 Deepak Kumar Jha
Deepak Kumar Jha Subhadarshini Parida
Subhadarshini Parida Seema Pradhan
Seema Pradhan Nrisingha Dey
Nrisingha Dey Shuvobrata Majumder
Shuvobrata Majumder- 1Department of Plant & Microbial Biotechnology, BRIC-Institute of Life Sciences (ILS), NALCO Square, Bhubaneswar, Odisha, India
- 2BRIC-Regional Centre for Biotechnology, Faridabad, India
Tossa jute (Corchorus olitorius) dominates global jute cultivation but has a high lignin content (13-14%) in its fibres, making them coarse and limiting their industrial applications. Reducing the lignin content requires a deeper understanding of the lignification process and the associated genes. Laccase (EC 1.10.3.2) is a key enzyme in the final step of lignin biosynthesis. A genome-wide analysis of the 361 Mb C. olitorius genome identified 46 laccase genes (ColLACs) from a total of 28,479 genes. In-silico analysis revealed that ColLAC genes are distributed across seven chromosomes, encode proteins ranging from 7.98 to 173.99 kDa, with 74 to 1548 amino acids and 10 conserved motifs. Additionally, 48.83% of ColLACs are predicted to be transmembrane proteins. Phylogenetic analysis classified them into eight groups, with GO term assignments suggesting their involvement in lignification. Tissue-specific expression analysis showed that 43.47% of ColLAC genes are predominantly expressed in roots, aligning with RNA-seq data. ColLAC gene expression varied across the developmental stages, from seedling to fibre harvest, and was influenced by heavy metal copper and abscisic acid (ABA) treatments. This variation correlated with upstream cis-acting elements. Ath-miR397 target sites were identified in 14 ColLAC genes, indicating potential post-transcriptional regulation. Further expression analysis in X-ray-induced bfs (bast fibre-shy) mutant tossa jute lines suggested that ColLAC34 is involved in both lignification and structural development, while ColLAC22, ColLAC40, and ColLAC46 play key roles in lignification. This study presents the first comprehensive genome-wide identification and characterization of the LAC gene family in jute. Understanding ColLAC functions could facilitate the development of low-lignin jute fibres, meeting the growing industrial demand for high-quality natural fibres of jute.
1 Introduction
Lignocellulosic jute (Corchorus sp.) fibres have multiple commercial applications industrially and domestically in paper, packaging, and fabrics, producing a variety of sustainable products. The fibres are bast or stem fibres, specifically the phloem tissue, extracted at around 120 days of plant age. Jute plants, valued as the most significant among stem fibre crops in the world, create a soaring global demand for their long, lustrous, strong, biodegradable/organic golden fibres. The only limitation of these fibres is their high lignin content, which makes them rough, rigid, and prone to discoloration upon storage. This makes them unsuitable for applications where softness and smoothness are priorities, and low lignin bast fibres such as flax (Linum usitatissimum) or lignin-free flower fibres such as cotton (Gossypium herbaceum) are preferred.
Commercially there are only two cultivated jute species and both of them are lignin rich. Lignin content in the fibres of white jute (Corchorus capsularis) was found to be 12-13% and that of tossa jute (C. olitorius) 13-14% (Majumder et al., 2020a). Economically, there is a persistent and heavy demand for low lignin jute fibres, which necessitates low-lignin fibre production. This can only be achieved by acquiring detailed knowledge of the lignification process and the associated genes in the jute plant system.
Lignin is a complex aromatic polymer that provides structural support, facilitates water and nutrient transport, and contributes to plant defence mechanisms (Dixon and Barros, 2019). It is primarily composed of three monolignol-derived subunits: p-hydroxyphenyl (H) from p-coumaryl alcohol, guaiacyl (G) from coniferyl alcohol, and syringyl (S) from sinapyl alcohol (Boerjan et al., 2003). The lignin biosynthesis pathway is intricate and partially unresolved, with certain enzymatic steps yet to be fully characterized (Goujon et al., 2003). In white jute, the expression of key lignin biosynthesis genes, including PAL1 (phenylalanine ammonia-lyase; EC 4.3.1.24), C4H1 (cinnamate 4-hydroxylase; EC 1.14.13.11), 4CL1 (4-coumarate:CoA ligase; EC 6.2.1.12), CCoAoMT2 (caffeoyl-CoA O-methyltransferase; EC 2.1.1.104), CCR1 (cinnamoyl-CoA reductase; EC 1.2.1.44), F5H1 (ferulate 5-hydroxylase; EC 1.14.-.-), COMT1 (caffeic acid O-methyltransferase; EC 2.1.1.68), and CAD7 (cinnamyl alcohol dehydrogenase; EC 1.1.1.195), has been reported by Chakraborty et al. (2015).
In our previous qRT-PCR-based gene expression study, we re-examined the expression of eight key lignin biosynthesis genes—PAL1, C4H1, 4CL1, CCoAoMT2, CCR1, F5H1, COMT1, and CAD7—in jute phloem tissue at different developmental stages. The analysis spanned from 30 days after sowing (DAS), when lignification began, to fibre harvest at 120 DAS, using ubiquitin1 and beta-tubulin as internal control genes (Parida et al., 2024b). However, information on several crucial genes in the jute lignin pathway, such as peroxidase (PRX) and laccase (LAC), remains unexplored. Among the lignin biosynthesis pathway genes, a systematic gene family analysis has only been conducted on CCoAoMT2 (Akhter et al., 2022). This study presents the first comprehensive genome-wide identification and characterization of the LAC gene family in tossa jute (C. olitorius).
LAC is intrinsic to the final stage of lignin polymerization (Boerjan et al., 2003). The LAC gene products comprise a heterogeneous collection of multicopper oxidases capable of facilitating one electron oxidations (Berthet et al., 2012). Multiple investigations have suggested the potential function of LAC in lignin polymerisation (Sterjiades et al., 1993). Recently, our group conducted a genome-wide study on this enzyme family in the white jute system and reported the active presence of 34 CcaLAC genes (Parida et al., 2024a). Investigations made in the past have reported many LAC genes in Arabidopsis thaliana (17) (McCaig et al., 2005), Populus trichocarpa (49) (Lu et al., 2013), Gossypium hirsutum (84) (Balasubramanian et al., 2016) Linum usitatissimum (45) (Le Roy et al., 2017), Punica granatum (57) (Shi et al., 2023), Glycine max (93) (Wang et al., 2019) and Eucalyptus grandis (54) (Arcuri et al., 2020) along with their involvement in the lignification process of the respective plants. Knockout experiments of LAC in Arabidopsis (Berthet et al., 2011) and cotton (Yang et al., 2024) have confirmed their involvement in lignification, fibre development, and stem structure.
To understand the functionality of LAC genes in jute plant development and fibre lignification, detailed analysis of these genes is necessary, and knockout lines could serve as valuable models for such studies. While knockout lines are available for all 17 LAC genes in Arabidopsis, similar resources were not available for jute. Creating knockout lines in jute is challenging without first identifying the target genes. As an alternative, X-ray-irradiated mutant jute could help identify key LAC genes that are directly involved in growth and development. One such mutant jute variety is the bfs (bast fibre-shy) tossa, which lacks the ability of differentially developing secondary phloem fibres (SPF) and secondary xylem (wood), but can still develop fibre with some lignin content (Kundu et al., 2012). It matures earlier, produces significantly fewer bast fibres and wood, and yields lower-quality fibres compared to its wild-type control, JRO632 (Kundu et al., 2012). In this study, we used the bfs mutant and its control, JRO632, to examine the expression of different LAC genes of C. olitorius (ColLACs).
The expression of LAC genes varies across different plant tissues, as documented in various studies. In Arabidopsis and white jute (C. capsularis), most LAC genes are predominantly expressed in stem tissue rather than in other tissues like leaves, and roots (Parida et al., 2024a). However, in the case of rice (Oryza sativa) during its vegetative growth stage and in Eucalyptus, LAC expression was mostly observed in roots (Liu et al., 2017; Arcuri et al., 2020). We used leaves, stem, and root tissues of tossa jute, and specifically examined phloem tissue at different time points in its development, from plantlets (30 DAS) to its fibre harvest stage (120 DAS), for LAC gene expression analysis.
Gene expression in plants is regulated by both intrinsic cellular mechanisms and external environmental factors. Pre- and post-transcriptional regulations are key processes that control gene expression. Pre-transcriptional regulation involves upstream cis-acting elements, which play a crucial role in gene expression under environmental stresses, developmental stages, hormonal signalling, and normal plant growth. Various cis-elements, such as MYB, MYC, ABRE, ERE, STRE, TGA, GT1, Sp1, GARE-motif, WUN-motif, GATA-motif, TCT-motif, P-box, W-box, G-box, Box-4, RY-element, and LAMP-element, have been identified as regulators of gene expression in plants (Ain-Ali et al., 2021; Jha et al., 2024). In-silico analysis can be used to identify the presence of these cis-elements in the promoter region of a gene, providing insights into its potential expression patterns and functional roles. However, no such study has yet been conducted on the LAC gene family in jute.
Post-transcriptional regulation in plants is predominantly controlled by miRNAs, which are small (21-nucleotide), non-coding RNAs that negatively regulate gene expression by targeting specific mRNAs for degradation or translational inhibition (Fang and Wang, 2021). Advanced in-silico analysis can predict whether a given mRNA is a potential target of miRNA-mediated post-transcriptional regulation. The miR397 family is well known for targeting LAC genes, thereby modulating lignin biosynthesis pathways in plants (Kozomara and Griffiths-Jones, 2011). However, in jute, the specific miRNAs involved in lignin pathway regulation and their role in targeting LAC genes remain to be explored. Understanding these regulatory mechanisms in jute will provide valuable insights into lignin biosynthesis and its potential genetic manipulation for fibre quality improvement.
Laccase (LAC) genes are known to respond to environmental stresses by increasing or decreasing their expression. They react to abiotic factors like heavy metals, drought, salinity, temperature changes, and oxidative stress, as well as biotic factors such as pathogens, insects, and fungi (Bai et al., 2023). However, LAC gene expression patterns in jute, under these stress conditions, remain unknown.
Our aim is to discover the gene family of ColLAC in C. olitorius and evaluate gene expression in different parts of the plant at various developmental stages. We seek to open new frontiers in identifying the LAC genes and their functionality in bast fibre production for subsequent functional investigations.
2 Materials and methods
2.1 The tossa jute genome sequence
For the in-silico analysis, the tossa jute genome sequences were downloaded from the Genome Warehouse (https://ngdc.cncb.ac.cn/gwh/Assembly/20707/show, accessed on December 12, 2023) following the method described by Zhang et al. (2021). The downloaded file contained DNA, mRNA (CDS), and protein sequences.
2.2 Plant material
Tossa jute cultivars JRO524, JRO632, and the bfs mutant were grown under greenhouse conditions (30 ± 2°C, 80% humidity, with a 13-hour light/11-hour dark cycle). Leaf, stem, and root tissues were collected from JRO632 plants at 60 DAS. Phloem tissue was sampled from JRO524, JRO632, and bfs plants at 30, 60, 90, and 120 DAS to assess ColLAC gene expression across different developmental stages. Stem samples from JRO524 plants subjected to abiotic stress at 60 DAS were also collected. All tissue samples were stored at -80°C until RNA isolation.
The bfs mutant displays a distinct phenotype characterised by stunted growth (Figure 1A), trilobed, split ribbon-like leaves (Figure 1B), and a spreading root architecture (Figure 1C). These traits make the bfs mutant easily distinguishable from the control cultivar, JRO632.

Figure 1. Tossa jute (Corchorus olitorius) cultivars JRO632 (control) and bfs (X-ray-irradiated mutant) used in the ColLAC gene expression study. Comparative images showing differences in (A) plant height, (B) leaf morphology, and (C) root architecture between the control and mutant cultivars.
2.3 Abiotic stress treatments
Abscisic acid (ABA) stress (0.15 mM) and copper heavy metal stress (0.10 mM CuSO4·H2O) were applied to healthy, 60-day-old JRO524 plants grown under greenhouse conditions, following the method described by Parida et al. (2024b). Stem tissue samples were harvested at various intervals (0 h, 2 h, 4 h, 8 h, 12 h, and 24 h). For each time point, three separate plants were randomly selected and used as biological replicates in the experiments.
2.4 Identification and sequence analysis of LAC genes in the tossa jute genome
LAC protein sequences of A. thaliana were retrieved from TAIR (Lamesch et al., 2012), Oryza sativa sequences were obtained from Oryzabase (Kurata and Yamazaki, 2006), and LAC sequences of Theobroma cacao were retrieved from the Phytozome database (Goodstein et al., 2012) to create a Hidden Markov Model (HMM) profile. A total of 99 LAC gene sequences were used to prepare the HMM profile using the HMMER tool (version 3.2) (Finn et al., 2011). In the HMMER tool, the E-value threshold was set to <1e-5, and the alignment parameters were kept at default settings.
The presence of specific domains (PF00394.19, PF07731.11, and PF07732.12) associated with the LAC protein family in the proposed ColLAC sequences was confirmed utilising NCBI CDD (Marchler-Bauer et al., 2015), SMART (Letunic et al., 2021), or HMM Scan (Finn et al., 2011). All sequence hits with redundancy cut-off values beyond 0.01 were omitted from the analysis, resulting in the inclusion of solely non-redundant hits (Jha et al., 2024). An examination of the physical and chemical properties of the ColLAC proteins was performed utilising the ProtParam program on the Expasy website (Wilkins et al., 1999). The transmembrane properties of the ColLAC proteins were assessed utilising the TMHMM-2.0 tool (Krogh et al., 2001).
2.5 Chromosomal mapping and phylogeny construction
The ColLAC genes were physically mapped using the MapInspect program version 1.0, based on their genomic coordinates (https://mapinspect.software.informer.com, accessed on December 10, 2023).
The LAC protein sequences from Arabidopsis (Arabidopsis thaliana), white jute (C. capsularis), and tossa jute (C. olitorius) were selected for phylogenetic analysis. Sequence alignment was performed using the MUSCLE algorithm (Edgar, 2004). Phylogenetic trees were constructed using the maximum likelihood method, employing MEGA software (version 10.0; Kumar et al., 2018). To ensure robustness, the analysis included 1000 bootstrap replications, applying gap deletions and a gamma (G) distribution rate for site variations (Jha et al., 2024).
2.6 Motif and gene structure determination
The conserved motifs in ColLAC proteins were examined using the MEME suite tool (Bailey et al., 2009). For motif prediction, the analysis was set to identify up to 10 motifs, with the motif width constrained between 6 and 50 amino acids (Chanwala et al., 2023). The Gene Structure Display Server (GSDS) program was used to determine the exon-intron structure of each ColLAC gene (Hu et al., 2015).
2.7 Gene duplication and synteny analysis
The analysis also included identifying mutations and duplication events in the ColLAC gene family. Gene duplication patterns were examined by performing a BLASTp search of ColLAC proteins against LAC proteins from Arabidopsis, rice, cacao (T. cacao), and white jute, using a cutoff e-value of 0.01.
For this analysis, Arabidopsis was chosen as a model plant, rice as a representative monocot with distinct traits from jute, and cacao due to its high genomic similarity to jute, as reported by Sarkar et al. (2017) and Zhang et al. (2021). Sequences with ≥80% similarity were considered for identifying gene duplications (Jha et al., 2021).
To assess evolutionary divergence, synonymous (Ks) and nonsynonymous (Ka) substitution rates were calculated using the PAL2NAL program (Suyama et al., 2006). The synteny relationships were analysed and visualized using TBtools software (Chen et al., 2020).
2.8 Cis-element analysis
The upstream regions (~2000 bp) of the ColLAC genes were extracted from the C. olitorius genome to analyse potential regulatory elements. The PlantCARE database (Rombauts et al., 1999) was used to identify these cis-regulatory elements. The primary focus of this analysis was to identify elements involved in responses to both biotic and abiotic stress, as well as those related to processes like cell cycle regulation, circadian rhythms, development, metabolism, and hormone-mediated signalling pathways.
2.9 Gene ontology term annotation
To investigate the functional roles of the ColLAC gene family, Gene Ontology (GO) terms were assigned. Protein sequences of ColLAC were analysed using BLASTp against the UniProt-SwissProt database (https://www.uniprot.org/uniprotkb?query=reviewed:true, accessed on January 14, 2025) to identify homologous proteins. The corresponding UniProt IDs were then submitted to the Gene Ontology (GO) resource (https://geneontology.org/, accessed on January 14, 2025) for GO term annotation, using AtLAC as a reference for functional categorization.
2.10 In-silico expression analysis of ColLAC genes
The coding sequences (CDS) of the 46 identified LAC genes from the C. olitorius genome-wide analysis were used as reference sequences for mapping short reads from jute tissue samples. Raw RNA-seq reads from leaves, roots, xylem (stem stick), and phloem (stem bast) tissues of C. olitorius were retrieved from the NCBI SRA database (BioProject PRJNA520880). For transcript quantification, the align_and_estimate_abundance.pl script was employed, and a gene expression matrix was generated using abundance_estimates_to_matrix.pl from the Trinity RNAseq suite (https://github.com/trinityrnaseq/trinityrnaseq; accessed on July 30, 2024). The read counts were TMM-normalised, log2-transformed, and visualised as a heatmap using MeV software (https://sourceforge.net/projects/mev-tm4/; accessed on July 30, 2024).
2.11 Identification of miRNA targets in ColLAC
The complete micro-RNA sequences of A. thaliana were acquired from PmiREN2.0 (Guo et al., 2022). The ColLAC sequences and the Arabidopsis miRNA sequences were subsequently submitted to psRNATarget to predict the target sites on the identified ColLAC genes (Dai and Zhao, 2011).
2.12 RNA isolation and cDNA synthesis
RNA was extracted from leaf, stem and root tissues using a commercially available RNA isolation kit (Macherey-Nagel, Germany). Following the manufacturer’s methodology, a tissue sample weighing 100 mg was pulverised into a fine powder using liquid nitrogen and a mortar-pestle. In order to eradicate any DNA contamination, a DNase treatment procedure was carried out in accordance with the instructions provided by the kit. The integrity of the RNA was evaluated by the utilisation of 2% agarose gel electrophoresis, while the RNA concentration was determined utilising a spectrophotometer (NanoDrop-2000c by ThermoScientific, USA). The synthesis of first-strand cDNA was performed using 1 μg of RNA and a first-strand cDNA synthesis kit (ThermoScientific, USA) according to the kit specified methodology.
2.13 PCR primers
The primers for this investigation were constructed using the PrimerQuest tool supplied by Integrated DNA Technologies (IDT), USA. The tool can be accessed at https://www.idtdna.com/PrimerQuest. The qRT-PCR primer design utilised default settings, which specified a melting temperature range of 59–62°C, a GC content range of 35–65%, a primer length range of 17–30 bp, and an amplicon size range of 75–150 bp. The comprehensive inventory of primers can be seen in Supplementary Table 1.
2.14 Gene expression analysis in qRT-PCR
The qRT-PCR analysis was conducted using the QuantStudio™ 5 machine (ThermoScientific, USA) which was equipped with QuantStudio™ Design and Analysis Software version 1.5.2. The recently produced complementary DNA (cDNA) was mixed with molecular-grade water in a 1:9 ratio before being utilised as a sample in qRT-PCR. The PCR cycle conditions used in the experiment were as follows: an initial denaturation step at 95°C for 2 minutes, followed by 40 cycles of denaturation at 95°C for 15 seconds, annealing at the temperature specific to the primer (as indicated in Supplementary Table 1) for 1 minute, and extension at 95°C for 15 seconds. The temperature range used to obtain the melt curve of the amplified PCR products was from 60°C to 95°C. The jute ubiquitin1 (UBI) gene, with the GenBank number GH985256, used as an internal control (Parida et al., 2024b). The expression levels of the mRNA transcript were determined and depicted using relative quantification (RQ) values, following the approach outlined by Livak and Schmittgen (2001). Three technical replicates were conducted for each biological replication in the qRT-PCR study, with a total of three biological replicates.
2.15 ColLACs gene expression analysis
Seven Arabidopsis genes implicated in the lignification process were selected based on prior research (Berthet et al., 2012). The genes include AtLAC2 (AT2G29130), AtLAC4 (AT2G38080), AtLAC5 (AT2G40370), AtLAC10 (AT5G01190), AtLAC11 (AT5G03260), AtLAC12 (AT5G05390), and AtLAC17 (AT5G60020). The AtLAC genes were utilised as queries in a BLAST search to identify their homologous genes in the tossa jute genome using TBtools software (Chen et al., 2020). Genes categorised as ColLAC, demonstrating over 60% similarity with AtLAC sequences, were considered homologous and selected for subsequent expression analysis via qRT-PCR (Jha et al., 2024). We analysed the expression of homologous ColLAC genes in the phloem tissue of white jute plants at different developmental stages (30 DAS, 60 DAS, 90 DAS, and 120 DAS) to explore the lignification process.
Homologs of the Arabidopsis laccase genes involved in the lignin pathway—ColLAC3, ColLAC22, ColLAC30, ColLAC32, ColLAC34, ColLAC38, ColLAC40, ColLAC42, and ColLAC46—were also examined in the phloem tissue of the tossa jute mutant bfs and its control, JRO632, at different developmental stages up to 120 DAS.
2.16 Statistical analysis
The average Ct values were calculated using Microsoft Excel. For detailed statistical analysis, GraphPad Prism (version 9.0) software (https://www.graphpad.com/features) was used. A one-way analysis of variance (ANOVA, F-test) was performed to evaluate the significance of ColLACs expression differences across different tissue samples (leaf, stem and root) and time points (30 DAS, 60 DAS, 90 DAS and 120 DAS), with a significance threshold of P ≤ 0.05. Prior to ANOVA, the data were tested for normality and homogeneity of variances to ensure compliance with statistical assumptions. Tukey’s post hoc test was then applied for multiple comparisons among the groups, specifically to compare ColLACs expression between JRO632 and bfs. Significant differences are indicated by asterisks, while non-significant values are denoted as ‘ns’. The expression data for ColLACs are presented as the mean of three biological replicates.
3 Results
3.1 Number of tossa jute laccase (ColLACs)
There were 46 laccase proteins found in the genome of tossa jute (C. olitorius), and they were called ColLAC1 to ColLAC46 according to their chromosomal locations (Table 1). The LAC genes that were discovered exhibited an inconsistent distribution throughout the seven chromosomes, as shown in Figure 2. Chromosome 1 has the most number of genes, namely 10 LAC genes. Chromosome 3 with 9 genes, chromosome 7 with 8 genes, chromosome 2 with 7 genes, and three LAC genes were detected on chromosomes 4, 5, and 6, respectively. Three LAC members ColLAC44, ColLAC45, and ColLAC46 were located on the scaffold.
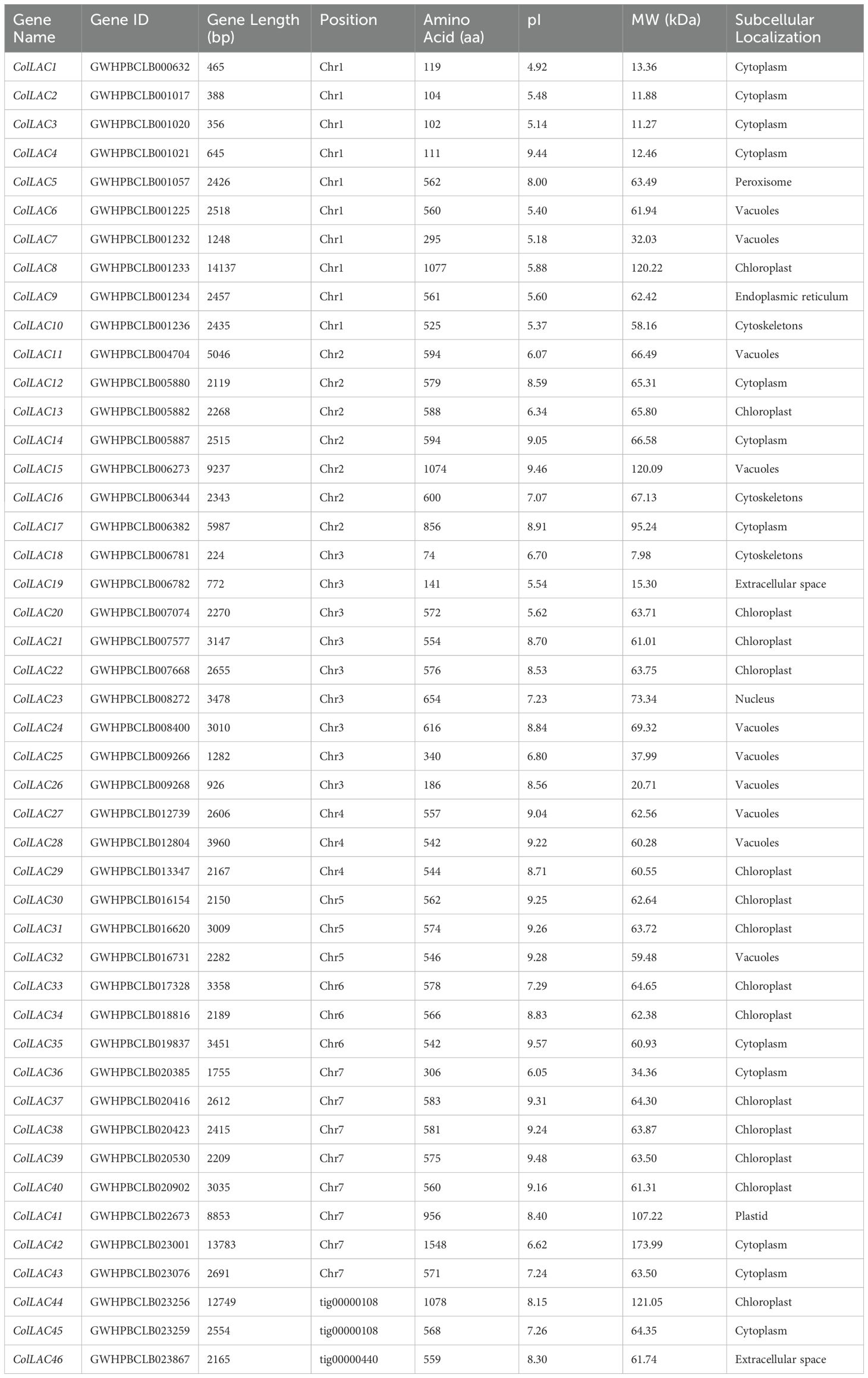
Table 1. Bioinformatics analysis and physicochemical properties of tossa jute laccases (ColLACs).

Figure 2. Chromosomal distribution of 43 laccase (ColLAC) genes in the genome of the tossa jute plant (C. olitorius). The remaining three genes, ColLAC44 and ColLAC45, were mapped to contig 000000108, and ColLAC46 was mapped to contig 000000440.
3.2 Laccase proteins
The length of these ColLAC proteins varied, with the smallest protein (ColLAC18) having a length of 7.98 kDa and the largest protein (ColLAC42) having a length of 173.99 kDa. The ColLAC proteins exhibited a maximum of 1548 amino acids, a minimum of 74 amino acids, and an average of around 554 amino acids (Table 1). The instability index of the discovered LAC proteins was projected to be less than 40, except for ColLAC1 (45.18), ColLAC2 (41.63), ColLAC4 (52.92), ColLAC19 (41.84), ColLAC42 (41.14), and ColLAC43 (43.48) (Supplementary Table 2). This suggests that most of the proteins are stable. Furthermore, the aliphatic amino acid index of ColLAC1 was the lowest at 72.02, whereas ColLAC4 had the highest value at 111.35. The overall mean hydropathy (GRAVY) score for all the ColLACs was negative (<0), demonstrating their hydrophilic characteristic (Kyte and Doolittle, 1982). The ColLAC proteins that were discovered were predicted to be located in different organelles of white jute, as shown in Supplementary Table 2. Our analysis revealed that 48.83% (21 out of 43) of ColLACs were identified as transmembrane proteins (Supplementary Figure 1).
3.3 Motif of laccase proteins
The found laccase proteins were anticipated to include 10 patterns, labelled motif-1 through motif-10 (Figure 3). ColLAC4, ColLAC18, ColLAC25, and ColLAC26 exhibited single motifs, but ColLAC1, ColLAC3, and ColLAC19 had double motifs. Moreover, the laccase proteins ColLAC8, ColLAC15, and ColLAC44 exhibited repetitions of certain patterns.
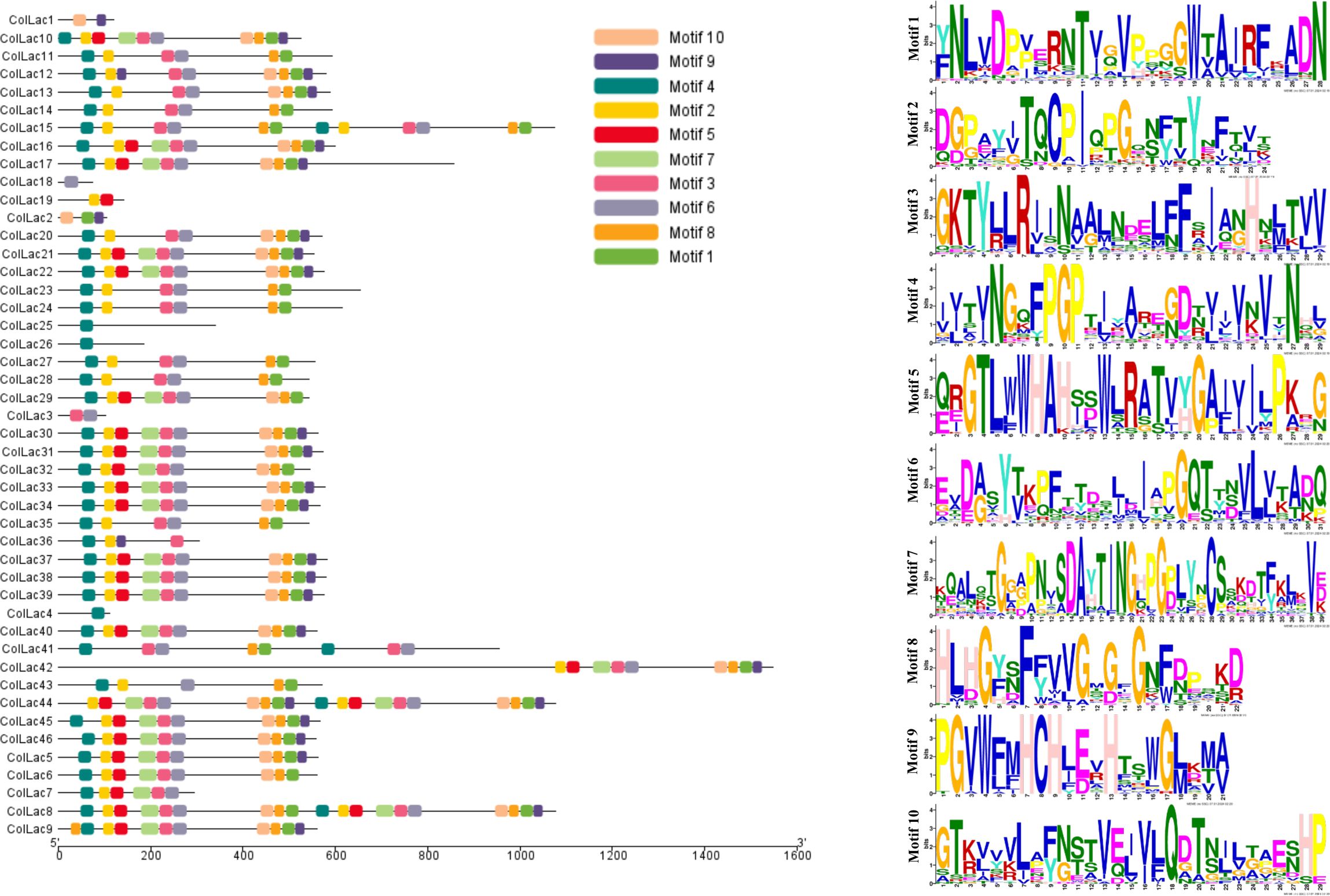
Figure 3. Distribution of conserved motifs in identified ColLAC proteins. Motifs are represented by distinct hues and their corresponding sequences.
3.4 Gene structure of ColLACs
The found ColLAC genes exhibited significant variability in exon-intron distribution, with exon counts ranging from 2 to 26. Only one exon was identified in the ColLAC18 gene, while the ColLAC42 gene had the greatest prediction of 26 exons (Figure 4).
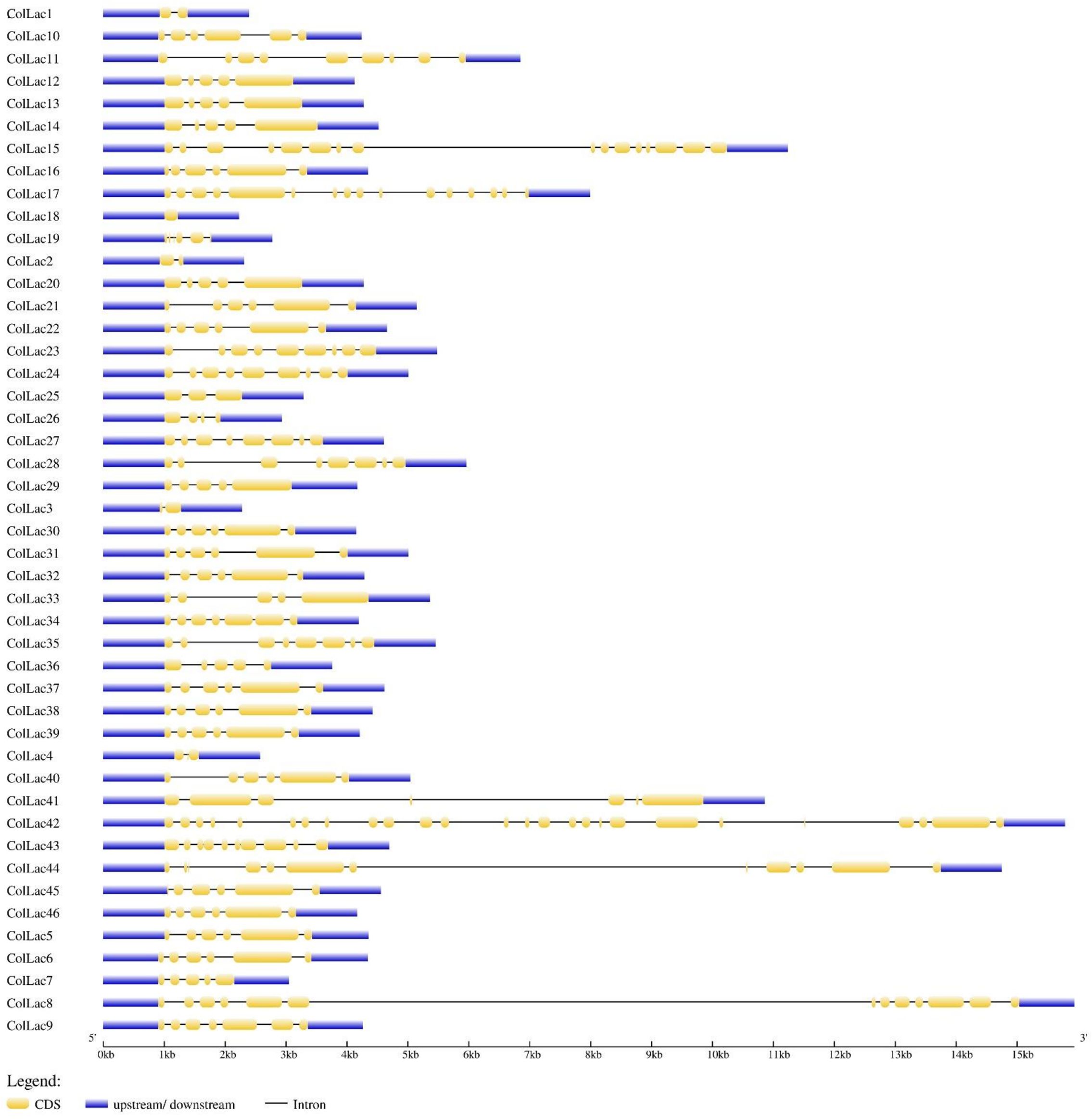
Figure 4. Gene architecture of 46 white jute laccase genes (ColLACs).
3.5 Phylogeny of ColLACs proteins
The phylogenetic analysis divided the identified laccase proteins of tossa jute into eight groups, designated as Group I to Group VIII (Figure 5). The majority of laccase proteins were clustered in Group IV, which contained 18 members. Other groups, such as Group V, included 11 laccase proteins, while Group I and Group III each had 5 members. Groups II, VII, and VIII each contained 2 members, and only 1 member was found in Group VI.
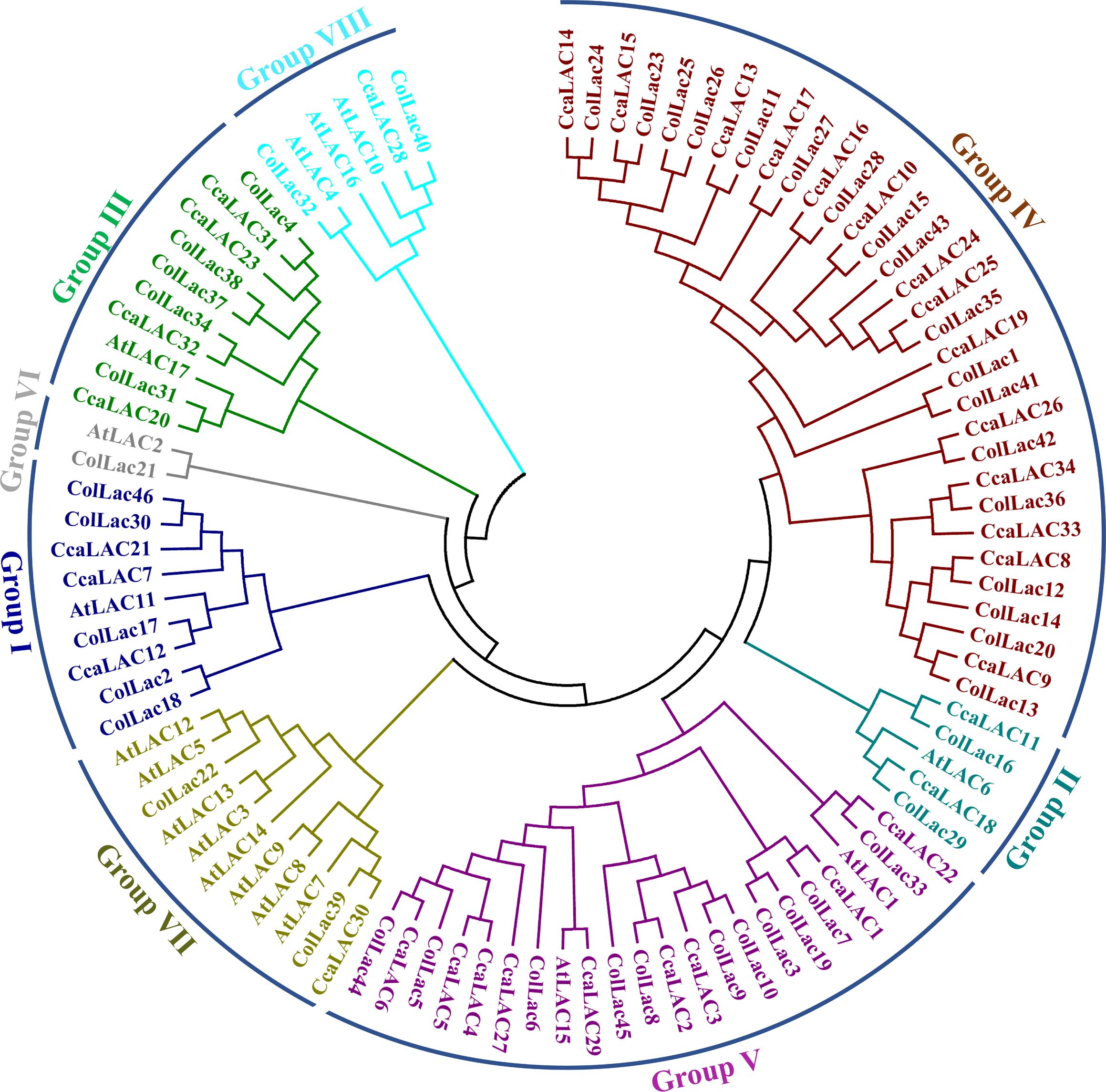
Figure 5. Phylogenetic tree of jute laccase proteins alongside laccase proteins from Arabidopsis (A. thaliana), white jute (C. capsularis), and tossa jute (C. olitorius). The phylogeny categorised the detected LAC members into eight categories.
3.6 Duplication of ColLAC genes
Gene duplication analysis revealed a total of 13 paralogous pairs, including 2 tandem duplications and 11 segmental duplications (Supplementary Table 3). Orthologous pairs were also identified: 5 pairs between C. capsularis and A. thaliana, 3 between C. capsularis and O. sativa, 26 between C. capsularis and T. cacao, and 55 pairs between C. olitorius and C. capsularis (Figure 6). Among species comparisons, the largest number of LAC orthologous pairs was found between C. olitorius and T. cacao, likely due to their close genomic relationship, as suggested by Sarkar et al. (2017).
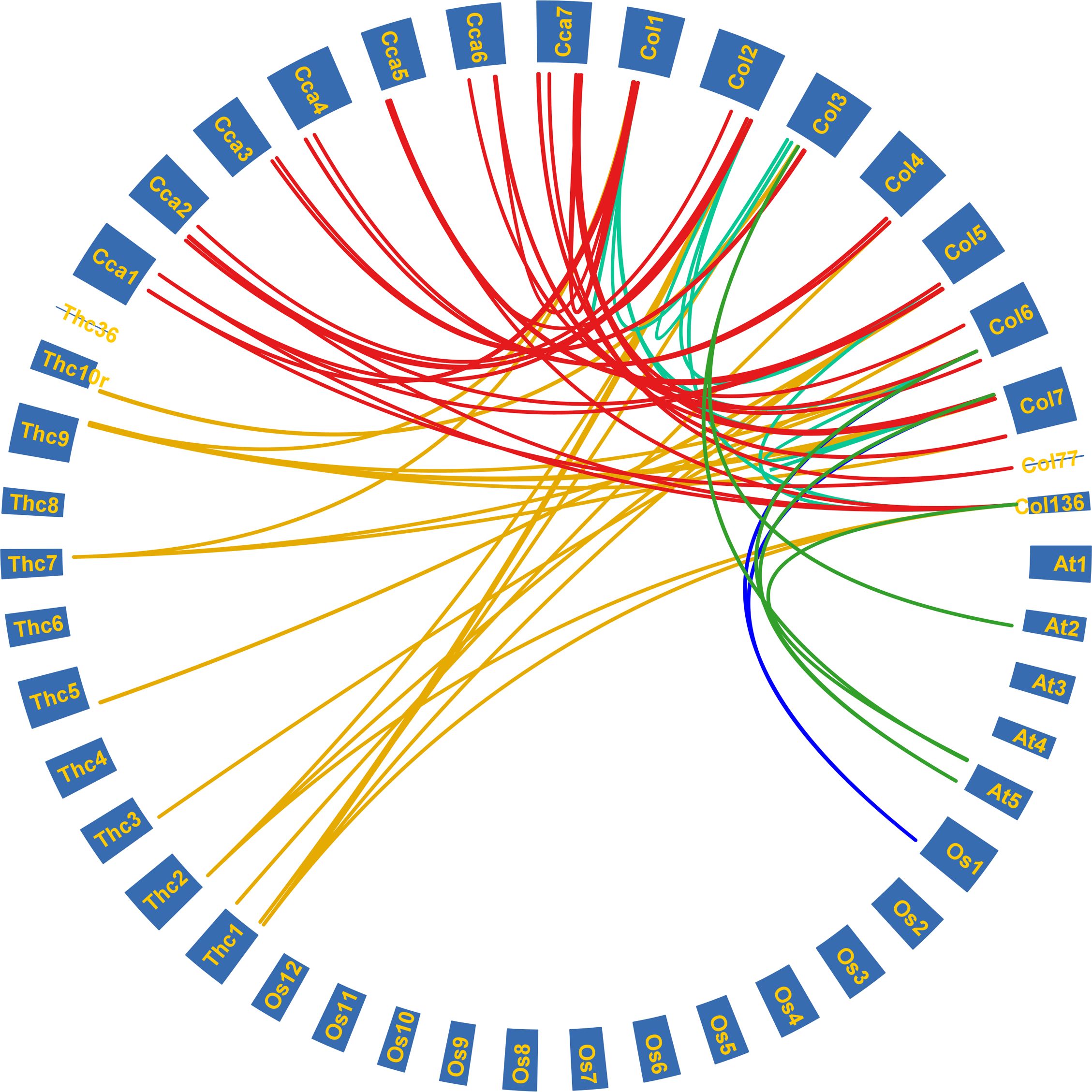
Figure 6. Syntenic relationships among the LAC genes of tossa jute (C. olitorius), Arabidopsis (A. thaliana), rice (O. sativa), cacao beans (T. cacao), and white jute (C. capsularis). Each coloured line signifies duplicated gene pairs across the species.
3.7 Cis-acting elements
The upstream regions of ColLAC genes were found to contain various predicted cis-elements (Figure 7). Among the 46 ColLAC members, MYB, MYC, and TATA-box elements were consistently identified. Additionally, a variety of cis-elements involved in different biological processes were detected (Ain-Ali et al., 2021; Jha et al., 2024). These included elements linked to stress responses (such as STRE, W-box, MYB, MYC, and WUN-motif), hormonal signalling (including ABRE, ERE, GARE-motif, P-box, and TGA element), and developmental regulation (such as G-box, GT1, RY-element, Sp1, GATA-motif, LAMP element, Box-4, and TCT-motif). These results suggest that the promoter regions of ColLAC genes may contribute to multiple biological functions.
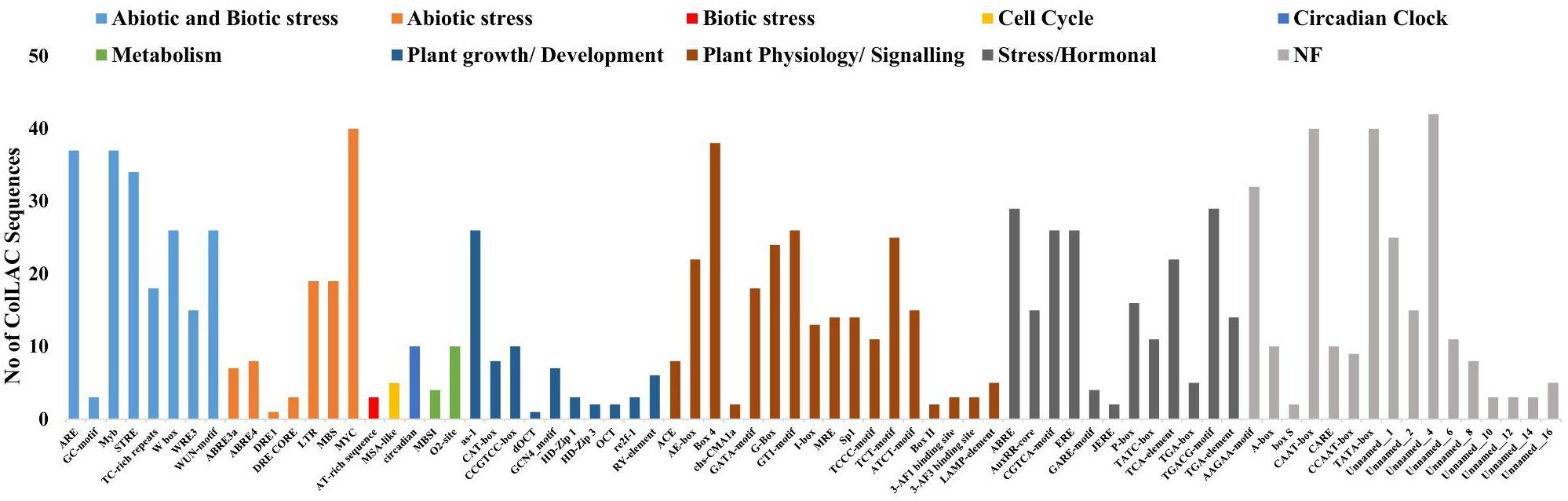
Figure 7. Cis-acting elements in the upstream regions of ColLACs.
Upstream regions of multiple ColLAC genes contain cis-acting elements associated with secondary wall formation (Supplementary Table 4). These include AC elements (ACE), G-box motifs, MYB Binding Sites (MBS), and other regulatory elements involved in hormonal signalling and plant cell wall development (Zhou et al., 2009; Zhao et al., 2010; Sun et al., 2022).
3.8 Functional role analysis of ColLAC genes through gene ontology (GO) term assignment
GO term assignment revealed that the ColLAC genes are associated with several biological processes, including the phenylpropanoid metabolic process (GO:0009698), phenylpropanoid biosynthetic process (GO:0009699), lignin metabolic process (GO:0009808), lignin biosynthetic process (GO:0009809), and secondary metabolite process (GO:0019748) (Supplementary Figure 2). Furthermore, the GO analysis indicated that ColLAC genes are involved in various molecular functions, such as oxidoreductase activity (GO:0016491), copper ion binding (GO:0005507), transition metal ion binding (GO:0046914), ion binding (GO:0043167), metal ion binding (GO:0046872) and catalytic activity (GO:0003824) (Supplementary Figure 2). These molecular functions are consistent with those typically associated with the laccase gene family.
3.9 Tissue-specific expression of ColLAC genes
The expression levels of all 46 ColLAC genes across three different tissues (leaf, stem, and root) were analysed using qRT-PCR. The mRNA transcript levels were presented as relative quantification (RQ) values, with root tissue serving as the reference point (set to an RQ value of 1). Two distinct expression patterns were identified: one group of ColLAC genes exhibited consistent expression across the leaf, stem, and root tissues (Figure 8), while another group showed significantly higher expression in root tissue compared to the leaf and stem (Figure 9).
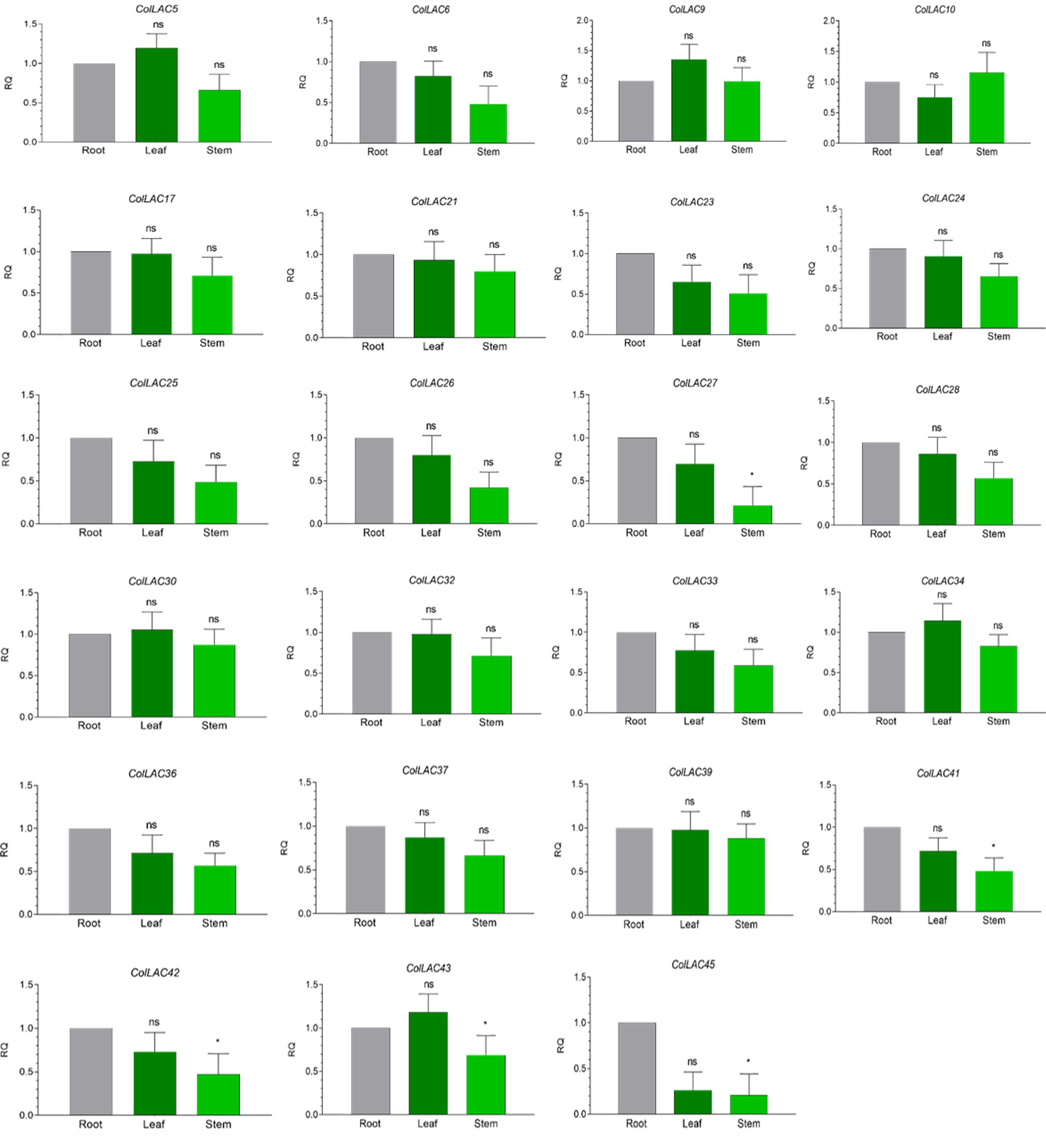
Figure 8. Tissue-specific consistent expression of laccase (ColLAC) genes. This figure illustrates the relative quantification (RQ) values of ColLAC gene expression in the leaf, stem, and root tissues of tossa jute. Asterisks indicate significant differences in P values from Tukey’s test, whereas non-significant values are labelled as ‘ns’.
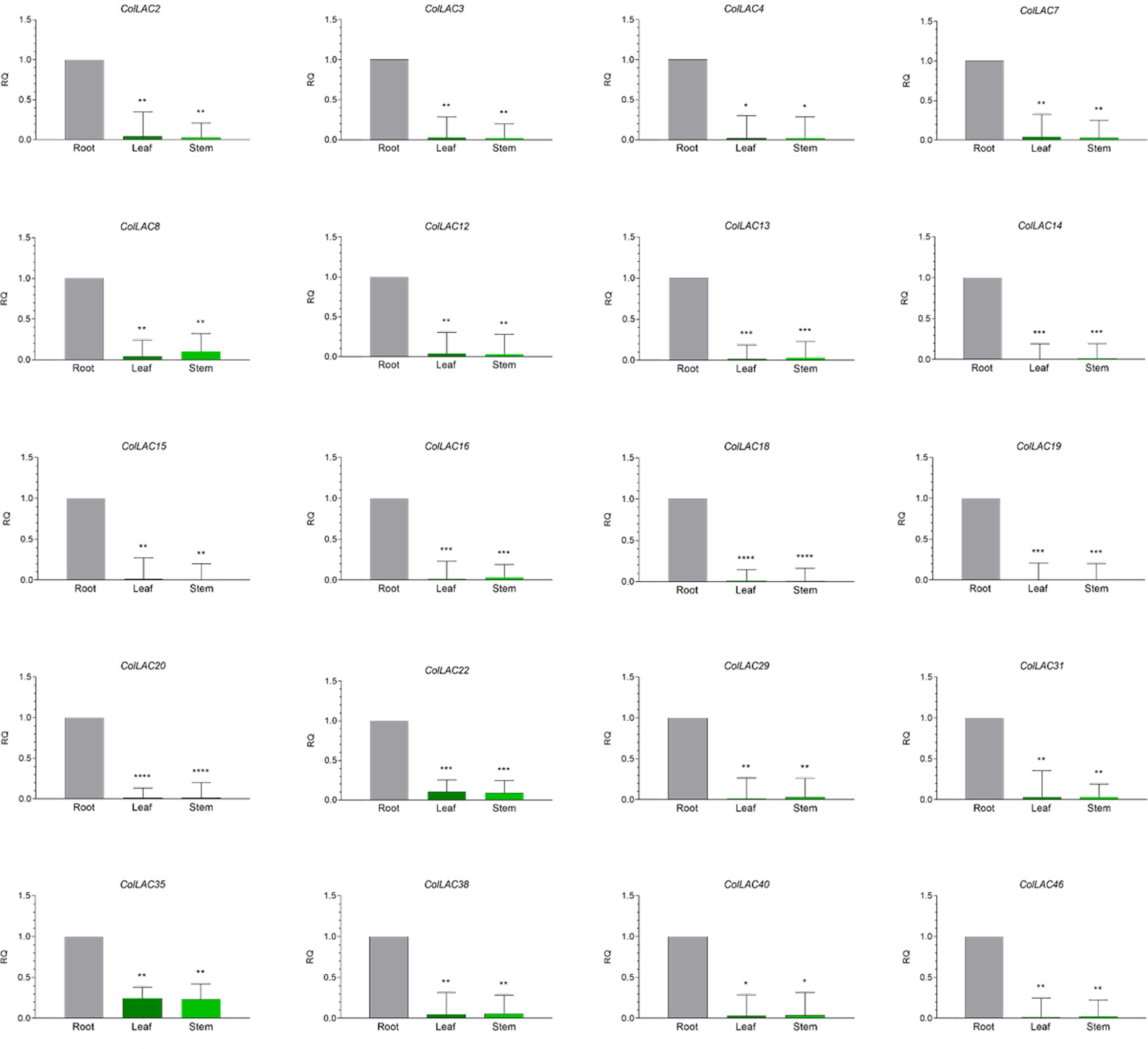
Figure 9. Tissue-specific "variable" expression of laccase (ColLAC) genes. This figure shows the relative quantification (RQ) values of ColLAC gene expression in the leaf, stem, and root tissues of tossa jute. Asterisks denote significant differences in P values from Tukey’s test.
Consistent expression of ColLAC genes was observed in ColLAC5, ColLAC6, ColLAC9, ColLAC10, ColLAC17, ColLAC21, ColLAC23, ColLAC24, ColLAC25, ColLAC26, ColLAC27, ColLAC28, ColLAC30, ColLAC32, ColLAC33, ColLAC34, ColLAC36, ColLAC37, ColLAC39, ColLAC41, ColLAC42, ColLAC43, and ColLAC45.
Significantly higher expression in root tissue compared to leaf and stem was observed in ColLAC2, ColLAC3, ColLAC4, ColLAC7, ColLAC8, ColLAC12, ColLAC13, ColLAC14, ColLAC15, ColLAC16, ColLAC18, ColLAC19, ColLAC20, ColLAC22, ColLAC29, ColLAC31, ColLAC35, ColLAC38, ColLAC40, and ColLAC46.
3.10 In-silico expression analysis of ColLACs
Publicly available transcriptomic data from BioProject PRJNA520880 on various tissues of tossa jute were used for in-silico expression analysis of ColLAC genes. The expression levels of all 46 ColLAC genes in leaf, phloem, xylem, and root tissues were analysed and visualised through a heatmap (Figure 10). Nine ColLAC genes were randomly selected from the heatmap for validation of their expression via qRT-PCR. The qRT-PCR results showed a similar expression pattern to that observed in the transcriptomic heatmap (Supplementary Figure 3). In the heatmap, most ColLAC genes exhibited higher expression in root tissue compared to leaf, phloem, and xylem.

Figure 10. In-silico expression study of laccase genes in tossa jute (ColLACs) across leaf, phloem, xylem, and root tissues. Heat map derived from publicly accessible transcriptomics data from BioProject PRJNA520880.
3.11 ColLAC gene expression at different developmental stages
Nine genes from the lignin biosynthesis pathway in Arabidopsis were selected, and their homologous counterparts in jute, ColLAC3, ColLAC22, ColLAC30, ColLAC32, ColLAC34, ColLAC38, ColLAC40, ColLAC42, and ColLAC46, were identified for expression analysis at different developmental stages (30 DAS, 60 DAS, 90 DAS, and 120 DAS) in two tossa jute cultivars—JRO632 (control) and bfs (mutant). The expression of these genes was analysed in the phloem tissue, as jute produces phloem fibres, and the findings could provide insights into the lignification process of the fibres.
In both cultivars, JRO632 and bfs, the highest expression of ColLAC genes was observed at 90 DAS. Significant differences in ColLAC expression between JRO632 and bfs were detected at 60 DAS and 90 DAS, while differences at 120 DAS were not statistically significant (Figure 11). Among all the genes, ColLAC34 exhibited the most significant expression differences at all time points between the two cultivars. The expression pattern of ColLAC34 suggests it may play a role in these developmental processes, as it exhibited significantly lower expression in the bfs mutant. However, further studies are required to fully understand its function in tossa jute.
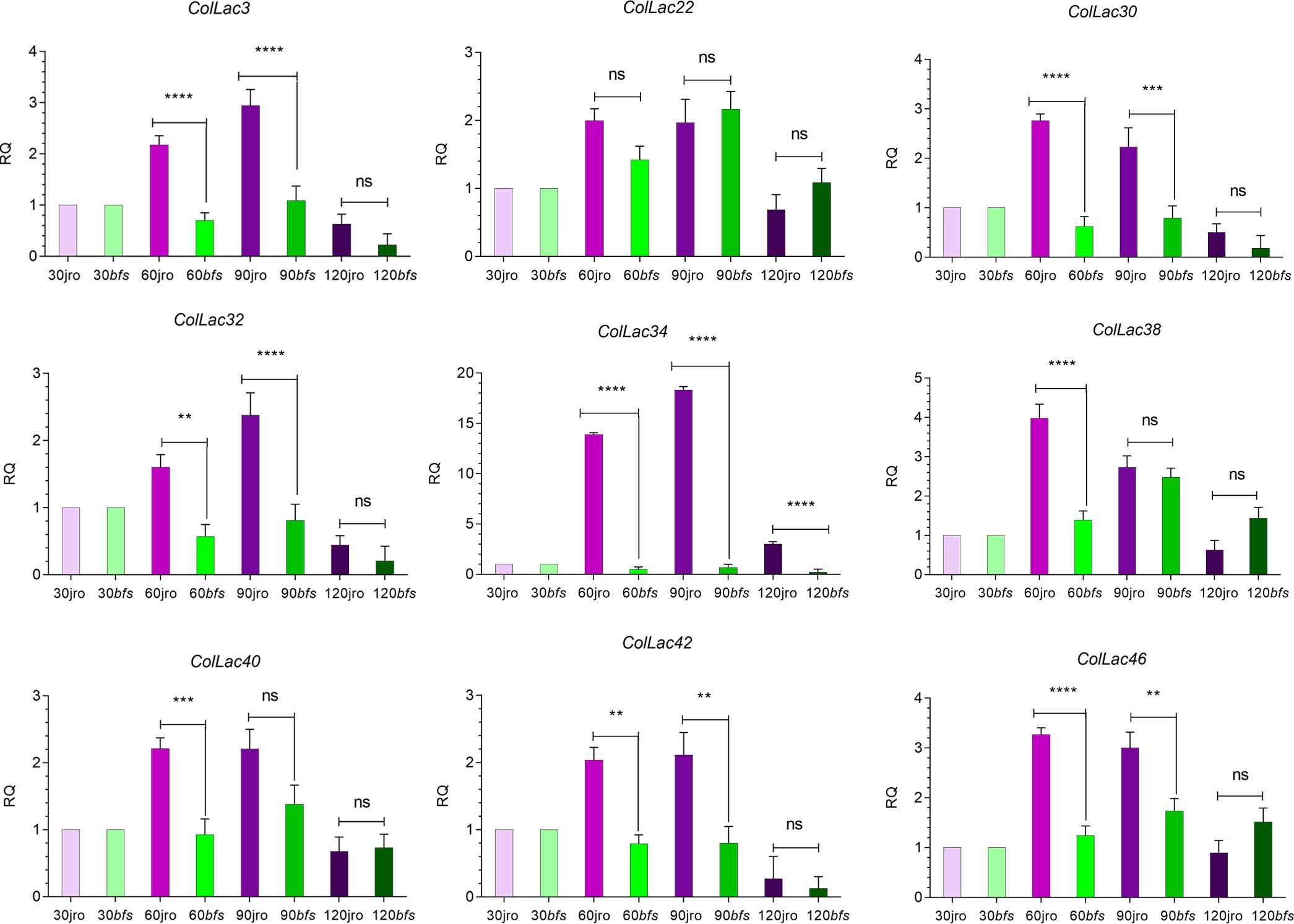
Figure 11. Expression profiles of ColLAC genes at different developmental stages (30, 60, 90, and 120 days after sowing, DAS) in tossa jute cultivars JRO632 (control) and bfs (mutant). Significant differences (P ≤ 0.05) are marked by asterisks, while non-significant values are labelled as ‘ns’.
3.12 Expression of ColLAC under copper stress
The expression of ColLAC3, ColLAC22, ColLAC30, ColLAC32, ColLAC34, ColLAC38, ColLAC40, ColLAC42, and ColLAC46—homologs of Arabidopsis laccase (AtLAC) genes involved in the lignin pathway—was examined in plants subjected to copper heavy metal stress (using 0.10 mM CuSO4·H2O). With the exception of ColLAC30, all other ColLAC genes showed peak expression at 8 hours of copper treatment (Figure 12). Although ColLAC30 exhibited a minimal fold change compared to the other ColLAC genes, none of the tested genes showed significant expression differences at 24 hours compared to the starting point (0-hour samples). Furthermore, aside from ColLAC3, ColLAC22, and ColLAC40, no significant expression was observed at the 12-hour mark. Our findings suggest that ColLAC genes respond predominantly within the first 12 hours of copper stress, with the highest expression levels occurring at 8 hours.
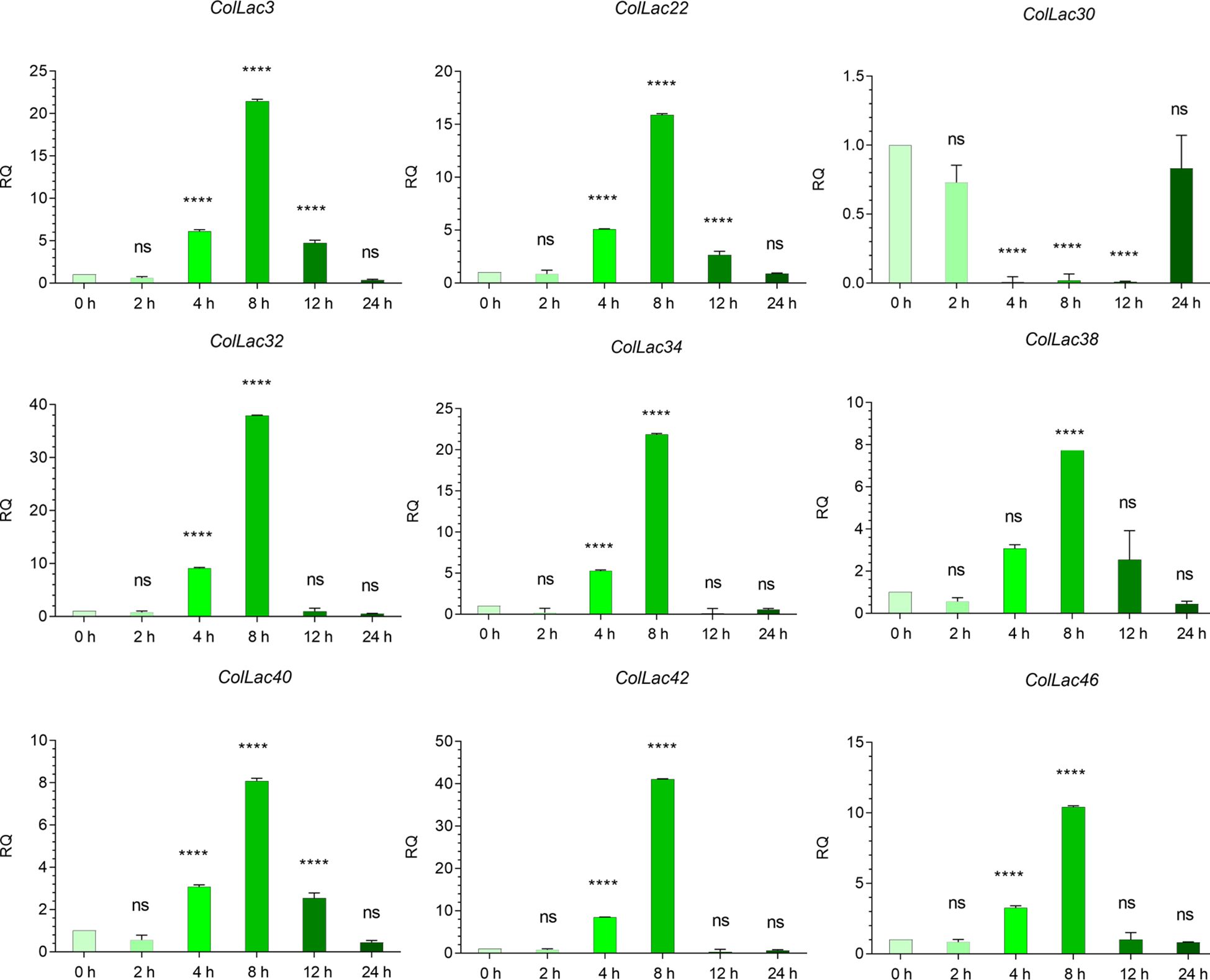
Figure 12. Expression profiles of ColLAC genes under copper stress (0.10 mM CuSO4·H2O treatment). Tukey’s multiple comparison test. Significant differences (P ≤ 0.05) are indicated by asterisks, while non-significant results are labelled as ‘ns’.
3.13 Expression of ColLAC under ABA hormonal stress
The expression levels of AtLAC-homologous ColLAC genes, including ColLAC3, ColLAC22, ColLAC30, ColLAC32, ColLAC34, ColLAC38, ColLAC40, ColLAC42, and ColLAC46, were analysed under ABA treatment. With the exception of ColLAC30, a significant increase in gene expression was observed at 4 hours, reaching its peak at this time point (Figure 13). For ColLAC30, the highest expression was recorded at 8 hours. With the exception of ColLAC30, all the ColLAC genes exhibited a U-shaped expression pattern, with elevated levels at 4 and 24 hours, and reduced expression at 8 and 12 hours. In contrast, ColLAC30 showed a gradual decline in expression from 8 to 24 hours.
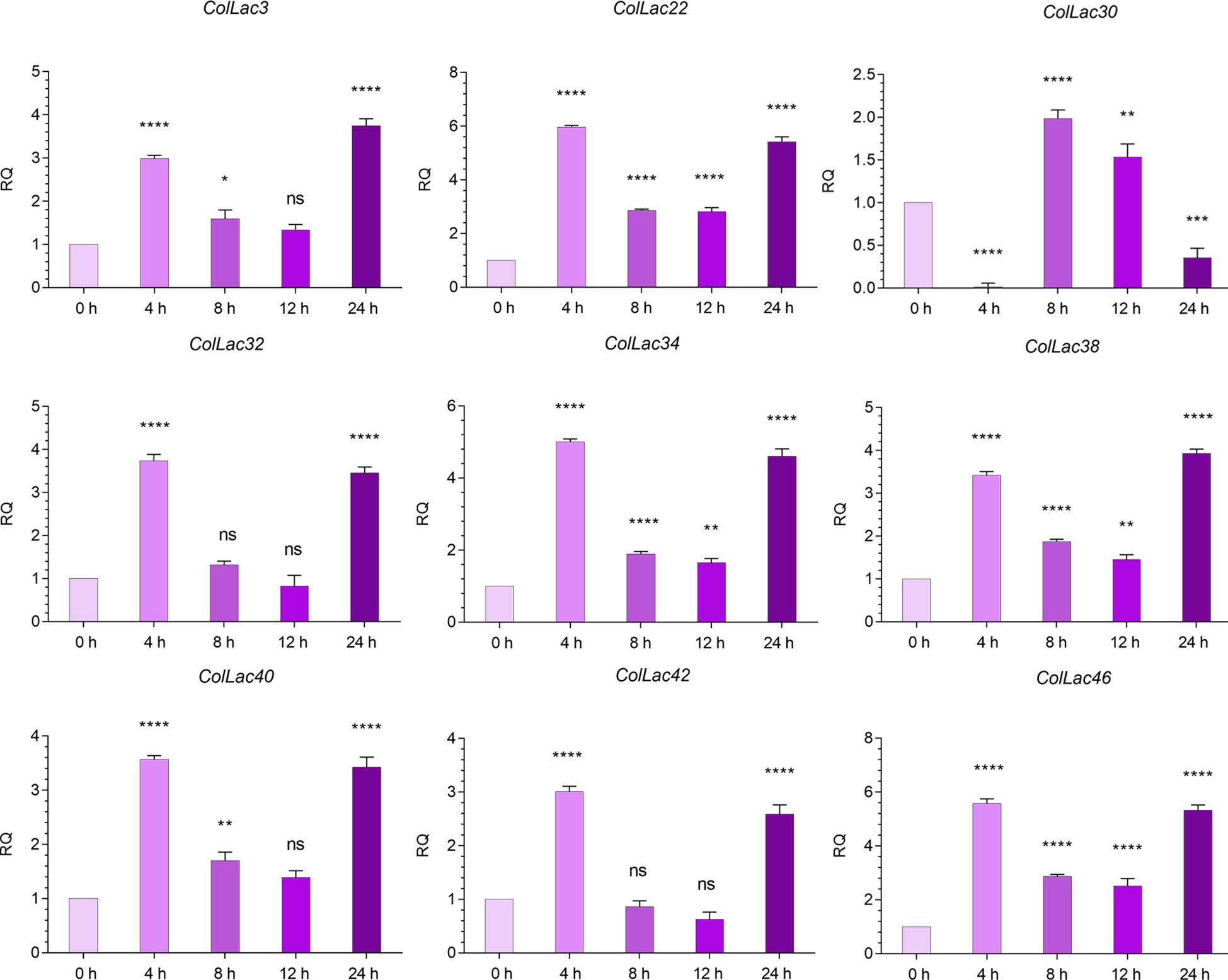
Figure 13. Expression profiles of ColLAC genes under ABA hormonal stress (using 0.15 mM ABA treatment). Significant differences (P ≤ 0.05) are indicated by asterisks, while non-significant results are labelled as ‘ns’.
3.14 ColLAC targeted by miRNA
The laccase genes in tossa jute (ColLACs) were identified as potential target sites for lignin-related miRNAs, particularly miR397 (Supplementary Table 5). Specifically, Ath-miR397a was predicted to target 14 ColLAC genes, including ColLAC3, ColLAC8, ColLAC9, ColLAC17, ColLAC30, ColLAC31, ColLAC32, ColLAC33, ColLAC34, ColLAC38, ColLAC39, ColLAC40, ColLAC42, and ColLAC46. In comparison, Ath-miR397b was predicted to target 9 ColLAC genes: ColLAC3, ColLAC30, ColLAC31, ColLAC32, ColLAC33, ColLAC34, ColLAC39, ColLAC40, and ColLAC46 (Figure 14). Among these, eight genes—ColLAC3, ColLAC30, ColLAC31, ColLAC32, ColLAC33, ColLAC34, ColLAC40, and ColLAC46—were commonly targeted by both Ath-miR397a and Ath-miR397b.
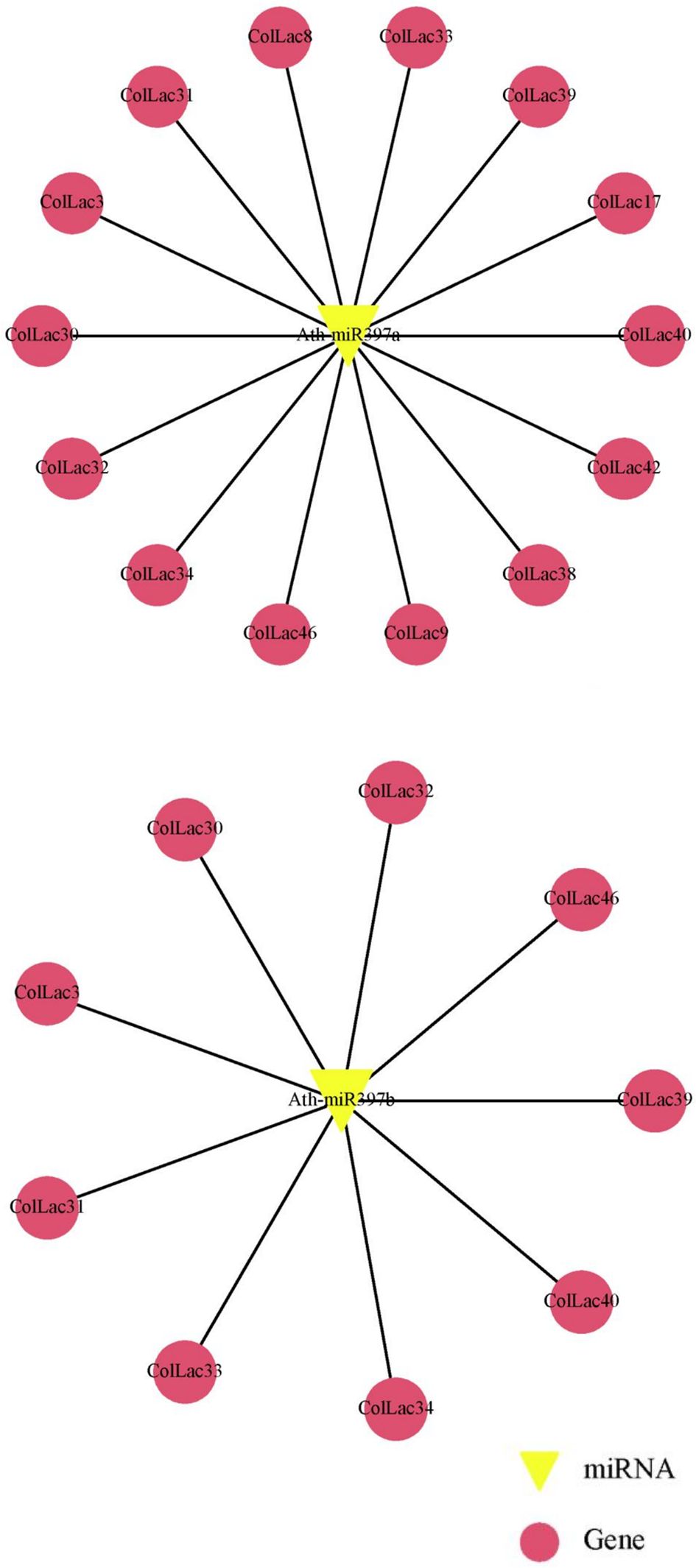
Figure 14. Prediction of miR397 target sites in ColLAC genes. Target sites for Arabidopsis miR397 (Ath-miR397a and Ath-miR397b) identified across multiple ColLAC genes.
4 Discussion
Studies on different plant laccases and their functionality are definitely not scarce. In Arabidopsis, mutations in AtLAC4 and AtLAC17 individually reduced lignin content by 10%, while double mutations in both genes (AtLAC4 and AtLAC17) led to a 39% reduction (Berthet et al., 2011). TaLAC4 silencing resulted in decreased lignin deposition in wheat (Triticum aestivum) stems (Soni et al., 2020). Overexpression of PbLAC1 increased lignin content and promoted cell wall growth in pear (Pyrus bretschneideri) (Cheng et al., 2019). Similarly, MsLAC1 overexpression resulted in higher lignin content in silvergrass (Miscanthus sp.) (He et al., 2019). Downregulation of GhLAC1 led to increased fibre initials and reduced fibre lengths in cotton (Gossypium herbaceum) (Hu et al., 2020). Functional studies on jute LAC genes were lacking until recently, when we reported the presence of 34 C. capsularis laccase (CcaLACs) genes (Parida et al., 2024a).
In white jute (C. capsularis), 11.76% of the CcaLAC genes (4 out of 34) were predominantly expressed in the roots. However, in tossa jute (C. olitorius), we observed a significantly higher percentage, with 43.47% of the ColLAC genes (20 out of 46) showing predominant expression in the roots (Figure 9). Additionally, 50% of the ColLAC genes (23 out of 46) exhibited no specific tissue preference (Figure 8), while 6.52% (3 out of 46) had undetectable expression in the tested tissues—leaves, stems, and roots. The high root-specific expression of ColLAC genes, compared to the CcaLAC genes, caught our attention.
To ensure accuracy, we repeated the tissue-specific experiments, validating the expression of ColLAC genes in two tossa jute cultivars, JRO524 and JRO632, and consistently found similar results. Similar root-specific expression patterns have been observed in other species, such as Eucalyptus laccases (EgrLACs), where 58% of the EgrLAC genes showed root predominance (Arcuri et al., 2020). Rice laccases (OsLACs) also demonstrated high expression in root tissues during vegetative stage (Liu et al., 2017), aligning with our findings in tossa jute ColLACs. These results suggest that LAC gene expression in jute is species-specific, with significant differences between them. The detailed functionality study of laccases in roots is yet to be explored, but their presence strongly suggests a contribution to lignification.
To identify the LAC genes involved in tossa jute lignification, we selected a few homologous genes well studied as key players in Arabidopsis lignification. The rationale for this approach is based on the fact that Arabidopsis is an ideal model for plant lignification studies (Vanholme et al., 2010). There is extensive research available on Arabidopsis LAC genes (AtLAC2, AtLAC4, AtLAC5, AtLAC10, AtLAC11, AtLAC12, and AtLAC17) confirming their involvement in lignin biosynthesis (Berthet et al., 2011). In this study the protein sequences of Arabidopsis LAC genes were compared with those of tossa jute; after which the two most similar jute genes were selected based on sequence homology for further analysis across different developmental stages.
The sequence similarity analysis revealed that AtLAC2 shared homology with ColLAC3 (76.74%) and ColLAC38 (74.32%), AtLAC4 with ColLAC40 (78.41%) and ColLAC32 (75.81%), AtLAC5 with ColLAC22 (80.62%) and ColLAC42 (66.26%), AtLAC10 with ColLAC32 (73.82%) and ColLAC40 (73.56%), AtLAC11 with ColLAC46 (80.10%) and ColLAC30 (79.85%), AtLAC12 with ColLAC22 (80.85%) and ColLAC42 (65.91%), AtLAC15 with ColLAC1 (63.63%), and AtLAC17 with ColLAC34 (80.27%) and ColLAC38 (80.13%).
New insights about gene expression patterns were gained by studying these homologous genes at different development stages of the phloem tissue. Gene expression showed a steady rise from 30 DAS (when lignification begins) to 120 DAS (when the plants are harvested for lignocellulosic fibres). This steady rise suggests the contribution of LAC genes in the lignification process and structural development. To further investigate their potential role in structural development, we utilised the X-ray-irradiated mutant bfs (bast fibre-shy), which exhibits structural abnormalities (Kundu et al., 2012). Consequently, the ColLAC34 gene emerged as a candidate gene that could potentially have a dual role in lignification and structural buildout in tossa jute as it is homologous to AtLAC17. The AtLAC17 is a well-characterised lignification gene in Arabidopsis (Berthet et al., 2011). However, more detailed functional analyses are required to confirm its role (Figure 11).
Are the homologous LAC genes from tossa jute (ColLACs) influenced by environmental stresses? GO term assignment indicated that ColLAC genes participate in various molecular functions, including oxidoreductase activity (GO:0016491), copper ion binding (GO:0005507), transition metal ion binding (GO:0046914), ion binding (GO:0043167), metal ion binding (GO:0046872), and catalytic activity (GO:0003824) (Supplementary Figure 2). These molecular functions may also be associated with plant responses to environmental stress, which warrants further investigation. To investigate this, we applied two types of stress: heavy metal copper and ABA hormone treatment. The rationale for selecting these stresses stems from evidence in the literature suggesting that laccase activity is influenced by copper stress, as copper acts as a cofactor in the enzyme’s function. Additionally, drought, salinity, and cold stress like conditions can be mimicked by ABA hormone hence it is chosen to provide various abiotic stresses in plants (Liu et al., 2017; Sakata et al., 2014; Sharma et al., 2023; Turlapati et al., 2011).
There were some important changes in transcript expression, observed in stress conditions involving copper. These not only revealed the stress-responsive nature of these ColLAC genes but also confirmed that the in-silico-identified genes are indeed part of the laccase family. Under copper stress, the maximum expression of ColLAC mRNA transcripts was observed at 8 hours, with fold changes ranging from 8.08 to 41.11 (Figure 12). Variability in mRNA transcript levels was noted, with ColLAC42 showing the highest fold increase (41.11), followed by ColLAC32 (37.93), ColLAC34 (21.85), ColLAC3 (21.47), ColLAC22 (15.89), ColLAC46 (10.43), ColLAC40 (8.09), and ColLAC38 (8.08). ColLAC30, however, did not show significant expression.
Under ABA stress, transcript levels also increased, ranging from 2.98 to 5.97-fold (Figure 13). Although the fold changes under ABA stress were lower than those observed under copper stress, they were still statistically significant. Except for ColLAC3, ColLAC38, and ColLAC30, all tested ColLAC genes showed peak expression at 4 hours under ABA stress. The fold changes at this time point were as follows, in descending order: ColLAC22 (5.97), ColLAC46 (5.58), ColLAC34 (5.01), ColLAC32 (3.74), ColLAC40 (3.57), ColLAC38 (3.42), ColLAC42 (3.01), and ColLAC3 (2.98).
In comparison, the white jute CcaLAC28 and CcaLAC32 (homologous to AtLAC4 and AtLAC17, respectively) displayed a delayed response under copper and ABA stresses. For CcaLAC32, the highest expression under copper stress occurred at 8 hours, and for CcaLAC28, it was observed at 12 hours, in contrast to the 4-hour peak seen in tossa jute. Similarly, under ABA stress, the highest expression of LAC in white jute occurred at 24 hours, compared to 4 hours in tossa jute (Parida et al., 2024a). This shift in LAC gene expression under abiotic stress may be species-specific. However, the overall fold change ranges for LAC gene expression were similar in both species, white and tossa jute.
Gene expression is strongly influenced by upstream cis-acting elements. In order to identify the key cis-acting elements we analysed the 2 kb upstream sequences of all ColLAC genes. This search resulted in the discovery of MYB transcription factor (TF) binding sites in several of them (Supplementary Table 4). It has been reported that MYB TFs regulate LAC expression in various species such as Phyllostachys edulis (PeMYB4.1/20/85.2 regulates PeLAC20), Arabidopsis (AtMYB58 regulates AtLAC4), Pennisetum glaucum (PgMYB305 regulates PgLAC14) and Pyrus bretschneideri (PbMYB26 regulates PbLAC4) (Zhou et al., 2009; Bai et al., 2023). Such studies indicate the potential role of MYB TFs in regulating ColLAC gene expression. Other cis-acting elements, in addition to MYB, known to be involved in ABA responsiveness, were identified, these include MYC, W-box, and CAT-box (Fujita et al., 2011). This could explain the varied expression patterns of ColLACs under ABA stress (Figure 13).
Gene expression in plants is regulated not only at the transcriptional level but also post-transcriptionally, with microRNAs (miRNAs) playing a crucial role. Although miRNA-mediated gene regulation in jute remains largely unexplored, some studies have previously reported their involvement. For instance, miR-845b and the miR-166 superfamily were found to be expressed in jute under Macrophomina phaseolina fungal stress (Dey et al., 2016). In-silico analysis of the white jute genome has identified five miRNA superfamilies—miR1536, miR9567-3p, miR4391, miR11300, and miR8689—which may regulate multiple biological functions, including plant growth, cell cycle regulation, organelle synthesis, development, and responses to environmental stresses (Ahmed et al., 2021). Additionally, these miRNAs have the potential to regulate the phenylpropanoid pathway and secondary cell wall formation in white jute. However, the functional validation of these miRNAs remains incomplete, leaving their precise roles in jute unclear. Moreover, miRNAs specifically associated with stem and fibre development in jute are yet to be thoroughly investigated.
Flax, another economically important bast fibre crop, has been more extensively studied in this context. Several miRNAs related to stem and fibre development have been identified in flax, including miR162, miR172, miR395, miR397, and miR530, which show potential roles in stem development (Yu et al., 2021). Additionally, miR390 has been reported to be involved in phloem fibre intrusive growth. Notably, some of these flax miRNA families, particularly miR397, have also been observed in the jute genome, where they are predicted to target the laccase (LAC) gene family, a key player in lignification and fibre formation.
The involvement of microRNAs in regulating LAC genes has been reported in several plants where miR397 stood out as a well-documented negative regulator of LAC genes (Lu et al., 2013). LAC expression is regulated by miR397 in various species, including Arabidopsis, Populus, chickpea (Cicer arietinum), and rice (Wang et al., 2014; Lu et al., 2008; Sharma et al., 2023; Bakhshi et al., 2016). Here we identified in-silico miR397 target sites in the ColLAC genes (Supplementary Table 5).
A recent study on rapeseed (Brassica napus) demonstrated that miR397a regulates the BnaLAC2 gene, enhancing adaptation to low-temperature stress by modulating lignin remodelling and maintaining ROS (reactive oxygen species) homeostasis (Hussain et al., 2025). This finding suggests that the regulation of LAC genes extends beyond lignin biosynthesis, potentially influencing various plant developmental processes and stress response mechanisms. In tossa jute, the role of miRNA-LAC interactions remains unexplored. Future functional validation of these regulatory mechanisms will provide deeper insights into how microRNAs fine-tune LAC gene expression, thereby shaping both lignification dynamics and stress resilience in this economically significant fibre crop.
A part of the study was also dedicated to the analysis of tissue-specific expression patterns of ColLACs across leaves, stems, and roots (Figures 8, 9). However, what about their cellular expression patterns? An in-silico analysis was conducted to investigate ColLAC expression in various cellular organelles (Supplementary Table 2). The 46 ColLAC genes were found to be expressed across nine organelles: chloroplast (15 ColLAC genes expressed), cytoplasm (12), vacuoles (10), cytoskeleton (3), extracellular space (2), plastid (1), peroxisome (1), endoplasmic reticulum (1), and nucleus (1). Since many of these are membrane-bound organelles, this suggests that ColLACs may possess structures that enable them to be anchored in membranes. Our findings support this possibility, as transmembrane helices were identified in 45.65% of ColLACs (21 out of 46 genes) (Supplementary Figure 1). This insight could be valuable for future structural studies on ColLACs, as currently, very limited structural information is available for LAC proteins in any plant species.
Significant advancements in jute biotechnology have been reported in recent years. These include the development of jute with properties of insect-resistance (Majumder et al., 2018a, 2020b), fungus- and herbicide-resistance (Majumder et al., 2018b), multi-trait jute (Majumder et al., 2024a), and low-lignin jute (Nath et al., 2021). Additionally, cutting-edge gene editing and silencing techniques have been adapted for jute improvement, such as CRISPR/Cas9 technology, virus-induced gene silencing (VIGS), and RNA interference (RNAi) (Jiang et al., 2024; Li et al., 2024; Nath et al., 2021). Stable and efficient gene transformation methods, for both white and tossa jute species, have also been developed (Majumder et al., 2024b). Furthermore, the AtSUC2 promoter, which enables phloem-specific expression of genes in jute fibres, has been successfully validated (Majumder et al., 2020c). These advancements provide a solid groundwork for the further characterization and functional validation of ColLACs genes.
In the near future, overexpression, RNA interference (RNAi), and CRISPR/Cas9-mediated knockout of ColLAC34 in jute could provide definitive evidence of its role in both lignification and structural development. Additionally, ColLAC22, ColLAC40, and ColLAC46 are likely key regulators of lignin biosynthesis. The first crucial step in this direction was taken in the present study through a comprehensive whole-genome identification of laccase (ColLAC) genes in tossa jute, laying the foundation for future functional validation and biotechnological applications.
5 Conclusion
Laccase (LAC) contributes to lignification as a key player in the final steps of the pathway. The tossa jute genome holds 46 LAC genes (ColLACs). Among these 46, nine genes (ColLAC3, ColLAC22, ColLAC30, ColLAC32, ColLAC34, ColLAC38, ColLAC40, ColLAC42, and ColLAC46) show strong similarity to Arabidopsis LAC genes, that are known to be involved in lignin biosynthesis. GO term assignment suggests their likely involvement in the lignification process, and phenylpropanoid pathways. These ColLAC genes were analysed and their expression patterns were studied in the plant tissues, at different stages of plant growth, and under various abiotic stresses, such as copper exposure and ABA treatment. Their significant expression levels suggest that these genes play crucial roles in tossa jute’s lignification process. A deeper dive into this wealth of knowledge could open new possibilities in the future, such as targeted regulation of their expression in jute stems to develop low-lignin jute fibres, enabling the production of more diverse biodegradable products, and enhancing the economic value of jute.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
DJ: Formal analysis, Investigation, Methodology, Writing – original draft. SuP: Formal analysis, Investigation, Methodology, Writing – original draft. SeP: Formal analysis, Data curation, Writing – review & editing. ND: Writing – review & editing, Supervision. SM: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Department of Biotechnology, Government of India, New Delhi, under the project titled “Metabolic engineering of jute stem for lowering its lignin content and improving its fibre quality” (project number BT/HRD/MKYRFP/50/17/2021, approved on February 2, 2022).
Acknowledgments
We express our profound appreciation to the Director of the ICAR-Central Research Institute for Jute and Allied Fibres (CRIJAF), Kolkata, India, for supplying the jute seeds. We express our gratitude to Prof. Swapan K. Datta and Dr. Karabi Datta from the Department of Botany, University of Calcutta, Kolkata, India, for their invaluable recommendations. We express our gratitude to the late Dr. Ajay Parida, former Director, and Dr. Debasis Dash, Director of BRIC-Institute of Life Sciences, Bhubaneswar, Odisha, India, for their steadfast support and vital resources indispensable to the project’s success. We sincerely appreciate Mrs. Soma Roy from Bioingene.com for her insightful comments and suggestions as an internal reviewer.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1568674/full#supplementary-material
Supplementary Figure 1 | Detection of transmembrane domains in ColLAC proteins.
Supplementary Figure 2 | Gene Ontology (GO) term classification of the ColLAC gene family.
Supplementary Figure 3 | Validation of tissue-specific transcriptomics data through qRT-PCR. Asterisks denote significant differences in P values from Tukey’s test, while non-significant values are marked as ‘ns’. The data represent three biological replicates.
Supplementary Table 1 | List of Primers.
Supplementary Table 2 | Protein features and chromosomal details of the 46 ColLAC members identified in Corchorus olitorius.
Supplementary Table 3 | List of orthologous and paralogous pairs of ColLAC genes.
Supplementary Table 4 | List of cis-acting elements in the upstream regions of ColLAC genes.
Supplementary Table 5 | miRNA targets in ColLAC genes.
References
Ahmed, M., Ahmed, F., Ahmed, J., Akhand, M. R. N., Azim, K. F., Imran, M. A. S., et al. (2021). In silico identification of conserved miRNAs in the genome of fibre biogenesis crop Corchorus capsularis. Heliyon 7, e06705. doi: 10.1016/j.heliyon.2021.e06705
Ain-Ali, Q.-U., Mushtaq, N., Amir, R., Gul, A., Tahir, M., Munir, F. (2021). Genome-wide promoter analysis, homology modeling and protein interaction network of Dehydration Responsive Element Binding (DREB) gene family in Solanum tuberosum. PloS One 16, e0261215. doi: 10.1371/journal.pone.0261215
Akhter, S., Sami, A. A., Toma, T. I., Jahan, B., Islam, T. (2022). Caffeoyl-CoA 3-O methyltransferase gene family in jute: Genome-wide identification, evolutionary progression and transcript profiling under different quandaries. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1035383
Arcuri, M. L. C., Fialho, L. C., Nunes-Latiz, A. V., Fuchus-Ferraz, P. M. C., Wolf, I. R., Valente, G. T., et al. (2020). Genome-wide identification of multifunctional laccase gene family in Eucalyptus grandis: potential targets for lignin engineering and stress tolerance. Trees 34, 745–758. doi: 10.1007/s00468-020-01954-3
Bai, Y., Ali, S., Liu, S., Zhou, J., Tang, Y. (2023). Characterization of plant laccase genes and their functions. Gene 852, 147060. doi: 10.1016/j.gene.2022.147060
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Bakhshi, B., Mohseni Fard, E., Nikpay, N., Ebrahimi, M. A., Bihamta, M. R., Mardi, M., et al. (2016). MicroRNA signatures of drought signaling in rice root. PloS One 11, e0156814. doi: 10.1371/journal.pone.0156814
Balasubramanian, V. K., Rai, K. M., Thu, S. W., Hii, M. M., Mendu, V. (2016). Genome-wide identification of multifunctional laccase gene family in cotton (Gossypium spp.); expression and biochemical analysis during fibre development. Sci. Rep. 6, 34309. doi: 10.1038/srep34309
Berthet, S., Demont-Caulet, N., Pollet, B., Bidzinski, P., Cezard, L., Le Bris, P., et al. (2011). Disruption of LACCASE4 and 17 results in tissue-specific alterations to lignification of Arabidopsis thaliana stems. Plant Cell 23, 1124–1137. doi: 10.1105/tpc.110.082792
Berthet, S., Thevenin, J., Baratiny, D., Demont-Caulet, D., Debeaujon, I., Bidzinski, P., et al. (2012). Role of plant laccases in lignin polymerization, Editor(s): Lise Jouanin, Catherine Lapierre. Adv. Botanical Res. 61, 145–172. doi: 10.1016/B978-0-12-416023-1.00005-7
Boerjan, W., Ralph, J., Baucher, M. (2003). Lignin biosynthesis. Annu. Rev. Plant Biol. 54, 519–546. doi: 10.1146/annurev.arplant.54.031902.134938
Chakraborty, A., Sarkar, D., Satya, P., Karmakar, P. G., Singh, N. K. (2015). Pathways associated with lignin biosynthesis in lignomaniac jute fibres. Mol. Genet. Genomics 290, 1523–1542. doi: 10.1007/s00438-015-1013-y
Chanwala, J., Khadanga, B., Jha, D. K., Sandeep, I. S., Dey, N. (2023). MYB transcription factor family in pearl millet: Genome-wide identification, evolutionary progression and expression analysis under abiotic stress and phytohormone treatments. Plants 12, 355. doi: 10.3390/plants12020355
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cheng, X., Li, G., Ma, C., Abdullah, M., Zhang, J., Zhao, H., et al. (2019). Comprehensive genome-wide analysis of the pear (Pyrus bretschneideri) laccase gene (PbLAC) family and functional identification of PbLAC1 involved in lignin biosynthesis. PloS One 14, e0210892. doi: 10.1371/journal.pone.0210892
Dai, X., Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. doi: 10.1093/nar/gkr319
Dey, P., Biswas, C., Karmakar, P. G. (2016). Identification and characterization of differentially expressed novel miRNAs (21–24 nt) in a Macrophomina phaseolina resistant RIL line of jute (Corchorus capsularis L.). Physiol. Mol. Plant Pathol. 94, 62–66. doi: 10.1016/j.pmpp.2016.04.005
Dixon, R. A., Barros, J. (2019). Lignin biosynthesis: old roads revisited and new roads explored. Open Biol. 9, 190215. doi: 10.1098/rsob.190215
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fang, L., Wang, Y. (2021). MicroRNAs in woody plants. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.686831
Finn, R. D., Clements, J., Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Fujita, Y., Fujita, M., Shinozaki, K., Fujita, Y., Fujita, M., Shinozaki, K., et al. (2011). ABA-mediated transcriptional regulation in response to osmotic stress in plants. J. Plant Res. 124, 509–525. doi: 10.1007/s10265-011-0412-3
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Goujon, T., Sibout, R., Eudes, A., MacKay, J., Jouanin, L. (2003). Genes involved in the biosynthesis of lignin precursors in Arabidopsis thaliana. Plant Physiol. Bioch. 41, 677–687. doi: 10.1016/S0981-9428(03)00095-0
Guo, Z., Kuang, Z., Zhao, Y., Deng, Y., He, H., Wan, M., et al. (2022). PmiREN2.0: from data annotation to functional exploration of plant microRNAs. Nucleic Acids Res. 50, D1475–D1482. doi: 10.1093/nar/gkab811
He, F., Machemer-Noonan, K., Golfier, P., Unda, F., Dechert, J., Zhang, W., et al. (2019). The in vivo impact of MsLAC1, a Miscanthus laccase isoform, on lignification and lignin composition contrasts with its in vitro substrate preference. BMC Plant Biol. 19, 552. doi: 10.1186/s12870-019-2174-3
Hu, B., Jin, J., Guo, A.-Y., Zhang, H., Luo, J., Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Hu, Q., Xiao, S., Guan, Q., Tu, L., Sheng, F., Du, X., et al. (2020). The laccase gene GhLac1 modulates fibre initiation and elongation by coordinating jasmonic acid and flavonoid metabolism. Crop J. 8, 522–533. doi: 10.1016/j.Cj.2019.11.006
Hussain, M. A., Huang, Y., Luo, D., Mehmood, S. S., Raza, A., Zhang, X., et al. (2025). Integrative analyses reveal Bna-miR397a–BnaLAC2 as a potential modulator of low-temperature adaptability in Brassica napus L. Plant Biotechnol. J. doi: 10.1111/pbi.70017
Jha, D. K., Chanwala, J., Barla, P., Dey, N. (2024). Genome-wide identification of bZIP gene family in Pearl millet and transcriptional profiling under abiotic stress, phytohormonal treatments; and functional characterization of PgbZIP9. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1352040
Jha, D. K., Chanwala, J., Sandeep, I. S., Dey, N. (2021). Comprehensive identification and expression analysis of GRAS gene family under abiotic stress and phytohormone treatments in Pearl millet. Funct. Plant Biol. 48, 1039–1052. doi: 10.1071/FP21051
Jiang, S., Li, Q., Meng, X., Huang, M., Yao, J., Wang, C., et al. (2024). Development of an Agrobacterium-mediated CRISPR/Cas9 gene editing system in jute (Corchorus capsularis). Crop J. 12, 1266–1270. doi: 10.1016/j.cj.2024.06.002
Kozomara, A., Griffiths-Jones, S. (2011). miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39, D152–D157. doi: 10.1093/nar/gkq1027
Krogh, A., Larsson, B., Von Heijne, G., Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547. doi: 10.1093/molbev/msy096
Kundu, A., Sarkar, D., Mandal, N. A., Sinha, M. K., Mahapatra, B. S. (2012). A secondary phloic (bast) fibre-shy (bfs) mutant of dark jute (Corchorus olitorius L.) develops lignified fibre cells but is defective in cambial activity. Plant Growth Regul. 67, 45–55. doi: 10.1007/s10725-012-9660-z
Kurata, N., Yamazaki, Y. (2006). Oryzabase. An integrated biological and genome information database for rice. Plant Physiol. 140, 12–17. doi: 10.1104/pp.105.063008
Kyte, J., Doolittle, R. F. (1982). A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132. doi: 10.1016/0022-2836(82)90515-0
Lamesch, P., Berardini, T. Z., Li, D., Swarbreck, D., Wilks, C., Sasidharan, R., et al. (2012). The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210. doi: 10.1093/nar/gkr1090
Le Roy, J., Blervacq, A.-S., Créach, A., Huss, B., Hawkins, S., Neutelings, G. (2017). Spatial regulation of monolignol biosynthesis and laccase genes control developmental and stress-related lignin in flax. BMC Plant Biol. 17, 124. doi: 10.1186/s12870-017-1072-9
Letunic, I., Khedkar, S., Bork, P. (2021). SMART: recent updates, new developments and status in 2020. Nucleic Acids Res. 49, D458–D460. doi: 10.1093/nar/gkaa937
Li, Q., Chen, S., Chen, L., Zhuang, L., Wei, H., Jiang, S., et al. (2024). Cloning and functional mechanism of the dwarf gene gba affecting stem elongation and cellulose biosynthesis in jute (Corchorus olitorius). Plant J. 118, 2003–2019. doi: 10.1111/tpj.16724
Liu, Q., Luo, L., Wang, X., Shen, Z., Zheng, L. (2017). Comprehensive analysis of rice laccase gene (OsLAC) family and ectopic expression of OsLAC10 enhances tolerance to copper stress in Arabidopsis. Int. J. Mol. Sci. 18, 209. doi: 10.3390/ijms18020209
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, S., Li, Q., Wei, H., Chang, M. J., Tunlaya-Anukit, S., Kim, H., et al. (2013). Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc. Natl. Acad. Sci. U.S.A. 110, 10848–10853. doi: 10.1073/pnas.1308936110
Lu, S., Sun, Y. H., Chiang, V. L. (2008). Stress-responsive microRNAs in populus. Plant J. 55, 131–151. doi: 10.1111/j.1365-313X.2008.03497.x
Majumder, S., Datta, K., Datta, S. K. (2024a). Pyramided transgenic jute (Corchorus capsularis) with biotic stress resistance and herbicide tolerance. Ind. Crops Prod. 208, 117776. doi: 10.1016/j.indcrop.2023.117776
Majumder, S., Datta, K., Sarkar, C., Saha, S. C., Datta, S. K. (2018b). The development of Macrophomina phaseolina (fungus) resistant and glufosinate (herbicide) tolerant transgenic jute. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00920
Majumder, S., Datta, K., Satpathy, S., Datta, S. K. (2020b). Development and evaluation of lepidopteran insect resistant jute expressing the fused Bt—Cry1Ab/Ac toxin driven by CaMV35S promoter. Ind. Crops Prod. 156, 112873. doi: 10.1016/j.indcrop.2020.112873
Majumder, S., Parida, S., Dey, N. (2024b). Protocol for imbibed seed piercing for Agrobacterium-mediated transformation of jute. STAR Protoc. 5, 102767. doi: 10.1016/j.xpro.2023.102767
Majumder, S., Saha, P., Datta, K., Datta, S. K. (2020a). “Fibre crop, jute improvement by using genomics and genetic engineering,” in Advancement in Crop Improvement Techniques. Eds. Tuteja, N., Tuteja, R., Passricha, N., Saifi, S. K. (Woodhead Publishing with Elsevier Inc, Amsterdam, Netherlands), 363–383. doi: 10.1016/B978-0-12-818581-0.00022-X
Majumder, S., Sarkar, C., Datta, K., Datta, S. K. (2020c). Establishment of the imbibed seed piercing method for Agrobacterium–mediated transformation of jute and flax bast fibre crops via phloem—specific expression of the β—glucuronidase Gene. Ind. Crops Prod. 154, 112620. doi: 10.1016/j.indcrop.2020.112620
Majumder, S., Sarkar, C., Saha, P., Gotyal, B. S., Satpathy, S., Datta, K., et al. (2018a). Bt jute expressing fused δ-endotoxin Cry1Ab/Ac for resistance to Lepidopteran pests. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.02188
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222D226. doi: 10.1093/nar/gku1221
McCaig, B., Meagher, R., Dean, J. (2005). Gene structure and molecular analysis of the laccase-like multicopper oxidase (LMCO) gene family in Arabidopsis thaliana. Planta 221, 619–636. doi: 10.1007/s00425-004-1472-6
Nath, M., Chowdhury, F. T., Ahmed, S., Das, A., Islam, M. R., Khan, H. (2021). Value addition to jute: assessing the effect of artificial reduction of lignin on jute diversification. Heliyon. 7 (3), e06353. doi: 10.1016/j.heliyon.2021.e06353
Parida, S., Datta, S. K., Dey, N., Majumder, S. (2024b). Selection of stable housekeeping genes for qPCR in biotic and abiotic stressed jute and their application in lignin pathway analysis. Plant Stress 13, 100525. doi: 10.1016/j.stress.2024.100525
Parida, S., Jha, D. K., Kumari, K., Pradhan, S., Dey, N., Majumder, S. (2024a). Genome-wide identification of the laccase gene family in white jute (Corchorus capsularis): Potential targets for lignin engineering in bast fibre. bioRxiv. 2024, doi: 10.1101/2024.07.17.603856
Rombauts, S., Déhais, P., Van Montagu, M., Rouzé, P. (1999). PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 27, 295–296. doi: 10.1093/nar/27.1.295
Sakata, Y., Komatsu, K., Takezawa, D. (2014). “ABA as a Universal Plant Hormone,” in Progress in Botany, vol. 75 . Eds. Lüttge, U., Beyschlag, W., Cushman, J. (Springer, Berlin, Heidelberg). doi: 10.1007/978-3-642-38797-5_2
Sarkar, D., Mahato, A. K., Satya, P., Kundu, A., Singh, S., Jayaswal, P. K., et al. (2017). The draft genome of Corchorus olitorius cv. JRO-524 (Navin). Genom. Data 12, 151–154. doi: 10.1016/j.gdata.2017.05.007
Sharma, N. K., Yadav, S., Gupta, S. K., Irulappan, V., Francis, A., Senthil-Kumar, M., et al. (2023). MicroRNA397 regulates tolerance to drought and fungal infection by regulating lignin deposition in chickpea root. Plant Cell Environ. 46, 3501–3517. doi: 10.1111/pce.14666
Shi, J., Yao, J., Tong, R., Wang, S., Li, M., Song, C., et al. (2023). Genome-wide identification of laccase gene family from Punica granatum and functional analysis towards potential involvement in lignin biosynthesis. Horticulturae 9, 918. doi: 10.3390/horticulturae9080918
Soni, N., Hegde, N., Dhariwal, A., Kushalappa, A. C. (2020). Role of laccase gene in wheat NILs differing at QTL-Fhb1 for resistance against Fusarium head blight. Plant Sci. 298, 110574. doi: 10.1016/j.plantsci.2020.110574
Sterjiades, R., Dean, J. F. D., Gamble, G., Himmelsbach, D. S., Eriksson, K. E. L. (1993). Extracellular laccases and peroxidases from sycamore maple (Acer pseudoplatanus) cell-suspension cultures: Reactions with monolignols and lignin model compounds. Planta 190, 75–87. doi: 10.1007/BF00195678
Sun, Q., Xi, Y., Lu, P., Lu, Y., Wang, Y., Wang, Y. (2022). Genome-wide analysis of the G-box regulating factors protein family reveals its roles in response to Sclerotinia sclerotiorum infection in rapeseed (Brassica napus L.). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.986635
Suyama, M., Torrents, D., Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Turlapati, P. V., Kim, K. W., Davin, L. B., Lewis, N. G. (2011). The laccase multigene family in Arabidopsis thaliana: Towards addressing the mystery of their gene function(s). Planta 233, 439–470. doi: 10.1007/s00425-010-1298-3
Vanholme, R., Acker, R. V., Boerjan, W. (2010). Potential of Arabidopsis systems biology to advance the biofuel field. Trends Biotechnol. 28, 543–547. doi: 10.1016/j.tibtech.2010.07.008
Wang, C.-Y., Zhang, S., Yu, Y., Luo, Y.-C., Liu, Q., Ju, C., et al. (2014). MiR397b regulates both lignin content and seed number in Arabidopsis via modulating a laccase involved in lignin biosynthesis. Plant Biotechnol. J. 12, 1132–1142. doi: 10.1111/pbi.12222
Wang, Q., Li, G., Zheng, K., Zhu, X., Ma, J., Wang, D., et al. (2019). The soybean laccase gene family: Evolution and possible roles in plant defense and stem strength selection. Genes 10, 701. doi: 10.3390/genes10090701
Wilkins, M. R., Gasteiger, E., Bairoch, A., Sanchez, J. C., Williams, K. L., Appel, R. D., et al. (1999). Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552. doi: 10.1385/1-59259-584-7:531
Yang, Y., Zhou, X., Zhu, X., Ding, B., Jiang, L., Zhang, H., et al. (2024). GhMYB52 like: A key factor that enhances lint yield by negatively regulating the lignin biosynthesis pathway in fibres of upland cotton (Gossypium hirsutum L.). Int. J. Mol. Sci. 25, 4921. doi: 10.3390/ijms25094921
Yu, Y., Chen, H., Yang, Y., Lou, D., Liang, C., Yuan, H., et al. (2021). Identification and characterization of differentially expressed microRNAs and target gene related to flax stem development. J. Nat. Fibres. 19, 5974–5990. doi: 10.1080/15440478.2021.1902902
Zhang, L., Ma, X., Zhang, X., Xu, Y., Ibrahim, A. K., Yao, J., et al. (2021). Reference genomes of the two cultivated jute species. Plant Biotechnol. J. doi: 10.1111/pbi.13652
Zhao, Q., Wang, H., Yin, Y., Xu, Y., Chen, F., Dixon, R. A. (2010). Syringyl lignin biosynthesis is directly regulated by a secondary cell wall master switch. Proc. Natl. Acad. Sci. U.S.A. 107, (32) 14496–14501. doi: 10.1073/pnas.1009170107
Keywords: jute, laccase, whole genome search, microRNA, abiotic stress, lignin pathway
Citation: Jha DK, Parida S, Pradhan S, Dey N and Majumder S (2025) Genome-wide analysis of the laccase gene family in tossa jute (Corchorus olitorius): insights into stem development, lignification, and responses to abiotic stress. Front. Plant Sci. 16:1568674. doi: 10.3389/fpls.2025.1568674
Received: 30 January 2025; Accepted: 07 April 2025;
Published: 09 May 2025.
Edited by:
Peng Wang, Jiangsu Province and Chinese Academy of Sciences, ChinaReviewed by:
Deepu Pandita, Government department of School Education, IndiaXin Ming, Northeast Agricultural University, China
Copyright © 2025 Jha, Parida, Pradhan, Dey and Majumder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuvobrata Majumder, c2h1dm9AaWxzLnJlcy5pbg==
†Present address: Nrisingha Dey, Ramakrishna Mission Vivekananda Educational And Research Institute, Kolkata, West Bengal, India
‡These authors have contributed equally to this work