 Pradeep Sharma
Pradeep Sharma Shefali Mishra
Shefali Mishra Ratan Tiwari
Ratan Tiwari- Crop Improvement Division, ICAR-Indian Institute of Wheat and Barley Research, Karnal, India
Introduction: MicroRNAs (miRNAs) are a class of 20- to 24-nucleotide endogenous small RNAs that regulate gene expression post-transcriptionally, playing vital roles in plant development and stress responses. Among abiotic stresses, drought stress (DS) is one of the most critical factors affecting wheat yield worldwide. Understanding miRNA-mediated regulatory mechanisms under drought stress conditions is crucial for improving drought tolerance in wheat.
Methods: To identify drought-responsive miRNAs in wheat, small RNA libraries were constructed from drought-tolerant (NI5439) and drought-susceptible (WL711) genotypes subjected to both control and drought-stress conditions. High-throughput sequencing was used to identify known and novel miRNAs. The family distribution of miRNAs, target prediction, pathway analysis, and differential expression analysis were conducted. A heat map was generated for the top 50 up- and downregulated miRNAs, and novel miRNAs were validated through qRT-PCR.
Results and discussion: A total of 306 known and 58 novel miRNAs were identified across the two wheat genotypes. The identified miRNAs belonged to over 18 families, with miR9662a-3p being the most abundant. Most identified miRNAs were 21 nucleotides in length. A total of 2,300 target genes were predicted for the known miRNAs. Pathway analysis revealed that target genes were involved in key biological processes including signal transduction, transport, organelle localization, DNA methylation, histone and chromatin modification, and plant development. Ten novel miRNAs were validated using qRT-PCR, confirming their differential expression under drought stress. The findings significantly expand the repertoire of drought-responsive and novel miRNAs in wheat. These miRNAs and their target genes provide valuable insights into the molecular mechanisms underlying drought tolerance. The validated novel miRNAs represent potential targets for genetic manipulation to enhance drought resilience in wheat cultivars.
Conclusion: This study provides a comprehensive miRNA expression profile in wheat under drought conditions and highlights several novel miRNAs that are differentially expressed between tolerant and susceptible genotypes. The integration of sequencing, computational analysis, and qRT-PCR validation strengthens the utility of these findings for future functional genomics studies and breeding programs aimed at developing drought-tolerant wheat varieties.
1 Introduction
Climate change, primarily driven by global warming, has emerged as a significant threat to ecosystems and food security worldwide. Rising temperatures, erratic precipitation patterns, and increased frequency of extreme weather events have intensified challenges in global agriculture, necessitating urgent strategies for climate-resilient food production. Wheat (Triticum aestivum L.), one of the most widely cultivated cereal crops, serves as a fundamental source of calories and nutrition for billions of people. However, despite advancements in wheat production over the past decade, global consumption has outpaced supply, exacerbating the demand-supply gap (Parmar et al., 2020; Kaur et al., 2023a; Zhao et al., 2024).
Drought stress at the reproductive stage poses a significant threat to wheat productivity by disrupting key physiological and molecular processes that are essential for grain development. It induces oxidative stress due to excessive accumulation of reactive oxygen species (ROS), leading to cellular damage, impaired photosynthetic efficiency, and premature leaf senescence during the grain-filling stage, ultimately reducing biomass accumulation and grain yield (Nelson et al., 2014; Kim et al., 2020; Berahim et al., 2021; Ahmad et al., 2023). To mitigate these detrimental effects, breeding programs have focused on developing drought-tolerant wheat varieties with enhanced physiological adaptability and stress-responsive molecular mechanisms (Wang et al., 2023). However, the intricate nature of drought responses necessitates a deeper understanding of the regulatory pathways involved in stress adaptation.Phytohormones play a crucial role in modulating wheat’s response to drought stress at the reproductive stage. Abscisic acid (ABA) and jasmonic acid (JA) regulate stomatal closure to minimize water loss while activating stress-responsive gene networks through the mitogen-activated protein kinase (MAPK) signaling pathway (Sharma et al., 2023). Additionally, JA influences ABA biosynthesis and degradation, creating a dynamic hormonal interplay that is critical for drought adaptation (Wang et al., 2022). However, excessive accumulation of ABA and JA can accelerate leaf senescence, impairing photosynthetic activity and reducing overall grain yield (Liu et al., 2023). In contrast, brassinosteroids (BRs) contribute to osmotic regulation, while cytokinins (CTKs) delay senescence and counteract the negative effects of ABA and JA, thereby enhancing stress resilience in wheat (Chen et al., 2023; Sun et al., 2023). Understanding these complex hormonal interactions is essential for developing strategies to improve wheat’s reproductive-stage drought tolerance and ensure sustainable yield production under water-limited conditions.
Recent advancements in molecular biology have highlighted the role of microRNAs (miRNAs) as key post-transcriptional regulators in plant stress responses. miRNAs are small, non-coding RNA molecules that regulate gene expression by targeting specific mRNAs for degradation or translational repression. Emerging evidence suggests that miRNAs play a crucial role in modulating drought tolerance by influencing stress-responsive pathways such as transcriptional regulation, hormone signaling, and antioxidant defense mechanisms (Sunkar et al., 2012a; Liu et al., 2023). High-throughput sequencing technologies have facilitated the identification of numerous stress-inducible miRNAs in wheat, including miR156, miR166, miR169, miR172, and miR399, which target key genes involved in stress adaptation (Ramachandran et al., 2020; Saroha et al., 2024). Despite growing knowledge of miRNA-mediated stress regulation, the molecular mechanisms underlying miRNA-mRNA interactions in wheat drought responses remain largely unexplored. Functional enrichment analyses and genome-wide expression studies suggest that miRNAs play a significant role in coordinating complex regulatory networks to enhance drought resilience (Mishra et al., 2023a). However, a comprehensive understanding of these regulatory pathways is still lacking, particularly during the critical grain-filling stage when drought stress has the most profound impact on yield formation.
This study aims to elucidate the role of drought-responsive miRNAs in wheat by identifying key miRNA-mRNA regulatory networks involved in drought adaptation. By integrating high-throughput sequencing, transcriptome analysis, and functional validation approaches, we seek to uncover novel miRNA-mediated mechanisms that contribute to drought tolerance in wheat. The findings of this research will provide valuable insights for breeding climate-resilient wheat varieties with enhanced drought tolerance, ensuring sustainable wheat production in the face of global climate change.
2 Materials and methods
2.1 Stress treatment, tissue collection, and root phenotyping
The study utilized two contrasting wheat genotypes for drought stress, NI5439 as tolerant (T) and WL711 as susceptible (S), to evaluate their performance under control (C) and drought stress (D) conditions (Kaur et al., 2017). Both genotypes were grown in cylindrical mud pots measuring 1.05 m in length and 0.18 m in diameter. The columns were filled with a homogenized mixture of soil, sand, and vermicompost in a 3:1:1 ratio, respectively. Plants in the well-watered treatment were maintained under normal environmental conditions, whereas drought-treated plants were placed in an area covered with a transparent sheet to simulate drought stress. Initially, three germinated seeds were planted per pot, with only one healthy seedling retained after 15 days. Before the onset of drought stress, columns were irrigated twice daily to maintain optimal soil moisture. Drought stress was initiated at the Z24 stage of Zadok’s scale (main shoot with four tillers), and root tissues were collected at the Z37 stage (flag leaf just visible). These root samples were immediately flash-frozen in liquid nitrogen and stored at −80°C for subsequent analyses (Zadoks and Board, 1999). The root systems were carefully extracted by breaking the pots and sectioned into four depths: 0–30 cm, 30–60 cm, 60–90 cm, and 90–120 cm, following the approach of Narayanan et al. (2014). Roots were gently washed using a low-pressure water fountain over a 1.5 m sieve to minimize damage. The cleaned root samples were then preserved in 70% ethanol and scanned using a document scanner. Root volume and other traits like root length and diameter were quantified using WinRHIZO® software, which provides an accurate digital analysis of root morphological parameters (Singh et al., 2011).
2.2 RNA extraction, construction of small RNA libraries and deep sequencing
Total RNA was extracted using the TRIzol method, and its quality was assessed via NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, USA).Small RNAs were isolated in triplicate from frozen root tissues using the mirVana™ miRNA isolation kit (Ambion), following the manufacturer’s protocol. The small RNA fractions from the three replicates were pooled together for library construction. The preparation of small RNA libraries was performed according to the protocol by (Lu et al., 2007), with minor modifications. Small RNAs were sequentially ligated with 3’ and 5’ adapters (Supplementary Table 1). Following this, reverse transcription was performed using an RT primer, and PCR amplified the resulting cDNA. The integrity and quantity of the constructed libraries were assessed using the RNA Integrity Number (RIN) on an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). The final libraries were submitted for high-throughput sequencing at SciGenome Labs (India) using the Illumina MiSeq platform (Illumina, USA). The libraries were labelled as Tolerant Control (TC), Tolerant Drought (TD), Susceptible Control (SC) and Susceptible Drought (SD). The raw data have been submitted to NCBI and the accession number is PRJNA1012115.
2.3 Computational analysis of small RNA sequencing data
Sequencing reads with a Phred quality score > 30 were retained and processed using Cutadapt v1.3 to remove adapter sequences. Non-coding small RNAs, including siRNAs, snRNAs, snoRNAs, piRNAs, tRNAs, and rRNAs, were filtered out by mapping them to respective databases (Supplementary Table 2) using Bowtie2 v2.1.0 (Langmead and Salzberg, 2012). The remaining non-redundant reads (17–35 nucleotides) were used for identifying both conserved and novel miRNAs in wheat. For conserved miRNAs, reads were aligned to the Triticum aestivum reference genome using Bowtie v1.2.3 (Langmead et al., 2009) and compared with known miRNAs in miRBase v22 (Kozomara et al., 2019). Initially, reads were mapped to mature miRNAs, followed by precursor sequences to ensure accuracy. Differential expression analysis was performed using DESeq2 in R v4.0.0 (Love et al., 2014) to identify significantly differentially expressed miRNAs.
For novel miRNA identification, miRDeep2 v2.0.0.7 (Friedländer et al., 2012) was used with the Triticum aestivum genome as a reference (Ensembl Plants Release 60: https://ftp.ebi.ac.uk/ensemblgenomes/pub/release-60/plants/fasta/triticum_aestivum/ncrna/). High-confidence novel miRNAs were identified based on (Meyers et al., 2008) criteria, including a 3’ two-nucleotide overhang, no more than four mismatches with the complementary precursor arm, and minimal asymmetric bulges (one or two bases) in the miRNA/miRNA* duplex. Secondary structure prediction was performed using MFOLD (Zuker, 2003) to confirm the characteristic stem-loop hairpin structure of miRNA precursors. Potential target genes for novel miRNAs were predicted using miRanda, which identifies mRNA targets based on sequence complementarity. Thisworkflow, integrating updated genome references and bioinformatics tools, ensures high-precision miRNA discovery and expression analysis, providing insights into their regulatory roles in gene silencing and stress response pathways in wheat.
2.4 Gene ontology analysis
To understand the functional roles of differentially expressed genes (DEGs) in wheat under drought stress, Gene Ontology (GO) enrichment analysis was performed. The DEGs were annotated using the BLAST2GO tool (Conesa et al., 2005) by mapping sequences against the non-redundant (NR) database of the National Center for Biotechnology Information (NCBI). The identified genes were classified into three major GO categories: biological process (BP), molecular function (MF), and cellular component (CC). The functional enrichment analysis was conducted using AgriGO v2.0, a specialized tool for plant GO analysis (Tian et al., 2017), to determine significantly overrepresented GO terms. The statistical significance of enrichment was assessed using Fisher’s exact test with a false discovery rate (FDR) < 0.05 for multiple testing corrections. Figure 1 presents a summary of the miRNA analysis conducted under stress conditions, highlighting the key findings and insights from the study.
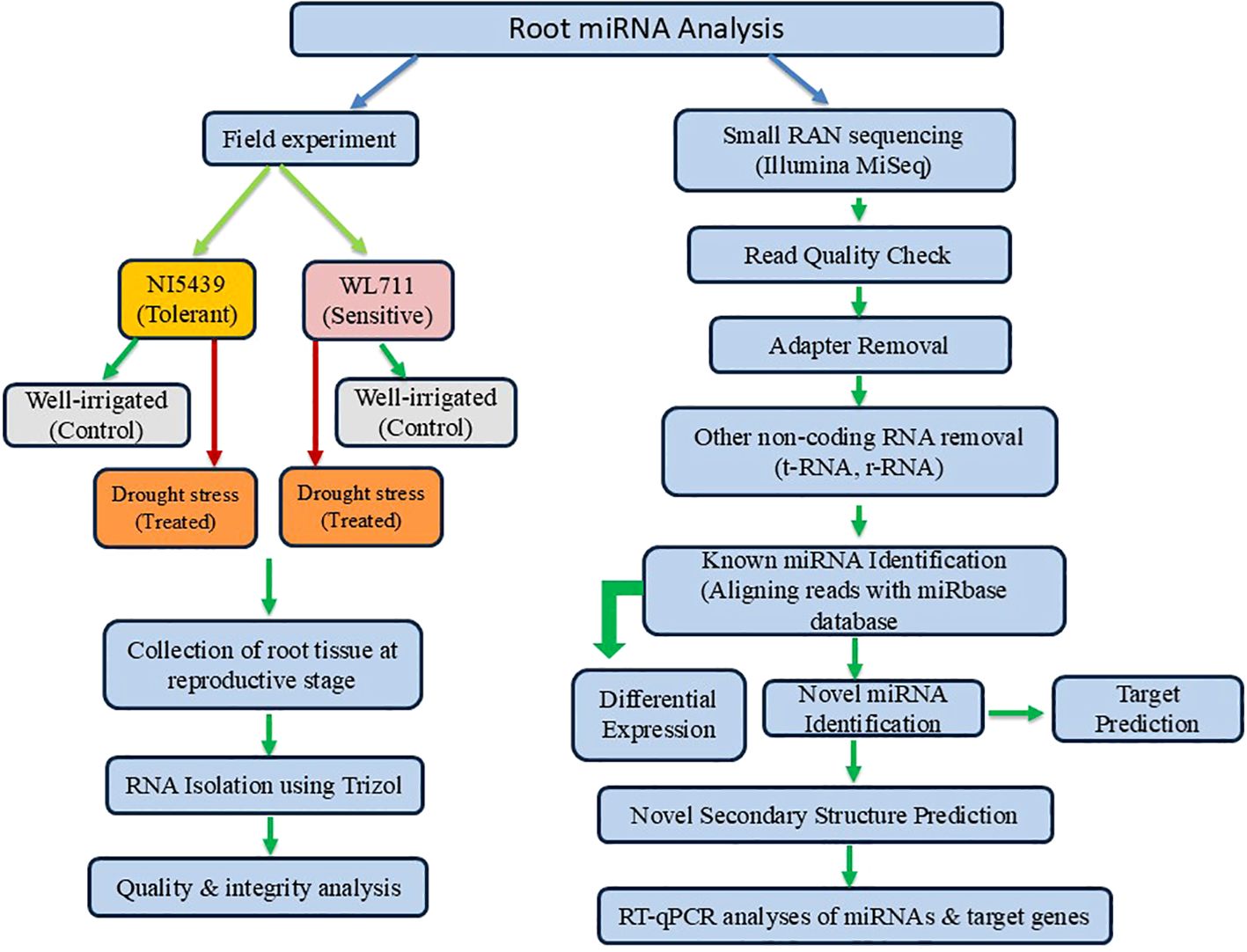
Figure 1. Summary of the approach employed for the identification, characterization, and functional validation of known and novel drought-responsive miRNAs in wheat root tissues at the reproductive stage.
2.5 Gene regulatory network analysis
Cytoscape (version 3.2.1)tool was used for analysis of gene network analysis of differential expressed genes. For network analysis, top 100 upregulated and downregulated genes each were considered. ARACNE (Algorithm for the Reconstruction of Accurate Cellular Networks) and Network Analyzer plug-in were used for analyzing the network of all the four sets of DEGs. On the basis of high degree and betweenness, hub genes were selected.
2.6 Validation and expression profiling of miRNAs and their target genes by quantitative PCR
Small RNA cDNA (srcDNA) library was constructed according to the protocol of (Bailey et al., 2013). Briefly, small RNAs were polyadenylated at 37°C for 45 min in 50 μl reaction volume containing 0.3 μg ofmall RNA, 0.1 U E. coli poly(A) polymerase, 1X E. coli poly(A) polymerase reaction buffer [50 mM Tris-HCl, 250 mM NaCl, 10 mM MgCl2, pH 7.9 at 25°C] and 1 mM ATP. Then, the poly(A)-tailed small RNA samples were purified to remove unincorporated ATP by using a purification cartridge provided in mirVana probe and marker kit as per manufacturer’s protocols. The purified poly(A)-tailed small RNA samples were stored at –70°C. The srcDNA libraries were generated by mixing 500 ng of poly(A)-tailed small RNA and 1 μg of RTQ primer in a 26 μl reaction volume. The reaction mixture was incubated at 65°C for 10 min followed by addition of 0.2 U M-MuLV reverse transcriptase, 1XM-MuLV reverse transcriptase reaction buffer [50 mM Tris-HCl, 75 mM KCl, 3 mM MgCl2, 10 mM DTT, pH 8.3 at 25°C] and 1 mM dNTP mix in a final reaction volume of 40 μl. The reverse transcription was carried out at 37°C for 60 min followed by inactivation of the enzyme at 70°C for 15 min. About 1 μl of 5 U RNaseH was added to remove poly(A)-tailed small RNAs. The samples were purified by using the QIAquick PCR purification kit in 50 μl of final volume.
The qRT-PCR was performed using 0.3 μg of srcDNA, 1X SYBR green/fluorescein qPCR master mix, 1 μM RTQ-UNIr primer, and 1 μM miRNA-specific forward primer (Supplementary Table 3). The PCR reactions were performed in triplicate for each gene. The thermal cycling PCR reactions were performed with the following profile: 95°C for 5 min, 40 cycles of 15 sec denaturing at 94°C, 30-sec annealing at 55°C and 30-sec extension at 72°C, and finally a melt curve step from 65°C to 95°C with a rise of 0.5°C for 5 sec. The U6 snRNA was used as a reference gene for all the samples amplified. Relative quantification of expression for each miRNA was analyzed using the comparative CT method as described by (Livak and Schmittgen, 2001). Ten novel miRNAs viz. #ps_55, #ps _199, #ps _45 and #ps_160, #ps_19, #ps_91, #ps_187, #ps_103, #ps_74, #ps_47, #ps_89, #ps_157, #ps_55, #ps_121 and its targets TaDRA1, TaDRA2, TaDRA3, TaDRA4, TaDRA5, TaDRA6, TaDRA7, TaDRA8, TaDRA9, TaDRA10, TaDRA11, TaDRA12, TaDRA13 and TaDRA14 were chosen at random to validate them under drought stress in wheat respectively. The real ids of miRNAs and its targets were mentioned in (Supplementary Table 4).
3 Results
3.1 Phenotyping under drought stress
Two wheat genotypes that were already known for their distinct behavior in drought stress were grown in fields under irrigated and drought conditions. Root samples were collected for small RNA library preparation at the booting stage of growth and development. The pooled analysis of variance revealed that both genotype and stress conditions differed significantly for all the traits studied. Based on LSD, the two genotypes differed in root length, surface area, and Length perVolume whereas stress conditions were significantly different for root length, surface area, and root volume.The two genotypes under normal conditions differed significantly for root length, surface area, and Length perVolume (Figure 2). However, under drought stress conditions, the two genotypes differed in total length and Length perVolume.Genotype 2 registered more reduction than genotype 1 in all the traits except diameter (Figure 2). Under normal conditions, the root length had a significant positive correlation with surface area and Length perVolume and the root volume had a significant positive correlation with surface area and average diameter. However, under drought-stressconditions, all the traits were interlinked positively.

Figure 2. (A) Phenotyping of contrasting wheat genotypes under drought stress conditions to evaluate variations in root traits. (B) Correlation Matrix of Root Morphological Traits in Wheat under Drought Stress. L (cm) – Root Length, SA (cm²) – Root Surface Area, AD (mm) – Root Average Diameter, LPL (cm/m³) – Root Length Per Unit Volume, RV (cm³) – Root Volume. The figure represents a Pearson correlation matrix for root traits in wheat, with color intensity indicating correlation strength (red = positive, blue = negative).
3.2 Analysis of the small RNA libraries
Small RNA libraries were generated from the root tissues of two wheat genotypes, one exhibiting drought tolerance and the other drought sensitivity, cultivated under both normal and drought-stressed conditions. Sequencing produced approximately 200 million raw reads (200,780,257). Specifically, 62,320,791 reads were obtained from the drought-tolerant genotype under normal conditions, 73,006,712 from the same genotype under drought stress, 24,620,142 from the drought-sensitive genotype under normal conditions, and 40,832,612 from the drought-sensitive genotype under drought stress (Table 1). Raw sequencing reads underwent preprocessing to remove adapter sequences, yielding clean, non-redundant reads of 10,825,961, 6,203,540, 4,552,914, and 8,142,717 for the respective libraries. Non-coding RNAs, including siRNAs, snRNAs, snoRNAs, piRNAs, tRNAs, and rRNAs, were filtered out, leaving unannotated reads ranging from 17 to 35 nucleotides in length. These unannotated sequences were then analyzed to identify both conserved and novel miRNAs in wheat, offering valuable insights into miRNA-based regulatory mechanisms in response to drought stress.
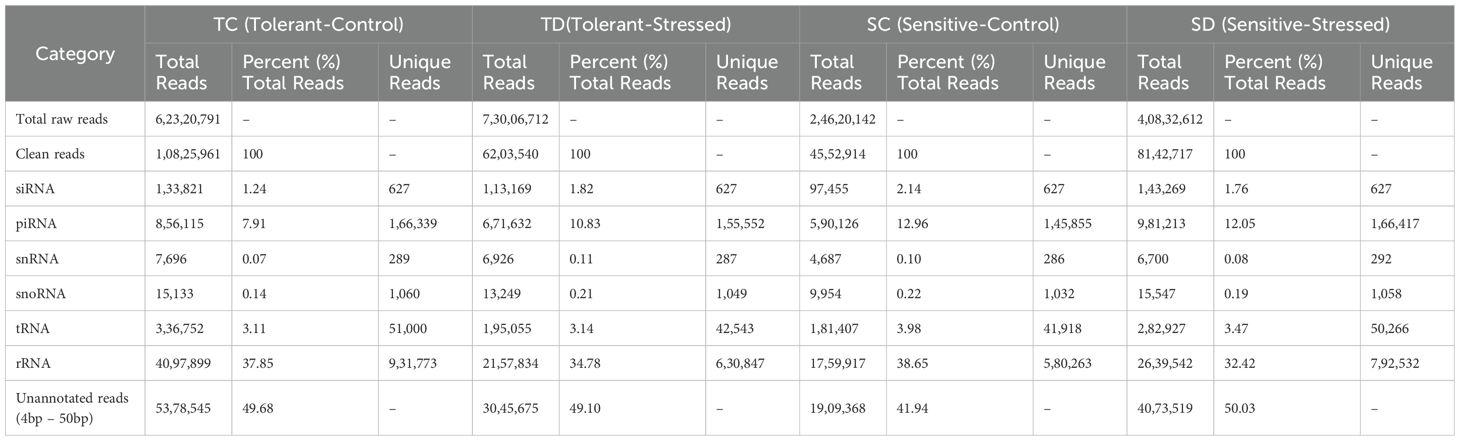
Table 1. Summary of small RNA sequencing data in the four libraries ofwheat.
3.3 Identification of conserved miRNAs
The identification of conserved miRNAs was conducted by aligning 17–35 bp unique reads to a wheat reference genome. Initially, these reads were mapped to mature miRNAs, and any unmapped sequences were subsequently aligned with precursor sequences. This approach led to the identification of 150, 132, 146, and 167 previously characterized miRNAs, distributed across 86 families, in the tolerant-control (TC), tolerant-stressed (TD), sensitive-control (SC), and sensitive-stressed (SD) libraries, respectively (Table 2). Among these, 119 miRNAs were found to be shared across all libraries analyzed in this study (Figure 3). Additionally, several miRNAs were uniquely expressed under individual conditions, such as 8 in TC, 3 in SC, 15 in SD, and 1 in TD—suggesting condition-specific regulatory roles. Shared miRNAs between pairs and triplets of conditions also varied, with notable overlap between SC and SD (10 miRNAs) and between SC and TC (9 miRNAs). These results highlight both common and condition-specific miRNA-mediated regulatory responses under the tested stress conditions.
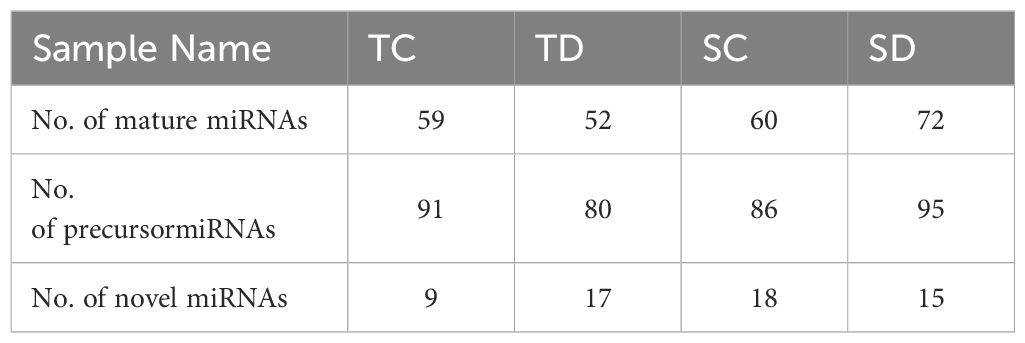
Table 2. Summary of conserved and novel miRNA in wheat under drought stress.
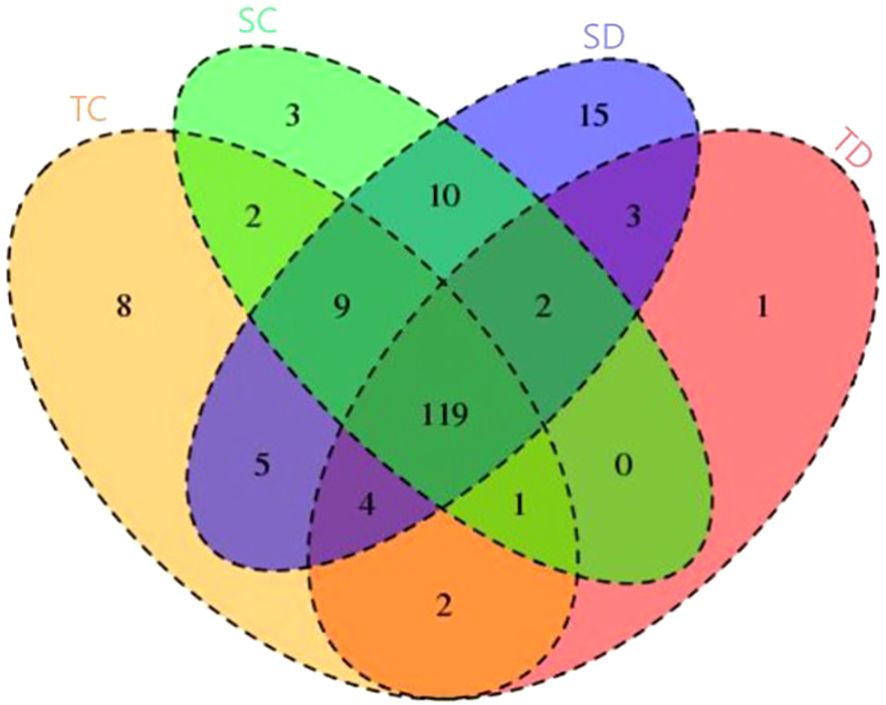
Figure 3. Venn diagram represents distribution of conserved miRNA among four libraries. illustrates the distribution of differentially expressed genes (DEGs) across four experimental conditions: TC (yellow), SC (green), SD (blue), and TD (red). Each section represents unique or shared DEGs among the conditions. The numbers indicate the count of DEGs specific to one condition or shared among multiple conditions.
Notably, two miRNAs (tae-miR1119 and tae-miR9773) were exclusively detected under control conditions in both genotypes, whereas three miRNAs (tae-miR1129, tae-miR9660, and tae-miR9661) were specifically recovered under drought stress. These five miRNAs are likely to play a regulatory role in drought stress response. Additionally, 12 genotype-specific miRNAs were identified, with two (tae-miR1125 and tae-miR5049) associated with the drought-tolerant genotype, while the remaining ten (tae-miR1136, tae-miR9652, tae-miR9657b, tae-miR9657c, tae-miR9663, tae-miR9666a, and tae-miR9670) were linked to the drought-sensitive genotype.
The number of miRNA members varied across different families. The tae-miR159 and tae-miR9662 families exhibited the highest diversity, with an average of 20 members each. These were followed by tae-miR9653, tae-miR167, tae-miR1130, tae-miR9672, and tae-miR9657, each containing more than 10 members. The most highly conserved miRNA identified across all wheat libraries was tae-miR9662a-3p, along with its precursor tae-MIR9662a (Supplementary Table 5). The second and third most abundant miRNAs belonged to the conserved MIR159 family, specifically tae-miR159a and tae-miR159b. In terms of length distribution, the most prevalent class of conserved miRNAs was 21 nucleotides (28.57% on average), followed by the 19-nucleotide class (15.63% on average) (Figure 4).
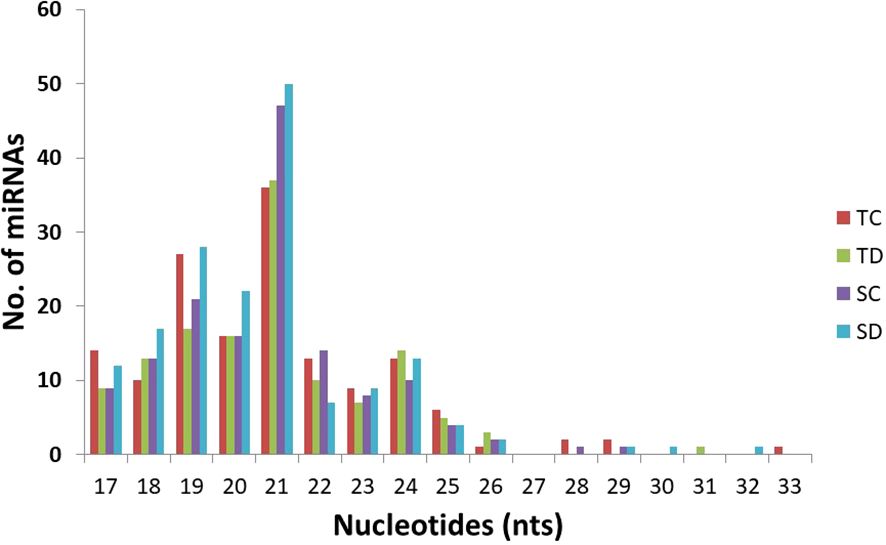
Figure 4. Length distribution of conserved miRNAs in the four libraries from wheat. TC: sRNA library from the drought-tolerant genotype NI-5439 without drought stress, TD: from NI-5439 with drought stress, SC: from the drought-sensitive genotype WL-711 without drought stress, SD: from WL-711 with drought stress.
3.4 Identification of Novel miRNAs
The discovery of novel miRNAs was achieved through secondary structure prediction, leading to the identification of 58 novel miRNAs in Triticum aestivum. These miRNAs were predicted from aligned sequencing data based on structural and genomic features. Among them, 9 novel miRNAs were detected in the control condition of the NI5439 genotype, while 10 were identified under drought stress. Similarly, in the WL711 genotype, 15 and 14 novel miRNAs were detected in control and drought-stressed samples, respectively (Table 3). Comprehensive characterization was conducted for each novel miRNA, including chromosomal localization, precursor sequence identification, secondary structure prediction, and mature miRNA sequence analysis. The secondary stem-loop structures were computationally predicted using an energy minimization approach. The minimal folding free energy (MFE) values of precursor miRNAs ranged from –46.71 to –13.47 kcal/mol, with an average of –33.73 ± 10.57 kcal/mol (Table 3), indicating their structural stability. The graphical representation of these structures is provided in Supplementary Figure 1a-d.
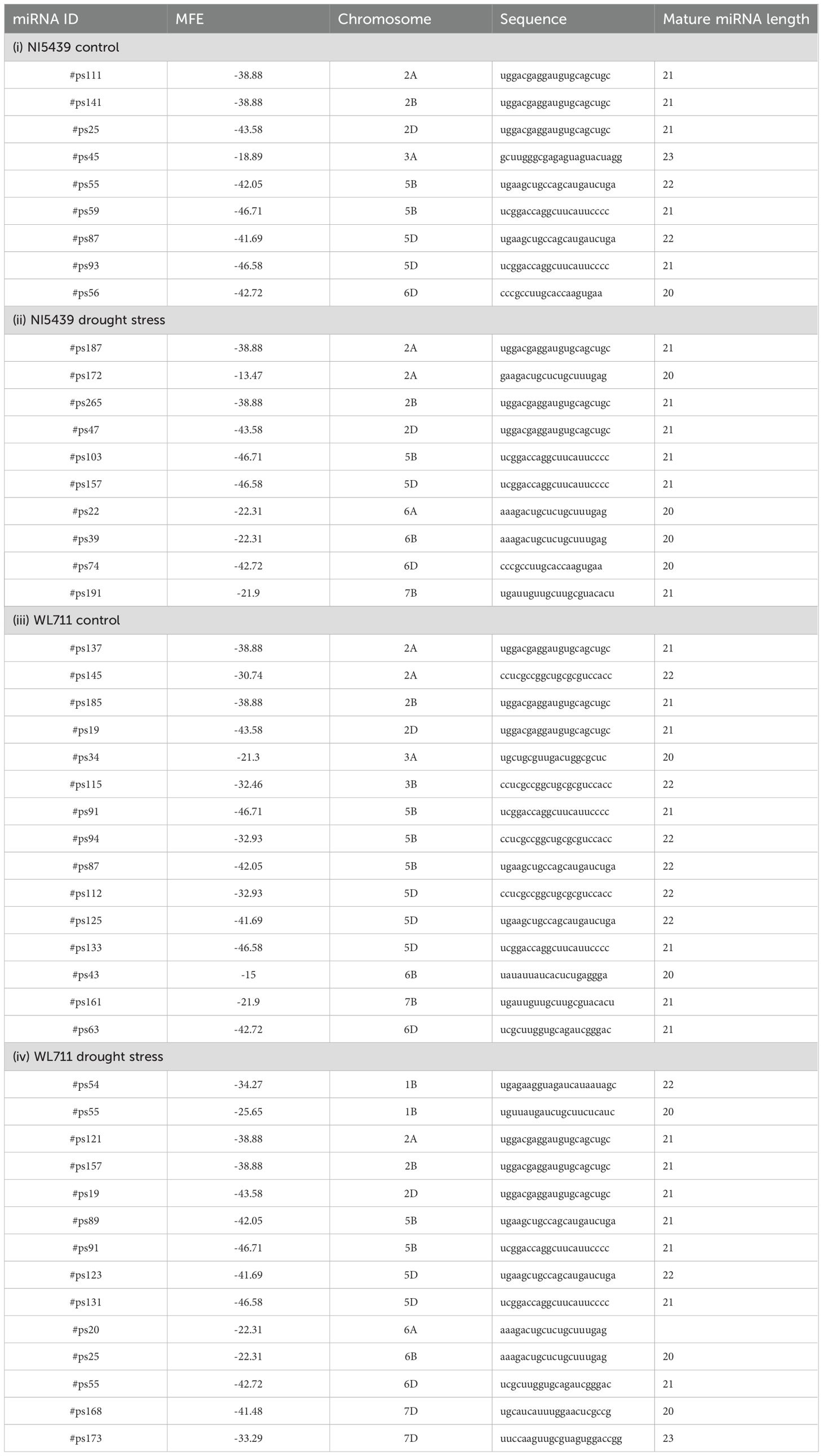
Table 3. Novel miRNAs identified by reference genome of Triticum aestivum in NI5439 and WL711 samples under control and drought stress condition.
Interestingly, two novel miRNAs, #ps19 and #ps91, were consistently detected in both libraries of the drought-sensitive genotype, suggesting their potential involvement in drought stress regulation. The most prevalent length among the novel miRNAs was 21 nucleotides, with a range spanning from 18 to 23 nucleotides. The GC content of these novel miRNAs averaged 55.67 ± 13.16%, indicative of their potential stability and functional relevance. Regarding chromosomal distribution, 13 loci were mapped to genome A, whereas genome B and genome D harbored 26 and 20 loci, respectively. Notably, the highest number of miRNA loci detected on a single chromosome was 8, found on chromosomes 5B and 5D, highlighting possible regions of miRNA enrichment in the wheat genome. These findings provide new insights into the regulatory landscape of wheat miRNAs, particularly under drought stress conditions.
3.5 Gene ontology
To annotate and analyze the functional roles of predicted miRNA target genes in wheat, a total of 4,551 target transcripts (740 in TC, 894 in TD, 1,340 in SC, and 1,577 in SD) were subjected to Gene Ontology (GO) analysis. These transcripts, associated with genes of known functions, were classified into biological processes, cellular components, and molecular functions based on their GO annotations (Figure 5).
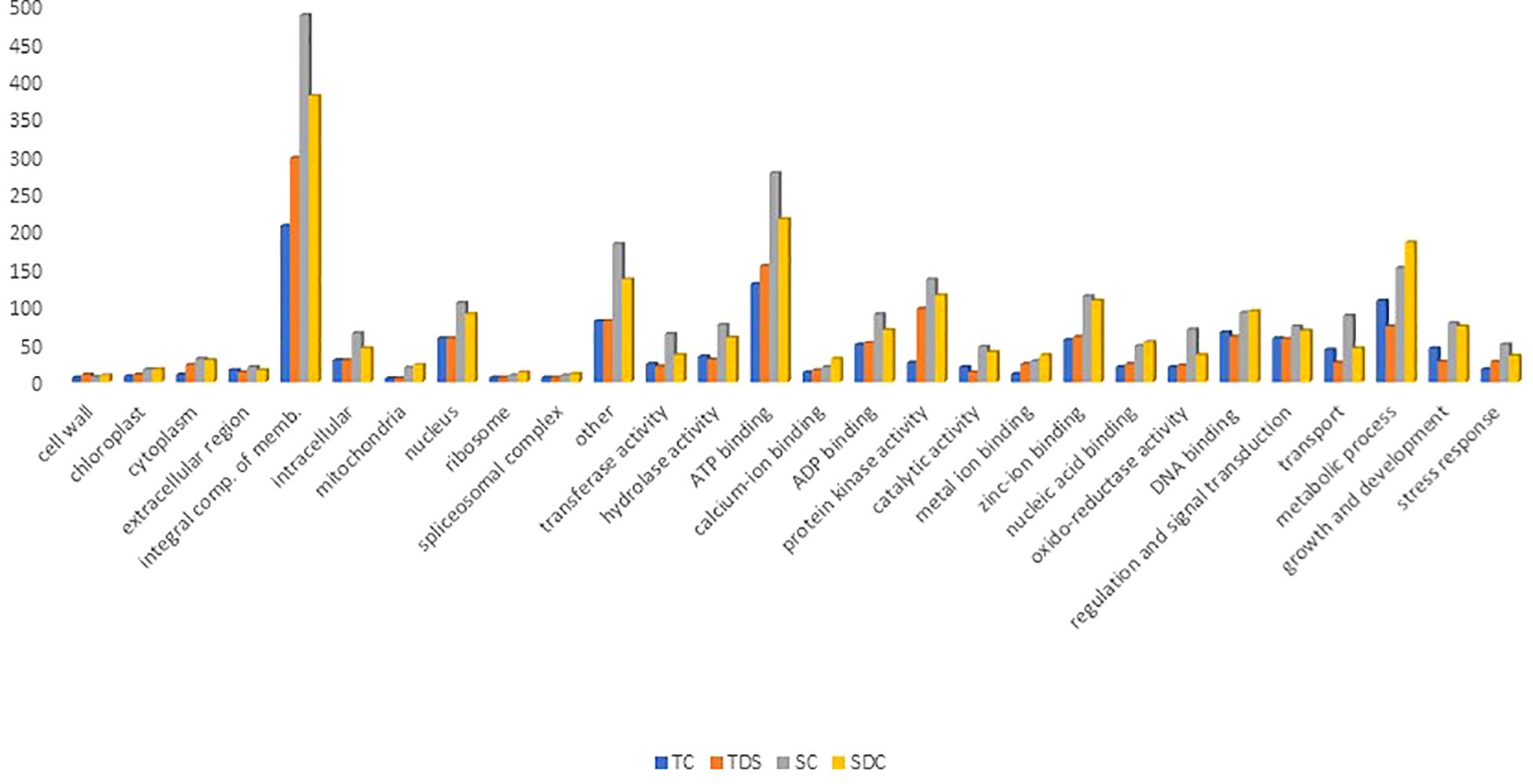
Figure 5. Gene ontology analysis of pooled libraries under control and stress conditions.
In the molecular function category, the predicted miRNA targets were primarily linked to DNA, nucleic acid, and ion binding (183 terms in TC, 167 in TD, 302 in SC, and 322 in SD), catalytic activity (20 in TC, 13 in TD, 47 in SC, and 40 in SD), transferase activity (24 in TC, 21 in TD, 64 in SC, and 36 in SD), protein kinase activity (26 in TC, 97 in TD, 136 in SC, and 115 in SD), and oxidoreductase activity (20 in TC, 22 in TD, 70 in SC, and 36 in SD) (Figure 5; Supplementary Table 6a-d). In the biological processes category, many target transcripts were associated with stress response and defense mechanisms (17 terms in TC, 27 in TD, 50 in SC, and 35 in SD) and regulatory functions, including metabolic processes, growth and development, signal transduction, transcriptional regulation, and photosynthesis (Figure 5; Supplementary Table 6a-d). Similar patterns have been observed in rice and maize (Sunkar et al., 2012b; Božić et al., 2024). The cellular component category revealed that most target genes were associated with membrane components, while fewer were linked to organelles such as chloroplasts, mitochondria, ribosomes, and spliceosomes. The lower representation of chloroplast-related genes suggests a suppression of photosynthetic activity under stress, supported by the decline in photosynthesis-related GO terms in the biological process category. Interestingly, in resistant plants, plastid-associated terms increased under the cellular component category, although photosynthetic activity remained suppressed.
3.6 Gene regulating network of miRNAs
The identification of miRNA-regulated target genes is essential for elucidating the functionalroles of miRNAs in plants, particularly in response to environmental stress. Computational target prediction revealed 314 putative target genes for differentially expressed miRNAs in wheat, spanning various biological and molecular functions. In the tolerant-control (TC) library, five miRNAs were predicted to regulate 41 genes, while 13 novel miRNAs were associated with the regulation of 60 genes in the tolerant-drought (TD) library (Figure 6). Similarly, in the sensitive-control (SC) library, 11 miRNAs were linked to 78 target genes, whereas in the sensitive-drought (SD) library, eight novel miRNAs were predicted to influence the expression of 134 genes under both control and drought stress conditions (Supplementary Figure 2a-c).
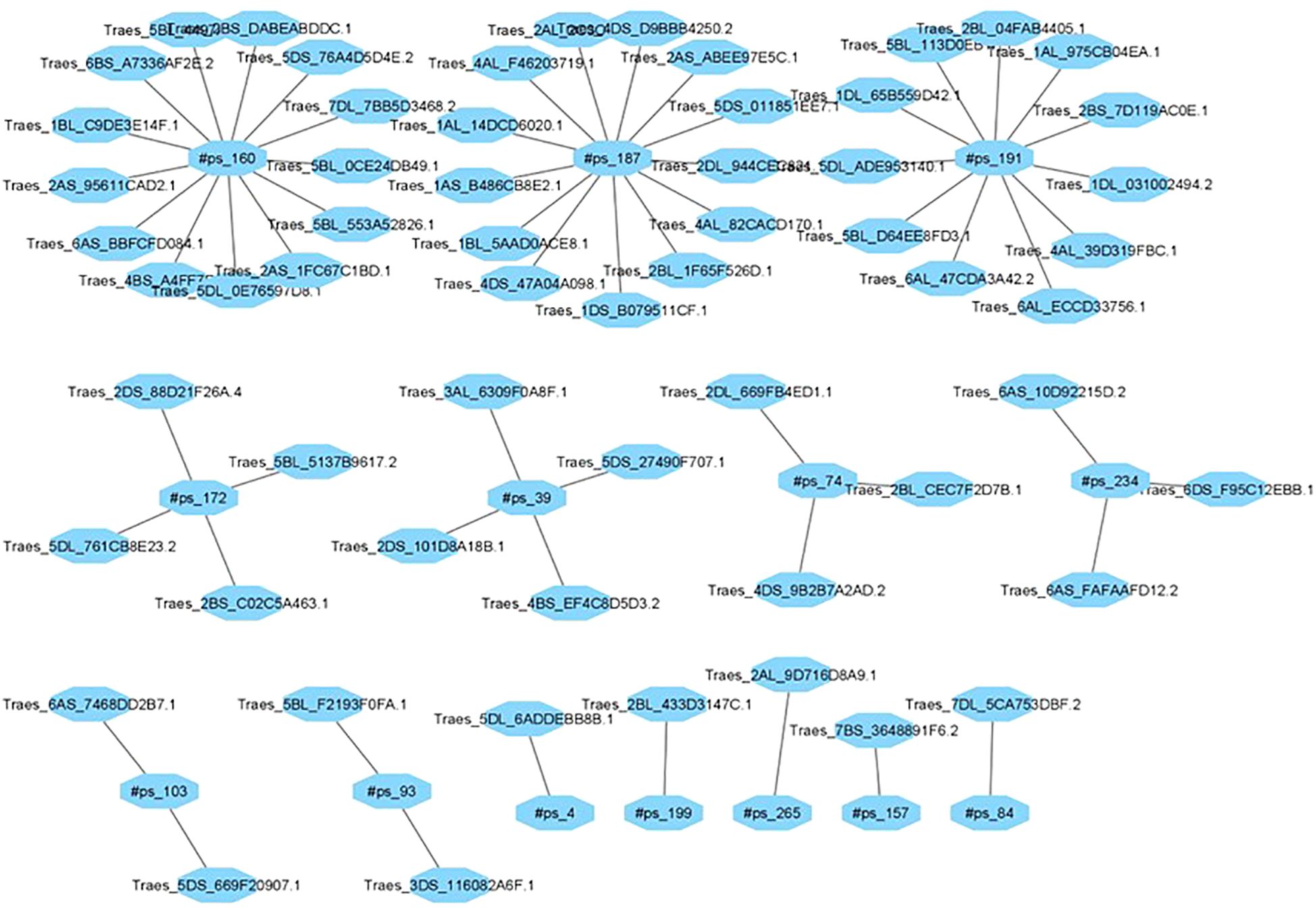
Figure 6. Network of miRNA targets in wheat genes under drought stress in the TD library.
The predicted target genes exhibit a broad spectrum of functional categories, underscoring their pivotal roles in plant growth, development, and stress adaptation. Notably, several identified miRNAs target key transcription factor (TF) families, including MYB, NAC, WRKY, and bZIP, which serve as master regulators of gene expression networks balancing abiotic stress responses, hormone signaling, and developmental processes. The enrichment of TFs among miRNA targets suggests a hierarchical regulatory mechanism wherein miRNAs modulate multiple downstream genes, thereby influencing extensive biological pathways. Additionally, a subset of target genes encodes proteins associated with phytohormonal signaling, such as auxin response factors (ARFs) and abscisic acid (ABA)-responsive elements, which are integral to stress tolerance, growth regulation, and developmental plasticity under drought stress. The identification of histone-modifying enzymes, including histone deacetylases (HDACs) and methyltransferases, implies a potential role for miRNAs in chromatin remodeling and epigenetic regulation, thereby contributing to transcriptional stability and adaptive gene expression in response to environmental stressors. Furthermore, several miRNAs were predicted to regulate genes involved in ion transport and homeostasis, such as potassium and calcium transporters, which are essential for maintaining osmotic balance and intracellular signaling under drought conditions. The identification of miRNA-targeted proteases suggests a role in stress-induced proteolysis, which may facilitate cellular adaptation by regulating protein turnover under adverse conditions. Additionally, the modulation of kinase-mediated phosphorylation cascades by miRNAs indicates their involvement in signal transduction pathways, influencing cellular responses to external stimuli. Collectively, these findings highlight the intricate miRNA-mediated regulatory networks that fine-tune gene expression, thereby enhancing wheat’s adaptive potential to drought stress. A deeper understanding of these interactions could provide a foundation for developing stress-resilient wheat cultivars through targeted genetic and biotechnological approaches.
3.7 In silico expression analysis of miRNAs
The differential expression patterns of conserved miRNAs in both wheat genotypes were analyzed to assess their regulatory roles under drought stress conditions. Hierarchical clustering of all samples was performed to visualize expression trends, with the heatmap representing distinct expression profiles across different experimental conditions (Figure 7).
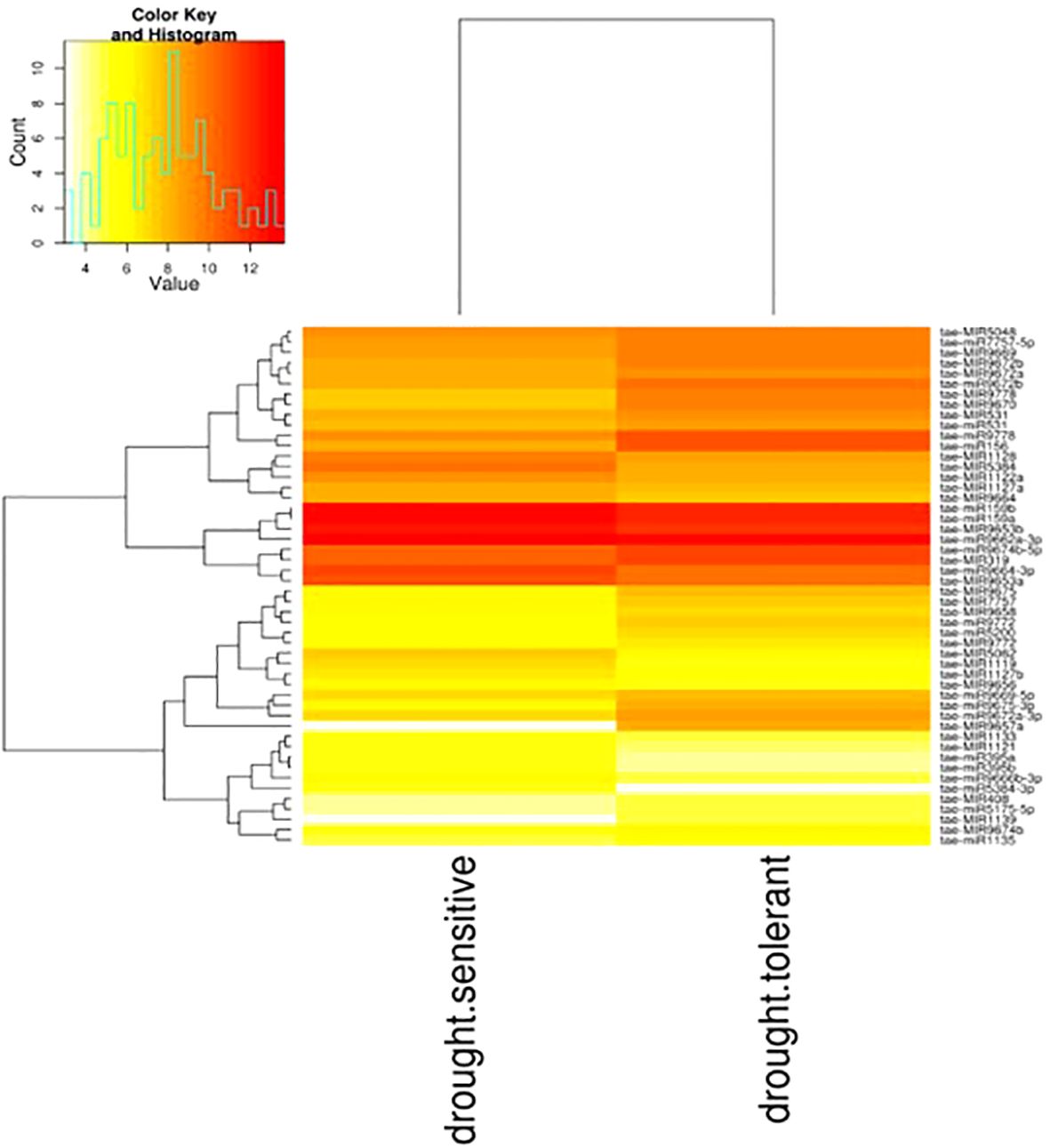
Figure 7. Visualizing differential expression between sample drought-tolerant and drought-sensitive using g-plot heatmap from Wheat. The plot shows the hierarchical clustering of sample both samples. (Yellow shows low-level expression; Red shows high-level expression).
A total of 18 mature miRNAs exhibited significant differential expression based on fold change criteria (<1 or >1), indicating their potential involvement in drought stress response. Among them, six miRNAs—tae-miR395a, tae-miR395b, tae-miR5049-3p, tae-miR5384-3p, tae-miR9664-3p, and tae-miR9666b-3p—were significantly upregulated during drought stress, suggesting their potential role in activating stress-responsive pathways (Table 4). These miRNAs may be involved in regulating sulfur metabolism, oxidative stress responses, and transcriptional regulation under water-limiting conditions.
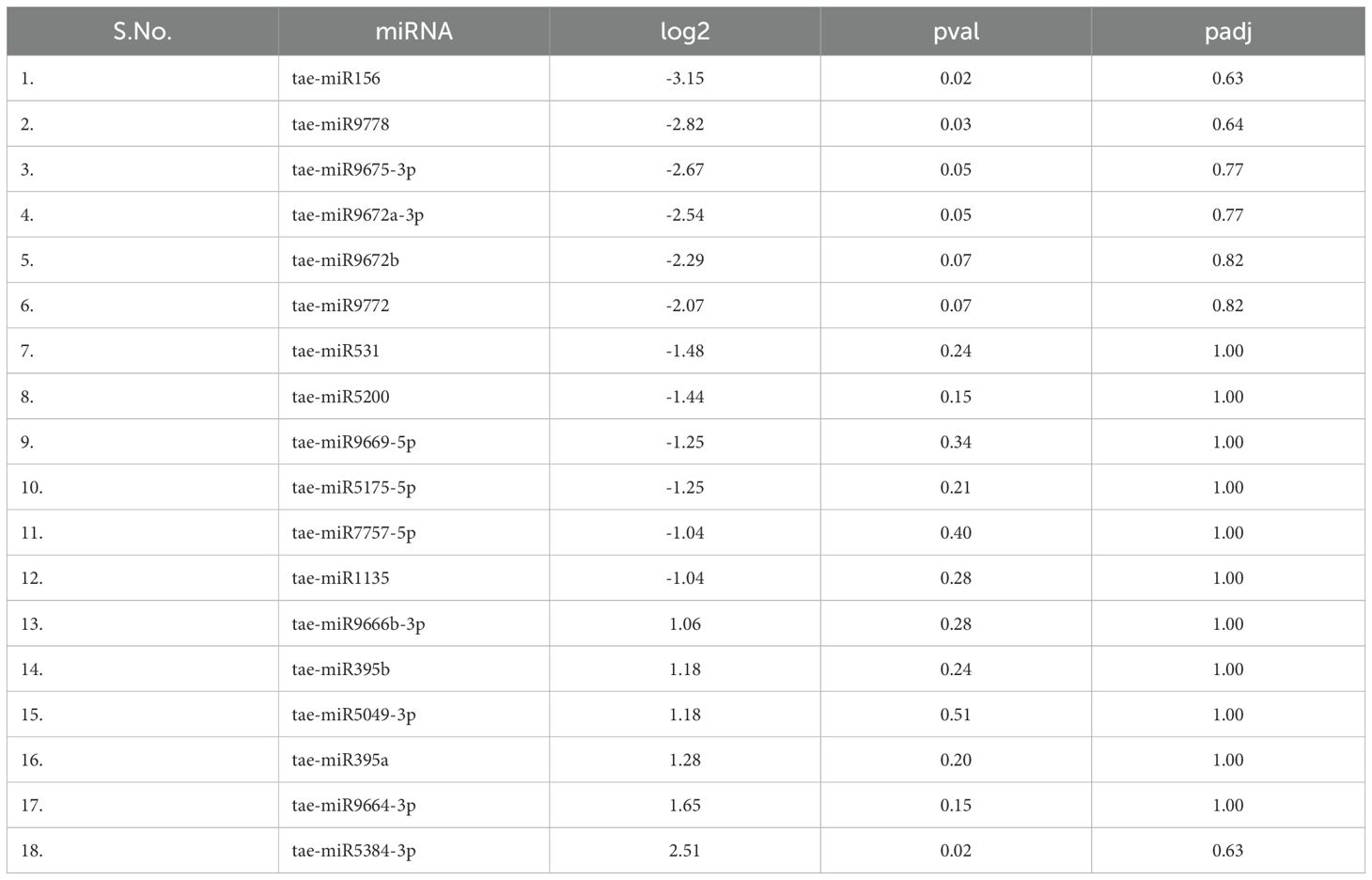
Table 4. Comparative expression Profile of miRNAs in contrasting wheat genotypes.
Conversely, two-thirds of the differentially expressed miRNAs were downregulated under drought stress, including tae-miR156, tae-miR1135, tae-miR531, tae-miR5175-5p, tae-miR5200, tae-miR7757-5p, tae-miR9669-5p, tae-miR9672a-3p, tae-miR9672b, tae-miR9675-3p, tae-miR9772, and tae-miR9778. The repression of these miRNAs may be linked to the modulation of stress-adaptive processes such as leaf morphogenesis, hormone signaling, and secondary metabolite biosynthesis, which are critical for drought tolerance.These results suggest that specific miRNAs play contrasting roles in drought stress adaptation by either enhancing or suppressing gene expression networks associated with plant survival under water-deficient conditions. The observed differential expression patterns provide valuable insights into the complex regulatory mechanisms governing stress responses in wheat, offering potential targets for improving drought resilience through genetic or biotechnological approaches.
3.8 Validation of novel miRNAs
High-throughput deep sequencing of root tissues from contrasting wheat genotypes revealed several putative novel miRNAs involved in drought stress response. To validate these findings, plants were cultivated under both control and drought conditions, followed by qPCR-based expression analysis in root tissues. A subset of ten novel miRNAs and their corresponding target genes were randomly selected for validation, including #ps_55, #ps_199, #ps_45, #ps_160, #ps_19, #ps_91, #ps_187, #ps_103, #ps_74, #ps_47, #ps_89, #ps_157, #ps_55, and #ps_121. Their predicted target genes (TaDRA1–TaDRA14) encode proteins involved in drought response and adaptation mechanisms, including stress-responsive transcription factors, osmotic regulation proteins, and key enzymes in antioxidant defense pathways.
Expression profiling in root tissues revealed genotype-specific regulatory patterns of the novel miRNAs, with distinct expression dynamics between the drought-tolerant (NI5439) and drought-sensitive (WL711) genotypes (Figures 8A, B). Notably, #ps_199 exhibited significantly higher expression in the root tissues of the NI5439 genotype compared to other miRNAs, suggesting a potential role in root-specific drought response mechanisms. The NI5439 genotype displayed elevated expression levels of novel miRNAs relative to WL711, indicating a more robust miRNA-mediated regulatory network in drought adaptation.The identified miRNAs and their associated target genes likely participate in key physiological and molecular pathways, including hormone signaling, osmoprotectant biosynthesis, oxidative stress mitigation, and root system architecture modulation.
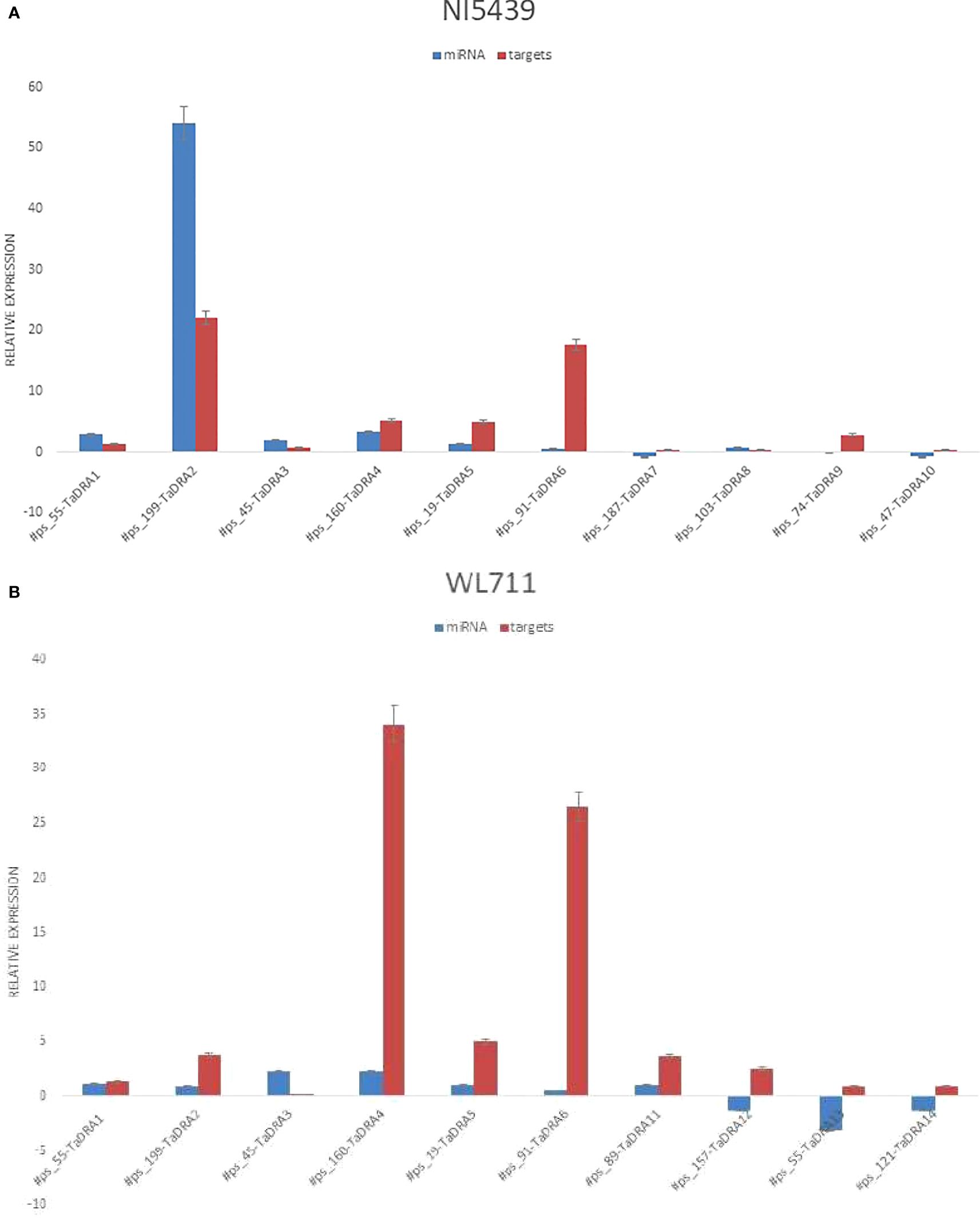
Figure 8. (A) Relative expression levels of miRNAs and their corresponding targets in root tissue of the drought-tolerant genotype NI5439. The orange colour represents the miRNA and blue represents the wheat genes. (B) Relative expression levels of miRNAs and their corresponding targets in the drought-susceptible genotype WL711 root tissue.
4 Discussion
Drought stress is one of the most significant abiotic factors limiting wheat productivity (Triticum aestivum L.). To cope with water-deficit conditions, plants have evolved complex regulatory mechanisms, including miRNA-mediated gene regulation, which plays a crucial role in modulating stress responses at the post-transcriptional level. This study provides a comprehensive analysis of drought-responsive miRNAs in wheat, identifying both conserved and novel miRNAs, along with their target genes and functional pathways. The findings were validated through expression profiling, functional annotation, and comparative analysis with prior studies, offering insights into miRNA-mediated drought tolerance mechanisms.
The identification of conserved and novel miRNAs is crucial for understanding the regulatory landscape of drought stress responses. In this study, multiple conserved miRNAs, including tae-miR159, tae-miR395, tae-miR156, tae-miR398, and tae-miR319, were differentially expressed in response to drought stress. These findings align with prior research in wheat and other cereals, where these miRNAs have been implicated in drought tolerance mechanisms (Sunkar et al., 2007; Akdogan et al., 2016; Zhao et al., 2023a). The upregulation of tae-miR395 in this study is consistent with previous reports highlighting its role in sulfur metabolism and stress adaptation (Zhou et al., 2010; Wang et al., 2013). Similarly, tae-miR398, known to regulate Cu/Zn superoxide dismutase (CSD) genes involved in ROS detoxification, was significantly upregulated, reinforcing its importance in oxidative stress mitigation (Gupta et al., 2014). In addition to conserved miRNAs, 59 novel miRNAs were identified, with several exhibiting genotype-specific expression patterns. Notably, #ps_91 was highly expressed in the drought-tolerant genotype, suggesting their potential role in stress resilience. Similar genotype-dependent miRNA expression has been reported in wheat, barley, and soybean, supporting the hypothesis that novel miRNAs contribute to adaptive stress responses (Gómez-Martín et al., 2023; Božić et al., 2024).
Hierarchical clustering and differential expression analysis revealed that several miRNAs exhibited significant upregulation or downregulation in response to drought stress. A total of 18 mature miRNAs were significantly expressed based on fold-change criteria. Among them, six miRNAs (tae-miR395a, tae-miR395b, tae-miR5049-3p, tae-miR5384-3p, tae-miR9664-3p, and tae-miR9666b-3p) were upregulated, whereas 12 miRNAs, including tae-miR156, tae-miR1135, tae-miR531, and tae-miR5200, were downregulated under drought conditions.The upregulation of tae-miR395 has been linked to enhanced sulfur metabolism and secondary metabolite biosynthesis, essential for stress adaptation (Kawakshima et al., 2009; Zhang J et al., 2022). The downregulation of tae-miR156 is of particular interest, as it has been previously associated with enhanced shoot growth and delayed flowering under stress conditions (Xu et al., 2021). These findings are consistent with prior studies in Arabidopsis and Oryza sativa, where miR156 negatively regulates SPL transcription factors, thereby influencing drought responses (Wang et al., 2011; Trindade et al., 2010). In contrast to previous studies where miR319 was downregulated under drought stress in wheat (Akdogan et al., 2016), this study observed its significant upregulation, particularly in tolerant genotypes. Given its role in targeting TCP transcription factors, this suggests that miR319 might be involved in modifying leaf morphology and cell wall biosynthesis to counteract stress effects.
The identified miRNA-target interactions highlight a complex regulatory network governing stress adaptation in wheat, aligning with previous findings in other plant species. The results demonstrated that several miRNAs target transcription factor (TF) families, including MYB, NAC, WRKY, and bZIP, which are critical regulators of gene expression under abiotic stress. Similar miRNA-mediated regulation of TFs has been reported in Arabidopsis and rice, where these TF families modulate drought-responsive pathways by controlling stress-inducible gene expression (Zhao et al., 2019; Li et al., 2021). The presence of hormone-related target genes, such as auxin response factors (ARFs) and abscisic acid (ABA)-responsive elements, further supports the role of miRNAs in integrating hormonal signaling with stress responses. Studies in maize and barley have demonstrated that miRNAs modulate ABA signaling to fine-tune stomatal regulation and osmotic balance under water-deficit conditions (Guan et al., 2020).
Gene ontology (GO) and KEGG pathway analyses revealed that the miRNA target genes are involved in critical biological processes, including transcriptional regulation, hormone signaling, ion transport, osmotic balance, and ROS detoxification.Several miRNAs were found to target transcription factors, including members of the WRKY, NAC, MYB, and bZIP families. These transcription factors are well-documented regulators of abiotic stress responses (He et al., 2016; Zhang Y et al., 2022). The downregulation of tae-miR169, which targets NF-YA transcription factors, suggests its role in modulating drought-responsive genes, consistent with previous findings in wheat and rice (Zhao et al., 2007; Bakhshi et al., 2014). The involvement of miRNAs in hormone signaling pathways was also evident. tae-miR159 was found to regulate MYB transcription factors involved in ABA signaling, highlighting its role in stress-adaptive hormone responses (Reyes and Chua, 2007; Pandey et al., 2013). Similarly, tae-miR160, which targets auxin response factors (ARFs), exhibited significant expression changes, suggesting its role in modulating root development and drought adaptation (Ding et al., 2013). One of the most striking findings was the regulation of ROS scavenging and oxidative stress pathways. The upregulation of tae-miR398, which regulates superoxide dismutase (SOD), aligns with previous studies demonstrating miRNA-mediated control of oxidative stress responses in wheat and rice (Zhan and Meyers, 2023; Zhou et al., 2024). This supports the hypothesis that miRNA-regulated ROS detoxification plays a central role in drought tolerance.The identification of drought-responsive miRNAs provides a foundation for developing stress-resilient wheat varieties through molecular breeding and genetic engineering. The differential expression of miRNAs between drought-tolerant and drought-sensitive genotypes suggests that miRNAs could serve as biomarkers for selecting drought-adaptive traits in breeding programs.Several miRNAs identified in this study, particularly those regulating transcription factors, hormone signaling genes, and antioxidant enzymes, could be targeted for CRISPR/Cas9-based genome editing to enhance stress tolerance (Jaganathan et al., 2018; Tang and Chu, 2023). Based on our findings, we present a model illustrating miRNA-mediated gene regulation and stress responses in wheat under reproductive stage drought stress (Figure 9). Future studies should focus on functional validation of key miRNAs using transgenic approaches and miRNA knockout strategies to elucidate their precise roles in drought adaptation.Furthermore, integrating miRNA expression data with proteomics, metabolomics, and physiological measurements could provide a more comprehensive understanding of drought stress regulatory mechanisms in wheat. Multi-omics approaches will be crucial in unravelling the complex interplay between miRNAs and their targets, ultimately aiding in the development of climate-resilient crops (Zhao et al., 2023b).
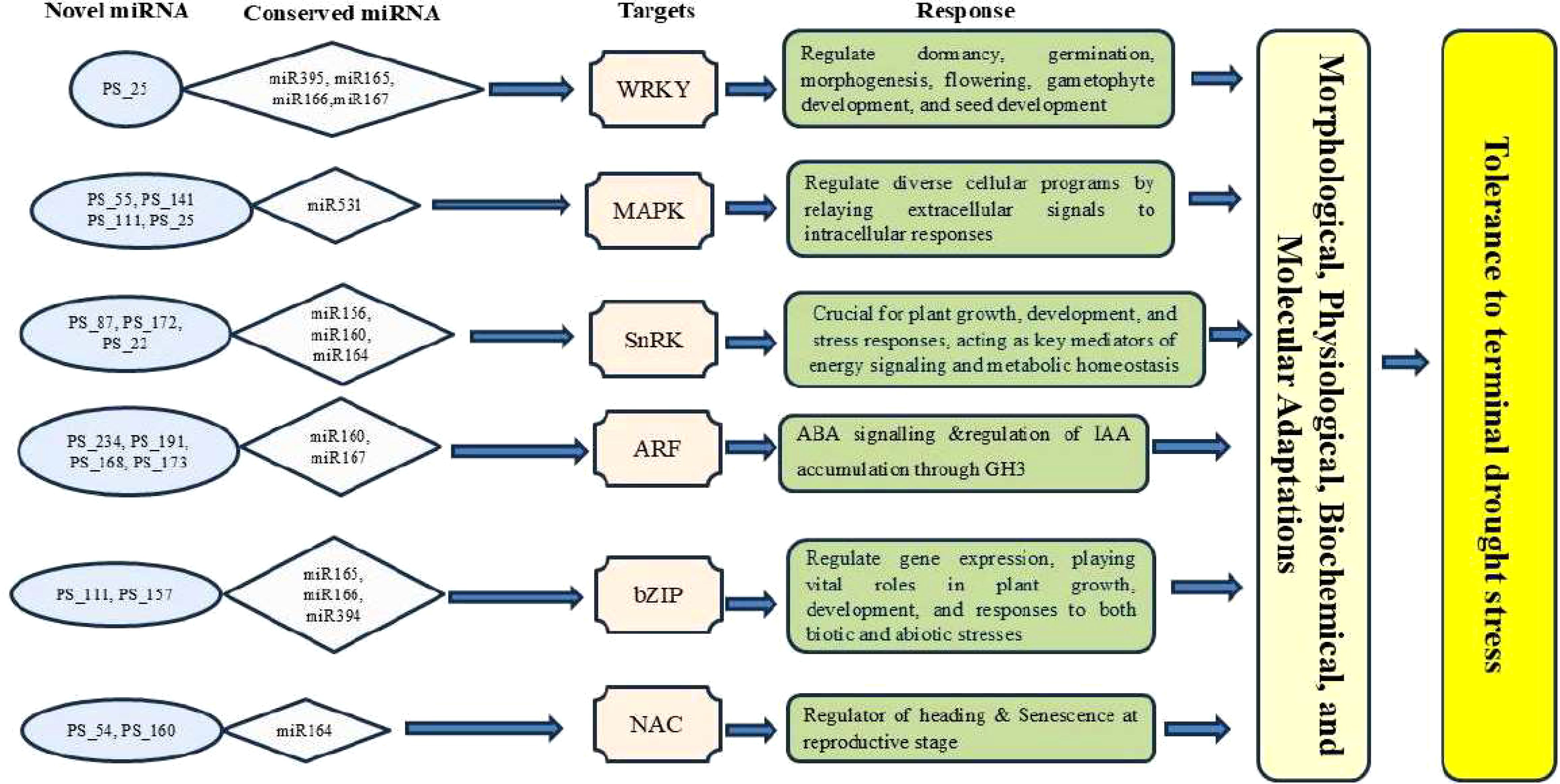
Figure 9. Illustrated depiction of the miRNA-mediated regulatory gene network involving known and novel miRNAs under drought stress at the reproductive stage in wheat.
5 Conclusion
In this study, four sRNA libraries were constructed from roots of drought-tolerant NI-5439 and drought-sensitive WL-711 wheat genotype from control and drought stress conditions at the booting stage. In total, 306 conserved and 58 novel microRNAs were identified from the four libraries. After computational expression analysis of mature miRNAs, 18 miRNAs showed significant changes in the expression after stress treatment. For the first time, 15 conserved miRNAs were emerged as drought-responsive in this study. The predicted targets of novel miRNAs were genes involved in gene silencing by RNA, DNA methylation, histone modification and chromatin modification. This study has significantly expanded the number of novel as well as drought-responsive miRNAs in wheat.
Data availability statement
The data have presented in the study are deposited in the NCBI, accession number is PRJNA1012115.
Author contributions
PS: Conceptualization, Data curation, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing. SM: Formal analysis, Investigation, Methodology, Software, Writing – original draft. AK: Validation, Writing – review & editing. OA: Writing – review & editing. RT: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received forthe research and/or publication of this article. PS acknowledge the financial assistance from Indian Council of Agricultural research, New Delhi and ICAR-IIWBR Core projects to carry out the experiments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1581542/full#supplementary-material
References
Ahmad, P., Abd Allah, E. F., Hashem, A., Sarwat, M., and Gucel, S. (2023). Oxidative stress and antioxidant defense in wheat under drought conditions: Molecular and physiological perspectives. Environ. Exp. Bot. 208, 105181. doi: 10.1016/j.envexpbot.2023.105181
Akdogan, G., Tufekci, E. D., Uranbey, S., and Unver, T. (2016). miRNA-based drought regulation in wheat. Funct. Integr. Genomics 16, 221–233. doi: 10.1007/s10142-016-0476-7
Bailey, J., Oliveri, A., and Levin, E. (2013). A PCR-based method for detection and quantification of small RNAs. Biochem. Biophys. Res. Commun. 23, 1–7. doi: 10.1016/j.bbrc.2006.10.105
Bakhshi, B., Salekdeh, G. H., Bihamta, M. R., and Tohidfar, M. (2014). Characterization of three key MicroRNAs in rice root architecture under drought stress using in silico analysis and quantitative real-time PCR. Biosci. Biotechnol. Res. Asia 11, 555–565. doi: 10.13005/bbra/1306
Berahim, Z., Ismail, M. R., and Rahman, M. A. (2021). Physiological and biochemical responses of wheat to drought stress: Insights into ROS scavenging and photosynthetic efficiency. J. Plant Physiol. 263, 153468. doi: 10.1016/j.jplph.2021.153468
Božić, M., Ignjatović Micić, D., Delić, N., and Nikolić, A. (2024). Maize miRNAs and their putative target genes involved in chilling stress response in 5-day old seedlings. BMC Genomics 25, 1–17. doi: 10.1186/s12864-024-10403-1
Chen, H., Sun, W., and Zhang, L. (2023). Role of brassinosteroids in wheat drought resilience: Osmotic regulation and stress tolerance. Plant Mol. Biol. 112, 431–445. doi: 10.1007/s11103-023-01459-5
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21 (18), 3674–3676. doi: 10.1093/bioinformatics/bti610
Ding, D., Zhang, L., Wang, H., Liu, Z., Zhang, Z., and Zheng, Y. (2013). Differential expression of miRNAs in response to salt stress in maize roots. Ann. Bot. 111, 767–779. doi: 10.1093/aob/mct043
Friedländer, M. R., MacKowiak, S. D., Li, N., Chen, W., and Rajewsky, N. (2012). MiRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52. doi: 10.1093/nar/gkr688
Gómez-Martín, C., Zhou, H., Medina, J. M., Aparicio-Puerta, E., Shi, B., and Hackenberg, M. (2023). Genome-wide analysis of microRNA expression profile in roots and leaves of three wheat cultivars under water and drought conditions. Biomolecules 13 (3), 440. doi: 10.3390/biom13030440
Guan, Q., Wu, J., Zhang, Y., Jiang, C., Liu, R., Chai, C., et al. (2020). Crosstalk between microRNAs and ABA signaling pathways in plant drought resistance. Cells 9, 1757. doi: 10.3390/cells9081757
Gupta, O. P., Meena, N. L., Sharma, I., and Sharma, P. (2014). Differential regulation of microRNAs in response to osmotic, salt, and cold stresses in wheat. Mol. Biol. Rep. 41 (7), 4623–4629. doi: 10.1007/s11033-014-3363-1
He, H., Dong, Q., Shao, Y., Jiang, H., Zhu, S., Cheng, B., et al. (2016). Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 35, 21–37. doi: 10.1007/s00299-015-1866-3
Jaganathan, D., Ramasamy, K., Sellamuthu, G., Jayabalan, S., and Venkataraman, G. (2018). CRISPR for crop improvement: An update review. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00985
Kawashima, C. G., Yoshimoto, N., Maruyama-Nakashita, A., Tsuchiya, Y. N., Saito, K., Takahashi, H., et al. (2009). Sulphur starvation induces the expression of microRNA-395 and one of its target genes but in different cell types. The Plant J. 57, 313–321. doi: 10.1111/j.1365-313X.2008.03690.x
Kaur, A., Gupta, O. P., Meena, N. L., Grewal, A., and Sharma, P. (2017). Comparative temporal expression analysis of micrornas and their target genes in contrasting wheat genotypes during osmotic stress. Appl. Biochem. Biotechnol. 181, 613–626. doi: 10.1007/s12010-016-2236-z
Kaur, R., Singh, J., and Dhillon, B. S. (2023a). Addressing climate challenges in wheat production: Advances in genetic and agronomic approaches. Agron. J. 115, 341–356. doi: 10.2134/agronj2022.05.0275
Kim, J. S., Kim, J., and Lee, H. (2020). Drought-induced oxidative stress and its impact on photosynthesis and growth in wheat: Mechanisms and mitigation strategies. Plant Physiol. Biochem. 150, 178–187. doi: 10.1016/j.plaphy.2020.03.005
Kozomara, A., Birgaoanu, M., and Griffiths-Jones, S. (2019). MiRBase: From microRNA sequences to function. Nucleic Acids Res. 47, D155–D162. doi: 10.1093/nar/gky1141
Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10 (3), R25. doi: 10.1186/gb-2009-10-3-r25
Li, H., Liu, H., Pei, T., Zhang, Z., Yang, Y., and Liu, X. (2021). MicroRNA-mediated hormone signaling and transcription factor regulatory networks in plant drought adaptation. Int. J. Mol. Sci. 22, 4694. doi: 10.3390/ijms22094694
Liu, Y., Shi, A., Chen, Y., Xu, Z., and Yao, Y. (2023). Beneficial microorganisms: Regulating growth and defense for plant welfare. Plant Biotechnol. J. 22, 14554. doi: 10.1111/pbi.14554
Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21. doi: 10.1186/s13059-014-0550-8
Lu, C., Meyers, B. C., and Green, P. J. (2007). Construction of small RNA cDNA libraries for deep sequencing. Methods 43, 110–117. doi: 10.1016/j.ymeth.2007.05.002
Meyers, B. C., Axtell, M. J., Bartel, B., Bartel, D. P., Baulcombe, D., Bowman, J. L., et al. (2008). Criteria for annotation of plant microRNAs. Plant Cell 20, 3186–3190. doi: 10.1105/tpc.108.064311
Mishra, P., Kumar, R., Singh, J., and Sharma, S. (2023a). Genome-wide expression studies reveal miRNA-mediated regulatory networks enhancing drought resilience in wheat. J. Exp. Bot. 74, 1956–1971. doi: 10.1093/jxb/erad065
Narayanan, S., Mohan, A., Gill, K. S., and Vara Prasad, P. V. (2014). Variability of root traits in spring wheat germplasm. PloS One 9 (6), e100317. doi: 10.1371/journal.pone.0100317
Nelson, G. C., Rosegrant, M. W., Koo, J., Robertson, R., Sulser, T., Zhu, T., et al. (2014). Climate change effects on agriculture: Economic impacts, adaptation, and mitigation. Food Policy 43, 18–28. doi: 10.1016/j.foodpol.2013.10.010
Pandey, R., Joshi, G., Bhardwaj, A. R., Agarwal, M., Katiyar-Agarwal, S., and Jagannath, A. (2013). A comprehensive genome-wide study on tissue-specific and abiotic stress-specific miRNAs in Glycine max. Mol. Biol. Rep. 40, 3101–3111. doi: 10.1007/s11033-012-2390-4
Parmar, S., Saharan, K., Kumar, P., and Singh, S. (2020). Climate change and its impact on wheat production: Strategies for mitigation and adaptation. Environ. Sci. Pollution Res. 27, 18192–18210. doi: 10.1007/s11356-020-08492-3
Ramachandran, S., Othman, S. M. I. S., Mustaffa, A. F., and Che-Othman, M. H. (2020). Overview of repressive miRNA regulation by short tandem target mimic (STTM): Applications and impact on plant biology. Plants 12, 669. doi: 10.3390/plants12030669
Reyes, J. L. and Chua, N. H. (2007). ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 49, 592–606. doi: 10.1111/j.1365-313X.2006.02980.x
Saroha, M., Arya, A., Singh, G., and Sharma, P. (2024). Genome-wide expression analysis of novel heat-responsive microRNAs and their targets in contrasting wheat genotypes at reproductive stage under terminal heat stress. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1328114
Sharma, P., Kumar, R., and Bhardwaj, R. (2023). MAPK signaling and hormonal crosstalk in drought stress tolerance of wheat: Mechanistic insights and applications. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1208759
Singh, V., van Oosterom, E. J., Jordan, D. R., Hunt, C. H., and Hammer, G. L. (2011). Genetic variability and control of nodal root angle in sorghum. Crop Sci. 51, 2011–2020. doi: 10.2135/cropsci2011.01.0038
Sun, J., Wang, P., and Li, X. (2023). Cytokinins and their role in mitigating drought-induced senescence in wheat: Molecular mechanisms and agricultural applications. J. Plant Growth Regul. 42, 1023–1038. doi: 10.1007/s00344-023-10782-6
Sunkar, R., Chinnusamy, V., Zhu, J., and Zhu, J. K. (2007). Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 12, 301–309. doi: 10.1016/j.tplants.2007.05.001
Sunkar, R., Kapoor, A., and Zhu, J. K. (2012a). Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 18, 2051–2065. doi: 10.1105/tpc.106.041673
Sunkar, R., Li, Y. F., and Jagadeeswaran, G. (2012b). Functions of microRNAs in plant stress responses. Trends Plant Sci. 17, 196–203. doi: 10.1016/j.tplants.2012.01.010
Tang, M. and Chu, C. (2023). MicroRNAs in crop improvement: Fine-tuners for complex traits. New Phytol. 237, 1688–1703. doi: 10.1111/nph.18600
Tian, T., Liu, Y., Yan, H., You, Q., Yi, X., Du, Z., et al. (2017). AgriGO v2.0: a GO analysis toolkit for the agricultural community update. Nucleic Acids Res. 45 (W1), W122–W129. doi: 10.1093/nar/gkx382
Trindade, I., Capitão, C., Dalmay, T., Fevereiro, M. P., and Santos, D. M. (2010). miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Plant Cell Environ. 33, 1582–1597. doi: 10.1111/j.1365-3040.2010.02165.x
Wang, Y., Chen, J., and Zhang, Q. (2022). The interplay between abscisic acid and jasmonic acid in drought adaptation: Regulatory networks and physiological implications. J. Exp. Bot. 73, 2857–2870. doi: 10.1093/jxb/erac024
Wang, J. W., Czech, B., and Weigel, D. (2011). miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 138, 738–749. doi: 10.1016/j.cell.2009.06.014
Wang, Y., Xu, L., Zhu, X., Zha, S., Yu, R., and Sun, L. (2013). Identification of microRNAs and their targets in wheat (Triticum aestivum L.) under phosphorus deprivation. J. Plant Physiol. 170 (18), 1668–1674. doi: 10.1016/j.jplph.2013.06.012
Wang, X., Zhang, H., Li, Y., and Zhao, C. (2023). Advances in breeding drought-tolerant wheat varieties: Molecular and physiological perspectives. Plant Physiol. Biochem. 201, 107611. doi: 10.1016/j.plaphy.2023.107611
Xu, M. Y., Zhang, L., Li, W. W., Hu, X. L., Wang, M. B., Fan, Y. L., et al. (2021). Stress-responsive miR156 improves drought resistance by modulating SPLs and metabolic processes in Oryza sativa. J. Exp. Bot. 72, 3754–3770. doi: 10.1093/jxb/erab017
Zadoks, J. C. and Board, E. (1999). Data sheet highlights close coupled pumps. World Pumps 1999, 9. doi: 10.1016/s0262-1762(99)80614-2
Zhan, J. and Meyers, B. C. (2023). Stress adaptation and innovations: Emerging roles of miRNAs in plant genomes. Trends Plant Sci. 28, 66–82. doi: 10.1016/j.tplants.2022.08.010
Zhang, Y., Wang, L., Li, Z., and Peng, Y. (2022). Genome-wide identification and characterization of the WRKY gene family in wheat (Triticum aestivum L.). BMC Genomics 23, 310. doi: 10.1186/s12864-022-08560-2
Zhang, J., Wang, Q., Wang, D., Du, Y., Zhang, G., and Yang, L. (2022). miR395 promotes sulfur metabolism and secondary metabolism to enhance stress adaptation in wheat. Int. J. Mol. Sci. 23, 2054. doi: 10.3390/ijms23042054
Zhao, B., Ge, L., Liang, R., Li, W., Ruan, K., Lin, H., et al. (2007). Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol. Biol. 8, 52. doi: 10.1186/1471-2199-8-52
Zhao, Y., Liu, H., Wang, X., and Li, Z. (2024). Impact of global warming on wheat productivity: Current trends and future projections. Nat. Climate Change 14, 45–57. doi: 10.1038/s41558-024-01764-9
Zhao, Y., Wang, Y., Xu, J., and Li, X. (2023a). The role of conserved miRNAs in drought stress responses in wheat and cereals: Regulatory mechanisms and adaptive significance. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1189472
Zhao, Y., Xu, Z., Mo, Q., Zou, C., Li, W., Xu, Y., et al. (2019). Combined small RNA and degradome sequencing reveals microRNA regulatory network with MYB, NAC, WRKY, and bZIP transcription factors in maize. Sci. Rep. 9, 16328. doi: 10.1038/s41598-019-52856-1
Zhao, Y., Zhang, Y., and Wang, M. (2023b). Multi-omics approaches reveal the regulatory network of microRNAs in wheat under drought stress. Plant Sci. 327, 111543. doi: 10.1016/j.plantsci.2023.111543
Zhou, L., Liu, Y., Liu, Z., Kong, D., Duan, M., and Luo, L. (2010). Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J. Exp. Bot. 61, 4157–4168. doi: 10.1093/jxb/erq237
Zhou, M., Chen, H., Wei, D., Ma, H., and Lin, J. (2024). miR398 modulates plant responses to drought stress through regulation of SOD and other ROS-scavenging enzymes in rice. Plant Sci. 315, 111123. doi: 10.1016/j.plantsci.2023.111123
Keywords: wheat, miRNA, transcripts, abiotic stress, drought, DEG, reproductive stage
Citation: Sharma P, Mishra S, Kaur A, Ahlawat OP and Tiwari R (2025) Novel and conserved drought-responsive microRNAs expression analysis in root tissues of wheat (Triticum asetivum L.) at reproductive stage. Front. Plant Sci. 16:1581542. doi: 10.3389/fpls.2025.1581542
Received: 22 February 2025; Accepted: 21 April 2025;
Published: 20 May 2025.
Edited by:
Simardeep Kaur, The ICAR Research Complex for North Eastern Hill Region (ICAR RC NEH), IndiaReviewed by:
Subodh Kumar Sinha, Indian Council of Agricultural Research, IndiaSundeep Kumar, Indian Council of Agricultural Research (ICAR), India
Copyright © 2025 Sharma, Mishra, Kaur, Ahlawat and Tiwari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pradeep Sharma, UHJhZGVlcC5TaGFybWFAaWNhci5vcmcuaW4=