 Qingqing Ye
Qingqing Ye Na Zhang2†
Na Zhang2† Wei Zhou
Wei Zhou Zhikun Wu
Zhikun Wu- 1Department of Pharmacy, Guizhou University of Traditional Chinese Medicine, Guiyang, China
- 2Department of Pharmacy, Tongren Hospital of Traditional Chinese Medicine, Tongren, China
- 3Germplasm Bank of Wild Species & Yunnan Key Laboratory of Crop Wild Relatives Omics, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China
Introduction: Indigofera stachyodes Lindl. is a perennial shrub belonging to the Fabaceae family that has been traditionally utilized as a medicinal plant by ethnic minority groups in Guizhou Province, China. This species exhibits significant ethnopharmacological value in local traditional medicine systems. The plant predominantly inhabits karst mountainous regions characterized by frequent drought stress, which represents a typical harsh habitat for plant growth. Notably, drought conditions particularly impair the establishment and development of I. stachyodes seedlings. However, the molecular mechanisms underlying its drought tolerance and adaptive responses remain largely unexplored, warranting further investigation at the molecular level.
Methods: We conducted pot-based water control experiments to subject I. stachyodes seedlings to drought stress treatments (CK, T0, T2). Root tissues from each treatment group were analyzed using transcriptomics (RNA-seq) and metabolomics (LC-MS/GC-MS) approaches to identify differentially expressed genes (DEGs) and differentially expressed metabolites (DEMs). Through integrated analysis of DEGs and DEMs, we performed KEGG pathway enrichment and constructed co-expression networks to elucidate the molecular mechanisms underlying drought stress responses in the roots of I. stachyodes seedlings.
Results: A total of 11,509 DEGs were detected in the transcriptome. Among them, the CK vs T0 group shared 7,191 DEGs, the CK vs T2 group shared 1,264 DEGs, and the T2 vs T0 group shared 3,054 DEGs. In the metabolome, a total of 622 metabolites were detected. Among them, the CK vs T0 group shared 187 DEMs, the CK vs T2 group shared 127 DEMs, and the T2 vs T0 group shared 86 DEMs. The transcriptome-metabolome analysis revealed that the roots of I. stachyodes seedlings regulate metabolic balance through the phenylpropanoid biosynthesis pathway and the flavonoid biosynthesis pathway when subjected to varying degrees of drought stress. Metabolites such as p-coumaric acid, sinapine malate, eugenol, coumestrol, medicarpin, prunin, isosakuranetin, vitexin, gallocatechin, catechin, garbunzol and dihydromyricetin, along with genes including PAL, C4H, COMT, 4CL, CHS, DFR, HIDH, I2’H, IF7GT, IF7MAT, IFR, VR, PTS and IFS are potential key substances that enable the roots of I. stachyodes seedlings to resist drought stress.
Discussion: These results elucidate that the roots of I. stachyodes seedlings can resist drought stress and adapt to drought environments by regulating the expression of genes and the synthesis of metabolites in the flavonoid and phenylpropanoid metabolic pathways, providing a foundation to facilitate the domestication of wild I. stachyodes.
1 Introduction
Indigofera stachyodes Lindl., a species of the genus Indigofera (family Fabaceae), is primarily distributed in the karst mountainous regions of Guizhou, Yunnan, and Guangxi in China, where the plant has been traditionally used as a medicinal resource by ethnic minority communities. Its dried root is a renowned medicinal material known as “Blood Ginseng” (Xue Renshen), one of the top ten Miao ethnic medicines in Guizhou Province. It possesses the effects of tonifying deficiency, promoting blood circulation, securing collapse, draining dampness, nourishing yin, and resolving phlegm. It is primarily used to treat conditions such as chronic diarrhea due to physical weakness, metrorrhagia and metrostaxis, rheumatic arthralgia, cold and fever, cough, non-healing ulcers, traumatic injuries, and infantile malnutrition (Chen et al., 2014). In addition, Qijiao Shengbai capsule, which is produced using I. stachyodes as the main ingredient, has shown clinical efficacy in treating leukemia (Zhao et al., 2020). Currently, the medicinal material derived from I. stachyoides is predominantly obtained from wild sources. The environment in which these wild populations exist has a direct impact on their survival and expansion. Furthermore, the ability of seedlings to adapt to the ecological conditions of their natural habitat significantly influences both their survival rates and the overall development of the plant population.
Water is essential for plant growth, development, and physiological metabolism. However, approximately one-third of the world’s land area and half of China’s territory are in arid or semi-arid conditions (Wen et al., 2020). Plants undergo phenotypic, biochemical, and molecular-level changes when facing drought stress. In severe cases, this can lead to the cessation of photosynthesis, metabolic disorders, and even plant death (Jaleel et al, 2008). Therefore, research on plant adaptation to water environments is of great significance. During medicinal plant growth, root systems experience drought stress when water supply-demand balance is disrupted. The roots are the most sensitive and the first organ to respond to such conditions (Maurel and Nacry, 2020). Recent studies on medicinal plant responses to drought stress have primarily investigated root systems. Representative examples include: PEG-6000-induced stress effects on tanshinone accumulation in Salvia miltiorrhiza hairy roots (Sheng and Chen, 2013); effect of drought stress on root morphology and physiological characteristics of Malus micromalus cv. ‘Ruby’ (Zheng et al, 2020); and transcriptome sequencing-based analysis of gene expression regulation in Glycyrrhiza uralensis roots under moderate drought stress (Zhang et al., 2015).
With the rapid development of next-generation DNA sequencing technologies and systems biology, multi-omics approaches have become indispensable research methodologies. These techniques provide systematic and cellular-level insights into the dynamic changes occurring during plant growth and development. Among them, transcriptomics and metabolomics stand out as relatively mature, in-depth, and technically accessible platforms (Wu et al., 2023). Transcriptomics enables high-throughput acquisition of RNA-level gene expression information, revealing intrinsic connections between gene expression patterns and biological phenomena. Metabolomics serves as the critical bridge between genotype and phenotype, providing systematic analysis of all low-molecular-weight metabolites in tissues, organs, or cells to identify significantly differential metabolites with biological importance. Integrated transcriptomic-metabolomic analysis allows comprehensive investigation of plant physiological changes from both causal (transcriptional regulation) and consequential (metabolic output) perspectives. This approach facilitates the identification of key pathways associated with metabolic alterations and subsequent construction of core regulatory networks, thereby elucidating their underlying mechanisms (Wang et al., 2015; Duan et al., 2016; Xue et al., 2022). Currently, integrated transcriptomic-metabolomic analysis has been widely applied in plant abiotic stress research. Representative studies include the mechanistic elucidation of drought resistance in Gossypium hirsutum through integrated transcriptomic-metabolomic profiling (Zhang, 2018), an investigation into the drought resistance of tartary buckwheat based on metabolomics and transcriptomics (Ouyang, 2020), and the phosphorus-mediated modulation of ionomic-metabolic networks under drought stress in Triticum aestivum seedlings (Li et al., 2022).
Therefore, to examine the effects of drought stress on the biosynthesis of root secondary metabolites in I. stachyodes seedlings, this study performed integrated transcriptomic and metabolomic analyses on root samples subjected to drought stress. The objective is to elucidate the response mechanisms from the secondary metabolites in I. stachyodes roots under drought conditions, thereby providing foundational data to facilitate the domestication of wild I. stachyodes.
2 Materials and methods
2.1 Plant material
The seeds utilized in this study were collected from the Yue-liang River in Liuzhi, Guizhou Province, China. They were then germinated under controlled conditions within growth chambers and subsequently cultivated in a greenhouse with a photoperiod of 16 hours light and 8 hours dark. After reaching stable growth stage, uniformly developed seedlings were selected for pot-based drought stress experiments. Three gradients of field soil water holding capacity were established: 0–5% relative field capacity (extreme drought, T0), 40–45% relative field capacity (moderate drought, T2), and 80–85% relative field capacity (CK, control group). Soil moisture content was measured using the weighing method. The water stress treatment lasted for 30 days. After the experiment, root samples from seedlings in the T0, T2, and CK groups were randomly collected, with three replicates per treatment. The samples were immediately frozen in liquid nitrogen, stored in cryotubes, and preserved at −80°C for subsequent RNA and metabolite extraction.
2.2 Transcriptome sequencing and data analysis
RNA extraction was performed using the tengen biotech RNA extraction kit (DP441) according to the manufacturer’s protocol. The cDNA library was constructed and subjected to quality control using illumina’s NEBNext® Ultra™ RNA library prep kit. After quality inspection, sequencing was carried out on the illumina platform. High-quality clean reads were obtained from the raw reads using the fastp tool to remove low-quality bases and N bases. Trinity and corset software were used to assemble and cluster transcripts to remove redundancy. Subsequently, the corresponding amino acid sequences were predicted from the processed transcripts.
DIAMOND software was then utilized to align the redundant transcript sequences against seven databases: kyoto encyclopedia of genes and genomes (KEGG), non-redundant protein sequence database (NR), curated protein sequence database (Swiss-Prot), gene ontology (GO), clusters of orthologous groups (COG/KOG), translated EMBL nucleotide sequence database (TrEMBL), and protein family database (PFAM) to generate functional annotations for the transcripts. The expression levels of transcripts were calculated using RNA-Seq by expectation-maximization (RSEM) software and quantified based on fragments per kilobase of transcript per million mapped reads (FPKM) values. Differential expression analysis between groups was performed using DESeq2 (Love et al., 2014; Huson and Buchfink, 2015; Varet et al., 2016). Differentially expressed genes (DEGs) were identified based on the thresholds of |log2fold change (FC)| ≥ 1 and FDR < 0.05. Subsequently, GO and KEGG enrichment analyses were conducted to determine the main biological functions and pathways associated with the DEGs.
2.3 Metabolomic profiling and data analysis
Samples from each treatment group were ground into powder and extracted with methanol. The extracts were vortexed, centrifuged, and filtered to obtain the supernatant, which was stored for further analysis. The samples were analyzed using an ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) system equipped with a triple quadrupole detector. The UPLC conditions included optimized parameters for column type, mobile phase composition, elution gradient, flow rate, column temperature, and injection volume. The MS/MS analysis was performed with optimized ion source settings, including temperature, voltage, and gas flow rates. Qualitative and quantitative analyses of metabolites were conducted using the metabolomics workbench database (MWDB) and multi-reaction monitoring following standard protocols. To monitor technical variability during metabolomic profiling, a pooled quality control (QC) sample was prepared by combining equal aliquots (10 μL) from all experimental samples. The QC sample was injected repeatedly (n=5) at the beginning of the analytical sequence to equilibrate the system, followed by periodic injections after every 10 experimental samples throughout the LC-MS/MS run. This QC strategy enabled comprehensive monitoring of instrument stability, evaluation of data reproducibility, correction of batch effects, and filtration of technical noise.
Principal component analysis (PCA) and orthogonal partial least squares discriminant analysis (OPLS-DA) were performed on the metabolite data using the statistical functions in R software. These analyses aimed to identify differences in metabolites among samples, assess variation within groups, filter out noise, highlight inter-group differences, and facilitate the screening of differentially expressed metabolites (DEMs). The relative content of metabolites was normalized using the unit variance scaling method (UV). Hierarchical cluster analysis (HCA) was then performed, and heatmaps were generated using the pheatmap software package.
DEMs were screened by combining FC values and variable importance in projection (VIP) values from the OPLS-DA model, with thresholds set at |log2FC| ≥ 1, P < 0.05, and VIP > 1. Subsequently, KEGG enrichment analysis was performed on the identified DEMs to explore their associated biological pathways.
2.4 Integrated analysis of transcriptome and metabolome data
The results of metabolomic DEMs and transcriptomic DEGs were subjected to co-expression analysis and mapped onto KEGG pathways to identify biological pathways simultaneously affected at both transcriptional and metabolic levels under drought stress in I. stachyodes seedlings. This integrated approach comprehensively reveals the intrinsic connections between molecular regulation and physiological metabolic changes in the root system under drought conditions.
3 Results
3.1 Transcriptomic sequencing and analysis
The sequencing generated a total of 416,171,080 clean reads, corresponding to 62.42 Gb of high-quality clean bases. All libraries exhibited Q30 scores above 92.91%, with GC content ranging from 42.06% to 44.30%. De novo assembly using Trinity software produced 176,876,362 mapped reads, with mapping rates exceeding 80.66% (Supplementary Table S1). These results demonstrate high sequencing data saturation and excellent assembly performance. Pairwise correlation analysis revealed high reproducibility among replicates, with Pearson correlation coefficients ranging from 0.73 to 0.99 (Supplementary Figure S1). These results demonstrate strong inter-sample consistency, confirming the technical reliability of our transcriptomic datasets for subsequent analyses.
3.2 Analysis of DEGs between different treatments
The results show that a total of 11,509 DEGs were identified, including 5,835 up-regulated and 5,674 down-regulated genes. Specifically, the CK vs T0 group contained 7,191 DEGs (3,769 up-regulated and 3,422 down-regulated), the CK vs T2 group had 1,264 DEGs (498 up-regulated and 766 down-regulated), and the T2 vs T0 group exhibited 3,054 DEGs (1,568 up-regulated and 1,486 down-regulated). (Supplementary Table S2). Venn diagram analysis (Figure 1) revealed 67 common DEGs shared among all three treatment groups, while each comparison group exhibited unique DEG profiles: 4,654 (CK vs T0), 417 (CK vs T2), and 917 (T2 vs T0) group-specific DEGs. These results demonstrate distinct transcriptional reprogramming in I. stachyodes seedling roots under progressive drought stress intensities, with the number of responsive DEGs increasing significantly as drought severity escalates.
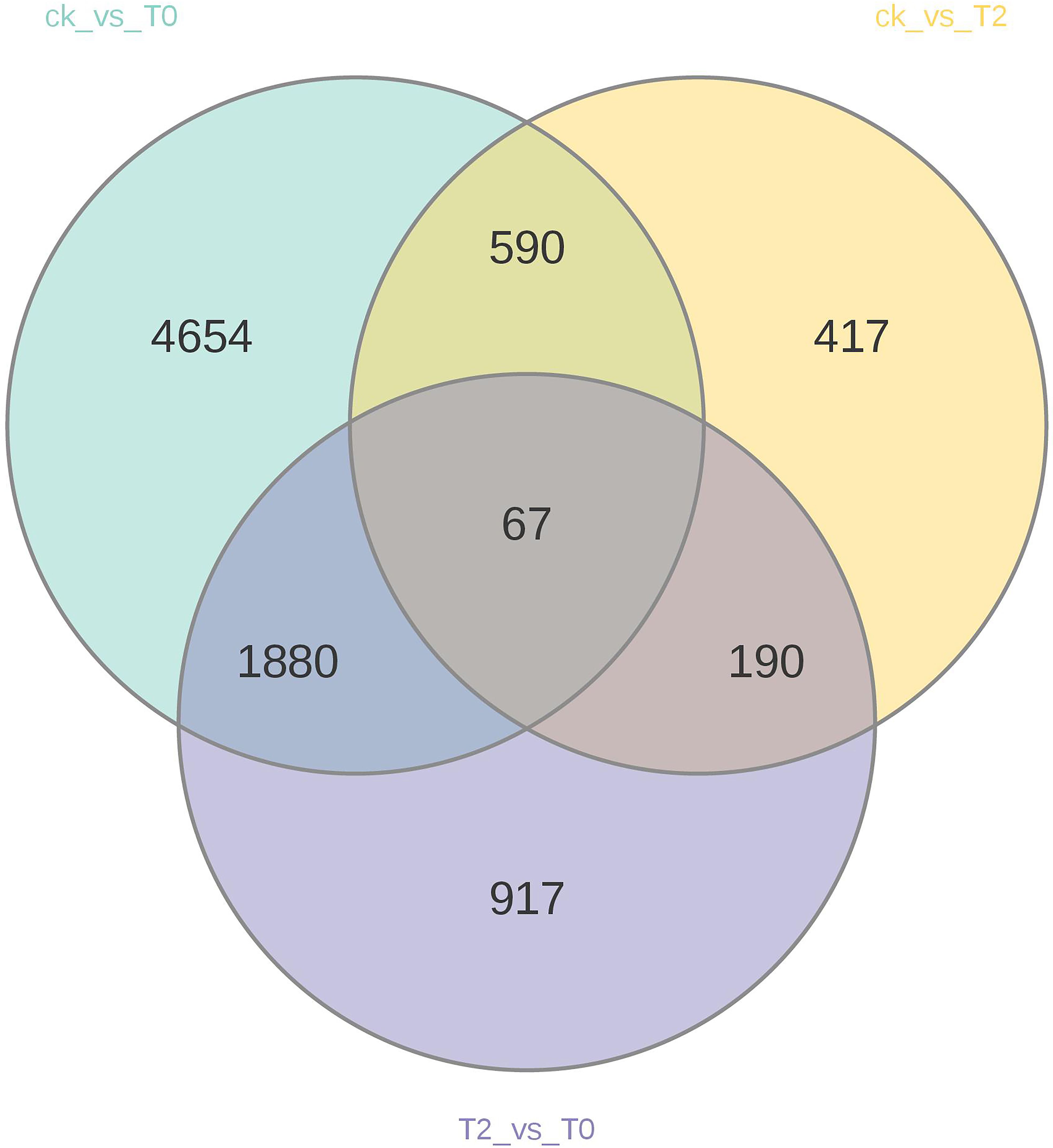
Figure 1. Venn diagram of differentially expressed genes (DEGs) analysis in each group.
HCA (Figure 2) revealed distinct expression patterns of DEGs across water stress treatments. Based on the expression levels of DEGs in each group under different degrees of drought treatment, the three treatment groups can be divided into two categories: T2 and CK are clustered into one category, while T0 forms a separate category. It is evident that the DEGs in the roots of I. stachyodes seedlings exhibited significant differences under varying degrees of drought stress.
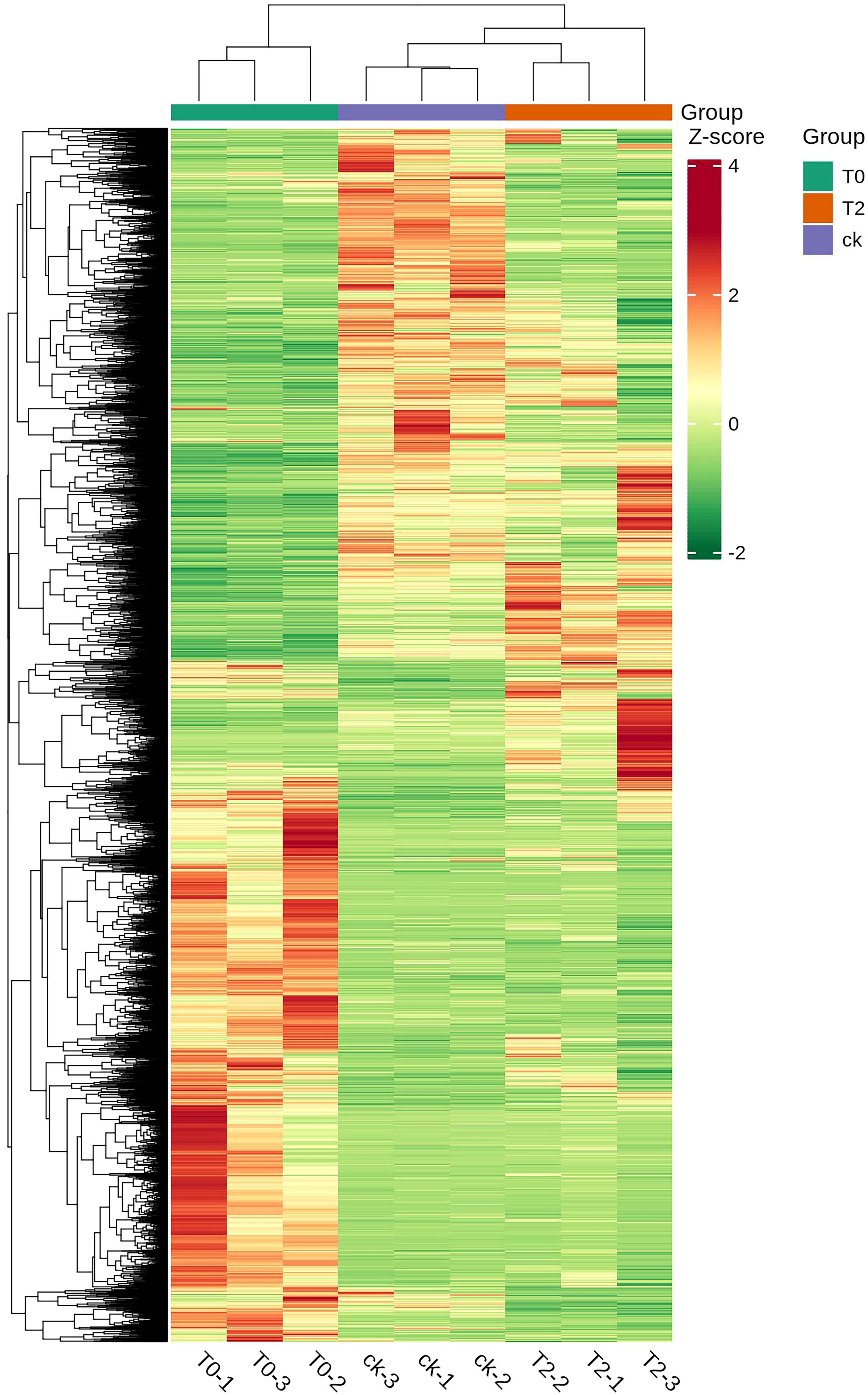
Figure 2. Hierarchical cluster analysis (HCA) of DEGs expression in each group.
3.3 GO functional enrichment analysis of DEGs
GO functional enrichment analysis revealed that in the CK vs T0 comparison group, a total of 4,965 DEGs were enriched, with 431 DEGs showing significant enrichment (P<0.05). These included 231 DEGs involved in biological processes, 172 in molecular functions, and 28 in cellular components. Among the top 20 most significantly enriched terms (lowest P-values), 9 were associated with biological processes-primarily related to phenylpropanoid and flavonoid biosynthesis and metabolic pathways; 5 were linked to cellular components-mainly ribosomal subunits; and 6 were connected to molecular functions predominantly enzyme activity-related terms.
The GO enrichment analysis identified 2,422 DEGs in the CK vs T2 comparison group, with 252 DEGs showing significant enrichment (P<0.05). These included 162 DEGs involved in biological processes, 11 in cellular components, and 79 in molecular functions. Among the top 20 most significantly enriched terms (lowest P-values), analysis revealed: 10 terms associated with biological processes-primarily cellular response to hypoxia; 1 term linked to cellular components-specifically the anchored component of membrane; and 9 terms connected to molecular functions-mainly inhibition of peptidase and endopeptidase activities.
GO functional enrichment analysis of the T2 vs T0 comparison identified 3,933 DEGs, with 326 showing significant enrichment (P<0.05). These included 186 DEGs associated with biological processes, 120 with molecular functions, and 20 with cellular components. Among the top 20 most significantly enriched terms (lowest P-values), analysis revealed: 8 biological processes-primarily cellular response to oxygen levels; 6 cellular components-mainly ribosomal subunits; and 6 molecular functions-predominantly translation elongation factor activity and water channel activity.
3.4 KEGG pathway enrichment analysis of DEGs
KEGG enrichment analysis revealed that the DEGs in the CK vs T0 comparison group were mapped to 140 metabolic pathways, with 27 pathways showing significant enrichment (P<0.05). The top 20 most significantly enriched pathways (Figure 3A) included: ribosome biogenesis in eukaryotes, phenylpropanoid biosynthesis, isoquinoline alkaloid biosynthesis, flavonoid biosynthesis, phenylalanine metabolism, and isoflavonoid biosynthesis. These results demonstrate that drought stress induces substantial metabolic reprogramming in I. stachyodes seedling roots, particularly affecting flavonoid metabolism, phenylpropanoid pathways, and phenylalanine-derived secondary metabolite biosynthesis.
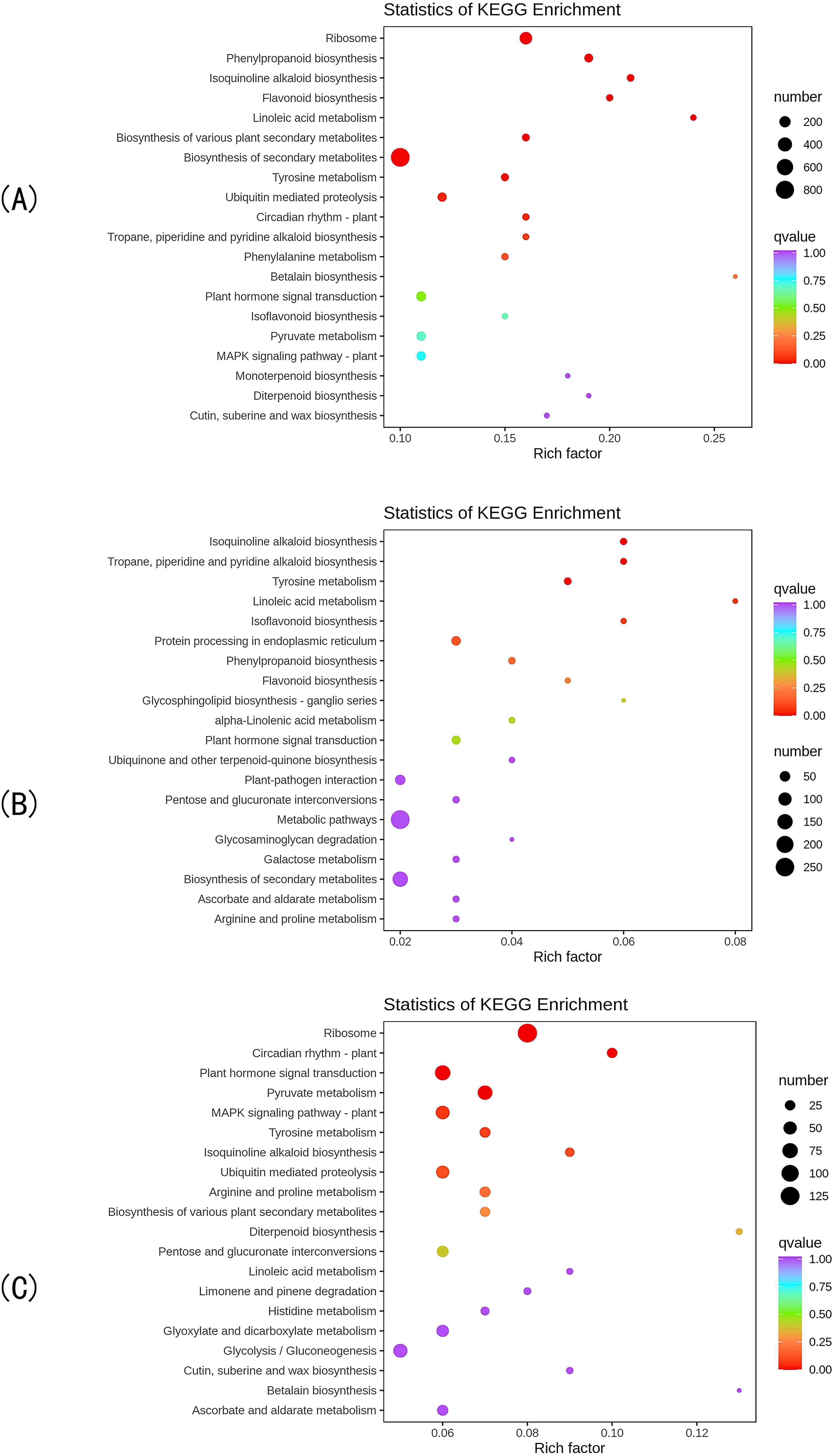
Figure 3. The top 20 pathways with the smallest KEGG enrichment P-values of DEGs in the CK vs T0 group (A), CK vs T2 group (B), and T2 vs T0 group (C).
KEGG pathway analysis identified 123 metabolic pathways enriched with DEGs in the CK vs T2 comparison, among which 20 pathways showed significant enrichment (P<0.05). The top 20 most significantly enriched pathways (Figure 3B) primarily included: isoflavonoid biosynthesis, isoquinoline alkaloid biosynthesis, flavonoid biosynthesis, phenylpropanoid biosynthesis, and linoleic acid metabolism. These findings indicate that moderate drought stress triggers substantial metabolic reprogramming in I. stachyodes seedling roots, particularly affecting the biosynthesis pathways of flavonoids, isoflavonoids, alkaloids, and fatty acid derivatives.
KEGG pathway enrichment analysis revealed 136 metabolic pathways associated with DEGs in the T2 vs T0 comparison group, with 22 pathways demonstrating significant enrichment (P<0.05). The top 20 most significantly enriched pathways (Figure 3C) included: ribosome, plant hormone signal transduction, pyruvate metabolism, isoquinoline alkaloid biosynthesis, and ubiquitin mediated proteolysis. These results demonstrate that progressive drought stress induces substantial metabolic reprogramming in I. stachyodes seedling roots, particularly affecting: translational machinery (ribosomal pathways), stress hormone signaling networks, central carbon metabolism, and specialized metabolite biosynthesis. The coordinated regulation of these pathways suggests a hierarchical adaptive strategy in response to increasing drought severity.
3.5 Metabolomics PCA and OPLS-DA
PCA results derived from different drought stress treatments and QC sample revealed minimal variation among biological replicates within each treatment group, while showing distinct separation trends between different treatment groups. These findings demonstrate significant metabolic differences among drought stress treatments. Pairwise comparative analysis further indicated clear separation along both PC1 and PC2 axes between treatment groups, confirming that drought stress significantly alters the metabolic profile of I. stachyodes (Figure 4A). After filtering out noise unrelated to classification information using the OPLS-DA model, the sample groups exhibited clear separation trends in the OPLS-DA score plot (Figures 4B, C, D). In plot B, all samples of the CK group were distributed on the left side, while those of the T0 group were entirely located on the right. In plot C, all CK group samples were clustered on the left, whereas the T2 group samples were entirely positioned on the right. Similarly, in plot D, all T0 group samples were distributed on the left, and the T2 group samples were entirely located on the right. These results indicate significant differences in metabolites among the sample groups and demonstrate the reliability of the OPLS-DA model, which can be effectively used for screening DEMs.
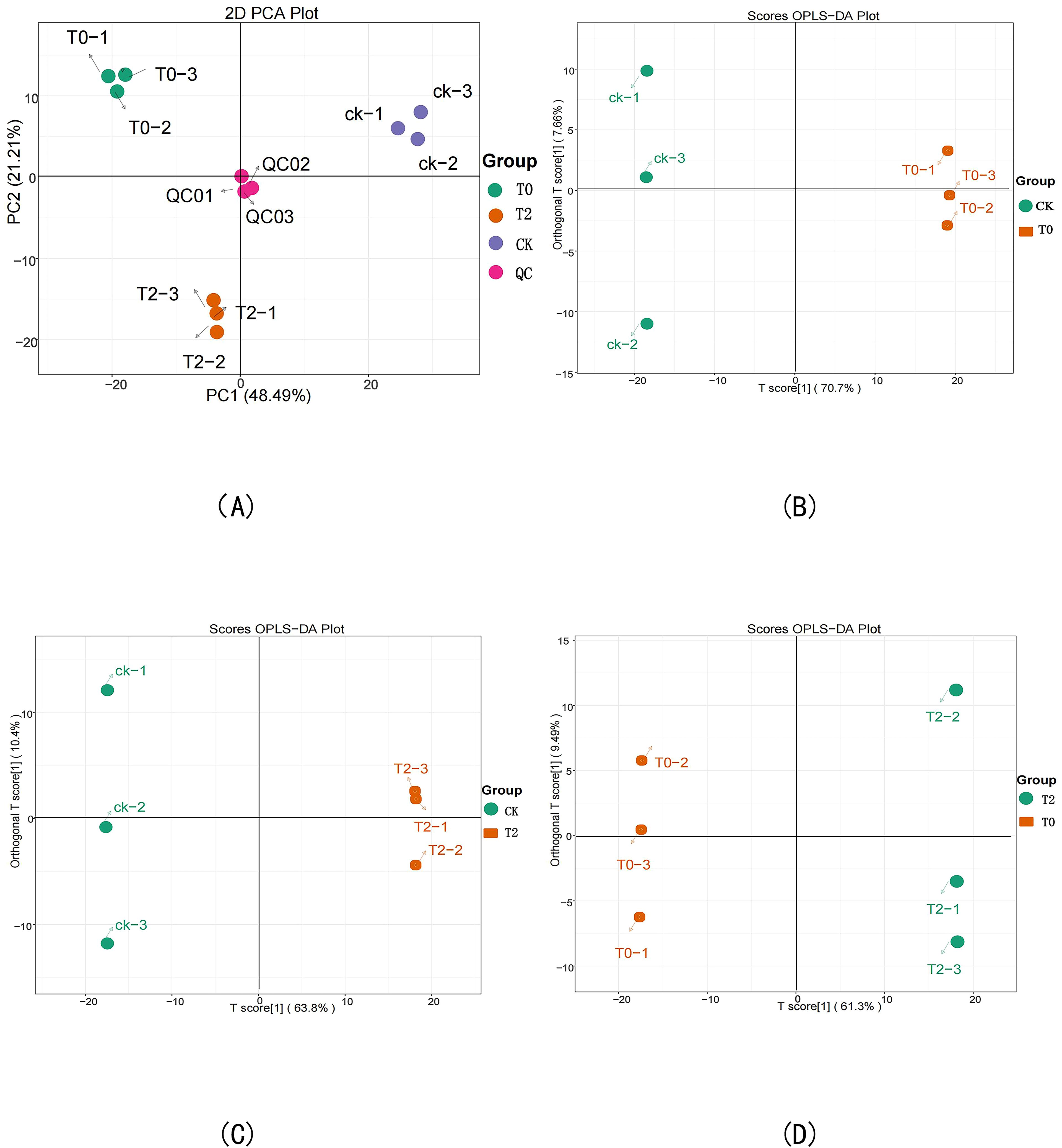
Figure 4. Principal component analysis (PCA) of sample metabolome test results (A), OPLS-DA score plots of CK vs T0 (B), CK vs T2 (C), and T2 vs T0 (D).
3.6 Metabolite detection results
In this study, a total of 622 metabolites were detected using the UPLC-MS/MS platform and the database established by Wuhan metware biotechnology Co., Ltd. (Figure 5). These metabolites were classified into 8 categories, with their respective proportions as follows: flavonoids (44.05%, 274/622), phenolic acids (25.4%, 158/622), alkaloids (13.18%, 82/622), lignans and coumarins (4.5%, 28/622), terpenoids (4.18%, 26/622), other compounds (3.7%, 23/622), quinones (3.05%, 19/622), and tannins (1.93%, 12/622).
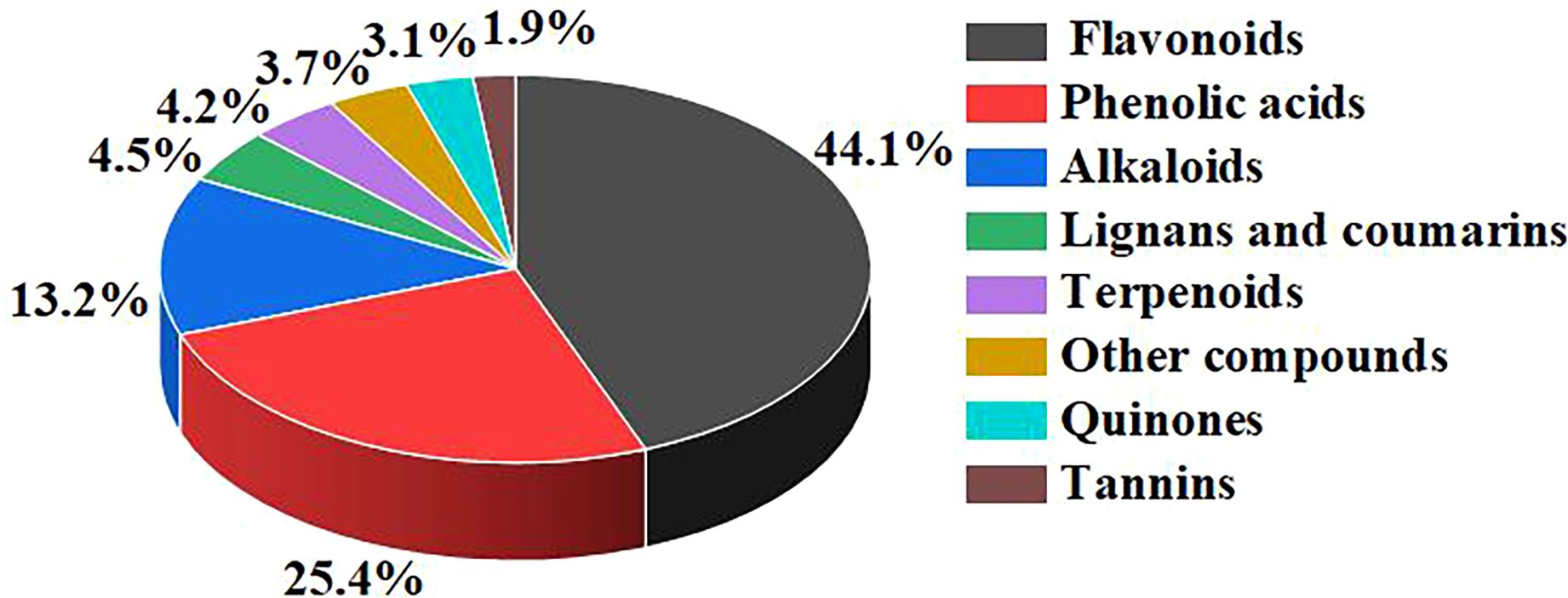
Figure 5. Metabolite classification map of sample metabolome test results.
3.7 Analysis of DEMs between treatments
Using the screening criteria of VIP ≥ 1 and P < 0.05 for DEMs, pairwise comparative analyses between groups were conducted, yielding the following results (Table 1): a total of 127 DEMs were detected in the CK vs T2 group, including 37 up-regulated and 90 down-regulated metabolites. Among the up-regulated DEMs, there were 13 flavonoids, 16 phenolic acids, 4 alkaloids, 2 lignans and coumarins, and 2 quinones. The down-regulated DEMs mainly included 40 flavonoids, 24 phenolic acids, 6 alkaloids, and 9 terpenoids. A total of 187 DEMs were detected in the CK vs T0 group, including 65 up-regulated and 122 down-regulated metabolites. The up-regulated DEMs mainly included 16 flavonoids, 25 phenolic acids, 12 alkaloids, 6 lignans, and 6 coumarins. The down-regulated DEMs mainly consisted of 66 flavonoids, 31 phenolic acids, and 7 alkaloids. A total of 86 DEMs were detected in the T2 vs T0 group, including 39 up-regulated and 47 down-regulated metabolites. Among the up-regulated DEMs, there were mainly 9 flavonoids, 10 phenolic acids, and 9 alkaloids. The down-regulated DEMs mainly consisted of 32 flavonoids, 7 phenolic acids, and 3 alkaloids.
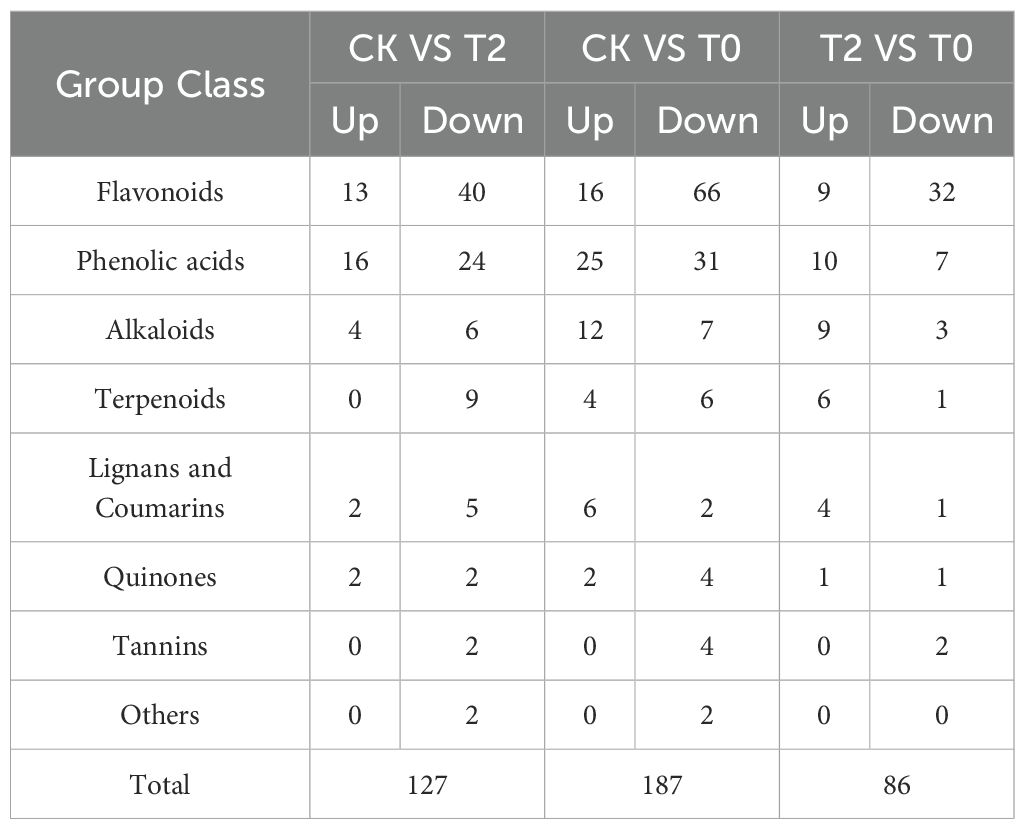
Table 1. Comparison of differentially expressed metabolites (DEMs) between groups.
Analysis of DEMs revealed that drought stress primarily affected the accumulation of flavonoids, phenolic acids, and alkaloids in the roots of I. stachyodes seedlings. The comparative results of DEMs between treatment groups showed that the number of down-regulated metabolites exceeded that of up-regulated metabolites under drought stress, with flavonoids and phenolic acids being the predominant affected classes. These findings demonstrate that drought stress predominantly influences the biosynthesis of flavonoids and phenolic acids in I. stachyodes seedlings.
3.8 KEGG metabolic pathway enrichment analysis
KEGG pathway enrichment analysis of significant DEMs between groups revealed that the CK vs T0 group showed predominant enrichment in several metabolic pathways (Figure 6A). These included isoflavonoid biosynthesis, biosynthesis of secondary metabolites, ubiquinone and other terpenoid-quinone biosynthesis, tyrosine metabolism, sphingolipid metabolism, flavonoid biosynthesis, and phenylpropanoid biosynthesis. These findings suggest that drought stress primarily affects these key metabolic pathways in I. stachyodes seedlings.
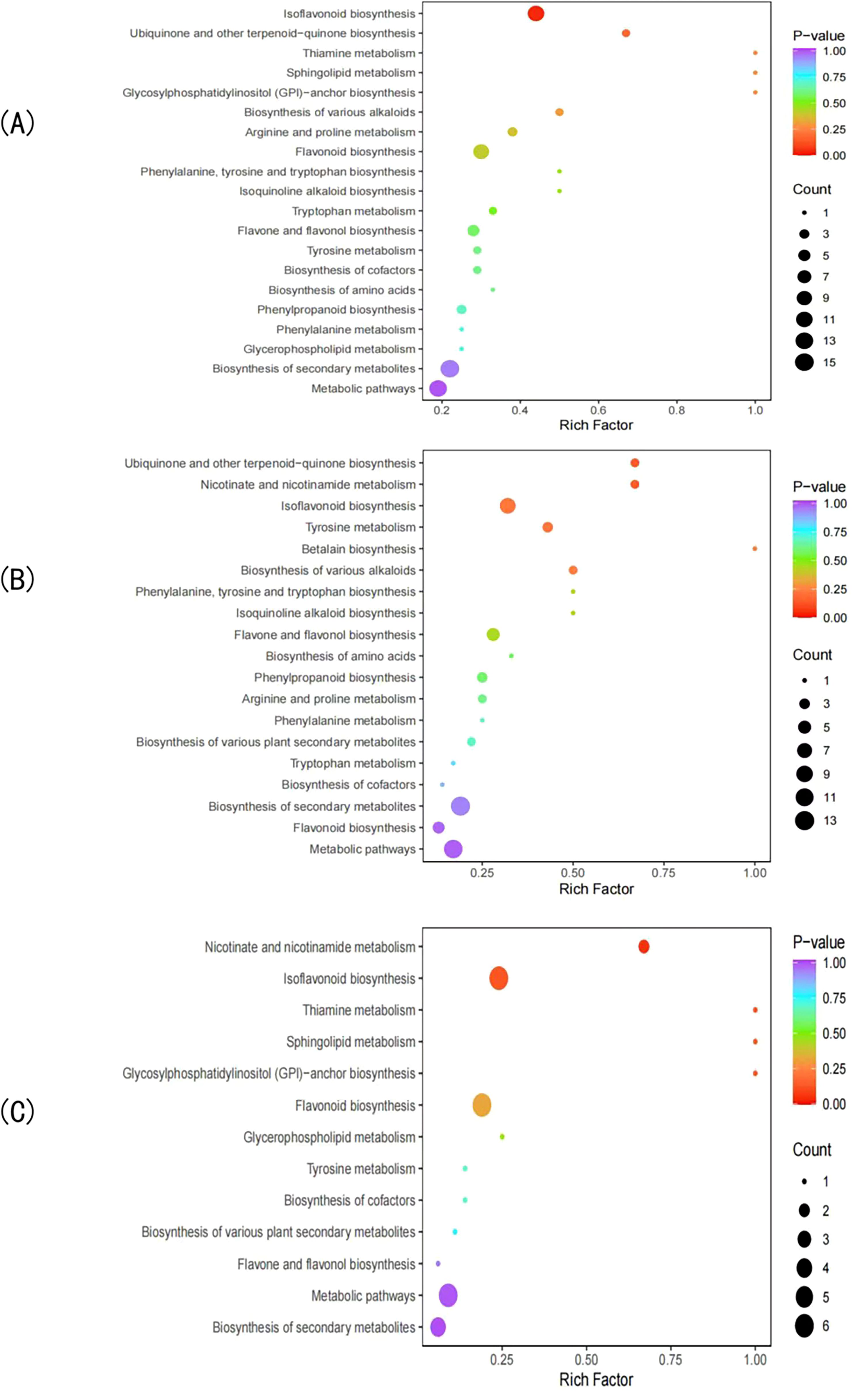
Figure 6. KEGG enrichment analysis of DEMs in the CK vs T0 (A), CK vs T2 (B), and T2 vs T0 (C) groups.
The KEGG pathway enrichment analysis of significant DEMs in the CK vs T2 group revealed predominant enrichment in several key metabolic pathways (Figure 6B). These included ubiquinones and other terpenoid-quinone biosynthesis, nicotinate and nicotinamide metabolism, isoflavonoid biosynthesis, tyrosine metabolism, betalain biosynthesis, and biosynthesis of various alkaloids. These results indicate that prolonged drought stress significantly impacts these specific metabolic networks in I. stachyodes, particularly affecting redox-related pathways (ubiquinone biosynthesis), secondary metabolite production (isflavonoids and alkaloids), and nitrogen metabolism (nicotinamide and tyrosine pathways).
The KEGG pathway enrichment analysis of significant DEMs in the T2 vs T0 group showed predominant enrichment in several crucial metabolic pathways (Figure 6C). These included nicotinate and nicotinamide metabolism, isoflavonoid biosynthesis, tyrosine metabolism, sphingolipid metabolism, and glycosylphosphatidylinositol-anchor biosynthesis. These findings suggest that the transition from moderate (T0) to prolonged (T2) drought stress in I. stachyodes primarily affects pathways related to: energy metabolism and redox homeostasis (nicotinate/nicotinamide), secondary metabolite production (isflavonoids), amino acid metabolism (tyrosine), and membrane structure and signaling (sphingolipids and GPI-anchored proteins).
3.9 Association analysis of transcriptome and metabolome
Comparative analysis of DEGs and DEMs between drought-stressed groups (T0, T2) and the control group (CK) revealed that the CK vs T0 comparison exhibited the highest numbers of both DEGs and DEMs. Consequently, we performed integrated transcriptome-metabolome association analysis specifically for the CK vs T0 group. The results demonstrated that drought stress-induced DEGs in I. stachyodes seedling roots were predominantly associated with DEMs of flavonoids, phenolic acids, alkaloids, and lignins (Supplementary Figure S2). Both DEGs and DEMs were significantly enriched in key metabolic pathways including isoflavonoid biosynthesis, phenylpropanoid biosynthesis, and flavonoid biosynthesis (Figure 7), with the isoflavonoid and flavonoid biosynthesis pathways showing particularly prominent differential expression patterns. Based on these findings, we focused our subsequent analysis on the flavonoid biosynthesis-related metabolic pathways.
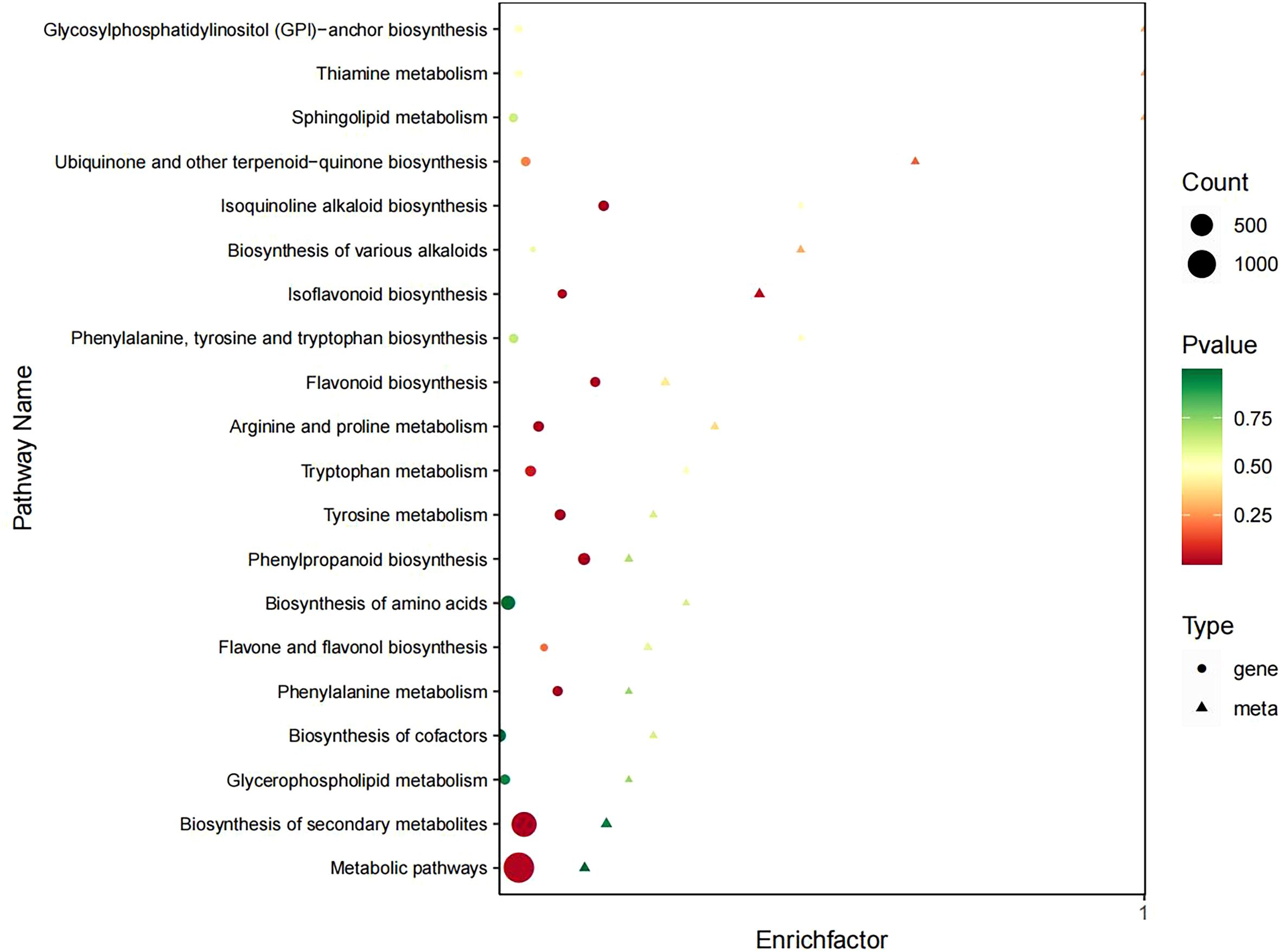
Figure 7. Pathway enrichment analysis of DEGs and DEMs.
3.9.1 Analysis of the phenylpropanoid biosynthesis metabolic pathway
In the phenylpropanoid biosynthesis pathway (Figure 8), phenylalanine serves as the substrate. It is significantly down-regulated by phenylalanine ammonia-lyase (PAL) to generate the intermediate product cinnamic acid. Subsequently, the pathway is regulated by genes such as trans-cinnamate 4-monooxygenase (C4H), which is significantly down-regulated, caffeic acid 3-O-methyltransferase (COMT), cinnamoyl-CoA reductase (CCR) and cinnamyl-alcohol dehydrogenase (CAD), which are significantly up-regulated/down-regulated. These changes led to a significant up-regulation in the contents of p-coumaric acid, sinapine malate, and eugenol compounds. Moreover, the flavonoid biosynthesis pathway is significantly affected by the down-regulation of 4-coumarate-CoA ligase (4CL) using cinnamic acid as a substrate, making it an upstream regulator in the flavonoid biosynthesis pathway. The produced cinnamic acid serves as a key precursor involved in the flavonoid biosynthesis pathway.
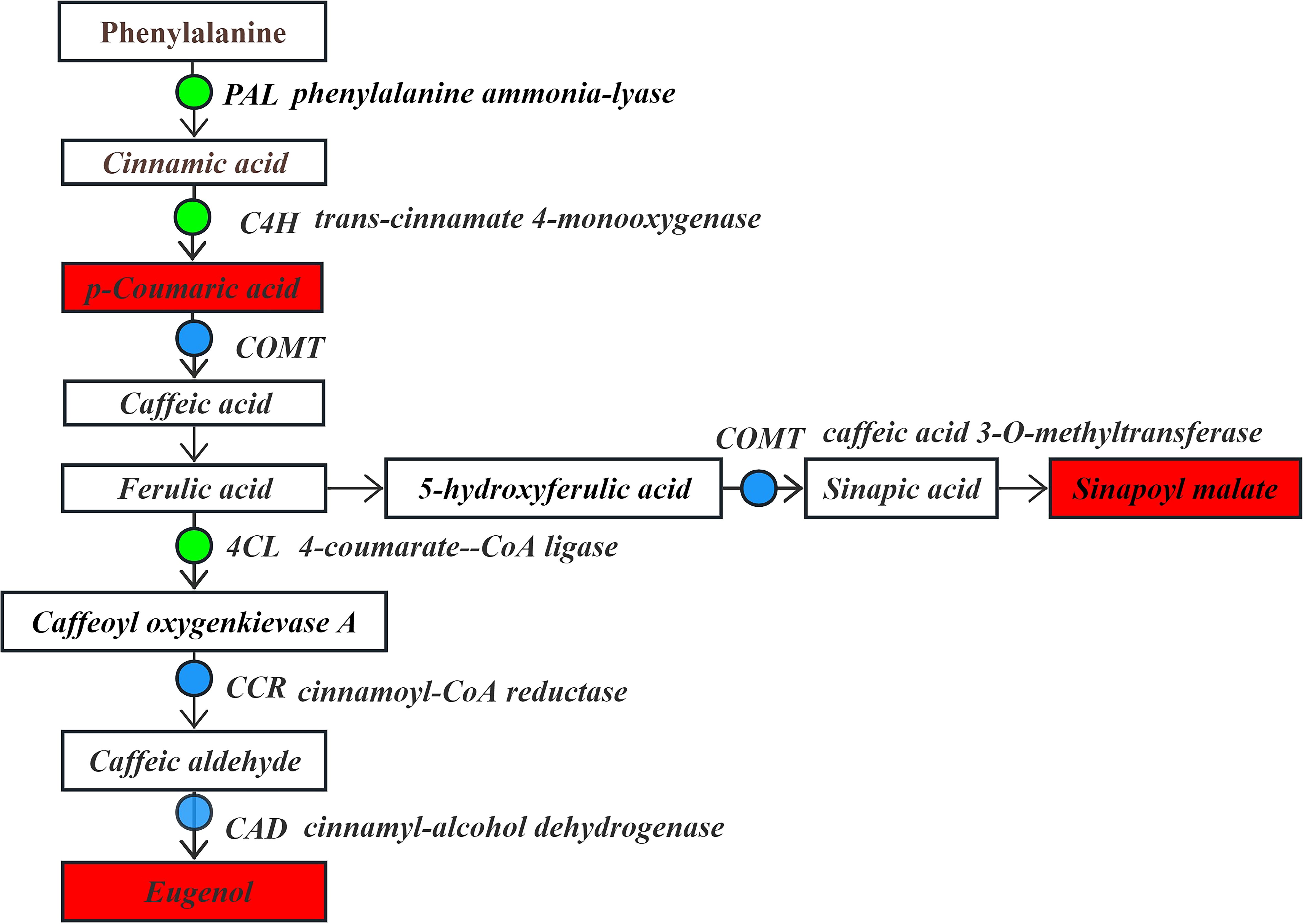
Figure 8. The expression levels of differential DEGs and DEMs in the phenylpropanoid biosynthesis pathway.
Note: The boxes represent DEMs, and the dots represent DEGs. Green indicates significantly downregulated, red indicates significantly upregulated, and blue indicates both up-regulation and down-regulation.
3.9.2 Analysis of the flavonoid biosynthetic metabolic pathway
Under drought stress, the flavonoid biosynthesis pathway (Figure 9) in the roots of I. stachyodes seedlings utilized cinnamoyl-CoA as the substrate. Chalcone synthase (CHS), which was significantly up-regulated/down-regulated, catalyzed the production of pinocembrin chalcone, indirectly leading to a significant down-regulation in the content of pinocembrin. Using the intermediate products cinnamoyl-CoA as substrates, the significant down-regulation by C4H eads to the production of p-coumaroyl-CoA. This compound is then significantly up-regulated/down-regulated by CHS to form isoliquiritigenin and naringenin chalcone, which are further converted into liquiritigenin and naringenin. Using liquiritigenin as an intermediate, the content of garbunzol is significantly up-regulated. In contrast, the contents of phloridzin chalcone, which are involved in the naringenin chalcone pathway, were significantly down-regulated.
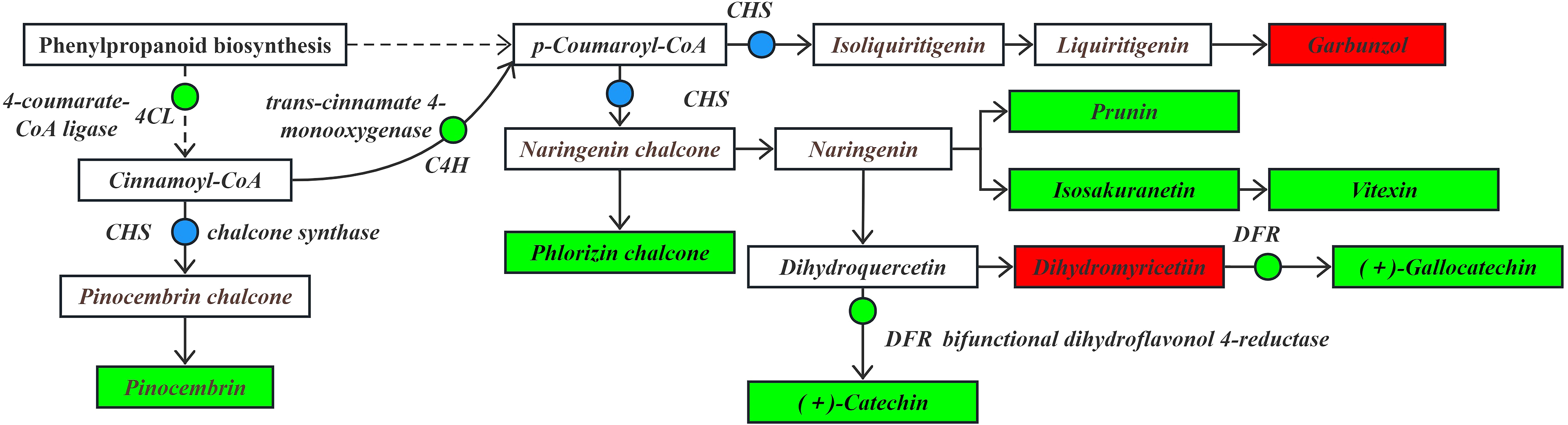
Figure 9. The expression levels of DEGs and DEMs in the flavonoid biosynthetic pathway.
In addition, in the apigenin synthesis pathway using naringenin as a substrate, the content of vitexin prunin, and isosakuranetin was significantly down-regulated. In the metabolic pathway where naringenin is used as a substrate to synthesize eriodictyol, and eriodictyol is further converted into dihydroquercetin, the content of dihydromyricetin was detected to be significantly up-regulated. However, through the significant down-regulation by dihydroflavonol 4-reductase (DFR), the content of gallocatechin was significantly down-regulated. Additionally, the content of catechin, which is directly produced from dihydroquercetin through the down-regulation of DFR, was also significantly down-regulated. Catechin and gallocatechin in flavonoids are among the key components for quality control of the I. stachyodes (Fu et al., 2013; Zhu et al., 2016). According to the results of this study, the content of catechin in the roots of I. stachyodes seedlings was significantly down-regulated under drought stress. Therefore, attention should be paid to soil moisture management during the transplanting of I. stachyodes seedlings.
Note: The boxes represent DEMs, and the dots represent DEGs. Green indicates significantly downregulated, red indicates significantly upregulated, and blue indicates both up-regulation and down-regulation.
3.9.3 Analysis of the isoflavone biosynthetic metabolic pathway
Under drought stress, the isoflavone biosynthetic pathway (Figure 10) in the roots of I. stachyoides seedlings exhibited significant metabolic reprogramming when liquiritigenin from the flavonoid pathway served as the precursor. Key enzymatic perturbations included substantial down-regulation of both 2-hydroxyisoflavanone synthase (IFS) and 2-hydroxyisoflavone dehydratase (HIDH), resulting in the accumulation of 2,7,4’-trihydroxy-isoflavone and subsequent production of daidzein. These modifications triggered a marked increase in coumestrol content while significantly reducing isosakuranetin levels. When daidzein functioned as an intermediate substrate, the pathway showed pronounced suppression of formononetin-mediated isoflavone 2’-hydroxylase (I2’H) activity, leading to significantly increase 2’-hydroxydaidzein accumulation. Furthermore, daidzein is metabolized through the coordinated action of significantly up-regulated isoflavone 7-O-glucosyltransferase (IF7GT) and down-regulated isoflavone 7-O-glucoside-6″-O-malonyltransferase (IF7MAT), ultimately causing a substantial down-regulation in the production of daidzein 7-O-glucoside-6″-O-malonate. The daidzein-derived formononetin was subjected to significant up-regulation by IF7GT, resulting in a marked decrease in the production of formononetin 7-O-glucoside. Subsequently, the significantly down-regulated activity of IF7MAT led to a substantial reduction in the accumulation of formononetin 7-O-glucoside-6″-O-malonate. Using the intermediate formononetin as substrate, significant down-regulation of I2’H activity led to the production of 2’-hydroxymethy-formononetin. Subsequent coordinated enzymatic modifications including significant down-regulation of both 2’-hydroxyisoflavone reductase (IFR) and vestitone reductase (VR), along with significant up-regulation/down-regulation of pterocarpan synthase (PTS) collectively resulted in markedly increased accumulation of medicarpin.
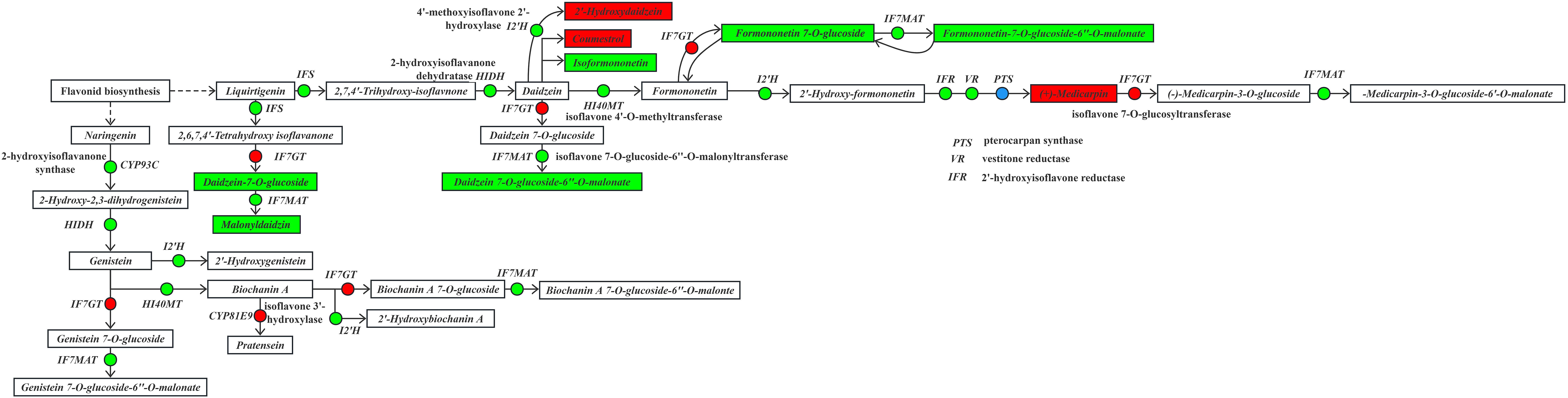
Figure 10. The expression levels of DEGs and DEMs in the isoflavone biosynthesis pathway.
In the isoflavone biosynthetic pathway, an alternative branch utilizing liquiritigenin as substrate proceeds through 6,7,4’-trihydroxyflavanone. Despite significant down-regulation of IFS activity, the production of 2,6,7,4’-tetrahydroxy-isoflavone remained unchanged. The daidzein generated through this pathway was subjected to significant up-regulation by IF7GT, resulting in markedly decreased accumulation of daidzein-7-O-glucoside. Subsequently, the significantly down-regulated activity of IF7MAT led to substantially reduced formation of malonyldaidzin.
Note: The boxes represent DEMs, and the dots represent DEGs. Green indicates significantly downregulated, red indicates significantly upregulated, and blue indicates both up-regulation and down-regulation.
3.9.4 Analysis of flavone and flavonol biosynthesis metabolic pathways
No significant enrichment of DEGs was detected in this pathway, but the contents of isovitexin and vitexin, luteolin 7-O-β-D-glucosidic acid, astragalin and trifolin produced by apigenin, luteolin and kaempferol as substrates were significantly down-regulated (Supplementary Figure S3).
4 Discussions
Our previous studies have shown that under simulated drought stress on the growth of I. stachyodes seedlings, as the concentration of PEG-6000 increased, the shoot length, primary root length, number of fibrous roots, and fresh weight of I. stachyodes seedlings initially increased and then decreased (Yang et al., 2024a). Under extreme drought conditions, the malondialdehyde (MDA) content and free proline (Pro) content in I. stachyodes seedlings were the lowest, while catalase (CAT) activity was the strongest (Yang et al., 2024b). This indicates that mild drought can, to some extent, promote the growth and development of I. stachyodes seedlings, but intensified drought will inhibit their growth or even lead to death. In this study, we aim to explore the molecular-level response mechanisms of I. stachyodes seedlings to drought stress using transcriptomics and metabolomics.
Flavonoids are a major class of plant secondary metabolites with diverse biological functions, including antioxidant activity, free radical scavenging, anti-inflammatory effects, antiviral properties, and enhanced stress resistance (Nakabayashi et al., 2014). Structurally, flavonoids can be categorized into subgroups such as flavones, isoflavones, flavanones, and flavonols (Schijlen et al., 2004). Under drought stress, plants experience a significant accumulation of reactive oxygen species (ROS), leading to oxidative damage to cellular membranes, proteins, and nucleic acids. To counteract this, plants up-regulate flavonoid biosynthesis, which mitigates ROS-induced toxicity through their potent antioxidant capacity (Ahmed et al., 2021; Shomali et al., 2022). This protective mechanism enhances drought tolerance by reducing oxidative stress and maintaining cellular homeostasis (Foyer et al., 1994; Liu et al., 2024). For instance: in citrus, increased flavonol and anthocyanin biosynthesis has been shown to significantly boost antioxidant activity and drought resilience (Zandalinas et al., 2017). Scutellaria baicalensis exhibits elevated flavonoid accumulation and enhanced antioxidant defenses under drought, improving its stress tolerance (Zhang, 2010; Guan et al., 2022). Studies on Solanum tuberosum (Watkinson et al., 2006) and Epimedium koreanum (Wang, 2024) further support a strong correlation between flavonoid content and drought resistance. These findings align with the observed changes in flavonoid profiles in the roots of I. stachyodes seedlings under drought stress. Beyond direct ROS scavenging, flavonoids also contribute to drought adaptation by modulating phytohormone signaling, improving photosynthetic efficiency, and optimizing water uptake and utilization (Zhuang et al., 2023).
Based on the analysis of the flavonoid biosynthesis pathway, it was found that in the roots of I. stachyodes seedlings, the modulation of genes such as CHS, C4H, and 4CL led to a significant down-regulation of most metabolites, including pinocembrin, gallocatechin, and catechin. Only the contents of garbunzol and dihydromyricetin showed a significant up-regulation. This selective accumulation phenomenon is consistent with the findings reported by in rice (Jayaraman et al., 2022). Previous studies have demonstrated that dihydromyricetin is a potent antioxidant capable of effectively scavenging ROS. Under drought conditions, plants may enhance their resistance to oxidative stress and improve drought tolerance by preferentially up-regulating such flavonoids with strong antioxidant activity (Huang et al., 2020). Furthermore, although flavanol metabolites such as gallocatechin and catechin exhibit strong antioxidant activity, they remained in a down-regulated state. This phenomenon may be associated with the suppression of this biosynthetic branch to conserve carbon sources and energy under extreme drought conditions. This finding aligns with the proposal that plants undergo metabolic reprogramming to cope with environmental stress under drought conditions (Kumar et al., 2021).
The isoflavonoid biosynthesis pathway displayed a similar accumulation pattern. Through regulation of key genes including IFS, HIDH and I3’H, the contents of major metabolites such as malonyldaidzin, formononetin 7-O-glucoside and daidzein-7-O-glucoside were significantly down-regulated, while only coumestrol, and medicarpin showed significant up-regulation. Studies have revealed that coumestrol enhances drought tolerance by activating the ABA signaling pathway to induce stomatal closure and reduce water loss (Tripathi et al., 2016). Medicarpin improves drought resistance through enhancing cell wall lignification to maintain water retention capacity under drought stress (Wang et al., 2022). As a crucial hydroxylated isoflavonoid derivative in leguminous plants, 2’-hydroxydaidzein is derived from daidzein hydroxylation. The introduction of a 2’-hydroxyl group significantly augments its free radical scavenging capacity, thereby enabling plants to effectively mitigate oxidative damage induced by drought stress (Fischer et al., 1990; Shao, 2020). These compounds participate in drought response by modulating cellular osmotic pressure or functioning as signaling molecules, exhibiting mechanisms analogous to Gentiana rhodantha’s response to drought stress (Ye et al., 2025). From this, it can be inferred that under drought stress, I. stachyodes adopts a regulatory strategy by prioritizing the synthesis of highly active defense compounds while suppressing energy-demanding secondary metabolic pathways, thereby redirecting carbon and nitrogen resources to maintain ribosomal function and enhance cell wall lignification. This metabolic reprogramming ensures the preservation of essential physiological functions under drought conditions. This regulatory mechanism shows similarities to the abiotic stress response strategies in woody plants (Yan et al., 2024) and wheat (Hu et al., 2018; Riaz et al., 2023).
Through combined transcriptomic and metabolomic analysis, it was found that drought stress primarily affects the flavonoid biosynthesis pathway. In response to the drought stress, some genes in the phenylpropanoid/flavonoid pathway such as PAL, C4H, CCR, and DFR, and their metabolites, i.e., dihydromyricetin, catechin, coumestrol, and formononetin 7-O-glucoside, experienced significant changes in expression and synthesis, respectively. This finding is consistent with previous studies on Gossypium hirsutum and Ammopiptanthus mongolicus under drought stress (Wu et al., 2014; Zhang, 2018). Through homology alignment analysis, these genes were found to have functionally conserved homologs in model plants such as Arabidopsis thaliana. For instance, PAL serves as the rate-limiting enzyme in the plant phenylpropanoid pathway and is extensively involved in flavonoid biosynthesis. Research has demonstrated that PAL exhibits clear homology with AtPAL1/2, with its function and regulatory mechanisms being highly conserved among plants (Dreßen et al., 2017; Chai et al., 2024). In this study, the key phenylpropanoid pathway enzyme PAL was significantly down-regulated to produce cinnamic acid, which serves as a crucial precursor for flavonoid biosynthesis. This regulatory pattern resembles that of the homologous genes AtPAL1/2 in A. thaliana, demonstrating the highly conserved role of PAL in plant flavonoid biosynthesis (Huang et al., 2010).
Under drought stress, plants employ integrated physiological, biochemical, and molecular strategies to mitigate stress-induced damage and enhance survival. These adaptive responses can be broadly classified into: morphological modifications, metabolic reprogramming and molecular signaling regulation (Li, 2014). Among these, lignin deposition serves as a critical defense mechanism, bridging structural reinforcement and metabolic adaptation (Han et al., 2022). As a major component of the secondary cell wall, lignin enhances mechanical strength and confers resistance to abiotic stresses. Its biosynthesis involves two key reductases: CCR and CAD. Under drought conditions, coordinated up-regulation of CCR and CAD promotes lignin polymerization, leading to: enhanced cell wall lignification, reducing water loss through cuticular reinforcement, improved vascular bundle rigidity, maintaining hydraulic conductivity under water deficit, increased root mechanical strength, facilitating soil penetration and water uptake (Sun et al., 2022; Wu et al., 2022). Evidence from model and crop species in A. thaliana, drought-induced CCR expression and CmCAD2/CmCAD3 up-regulation catalyze the conversion of cinnamaldehyde to cinnamyl alcohol, accelerating lignin monomer polymerization. This process preserves root structural integrity and xylem function under stress (Liu et al., 2020). Populus davidiana exhibits drought-responsive CCR/CAD up-regulation, reinforcing root cell walls to mitigate dehydration and maintain stability (Yang et al., 2023). In Cucumis melo, drought-triggered CAD overexpression enhances lignin biosynthesis, directly correlating with improved stress tolerance (Liu, 2019). In this study, significant CCR and CAD expression was detected in the phenylpropanoid biosynthesis pathway of I. stachyodes, suggesting a conserved lignin-mediated drought resistance mechanism.
5 Conclusion
In this study, metabolomics-transcriptomics technology was used to investigate the root response of I. stachyoides seedling under drought stress. It was found that under the condition of 0-5% relative field water, the number of detected genes and metabolites was the highest, and they were mainly enriched in the phenylpropanoid and flavonoid biosynthesis pathways. The phenylpropanoid and flavonoid biosynthesis pathways are the secondary metabolite synthesis pathways that actively respond to drought stress in the seedling roots. Metabolites such as p-coumaric acid, sinapine malate, eugenol, coumestrol, isosakuranetin, 2’-hydroxydaidzein, daidzein 7-O-glucoside-6″-O-malonate, formononetin 7-O-glucoside, 7-O-glucoside-6″-O-malonate, medicarpin, daidzein-7-O-glucoside, malonyldaidzin, pinocembrin, phloridzin chalcone, prunin, isosakuranetin, vitexin, gallocatechin, catechin, garbunzol and dihydromyricetin, as well as genes including PAL, C4H, COMT, 4CL, CHS, DFR, HIDH, I2’H, IF7GT, IF7MAT, IFR, VR, PTS and IFS, are potential key substances for the roots of I. stachyoides seedlings to resist drought stress. Furthermore, as one of the effective components for the quality control of its medicinal materials, the content of catechins was significantly down-regulated under the condition of 0-5% relative field water holding capacity. Therefore, to improve the catechin content during the cultivation of I. stachyoides, attention should be paid to controlling soil moisture to avoid prolonged moderate or extreme drought conditions, as extreme drought can negatively affect the normal growth of I. stachyoides. At present, there are few studies on the molecular response mechanisms of drought treatment in I. stachyoides seedlings, and the available genetic information for reference is limited, which restricts the exploration of functional genes in I. stachyoides to some extent. In this study, the transcriptional and metabolic information of I. stachyoides roots under drought treatment was obtained, and the DEGs and DEMs in the flavonoid biosynthesis pathway were preliminarily investigated. This provides a reference for understanding the physiological responses of the seedlings to drought stress, and foundational data to facilitate the domestication of wild I. stachyodes.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
QY: Conceptualization, Data curation, Formal Analysis, Methodology, Writing – original draft. NZ: Investigation, Methodology, Software, Writing – original draft. XT: Investigation, Writing – original draft. LY: Investigation, Writing – original draft. ND: Writing – review & editing. WZ: Writing – review & editing, Methodology, Funding acquisition. ZW: Writing – review & editing, Conceptualization, Funding acquisition, Methodology.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the Science and Technology Plan Project of Guizhou Province ((2022)-4016), the National Wild Plant Germplasm Resource Center for Guizhou University of Traditional Chinese Medicine (ZWGX-2405), the Top-notch Young Talents Project of Yunnan Province (YNWR-QNBJ-2019-203), the Key Basic Research Program of Yunnan Province (202201AS070057), and the Southeastern Guizhou Prefectural Key Technology R&D Program [(2023) 08].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1607789/full#supplementary-material
References
Ahmed, U., Rao, M. J., Qi, C., Xie, Q., Noushahi, H. A., Yaseen, M., et al. (2021). Expression profiling of flavonoid biosynthesis genes and secondary metabolites accumulation in populus under drought stress. Molecules. 26, 5546. doi: 10.3390/molecules26185546
Chai, P., Cui, M., Zhao, Q., Chen, L., Guo, T., Guo, J., et al. (2024). Genome-wide characterization of the phenylalanine ammonia-lyase gene family and their potential roles in response to Aspergillus flavus L. infection in cultivated peanut (Arachis hypogaea L.). Genes 15, 265. doi: 10.3390/genes15030265
Chen, T. Q., Wen, G., and Wei, S. H. (2014). Investigation on wild resources of Miao medicinal plant Indigofera stachyoides in Guizhou. J. Anhui Agric. Sci. 42, 3235–3237. doi: 10.13989/j.cnki.0517-6611.2014.11.066
Dreßen, A., Hilberath, T., Mackfeld, U., Billmeier, A., Rudat, J., and Pohl, M. (2017). Phenylalanine ammonia lyase from Arabidopsis thaliana (AtPAL2): A potent MIO-enzyme for the synthesis of non-canonical aromatic alpha-amino acids: Part I: Comparative characterization to the enzymes from Petroselinum crispum (PcPAL1) and Rhodosporidium toruloides (RtPAL). J. Biotechnol. 258, 148–157. doi: 10.1016/j.jbiotec.2017.04.005
Duan, L. X., Dai, Y. T., Sun, C., and Chen, S. L. (2016). Metabolomics research of medicinal plants. Chin. J. Chin. Mater. Med. 41, 4090–4095. doi: 10.4268/cjcmm20162202
Fischer, D., Ebenau-Jehle, C., and Grisebach, H. (1990). Phytoalexin synthesis in soybean: Purification and characterization of NADPH: 2′-hydroxydaidzein oxidoreductase from elicitor-challenged soybean cell cultures. Arch. Biochem. biophysics 276, 390–395. doi: 10.1016/0003-9861(90)90737-J
Foyer, C., Maud Lelandais, H., Karl, J., and Kunert (1994). Photooxidative stress in plants. Physiologia Plantarum. 92, 696–717. doi: 10.1111/j.1399-3054.1994.tb03042.x
Fu, J., Liang, G. Y., Zhang, J. X., Zhang, Y. P., Liu, L., Pan, W. D., et al. (2013). Chemical constituents in Indigofera stachyoides. Chin. Tradit. Pat. Med. 28, 265–268. doi: 10.7501/j.issn.1674-5515.2013.03.001
Guan, R. W., Guo, R. Q., Lin, H. B., and Lin, J. Q. (2022). Effects of drought and salt stress on flavonoids in Scutellaria baicalensis based on plant metabolomics. Chin. Tradit. Herb. Drugs 53, 1504–1511. doi: 10.7501/j.issn.0253-2670.2022.05.026
Han, X., Zhao, Y., Chen, Y., Xu, J., Jiang, C., Wang, X., et al. (2022). Lignin biosynthesis and accumulation in response to abiotic stresses in woody plants. Forestry Res. 2, 9. doi: 10.48130/FR-2022-0009
Hu, L., Xie, Y., Fan, S., Wang, Z., Wang, F., Zhang, B., et al. (2018). Comparative analysis of root transcriptome profiles between drought-tolerant and susceptible wheat genotypes in response to water stress. Plant Sci. 272, 276–293. doi: 10.1016/j.plantsci.2018.03.036
Huang, J., Gu, M., Lai, Z., Fan, B., Shi, K., Zhou, Y. H., et al. (2010). Functional analysis of the Arabidopsis PAL gene family in plant growth, development, and response to environmental stress. Plant Physiol. 153, 1526–1538. doi: 10.1104/pp.110.157370
Huang, J., He, Z., Cheng, R., Cheng, Z., Wang, S., Wu, X., et al. (2020). Assessment of binding interaction dihydromyricetin and myricetin with bovine lactoferrin and effects on antioxidant activity. Spectrochimica acta. Part A Mol. biomolecular spectroscopy., 243, 118731. doi: 10.1016/j.saa.2020.118731
Huson, D. H. and Buchfink, B. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Jaleel, C. A., Gopi, R., Sankar, B., Gomathinayagam, M., and Panneerselvam, R. (2008). Differential responses in water use efficiency in two varieties of Catharanthus roseus under drought stress. C R Biol. 331, 42–47. doi: 10.1016/j.crvi.2007.11.003
Jayaraman, K., Sevanthi, A. M., Raman, V. K., and Mandal, P. K. (2022). Expression profiling of flavonoid biosynthesis genes in association with accumulation of flavonoid compounds in rice under water deficit stress. J. Environ. Biol. 43, 498–505. doi: 10.22438/jeb/43/4/MRN-2065
Kumar, M., Kumar Patel, M., Kumar, N., Bajpai, A. B., and Siddique, K. H. M. (2021). Metabolomics and molecular approaches reveal drought stress tolerance in plants. Int. J. Mol. Sci. 22, 9108. doi: 10.3390/ijms22179108
Li, G., Bai, Y., Jia, Z. Y., Ma, Z. Y., Zhang, X. C., Li, C. Y., et al. (2022). Phosphorus altered the response of ionomics and metabolomics to drought stress in wheat seedlings. Sci. Agric. Sin. 5, 280–294. doi: 10.3864/j.issn.0578-1752.2022.02.004
Li, J. (2014). Progress on drought stress adaptation mechanisms of plant. Guangdong Agric. Sci. 41, 154–159. doi: 10.16768/j.issn.1004-874x.2014.19.005
Liu, R., Zhao, L., Ye, G. S., and Ma, Y. H. (2024). Correlation of HrANR genes and flavonoid accumulation with drought resistance in sea buckthorn (Hippophae rhamnoides subsp. sinensis). Guihaia. 44, 235–244. doi: 10.11931/guihaia.gxzw202212006
Liu, W. (2019). Functional research of CmCADs in lignin biosynthesis in oriental melon (Cucumis melo L.) under abiotic stresses (China: (Liaoning: Shenyang Agricultural University).
Liu, W., Jiang, Y., Wang, C., Zhao, L., Jin, Y., Xing, Q., et al. (2020). Lignin synthesized by CmCAD2 and CmCAD3 in oriental melon (Cucumis melo L.) seedlings contributes to drought tolerance. Plant Mol. Biol. 103, 689–704. doi: 10.1007/s11103-020-01018-7
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Maurel, C. and Nacry, P. (2020). Root architecture and hydraulics converge for acclimation to changing water availability. Nat. Plants. 6, 744–749. doi: 10.1038/s41477-020-0684-5
Nakabayashi, R., Yonekura-Sakakibara, K., Urano, K., Suzuki, M., Yamada, Y., Nishizawa, T., et al. (2014). Enhancement of oxidative and drought tolerance in Arabidopsis by overaccumulation of antioxidant flavonoids. Plant J. 77, 367–379. doi: 10.1111/tpj.12388
Ouyang, J. Y. (2020). Study on drought resistance of tartary buckwheat based on metabonomics and transcriptomics (China: (Sichuan: Chengdu University).
Riaz, M. W., Yousaf, M. I., Hussain, Q., Yasir, M., Sajjad, M., and Shah, L. (2023). Role of lignin in wheat plant for the enhancement of resistance against lodging and biotic and abiotic stresses. Stresses. 3, 434–453. doi: 10.3390/stresses3020032
Schijlen, E. G., RicdeVos, C. H., Vantunen, A. J., and Bovy, A. G. (2004). Modification of flavonoid biosynthesis in crop plants. Phytochemistry. 65, 2631–2648. doi: 10.1016/j.phytochem.2004.07.028
Shao, J. (2020). Synthesis and in vitro antioxidant activity study of hydroxylated of derivatives daidzein (China: (Gansu: Lanzhou University).
Sheng, D. F. and Chen, L. (2013). Effects of PEG-6000 stress on tanshinones accumulation in hairy roots of Salvia miltiorrhiza. Chin. Tradit. Herb. Drugs 44, 1181–1185. doi: 10.7501/j.issn.0253-2670.2013.09.022
Shomali, A., Das, S., Arif, N., Sarraf, M., Zahra, N., Yadav, V., et al. (2022). Diverse physiological roles of flavonoids in plant environmental stress responses and tolerance. Plants. 11, 3158. doi: 10.3390/plants11223158
Sun, X. C., Yuan, J. J., Li, J. P., Chen, Y. H., Zhang, B. Y., and Wang, H. Z. (2022). Genetic adaptation under drought stress for the synthesis of phenylpropanoid, sesquiterpenoids and triterpenoids in Codonopsis pilosula. Chin. Traditional Patent Med. 44, 3902–3909. doi: 10.3969/j.issn.1001-1528.2022.12.026
Tripathi, P., Rabara, R. C., Reese, R. N., Miller, M. A., Rohila, J. S., Subramanian, S., et al. (2016). A toolbox of genes, proteins, metabolites and promoters for improving drought tolerance in soybean includes the metabolite coumestrol and stomatal development genes. BMC Genomics 17, 102. doi: 10.1186/s12864-016-2420-0
Varet, H., Brillet-Guéguen, L., Coppée, J. Y., and Dillies, M. A. (2016). Sartools: A DESeq 2- and edger-based r pipeline for comprehensive differential analysis of RNA-seq data. PloS One 11, e0157022. doi: 10.1371/journal.pone.0157022
Wang, D. T. (2024). Effect of soil moisture change on the quality of Epimedium koreanum during harvesting (China: (Jilin: Jilin Agricultural University).
Wang, Y. L., Huang, L. Q., Yuan, Y., and Zha, L. P. (2015). Research advances on analysis of medicinal plants transcriptome. Chin. J. Chin. Mater. Med. 40, 2055–2061. doi: 10.4268/cjcmm20151101
Wang, X., Zhou, Q., Wang, X., Song, S., Liu, J., and Dong, S. (2022). Mepiquat chloride inhibits soybean growth but improves drought resistance. Front. Plant science. 13. doi: 10.3389/fpls.2022.982415
Watkinson, J. I., Hendricks, L., Sioson, A. A., Vasquez-Robinet, C., Stromberg, V., Heath, L. S., et al. (2006). Accessions of Solanum tuberosum ssp. andigena show differences in photosynthetic recovery after drought stress as reflected in gene expression profiles. Plant Science., 171, 745-758. doi: 10.1016/j.plantsci.2006.07.010
Wen, Q., Zhao, W. B., Zhang, Y. J., Liang, T. N., Zhang, Y. X., Li, L. L., et al. (2020). Research progress of plant response to drought stress. Jiangsu Agric. Sci. 48, 11–15. doi: 10.15889/j.issn.1002-1302.2020.12.003
Wu, H. X., Lai, M., Sun, Y. Q., and Li, G. L. (2022). Physiological responses and proteomic analysis of sugar beet seedling leaves under drought stress. Plant Physiol. J. 58, 835–843. doi: 10.13592/j.cnki.ppj.100065
Wu, Y., Wei, W., Pang, X., Wang, X., Zhang, H., Dong, B., et al. (2014). Comparative transcriptome profiling of a desert evergreen shrub, Ammopiptanthus mongolicus, in response to drought and cold stresses. BMC Genomics 15, 671. doi: 10.1186/1471-2164-15-671
Wu, D., Zhang, Y. Y., Lin, N., Li, Y., Zhang, J. Y., and Wei, Y. C. (2023). Transcriptional regulation mechanism of differential accumulation of flavonoids in leaves and roots of Sarcandra glabra based on metabonomics and transcriptomics. Chin. J. Chin. Mater. Med. 48, 5767–5778. doi: 10.19540/j.cnki.cjcmm.20230804.101
Xue, S. Y., Zhu, T., Li, B. B., and Li, T. T. (2022). Application research of combined transcriptome with metabolome in plants. J. Shanxi Agric. Univ. (Nat. Sci. Ed.). 42, 1–13. doi: 10.13842/j.cnki.issn1671-8151.202201013
Yan, T., Shu, X., Ning, C., Li, Y., Wang, Z., Wang, T., et al. (2024). Functions and Regulatory Mechanisms of bHLH Transcription Factors during the Responses to Biotic and Abiotic Stresses in Woody Plants. Plants (Basel Switzerland). 13, 2315. doi: 10.3390/plants13162315
Yang, W. H., Fu, J., Zhang, N., and Wu, Z. K. (2024a). The impact of drought stress on seed germination and seedling growth of Indigofera stachyodes after cold storage. Tillage Cultivation. 44, 12–16. doi: 10.13605/j.cnki.52-1065/s.2024.06.013
Yang, M., Wang, L., Wang, X., Li, Y., and Huang, H. (2023). Transcriptomic response to drought stress in Populus davidiana Dode. Forests. 14, 1465. doi: 10.3390/f14071465
Yang, W. H., Zhang, N., and Wu, Z. K. (2024b). Effects of calcium stress and drought stress on the growth of Indigofera stachyodes seedlings. Tillage Cultivation. 44, 17–24. doi: 10.13605/j.cnki.52-1065/s.2024.01.011
Ye, Q. Q., Wu, Z. K., and Ren, D. Q. (2025). Metabolomics study on Gentiana rhodantha with different shading treatments. Tillage Cultivation. 45, 19-23, 30. doi: 10.13605/j.cnki.52-1065/s.2025.02.012
Zandalinas, S. I., Sales, C., Beltrán, J., Gómez-Cadenas, A., and Arbona, V. (2017). Activation of secondary metabolism in citrus plants is associated to sensitivity to combined drought and high temperatures. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01954
Zhang, R. (2010). Impacts, mechanisms and possible application of water deficit on levels of flavonoids content in Scutellaria baicalensis Georgi (China: (shanghai: Fudan University).
Zhang, C. Y. (2018). Study on the molecular mechanism of drought resistance in Gossypium hirsutum races based on transcriptomics and metabonomics (China: (Beijing: Chinese Academy of Agricultural Sciences).
Zhang, C. R., Sang, X. Y., Qu, M., Tang, X. M., Cheng, X. X., Pan, L. M., et al. (2015). De novo sequencing and analysis of root transcriptome to reveal regulation of gene expression by moderate drought stress in Glycyrrhiza uralensis. Chin. J. Chin. Mater. Med. 40, 4817–4823. doi: 10.4268/cjcmm20152417
Zhao, T. D., Tian, S. T., Hou, L., Zhang, L., Ge, N., Xia, X. J., et al. (2020). Clinical study on miao ethnomedicine qijiao shengbai capsules for prevention and treatment of chemotherapy-induced leukopenia. World J. Integr. Tradit. West. Med. 15, 1547–1552. doi: 10.13935/j.cnki.sjzx.200839
Zheng, M., Guo, Y., and Wang, L. M. (2020). Effect of drought stress on root morphology and physiological characteristics of Malus micromalus cv. ‘Ruby’. J. Agric. Sci. Technol. 22, 24–30. doi: 10.13304/j.nykjdb.2019.0143
Zhu, X. Y., Luo, H. C., He, M. Q., Hu, Z. Y., Liang, Y., and Hao, X. Y. (2016). Study on the quality standard of miao medicine Indigofera stachyoides. China Pharm. 27, 3829–3831. doi: 10.6039/i.issn1001-0408.2016.27.28
Keywords: Indigofera stachyodes, drought stress, transcriptome, metabolome, karst mountainous regions
Citation: Ye Q, Zhang N, Tan X, Yang L, Ding N, Zhou W and Wu Z (2025) Transcriptomic and metabolomic analyses of root responses in Indigofera stachyodes seedlings under drought stress: a medicinal plant native to karst mountainous regions. Front. Plant Sci. 16:1607789. doi: 10.3389/fpls.2025.1607789
Received: 08 April 2025; Accepted: 11 June 2025;
Published: 01 July 2025.
Edited by:
Dilip Kumar, Academy of Sciences of the Czech Republic (ASCR), CzechiaReviewed by:
Shen Rao, Wuhan Polytechnic University, ChinaEstefania Elorriaga, North Carolina State University, United States
Copyright © 2025 Ye, Zhang, Tan, Yang, Ding, Zhou and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhikun Wu, NDczOTA5MzNAcXEuY29t; Wei Zhou, emhvdXdlaUBtYWlsLmtpYi5hYy5jbg==
†These authors have contributed equally to this work