 N. D. Vinay1
N. D. Vinay1 Kalpana Singh2
Kalpana Singh2 Ranjith Kumar Ellur3
Ranjith Kumar Ellur3 Viswanathan Chinnusamy4
Viswanathan Chinnusamy4 Sarika Jaiswal2
Sarika Jaiswal2 Mir Asif Iquebal2
Mir Asif Iquebal2 Anilabha Das Munshi1
Anilabha Das Munshi1 Hideo Matsumura5
Hideo Matsumura5 G. Boopalakrishnan1
G. Boopalakrishnan1 Gograj Singh Jat1Chittaranjan Kole6
Gograj Singh Jat1Chittaranjan Kole6 Ambika Baladev Gaikwad7Dinesh Kumar2
Ambika Baladev Gaikwad7Dinesh Kumar2 Shyam Sundar Dey1
Shyam Sundar Dey1 Tusar Kanti Behera1,8*
Tusar Kanti Behera1,8*- 1Division of Vegetable Science, Indian Council of Agricultural Research (ICAR)-Indian Agricultural Research Institute, New Delhi, India
- 2Division of Agricultural Bioinformatics, Indian Council of Agricultural Research (ICAR)-Indian Agricultural Statistics Research Institute, New Delhi, India
- 3Division of Genetics, Indian Council of Agricultural Research (ICAR)-Indian Agricultural Research Institute, New Delhi, India
- 4Division of Plant Physiology, Indian Council of Agricultural Research (ICAR)-Indian Agricultural Research Institute, New Delhi, India
- 5Gene Research Centre, Shinshu University, Ueda, Japan
- 6Foundation for Science and Society, Kolkata, India
- 7Division of Genomic Resources, Indian Council of Agricultural Research (ICAR)-National Bureau of Plant Genetic Resources, New Delhi, India
- 8Indian Council of Agricultural Research (ICAR)-Indian Institute of Vegetable Research, Varanasi, Uttar Pradesh, India
Introduction: Momordica balsamina is the closest wild species that can be crossed with an important fruit vegetable crop, Momordica charantia, has immense medicinal value, and placed under II subclass of primary gene pool of bitter gourd. M. balsamina is tolerant to major biotic and abiotic stresses. Genome characterization of Momordica balsamina as a wild relative of bitter gourd will contribute to the knowledge of the gene pool available for improvement in bitter gourd. There is potential to transfer gene/s related to biotic resistance and medicinal importance from M. balsamina to M. charantia to produce high-quality, better yielding and stress tolerant bitter gourd genotypes.
Methods: The present study provides the first and high-quality chromosome-level genome assembly of M. balsamina with size 384.90 Mb and N50 30.96 Mb using sequence data from 10x Genomics, Nanopore, and Hi-C platforms.
Results: A total of 6,32,098 transposons elements; 2,15,379 simple sequence repeats; 5,67,483 transcription factor binding sites; 3,376 noncoding RNA genes; and 41,652 protein-coding genes were identified, and 4,347 disease resistance, 67 heat stress–related, 05 carotenoid-related, 15 salt stress–related, 229 cucurbitacin-related, 19 terpenes-related, 37 antioxidant activity, and 06 sex determination–related genes were characterized.
Conclusion: Genome sequencing of M. balsamina will facilitate interspecific introgression of desirable traits. This information is cataloged in the form of webgenomic resource available at http://webtom.cabgrid.res.in/mbger/. Our finding of comparative genome analysis will be useful to get insights into the patterns and processes associated with genome evolution and to uncover functional regions of cucurbit genomes.
1 Introduction
Momordica balsamina (2n = 2x = 22), commonly referred to as Balsam apple, Southern Balsam pear, or African pumpkin, is a wild plant belonging to the Momordica genus within the Cucurbitaceae family (Bharathi and John, 2013). It is native to tropical regions of Africa, Asia, and Australia (Jeffrey, 1967; Mishra et al., 1986). M. balsamina has an annual to perennial life cycle and grows as a trailing herb (John, 2005; Behera et al., 2010). It grows better in hot, humid climates and prefers acidic soil (pH 5.0–6.5) (Mishra et al., 1986). Ellipsoid-shaped immature fruits of M. balsamina are rich in essential vitamins (A and C) and vital minerals (iron and calcium) (Wehner et al., 2020). Additionally, its leaves are abundant in carotenoids (Mashiane et al., 2022). These nutritionally and medicinally enriched fruits and leaves are consumed in rural areas of Africa and Asia (Flyman and Afolayan, 2007; Bharathi and John, 2013). It is one of the four Momordica species cultivated in India, primarily in the dry regions of the Northwest plains, Eastern Ghats, and Western Ghats (Peter and Abraham, 2007).
Balsam apple has the reputation of a “gifted plant” due to its richness in bioactive compounds, which offer diverse therapeutic benefits. These compounds exhibit wide spectrum of medicinal values, including anti-septic, anti-microbial, anti-bacterial, anti-viral (including anti-HIV), anti-inflammatory, anti-plasmodial, antioxidant, and analgesic properties (Hassan and Umar, 2006; Thakur et al., 2009). The extensive range of medicinal properties exhibited by M. balsamina can be attributed to its diverse array of terpenoid compounds, such as balsaminol, balsaminoside, balsaminagenins, karavilagenin, cucurbalsaminol, and balsaminapentaol (Ramalhete et al., 2009; Ramalhete et al., 2010; Ramalhete et al., 2011a; Ramalhete et al., 2011b). Numerous researches have been conducted on these compounds in order to highlight their potential medical uses. Additionally, “cucurbitacins” derived from M. balsamina were found to have selective antiproliferative activity against multidrug resistant cancer cells (Ramalhete et al., 2022). Furthermore, Balsam apple contains ribosomal-inactivating proteins (RIPs) such as Momordin II and Balsamin, which possess remarkable antiviral, anticancer, and antibacterial properties. These RIPs have found practical applications in the development of commercial drug preparations (Khare, 2007; Kaur et al., 2012; Ajji, 2016; Ajji et al., 2017). The findings from these aforementioned studies justify the immense potential of M. balsamina within the pharmaceutical industry, thus making it a subject of intense scientific research in the field of cucurbitaceous vegetable crops.
Momordica charantia, commonly known as Bitter gourd, is the most widely cultivated vegetable within the Momordica genus, renowned for its distinctive bitter taste, attributed by cucurbitane-type tri-terpenoids (Chen et al., 2005). The fruits of Bitter gourd are abundant in vitamin C and iron and exhibit high antioxidant activity (Behera et al., 2010). Beyond its culinary use, it finds extensive application in traditional medicine, alleviating stomach pain, anemia, malaria, coughs, and fever, and it is a renowned source of anti-diabetic drug in pharmaceutical industry (Tan et al., 2008; Krawinkel et al., 2018). Despite its biological and economic significance, the crop improvement and varietal development program in Bitter gourd have been hindered by the limited genetic diversity found in natural populations (Dhillon et al., 2016). Furthermore, bitter gourd, being a crop of tropics and subtropics, is affected by various biotic and abiotic stresses. To overcome these obstacles, there is a critical need for diverse and valuable genetic resources to facilitate the development of elite high-yielding and resilient bitter gourd varieties (Cui et al., 2020).
Among the seven Momordica species found in India, M. charantia and M. balsamina are the only two species with monoecious sex expression. These two species share same basic chromosome number of x = 11 and exhibit similar frequencies and ranges of bivalent and chiasmata formation. This high karyo-morphological similarity indicates a close ancestral relationship between these two species (Trivedi and Roy, 1972; Singh, 1990; Bharathi et al., 2011). M. balsamina, in particular, is considered the closest wild relative that can be crossed with Bitter gourd, falling under the II subclass of the primary gene pool of Bitter gourd (Bharathi et al., 2012). M. balsamina also possesses a high level of tolerance to like pests such as ladybird beetle (Epilacna septima), pumpkin caterpillar (Margaronia indica), red pumpkin beetle (Aulocophora fevicoli), gall fly (Lasioptera falcata), root-knot nematode (Meladogyne incognita), and diseases such as yellow mosaic and little leaf disease, making it an invaluable genetic resource for the improvement of M. charantia (Rathod et al., 2021). Hence, in addition to medicinal attributes, M. balsamina can serve as a potent genetic source of biotic stress resistance.
Interspecific hybridization has proven to be a successful method for harnessing natural genetic variation and transferring desirable genes from wild relatives to cultivated crops (Bowley and Taylor, 1987; Dempewolf et al., 2017). In the Cucurbitaceae family, successful inter-specific hybrids have been developed within and between wild and cultivated taxa (Weeden and Robinson, 1986; Singh, 1991; Robinson and Decker-Walters, 1997). Likewise, there is great potential for the transfer of beneficial genes from M. balsamina to M. charantia for the genetic improvement of Bitter gourd. Previous studies have reported partial cross-compatibility between M. charantia and M. balsamina, resulting in progenies exhibiting normal meiosis (Singh, 1990; Bharathi et al., 2012). Recently, a detailed study on crossability involving 116 diverse Bitter gourd genotypes demonstrated success in six cross-combinations (Rathod et al., 2021). The study also confirmed the partial introgression of chromosome segments from M. balsamina into the Bitter gourd genome through morpho-cytological and molecular analysis of interspecific hybrids between M. charantia cv. Pusa Aushadhi × M. balsamina and their advanced generations (F2 and backcross generations). These findings suggest the possibility of transferring genes or traits related to biotic resistance and medicinal properties from M. balsamina to M. charantia, producing high-quality and resistant Bitter gourd varieties.
The era of genomics-assisted vegetable breeding commenced with the completion of the cucumber whole genome assembly in 2009 (Huang et al., 2009). In 2016, the first draft genome of Bitter gourd was published (Urasaki et al., 2017), followed by subsequent high-quality, chromosome-level assemblies (Cui et al., 2020; Matsumura and Urasaki, 2020). With advancements in sequencing technologies and bioinformatics tools, genomic data for flowering plants has been expanding rapidly (Chen et al., 2018), and genome assemblies for most cultivated cucurbits are now available in the public domain. Presently, there is a focus on genome characterization of closely related cross-compatible crop wild relatives (CWRs).
CWRs serve as a dynamic gene pool to access vital genetic diversity needed for crop improvement. Earlier, molecular techniques were used to characterize CWR (Dillon et al., 2007a; Sotowa et al., 2013). Now, advanced next-generation sequencing (NGS) platforms can be utilized for genome characterization of CWR to study phylogeny and discover useful genes in order to support agriculture and food security (Brozynska et al., 2016). Several wild relatives of tomato (Sato et al., 2012), brinjal (Gramazio et al., 2019), potato (Aversano et al., 2015), and sweet potato (Wu et al., 2018) have already been sequenced. In the current study, we present first high-quality genome assembly of M. balsamina a, close relative of bitter gourd that can be a vital genetic resource to improve medicinal value and stress resistance in bitter gourd.
2 Material and method
2.1 Sample collection and DNA extraction
Young leaf samples of M. balsamina (IC-467683) weighing around 10 g were collected for DNA isolation from 30-day-old seedlings at the active vegetative stage during the early morning hours. The collected leaf samples were packed immediately in aluminium foil, frozen into liquid nitrogen and stored at −80°C. Total DNA was isolated using the modified cetyl trimethyl ammonium bromide (CTAB) method (Saghai-Maroof et al., 1984). The genomic DNA samples were adjusted to 50 ng DNA/µL and stored at 4°C until used for sequencing. The quality and quantity of the extracted DNA were estimated with an Eppendorf Biospectrometer confirmed by running on 0.8% w/v agarose gel.
2.2 10x genomics sequencing and library preparation
High-molecular weight DNA (1.25 ng) was loaded onto a Chromium Controller chip, along with 10x Chromium reagents and gel beads following manufacturers recommended protocols. Initial library construction occurred within droplets containing Gel Beads-in-Emulsion (GEMs) beads with unique barcodes. The library construction incorporated a unique barcode adjacent to read one. All molecules within a GEM got tagged with the same barcode. However, because of the limiting dilution of the genome (roughly 300 haploid genome equivalents), the probability that two molecules from the same region of the genome were partitioned in the same GEM was minimal. Thus, the barcodes were used to associate short reads with their source long molecule statistically. The resulting library was sequenced on Illumina HiSeq X Ten sequencer (San Diego, CA, USA) as per the manufacturer’s protocol to produce 2 × 150 paired-end sequences. The entire process was performed on four replicates; thus, four pair-end libraries were prepared.
2.3 NanoPore sequencing and library preparation
First, 05-µg genomic DNA was sheared to approximately 15,000 bp by centrifugation at 5,200 rpm in a gTUBE. DNA was repaired with damage repair reagent and end-repaired using end-repair mix before ligation to nanopore blunt end adapter. Unligated material was digested with Exo III and Exo VII. Then, 12–25 Kb library fragments were purified via two consecutive Ampure cleanups, and size selection was done on Blue Pippin (SageScience, Beverley, MA, USA) with a 0.75% agarose cassette. An aliquot of 20 picomol of the final library was loaded onto the flow cell and sequenced on machine MinION (Oxford Nanopore Technologies, Oxford Science Park, United Kingdom) using Oxford Nanopore sequencing kit 2.0 and improved instrument workflow (Instrument Control Software 4.0).
2.4 Hi-C sequencing and library preparation
Fresh and young leaf samples were collected and cross-linked for 10 min with a 1% final concentration of fresh formaldehyde and quenched with a 0.2 M final concentration of glycine for 5 min. The cross-linked cells were subsequently lysed in lysis buffer. The extracted nuclei were re-suspended with a 150-µL 0.1% Sodium dodecyl sulfate (SDS) and incubated at 65°C for 10 min. Furthermore, they were quenched by adding 120 µL of water and 30 µL of 10% Triton X-100 and incubated at 37°C for 15 min. The DNA in the nuclei was digested by adding 30 µL of 10x NEB buffer 2.1 and 150 U of Mbol and incubated at 37°C for 12h. This was followed by inactivation of Mbol enzyme at 65°C for 20 min and filling of cohesive ends by adding 1 µL of each 10 mM deoxythymidine triphosphate (dTTP), deoxyadenosine triphosphate (dATP), and deoxyguanosine triphosphate (dGTP), 2 µL of 5 mM biotin-14-deoxycytidine triphosphate (dCTP), and 4 µL (40 U) Klenow and after that incubated at 37°C for 2h. To start proximity ligation, 120 pL 10x blunt-end ligation buffer, 100 pL 10% Triton X-100, and 20U T4 DNA ligase were added and held at 16°C for 4h. This was followed by reversing of the cross-linking with 200 ug/mL proteinase K (Thermo Fisher Scientific) at 65°C for 12h. Furthermore, chromatin DNA manipulations were performed using a method described by Belaghzal et al. (2017), followed by DNA purification using QIAamp DNA Mini Kits (Qiagen) and shearing of purified DNA in length of 400 bp. Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific) was used to pull down point ligation junctions. NEB Next Ultra II DNA library Prep Kit for Illumina (NEB) was used to prepare Hi-C library for Illumina sequencing. The final library was sequenced on the Illumina HiSeq X Ten platform (San Diego, CA, USA) as per the manufacturer’s protocol with 2 × 150 paired-end mode.
2.5 Data pre-processing and genome assembly
All the raw reads of 10x Genomics, Nanopore and HiC libraries used in the present study have been submitted in National Center for Biotechnology Information (NCBI) with SRA IDs SRR21495983, SRR21495982, and SRR21495981, respectively. Figure 1 shows the outline followed during the present study. Prior to assembly, reads of these libraries were cleaned using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc: Andrews, 2010) by removing low quality reads at < 20 phred score, followed by adapter cleaning using TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). De-novo genome assembly was performed using all the 10x Genomics libraries of four replicates using Supernova v2.1.1 (Weisenfeld et al., 2017). After this, Nanopore libraries were mapped on de-novo genome assembly for further scaffolding using npScarf (Cao et al., 2017). Finally, HiC libraries were mapped on improved genome assembly using Juicerv1.5 (Durand et al., 2016) to obtain the de-duplicated alignment file. Furthermore, scaffolding, editing, and polishing of assembly was performed using 3dDNA v180419 (Dudchenko et al., 2017). Finally, identification of chromosomes and editing of miss-assembly was performed using JuiceBox v1.11.08 (Robinson et al., 2018) to construct contact maps for chromosomes. Genome polishing was performed on final assembly using Pilon (Walker et al., 2014).
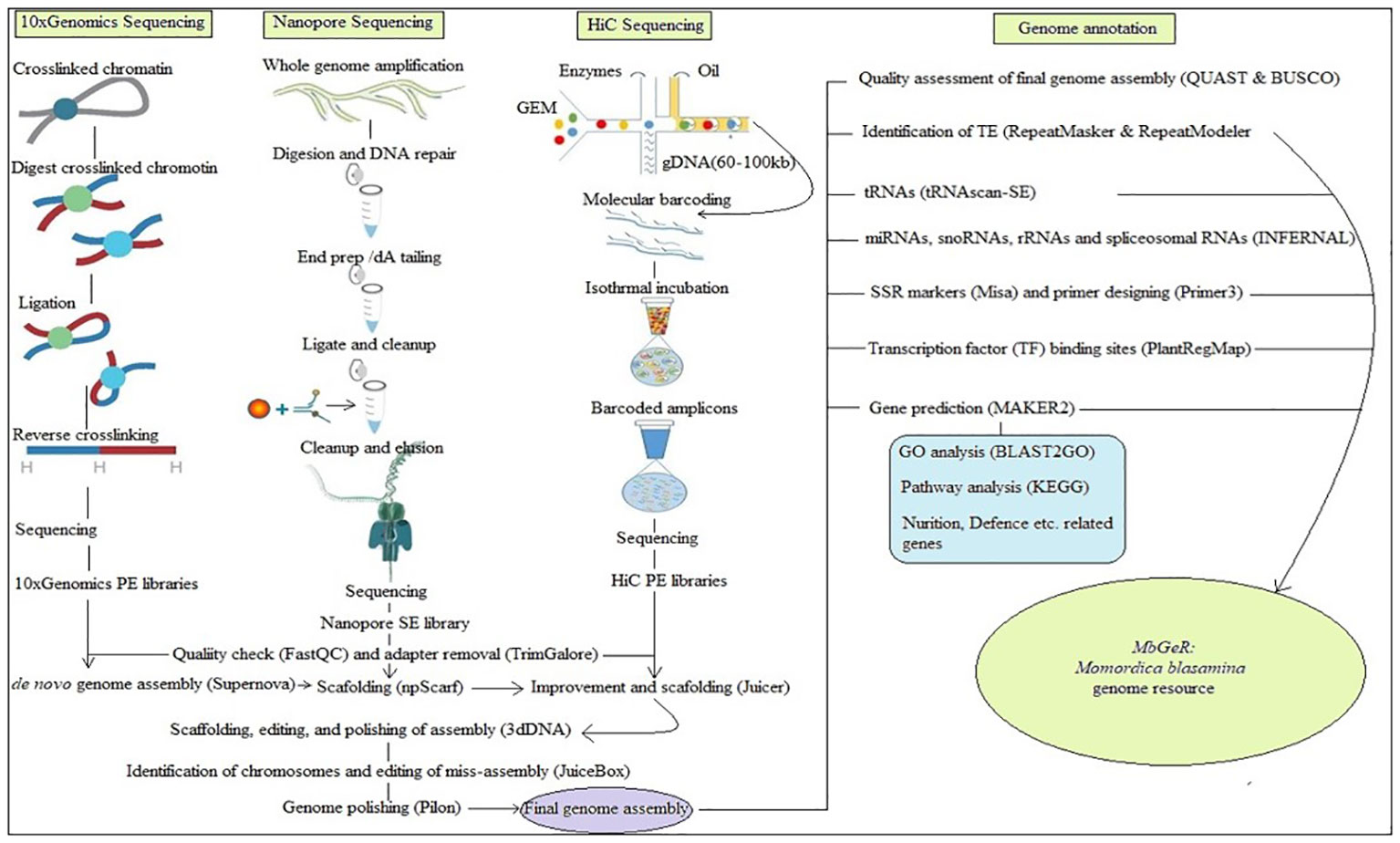
Figure 1 Over-view of pipeline adopted in the study.
2.6 Validation of chromosome level assembly
To assess the quality of the assembled genome, assembly statistics were calculated using QUAST (Gurevich et al., 2013). Furthermore, validation of assembly was performed using BUSCO (Simao et al., 2015) to find the completeness and contamination within genome assembly. A comparative study of M. balsamina genome assembly with other related species, such as Momordica charantia, Citrullus lanatus, Cucumis sativus, and Cucumis melo was also performed.
2.7 Genome annotation
For genome annotation, a series of bioinformatics tools were employed. First, repeat regions of the assembled genome were masked using RepeatMasker v4.1.0 (http://www.repeatmasker.org/RMDownload.html). This was followed by the identification of transposable elements (TEs) using RepeatModeler (http://www.repeatmasker.org/RepeatModeler/) to find LINE, SINE, Simple Repeats, LTR elements, DNA elements, and so forth. The ncRNA-encoding genes were also identified from the assembled genome. Furthermore, tRNAs were identified using tRNA scan-SE v.1.3.1 (Chan and Lowe, 2019) with < 1 false positive per 15 gigabases. Other ncRNAs, such as microRNAs, snRNAs, rRNAs, and spliceosomal RNAs, were also identified using INFERNAL v1.1.4 (Nawrocki and Eddy, 2013) at default parameters., Protein-encoding genes were predicted using SEQing v0.1.45 (Lewinski et al., 2020), which is an automated pipeline of self-trained hidden Markov models (HMM) models and transcriptomic data for gene prediction by Glimmer HMM, SNAP, and AUGUSTUS and combining their results by MAKER2 in association with transcriptomic evidence of Momordica charantia. Finally, the predicted genes passed through Cluster Database at High Identity with Tolerance (CD-HIT) (Limin et al., 2012), clustering at 90% sequence similarity to extract non-redundant genes. Extraction of Single Sequence Repeat (SSR) markers was performed using MIcroSAtellites (MISA) (Beier et al., 2017), considering mononucleotide repeats motif with at least 10 repeats, dinucleotide with six, tri-, tetra-, penta-, and hexa-nucleotide with five repeats (Thiel et al., 2003). Compound microsatellites were defined as those with the interval between two repeats motifs ≤100 nucleotides in the previous reports (Zhao et al., 2017). Furthermore, primers were also designed for each of the SSR makers using Primer3 (Untergasser et al., 2012) with parameters 18–27 bp primer length, 57°C–63°C melting temperature, 30%–70% GC content, and 100–300 bp product size. Transcription factor (TF) binding sites were extracted using PlantRegMap (Jin et al., 2017).
2.8 Functional annotation of protein-coding genes
The predicted protein-coding genes were mapped against the NR database (updated May 2020) and the plant TF database (version 5.0) using NCBI blast (version 2.2.29+) (Lipman and Pearson, 1985) for functional annotation. Furthermore, gene ontology (GO) analysis was performed on predicted genes using Blast2GO (Conesa et al., 2005). Pathway analysis was performed using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Erxleben and Grüning, 2020).
2.9 Disease resistant, defence, stress, and sex expression–related genes
Disease resistant genes were identified by mapping proteins against the PRGDB database v.4.0 (García et al., 2021) with e-value cutoff of 1e-10 using BLAST (NCBI 2.2.29+) (Lipman and Pearson, 1985). Along with resistance genes, genes related to salt stress, heat stress and sex expression were also extracted.
2.10 Orthologous genes, phylogenetic, and synteny analysis
M. balsamina genes, orthologous with M. charantia, Citrullus lanatus, Cucumis sativus, and Cucumis melo, were predicted using OrthoMCL (Chen et al., 2006) based on a Markov Cluster algorithm to group (putative) orthologs utilizing all-against-all BLAST (Lipman and Pearson,1985) comparisons among protein sequences of considered species. For the detection of synteny between M. balsamina genome assembly and four other genome assemblies of abovementioned species was performed by SyMAP v4.2 (Soderlund et al., 2011). Synteny blocks shown as colored ribbons between the chromosomes arranged in circle were visualized in the circular plots using Circos (Krzywinski et al., 2009). Furthermore, micro-synteny, a synteny in small regions, identified between two or more genomic regions was performed between of M. balsamina and M. charantia genomes using McScan python version (Tang and Krishnakuar, 2015). Furthermore, a phylogenetic study was also performed among genome assemblies of M. balsamina, M. charantia, Cucumis melo, Citrullus lanatus, and Cucumis sativus. First, a multi-sequence alignment (MSA) was performed among genome assemblies using Multiple Alignment using Fast Fourier Transform (MAFFT) (Katoh et al., 2002). Later, a distance matrix was calculated among assemblies using MSA with Poisson correction method, >70% site coverage, and <30% alignment gaps, missing data, and ambiguous bases by ClustalW2 (Thompson et al., 1994). Finally, a phylogenetic tree was constructed using Neighbor-Joining method by ClustalW2.
2.11 Development of M. balsamina genomic resource
A web-genomic resource for M. balsamina, named MbGeR, was developed using all the results obtained from the genomic data analyses performed in the present study. MbGeR catalogs the information related to molecular markers such as SSRs, transposons elements (TEs), TF sites, ncRNAs and genes. It is based on a three-tier architecture, namely, client tier, middle tier, and database tier, developed using PHP, MySQL, HTML, and Apache. Web pages are developed using PHP and HTML in order to browse MbGeR and put up queries by users in client tier. All the information regarding transcripts, Differentially Expressed Genes (DEGs), markers, and so forth. are placed in different tables in MySQL database in the database tier. The scripting of client query page was done in PHP and HTML for execution and fetching in the middle tier. The web hosting was performed using Apache server. The bitter melon web resources are available at http://webtom.cabgrid.res.in/mbger/.
3 Result
3.1 Data pre-processing, genome assembly, and comparative analysis
In the present study, the whole genome of M. balsamina was assembled using reads obtained from three different platforms: Oxford Nanopore, 10 X and Hi-C. A combination of multiple technologies is reported to improve the quality and completeness of genome assembly (Wang et al., 2023). An average of 27,767,526; 2,331,456; and 168,098,715 reads were accessed in 10x Genomics, Nanopore, and Hi-C libraries, respectively after pre-processing and quality check. Supplementary Table S1 shows the detailed read statistics in different replicates and their average length in all three libraries. GC% was 39 for 10x Genomics and Hi-C read libraries, while Nanopore reads had 35% GC content.
De-novo genome assembly was generated using 10x Genomics libraries followed by mapping of Nanopore libraries onto de-novo genome assembly for further scaffolding. The nanopore raw read size ranged from 1000 bp to 222917 bp, with N50 (minimum length representing half of the total length of the assembly) as 26.08 Kb and 15.29 Mb for raw reads and scaffolds, respectively. Then, reads from HiC libraries were used for chromosome-level scaffolding, which is considered as the best choice for capturing the longest range DNA connectedness (Wang et al., 2023).
The genome assembly of M. balsamina and its assessment was found to have 3,710 scaffolds of 384,902,967 bp length and N50 of 30,984,295 bp (Table 1). BUSCO analysis, which uses universal single-copy orthologs, is considered as high-resolution quantifications of genomes, which facilitate informative comparisons and provides suggestions for improvements to assemblies or annotations (Simao et al., 2015). Assessment of this generated assembly shows 2,266 (97.4%) of 2,326 BUSCO to be complete and single copy (Table 1). The comparative statistics of M. balsamina assembly with other assemblies of related species showed the assembly size to be comparable with others while the N50 value (30.96 mb) was much improved than other assemblies (Table 2).
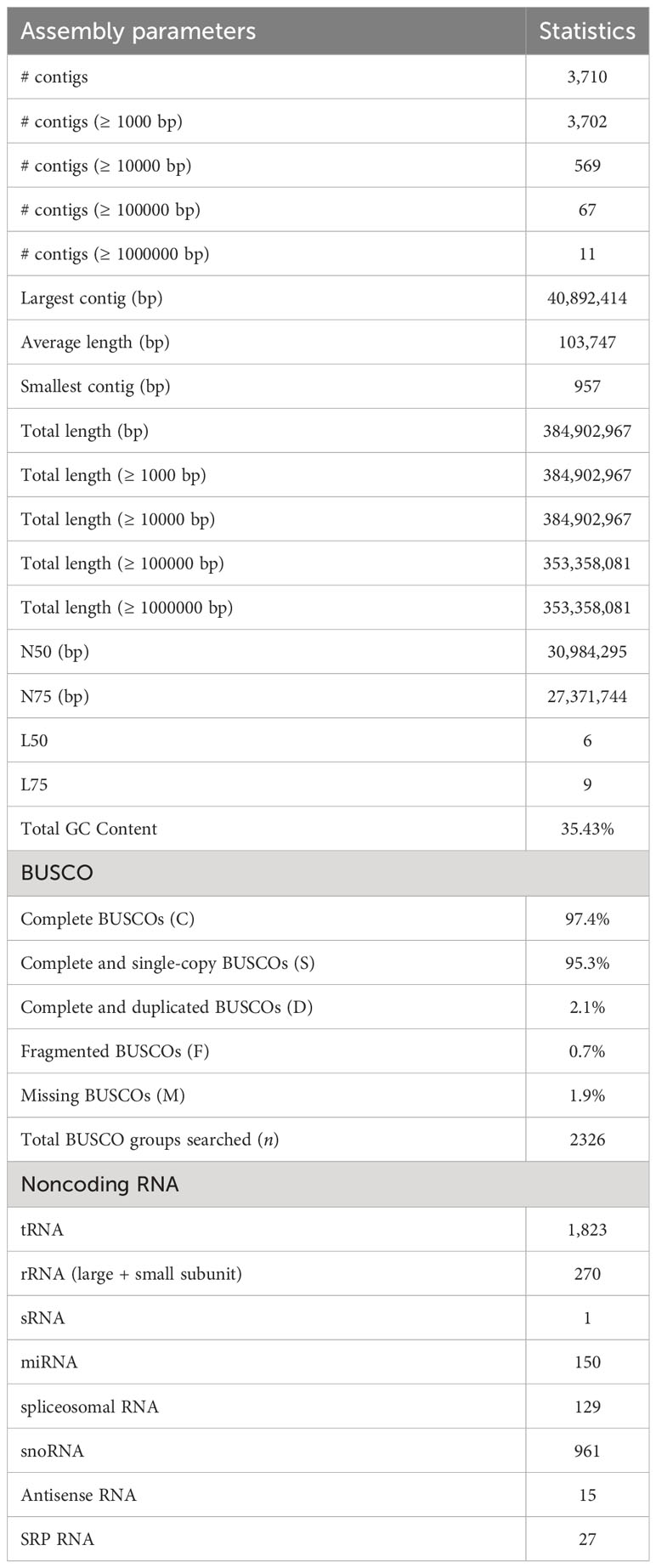
Table 1 M. balsamina assembly statistics.
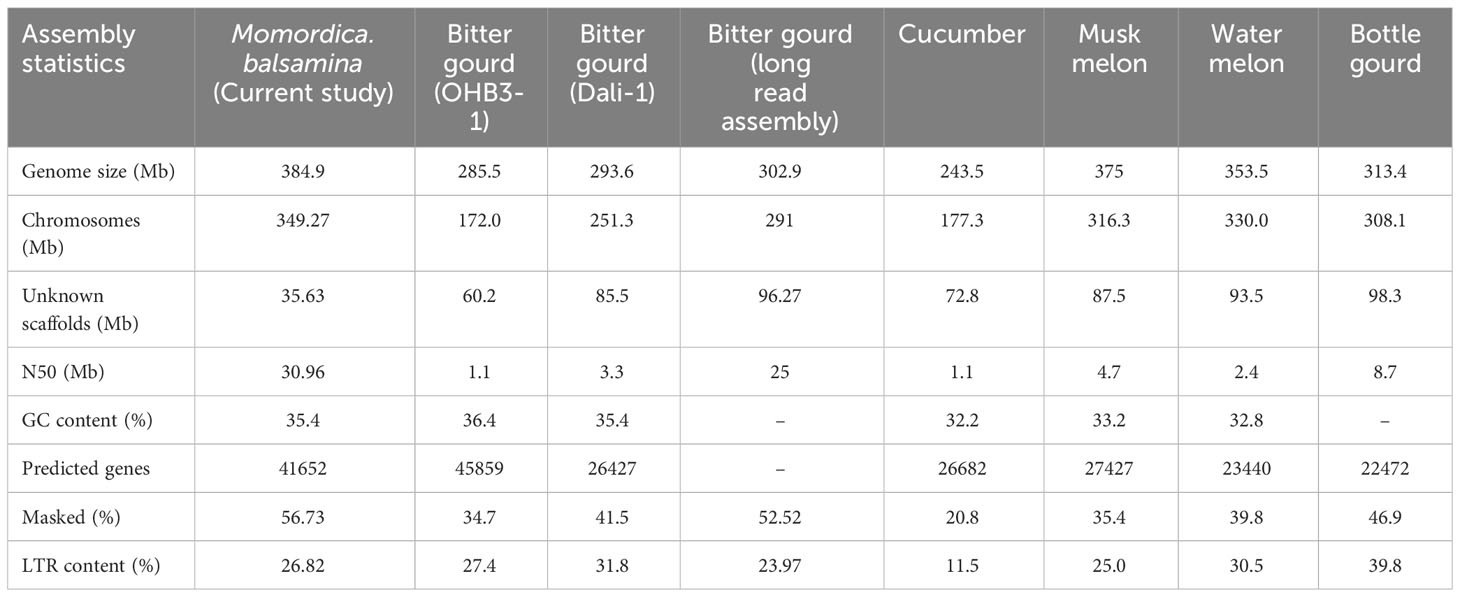
Table 2 Comparative statistics of M.balsamina genome assembly with genome assemblies of related species.
3.2 Annotation of genome assembly
Genome annotation is crucial to facilitate the utilization of assembled genomes in genetic studies. In the current study, homology-based inference, in-silico prediction techniques and merged transcriptomics data (of Momordica charantia) are merged into a single concordant annotation (Yandell and Ence, 2012). Genome annotation was done to identify TEs, ncRNA encoding genes, tRNAs, ncRNAs, SSR makers, TF binding sites and protein-encoding genes in the assembled genome.
Out of the total 384,902,967 bp length of 3,710 scaffolds of the assembled genome, 218,862,155 (56.73%) bases were masked. Frequencies of various classes of predicted TEs in genome assembly are delineated in Table 3. A significant proportion of TE class belonged to LTR elements, while 22.29% were found to be unclassified. The frequency of SINEs was the least (0.05%), while it was 3.02% for SINEs. A sum of 567,483 TF binding sites were predicted in M. balsamina genome and Figure 2A is showing chromosome wide distribution of TF binding sites. Maximum number of TF binding sites were observed in chromosome number 2 (~12%), followed by chromosome number 1 (~9%) and chromosome number 11 (~9%). Almost ~12% of TF binding sites were associated with the remaining unknown scaffolds (Figure 2A).
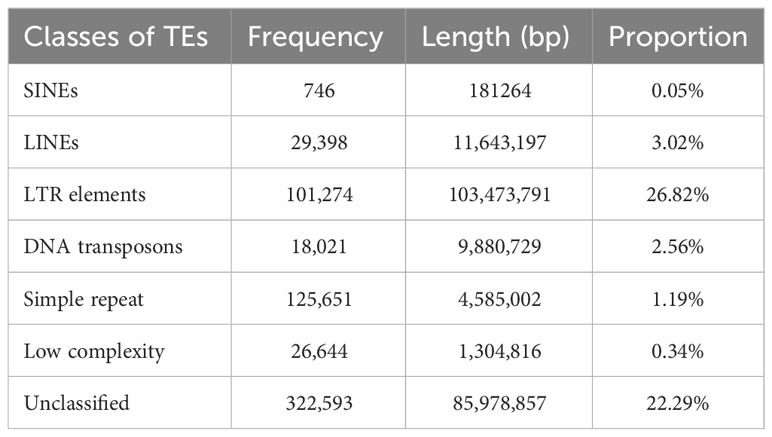
Table 3 Frequencies and proportion of various classes of TEs predicted in M. balsamina assembly.
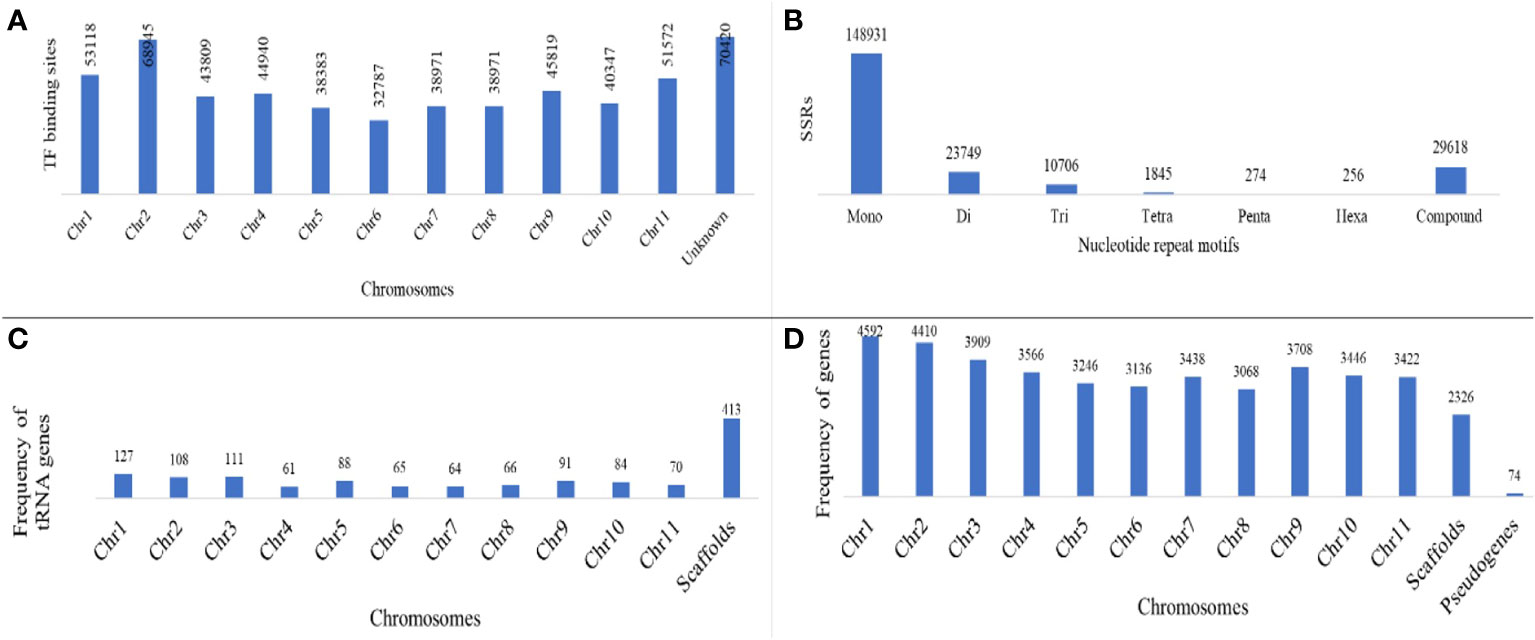
Figure 2 (A) Distribution of TF binding sites on different chromosomes; (B) frequency of SSRs of different nucleotide repeat motifs; (C) frequency of predicted tRNA genes and (D) protein coding genes distributed over chromosomes along with pseudogenes predicted in M. balsamina assembly.
A total of 2,15,379 SSR markers were mined from the assembled genome. The highest number of SSR belonged to motif type mono-nucleotide (~69%), followed by di (~13%) and tri (~6%). A total of 29,618 (~9%) SSRs were compound type (Figure 2B). A total 3,376 different non-coding RNA genes were predicted in M. balsamina assembly, out of which 1,823 tRNA, 270 rRNA, 150 microRNA, 961 snoRNA, 27 SRP RNA, and 129 spliceosomal RNA genes were predicted (Table 1). Out of the total 1,823 predicted tRNA genes in M. balsamina assembly, their frequency distribution over chromosome 1 was highest, followed by chromosomes 3 and 2. A minimum number of tRNA genes were observed in chromosome 4 (Figure 2C). Apart from the chromosomes, higher number of tRNA genes were found localized on unknown scaffolds. Figure 2D shows the frequencies of protein-coding genes distributed over various chromosomes along with 74 pseudogenes predicted in M. balsamina assembly. It was observed that a higher number of protein-coding genes were found on chromosomes 1 (4,592), followed by chromosome 2 (4,410) and 3 (3,909).
3.3 Functional annotation of protein-coding genes
Functional annotation of protein-coding genes yielded a total of 33,450 genes that were annotated with NR database. GO analysis of these annotated genes showed 52 GO terms to be associated with 20,525 genes, of which 16, 12, and 25 were from cellular component, molecular function, and biological process classes, respectively. The GO terms were categorized into three classes, namely, molecular function, biological functions and cellular components. Figure 3A shows the GO terms associated with more than five protein-coding genes predicted in M. balsamina assembly. It was found that the GO terms named binding activities (11,892) followed by the catalytic activities (9,604) and transporter activities (889) were associated with most genes in molecular function class. In biological processes, cellular processes GO term (8,650) was the most frequent in genes, followed by metabolic processes (8,458) and biological regulations (1,252). Cell (5,026), cell part (5,026), and membrane (4,850) GO terms were the most frequent in cellular component class (Figure 3A). Figure 3B shows the top 10 KEGG pathways associated with 3,414 annotated genes in M. balsamina assembly. It was found that metabolic pathways (>1,500 genes involved) were the most abundant pathway, followed by biosynthesis of secondary metabolites (~700 genes involved) and microbial metabolism (~250 genes involved) in diverse environments.
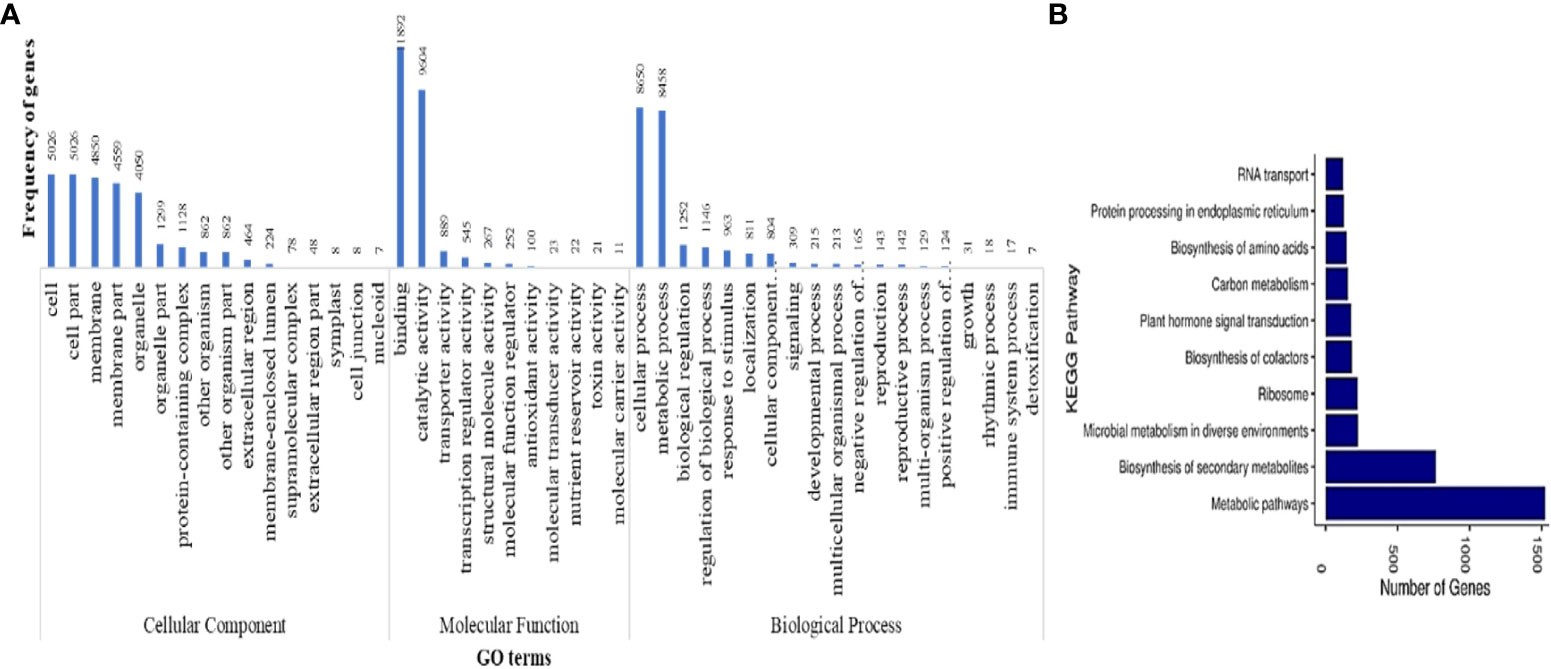
Figure 3 (A) GO terms associated with predicted protein coding genes and (B) top 10 KEGG pathways associated with annotated protein coding genes in M. balsamina assembly.
3.4 Genes related to plant defence, medicinal properties, and sex expression
M. balsamina is well-known for its biotic and abiotic stress tolerance and medicinal properties. In the M. balsamina assembly, a total of 4,347 important disease resistance genes (R genes) were identified, out of which 1,174 genes encoded for nucleotide-binding site–leucine-rich repeat (NBS-LRR) domains along with 858 RLP and 273 RLK encoding genes, which are well known in resistance response in plants. In our study, we identified 67 heat stress-related genes, including a total of 34 heat stress factor genes (HSFs), contribute to thermo-tolerance through the regulation of heat shock proteins (HSPs). In addition, 29 HSP genes, predominantly encoding HSP70, and 17 small heat proteins (HSP20) were identified in the M. balsamina assembly. Similarly, 15 genes encoding proteins related to salinity tolerance in the M. balsamina assembly, including alkaline ceramidase (ACER), S-acyltransferase, salt stress root protein RS1-like, and protein RICE SALT SENSITIVE 3 isoforms were identified. Cucurbit crops are considered as models for deciphering the mechanism of sex determination in monoecious plant species and ethylene is considered to be the core regulator. To shed more light on this, in the current study, 06 genes related to ethylene biosynthesis were extracted. M. balsamina contains a diverse array of Cucurbitacin terpenoid compounds exhibiting anti-septic, anti-microbial, anti-bacterial, anti-viral (including anti-HIV), anti-inflammatory, anti-plasmodial, antioxidant, and analgesic properties (Thakur et al., 2009; Ramalhete et al., 2022). The genes related to terpenoid biosynthesis were searched in the genome to elucidate the mechanism behind the medicinal property exhibited by this species. Thirty-seven antioxidant activity related and 229 genes related to the biosynthesis of cucurbitacin, the key factors behind medicinal attributes of the M. balsamina, were detected. Table 4 shows the frequencies of genes extracted with provided functions. GO terms of pathogenesis-related genes, heat tolerant genes, salt tolerance-related genes, sex determination-related genes, triterpenoid-related genes, cucurbitin-related genes, nutrition-related genes, and phloem-related genes are graphically represented in Supplementary Figure S1.
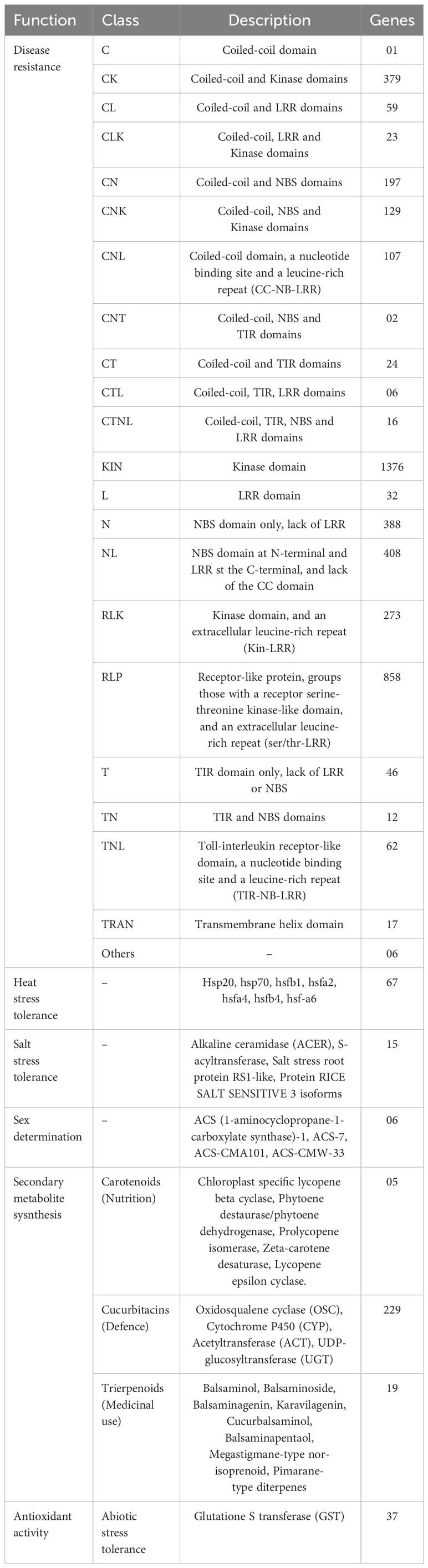
Table 4 Frequencies of genes associated with disease resistance, defence, salt stress, heat stress, sex determination, and secondary metabolite synthesis identified in M. balsamina assembly.
3.5 Orthologous genes, phylogenetic, and synteny analysis
Comparative genetic parameters such as orthology, synteny, and phylogeny were utilized in the study to understand the genome composition, evolution and relatedness among the members of a family or clade at the nucleotide/molecular level. A total of 1,542 genes of M. balsamina were found orthologous with other related species considered in the present study. Frequencies of these genes are provided in Table 5 along with the species with which these are found orthologous. The unique and overlapping M. balsamina genes found orthologous in other related species are delineated in Figure 4A. It is observed that 165, 159, 953, and136 M. balsamina genes were orthologous in Cucumis melo, Citrullus lanatus, M. charantia, and Cucumis sativus, respectively, only and the rest of the genes were orthologous in more than two species.
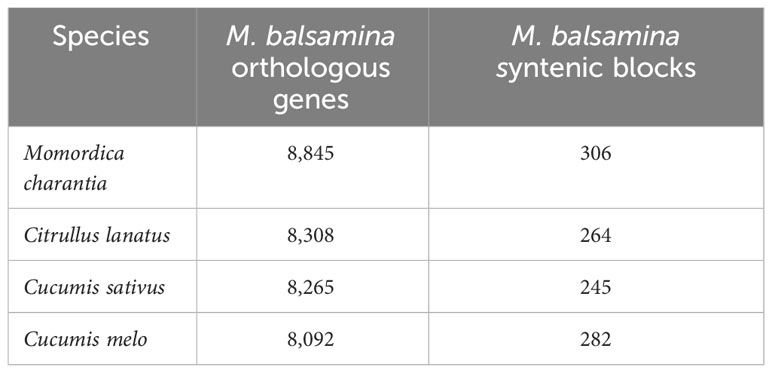
Table 5 Frequencies of M. balsamina orthologous genes and syntenic blocks found in other related species.
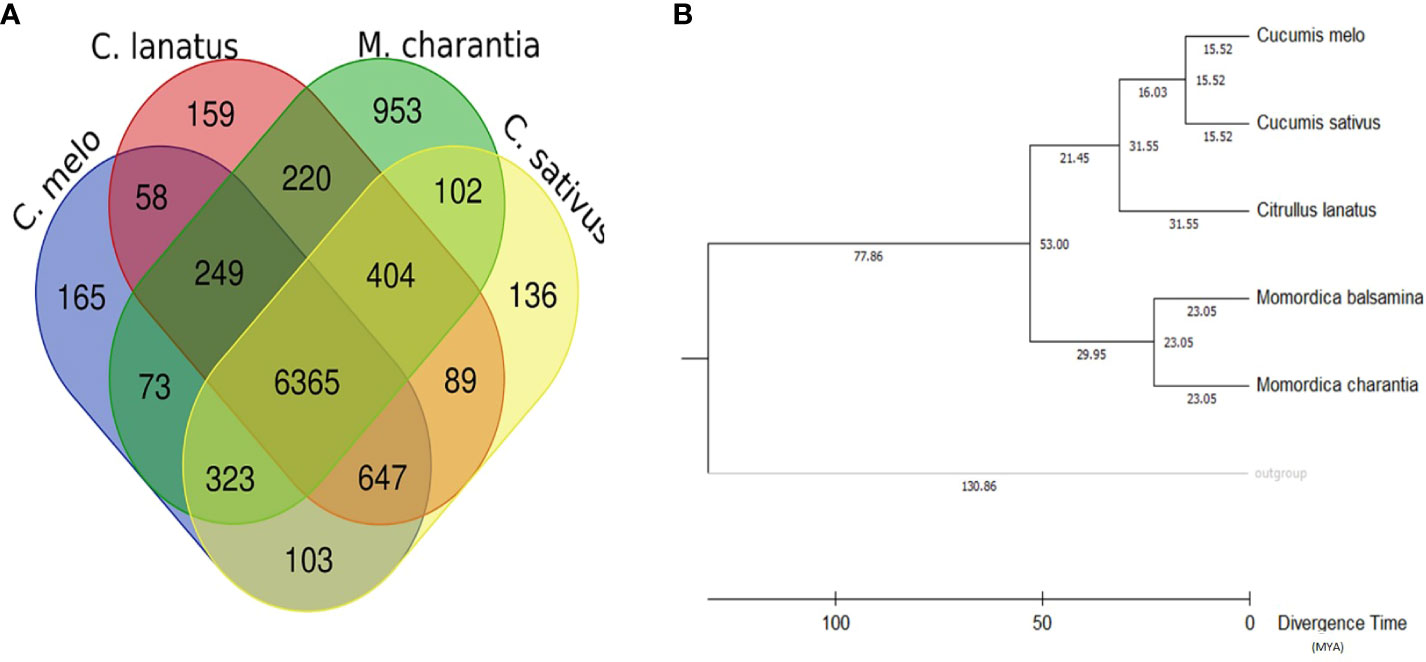
Figure 4 (A) Unique and overlapping M. balsamina genes found orthologous in other related species (Cucumis sativus, Cucumis melo, Citrullus lanatus, and M. charantia); (B) rooted phylogenetic tree represented in terms of divergence time (MYA: million years ago) based on whole genome assemblies of M. balsamina and other related species (Cucumis sativus, Cucumis melo, Citrullus lanatus, and M. charantia).
The syntenic relationship analyses of M. balsamina with other species were performed. In the synteny analysis, the sequences of related species were aligned, and conserved genes between the two genomes were identified as anchors, and then regions with more than seven anchors connecting two species were considered as synteny blocks. Frequencies of orthologous genes and syntenic blocks of M. balsamina with related species, M. charantia, Citrullus lanatus, Cucumis sativus, and Cucumis melo were found to be (8845, 306), (8308, 264), (8265, 245), and (8092, 282), respectively (Table 5). Also, the diagrammatic representation of syntenic blocks in the form of Circos figures is provided for synteny between M. balsamina and Cucumis sativus, M. balsamina and Cucumis melo, M. balsamina and Citrullus lanatus, M. balsamina and M. charantia (all scaffolds), and M. balsamina and M. charantia (scaffolds >100Mb), respectively (Supplementary Figures S2A–E). A general absence of a one-to-one relationship in the chromosomes between the Momordica balsamina and other cucurbit genomes was observed. However, syntenic loci of one chromosome of Momordica balsamina chromosome exhibited a syntenic relationship between one or two chromosomes of studied cucurbits. Momoridica balsamina Chr11 was syntenic to Chr6 and Chr7 of Cucumis sativus and Chr5 was syntenic to Chr3 and Chr4 of C. sativus. Similarly, Chr8 of Momordica balsamina was syntenic to Chr 11 of C. melo. Furthermore, Chr7 was colinear to Chr 2 and 12 of Melon. Synteny between M. balsamina Chr 7 and Chr2 of watermelon was observed. Furthermore, Chr 5 was syntenic to Chr5 and Chr7 of watermelon.
Maximum number of genes on each chromosome of M. Balsamina found homologous with genes on corresponding scaffolds of M. charantia are shown in Supplementary Table 2. In addition, the Supplementary Figures S3A–K show homologous genes on chromosomes 1–11 of M. balsamina with syntenic relationship with corresponding scaffolds M. charantia. The rooted phylogenetic tree was constructed to represent the phylogenetic relationship of M. balsamina with other related species, namely, M. charantia, Cucumis melo, Cucumis sativus, and Citrullus lanatus (Figure 4B). M. balsamina was observed to be more closely related to M. charantia.
3.6 Development of M. balsamina web-genomic resource
A web genomic resource for M. balsamina, named MbGeR, was developed from the output obtained after genomic data analyses of M. balsamina genome in the present study. Its web interface includes a home page with an introduction to MbGeR with horizontal and vertical tabs including statistics, SSRs, TEs, TF sites, ncRNAs, genes and team, each of which is linked to their respective pages (Figure 5). The statistics page provides summary statistics of data provided in genome resources in the form of histograms. Users are provided with flexible options to select SSR data on the desired 11 chromosomes of M. balsamina along with desired motifs on SSRs page. Users can choose TEs from the TEs page according to their desired types and chromosome numbers. TF sites provide options to choose TF binding sites on the desired chromosome. On the ncRNAs page, users can select non-coding RNAs among the various types. Gene’s page has two options: (i) selection of chromosomes for all genes extracted from the genome and (ii) choice of extracted genes associated with a certain function. Once the desired options are submitted on each of the mentioned the page, the output is displayed in tabular form in desired combinations of options. The Team page provides information and hyperlinked profiles of the team members involved in the study. The bitter gourd web resources, MbGeR is available for non-commercial use for research community at http://webtom.cabgrid.res.in/mbger/.
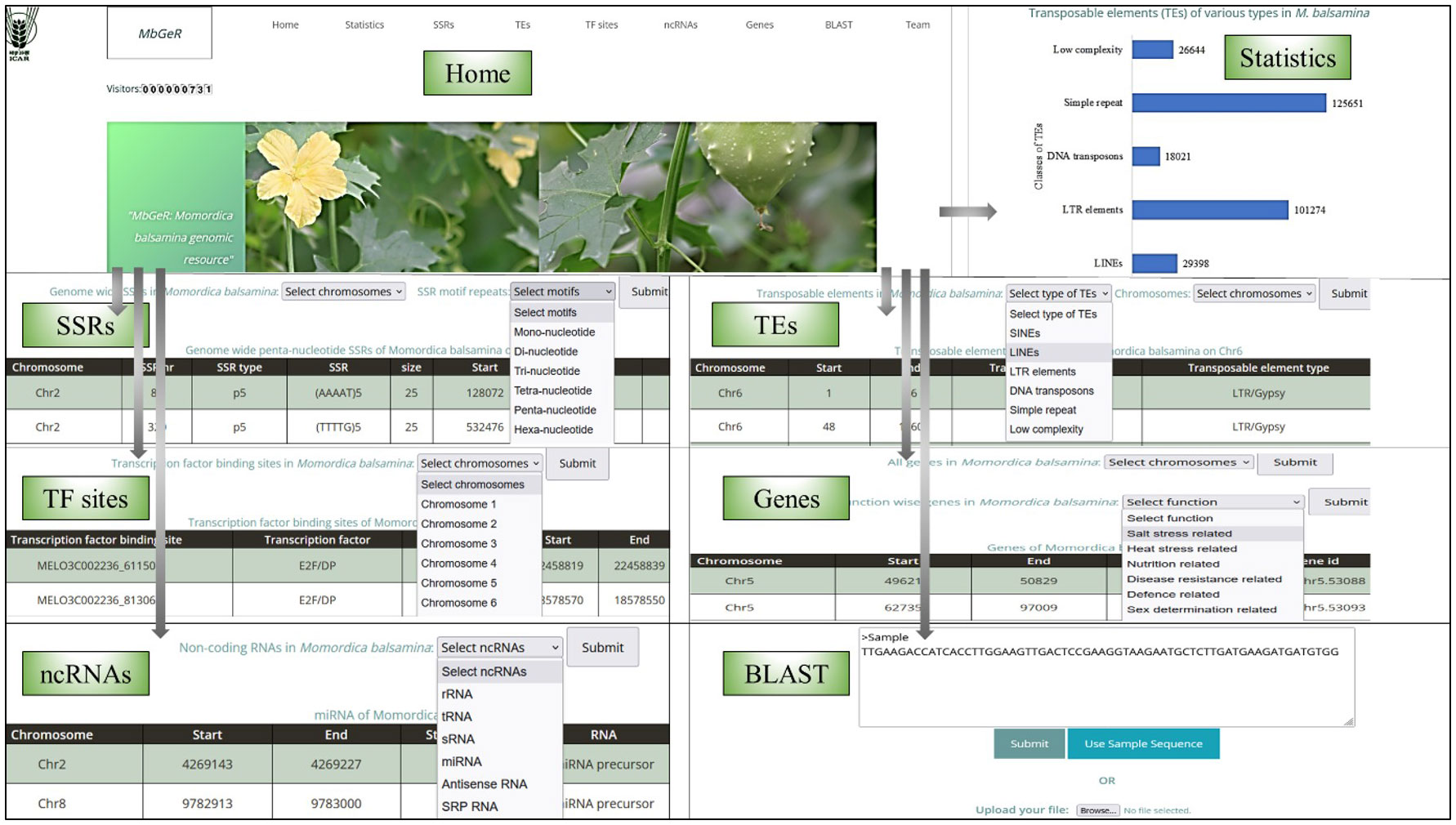
Figure 5 Various interfaces of Momordica balsamina web-genomic resource (MbGeR).
4 Discussion
CWRs are the primary source of diversity for utilization in crop improvement. Specifically, in crops with narrow genetic bases, the lack of diversity becomes the major bottlenecks in breeding program. To address the issue, close wild relatives inter-fertile with the cultivated crop species can be used as extended gene pool in crop improvement (Brozynska et al., 2016). CWRs evolve continuously in the natural environment and, hence, serve as a dynamic resource to access desirable genes to overcome several challenges in agriculture posed by increasing human population and climate change. Several workers have documented the wide-scale use of CWR to enhance agriculture production (Maxted et al., 2012; Fitzgerald, 2013; Dempewolf et al., 2014; Kell et al., 2015). It was estimated that about 30% of increased crop yields in the late 20th century can be attributed to the use of CWR in plant breeding programs (Pimentel et al., 1997). Hence, there is an increased need for the conservation and characterization of wild germplasm to utilize in crop improvement programmes. Molecular tools [e.g., simple sequence repeat (SSR) markers or microsatellites] were used in the past to characterize the CWR and to establish the relationship between wild and domesticated species (Dillon et al., 2007a; Sotowa et al., 2013). Recent DNA sequencing technology advancements increase opportunities to understand species at the whole-genome level (Edwards and Henry, 2011). Hence, genomic tools serve as the best strategy to characterize CWR and elucidate phylogenetic relationships between species, so that wild genetic diversity can be used in crop improvement (Kasem et al., 2010).
M. balsamina, Balsam apple is the closest wild species with cross-compatibility with M. charantia, exhibits greater tolerance to biotic stress, and possesses medicinal qualities (Rathode et al., 2021). Therefore, it is an ideal candidate species for harnessing natural variation within the primary gene pool and transferring desirable genes to cultivated M. charantia. Hence, genome characterization of this species proved vital for its usage in future breeding programs. In this study, we present the world’s first high-quality chromosome-level genome assembly of M. balsamina, with a genome size estimate of 384.90 Mb and an N50 of 30.96 Mb. This study used reads from multiple platforms (Oxford Nanopore, 10 X and Hi-C), which facilitates chromosome-level scaffolding with improved base accuracy. This assembly will facilitate targeted gene introgression between M. balsamina and M. charantia, enhancing tolerance and medicinal properties. Furthermore, this assembly, a combination of multiple technologies, can be used to improve further the quality and completeness of genome assembly of related species (Wang et al., 2023).
Approximately 89.44% (345 Mb) of the assembly was anchored on 11 chromosomes, while the remaining scaffolds remained un-localized. The quality of this assembly, based on the N50 value, surpasses that of previously published assemblies for other members of the Cucurbitaceae family, such as cucumber (Huang et al., 2009), melon (Garcia-Mas et al., 2012), watermelon (Guo et al., 2013), and bitter gourd (Cui et al., 2020 and Matsumura and Urasaki, 2020). Additionally, the BUSCO analysis revealed that the M. balsamina assembly contains 97.4% conserved core genes, a higher percentage compared to other M. charantia assemblies [M. cDali-11 (95.9%), TR (95.5%), and OHB3-1 (82.20%)] and related species: C. lanatus (86.50%), C. melo (86.9%), Cucurbita pepo (92.8%), C. sativus (94.8%), and Lagenaria siceraria (88.2%) assemblies (Cui et al., 2020). Gene space completeness as measured by single-copy standards, including universal single-copy orthologs (BUSCOs) and core gene families (CoreGFs) are widely used for evaluating genome assembly and annotation for its completeness and quality (Vaattovaara et al., 2019). Using estimates of gene content from hundreds of species and guided by evolution, BUSCO assessments provide comprehensible metrics to assess the completeness of genome and. hence it is considered high-resolution quantifications of the genomes (Simao et al., 2015). Therefore, with a high BUSCO score (97.4%), this assembly provides a comprehensive representation of the M. balsamina genome and serves as a valuable reference for studying the genome architecture and evolution of related cucurbits, including its closest cultivated species, M. charantia. The assembled genome of M. balsamina will aid in the identification of a greater number of genome-wide markers, allowing for the specific and accurate tracing of introgressed segments, which is crucial in interspecific introgression breeding, as reported by Qin et al. (2021). The assembly also revealed the presence of 632,098 TEs; 215,379 SSRs; 3,376 noncoding RNAs (ncRNAs); 567,483 TF binding sites; and 41,652 protein-coding genes. Many of these genes are associated with disease resistance (4421), heat stress tolerance (67), salt stress tolerance (15), carotenoid biosynthesis (05), cucurbitacin biosynthesis (229), terpenes related (19). antioxidant activity (37), and sex determination (06). Identifying these genes provides insights into the defence mechanisms, nutritional properties, and stress responses of M. balsamina.
TEs are well recognized for their role in genome evolution and regulation, providing alternative promoters, novel exons, neo-functionalization, and extensive rearrangements (Hoen & Bureau, 2015). A Comparison of our study’s assembly with recent studies on M. charantia assemblies by Cui et al., 2020, and Matsumura and Urasaki, 2020, revealed an improvement in the genome size of approximately 95 Mb and 84 Mb, respectively. This enhancement could be attributed to a higher repeat content in the M. balsamina genome than M. charantia. Our findings supported this hypothesis, as we observed that 56.73% (218.86 Mb) of the M. balsamina assembly was masked as TEs, which was higher than the percentages reported for M. charantia (52.52%), cucumber (20.8%), watermelon (39.8%), and muskmelon (35.4%) assemblies. LTR repeat content (26.82%) was the most abundant in M. balsamina genome. Higher LTR repeats are a common feature of cucurbit genomes evident from genomes of cucumber, melon, and watermelon (Huang et al., 2009; Garcia-Mas et al., 2012; Guo et al., 2013). In addition to this, the findings of the current experiment support the results of the previous studies on genome characterization of bitter gourd done by Urasaki et al., 2017 and Cui et al., 2020, which reported a higher accumulation of repeat content in the Momordica genus compared to Cucumis and Citrullus, particularly LTR repeats. However, LTR repeat content in the M. balsamina genome was less than in the Watermelon (30.5) and Bottle gourd (39.8). Earlier studies also speculated a differential rate of LTR retro transposon accumulation in the cucurbits as the reason behind the difference in the genome size among cucurbits. For instance, a higher accumulation of LTR retrotransposons is found in sponge gourd (Wu et al., 2020) and watermelon genome (Guo et al., 2013) than in cucumber (Huang et al., 2009). Hence, with absence of WGD (whole genome duplication) in cucurbits, TE might be playing vital role in genome expansion (Wu et al., 2020).
In our study, 3,376 noncoding RNA genes were annotated in the M. balsamina assembly. Extracted miRNAs, tRNAs, rRNAs, and other noncoding genes can be important resources for further studies. Additionally, we predicted 41,652 protein-coding genes in the M. balsamina assembly, a number comparable to the M. charantia OHB3-1 assembly (45859) by Urasaki et al. (2017), and significantly higher than the assemblies of M. charantia Dali-1 (26,427) by Cui et al., 2020, cucumber (26,682) by Huang et al. (2009), melon (27,427) by Garcia-Mas et al. (2012), and watermelon (23,440) by Guo et al. (2013). The variation in gene numbers could be attributed to the utilization of different transcript information during the annotation of genome assemblies or the loss of genetic diversity due to the domestication of cucurbits. Functional annotation of the protein-coding genes in our study revealed the presence of essential genes associated with detoxification, antioxidant activity, toxin activity, response to stimuli, immune system processes, defence, nutrient reservoir activity, and nutritional properties. These genes were also associated with pathways such as biosynthesis of secondary metabolites, plant hormone signal transduction, and protein processing in the endoplasmic reticulum.
M. balsamina is resistant to significant pest and diseases affecting cucurbits (Rathod et al., 2021). To understand the molecular basis for pest and pathogen resistance three major classes of R/resistance genes were searched in the genome. In the M. balsamina assembly, we identified 4,347 disease resistance genes (R genes), out of which 1,174 genes encoded NBS–LRR domains. These genes were grouped into two subfamilies based on the presence of either the toll/interleukin-1 receptor (TIR) domain or the coiled-coil (CC) domain at the N-terminal region, as described by Tameling et al. (2002). Additionally, we identified 858 RLP and 273 RLK encoding genes involved in conferring resistance response. These genes, such as Cf family proteins in tomatoes conferring resistance against Cladosporium fulvum fungus (Jones et al., 1994; Thomas et al., 1997) and HcrVf2 in apples conferring apple scab resistance (Belfanti et al., 2004), were found in lower numbers compared to melon and cucumber. The number of R genes identified in M. balsamina was much higher than reported in bottle gourd, watermelon, cucumber, and melon. However, cucurbits generally have fewer NBS-LRR encoding genes than Arabidopsis (Baumgarten et al., 2003) and rice (Goff et al., 2002). Only 61 NBS containing resistance were found in the cucumber genome (Huang et al., 2009). Likewise, out of 411 genes associated with disease resistance in melon only 81 disease resistance genes encoded NBS, the LRR and the TIR domains (Garcia-Mas et al., 2012). Similarly, only 44 NBS-LRR genes were found in watermelon genome (Guo et al., 2013). So, in general, Cucurrbitaceae genomes possess comparatively a smaller number of R genes encoding NBS-LRR proteins (Lin et al., 2013). Hence, other mechanisms might be involved in stress response. For instance, in cucumber and LOX gene family expansion is speculated as the possible complementary mechanism to cope with pathogen invasion (Huang et al., 2009). However, in M. balsamina, it seems the defence mechanisms works through the involvement of “R” genes like the majority of crop plants. The variation in the number of R genes in cucurbits suggests that they are not conserved, and the differential expansion of NBS-encoding families could be attributed to segmental and whole-genome duplications during the evolution of plant species, as suggested by Wang et al. (2009). The higher number of R genes in the M. balsamina assembly suggests their potential use in improving resistance to a wide variety of prevalent biotic stresses in its closest relative, M. charantia.
In our study, we identified a total of 34 HSFs in the M. balsamina assembly, which was higher than the numbers reported for rice (25) by Chauhan et al. (2011), Arabidopsis (21) by Nover et al. (2001), and cucumber (23) by Chen et al. (2021). Among these genes, the primary heat stress factors identified were HSFB1 (01), HSFA2 (03), HSFA4 (04), HSFB4 (04), and HSF-A6 (04), which contribute to thermo-tolerance through regulating HSPs as described by Ohama et al. (2017). Additionally, we identified 29 HSP genes, predominantly encoding HSP70, and 17 small heat proteins (HSP20) in the M. balsamina assembly. HSPs play an essential role in the regulation of HSFs and, subsequently, the expression of heat-responsive genes associated with heat tolerance. HSP20 has been reported to contribute to heat stress tolerance in melon (Zheng et al., 2021), watermelon (He et al., 2019), cucumber (Chen et al., 2021), and pumpkin (Hu et al., 2021). Over-expression of HSP70 has also been reported to significantly increase heat tolerance in watermelon, cabbage, and chilli (Park et al., 2013; Guo et al., 2015; Usman et al., 2015; Zhao et al., 2018; He et al., 2019). Therefore, the thermo-tolerance capacity of M. balsamina can be attributed to the identified important HSPs, which can be further functionally validated for future use. Similarly, we identified 15 genes encoding proteins related to salinity tolerance in the M. balsamina assembly, including ACER, S-acyltransferase, salt stress root protein RS1-like, and protein RICE SALT SENSITIVE 3 isoforms. These proteins have previously been reported to play a role in salinity tolerance in Arabidopsis by Wu et al. (2015) and in wheat by Kang et al. (2012). However, their role in salt tolerance in cucurbits has yet to be well documented. These identified genes with a possible role in salt tolerance can be further studied to understand the detailed physiological and molecular network associated with salt tolerance and improve the salt tolerance of related species through inter-specific introgression. Additionally, we found 37 glutathione S-transferase (GST) family genes in M. balsamina, which are vital antioxidant enzymes involved in reducing the damage caused by reactive oxygen species during abiotic stress (salt, drought, and cold) tolerance mechanisms (Venkateswarlu et al., 2012; Chan and Lam, 2014; Islam et al., 2019; and Song et al., 2021). GSTs are also involved in detoxification processes and protection against damage from various environmental factors (Dixon et al., 1998; (Esmaeili et al., 2009). The large number of identified GST family genes in M. balsamina suggests its high tolerance to abiotic stress, which can be harnessed to improve abiotic stress tolerance in M. charantia.
In the M. balsamina assembly, we identified five genes related to carotenoid biosynthesis, including chloroplast-specific lycopene beta-cyclase, phytoene desaturase/phytoene dehydrogenase, pro-lycopene isomerase, zeta-carotene desaturase, and lycopene epsilon cyclase. The overexpression of one or more carotenoid biosynthesis genes to produce carotene-rich varieties has been successfully employed in advanced vegetable improvement programs for crops such as tomatoes (Fraser et al., 2001), carrot (Fraser and Bramley, 2004), and potatoes (Diretto et al., 2007). Carotenoids contribute to color, serve as precursors of vitamin A, and have various health benefits, including reducing the risk of cancers and cardiovascular diseases (Paine et al., 2005; Aluru et al., 2008). Therefore, the transfer of these carotenoid biosynthesis genes from M. balsamina to M. charantia could be utilized to improve its nutritional value. Furthermore, we identified 229 genes related to cucurbitacin biosynthesis in the M. balsamina assembly. Cucurbitacins are signature bioactive compounds of the Cucurbitaceae family and confer a bitter taste to cucurbits (Chen et al., 2005). The identified genes encoding enzymes such as oxidosqualene cyclase, cytochromes P450, and acyltransferases are essential for cucurbitacin biosynthesis. Similar pathways and mechanisms are involved in the production of terpenoids across the genera of the Cucurbitaceae family (Huang et al., 2009; Shang et al., 2014). Moreover, we identified 19 genes related to the biosynthesis of other triterpenoids in the M. balsamina assembly. These triterpenoids have diverse medicinal properties, namely, anticancer, antidiabetic, anti-HIV, antimalarial, anti-inflammatory, and antimicrobial activities (Ramalhete et al., 2022). Many of these triterpenoids such as balsaminol, balsaminoside, balsaminagenin, karavilagenin, cucurbalsaminol, and balsaminapentaol (Ramalhete et al., 2009a; Ramalhete et al., 2009; Ramalhete et al., 2010; Ramalhete et al., 2011a; and Ramalhete et al., 2011b) have been previously isolated from M. balsamina, highlighting its potential as a source of bioactive compounds. These results confirm the value of M. baslamina in terms of its nutritional and therapeutic properties.
M. balsamina is a monoecious plant with separate male and female flowers on the same plant. Sex determination and expression in cucurbits have been extensively studied, and various phytohormones and their cross talk have been identified as key regulators (Chen et al., 2016; Wang et al., 2019). Ethylene, in particular, is considered a core regulator of sex expression in cucurbits (Yin and Quinn, 1995; Boualem et al., 2015; Chen et al., 2016). In the M. balsamina assembly, we identified six genes related to ethylene biosynthesis, including ACS (1-aminocyclopropane-1-carboxylate synthase) ACS-7, ACS-CMA101, and ACS-CMW-33 genes. These genes are involved in the production of ethylene, which regulates sex expression in cucurbits. In Cucumis sativus, ACS-1 is encoded by the F locus and is known to promote female sex expression by suppressing stamen development in bisexual flower primordial (Trebitsh et al., 1997; Mibus and Tatlioglu, 2004). Likewise, ACS-7 is encoded by A locus (orthologue of the cucumber M gene) and is known to promote femaleness in monoecious melon lines, and a miss-sense mutation in CmACS-7 led to andromonoecy, the predominant sex type of commercial melon (Boualem et al., 2008; Boualem et al., 2009). Similarly, two genes (MOMC46_189, MOMC518_1) encoding CmAcs-7 like protein and a gene (MOMC3_649) encoding CmACS 11 like protein were identified in M. charantia (Urasaki et al., 2017). ACS encoding genes for sex determination in M. balsamina and M. charantia were found orthologous by synteny analysis as well. This suggests the possible involvement ethylene regulated sex expression like all other cucurbits in Momoridica genus. The orthologous relationship of these ACS genes with those identified in M. charantia and other cucurbits suggests a highly conserved nature of sex-regulating genes across the Cucurbitaceae family. Additionally, our study revealed a high number of conserved genes (approximately 8,500) between M. balsamina and M. charantia, Cucumis sativus, Cucumis melo, and Citrullus lanatus, indicating a substantial level of genetic similarity and potential for comparative genomics studies among cucurbits.
Comparative plant genomics investigates the distinctiveness and differences among plant genomes. By comparing the genomes of closely and distantly related species, researchers can gain insights into the patterns and processes associated with plant genome evolution and identify functional regions within genomes (Caicedo and Purugganan, 2005). In this particular study, we conducted a genome comparison of Momordica balsamina with other related cucurbit species, namely, Momordica charantia (Bitter gourd), Cucumis sativus (Cucumber), Cucumis melo (Musk melon), and Citrullus lanatus (Watermelon), in order to identify syntenic and phylogenetic relationships. Our analysis revealed that Momordica balsamina shared the highest number of orthologous pairs (8,845) with Momordica charantia, followed by 8,265 orthologous pairs between Momordica balsamina and Cucumis sativus. Previous research by Garcia-Mas et al. (2012) identified 19,377 one-to-one ortholog pairs between Cucumis melo and Cucumis sativus.
Furthermore, we detected paralogous and orthologous relationships between the five studied Cucurbitaceae genomes, which can serve as a guide for translational research and facilitate the study of conserved economic traits. By utilizing conserved BUSCO genes (orthologous genes), we identified the evolutionary relationship between Momordica balsamina, Momordica charantia, Citrullus lanatus, Cucumis sativus, and Cucumis melo. Phylogenetic analysis done using Vitis vinifera as an outgroup classified Momordica balsamina and Momordica charantia to the same clade, indicating a close genetic relationship between these two species with a speciation/separation event estimated to have occurred 23 million years ago. Additionally, Momordica was found to be closer to Citrullus (Watermelon) than to Cucumis, suggesting a divergence around 53 million years ago. Previous studies by Urasaki et al. (2017); Jobst et al. (1998), and Schaefer et al. (2009) also reported a closer genetic association between bitter gourd and watermelon compared to cucumber or melon in phylogenetic and genetic analyses.
We performed synteny analysis to elucidate variations at the nucleotide level arising from mutations, duplications, chromosomal rearrangements, and gene family expansion or loss (Alkan et al., 2011). Synteny blocks, which identify regions of chromosomes shared between genomes that have a common order of homologous genes from a common ancestor, were identified to shed light on evolutionary relationships between species (Vergara and Chen, 2010). Previous synteny analysis in members of the Cucurbitaceae family helped to clarify the reason behind differences in basic chromosome number between Cucumis sativus and C. melo (Huang et al., 2009; Li et al., 2011). In our current study, we found the highest number of syntenic blocks between Momordica balsamina and Momordica charantia (306), followed by 282 syntenic blocks between Momordica balsamina and Citrullus lanatus, indicating a high level of synteny between M. balsamina and M. charantia, followed by watermelon (Citrullus lanatus). Previous synteny analyses also reported a high level of co-linearity between Momordica and Citrullus genomes (Urasaki et al., 2017; Cui et al., 2020). Our findings revealed a general absence of one-to-one relationships in the chromosomes between Momordica balsamina and other cucurbit genomes. This observation aligns with most of the synteny analyses conducted in cucurbits (Matsusmura and Urasaki, 2020); Wu et al., 2020, except for the study by Wu et al. (2017), which identified chromosome-level synteny between bottle gourd and melon (C. melo) and watermelon (Citrullus lanatus) genomes. The findings of our study, along with the synteny analysis by Matsusmura and Urasaki (2020), support the fact that most Cucurbitaceae genomes belong to a different clade than the genus Momordica (Renner and Schaefer, 2016). Therefore, the absence of one-to-one chromosome synteny between Momordica (balsamina and charantia) and other cucurbits may be attributed to higher structural re-arrangement in chromosomes after speciation.
In addition to the genome comparison and synteny analysis, we identified 215,379 SSRs and 567,483 TF binding sites (TFBSs). These data were incorporated into a genomic web resource called MbGeR, developed to provide access to the data extracted during this study. Characterizing the M. balsamina genome contributes to our understanding of the available gene pool that can be utilized to improve M. charantia through advanced plant breeding techniques. Due to the significant therapeutic values, resilience to biotic and abiotic stress and nutritional value of M. balsamina, this study offers valuable insights and a high-quality assembly and annotation of its genome, thereby assisting in the development of high-yielding and resistant varieties of this promising vegetable crop.
5 Conclusion
M. balsamina is the closest wild species of M. charantia, with higher resilience to biotic and abiotic stresses and greater medicinal and nutritional qualities. The present study provides the first high-quality chromosome-level genome assembly of M. balsamina with size 384.90 Mb and N5030.96 Mb using sequence data from 10x Genomics, Nanopore, and Hi-C platforms. Annotation of the provided assembly identified 215,379 SSRs; 632,098 TEs; 567,483 TF binding sites; 3,376 noncoding RNAs (tRNA, miRNA, snoRNA, and so forth) genes, and 41,652 protein coding genes. A sum of 4,347 disease resistance, 67 heat stress–related, 15 salt stress related, 229 cucurbitacin related, 19 terpenes related, 37 antioxidant activity, 05 carotenoid related, and 06 sex determination related genes were identified in M. balsamina assembly. Because of stress tolerance and better therapeutic values, M. balsamina will serve as a potential genomic resource, and provided assembly will help to boost the targeted gene introgression between M. balsamina and M. charantia species in developing high-yielding climate-smart and stress-resilient crop varieties. In addition, this high-quality genome assembly done using reads from multiple sequencing platforms can be used to improve further the quality and completeness of genome assembly of related species. The SSR markers obtained in this study would assist in linkage mapping, QTL and gene discovery, population genetics, evolutionary studies and gene regulation. The provided assembly will also help in identifying a higher number of genome-wide markers with greater specificity and accuracy to trace the introgressed segments during advanced breeding programs to improve resistance and medicinal values to high-yielding M. charantia varieties, which is significantly lost due to domestication of bitter gourd. Furthermore, the finding of comparative genome analysis (phylogeny and synteny) will be helpful to get insights into the patterns and processes associated with genome evolution and to uncover functional regions of cucurbit genomes.
Data availability statement
Whole genome sequencing of Momordica balsamina: BioProject: PRJNA877043: First and high-quality assembly of Momordica balsamina, a potential genetic resource to improve tolerance and medicinal properties in bitter gourd, BioSample: SAMN30678163: Momordica balsamina genome; SRA: SRR21495983, SRR21495982 and SRR21495981. The assembly is submitted in NCBI with ID: SUB13995037.
Author contributions
VN: Writing – original draft, Investigation. KS: Data curation, Writing – original draft. RE: Supervision, Writing – review & editing. VC: Writing – review & editing, Resources. SJ: Writing – review & editing, Data curation, Writing – original draft. MI: Data curation, Writing – review & editing. AM: Writing – review & editing, Resources, Supervision. HM: Supervision, Writing – review & editing. BG: Writing – review & editing, Investigation. GJ: Writing – review & editing. CK: Writing – review & editing, Supervision. AG: Writing – review & editing, Supervision. DK: Supervision, Writing – review & editing, Data curation, Investigation, Writing – original draft. SD: Supervision, Writing – review & editing, Data curation, Investigation, Writing – original draft. TB: Resources, Supervision, Writing – original draft, Writing – review & editing, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The research work was funded by the NAHEP-CAAST programme of Indian Council of Agricultural Research (ICAR). This work was also supported by the Indian Council of Agricultural Research, Ministry of Agriculture and Farmers’ Welfare, Govt. of India, who provided financial assistance in the form of a CABin grant (F. no. Agril. Edn.4-1/2013-A&P), as well as Advanced Super Computing Hub for Omics Knowledge in Agriculture (ASHOKA) facility at ICAR-IASRI, New Delhi, India.
Acknowledgments
Authors are thankful to the ICAR-Indian Agricultural Research Institute, New Delhi for providing financial support and conduct of the research program of the PhD student, Mr. VN. We are thankful to the Indian Council of Agricultural Research, Ministry of Agriculture and Farmers’ Welfare, Govt. of India for Advanced Super Computing Hub for Omics Knowledge in Agriculture (ASHOKA) facility at ICAR-IASRI, New Delhi, India created under National Agricultural Innovation Project, funded by World Bank at ICAR-IASRI, New Delhi.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1258042/full#supplementary-material
References
Ajji, P. K. (2016). Functional characterization of a novel ribosome inactivating protein from Momordica balsamina (Deakin University, Australia: Doctoral dissertation, Deak in University). Available at: https://dro.deakin.edu.au/eserv/DU:30103049/ajji-functionalcharacterization-2017.pdf.
Ajji, P. K., Binder, M. J., Walder, K., Puri, M. (2017). Balsamin induces apoptosis in breast cancer cells via DNA fragmentation and cell cycle arrest. Mol. Cell. Biochem. 432 (1), 189–198. doi: 10.1007/s11010-017-3009-x
Alkan, C., Coe, B. P., Eichler, E. E. (2011). Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12 (5), 363–376. doi: 10.1038/nrg2958
Aluru, M., Xu, Y., Guo, R., Wang, Z., Li, S., White, W., et al. (2008). Generation of transgenic maize with enhanced provitamin A content. J. Exp. Botany. 59 (13), 3551–3562. doi: 10.1093/jxb/ern212
Andrews, S. (2010) FASTQC. A quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Aversano, R., Contaldi, F., Ercolano, M. R., Grosso, V., Iorizzo, M., Tatino, F., et al. (2015). The Solanum commersonii genome sequence provides insights into adaptation to stress conditions and genome evolution of wild potato relatives. Plant Cell. 27 (4), 954–968. doi: 10.1105/tpc.114.135954
Baumgarten, A., Cannon, S., Spangler, R., May, G. (2003). Genome-level evolution of resistance genes in Arabidopsis thaliana. Genetics 165 (1), 309–319. doi: 10.1093/genetics/165.1.309
Behera, T. K., Behera, S., Bharathi, L. K., John, K. J., Simon, P. W., Staub, J. E. (2010). Bitter gourd: botany, horticulture, breeding. Hortic. Rev. 37, 101–141. doi: 10.1002/9780470543672.ch2
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33 (16), 2583–2585. doi: 10.1093/bioinformatics/btx198
Belaghzal, H., Dekker, J., Gibcus, J. H. (2017). Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods 123, 56–65. doi: 10.1016/j.ymeth.2017.04.004
Belfanti, E., Silfverberg-Dilworth, E., Tartarini, S., Patocchi, A., Barbieri, M., Zhu, J., et al. (2004). The HcrVf2 gene from a wild apple confers scab resistance to a transgenic cultivated variety. Proc. Natl. Acad. Sci. 101 (3), 886–890. doi: 10.1073/pnas.0304808101
Bharathi, L. K., John, K. J. (2013). Momordica Genus in Asia-An Overview Vol. p (New Delhi: Springer), 147). doi: 10.1007/978-81-322-1032-0
Bharathi, L. K., Munshi, A. D., Behera, T. K., Vinod, J. K.J., Das, A. B., Bhat, K. V., et al. (2012). Production and preliminary characterization of inter-specific hybrids derived from Momordica species. Curr. Sci. 103 (2), 178–186.
Bharathi, L. K., Munshi, A. D., Chandrashekaran, S., Behera, T. K., Das, A. B., John, K. J. (2011). Cytotaxonomical analysis of Momordica L. (Cucurbitaceae) species of Indian occurrence. J. Genet. 90 (1), 21–30.
Boualem, A., Fergany, M., Fernandez, R., Troadec, C., Martin, A., Morin, H., et al. (2008). A conserved mutation in an ethylene biosynthesis enzyme leads to andromonoecy in melons. Science 321 (5890), 836–838. doi: 10.1126/science.1159023
Boualem, A., Troadec, C., Camps, C., Lemhemdi, A., Morin, H., Sari, A., et al. (2015). A cucurbit androecy gene reveals how unisexual flowers develop and dioecy emerges. Science 350 (6261), 688–691. doi: 10.1126/science.aac8370
Boualem, A., Troadec, C., Kovalski, I., Sari, M. A., Perl-Treves, R., Bendahmane, A. (2009). A conserved ethylene biosynthesis enzyme leads to andromonoecy in two Cucumis species. PloS One 4 (7), e6144. doi: 10.1371/journal.pone.0006144
Bowley, S. R., Taylor, N. L. (1987). “Introgressive hybridization,” in CRC handbook of plant science in agriculture, vol. 1 . Ed. Christie, B. R. (Boca Raton: CRC Press), 23–59.
Brozynska, M., Furtado, A., Henry, R. J. (2016). Genomics of crop wild relatives: expanding the gene pool for crop improvement. Plant Biotechnol. J. 14 (4), 1070–1085. doi: 10.1111/pbi.12454
Caicedo, A. L., Purugganan, M. D. (2005). Comparative plant genomics. Frontiers and prospects. Plant Physiol. 138 (2), 545–547. doi: 10.1104/pp.104.900148
Cao, M. D., Nguyen, S. H., Ganesamoorthy, D., Elliott, A. G., Cooper, M. A., Coin, L. J. (2017). Scaffolding and completing genome assemblies in real-time with nanopore sequencing. Nat. Commun. 8 (1), 1–10. doi: 10.1038/ncomms14515
Chan, C., Lam, H. M. (2014). A putative lambda class glutathione S-transferase enhances plant survival under salinity stress. Plant Cell Physiol. 55 (3), 570–579. doi: 10.1093/pcp/pct201
Chan, P. P., Lowe, T. M. (2019). tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol. Biol. (Clifton NJ) 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Chauhan, H., Khurana, N., Agarwal, P., Khurana, P. (2011). Heat shock factors in rice (Oryza sativa L.): genome-wide expression analysis during reproductive development and abiotic stress. Mol. Genet. Genomics 286 (2), 171–187. doi: 10.1007/s00438-011-0638-8
Chen, J. C., Chiu, M. H., Nie, R. L., Cordell, G. A., Qiu, S. X. (2005). Cucurbitacins and cucurbitane glycosides: structures and biological activities. Natural product Rep. 22 (3), 386–399. doi: 10.1039/B418841C
Chen, F., Dong, W., Zhang, J., Guo, X., Chen, J., Wang, Z., et al. (2018). The sequenced angiosperm genomes and genome databases. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00418
Chen, F., Mackey, A. J., Stoeckert, C. J., Jr., Roos, D. S. (2006). OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34 (suppl_1), D363–D368. doi: 10.1093/nar/gkj123
Chen, H., Sun, J., Li, S., Cui, Q., Zhang, H., Xin, F., et al. (2016). An ACC oxidase gene essential for cucumber carpel development. Mol. Plant 9 (9), 1315–1327. doi: 10.1016/j.molp.2016.06.018
Chen, X., Wang, Z., Tang, R., Wang, L., Chen, C., Ren, Z. (2021). Genome-wide identification and expression analysis of Hsf and Hsp gene families in cucumber (Cucumis sativus L.). Plant Growth Regul. 95 (2), 223–239. doi: 10.1007/s10725-021-00739-z
Conesa, A., Gotz, S., García-Gomez, J. M., Terol, J., Talón, M., Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21 (18), 3674–3676. doi: 10.1093/bioinformatics/bti610
Cui, J., Yang, Y., Luo, S., Wang, L., Huang, R., Wen, Q., et al. (2020). Whole-genome sequencing provides insights into the genetic diversity and domestication of bitter gourd (Momordica spp.). Horticulture Res. 7 (1), 85. doi: 10.1038/s41438-020-0305-5
Dempewolf, H., Baute, G., Anderson, J., Kilian, B., Smith, C., Guarino, L. (2017). Past and future use of wild relatives in crop breeding. Crop Sci. 57 (3), 1070–1082. doi: 10.2135/cropsci2016.10.0885
Dempewolf, H., Eastwood, R. J., Guarino, L., Khoury, C. K., Müller, J. V., Toll, J. (2014). Adapting agriculture to climate change: a global initiative to collect, conserve, and use crop wild relatives. Agroecology Sustain. Food Syst. 38 (4), 369–377. doi: 10.1080/21683565.2013.870629
Dhillon, N. P., Sanguansil, S., Schafleitner, R., Wang, Y. W., McCreight, J. D. (2016). Diversity among a wide Asian collection of bitter gourd landraces and their genetic relationships with commercial hybrid cultivars. J. Am. Soc. Hortic. Sci. 141 (5), 475–484. doi: 10.21273/JASHS03748-16
Dillon, S. L., Lawrence, P. K., Henry, R. J., Price, H. J. (2007). Sorghum resolved as a distinct genus based on combined ITS1, ndh F and Adh 1 analyses. Plant Systematics Evol. 268, 29–43. doi: 10.1007/s00606-007-0571-9
Diretto, G., Al-Babili, S., Tavazza, R., Papacchioli, V., Beyer, P., Giuliano, G. (2007). Metabolic engineering of potato carotenoid content through tuber-specific overexpression of a bacterial mini-pathway. PloS One 2 (4), e350. doi: 10.1371/journal.pone.0000350
Dixon, D. P., Cummins, I., Cole, D. J., Edwards, R. (1998). Glutathione-mediated detoxification systems in plants. Curr. Opin. Plant Biol. 1 (3), 258–266. doi: 10.1016/S1369-5266(98)80114-3
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the Aedes aEgypti genome using Hi-C yields chromosome-length scaffolds. Science 356 (6333), 92–95. doi: 10.1126/science.aal3327
Durand, N. C., Shamim, M. S., Machol, I., Rao, S. S., Huntley, M. H., Lander, E. S., et al. (2016). Juicer provides a one-click system for analysing loop-resolution Hi-C experiments. Cell Syst. 3 (1), 95–98. doi: 10.1016/j.cels.2016.07.002
Edwards, M. A., Henry, R. J. (2011). DNA sequencing methods contributing to new directions in cereal research. J. Cereal Sci. 54 (3), 395–400. doi: 10.1016/j.jcs.2011.07.006
Erxleben, A., Grüning, B. (2020) Genome Annotation (Galaxy Training Materials). Available at: https://training.galaxyproject.org/training-material/topics/genome-annotation/tutorials/genome-annotation/tutorial (Accessed May 24 2022).
Esmaeili, M., Shahrtash, M., Moosavi, F., Mohsenzadeh, S., Mohabatkar, H. (2009). Plant glutathione S-transferase function. Paper Presentation Proc. 6th Natl. Biotechnol. Congress Iran Tehran Iran.
Fraser, P. D., Bramley, P. M. (2004). The biosynthesis and nutritional uses of carotenoids. Prog. Lipid Res. 43 (3), 228–265. doi: 10.1016/j.plipres.2003.10.002
Flyman, M. V., Afolayan, A. J. (2007). Proximate and mineral composition of the leaves of Momordica balsamina L.: an under-utilized wild vegetable in Botswana. Int. J. Food Sci. Nutr. 58 (6), 419–423. doi: 10.1080/09637480701253417
Fraser, P. D., Romer, S., Kiano, J. W., Shipton, C. A., Mills, P. B., Drake, R., et al. (2001). Elevation of carotenoids in tomato by genetic manipulation. J. Sci. Food Agric. 81 (9), 822–827. doi: 10.1002/JSFA.908
García, J. C., Guadagno, A., Paytuvi-Gallart, A., Saera-Vila, A., Amoroso, C. G., D’Esposito, D., et al. (2021). PRGdb 4.0: an updated database dedicated to genes involved in plant disease resistance process. Nucleic Acids Res. 50 (D1), D1483–D1490. doi: 10.1093/nar/gkab1087
Garcia-Mas, J., Benjak, A., Sanseverino, W., Bourgeois, M., Mir, G., González, V. M., et al. (2012). The genome of melon (Cucumis melo L.). Proc. Natl. Acad. Sci. 109 (29), 11872–11877. doi: 10.1073/pnas.1205415109
Goff, S. A., Ricke, D., Lan, T. H., Presting, G., Wang, R., Dunn, M., et al. (2002). A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296 (5565), 92–100. doi: 10.1126/science.1068275
Gramazio, P., Yan, H., Hasing, T., Vilanova, S., Prohens, J., Bombarely, A. (2019). Whole-genome resequencing of seven eggplant (Solanum melongena) and one wild relative (S. incanum) accessions provides new insights and breeding tools for eggplant enhancement. Front. Plant science. 1220. doi: 10.3389/fpls.2019.01220
Guo, M., Lu, J. P., Zhai, Y. F., Chai, W. G., Gong, Z. H., Lu, M. H. (2015). Genome-wide analysis, expression profile of heat shock factor gene family (CaHsfs) and characterisation of CaHsfA2 in pepper (Capsicum annuum L.). BMC Plant Biol. 15 (1), 151. doi: 10.1186/s12870-015-0512-7
Guo, S., Zhang, J., Sun, H., Salse, J., Lucas, W. J., Zhang, H., et al. (2013). The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 45 (1), 51–58. doi: 10.1038/ng.2470
Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29 (8), 1072–1075. doi: 10.1093/bioinformatics/btt086
Hassan, L. G., Umar, K. J. (2006). Nutritional value of Balsam Apple (Momordica balsamina L.) leaves. Pakistan J. Nutr. 5 (6), 522–529. doi: 10.3923/pjn.2006.522.529
He, Y., Fan, M., Sun, Y., Li, L. (2019). Genome-wide analysis of watermelon HSP20s and their expression profiles and subcellular locations under stresses. Int. J. Mol. Sci. 20 (1), 12. doi: 10.3390/ijms20010012
Hoen, D. R., Bureau, T. E. (2015). Discovery of novel genes derived from transposable elements using integrative genomic analysis. Mol. Biol. Evol. 32 (6), 1487–1506. doi: 10.1093/molbev/msv042
Hu, Y., Zhang, T., Liu, Y., Li, Y., Wang, M., Zhu, B., et al. (2021). Pumpkin (Cucurbita moschata) HSP20 Gene Family Identification and Expression under Heat Stress. Front. Genet. 2062. doi: 10.3389/fgene.2021.753953
Huang, S., Li, R., Zhang, Z., Li, L. I., Gu, X., Fan, W., et al. (2009). The genome of the cucumber, Cucumis sativus L. Nat. Genet. 41 (12), 1275–1281. doi: 10.1038/ng.475
Islam, S., Sajib, S. D., Jui, Z. S., Arabia, S., Islam, T., Ghosh, A. (2019). Genome-wide identification of glutathione S-transferase gene family in pepper, its classification, and expression profiling under different anatomical and environmental conditions. Sci. Rep. 9 (1), 1–15. doi: 10.1038/s41598-019-45320-x
Jeffrey, C. (1967). “Cucurbitaceae,” in Flora of tropical East Africa. Eds. Milne-Redhead, C. E., Polhill, R. M. (London, UK: Crown Agents for Overseas Governments and Administrations), 1–156.
Jin, J., Tian, F., Yang, D. C., Meng, Y. Q., Kong, L., Luo, J., et al. (2017). PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D1040–D1045. doi: 10.1093/nar/gkw982
Jobst, J., King, K., Hemleben, V. (1998). Molecular evolution of the internal transcribed spacers (ITS1 and ITS2) and phylogenetic relationships among the species of the family Cucurbitaceae. Mol. Phylo. Evol. 9, 204–219. doi: 10.1006/mpev.1997.0465
John, K. J. (2005). Studies on ecogeography and genetic diversity of the genus Momordica L. @ in India (Kottayam, Kerala: Dissertation, Mahatma Gandhi University).
Jones, D. A., Thomas, C. M., Hammond-Kosack, K. E., Balint-Kurti, P. J., Jones, J. D. (1994). Isolation of the tomato Cf-9 gene for resistance to Cladosporium fulvum by transposon tagging. Science 266 (5186), 789–793. doi: 10.1126/science.7973631
Kang, G., Li, G., Zheng, B., Han, Q., Wang, C., Zhu, Y., et al. (2012). Proteomic analysis on salicylic acid-induced salt tolerance in common wheat seedlings (Triticum aestivum L.). Biochim. Biophys. Acta (BBA)-Proteins Proteomics. 1824 (12), 1324–1333. doi: 10.1016/j.bbapap.2012.07.012
Kasem, S., Waters, D. L., Rice, N., Shapter, F. M., Henry, R. J. (2010). Whole grain morphology of Australian rice species. Plant Genet. Resour. 8 (1), 74–81. doi: 10.1017/S1479262109990189
Katoh, K., Misawa, K., Kuma, K. I., Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30 (14), 3059–3066. doi: 10.1093/nar/gkf436
Kaur, I., Yadav, S. K., Hariprasad, G., Gupta, R. C., Srinivasan, A., Batra, J. K., et al. (2012). Balsamin, a novel ribosome-inactivating protein from the seeds of Balsam apple Momordica balsamina. Amino Acids 43 (2), 973–981. doi: 10.1007/s00726-011-1162-1
Kell, S., Qin, H., Chen, B., Ford-Lloyd, B., Wei, W., Kang, D., et al. (2015). China’s crop wild relatives: diversity for agriculture and food security. Agriculture Ecosyst. Environ. 209, 138–154. doi: 10.1016/j.agee.2015.02.012
Khare, C. (2007). “Momordica balsamina Linn,” in Indian Medicinal Plants. Ed. Khare, C. (New York, NY: Springer). doi: 10.1007/978-0-387-70638-2_1027
Krawinkel, M. B., Ludwig, C., Swai, M. E., Yang, R. Y., Chun, K. P., Habicht, S. D. (2018). Bitter gourd reduces elevated fasting plasma glucose levels in an intervention study among prediabetics in Tanzania. J. ethnopharmacology 216, 1–7. doi: 10.1016/j.jep.2018.01.016
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19 (9), 1639–1645. doi: 10.1101/gr.092759.109
Lewinski, M., Bramkamp, Y., Köster, T., Staiger, D. (2020). SEQing: web-based visualization of iCLIP and RNA-seq data in an interactive python framework. BMC Bioinf. 21 (1), 113. doi: 10.1186/s12859-020-3434-9
Li, D., Cuevas, H. E., Yang, L., Li, Y., Garcia-Mas, J., Zalapa, J., et al. (2011). Syntenic relationships between cucumber (Cucumis sativus L.) and melon (C. melo L.) chromosomes as revealed by comparative genetic mapping. BMC Genomics 12 (1), 1–14. doi: 10.1186/1471-2164-12-396
Limin, F., Niu, B., Zhu, Z., Wu, S., Li, W. (2012). CD-HIT: accelerated for clustering the next generation sequencing data. Bioinformatics 28 (23), 3150–3152. doi: 10.1093/bioinformatics/bts565
Lin, X., Zhang, Y., Kuang, H., Chen, J. (2013). Frequent loss of lineages and deficient duplications accounted for low copy number of disease resistance genes in cucurbitaceae. BMC Genomics 14, 1–13.
Lipman, D. J., Pearson, W. R. (1985). Rapid and sensitive protein similarity searches. Science 227 (4693), 1435–1441. doi: 10.1126/science.2983426
Mashiane, P., Shoko, T., Manhivi, V., Slabbert, R., Sultanbawa, Y., Sivakumar, D. (2022). A Comparison of bioactive metabolites, antinutrients, and bioactivities of african pumpkin leaves (Momordica balsamina L.) cooked by different culinary techniques. Molecules 27 (6), 1901. doi: 10.3390/molecules27061901
Matsumura, H., Urasaki, N. (2020). “Genome sequence of bitter Gourd and Its Comparative Study with Other Cucurbitaceae Genomes,” in The Bitter Gourd Genome. Compendium of plant Genomes. Eds. Kole, C., Matsumura, H., Behera, T. (Cham: Springer), 113–123). doi: 10.1007/978-3-030-15062-4_10
Maxted, N., Kell, S., Ford-Lloyd, B., Dulloo, E., Toledo, Á. (2012). Toward the systematic conservation of global crop wild relative diversity. Crop Sci. 52 (2), 774–785. doi: 10.2135/cropsci2011.08.0415
Mibus, H., Tatlioglu, T. (2004). Molecular characterization and isolation of the F/f gene for femaleness in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 109 (8), 1669–1676. doi: 10.1007/s00122-004-1793-7
Mishra, K. C., Sahu, P. R., Jha, U. C. (1986). Balsam apple for your vegetable garden. Indian Horticulture J. 13.
Nawrocki, E. P., Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29 (22), 2933–2935. doi: 10.1093/bioinformatics/btt509
Nover, L., Bharti, K., Döring, P., Mishra, S. K., Ganguli, A., Scharf, K. D. (2001). Arabidopsis and the heat stress transcription factor world: how many heat stress transcription factors do we need? Cell Stress chaperones 6 (3), 177. doi: 10.1379/1466-1268(2001)006<0177:aathst>2.0.co;2
Ohama, N., Sato, H., Shinozaki, K., Yamaguchi-Shinozaki, K. (2017). Transcriptional regulatory network of plant heat stress response. Trends Plant Sci. 22 (1), 53–65. doi: 10.1016/j.tplants.2016.08.015
Paine, J. A., Shipton, C. A., Chaggar, S., Howells, R. M., Kennedy, M. J., Vernon, G., et al. (2005). Improving the nutritional value of Golden Rice through increased pro-vitamin A content. Nat. Biotechnol. 23 (4), 482–487. doi: 10.1038/nbt1082
Park, H. J., Jung, W. Y., Lee, S. S., Song, J. H., Kwon, S. Y., Kim, H., et al. (2013). Use of heat stress responsive gene expression levels for early selection of heat tolerant cabbage (Brassica oleracea L.). Int. J. Mol. Sci. 14 (6), 11871–11894. doi: 10.3390/ijms140611871
Peter, K. V., Abraham, Z. (2007). Biodiversity in horticultural crops Vol. 1 (New Delhi, India: Daya Publisher).
Pimentel, D., Wilson, C., McCullum, C., Huang, R., Dwen, P., Flack, J., et al. (1997). Economic and environmental benefits of biodiversity. BioScience 47 (11), 747–757. doi: 10.2307/1313097
Qin, X., Zhang, Z., Lou, Q., Xia, L., Li, J., Li, M., et al. (2021). Chromosome-scale genome assembly of Cucumis hystrix—a wild species interspecifically cross-compatible with cultivated cucumber. Horticulture Res. 8 (1), 40. doi: 10.1038/s41438-021-00475-5
Ramalhete, C., da Cruz, F. P., Lopes, D., Mulhovo, S., Rosario, V. E., Prudêncio, M., et al. (2011a). Triterpenoids as inhibitors of erythrocytic and liver stages of Plasmodium infections. Bioorganic medicinal Chem. 19 (24), 7474–7481. doi: 10.1016/j.bmc.2011.10.044
Ramalhete, C., Gonçalves, B. M., Barbosa, F., Duarte, N., Ferreira, M. J. U. (2022). Momordica balsamina: phytochemistry and pharmacological potential of a gifted species. Phytochem. Rev. 21 (2), 617–646. doi: 10.1007/s11101-022-09802-7
Ramalhete, C., Lopes, D., Molnár, J., Mulhovo, S., Rosário, V. E., Ferreira, M. J. U. (2011b). Karavilagenin C derivatives as antimalarial. Bioorganic medicinal Chem. 19 (1), 330–338. doi: 10.1016/j.bmc.2010.11.015
Ramalhete, C., Lopes, D., Mulhovo, S., Molnar, J., Rosário, V. E., Ferreira, M. J. U. (2010). New antimalarial with a triterpenic scaffold from Momordica balsamina. Bioorganic medicinal Chem. 18 (14), 5254–5260. doi: 10.1016/j.bmc.2010.05.054
Ramalhete, C., Mansoor, T. A., Mulhovo, S., Molnár, J., Ferreira, M. J. U. (2009). Cucurbitane-type triterpenoids from the African plant Momordica balsamina. J. Natural products 72 (11), 2009–2013. doi: 10.1021/np900457u
Rathod, V., Behera, T. K., Munshi, A. D., Gaikwad, A. B., Singh, S., Vinay, N. D., et al. (2021). Developing partial interspecific hybrids of Momordica charantia× Momordica balsamina and their advance generations. Scientia Hortic. 281, 109985. doi: 10.1016/j.scienta.2021.109985
Renner, S. S., Schaefer, H. (2016). “Phylogeny and Evolution of the Cucurbitaceae,” in Genetics and Genomics of Cucurbitaceae. Plant Genetics and Genomics: Crops and Models, vol. 20 . Eds. Grumet, R., Katzir, N., Garcia-Mas, J. (Cham: Springer). doi: 10.1007/7397_2016_14
Robinson, R. W., Decker-Walters, D. S. (1997). “Interspecific hybridization,” in Cucurbits. Eds. Robinson, R., Decker-Walters, D. S., (Oxon, U.K: CAB Intl.) 51–55. doi: 10.1073/pnas.81.24.8014
Robinson, J. T., Turner, D., Durand, N. C., Thorvaldsdottir, H., Mesirov, J. P., Aiden, E. L. (2018). Juicebox. js provides a cloud-based visualization system for Hi-C data. Cell Syst. 6 (2), 256–258. doi: 10.1016/j.cels.2018.01.001
Saghai-Maroof, M. A., Jorgensen, R. A., Allard, R. W. (1984). Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location and population dynamics. Proc. Natl. Acad. Sci. U.S.A. 81, 8014–8018.
Sato, S., Tabata, S., Hirakawa, H., Asamizu, E., Shirasawa, K., Isobe, S., et al. (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485 (7400), 635–641. doi: 10.1038/nature11119
Schaefer, H., Heibl, C., Renner, S. S. (2009). Gourds afloat: a dated phylogeny reveals an Asian origin of the gourd family (Cucurbitaceae) and numerous oversea dispersal events. Proc. R. Soc. B: Biol. Sci. 276 (1658), 843–851. doi: 10.1098/rspb.2008.1447
Shang, Y., Ma, Y., Zhou, Y., Zhang, H., Duan, L., Chen, H., et al. (2014). Biosynthesis, regulation, and domestication of bitterness in cucumber. Science 346 (6213), 1084–1088. doi: 10.1126/science.1259215
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31 (19), 3210–3212. doi: 10.1093/bioinformatics/btv351
Singh, A. K. (1990). “Cytogenetics and evolution in the cucurbitaceae,” in Biology and Utilization of Cucurbitaceae. Eds. Bates, D. M., Robinson, R. W., Jeffrey, C. (Ithaca, New York, London: Comstock Publishing Associates, Cornell University Press), 10–28.
Singh, B. P. (1991). Interspecific hybridization in between new and old-world species of Luffa and its phylogenetic implication. Cytologia 56 (3), 359–365. doi: 10.1508/cytologia.56.359
Soderlund, C., Bomhoff, M., Nelson, W. M. (2011). SyMAP v3. 4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 39 (10), e68–e68. doi: 10.1093/nar/gkr123
Song, W., Zhou, F., Shan, C., Zhang, Q., Ning, M., Liu, X., et al. (2021). Identification of Glutathione S-Transferase Genes in Hami Melon (Cucumis melo var. saccharinus) and Their Expression Analysis under Cold Stress. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.672017
Sotowa, M., Ootsuka, K., Kobayashi, Y., Hao, Y., Tanaka, K., Ichitani, K., et al. (2013). Molecular relationships between Australian annual wild rice, Oryza meridionalis, and two related perennial forms. Rice 6, 1–19. doi: 10.1186/1939-8433-6-26
Tameling, W. I., Elzinga, S. D., Darmin, P. S., Vossen, J. H., Takken, F. L., Haring, M. A., et al. (2002). The tomato R gene products I-2 and MI-1 are functional ATP binding proteins with ATPase activity. Plant Cell 14 (11), 2929–2939. doi: 10.1105/tpc.005793
Tan, M., Ye, J., Turner, N., Hohnen-Behrens, C., Ke, C., Tang, C., et al. (2008). Antidiabetic activities of triterpenoids isolated from bitter melon associated with activation of the AMPK pathway. Chem. Biol. 15 (3), 263–273. doi: 10.1016/j.chembiol.2008.01.013
Tang, H., Krishnakuar, V., Li, J. (2015). jcvi: JCVI utility libraries. Zenodo. doi: 10.105281/zenodo31631
Thakur, G. S., Bag, M., Sanodiya, B. S., Bhadauriya, P., Debnath, M., Prasad, G. B. K. S., et al. (2009). Momordica balsamina: a medicinal and neutraceutical plant for health care management. Curr. Pharm. Biotechnol. 10 (7), 667–682. doi: 10.2174/138920109789542066
Thiel, T., Michalek, W., Varshney, R., Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Thomas, C. M., Jones, D. A., Parniske, M., Harrison, K., Balint-Kurti, P. J., Hatzixanthis, K., et al. (1997). Characterization of the tomato Cf-4 gene for resistance to Cladosporium fulvum identifies sequences that determine recognition specificity in Cf-4 and Cf-9. Plant Cell 9 (12), 2209–2224. doi: 10.1105/tpc.9.12.2209
Thompson, J. D., Higgins, D. G., Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 (22), 4673–4680. doi: 10.1093/nar/22.22.4673
Trebitsh, T., Staub, J. E., O’Neill, S. D. (1997). Identification of a 1-aminocyclopropane-1-carboxylic acid synthase gene linked to the female (F) locus that enhances female sex expression in cucumber. Plant Physiol. 113 (3), 987–995. doi: 10.1104/pp.113.3.987
TrimGalore (The Babraham Institute by @ FelixKrueger). Available at: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
Trivedi, R. N., Roy, R. P. (1972). Cytological studies in some species of Momordica. Genetica 43 (2), 282–291. doi: 10.1007/BF00123635
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res. 40 (15), e115–e115. doi: 10.1093/nar/gks596
Urasaki, N., Takagi, H., Natsume, S., Uemura, A., Taniai, N., Miyagi, N., et al. (2017). Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. DNA Res. 24 (1), 51–58. doi: 10.1093/dnares/dsw047
Usman, M. G., Rafii, M. Y., Ismail, M. R., Malek, M. A., Latif, M. A. (2015). Expression of target gene Hsp70 and membrane stability determine heat tolerance in chili pepper. J. Am. Soc Hortic. Sci. 140 (2), 144–150. doi: 10.21273/JASHS.140.2.144
Vaattovaara, A., Leppälä, J., Salojärvi, J., Wrzaczek, M. (2019). High-throughput sequencing data and the impact of plant gene annotation quality. J. Exp. Bot. 70 (4), 1069–1076.
Venkateswarlu, B., Shanker, A. K., Shanker, C., Maheswari, M. (2012). Crop stress and its management: perspectives and strategies (DORDRECHT, Netherlands: Springer Science & Business Media). doi: 10.1007/978-94-007-2220-0
Vergara, I. A., Chen, N. (2010). Large synteny blocks revealed between Caenorhabditis elegans and Caenorhabditis briggsae genomes using OrthoCluster. BMC Genomics 11 (1), 1–13. doi: 10.1186/1471-2164-11-516
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9 (11), e112963. doi: 10.1371/journal.pone.0112963
Wang, R., Jin, Q., Yao, C., Zhong, Y., Wu, T. (2019). RNA-Seq analysis of gynoecious and weak female cucumber revealing the cell cycle pathway may regulate sex determination in cucumber. Gene 687, 289–297. doi: 10.1016/j.gene.2018.11.071
Wang, X., Tang, H., Bowers, J. E., Paterson, A. H. (2009). Comparative inference of illegitimate recombination between rice and sorghum duplicated genes produced by polyploidization. Genome Res. 19 (6), 1026–1032. doi: 10.1101/gr.087288.108
Wang, J., Veldsman, W. P., Fang, X., Huang, Y., Xie, X., Lyu, A., et al. (2023). Benchmarking multi-platform sequencing technologies for human genome assembly. Briefings Bioinf. 24 (5), bbad300. doi: 10.1093/bib/bbad300
Weeden, N. F., Robinson, R. W. (1986). Allozyme segregation ratios in the interspecific cross Cucurbita maxima x C. Ecuadorensis suggest that hybrid breakdown is not caused by minor alterations in chromosome structure. Genetics 114 (2), 593–609. doi: 10.1093/genetics/114.2.593
Wehner, T. C., Naegele, R. P., Myers, J. R., Narinder, P. S., Crosby, K. (2020). Cucurbits. 2nd ed (Parlier, CA, USA: CABI). Available at: https://www.ars.usda.gov/research/publications/publication/?seqNo115=360003.
Weisenfeld, N. I., Kumar, V., Shah, P., Church, D. M., Jaffe, D. B. (2017). Direct determination of diploid genome sequences. Genome Res. 27 (5), 757–767.
Wu, S., Lau, K. H., Cao, Q., Hamilton, J. P., Sun, H., Zhou, C., et al. (2018). Genome sequences of two diploid wild relatives of cultivated sweet potato reveal targets for genetic improvement. Nat. Commun. 9 (1), 1–12. doi: 10.1038/s41467-018-06983-8
Wu, X., Li, J., Liu, Z., Yin, J., Chang, Y., Rong, C., et al. (2015). The Arabidopsis ceramidase AtACER functions in disease resistance and salt tolerance. Plant J. 81 (5), 767–780. doi: 10.1111/tpj.12769
Wu, S., Shamimuzzaman, M. D., Sun, H., Salse, J., Sui, X., Wilder, A., et al. (2017). The bottle gourd genome provides insights into Cucurbitaceae evolution and facilitates mapping of a Papaya ring-spot virus resistance locus. Plant J. 92 (5), 963–975. doi: 10.1111/tpj.13722
Wu, H., Zhao, G., Gong, H., Li, J., Luo, C., He, X., et al. (2020). A high-quality sponge gourd (Luffa cylindrica) genome. Horticulture Res. 7 (1), 128. doi: 10.1038/s41438-020-00350-9
Yandell, M., Ence, D. (2012). A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 13 (5), 329–342. doi: 10.1038/nrg3174
Yin, T., Quinn, J. A. (1995). Tests of a mechanistic model of one hormone regulating both sexes in Cucumis sativus (Cucurbitaceae). Am. J. Bot. 82 (12), 1537–1546. doi: 10.1002/j.1537-2197.1995.tb13856.x
Zhao, Q., Chen, W., Bian, J., Xie, H., Li, Y., Xu, C., et al. (2018). Proteomics and phosphoproteomics of heat stress-responsive mechanisms in spinach. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00800
Zhao, C., Qiu, J., Agarwal, G., Wang, J., Ren, X., Xia, H., et al. (2017). Genome-Wide Discovery of Microsatellite Markers from Diploid Progenitor Species, Arachis duranensis and A. ipaensis, and Their Application in Cultivated Peanut (A. hypogaea). Front. Plant Science. 8. doi: 10.3389/fpls.2017.01209
Zheng, Y., Chen, B., Zhi, C., Qiao, L., Liu, C., Pan, Y., et al. (2021). Genome-wide identification of small heat shock protein (HSP20) homologs in three cucurbit species and the expression profiles of CsHSP20s under several abiotic stresses. Int. J. Biol. Macromolecules 190, 827–836. doi: 10.1016/j.ijbiomac.2021.08.222
Keywords: cucurbits, genome assembly, genomic resources, medicinal value, Momordica balsamina, stress tolerance
Citation: Vinay ND, Singh K, Ellur RK, Chinnusamy V, Jaiswal S, Iquebal MA, Munshi AD, Matsumura H, Boopalakrishnan G, Jat GS, Kole C, Gaikwad AB, Kumar D, Dey SS and Behera TK (2024) High-quality Momordica balsamina genome elucidates its potential use in improving stress resilience and therapeutic properties of bitter gourd. Front. Plant Sci. 14:1258042. doi: 10.3389/fpls.2023.1258042
Received: 13 July 2023; Accepted: 29 December 2023;
Published: 24 January 2024.
Edited by:
Manohar Chakrabarti, The University of Texas Rio Grande Valley, United StatesReviewed by:
Aamir W. Khan, University of Missouri, United StatesDinakaran Elango, Iowa State University, United States
Copyright © 2024 Vinay, Singh, Ellur, Chinnusamy, Jaiswal, Iquebal, Munshi, Matsumura, Boopalakrishnan, Jat, Kole, Gaikwad, Kumar, Dey and Behera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tusar Kanti Behera, dHVzYXJAcmVkaWZmbWFpbC5jb20=