltd). doi: 10.1002/9781119048800.ch5
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11. doi: 10.1186/s13100-015-0041-9
Beaudoin, F., Wu, X., Li, F., Haslam, R. P., Markham, J. E., Zheng, H., et al. (2009). Functional characterization of the Arabidopsis beta-ketoacyl-coenzyme A reductase candidates of the fatty acid elongase. Plant Physiol. 150, 1174–1191. doi: 10.1104/pp.109.137497
Becerra, C., Jahrmann, T., Puigdomènech, P., and Vicient, C. M. (2004). Ankyrin repeat-containing proteins in Arabidopsis: Characterization of a novel and abundant group of genes coding ankyrin-transmembrane proteins. Gene 340, 111–121. doi: 10.1016/j.gene.2004.06.006
Bi, Q., Zhao, Y., Du, W., Lu, Y., Gui, L., Zheng, Z., et al. (2019). Pseudomolecule-level assembly of the Chinese oil tree yellowhorn (Xanthoceras sorbifolium) genome. Gigascience 8:giz070. doi: 10.1093/gigascience/giz070
Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M. C., Estreicher, A., Gasteiger, E., et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bremer, J., and Norum, K. R. (1982). Metabolism of very long-chain monounsaturated fatty acids (22:1) and the adaptation to their presence in the diet. J. Lipid Res 23, 243–256.
Campbell, M. S., Holt, C., Moore, B., and Yandell, M. (2014). Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinformatics 48, 4.11.1–4.11.39. doi: 10.1002/0471250953.bi0411s48
Cao, J., and Zhang, S. (2014). A bayesian extension of the hypergeometric test for functional enrichment analysis. Biometrics 70, 84–94. doi: 10.1111/biom.12122
Chen, N. (2004). Using repeat masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4:Unit 4.10. doi: 10.1002/0471250953.bi0410s05
Chen, N., Yu, K., Jia, R., Teng, J., and Zhao, C. (2020). Biocrust as one of multiple stable states in global drylands. Sci. Adv. 6:eaay3763. doi: 10.1126/sciadv.aay3763
Chin, C. S., Peluso, P., Sedlazeck, F. J., Nattestad, M., Concepcion, G. T., Clum, A., et al. (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054. doi: 10.1038/nmeth.4035
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: A computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Ding, L. N., Gu, S. L., Zhu, F. G., Ma, Z. Y., Li, J., Li, M., et al. (2020). Long-chain acyl-CoA synthetase 2 is involved in seed oil production in Brassica napus. BMC Plant Biol. 20:21. doi: 10.1186/s12870-020-2240-x
Drummond, A. J., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95. doi: 10.1126/science.aal3327
Durand, N. C., Robinson, J. T., Shamim, M. S., Machol, I., Mesirov, J. P., Lander, E. S., et al. (2016). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101. doi: 10.1016/j.cels.2015.07.012
Eddy, S. R. (2011). Accelerated Profile HMM Searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Edgar, R. C., and Myers, E. W. (2005). PILER: Identification and classification of genomic repeats. Bioinformatics 21(Suppl. 1) i152–i158. doi: 10.1093/bioinformatics/bti1003
EFSA Panel on Contaminants in the Food Chain [CONTAM], Knutsen, H. K., Alexander, J., Barregård, L., Bignami, M., Brüschweiler, B., et al. (2016). Erucic acid in feed and food. EFSA J. 14:e04593. doi: 10.2903/j.efsa.2016.4593
Ehrenreich, I. M. (2020). Evolution after genome duplication. Science 368, 1424–1425. doi: 10.1126/science.abc1796
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Fu, Y. J., Zu, Y. G., Wang, L. L., Zhang, N. J., Liu, W., Li, S. M., et al. (2008). Determination of fatty acid methyl esters in biodiesel produced from yellow horn oil by LC. Chromatographia 67, 9–14. doi: 10.1365/s10337-007-0471-8
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Group, T. A. P. (2016). An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.12385
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Guo, Y., Mietkiewska, E., Francis, T., Katavic, V., Brost, J. M., Giblin, M., et al. (2009). Increase in nervonic acid content in transformed yeast and transgenic plants by introduction of a Lunaria annua L. 3-ketoacyl-CoA synthase (KCS) gene. Plant Mol. Biol 69, 565–575. doi: 10.1007/s11103-008-9439-9
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K. Jr., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. doi: 10.1093/nar/gkg770
Han, Y., and Wessler, S. R. (2010). MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38:e199. doi: 10.1093/nar/gkq862
Haslam, T. M., and Kunst, L. (2013). Extending the story of very-long-chain fatty acid elongation. Plant Sci. 210, 93–107. doi: 10.1016/j.plantsci.2013.05.008
He, Z., Zhang, H., Gao, S., Lercher, M. J., Chen, W. H., and Hu, S. (2016). Evolview v2: An online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 44, W236–W241. doi: 10.1093/nar/gkw370
Hedges, S. B., Dudley, J., and Kumar, S. (2006). TimeTree: A public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972. doi: 10.1093/bioinformatics/btl505
Hoede, C., Arnoux, S., Moisset, M., Chaumier, T., Inizan, O., Jamilloux, V., et al. (2014). PASTEC: An automatic transposable element classification tool. PLoS One 9:e91929. doi: 10.1371/journal.pone.0091929
Huang, J., Yu, H., Guan, X., Wang, G., and Guo, R. (2016). Accelerated dryland expansion under climate change. Nat. Climate Change 6, 166–171. doi: 10.1038/nclimate2837
Huang, J., Zhao, X., Yu, H., Ouyang, Y., Wang, L., and Zhang, Q. (2009). The ankyrin repeat gene family in rice: Genome-wide identification, classification and expression profiling. Plant Mol. Biol. 71, 207–226. doi: 10.1007/s11103-009-9518-6
Jaillon, O., Aury, J. M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467. doi: 10.1038/nature06148
Jessen, D., Roth, C., Wiermer, M., and Fulda, M. (2015). Two activities of long-chain acyl-coenzyme A synthetase are involved in lipid trafficking between the endoplasmic reticulum and the plastid in Arabidopsis. Plant Physiol. 167, 351–366. doi: 10.1104/pp.114.250365
Joubès, J., Raffaele, S., Bourdenx, B., Garcia, C., Laroche-Traineau, J., Moreau, P., et al. (2008). The VLCFA elongase gene family in Arabidopsis thaliana: Phylogenetic analysis, 3D modelling and expression profiling. Plant Mol. Biol. 67, 547–566. doi: 10.1007/s11103-008-9339-z
Kageyama, Y., Deguchi, Y., Hattori, K., Yoshida, S., Goto, Y. I., Inoue, K., et al. (2021). Nervonic acid level in cerebrospinal fluid is a candidate biomarker for depressive and manic symptoms: A pilot study. Brain Behav. 11:e02075. doi: 10.1002/brb3.2075
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kawahara, Y., de la Bastide, M., Hamilton, J. P., Kanamori, H., McCombie, W. R., Ouyang, S., et al. (2013). Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice (N Y) 6:4. doi: 10.1186/1939-8433-6-4
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Lamesch, P., Berardini, T. Z., Li, D., Swarbreck, D., Wilks, C., Sasidharan, R., et al. (2012). The Arabidopsis information resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210. doi: 10.1093/nar/gkr1090
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Li, L., Stoeckert, C. J. Jr., and Roos, D. S. (2003). OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Liang, Q., Fang, H., Liu, J., Zhang, B., Bao, Y., Hou, W., et al. (2021). Analysis of the nutritional components in the kernels of yellowhorn (Xanthoceras sorbifolium Bunge) accessions. J. Food Compos Anal. 100:103925. doi: 10.1016/j.jfca.2021.103925
Liang, Q., Li, H., Li, S., Yuan, F., Sun, J., Duan, Q., et al. (2019). The genome assembly and annotation of yellowhorn (Xanthoceras sorbifolium Bunge. Gigascience 8:giz071. doi: 10.1093/gigascience/giz071
Li-Beisson, Y., Shorrosh, B., Beisson, F., Andersson, M. X., Arondel, V., Bates, P. D., et al. (2013). Acyl-lipid metabolism. Arabidopsis Book 11:e0161. doi: 10.1199/tab.0161
Lin, H., Shen, H., and Lee, Y. K. (2018). Cellular and molecular responses of dunaliella tertiolecta by expression of a plant medium chain length fatty acid specific acyl-acp thioesterase. Front. Microbiol. 9:619. doi: 10.3389/fmicb.2018.00619
Liu, F., Wang, P., Xiong, X., Zeng, X., Zhang, X., and Wu, G. (2021). A review of nervonic acid production in plants: Prospects for the genetic engineering of high nervonic acid cultivars plants. Front. Plant Sci. 12:626625. doi: 10.3389/fpls.2021.626625
Liu, H., Yan, X. M., Wang, X. R., Zhang, D. X., Zhou, Q., Shi, T. L., et al. (2021). Centromere-specific retrotransposons and very-long-chain fatty acid biosynthesis in the genome of yellowhorn (Xanthoceras sorbifolium. Sapindaceae), an oil-producing tree with significant drought resistance. Front. Plant. Sci. 12:766389. doi: 10.3389/fpls.2021.766389
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O., and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506. doi: 10.1093/nar/gki937
Lu, H., Rate, D. N., Song, J. T., and Greenberg, J. T. (2003). ACD6, a novel ankyrin protein, is a regulator and an effector of salicylic acid signaling in the Arabidopsis defense response. Plant Cell 15, 2408–2420. doi: 10.1105/tpc.015412
Lü, S., Song, T., Kosma, D. K., Parsons, E. P., Rowland, O., and Jenks, M. A. (2009). Arabidopsis CER8 encodes LONG-CHAIN ACYL-COA SYNTHETASE 1 (LACS1) that has overlapping functions with LACS2 in plant wax and cutin synthesis. Plant J. 59, 553–564. doi: 10.1111/j.1365-313X.2009.03892.x
Martínez, M., and Mougan, I. (1998). Fatty acid composition of human brain phospholipids during normal development. J. Neurochem. 71, 2528–2533. doi: 10.1046/j.1471-4159.1998.71062528.x
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Merrill, A. H. Jr., Schmelz, E. M., Wang, E., Dillehay, D. L., Rice, L. G., Meredith, F., et al. (1997). Importance of sphingolipids and inhibitors of sphingolipid metabolism as components of animal diets. J. Nutr. 127, 830s–833s. doi: 10.1093/jn/127.5.830S
Millar, A. A., and Kunst, L. (1997). Very-long-chain fatty acid biosynthesis is controlled through the expression and specificity of the condensing enzyme. Plant J. 12, 121–131. doi: 10.1046/j.1365-313x.1997.12010121.x
Mitchell, A., Chang, H. Y., Daugherty, L., Fraser, M., Hunter, S., Lopez, R., et al. (2015). The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 43, D213–D221. doi: 10.1093/nar/gku1243
Ortiz, R., Geleta, M., Gustafsson, C., Lager, I., Hofvander, P., Löfstedt, C., et al. (2020). Oil crops for the future. Curr. Opin. Plant Biol. 56, 181–189. doi: 10.1016/j.pbi.2019.12.003
Paul, S., Gable, K., Beaudoin, F., Cahoon, E., Jaworski, J., Napier, J. A., et al. (2006). Members of the Arabidopsis FAE1-like 3-ketoacyl-CoA synthase gene family substitute for the elop proteins of Saccharomyces cerevisiae. J. Biol. Chem. 281, 9018–9029. doi: 10.1074/jbc.M507723200
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Poulos, A. (1995). Very long chain fatty acids in higher animals—A review. Lipids 30, 1–14. doi: 10.1007/BF02537036
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl. 1) i351–i358. doi: 10.1093/bioinformatics/bti1018
Ruan, C. J., Yan, R., Wang, B. X., Mopper, S., Guan, W. K., and Zhang, J. (2017). The importance of yellow horn (Xanthoceras sorbifolia) for restoration of arid habitats and production of bioactive seed oils. Ecol. Eng. 99, 504–512. doi: 10.1016/j.ecoleng.2016.11.073
Sakai, H., Lee, S. S., Tanaka, T., Numa, H., Kim, J., Kawahara, Y., et al. (2013). Rice annotation project database (RAP-DB): An integrative and interactive database for rice genomics. Plant Cell Physiol. 54:e6. doi: 10.1093/pcp/pcs183
Sakamoto, H., Matsuda, O., and Iba, K. (2008). ITN1, a novel gene encoding an ankyrin-repeat protein that affects the ABA-mediated production of reactive oxygen species and is involved in salt-stress tolerance in Arabidopsis thaliana. Plant J. 56, 411–422. doi: 10.1111/j.1365-313X.2008.03614.x
Sakamoto, H., Sakata, K., Kusumi, K., Kojima, M., Sakakibara, H., and Iba, K. (2012). Interaction between a plasma membrane-localized ankyrin-repeat protein ITN1 and a nuclear protein RTV1. Biochem. Biophys. Res. Commun. 423, 392–397. doi: 10.1016/j.bbrc.2012.05.136
Statista (2022). Global production of vegetable oils from 2000/01 to 2020/21 (in million metric tons). Available online at: https://www.statista.com/statistics/263978/global-vegetable-oil-production-since-2000-2001 (accessed Jun 15, 2022).
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245. doi: 10.1007/978-1-4939-9173-0_14
Sharma, M., Gupta, S. K., and Mondal, A. K. (2012). “Production and trade of major world oil crops,” in Technological innovations in major world oil crops, Vol. 1, ed. S. K. Gupta (New York, NY: Springer New York), 1–15. doi: 10.1007/978-1-4614-0356-2_1
Slater, G. S., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31. doi: 10.1186/1471-2105-6-31
Song, W., Zhang, K., Xue, T., Han, J., Peng, F., Ding, C., et al. (2022). Cognitive improvement effect of nervonic acid and essential fatty acids on rats ingesting Acer truncatum Bunge seed oil revealed by lipidomics approach. Food Funct. 13, 2475–2490. doi: 10.1039/d1fo03671h
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: An updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
Taylor, D. C., Francis, T., Guo, Y., Brost, J. M., Katavic, V., Mietkiewska, E., et al. (2009). Molecular cloning and characterization of a KCS gene from Cardamine graeca and its heterologous expression in Brassica oilseeds to engineer high nervonic acid oils for potential medical and industrial use. Plant Biotechnol. J. 7, 925–938. doi: 10.1111/j.1467-7652.2009.00454.x
Terluk, M. R., Tieu, J., Sahasrabudhe, S. A., Moser, A., Watkins, P. A., Raymond, G. V., et al. (2022). Nervonic acid attenuates accumulation of very long-chain fatty acids and is a potential therapy for adrenoleukodystrophy. Neurotherapeutics 19, 1007–1017. doi: 10.1007/s13311-022-01226-7
Tomato Genome Consortium. (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641. doi: 10.1038/nature11119
Tuskan, G. A., Difazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The genome of black cottonwood. Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604. doi: 10.1126/science.1128691
Van de Peer, Y., Maere, S., and Meyer, A. (2009). The evolutionary significance of ancient genome duplications. Nat. Rev Genet 10, 725–732. doi: 10.1038/nrg2600
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, J., Guo, J., Zhang, Y., and Yan, X. (2020a). Integrated transcriptomic and metabolomic analyses of yellow horn (Xanthoceras sorbifolia) in response to cold stress. PLoS One 15:e0236588. doi: 10.1371/journal.pone.0236588
Wang, J., Zhang, Y., Yan, X., and Guo, J. (2020b). Physiological and transcriptomic analyses of yellow horn (Xanthoceras sorbifolia) provide important insights into salt and saline-alkali stress tolerance. PLoS One 15:e0244365. doi: 10.1371/journal.pone.0244365
Wang, N., Yang, Y., Moore, M. J., Brockington, S. F., Walker, J. F., Brown, J. W., et al. (2019). Evolution of portulacineae marked by gene tree conflict and gene family expansion associated with adaptation to harsh environments. Mol. Biol. Evol. 36, 112–126. doi: 10.1093/molbev/msy200
Wang, P., Xiong, X., Zhang, X., Wu, G., and Liu, F. (2022). A review of erucic acid production in Brassicaceae oilseeds: Progress and prospects for the genetic engineering of high and low-erucic acid rapeseeds (Brassica napus). Front. Plant Sci. 13:899076. doi: 10.3389/fpls.2022.899076
Wang, Y., Yang, X., Chen, Z., Zhang, J., Si, K., Xu, R., et al. (2022). Function and transcriptional regulation of CsKCS20 in the elongation of very-long-chain fatty acids and wax biosynthesis in Citrus sinensis flavedo. Hortic. Res. 9:uhab027. doi: 10.1093/hr/uhab027
Xu, G. C., Xu, T. J., Zhu, R., Zhang, Y., Li, S. Q., Wang, H. W., et al. (2019). LR_Gapcloser: A tiling path-based gap closer that uses long reads to complete genome assembly. Gigascience 8:giy157. doi: 10.1093/gigascience/giy157
Xu, Z., and Wang, H. (2007). LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Yang, R., Zhang, L., Li, P., Yu, L., Mao, J., Wang, X., et al. (2018). A review of chemical composition and nutritional properties of minor vegetable oils in China. Trends Food Sci. Tech. 74, 26–32. doi: 10.1016/j.tifs.2018.01.013
Yang, Z. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Ye, J., McGinnis, S., and Madden, T. L. (2006). BLAST: Improvements for better sequence analysis. Nucleic Acids Res. 34, W6–W9. doi: 10.1093/nar/gkl164
Yu, H., Fan, S., Bi, Q., Wang, S., Hu, X., Chen, M., et al. (2017). Seed morphology, oil content and fatty acid composition variability assessment in yellow horn (Xanthoceras sorbifolium Bunge) germplasm for optimum biodiesel production. Ind. Crops Prod. 97, 425–430. doi: 10.1016/j.indcrop.2016.12.054
Zhang, Z., Shrestha, J., Tateda, C., and Greenberg, J. T. (2014). Salicylic acid signaling controls the maturation and localization of the Arabidopsis defense protein ACCELERATED CELL DEATH6. Mol. Plant 7, 1365–1383. doi: 10.1093/mp/ssu072
Zhao, L., Haslam, T. M., Sonntag, A., Molina, I., and Kunst, L. (2019). Functional overlap of long-chain Acyl-CoA synthetases in Arabidopsis. Plant Cell Physiol. 60, 1041–1054. doi: 10.1093/pcp/pcz019
Zhao, L., Katavic, V., Li, F., Haughn, G. W., and Kunst, L. (2010). Insertional mutant analysis reveals that long-chain acyl-CoA synthetase 1 (LACS1), but not LACS8, functionally overlaps with LACS9 in Arabidopsis seed oil biosynthesis. Plant J. 64, 1048–1058. doi: 10.1111/j.1365-313X.2010.04396.x
Zhao, Y., Liu, X., Wang, M., Bi, Q., Cui, Y., and Wang, L. (2021). Transcriptome and physiological analyses provide insights into the leaf epicuticular wax accumulation mechanism in yellowhorn. Hortic. Res. 8:134. doi: 10.1038/s41438-021-00564-5
Keywords: yellowhorn (Xanthoceras sorbifolium), haplotype-resolved, genome assembly, transcriptome, fatty acids biosynthesis, adaptation
Citation: Liang Q, Liu JN, Fang H, Dong Y, Wang C, Bao Y, Hou W, Zhou R, Ma X, Gai S, Wang L, Li S, Yang KQ and Sang YL (2022) Genomic and transcriptomic analyses provide insights into valuable fatty acid biosynthesis and environmental adaptation of yellowhorn. Front. Plant Sci. 13:991197. doi: 10.3389/fpls.2022.991197
Received: 11 July 2022; Accepted: 15 August 2022;
Published: 06 September 2022.
Edited by:
Muhammad Ali Abid, Biotechnology Research Institute (CAAS), ChinaReviewed by:
Honglian Ye, University of California, Davis, United StatesLibing Wang, Chinese Academy of Forestry, China
Copyright © 2022 Liang, Liu, Fang, Dong, Wang, Bao, Hou, Zhou, Ma, Gai, Wang, Li, Yang and Sang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ke Qiang Yang, eWFuZ3dlcmVAMTI2LmNvbQ==; Ya Lin Sang, c2FuZ3lsQHNkYXUuZWR1LmNu
†These authors have contributed equally to this work