 Shruti Mahajan
Shruti Mahajan Manohar S. Bisht
Manohar S. Bisht Abhisek Chakraborty
Abhisek Chakraborty Vineet K. Sharma
Vineet K. Sharma- MetaBioSys Group, Department of Biological Sciences, Indian Institute of Science Education and Research Bhopal, Bhopal, Madhya Pradesh, India
Phyllanthus emblica or Indian gooseberry, commonly known as amla, is an important medicinal horticultural plant used in traditional and modern medicines. It bears stone fruits with immense antioxidant properties due to being one of the richest natural sources of vitamin C and numerous flavonoids. This study presents the first genome sequencing of this species performed using 10x Genomics and Oxford Nanopore Technology. The draft genome assembly was 519 Mbp in size and consisted of 4,384 contigs, N50 of 597 Kbp, 98.4% BUSCO score, and 37,858 coding sequences. This study also reports the genome-wide phylogeny of this species with 26 other plant species that resolved the phylogenetic position of P. emblica. The presence of three ascorbate biosynthesis pathways including L-galactose, galacturonate, and myo-inositol pathways was confirmed in this genome. A comprehensive comparative evolutionary genomic analysis including gene family expansion/contraction and identification of multiple signatures of adaptive evolution provided evolutionary insights into ascorbate and flavonoid biosynthesis pathways and stone fruit formation through lignin biosynthesis. The availability of this genome will be beneficial for its horticultural, medicinal, dietary, and cosmetic applications and will also help in comparative genomics analysis studies.
Introduction
Vitamin C, also known as ascorbic acid, is a vital vitamin due to its multifaceted roles in animals as well as plants, and is an essential component of the human diet (Gallie, 2013; Carr and Maggini, 2017). The prolonged deficiency of this vitamin causes scurvy which was infamously responsible for killing thousands of sailors in the medieval period, since humans and primates cannot synthesise vitamin C due to the absence of an enzyme gulono-lactone oxidase (GULO), which is responsible for the final conversion to ascorbic acid (Martini, 2003; Wheeler et al., 2015). Thus, they depend majorly on plants that are the dominant sources of vitamin C for animals (Wheeler et al., 2015).
Phyllanthus emblica is one of the richest sources of natural vitamin C, and is, also known as Indian gooseberry or amla. It is an economically important medicinal horticultural plant that belongs to the family Phyllanthaceae in order Malpighiales, and is widely used in pharmaceuticals, nutraceuticals, food industry, and cosmetics sectors with an estimated market of USD 49.34 billion by 2025 (Muzaffar et al., 2022). Genus Phyllanthus is the largest genus of its family with approximately 1,000 species of which several are used as ethnomedicinal herbs due to the presence of medicinal phytochemicals (Sarin et al., 2014; Mao et al., 2016; Geethangili and Ding, 2018). The morphological characteristics of P. emblica include a light grey stem with thin flaky bark, simple leaves, and greenish-yellow unisexual flowers that are arranged like female flowers on the top and male flowers on the lower side. The fruits are typical drupes of about 2 cm in diameter, also known as stone fruits, with seeds encased in a hard lignified endocarp known as stone that helps in seed protection and dispersal (Dardick and Callahan, 2014; Dasaroju and Gottumukkala, 2014).
The geographical distribution of this prominent ethnomedicinal herbal species spreads from tropical to warm temperate regions like India, China, Sri Lanka, Bangladesh, Indonesia, Thailand, etc., among which India is the top producers of amla with annual production of 1,275,660 metric tonnes (Mao et al., 2016; Department of Agriculture & Farmers Welfare et al., 2021). This plant has also been used in many traditional medicine systems like Indian Ayurveda, Traditional Chinese Medicine System, etc. and is now widely used in modern medicines (Mao et al., 2016; Gul et al., 2022). Extracts of almost all parts of this plant such as leaf, bark, seed, root and fruit show medicinal properties like anti-microbial, anti-viral, anti-inflammatory, anti-oxidant, anti-aging, anti-diabetic, hypolipidemic, hypoglycaemic, neuroprotective, anti-cancer, immunomodulatory and hepatoprotective, etc. due to the presence of various secondary metabolites (phytochemicals) like alkaloids, phenolic acids, hydrolysable tannins, flavonoids, etc. with significance to human health and diseases (Gantait et al., 2021; Gul et al., 2022; Saini et al., 2022; Yan et al., 2022). The clinical effectiveness of P. emblica has been confirmed in diseases like dyslipidaemia, type 2 diabetes, chronic periodontitis, symptomatic knee osteoarthritis, etc. (Gantait et al., 2021). Amla is used in treating COVID-19 patients where its consumption shortened the recovery time (Varnasseri et al., 2022). Additionally, its phytochemicals are reported as potential protease inhibitors of SARS-CoV-2 virus through in-silico evidences (Murugesan et al., 2021; Pandey et al., 2021). Its extracts are proven to have protective effects by maintaining gut microbiome homeostasis in vivo (Li X. et al., 2022; Luo et al., 2022). Along with its benefits in human health, it is also effective in aquaculture, dairy and poultry as feed additives (Nguse et al., 2022; Van Doan et al., 2022; Abo Ghanima et al., 2023).
Among the vitamin C-rich fruits, P. emblica is known to contain the highest content of vitamin C (up to 720mg/100g fruit) along with other phytochemicals, minerals and amino acids (Kubola et al., 2011; Chavhan, 2017; Abeysuriya et al., 2020; Gul et al., 2022). Plants produce this vitamin to protect them against biotic (pathogens) and abiotic stresses (heat or light), and is also needed for the biosynthesis of plant hormones, and plant pigments, and acts as a cofactor in the cell cycle and metabolism, etc. (Gallie, 2013). The ascorbate biosynthesis occurs in plants through four proposed pathways i.e., L-galactose (also known as Smirnoff-Wheeler pathway), galacturonate (uronic acid pathway), L-gulose and myo-inositol pathways (Fenech et al., 2019; Paciolla et al., 2019). Among these pathways, the Smirnoff-Wheeler (SW) pathway is considered as the most common pathway of ascorbate biosynthesis (Gómez-García and Ochoa-Alejo, 2016; Sodeyama et al., 2021). Various genome-wide studies revealed ascorbic acid biosynthesis pathways in Psidium guajava, Citrus sinensis, kiwifruits, etc., however the ascorbate biosynthesis pathways have not been examined in P. emblica (Xu et al., 2013; Feng et al., 2021; Liao et al., 2021; Han et al., 2022).
Despite being a pharmaceutically and nutritionally important plant, the genome sequence of P. emblica still remains unknown. However, transcriptome studies were carried out previously to explore a few biosynthesis pathways in P. emblica (Kumar et al., 2016; Xiong-fang et al., 2018). The number of chromosomes in P. emblica was first reported as 28 in 1943 (Perry, 1943). Several following studies reported the chromosome numbers ranging from 52 to 104, and the most recent study has reported the presence of 100 chromosomes in P. emblica (Ammal and Raghavan, 1958; Soontornchainaksaeng and Chaiyasut, 1999; Rahman et al., 2021). Thus, to gain genomic insights into the medicinal properties of P. emblica, we performed its genome sequencing and assembly using a hybrid approach that includes 10x Genomics and Oxford Nanopore Technology (ONT) long-read sequencing technologies along with transcriptomic sequencing using the Illumina technology. Further, we analysed the genes involved in vitamin C, lignin and flavonoid biosynthesis pathways. We also constructed a genome-wide phylogenetic tree of P. emblica with 26 plant species, which were further analysed for gene family expansion and contraction. Furthermore, this study performed a comprehensive comparative evolutionary genomic analysis across 19 plant species to uncover the genes with multiple signatures of adaptive evolution in P. emblica.
Materials and methods
DNA-RNA extraction, species identification and sequencing
The leaves sample from an individual plant located at the campus of Indian Institute of Science Education and Research Bhopal, India (23.2858° N, 77.2755° E) were used in this study (Supplementary Figure 1). The DNA was extracted from the leaves sample using Carlson lysis buffer except for the precipitation step that was carried out with 0.5X volume of NaCl ad 0.7X volume of isopropanol (Jaiswal et al., 2021). The extracted DNA was quantified, and quality was checked on Qubit 2.0 fluorometer and Nanodrop 8000 spectrophotometer, respectively. Species identification assay was performed using marker genes: Internal Transcribed Spacer (ITS) and Maturase K (MatK). The extracted DNA was utilised to prepare libraries for 10x Genomics and nanopore sequencing that were sequenced on Illumina NovaSeq 6000 and MinION Mk1C sequencers, respectively. The RNA extraction from leaf tissue was performed as per the protocol described by Kumar and Singh (2012) with a few modifications (Kumar and Singh, 2012). The RNA was used for preparing the library using TruSeq Stranded Total RNA Library Preparation kit (Illumina Inc., CA, USA) with Ribo-zero Plant workflow and sequenced on Illumina NovaSeq 6000 instrument for generating 150 bp paired-end reads. The detailed method of DNA and RNA extraction with library preparation and sequencing is discussed in Supplementary Text 1.
Genome assembly
The proc10xG set of python scripts (https://github.com/ucdavis-bioinformatics/proc10xG) was used to pre-process the 10x Genomics raw reads by removing the barcode sequences. The obtained reads were processed by SGA-preqc (paired-end mode) for genome size estimation which works on a k-mer distribution-based approach (Simpson and Durbin, 2012). For genome complexity assessment, these pre-processed reads were used by Jellyfish v2.2.10 and GenomeScope v2.0 for generating k-mer count histograms and calculating heterozygosity, respectively (Marçais and Kingsford, 2011; Ranallo-Benavidez et al., 2020).
Guppy v3.2.1 (Oxford Nanopore Technologies) was used to carry out base calling of nanopore raw reads. Adaptor removal was performed on this base called raw data using Porechop v0.2.4 (Oxford Nanopore Technologies). The pre-processed reads were utilised for genome assembly using three different assemblers: wtdbg v2.0.0, SMARTdenovo (https://github.com/ruanjue/smartdenovo), and Flye v2.9 (Kolmogorov et al., 2019; Ruan and Li, 2020). wtdbg v2.0.0 and Flye v2.9 were used with default settings whereas SMARTdenovo was used with zero as minimum read length (Kolmogorov et al., 2019; Ruan and Li, 2020). Quast v5.0.2 was employed to assess the genome assembly statistics (Gurevich et al., 2013). The genome assembly resulting from Flye v2.9 was considered for further analysis due to its better assembly statistics and assembled genome size (Kolmogorov et al., 2019). The assembly was polished three times by Pilon v1.24 using filtered reads. ARCS v1.2.2 and LINKS v2.0.0 (default settings) were employed for the first round of scaffolding using Longranger basic v2.2.2 (https://support.10xgenomics.com/genome-exome/software/pipelines/latest/installation) barcode filtered 10x Genomics linked reads and adaptor-removed nanopore reads, respectively (Walker et al., 2014; Warren et al., 2015; Yeo et al., 2018). After scaffolding, the quality-filtering of RNA-Seq paired-end raw reads was performed using Trimmomatic v0.39 with parameters- “LEADING:15 TRAILING:15 SLIDINGWINDOW:4:15 MINLEN:50” which were subsequently utilised by AGOUTI v0.3.3 for the second round of scaffolding (Bolger et al., 2014; Zhang et al., 2016).
Supernova v2.1.1 was used to perform de novo assembly of P. emblica with maxreads=all options with other default parameters (Weisenfeld et al., 2017). The obtained genome assembly was corrected by Tigmint v1.2.6 using Longranger basic v2.2.2 processed linked reads (Jackman et al., 2018). Further, the first round of scaffolding was performed with ARCS v1.2.2 and LINKS v2.0.0 using Longranger basic v2.2.2 processed linked reads and adaptor removed nanopore reads, respectively (Warren et al., 2015; Yeo et al., 2018). To increase the assembly contiguity, AGOUTI v0.3.3 was used with the pre-processed transcriptomic paired-end reads (quality-filtered) (Zhang et al., 2016).
RagTag v2.1.0 was used to merge both the assemblies obtained from Supernova and Flye assemblers using the patch command line utility (Alonge et al., 2021). RagTag uses the main assembly as reference and query assembly to fill the gap in the reference assembly. LR_Gapcloser was used to perform gap-closing of the assembly using pre-processed nanopore reads (Xu et al., 2019). Sealer v2.3.5 was used for gap-closing of the assembly using barcode-removed linked reads with 30-120 k-mer value and 10 bp interval (Paulino et al., 2015). The fixation of small indels, base errors, and local misassemblies was performed by Pilon v1.24 using pre-processed linked reads to provide the draft genome assembly of P. emblica (Walker et al., 2014). Obtained draft genome assembly was further length base filtered and scaffolds having length ≥5 kbp were retained. BUSCO v5.2.2 with embryophyta_odb10 single-copy orthologs dataset was employed to assess the completeness of genome assembly (Simão et al., 2015). For further assessment of the assembly quality, the barcode-filtered 10x Genomics reads, nanopore long reads and the quality-filtered transcriptomic reads were mapped onto the genome assembly using BWA-MEM v0.7.17, MiniMap2 v2.17 and HISAT2 v2.2.1, respectively, and SAMtools v1.13 “flagstat” was used to calculate the percentage of mapped reads (Li et al., 2009; Li, 2013; Kim et al., 2015; Li, 2018).
Annotation of genome and construction of gene set
For annotation of repeats in the final genome assembly, RepeatModeler v2.0.3 was used to generate a de novo repeat library (Flynn et al., 2020). The clustering of repeats was performed using CD-HIT-EST v4.8.1 with parameters - 8 bp seed size and 90% sequence identity to eliminate redundant sequences (Fu et al., 2012). The resultant repeat library was utilised by RepeatMasker v4.1.2 (http://www.repeatmasker.org) to soft-mask the final genome assembly of P. emblica.
The coding gene set was constructed on the resultant repeat-masked genome assembly using MAKER v3.01.04 pipeline which deploys approaches such as ab initio and evidence alignment for prediction (Campbell et al., 2014). The construction of de novo transcriptome assembly was performed by Trinity v2.14.0 (default parameters) using quality-filtered transcriptomic reads of P. emblica from this study and previously reported studies (Haas et al., 2013; Liu et al., 2018). The gene set was constructed with transcriptome assembly and protein sequences of species belonging to the same order Malpighiales (Populus trichocarpa and Manihot esculenta) that were used as EST and protein evidence, respectively. In the MAKER pipeline, AUGUSTUS v3.2.3 was used for ab initio gene prediction while empirical evidence alignments and alignment polishing were performed using BLAST and Exonerate v2.2.0 (https://github.com/nathanweeks/exonerate), respectively (Altschul et al., 1990; Stanke et al., 2006). Based on the length and Annotation Edit Distance (AED) of gene models, the final gene set was constructed by selecting genes with length (≥150bp) and AED values <0.5. The completeness of this final gene set (also termed a high-confidence gene set) was checked using BUSCO v5.2.2 with embryophyta_odb10 dataset (Simão et al., 2015).
Additionally, Barrnap v0.9 (https://github.com/tseemann/barrnap) and tRNAscan-SE v2.0.9 were used to perform de novo prediction of rRNA and tRNA, respectively (Chan and Lowe, 2019). Based on homology, miRNA gene sequences in the P. emblica genome were identified using miRbase database with e-value 10-9 and 80% identity (Griffiths-Jones et al., 2007).
Phylogenetic tree construction
The 26 plant species were selected from Ensembl plant release 54 for phylogenetic analysis considering the representation of each plant family among the selected species (Bolser et al., 2016). Besides the protein sequences of these selected 26 plant species, MAKER-derived protein sequences of P. emblica were used for phylogenetic tree construction. Among all the protein files, the longest isoform for each protein was selected and provided to OrthoFinder v2.5.4 to construct the set of orthologous genes (Emms and Kelly, 2019). KinFin v1.0 was used to extract fuzzy one-to-one orthologs protein sequences that were present in all 27 species (Laetsch and Blaxter, 2017). MAFFT v7.310 was used to individually align all the obtained fuzzy one-to-one orthologs which were filtered and concatenated using BeforePhylo v0.9.0 (https://github.com/qiyunzhu/BeforePhylo) (Katoh and Standley, 2013). These obtained protein sequences were used to construct a phylogenetic tree based on maximum likelihood using RAxML with the ‘PROTGAMMAAUTO’ amino acid substitution model and 100 bootstrap values (Stamatakis, 2014).
Amino acid sequences of MatK gene from 49 Phyllanthus species (top 49 species except P. emblica based on sequence length) and Zea mays (outgroup) obtained from UniProt database along with MatK protein sequence of P. emblica were used for the phylogenetic analysis. MAFFT v.7.310 was used to align these protein sequences, and RAxML v8.2.12 was used with 1000 bootstrap value and ‘PROTGAMMAAUTO’ amino acid substitution model to construct the MatK-based phylogenetic tree (Katoh and Standley, 2013; Stamatakis, 2014).
Gene family expansion and contraction analysis
The proteome files containing the longest isoform for every protein from selected 27 species along with generated species phylogenetic tree were provided to CAFE v5 to assess the evolution of gene families (Mendes et al., 2020). The species phylogenetic tree was adjusted to an ultrametric tree based on the calibration point of 120 million years between P. emblica and Beta vulgaris obtained from TimeTree database v5.0 (Kumar et al., 2022). BLASTP was performed on protein sequences of all 27 species in All-versus-All mode (Altschul et al., 1990). The BLASTP results were clustered using MCL v14-137 and gene families containing clade-specific genes and >100 gene copies for minimum of one species were eliminated. These resultant gene families and ultrametric species tree were used to analyse the evolution (expansion/contraction) of gene families using a two-lambda (λ) model where λ signifies a random birth-death parameter. Among the obtained contracted/expanded gene families, gene families with >10 genes were considered as highly contracted/expanded gene families.
Identification of signatures of adaptive evolution
The 19 plant species were selected for identification of genes with evolutionary signatures that included five species of order Malpighiales i.e., Linum usitatissimum, Manihot esculenta, Phyllanthus emblica, Populus trichocarpa and Ricinus communis along with Actinidia chinensis (order Ericales), Arabidopsis thaliana (order Brassicales), Coffea canephora (order Gentianales), Cucumis sativus (order Cucurbitales), Daucus carota (order Apiales), Eucalyptus grandis (order Myrtales), Ficus carica (order Rosales), Gossypium raimondii (order Malvales), Helianthus annuus (order Asterales), Olea europaea (order Lamiales), Pistacia vera (order Sapindales), Quercus lobata (order Fagales), Solanum lycopersicum (order Solanales) and Vitis vinifera (order Vitales). Orthologous gene sets were constructed by OrthoFinder v2.5.4 using the proteome files from 19 selected plant species. Orthogroups that contained protein sequences from all these selected species were retrieved and in case multiple protein sequences were present for a species, the longest isoform of that protein was selected and retained for further analysis.
Identification of genes with higher rate of evolution
The resulting orthogroups across 19 plant species were aligned individually using MAFFT v7.310 (Katoh and Standley, 2013). These obtained alignments were used to construct a phylogenetic tree for individual orthogroups using RAxML v8.2.12 with ‘PROTGAMMAAUTO’ amino acid substitution model and a bootstrap value of 100 (Stamatakis, 2014). R package “adephylo” was used to calculate root-to-tip branch length distance for genes of all species in the phylogenetic trees (Jombart et al., 2010). The genes of P. emblica with comparatively higher root-to-tip branch length distance values were extracted and listed as the genes with higher nucleotide divergence or rate of evolution.
Identification of P. emblica genes with unique amino acid substitutions
Using the multiple sequence alignments obtained from MAFFT v7.310, which were used for the identification of genes with a high rate of evolution, amino acid positions alike in all the species except P. emblica were extracted and labelled as genes with unique amino acid substitution. Ten amino acids around any gap were not included in this analysis. The functional impact of obtained genes showing amino acid substitution was evaluated using Sorting Intolerant From Tolerant (SIFT) with UniProt database (Ng and Henikoff, 2003).
Identification of positively selected genes
MAFFT v7.310 was used for individual alignment of nucleotide sequence of all orthologous gene sets across selected 19 species (Katoh and Standley, 2013). PAML v4.9a with “codeml” program based on a branch-site model used nucleotide alignments in PHYLIP format and a species phylogenetic tree of these 19 species (constructed using RAxML) to identify genes with positive selection (Yang, 2007). These obtained genes with their log likelihood values were further processed through likelihood-ratio tests and genes with FDR-corrected p-values of <0.05 were labelled as positively selected genes. Positively selected codon sites were identified using Bayes Empirical Bayes (BEB) analysis with criteria of >95% probability for the foreground lineage.
Genes with multiple signatures of adaptive evolution (MSA)
The high rate of evolution, unique amino acid substitution with functional impact and positive selection are the evolutionary signatures of adaptive evolution. P. emblica genes that showed at least two of these evolutionary signatures were considered as the genes with multiple signatures of adaptive evolution (MSA).
Functional annotation
The annotation of high-confidence gene sets of P. emblica was performed using NCBI non-redundant (nr) database, SWISS-PROT database and Pfam-A v32.0 database using BLASTP (10-5 e-value), BLASTP (10-5 e-value), and HMMER v3.3, respectively (Bairoch and Apweiler, 2000; Bateman et al., 2004; Finn et al., 2011). The coding genes including the genes with evolutionary signatures were functionally annotated using KAAS and eggNOG mapper (Moriya et al., 2007; Huerta-Cepas et al., 2017). Further, the considered contracted and expanded gene families of P. emblica were extracted and provided to KAAS v2.1 and eggNOG mapper v2.1.9 for functional annotation, respectively (Moriya et al., 2007; Huerta-Cepas et al., 2017). The functional annotation of contracted and expanded gene families was also checked manually on Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
Analysis of vitamin C biosynthesis genes
The protein sequences of all the available enzymes of all four proposed pathways of vitamin C biosynthesis for A. thaliana were downloaded from Swiss-Prot or NCBI database. The gene D-galactose reductase (GalUR) was not available for A. thaliana, thus, sequence from strawberry plant species was used. These protein sequences were matched against the protein sequences of P. emblica using BLASTP with e-value 10-9 (Altschul et al., 1990). The enzymes involved in galactose pathway from vitamin C-rich plants i.e., Actinidia chinensis (kiwi), Capsicum annuum (chilli pepper), Carica papaya (papaya), Citrus sinensis (sweet orange), Malpighia glabra (acerola), Myrciaria dubia (camu-camu), Solanum lycopersicum (tomato) and Vitis vinifera (grapes) along with Arabidopsis thaliana as an outgroup were also obtained from UniProt or NCBI databases. Six genes i.e., Mannose 1-phosphate guanylyl transferase (GMPP), GDP-D-Mannose 3’,5’-epimerase (GME), GDP-L-galactose phosphorylase (GGP/VTC2/VTC5) and L-galactose-1-phosphate phosphatase (GPP/VTC4), L-galactose dehydrogenase (GalDH) and L-galactono-1,4-lactone dehydrogenase (GLDH) were selected for phylogeny due to their sequence availability for all nine species. The phylogenetic tree was constructed using these six genes each from 10 selected species (including P. emblica) using RAxML (Stamatakis, 2014). Among the above expanded and contracted genes, we checked for the copy number of all the gene families involved in ascorbate biosynthesis and regeneration pathways in the P. emblica genome.
The distant orthologs of 20 genes involved in ascorbate biosynthesis and regeneration pathways were identified to elucidate their origin by HHblits web server with default parameters using UniRef30 database (Remmert et al., 2012). The top 10 hits considering unique genus for each gene were extracted and aligned using MAFFT v.7.310 (Katoh and Standley, 2013). These alignments were used to construct the phylogenetic trees for each of the genes using RAxML v8.2.12 (1000 bootstrap value and ‘PROTGAMMAAUTO’ amino acid substitution model) (Stamatakis, 2014).
Analysis of flavonoid biosynthesis pathway
The protein sequences of genes involved in flavonoid biosynthesis pathway for Manihot esculenta were downloaded from UniProt and NCBI databases and matched against the protein sequences of P. emblica using BLASTP (e-value 10-9) (Altschul et al., 1990).
Analysis of lignin biosynthesis pathway
The protein sequences of genes involved in lignin biosynthesis pathway for Arabidopsis thaliana were downloaded from UniProt and NCBI databases and matched against the protein sequences of P. emblica using BLASTP (e-value 10-9) (Altschul et al., 1990).
Analysis of gene structure
Exonerate v2.2.0 was used to examine the exon-intron structure of genes involved in ascorbate biosynthesis and regeneration pathways, flavonoid biosynthesis, and lignin biosynthesis.
Results
Species identification and sequencing
The species was confirmed using the sequencing of two DNA markers, ITS and MatK, that were aligned to P. emblica sequences available at NCBI-nt database with the highest identity of 100% and 99.89%, respectively. A total of 136 Gbp (~237x coverage) and 18.3 Gbp (~32x coverage) of genome sequence data were generated using third-generation sequencing technologies i.e., 10x Genomics and Oxford Nanopore Technology (ONT), respectively (Supplementary Table 1). Further, ~85 million transcriptomic reads from leaf tissue were used for analysis in this study. (Supplementary Table 2).
Genome assembly and annotation
We computationally estimated the genome size of P. emblica to be 579 Mbp. The final genome assembly had a size of 519 Mbp and consisted of 4,384 contigs with GC content of 33.49%, largest contig of 3.3 Mbp, and N50 of 597 Kbp (Supplementary Table 3). The heterozygosity was estimated to be 1.37%, which appears to be high given its small genome size. The 98.4% complete and 0.4% fragmented BUSCOs of this genome assembly indicated its completeness (Supplementary Table 4). Further, 96.8% of linked reads and 93.45% of nanopore long reads could be mapped on the final genome assembly. The repeats constituted 53.39% of the genome with 2,051 de novo repeat family sequences that were clustered into 1,803 repeat families. Among the interspersed repeats, 10.96% and 10.13% were predicted as Ty1/Copia and Gypsy/DIRS1 elements, respectively (Supplementary Table 5). A total of 815 transfer RNA (tRNA) and 141 ribosomal RNA (rRNA) genes were identified in the genome. The detailed information on 216 microRNAs (miRNA) of P. emblica genome is mentioned in Supplementary Table 6.
The de novo transcriptome assembly comprised of a total of 238,454 transcripts and these transcripts were used as EST (empirical evidence) in the MAKER pipeline. The high-confidence gene set constituted of 37,858 genes and had an 89.9% complete BUSCO score (Supplementary Table 4). Overall, ~ 96% (36,296 out of 37,858) high-confidence coding genes of P. emblica could be annotated using the three reference databases; NCBI-nr, Swiss-Prot, and Pfam-A (Supplementary Table 7). The functional annotations of coding genes of P. emblica are mentioned in Supplementary Tables 8–10.
Phylogenetic tree construction
We identified 145,194 orthogroups, of which 123 one-to-one fuzzy orthogroups were predicted from the selected 27 plant species. These selected concatenated protein sequence alignments of these fuzzy one-to-one orthogroups contained 104,108 alignment positions were used to construct a phylogenetic tree based on maximum likelihood using 26 eudicot species and Zea mays as an outgroup. The phylogenetic tree showed Populus trichocarpa and Manihot esculenta as the closest species to P. emblica as they belong to the same order Malpighiales. As per the phylogenetic tree, P. emblica diverged earlier (88 million years ago) than the other considered species of the order Malpighiales (Figure 1; Supplementary Text 2). Similarly, the phylogeny constructed with vitamin C biosynthesis genes followed the genome-wide phylogeny of P. emblica where the species like P. emblica and Malpighia glabra, Arabidopsis thaliana and Carica papaya and, Capsicum annuum and Solanum lycopersicum belonging to the same orders were sharing their common ancestral node (Figure 2). The phylogenetic tree of 50 Phyllanthus species using MatK indicated that P. emblica is evolutionarily closer to P. urinaria (Supplementary Figure 2), which has also been supported by other studies (Kathriarachchi et al., 2006; Bouman et al., 2021).
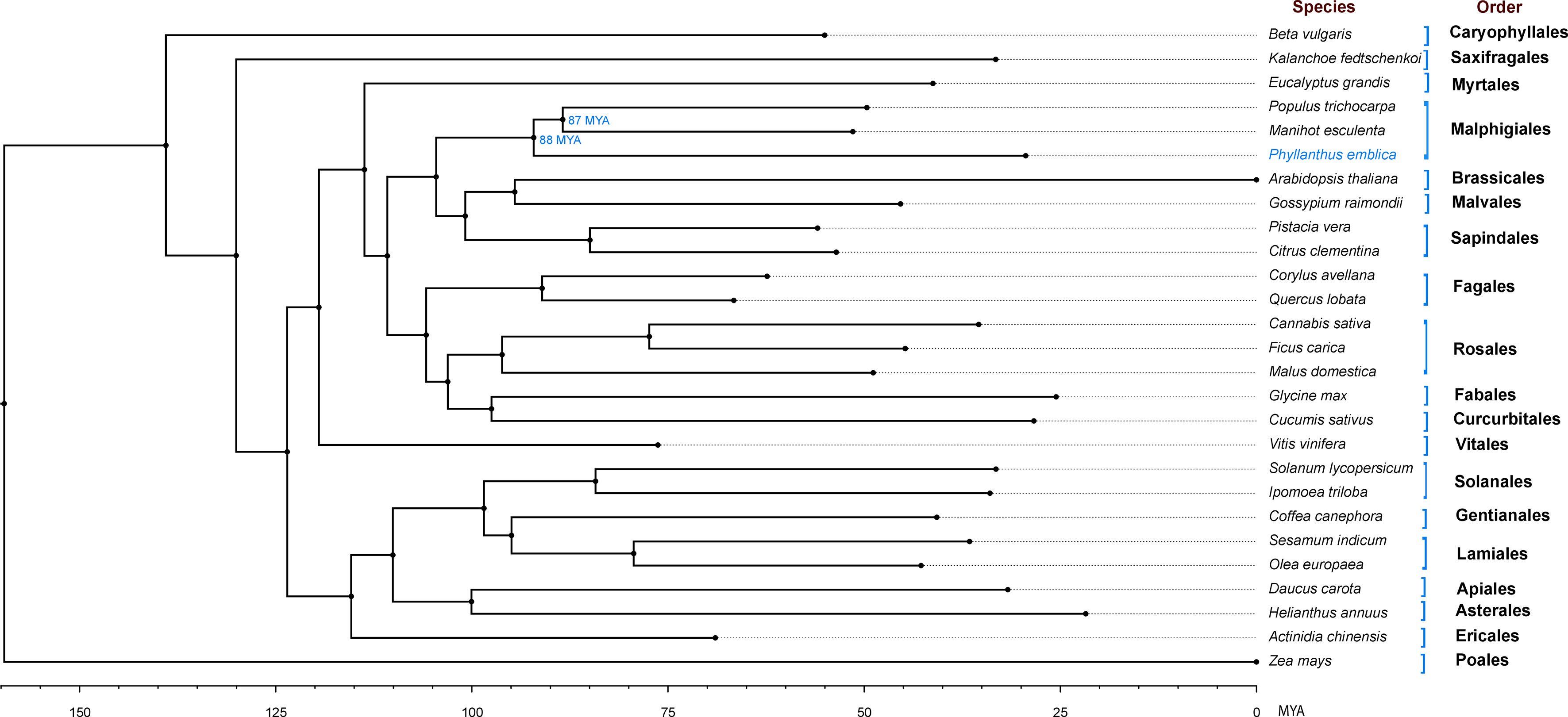
Figure 1 Genome-wide phylogeny of P. emblica with 26 other plant species Genome-wide phylogeny of P. emblica with 25 other eudicot species and a monocot species, Zea mays that was used as an outgroup. The indicated adjusted divergence time for Malpighiales order species were obtained from TimeTree database v5.0 (Kumar et al., 2022). The schematic representation method of the evolutionary time scale is similar to the previous studies (Teh et al., 2017; Xia et al., 2021).
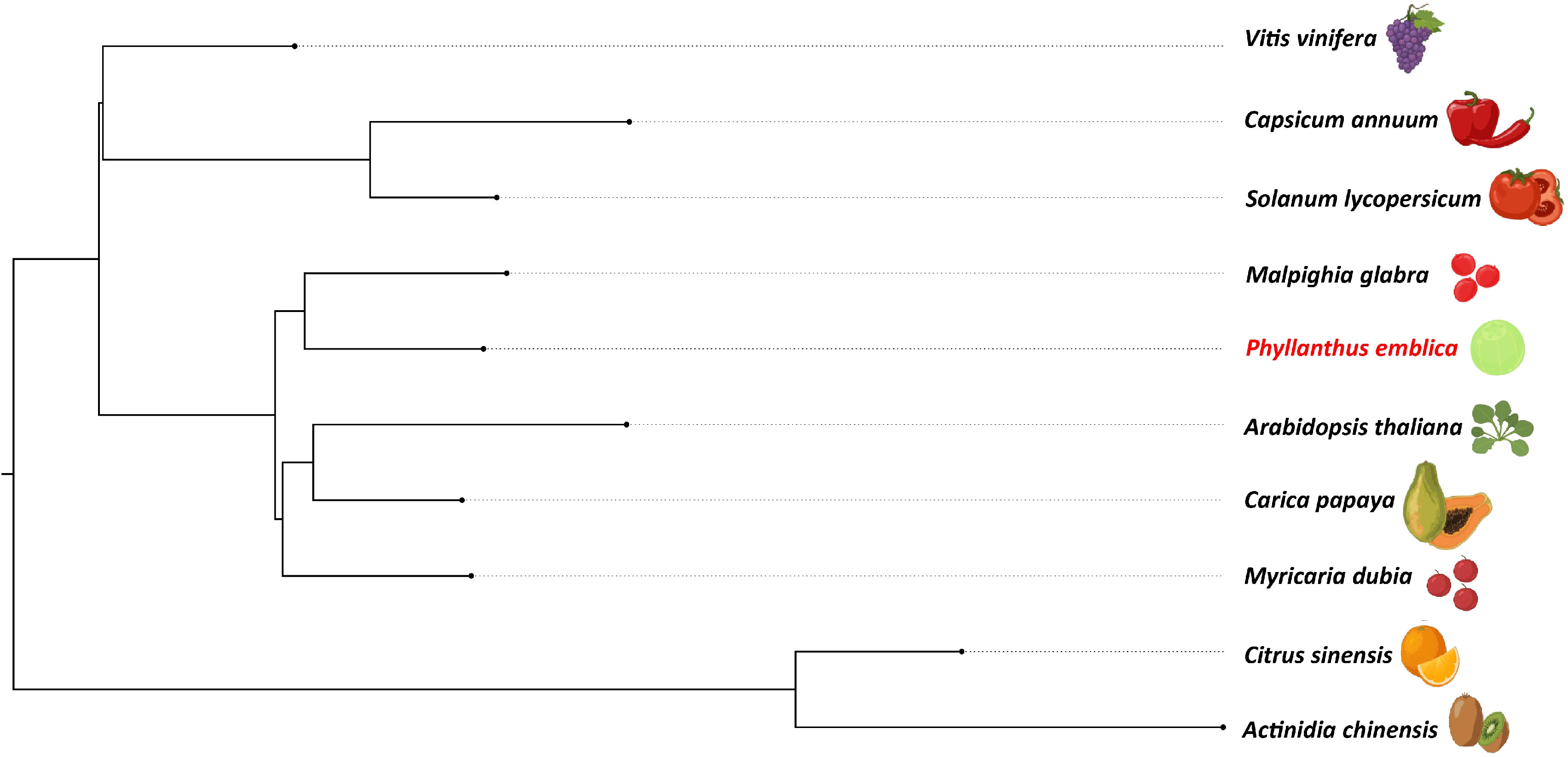
Figure 2 Vitamin C biosynthesis genes phylogeny of P. emblica with other vitamin C-rich fruits The phylogeny constructed for P. emblica, Actinidia chinensis, Capsicum annuum, Carica papaya, Citrus sinensis, Myrciaria dubia, Malpighia glabra, Solanum lycopersicum, Vitis vinifera and Arabidopsis thaliana using six genes of Smirnoff Wheeler pathway of ascorbate biosynthesis. The genes used were GDP-mannose pyrophosphorylase (GMPP), GDP-D-Mannose 3’,5’-epimerase (GME), GDP-L-galactose phosphorylase (GPP/VTC2), L-galactose-1-phosphate phosphatase (GPP/VTC4), L-galactose dehydrogenase (GalDH) and L-galactono-1,4-lactone dehydrogenase (GLDH).
Gene family expansion and contraction analysis
Gene family expansion and contraction analysis helps to identify the gene families that have increased or decreased in number in a given species. The analysis of adaptive evolution provides the clues of natural selection of specific phenotypic traits in a given species to cope with diverse environmental conditions that help in its survival. The gene expansion and contraction analysis showed a contraction of 1,048, and an expansion of 3,520 gene families in this species. Among these families, five and 42 gene families were found to be highly contracted and highly expanded, respectively. Among the 42 expanded gene families, 38 could be functionally annotated (Supplementary Table 11). The expanded gene families were majorly involved in lignin biosynthesis pathway, MAPK signaling pathway, transcription (as transcription factors), phenylpropanoid pathway, brassinosteroid biosynthesis, terpenoid biosynthesis, transportation (as transporters), plant hormone signal transduction, etc.
Identification of signatures of adaptive evolution
For evolutionary analysis, 7,864 orthogroups were obtained across 19 selected species. Among these orthogroups, 46 genes showed higher nucleotide divergence and 488 genes showed positive selection in P. emblica. The genes of P. emblica were present in ~35% (2,791 of 7,864) of the orthogroups showed unique amino acid substitutions. A total of 236 genes were identified as MSA genes in P. emblica. The MSA genes were found to be involved in physiological processes like plant growth, ROS regulation and detoxification, DNA damage response, immune signaling, abiotic stress response, pathogen resistance, response to hormones like ethylene, abscisic acid, gibberellin and cytokinin, and cell wall modification. The list and functional details of the MSA genes of P. emblica are mentioned in Supplementary Tables 12, 13.
Vitamin C biosynthesis pathway
Ascorbate, a non-enzymatic antioxidant, plays an important role in ROS detoxification and is a part of the ascorbate-glutathione pathway. P. emblica contains all the genes of the SW pathway similar to the other vitamin C-rich plant species like guava, kiwi, chilli pepper, etc., (Gómez-García and Ochoa-Alejo, 2016; Wang et al., 2018; Feng et al., 2021) (Figure 3). The gene structures of these genes are mentioned in Supplementary Table 14. The evolutionary analysis of six genes of SW pathway showed that P. emblica genes were phylogenetically closer to genes of Malpighia glabra, which also lies in the same order Malphigiales (Figure 2). Further, the genomic clues for presence of another pathway of ascorbate biosynthesis, i.e., galacturonate pathway were apparent from the presence of PME, PL, PG, GalUR and GLDH genes in P. emblica genome. This pathway is also proposed in tomatoes, strawberries, oranges, and grapes due to the presence of gene GalUR, which is the key gene of this pathway and was also present in P. emblica (Agius et al., 2003; Cruz-Rus et al., 2010; Cruz-Rus et al., 2011; Badejo et al., 2012; Xu et al., 2013). In addition, the genes MIOX and GulLDH involved in myo-inositol pathway were found, which supports the presence of the third pathway of ascorbate biosynthesis in P. emblica. However, the presence of the fourth pathway (L-gulose pathway) could not be confirmed due to lack of sufficient identification of genes involved in this pathway in P. emblica genome.
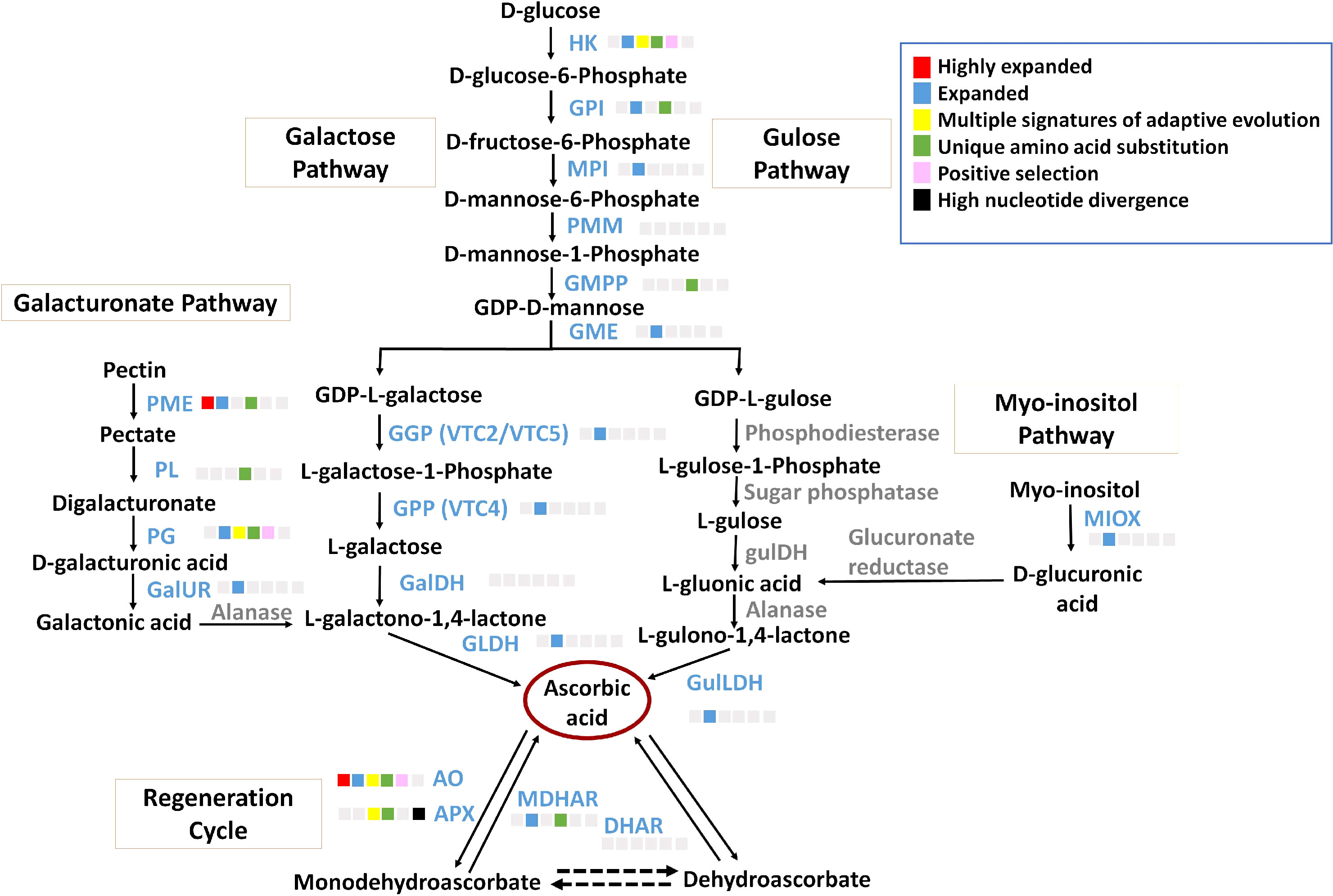
Figure 3 Ascorbate biosynthesis pathways The figure represents four proposed Ascorbate biosynthesis pathways i.e., Galactose pathway, Gulose pathway, Galacturonic acid pathway and Myo-inositol pathway and Ascorbate regeneration cycle. The enzymes of galactose pathway are HK, Hexokinase; GPI, Glucose 6-phosphate isomerase; MPI/PMI, Mannose 6-phosphate isomerase; PMM, Phosphomannomutase; GMPP, GDP-mannose pyrophosphorylase; GME, GDP-D-Mannose 3’,5’-epimerase; GGP/VTC2, GDP-L-galactose phosphorylase; GPP/VTC4, L-galactose-1-phosphate phosphatase; GalDH, L-galactose dehydrogenase; GLDH, L-galactono-1,4-lactone dehydrogenase. The Gulose pathway includes HK, GPI, MPI, PMM, GMPP, GME, Phosphodiesterase, Sugar phosphatase, L-gulose dehydrogenase, Aldonolactonase and Gulono-1,4-lactone dehydrogenase. The Galacturonic acid pathway includes PME, Pectin methylesterase; PL, Pectin lyase; PG, Polygalacturonse; GalUR, D-galacturonate reductase; Alanase, Aldono-lactonase; and GLDH. The Myo-inositol pathway includes MIOX, Myo-inositol oxidase; Glucuronate reductase; and GulLDH, Gulono-1,4-lactone dehydrogenase. The regeneration cycle of ascorbate includes AO, Ascorbate oxidase; APX, Ascorbate peroxidase; DHAR, Dehydroascorbate; and MDHAR, Monodehydroascorbate. The enzymes in blue colour and grey colour indicate presence and absence of their genes in P. emblica, respectively. The colour panel in front of each enzyme indicates the evolutionary signatures like highly expanded gene family, expanded gene family, MSA, unique amino acid substitution, positive selection and high nucleotide divergence shown by its gene in P. emblica.
Among the genes of all proposed ascorbate biosynthesis pathways, six genes HK, GPI, GMPP, PME, PL and PG were identified with unique amino acid substitutions. Gene family of pectin methylesterase (PME) involved in galacturonate pathway of ascorbate biosynthesis was highly expanded. Other genes of the galacturonate pathway i.e., polygalacturonase (PG) was found with MSA, and pectin lyase (PL) gene had unique amino acid substitutions. Along with these biosynthesis genes, MATE (Multidrug And Toxic compound Extrusion) gene family, which is a vacuolar ascorbate transporter, was also highly expanded in P. emblica (Hoang et al., 2021). The details of ascorbate biosynthesis and regeneration pathways are described in Figure 3 and Supplementary Table 15.
The phylogenetic relationships of 16 genes involved in ascorbate biosynthesis pathway with their distant orthologs are shown in Supplementary Figures 3–18. 10 out of 16 genes of ascorbate biosynthesis pathways had distant orthologs from other Malpighiales order members and among these genes, seven were phylogenetically closer to Malpighiales members. 11 genes had distant orthologs from monocot species. GME and GPI involved in the L-galactose pathway of ascorbate biosynthesis had algal and fungal orthologs, respectively. GMPP had orthology with algal and animal genes. MPI also had fungal and animal orthologs. GalDH and PMM had protozoan and animal orthologs along with a bacterial ortholog for GalDH. GalUR involved in the L-galacturonate pathway of ascorbate biosynthesis showed a fungal ortholog.
Glutathione metabolism and ascorbate-glutathione pathway
Glutathione is another non-enzymatic antioxidant that plays a key role in different environmental stresses mainly oxidative stress and is a part of the ascorbate-glutathione pathway (Hasanuzzaman et al., 2019; Dorion et al., 2021). A total of four genes (GPX, G6PD, gshA, and APX) involved in glutathione metabolism showed multiple signatures of adaptive evolution and along with these genes, GST showed positive selection in P. emblica. The functional details of these genes are mentioned in Supplementary Table 16.
The ascorbate-glutathione pathway, also known as ascorbate regeneration cycle, plays an important role in oxidative stress response by converting the oxidised ascorbate forms to ascorbate and vice versa via four enzymes i.e., ascorbate oxidase (AO), ascorbate peroxidase (APX), dehydroascorbate reductase (DHAR) and monodehydroascorbate reductase (MDHAR) (Chen et al., 2003; Li et al., 2017). All these genes of ascorbate regeneration pathway were found in P. emblica. Among the four genes, AO and APX showed multiple signatures of adaptive evolution, and MDHAR showed unique amino acid substitutions. The functional details of these genes are mentioned in Supplementary Table 16. The phylogenetic relationships of these genes with their distant orthologs are shown in Supplementary Figures 19–22. Three genes involved in the ascorbate regeneration pathway had orthologs from bryophyte species (APX and DHAR) and algal species (MDHAR). Also, DHAR had a phylogenetically closer ortholog from Malpighiales order.
Flavonoid biosynthesis pathway
All 15 key genes of flavonoid biosynthesis pathway were found in P. emblica genome (Figure 4), and their gene structures are mentioned in Supplementary Table 14. Seven of these genes contained unique amino acid substitutions. Flavonoid 3’5’-hydroxylase (F3’5’H) was among the genes with MSA, and flavonol synthase (FLS), flavone synthase (FS), isoflavone synthase (IFS), flavavone 3-hydroxylase (F3H), leucoanthocyanidin reductase (LDOX) and F3’5’H gene families were highly expanded. The detailed pathway of flavonoid biosynthesis is showed in Figure 4 and Supplementary Table 17.
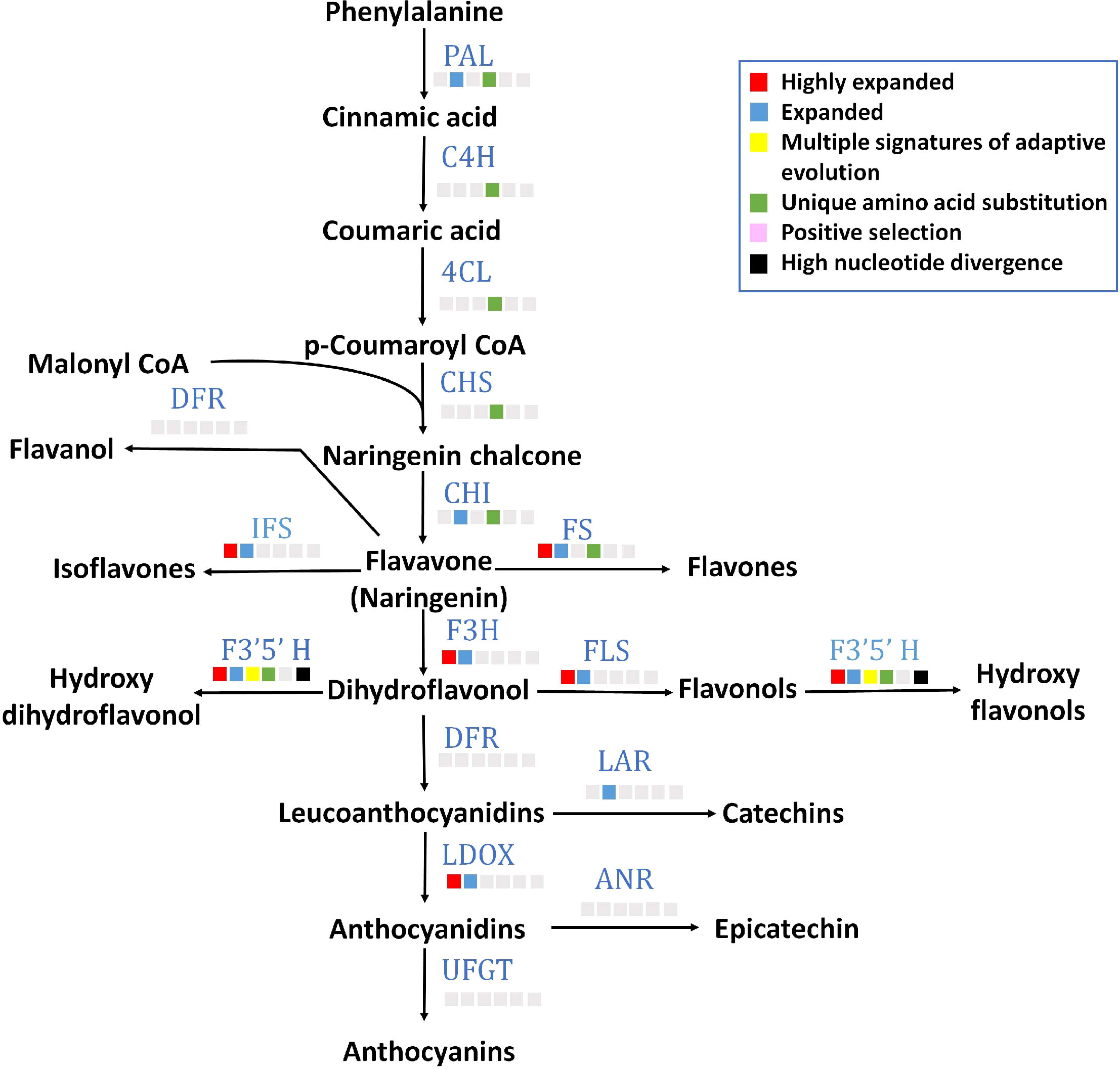
Figure 4 Flavonoid biosynthesis pathway The figure indicates the flavonoid biosynthesis pathway in P. emblica. The pathway includes enzymes PAL, phenylalanine ammonia lyase; C4H, cinnamate 4-hydroxylase;4CL, 4-coumaroyl CoA ligase; CHS, chalcone synthase; CHI, chalcone isomerase; FS, flavone synthase; DFR, dihydroflavonol 4-reductase; IFS, isoflavone synthase; F3’5’H, flavonol 3′5′ -hydroxylase; FLS, flavonol synthase; F3H, Flavanone 3-hydroxylase; LAR, leucoanthocyanidin reductase; LDOX, leucoanthocyanidin dioxygenase; ANR, anthocyanidin reductase; and UFGT, uridine diphosphate (UDP)-glucose flavonoid-3-O-glycosyltransferase. The blue colour of enzymes indicates presence of their genes in P. emblica. The colour panel in front of each enzyme indicates the evolutionary signatures like highly expanded gene family, expanded gene family, MSA, unique amino acid substitution, positive selection and high nucleotide divergence shown by its gene in P. emblica.
Lignified endocarp in this stone fruit
A lignified endocarp is a trait found in stone fruits like P. emblica. Lignin is important in stone cell formation in a drupe fruit that provides rigidity for seed protection and dispersal. Eight out of 13 genes (gene structures are mentioned in Supplementary Table 14) of lignin biosynthesis pathway including PAL, 4CL, C4H, PTAL, HCT, CAD, POD and LAC contained unique amino acid substitutions (Figure 5). Among these eight genes, CAD showed multiple signatures of adaptive evolution. Furthermore, six gene families C3H, HCT, COMT, CAD, POD and LAC were highly expanded. Moreover, the gene families of transcription factors MYB and LBD18 that are involved in lignin biosynthesis pathway were also found to be highly expanded (Supplementary Table 18).
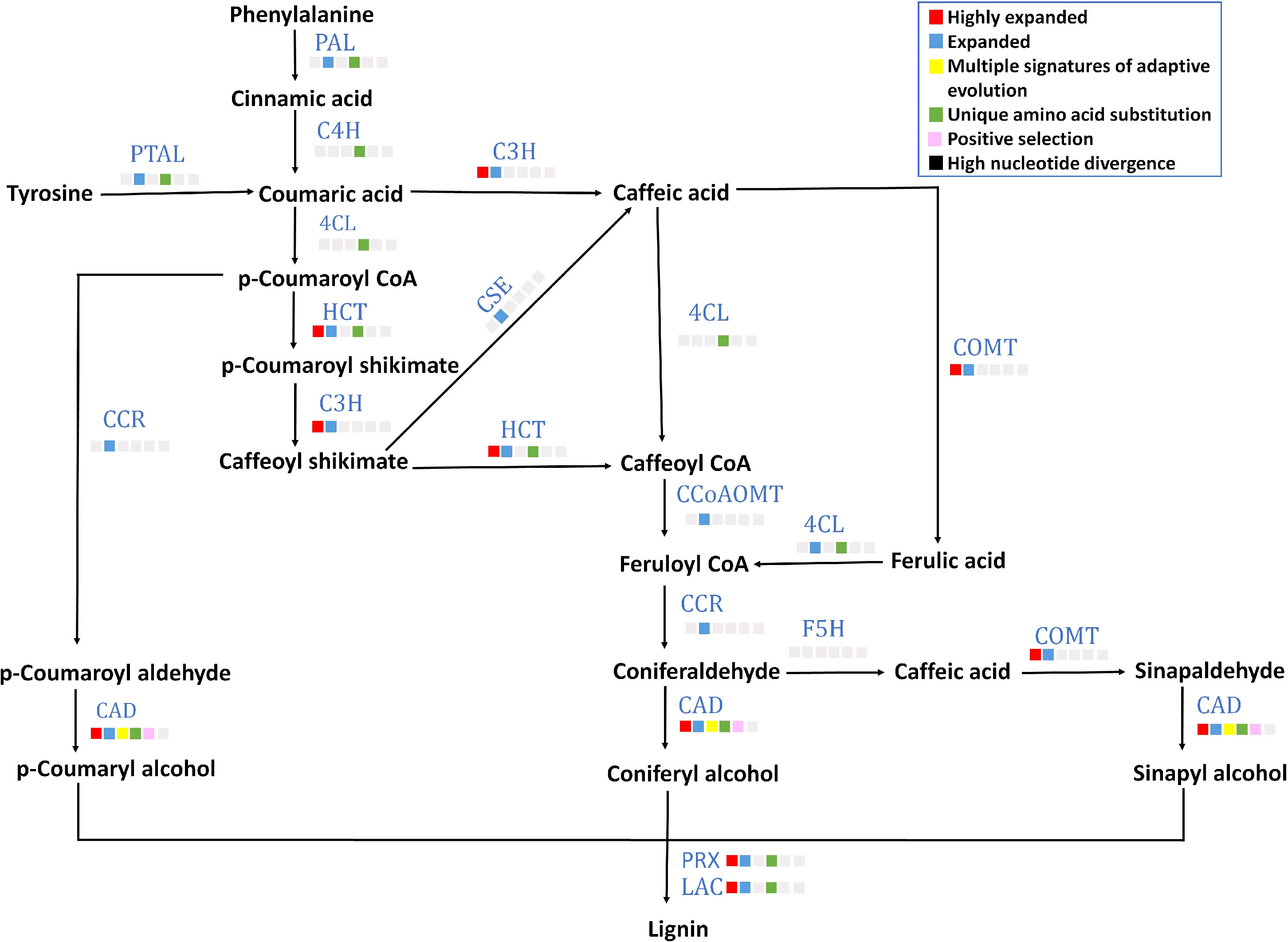
Figure 5 Lignin biosynthesis pathway The figure denotes lignin biosynthesis pathway in P. emblica. The pathway includes enzymes PAL, phenylalanine ammonia lyase; C4H, cinnamate 4-hydroxylase;4CL, 4-coumaroyl CoA ligase; PTAL, phenylalanine/tyrosine ammonia-lyase; C3H, 5-O-(4-coumaroyl)-D-quinate 3’-monooxygenase; HCT, shikimate O-hydroxycinnamoyl transferase; CSE, caffeoyl shikimate esterase; COMT, caffeic acid 3-O-methyltransferase; CCoAOMT, caffeoyl-CoA O-methyltransferase; CCR, cinnamoyl-CoA reductase; CAD, cinnamyl-alcohol dehydrogenase; F5H, ferulate-5-hydroxylase; PRX, peroxidase; and LAC, laccase. The blue colour of enzymes indicates presence of their genes in P. emblica. The colour panel in front of each enzyme indicates the evolutionary signatures like highly expanded gene family, expanded gene family, MSA, unique amino acid substitution, positive selection and high nucleotide divergence shown by its gene in P. emblica.
Plant growth, hormone and stress response
Among the 236 MSA genes, 36 genes were found to be involved in plant growth and development. These 36 genes are involved in cell division, flower development, seed development, seed germination, shoot development, cell elongation, sugar metabolism, cell wall biosynthesis, and root development, etc. (Supplementary Table 19). Phytohormones such as auxin, cytokinin, gibberellic acid, abscisic acid (ABA), ethylene, etc. help in plant growth, development, stress tolerance, etc. throughout the plant life. 20 MSA genes were responsible for plant hormone biosynthesis, signalling and response in plants. These include genes associated with plant hormone responses related to auxin, abscisic acid, ethylene, jasmonic acid, and cytokinin (Supplementary Table 19).
Plants have mechanisms for stress tolerance against abiotic and biotic stresses. 38 out of 236 MSA genes were associated with various responses against these stresses. Among these, 30 genes were associated with responses to abiotic stresses like salt, cold, heat, drought, etc., whereas 19 genes were associated with biotic stress tolerance. These genes are involved in stress responses like stress signal transduction, secondary metabolite biosynthesis, abscisic acid biosynthesis, ROS detoxification, stress-specific gene regulation, degradation of misfolded and damaged proteins, DNA damage response, cell wall modification, disease resistance, etc. (Supplementary Table 19). Also, six MSA genes were found to be involved in DNA damage repair mechanism in plants against environmental stresses (Supplementary Table 19).
ROS regulation and detoxification
Reactive oxygen species (ROS) are metabolic by-products produced in mitochondria, plastids and peroxisomes, which can cause irreversible DNA damage resulting into cell death. In plants, ROS not only cause harmful effects but also act as signalling molecules for plant growth and stress responses. 18 out of 236 MSA genes were associated with ROS response, regulation and detoxification. These genes are involved in porphyrin biosynthesis, ROS-induced responses, ROS scavenging, biosynthesis and protecting antioxidant enzymes, maintaining homeostasis, accumulation, and biosynthesis of antioxidants, and activation of Fe-S cluster (Supplementary Table 20).
Discussion
P. emblica or amla is a widely used medicinal plant with enormous antioxidant properties (Gul et al., 2022). To understand the genomic basis of these properties, we successfully constructed the first draft genome assembly of P. emblica using a hybrid sequencing approach using 10x Genomics, Illumina and ONT technologies. Despite a repetitive and highly heterozygous nature of this genome, implementation of a hybrid approach helped in constructing a high-quality genome assembly with N50 of ~0.6 Mbp and high BUSCO completeness (98.4%).
Further, this study is the first to resolve the genome-wide phylogenetic position of P. emblica with respect to 26 other plant species and found its early divergence from Manihot esculenta and Populus trichocarpa species of order Malpighiales, which was also confirmed by the adjusted time obtained from TimeTree database. Our phylogeny is also supported by the revised classification of order Malpighiales where Phyllanthaceae was separated as individual family from Euphorbiaceae in the Angiosperm Phylogeny Group Classification (APG III) (Group, 2009; Kawakita and Kato, 2017). The phylogeny of vitamin C biosynthesis genes also followed the genome-wide phylogeny. Phylogenetic analysis of genes involved in the L-galactose pathway of ascorbate biosynthesis showed that six out of 10 genes in P. emblica were closer to orthologs from other Malpighiales members. Further, key genomic insights were gained from the results of gene family expansion and contraction and from the genes with multiple signatures of adaptive evolution in P. emblica. The genes related to the biosynthesis of ascorbic acid, lignin and flavonoid were found to be evolutionarily selected in P. emblica.
Ascorbic acid is the major antioxidant in P. emblica and its fruit “amla” is one of the richest natural sources. A transcriptomic study of oranges had shown the attribution of different pathways of ascorbate biosynthesis in a tissue-specific as well as fruit developmental stage specific manner (Caruso et al., 2021). This could also be possible in P. emblica where the presence of three pathways of ascorbate biosynthesis is traced, and they could have roles in different stages and tissues. It was noted that the genes of one of the ascorbic acid biosynthesis pathways i.e., galacturonate pathway were found with MSA (PG), amino acid substitutions (PME, PL and PG) and highly expanded gene family (PME) in the P. emblica genome. The involvement of enzymes PME and PG in increased ascorbate production in tomatoes along with the role of PME in regulating ascorbate content through galacturonate pathway is shown in previous studies (Di Matteo et al., 2010; Badejo et al., 2012; Ruggieri et al., 2015; Rigano et al., 2018). Thus, it is tempting to speculate that the evolution of genes of galacturonate pathway could be associated with the high ascorbate production in P. emblica.
P. emblica is also rich in flavonoids that are synthesised in response to plant stress and contribute to its antioxidant property. The genes from PAL to CHI involved in the initial part of flavonoid biosynthesis pathway were found with unique amino acid substitutions, and the F3’5’H gene, which is previously reported to increase flavonoid accumulation, showed multiple signatures of adaptation (Wang et al., 2014; Nguyen et al., 2021). In addition to these, FLS, F3’5’H, FS, LDOX, F3H and IFS gene families were highly expanded which collectively indicates evolution of flavonoid biosynthesis genes in P. emblica. These genes are involved in biosynthesis of flavonoids such as isoflavones, flavones, anthocyanins and flavonols that have antioxidant properties and provide tolerance against various abiotic and biotic stresses (Verhoeyen et al., 2002; Agati et al., 2012). The evolutionary selection of these flavonoid associated genes might be responsible for the high antioxidant property and stress tolerance of P. emblica.
Being a stone fruit, a lignified endocarp is a trait found in the fruits of P. emblica, thus the evolution of lignin biosynthesis pathway was one of the major findings. Lignin is important for the stony seed coat formation in drupes that provides rigidity for its protection. The lignin biosynthesis genes were observed to be highly expanded and among the MSA genes in P. emblica genome, which hints towards the evolutionary significance of lignified endocarp in this stone fruit. The lignin biosynthesis gene families were also reported to be expanded in the other stone fruit genomes such as pear and Populus, which are economically important due to their fruit and wood, respectively, where lignin is the main content of pear’s stone cells and poplar’s wood (Wu et al., 2013; Li et al., 2022).
P. emblica also produces a large variety of secondary metabolites that provide tolerance against plant stresses. The expansion of gene families and MSA in genes involved in biosynthesis of various secondary metabolites and pathogen resistance against abiotic and biotic stresses were found that indicates the evolution of stress tolerance genes in this genome. Among the genes related to plant stress tolerance, the genes involved in ROS regulation and detoxification were also evolved in P. emblica.
Taken together, it is apparent that the adaptive evolution in genes involved in ascorbate biosynthesis, glutathione metabolism, flavonoid biosynthesis, and ROS detoxification are associated with the high antioxidant potential of P. emblica, which makes it a valuable herbal plant for use in traditional and modern medicine, horticulture, food and cosmetic products. Further, the high concentration of vitamin C in the amla fruit and the large production (up to 100 kg) of fruits per tree compared to other vitamin C rich fruits like Malpighia glabra (15-30 Kg/tree) and Myrciaria dubia (25-30 Kg/tree), makes it the perfect choice in switching from synthetic to natural supplementation of vitamin C (Rodrigues et al., 2001; Orwa et al., 2009; Carr and Vissers, 2013). Further, this plant also shows high genetic diversity and easy adaptation to various climatic zones and environmental conditions (Liu et al., 2020). The availability of the first draft genome of this economically important plant is likely to help in developing improved nutraceuticals, food, cosmetics and pharmaceutical products, and for further horticultural and genomic studies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, BioProject accession- PRJNA947813 and BioSample accession SAMN33867225.
Author contributions
VKS conceived and coordinated the project. SM performed sample collection, DNA-RNA extraction, prepared samples for sequencing, performed long read sequencing, species identification assays, functional annotation of gene sets, and constructed all the figures. MSB and AC designed computational framework of the study, and performed all the computational analyses presented in the study. SM, MSB, AC and VKS analysed the data and interpreted the results. SM and VKS wrote the first draft of manuscript. SM, AC, MSB and VKS wrote and prepared the final manuscript. All the authors have read and approved the final version of the manuscript.
Acknowledgments
SM and AC thank Council of Scientific and Industrial Research (CSIR) for fellowship. MSB thanks Ministry of Education, Govt. of India for Prime Minister Research Fellowship (PMRF). The authors also thank the sequencing facilities at Central Instrumentation Facility, IISER Bhopal and the intramural research funds provided by IISER Bhopal.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1210078/full#supplementary-material
References
Abeysuriya, H. I., Bulugahapitiya, V. P., Loku Pulukkuttige, J. (2020). Total vitamin C, ascorbic acid, dehydroascorbic acid, antioxidant properties, and iron content of underutilized and commonly consumed fruits in Sri Lanka. Int. J. Food Sci. 2020. doi: 10.1155/2020/4783029
Abo Ghanima, M. M., Aljahdali, N., Abuljadayel, D. A., Shafi, M. E., Qadhi, A., Abd El-Hack, M. E., et al. (2023). Effects of dietary supplementation of Amla, Chicory and Leek extracts on growth performance, immunity and blood biochemical parameters of broilers. Ital. J. Anim. Sci. 22 (1), 24–34. doi: 10.1080/1828051X.2022.2156932
Agati, G., Azzarello, E., Pollastri, S., Tattini, M. (2012). Flavonoids as antioxidants in plants: location and functional significance. Plant Sci. 196, 67–76. doi: 10.1016/j.plantsci.2012.07.014
Agius, F., González-Lamothe, R., Caballero, J. L., Muñoz-Blanco, J., Botella, M. A., Valpuesta, V. (2003). Engineering increased vitamin C levels in plants by overexpression of a D-galacturonic acid reductase. Nat. Biotechnol. 21 (2), 177–181. doi: 10.1038/nbt777
Alonge, M., Lebeigle, L., Kirsche, M., Aganezov, S., Wang, X., Lippman, Z. B., et al. (2021). Automated assembly scaffolding elevates a new tomato system for high-throughput genome editing. BioRxiv 2021, 2021.11.18.469135. doi: 10.1101/2021.11.18.469135
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J., (1990). Basic local alignment search tool. J. Mol. Biol. 215 (3), 403–410. doi: 10.1016/S0022-2836(05)80360-2
Ammal, E. J., Raghavan, R. S. (1958). “Polyploidy and vitamin C in Emblica officinalis Gaertn,” in Proceedings/Indian Academy of Sciences. (New Delhi: Springer India) 47, 312–314.
Badejo, A. A., Wada, K., Gao, Y., Maruta, T., Sawa, Y., Shigeoka, S., et al. (2012). Translocation and the alternative D-galacturonate pathway contribute to increasing the ascorbate level in ripening tomato fruits together with the D-mannose/L-galactose pathway. J. Exp. Bot. 63 (1), 229–239. doi: 10.1093/jxb/err275
Bairoch, A., Apweiler, R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28 (1), 45–48. doi: 10.1093/nar/28.1.45
Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., et al. (2004). The Pfam protein families database. Nucleic Acids Res. 32 (suppl_1), D138–D141. doi: 10.1093/nar/gkh121
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolser, D., Staines, D. M., Pritchard, E., Kersey, P. (2016). Ensembl plants: integrating tools for visualizing, mining, and analyzing plant genomics data. Plant Bioinf. 1374, 115–140. doi: 10.1007/978-1-4939-3167-5_6
Bouman, R. W., Keßler, P. J., Telford, I. R., Bruhl, J. J., Strijk, J. S., Saunders, R. M., et al. (2021). Molecular phylogenetics of Phyllanthus sensu lato (Phyllanthaceae): Towards coherent monophyletic taxa. Taxon 70 (1), 72–98. doi: 10.1002/tax.12424
Campbell, M. S., Holt, C., Moore, B., Yandell, M. (2014). Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinf. 48 (1), 4.11. 11–14.11. doi: 10.1002/0471250953.bi0411s48
Carr, A. C., Maggini, S. (2017). Vitamin C and immune function. Nutrients 9 (11), 1211. doi: 10.3390/nu9111211
Carr, A. C., Vissers, M. C. (2013). Synthetic or food-derived vitamin C—are they equally bioavailable? Nutrients 5 (11), 4284–4304. doi: 10.3390/nu5114284
Caruso, P., Russo, M. P., Caruso, M., Guardo, M. D., Russo, G., Fabroni, S., et al. (2021). A transcriptional analysis of the genes involved in the ascorbic acid pathways based on a comparison of the juice and leaves of navel and anthocyanin-rich sweet orange varieties. Plants 10 (7), 1291. doi: 10.3390/plants10071291
Chan, P. P., Lowe, T. M. (2019). tRNAscan-SE: searching for tRNA genes in genomic sequences. Gene Prediction 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Chavhan, A. (2017). Comparative studies on ascorbic acid content in various fruits, vegetables and leafy vegetables. Int. J. @ Life Sci. 5 (4), 667–671.
Chen, Z., Young, T. E., Ling, J., Chang, S.-C., Gallie, D. R. (2003). Increasing vitamin C content of plants through enhanced ascorbate recycling. Proc. Natl. Acad. Sci. 100 (6), 3525–3530. doi: 10.1073/pnas.0635176100
Cruz-Rus, E., Amaya, I., Amaya, I., Sanchez-Sevilla, J. F., Botella, M. A., Valpuesta, V. (2011). Regulation of L-ascorbic acid content in strawberry fruits. J. Exp. Bot. 62 (12), 4191–4201. doi: 10.1093/jxb/err122
Cruz-Rus, E., Botella, M. A., Valpuesta, V., Gomez-Jimenez, M. C. (2010). Analysis of genes involved in L-ascorbic acid biosynthesis during growth and ripening of grape berries. J. Plant Physiol. 167 (9), 739–748. doi: 10.1016/j.jplph.2009.12.017
Dardick, C., Callahan, A. M. (2014). Evolution of the fruit endocarp: molecular mechanisms underlying adaptations in seed protection and dispersal strategies. Front. Plant Sci. 5, 284. doi: 10.3389/fpls.2014.00284
Dasaroju, S., Gottumukkala, K. M. (2014). Current trends in the research of Emblica officinalis (Amla): A pharmacological perspective. Int. J. Pharm. Sci. Rev. Res. 24 (2), 150–159.
Department of Agriculture & Farmers Welfare, M. o. A. F. W, Government of India, India (2021) Area and Production of Horticulture crops for 2021-22 (3rd Advance Estimates). Available at: https://agricoop.nic.in/en/StatHortEst#gsc.tab=0.
Di Matteo, A., Sacco, A., Anacleria, M., Pezzotti, M., Delledonne, M., Ferrarini, A., et al. (2010). The ascorbic acid content of tomato fruits is associated with the expression of genes involved in pectin degradation. BMC Plant Biol. 10 (1), 1–11. doi: 10.1186/1471-2229-10-163
Dorion, S., Ouellet, J. C., Rivoal, J. (2021). Glutathione metabolism in plants under stress: Beyond reactive oxygen species detoxification. Metabolites 11 (9), 641. doi: 10.3390/metabo11090641
Emms, D. M., Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20 (1), 1–14. doi: 10.1186/s13059-019-1832-y
Fenech, M., Amaya, I., Valpuesta, V., Botella, M. A. (2019). Vitamin C content in fruits: Biosynthesis and regulation. Front. Plant Sci. 9, 2006. doi: 10.3389/fpls.2018.02006
Feng, C., Feng, C., Lin, X., Liu, S., Li, Y., Kang, M. (2021). A chromosome-level genome assembly provides insights into ascorbic acid accumulation and fruit softening in guava (Psidium guajava). Plant Biotechnol. J. 19 (4), 717–730. doi: 10.1111/pbi.13498
Finn, R. D., Clements, J., Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39 (suppl_2), W29–W37. doi: 10.1093/nar/gkr367
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117 (17), 9451–9457. doi: 10.1073/pnas.1921046117
Fu, L., Niu, B., Zhu, Z., Wu, S., Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28 (23), 3150–3152. doi: 10.1093/bioinformatics/bts565
Gallie, D. R. (2013). L-ascorbic acid: a multifunctional molecule supporting plant growth and development. Scientifica 2013, 1–24. doi: 10.1155/2013/795964
Gantait, S., Mahanta, M., Bera, S., Verma, S. K. (2021). Advances in biotechnology of Emblica officinalis Gaertn. syn. Phyllanthus emblica L.: a nutraceuticals-rich fruit tree with multifaceted ethnomedicinal uses. 3 Biotech. 11, 1–25. doi: 10.1007/s13205-020-02615-5
Geethangili, M., Ding, S.-T. (2018). A review of the phytochemistry and pharmacology of phyllanthus urinaria L. Front. Pharmacol. 9, 1109. doi: 10.3389/fphar.2018.01109
Gómez-García, M., Ochoa-Alejo, N. (2016). Predominant role of the l-galactose pathway in l-ascorbic acid biosynthesis in fruits and leaves of the Capsicum annuum L. chili pepper. Braz. J. Bot. 39 (1), 157–168. doi: 10.1007/s40415-015-0232-0
Griffiths-Jones, S., Saini, H. K., Saini, H. K., Van Dongen, S., Enright, A. J. (2007). miRBase: tools for microRNA genomics. Nucleic Acids Res. 36 (suppl_1), D154–D158. doi: 10.1093/nar/gkm952
Group, A. P. (2009). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 161 (2), 105–121. doi: 10.1111/j.1095-8339.2009.00996.x
Gul, M., Liu, Z.-W., Rabail, R., Faheem, F., Walayat, N., Nawaz, A., et al. (2022). Functional and nutraceutical significance of amla (Phyllanthus emblica L.): A review. Antioxidants 11 (5), 816. doi: 10.3390/antiox11050816
Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29 (8), 1072–1075. doi: 10.1093/bioinformatics/btt086
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8 (8), 1494–1512. doi: 10.1038/nprot.2013.084
Han, X., Zhang, Y., Zhang, Q., Ma, N., Liu, X., Tao, W., et al. (2022). Two haplotype-resolved, gap-free genome assemblies for Actinidia latifolia and Actinidia chinensis shed light on the regulatory mechanisms of vitamin C and sucrose metabolism in kiwifruit. Mol. Plant 16 (2), 452–470. doi: 10.1016/j.molp.2022.12.022
Hasanuzzaman, M., Bhuyan, M. B., Anee, T. I., Parvin, K., Nahar, K., Mahmud, J. A., et al. (2019). Regulation of ascorbate-glutathione pathway in mitigating oxidative damage in plants under abiotic stress. Antioxidants 8 (9), 384. doi: 10.3390/antiox8090384
Hoang, M. T. T., Almeida, D., Chay, S., Alcon, C., Corratge‐Faillie, C., Curie, C., et al. (2021). AtDTX25, a member of the multidrug and toxic compound extrusion family, is a vacuolar ascorbate transporter that controls intracellular iron cycling in Arabidopsis. New Phytol. 231 (5), 1956–1967. doi: 10.1111/nph.17526
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., Von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34 (8), 2115–2122. doi: 10.1093/molbev/msx148
Jackman, S. D., Coombe, L., Chu, J., Warren, R. L., Vandervalk, B. P., Yeo, S., et al. (2018). Tigmint: correcting assembly errors using linked reads from large molecules. BMC Bioinf. 19 (1), 1–10. doi: 10.1186/s12859-018-2425-6
Jaiswal, S. K., Mahajan, S., Chakraborty, A., Kumar, S., Sharma, V. K. (2021). The genome sequence of Aloe vera reveals adaptive evolution of drought tolerance mechanisms. Iscience 24 (2), 102079. doi: 10.1016/j.isci.2021.102079
Jombart, T., Balloux, F., Balloux, F., Dray, S. (2010). Adephylo: new tools for investigating the phylogenetic signal in biological traits. Bioinformatics 26 (15), 1907–1909. doi: 10.1093/bioinformatics/btq292
Kathriarachchi, H., Samuel, R., Hoffmann, P., Mlinarec, J., Wurdack, K. J., Ralimanana, H., et al. (2006). Phylogenetics of tribe Phyllantheae (Phyllanthaceae; Euphorbiaceae sensu lato) based on nrITS and plastid matK DNA sequence data. Am. J. Bot. 93 (4), 637–655. doi: 10.3732/ajb.93.4.637
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kawakita, A., Kato, M. (2017). Diversity of Phyllanthaceae plants. In: Kato, M, Kawakita, A (Eds) Obligate Pollination Mutualism, Ecological Research Monographs, (Tokyo: Springer), 81–116. doi: 10.1007/978-4-431-56532-1_4
Kim, D., Langmead, B., Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12 (4), 357–360. doi: 10.1038/nmeth.3317
Kolmogorov, M., Yuan, J., Lin, Y., Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37 (5), 540–546. doi: 10.1038/s41587-019-0072-8
Kubola, J., Siriamornpun, S., Meeso, N. (2011). Phytochemicals, vitamin C and sugar content of Thai wild fruits. Food Chem. 126 (3), 972–981. doi: 10.1016/j.foodchem.2010.11.104
Kumar, A., Kumar, S., Bains, S., Vaidya, V., Singh, B., Kaur, R., et al. (2016). De novo transcriptome analysis revealed genes involved in flavonoid and vitamin C biosynthesis in Phyllanthus emblica (L.). Front. Plant Sci. 7, 1610. doi: 10.3389/fpls.2016.01610
Kumar, A., Singh, K. (2012). Isolation of high quality RNA from Phyllanthus emblica and its evaluation by downstream applications. Mol. Biotechnol. 52 (3), 269–275. doi: 10.1007/s12033-011-9492-5
Kumar, S., Suleski, M., Craig, J. M., Kasprowicz, A. E., Sanderford, M., Li, M., et al. (2022). TimeTree 5: an expanded resource for species divergence times. Mol. Biol. Evol. 39 (8), msac174. doi: 10.1093/molbev/msac174
Laetsch, D. R., Blaxter, M. L. (2017). KinFin: software for Taxon-Aware analysis of clustered protein sequences. G3: Genes Genomes Genet. 7 (10), 3349–3357. doi: 10.1534/g3.117.300233
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997. doi: 10.48550/arXiv.1303.3997
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34 (18), 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, H., Huang, W., Wang, G.-L., Wang, W.-L., Cui, X., Zhuang, J. (2017). Transcriptomic analysis of the biosynthesis, recycling, and distribution of ascorbic acid during leaf development in tea plant (Camellia sinensis (L.) O. Kuntze). Sci. Rep. 7 (1), 1–11. doi: 10.1038/srep46212
Li, X., Lin, Y., Jiang, Y., Wu, B., Yu, Y. (2022). Aqueous Extract of Phyllanthus emblica L. Alleviates Functional Dyspepsia through Regulating Gastrointestinal Hormones and Gut Microbiome In Vivo. Foods 11 (10), 1491. doi: 10.3390/foods11101491
Li, C., Xing, H., Li, C., Ren, Y., Li, H., Wan, X.-Q., et al. (2022). Chromosome-scale genome assembly provides insights into the molecular mechanisms of tissue development of Populus wilsonii. Commun. Biol. 5 (1), 1125. doi: 10.1038/s42003-022-04106-0
Liao, G., Chen, L., He, Y., Li, X., Lv, Z., Yi, S., et al. (2021). Three metabolic pathways are responsible for the accumulation and maintenance of high AsA content in kiwifruit (Actinidia eriantha). BMC Genomics 22 (1), 1–11. doi: 10.1186/s12864-020-07311-5
Liu, X., Ma, H., Li, T., Li, Z., Wan, Y., Liu, X., et al. (2018). Development of novel EST-SSR markers for Phyllanthus emblica (Phyllanthaceae) and cross-amplification in two related species. Appl. Plant Sci. 6 (7), e01169. doi: 10.1002/aps3.1169
Liu, X., Ma, Y., Wan, Y., Li, Z., Ma, H. (2020). Genetic diversity of Phyllanthus emblica from two different climate type areas. Front. Plant Sci. 11, 580812. doi: 10.3389/fpls.2020.580812
Luo, X., Zhang, B., Pan, Y., Gu, J., Tan, R., Gong, P. (2022). Phyllanthus emblica aqueous extract retards hepatic steatosis and fibrosis in NAFLD mice in association with the reshaping of intestinal microecology. Front. Pharmacol. 13. doi: 10.3389/fphar.2022.893561
Mao, X., Wu, L.-F., Guo, H.-L., Chen, W.-J., Cui, Y.-P., Qi, Q., et al. (2016). The genus Phyllanthus: an ethnopharmacological, phytochemical, and pharmacological review. Evidence-Based Complement. Altern. Med 2016, 7584952. doi: 10.1155/2016/7584952
Marçais, G., Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27 (6), 764–770. doi: 10.1093/bioinformatics/btr011
Martini, E. (2003). How did Vasco da Gama sail for 16 weeks without developing scurvy? Lancet 361(9367), 1480. doi: 10.1016/S0140-6736(03)13131-5
Mendes, F. K., Vanderpool, D., Fulton, B., Hahn, M. W. (2020). CAFE 5 models variation in evolutionary rates among gene families.". Bioinformatics 36 (22-23), 5516–5518. doi: 10.1093/bioinformatics/btaa1022
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35 (suppl_2), W182–W185. doi: 10.1093/nar/gkm321
Murugesan, S., Kottekad, S., Crasta, I., Sreevathsan, S., Usharani, D., Perumal, M. K., et al. (2021). Targeting COVID-19 (SARS-CoV-2) main protease through active phytocompounds of ayurvedic medicinal plants–Emblica officinalis (Amla), Phyllanthus niruri Linn.(Bhumi Amla) and Tinospora cordifolia (Giloy)–A molecular docking and simulation study. Comput. Biol. Med. 136, 104683. doi: 10.1016/j.compbiomed.2021.104683
Muzaffar, K., Sofi, S. A., Makroo, H. A., Majid, D., Dar, B. (2022). Insight about the biochemical composition, postharvest processing, therapeutic potential of Indian gooseberry (amla), and its utilization in development of functional foods—A comprehensive review. J. Food Biochem. 46 (11), e14132. doi: 10.1111/jfbc.14132
Ng, P. C., Henikoff, S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31 (13), 3812–3814. doi: 10.1093/nar/gkg509
Nguse, M., Yang, Y., Fu, Z., Xu, J., Ma, L., Bu, D. (2022). Phyllanthus emblica (Amla) fruit powder as a supplement to improve preweaning dairy calves’ Health: effect on antioxidant capacity, immune response, and gut bacterial diversity. Biology 11 (12), 1753. doi: 10.3390/biology11121753
Nguyen, Y. T. H., Hoang, H. T. T., Mai, A. T. H., Nguyen, L. T. N., Nguyen, Q. H., Pham, N. T. T., et al. (2021). The Aconitum carmichaelii f3′ 5′ h gene overexpression increases flavonoid accumulation in transgenic tobacco plants. Horticulturae 7 (10), 384. doi: 10.3390/horticulturae7100384
Orwa, C., Mutua, A., Mutua, A., Kindt, R., Jamnadass, R., Simons, A. (2009). Agroforestree Database: a tree reference and selection guide. Version 4.
Paciolla, C., Fortunato, S., Dipierro, N., Paradiso, A., De Leonardis, S., Mastropasqua, L., et al. (2019). Vitamin C in plants: from functions to biofortification. Antioxidants 8 (11), 519. doi: 10.3390/antiox8110519
Pandey, K., Lokhande, K. B., Lokhande, K. B., Swamy, K. V., Nagar, S., Dake, M., et al. (2021). In silico exploration of phytoconstituents from Phyllanthus emblica and Aegle marmelos as potential therapeutics against SARS-CoV-2 RdRp. Bioinf. Biol. Insights 15, 11779322211027403. doi: 10.1177/11779322211027403
Paulino, D., Warren, R. L., Vandervalk, B. P., Raymond, A., Jackman, S. D., Birol, I. (2015). Sealer: a scalable gap-closing application for finishing draft genomes. BMC Bioinf. 16 (1), 1–8. doi: 10.1186/s12859-015-0663-4
Perry, B. A. (1943). Chromosome number and phylogenetic relationships in the Euphorbiaceae. Am. J. Bot. 30, 527–543. doi: 10.1002/j.1537-2197.1943.tb14796.x
Rahman, M. S., Sultana, S. S., Sultana, S. S., Hassan, M. A. (2021). Variable chromosome number and ploidy level of five phyllanthus species in Bangladesh. Cytologia 86 (2), 143–148. doi: 10.1508/cytologia.86.143
Ranallo-Benavidez, T. R., Jaron, K. S., Jaron, K. S., Schatz, M. C. (2020). GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11 (1), 1–10. doi: 10.1038/s41467-020-14998-3
Remmert, M., Biegert, A., Hauser, A., Söding, J. (2012). HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9 (2), 173–175. doi: 10.1038/nmeth.1818
Rigano, M. M., Lionetti, V., Raiola, A., Bellincampi, D., Barone, A. (2018). Pectic enzymes as potential enhancers of ascorbic acid production through the D-galacturonate pathway in Solanaceae. Plant Sci. 266, 55–63. doi: 10.1016/j.plantsci.2017.10.013
Rodrigues, R. B., De Menezes, H. C., Cabral, L. M., Dornier, M., Reynes, M. (2001). An Amazonian fruit with a high potential as a natural source of vitamin C: the camu-camu (Myrciaria dubia). Fruits 56 (5), 345–354. doi: 10.1051/fruits:2001135
Ruan, J., Li, H. (2020). Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17 (2), 155–158. doi: 10.1038/s41592-019-0669-3
Ruggieri, V., Sacco, A., Calafiore, R., Frusciante, L., Barone, A. (2015). Dissecting a QTL into candidate genes highlighted the key role of pectinesterases in regulating the ascorbic acid content in tomato fruit. Plant Genome 8 (2), plantgenome2014.2008.0038. doi: 10.3835/plantgenome2014.08.0038
Saini, R., Sharma, N., Oladeji, O. S., Sourirajan, A., Dev, K., Zengin, G. (2022). Traditional uses, bioactive composition, pharmacology, and toxicology of Phyllanthus emblica fruits: A comprehensive review. J. ethnopharmacol. 282, 114570. doi: 10.1016/j.jep.2021.114570
Sarin, B., Verma, N., Martín, J. P., Mohanty, A. (2014). An overview of important ethnomedicinal herbs of Phyllanthus species: present status and future prospects. Sci. World J. 2014. doi: 10.1155/2014/839172
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi: 10.1093/bioinformatics/btv351
Simpson, J. T., Durbin, R. (2012). Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 22 (3), 549–556. doi: 10.1101/gr.126953.111
Sodeyama, T., Nishikawa, H., Harai, K., Takeshima, D., Sawa, Y., Maruta, T., et al. (2021). The d-mannose/l-galactose pathway is the dominant ascorbate biosynthetic route in the moss Physcomitrium patens. Plant J. 107 (6), 1724–1738. doi: 10.1111/tpj.15413
Soontornchainaksaeng, P., Chaiyasut, K. (1999). Cytogenetic investigation of some Euphorbiaceae in Thailand. Cytologia 64 (3), 229–234. doi: 10.1508/cytologia.64.229
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34 (suppl_2), W435–W439. doi: 10.1093/nar/gkl200
Teh, B. T., Lim, K., Yong, C. H., Ng, C. C.Y., Rao, S. R., Rajasegaran, V., et al. (2017). The draft genome of tropical fruit durian (Durio zibethinus). Nat. Genet. 49 (11), 1633–1641. doi: 10.1038/ng.3972
Van Doan, H., Lumsangkul, C., Sringarm, K., Hoseinifar, S. H., Dawood, M. A., El-Haroun, E., et al. (2022). Impacts of Amla (Phyllanthus emblica) fruit extract on growth, skin mucosal and serum immunities, and disease resistance of Nile tilapia (Oreochromis niloticus) raised under biofloc system. Aquacult. Rep. 22, 100953. doi: 10.1016/j.aqrep.2021.100953
Varnasseri, M., Siahpoosh, A., Hoseinynejad, K., Amini, F., Karamian, M., Yad, M. J. Y., et al. (2022). The effects of add-on therapy of Phyllanthus emblica (Amla) on laboratory confirmed COVID-19 Cases: a randomized, double-blind, controlled trial. Complement. Therapies Med. 65, 102808. doi: 10.1016/j.ctim.2022.102808
Verhoeyen, M., Bovy, A., Collins, G., Muir, S., Robinson, S., De Vos, C., et al. (2002). Increasing antioxidant levels in tomatoes through modification of the flavonoid biosynthetic pathway. J. Exp. Bot. 53 (377), 2099–2106. doi: 10.1093/jxb/erf044
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9 (11), e112963. doi: 10.1371/journal.pone.0112963
Wang, Y.-S., Xu, Y.-J., Xu, Y.-J., Gao, L.-P., Yu, O., Wang, X.-Z., He, X.-J., et al. (2014). Functional analysis of flavonoid 3′, 5′-hydroxylase from tea plant (Camellia sinensis): critical role in the accumulation of catechins. BMC Plant Biol. 14, 1–14. doi: 10.1186/s12870-014-0347-7
Wang, J.-P., Yu, J.-G., Yu, J.-G., Li, J., Sun, P.-C., Wang, L., Yuan, J.-Q., et al. (2018). Two likely auto-tetraploidization events shaped kiwifruit genome and contributed to establishment of the Actinidiaceae family. Iscience 7, 230–240. doi: 10.1016/j.isci.2018.08.003
Warren, R. L., Yang, C., Vandervalk, B. P., Behsaz, B., Lagman, A., Jones, S. J., et al. (2015). LINKS: Scalable, alignment-free scaffolding of draft genomes with long reads. GigaScience 4 (1), s13742–13015-10076-13743. doi: 10.1186/s13742-015-0076-3
Weisenfeld, N. I., Kumar, V., Shah, P., Church, D. M., Jaffe, D. B. (2017). Direct determination of diploid genome sequences. Genome Res. 27 (5), 757–767. doi: 10.1101/gr.214874.116
Wheeler, G., Ishikawa, T., Pornsaksit, V., Smirnoff, N. (2015). Evolution of alternative biosynthetic pathways for vitamin C following plastid acquisition in photosynthetic eukaryotes. Elife 4, e06369. doi: 10.7554/eLife.06369.021
Wu, J., Wang, Z., Shi, Z., Zhang, S., Ming, R., Zhu, S., et al. (2013). The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 23 (2), 396–408. doi: 10.1101/gr.144311.112
Xia, Z., Huang, D., Zhang, S., Wang, W., Ma, F., Wu, B., et al. (2021). Chromosome-scale genome assembly provides insights into the evolution and flavor synthesis of passion fruit (Passiflora edulis Sims). Horticult. Res. 8. doi: 10.1038/s41438-020-00455-1
Xiong-fang, L., Tai-qiang, L., Zheng-hong, L., You-ming, W., Xiu-xian, L., Xu, Z., et al. (2018). Transcriptome analysis for Phyllanthus emblica distributed in dry-hot valleys in Yunnan, China. 林业科学研究 31 (5), 1–8. doi: 10.13275/j.cnki.lykxyj.2018.05.001
Xu, Q., Chen, L.-L., Ruan, X., Chen, D., Zhu, A., Chen, C., et al. (2013). The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 45 (1), 59–66. doi: 10.1038/ng.2472
Xu, G.-C., Xu, T.-J., Zhu, R., Zhang, Y., Li, S.-Q., Wang, H.-W., et al. (2019). LR_Gapcloser: a tiling path-based gap closer that uses long reads to complete genome assembly. GigaScience 8 (1), giy157. doi: 10.1093/gigascience/giy157
Yan, X., Li, Q., Jing, L., Wu, S., Duan, W., Chen, Y., et al. (2022). Current advances on the phytochemical composition, pharmacologic effects, toxicology, and product development of Phyllanthi Fructus. Front. Pharmacol. 13. doi: 10.3389/fphar.2022.1017268
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24 (8), 1586–1591. doi: 10.1093/molbev/msm088
Yeo, S., Coombe, L., Warren, R. L., Chu, J., Birol, I. (2018). ARCS: scaffolding genome drafts with linked reads. Bioinformatics 34 (5), 725–731. doi: 10.1093/bioinformatics/btx675
Keywords: Phyllanthus emblica, amla, medicinal plant, genome sequencing, antioxidant, vitamin C biosynthesis
Citation: Mahajan S, Bisht MS, Chakraborty A and Sharma VK (2023) Genome of Phyllanthus emblica: the medicinal plant Amla with super antioxidant properties. Front. Plant Sci. 14:1210078. doi: 10.3389/fpls.2023.1210078
Received: 21 April 2023; Accepted: 15 August 2023;
Published: 01 September 2023.
Edited by:
Mark Chapman, University of Southampton, United KingdomReviewed by:
Linchun Shi, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaAdriana Sacco, National Research Council (CNR), Italy
Copyright © 2023 Mahajan, Bisht, Chakraborty and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vineet K. Sharma, dmluZWV0a3NAaWlzZXJiLmFjLmlu