 Zhiyin Chen1,2,3,4*
Zhiyin Chen1,2,3,4* Wei Zhou1,2,3,4Zixu Wang1,2,3,4Ziyi Chen1,2,3,4Xinyuan You1,2,3,4Yihui Gong1,2,3,4*
Wei Zhou1,2,3,4Zixu Wang1,2,3,4Ziyi Chen1,2,3,4Xinyuan You1,2,3,4Yihui Gong1,2,3,4*- 1College of Agriculture and Biotechnology, Hunan University of Humanities, Science and Technology, Loudi, Hunan, China
- 2Key Laboratory of Characteristic Agricultural Resource Development and Quality Safety Control in Hunan Province, Hunan University of Humanities, Science and Technology, Loudi, Hunan, China
- 3Innovation and Entrepreneurship Education Center for Horticultural Production and Processing in Hunan Province, Hunan University of Humanities, Science and Technology, Loudi, Hunan, China
- 4Hunan University of Humanities, Science and Technology, Hunan Provincial Innovation and Entrepreneurship Demonstration Base, Loudi, Hunan, China
Introduction: Tea (Camellia sinensis) is a significant economic crop, and investigations into the structure and function of its mitochondrial genome are crucial for understanding the evolutionary history and genetic characteristics of this species. This study presents the first comprehensive analysis of the mitochondrial genome of the tea cultivar ’Zhuyeqi‘ (Camellia sinensis cv. ‘Zhuyeqi’), aiming to elucidate its genomic structural features, gene composition, and evolutionary patterns. The findings provide a theoretical foundation for genetic breeding and molecular biology research in tea plants.
Methods: High-throughput sequencing was employed to sequence the mitochondrial genome of ’Zhuyeqi‘. Bioinformatics approaches were utilized for genome assembly and annotation. Various analytical strategies, including identification of RNA editing sites, codon usage bias analysis, repeat sequence recognition, calculation of non-synonymous substitution rates (Ka) and synonymous substitution rates (Ks), comparative genomics, and collinearity analysis, were applied to comprehensively analyze the structural features and evolutionary dynamics of the mitochondrial genome.
Results and discussion: The mitochondrial genome of ‘Zhuyeqi’ consists of one circular chromosome and six linear chromosomes, with a total length of 911,255 bp and a GC content of 46%. Genome annotation identified 77 functional genes, including 38 protein-coding genes (PCGs). The study revealed heterogeneously distributed introns within genes such as trnM-CAT (5 copies) and nad1/2/5/7. RNA editing analysis identified 556 C-to-U editing sites, notably enriched in ccmFn (38 sites) and ccmB (34 sites). Codon usage bias analysis indicated that leucine (Leu, 10%) and arginine (Arg, 7%) were the most frequently used amino acids. Repeat sequence analysis showed that dispersed repeats (780, 72%) dominated, with satellite DNA exhibiting significant distribution biases on chr1 (11) and chr3 (5). Ka/Ks analysis revealed that 37 PCGs were under varying selective pressures (0.09–2.70), with rps4 (Pi=0.09) and atp8 (Pi=0.09) showing exceptionally high variability, while rps19 (Pi=0) was completely conserved. Comparative genomics uncovered 66 homologous segments (25,656 bp) between the mitochondrial and chloroplast genomes, containing 27 intact genes such as trnA-UGC, confirming horizontal gene transfer events. Collinearity analysis demonstrated a high degree of conservation in genomic structures between ‘Zhuyeqi’ and closely related Camellia species. This study lays an important theoretical foundation for further elucidating the structural characteristics and evolutionary mechanisms of the tea plant mitochondrial genome.
1 Introduction
Mitochondria, as the central organelles of energy metabolism in eukaryotes, have genome dynamics and recombination mechanisms that are closely linked to plant environmental adaptation and evolution. Compared to the highly conserved chloroplast genome, plant mitochondrial genomes are typically characterized by multichromosomal configurations, frequent homologous recombination mediated by repeat sequences, and inter-organelle gene transfer. These structural complexities pose significant challenges to understanding species evolutionary trajectories (Li et al., 2023b, 2024). For example, the mitochondrial genome of Assam tea (C. sinensis var. assamica) contains a high proportion of repetitive sequences, reaching 43.9%, with a large number of quadruplicate repeats, which is significantly higher than the 37.2% found in Chinese small-leaf tea (C. sinensis var. sinensis) (Li et al., 2023b). Additionally, the mitochondrial genome size of Dahongpao (C. sinensis cv. Dahongpao) reaches 1,082,025 bp, which is significantly larger than the 991,788 bp of Rougui (C. sinensis cv. Rougui), suggesting that differences in chromosomal structure may be related to ecological adaptation (Li et al., 2024). Although studies on model plant mitochondrial genomes have made significant progress, the assembly of the tea tree (Camellia sinensis) mitochondrial genome still faces technical bottlenecks due to high heterogeneity and complex recombination events. In particular, there is a notable gap in research on the relationship between dynamic chromosomal configurations in cultivated varieties and ecological adaptability (Bi et al., 2022; Li et al., 2023b).
The Camellia sinensis cv. ‘Zhuyeqi’ is a core germplasm resource in China. Its chloroplast genome has been shown to possess a typical quadripartite structure, with no inversions or rearrangements in the inverted repeat (IR) regions and a unique localization of the rps19 gene at the boundary between the large single-copy region and inverted repeat B (Chen et al., 2023). Notably, compared to the closely related species Camellia japonica, the IR region of Camellia sinensis cv. ‘Zhuyeqi’ has not undergone contraction. The distance of the rpl2 gene from the LSC boundary is 106 bp, which is significantly different from the 112 bp in C. sinensis var. pubilimba (Chen et al., 2023). This suggests that the dynamics of genome boundaries may influence the efficiency of gene transfer between organelles. However, its mitochondrial genome lacks high-precision assembly data, and research is urgently needed to clarify its multichromosomal dynamic balancing mechanisms, functional network of ribonucleic acid (RNA) editing sites, and regulatory relationships with stress response pathways. Comparative studies show that 139 RNA editing sites were identified in the mitochondria of the Assam tea cultivar (C. sinensis var. assamica cv. Duntsa), with U-to-C editing accounting for 68%, which is significantly higher than the 52% in the small-leaf variety (C. sinensis var. sinensis) (Zhang et al., 2022). However, whether Camellia sinensis cv. ‘Zhuyeqi’ has a similar editing preference remains unknown. Notably, comparative genomic studies of closely related species indicate a significant positive correlation between mitochondrial genome heterogeneity and the degree of tea plant variety differentiation. For example, 40–46 chloroplast homologous fragments have been identified between the mitochondrial genomes of Assam tea (C. sinensis var. assamica) and Chinese small-leaf tea (C. sinensis var. sinensis), accounting for 1.91%-2.45% of the total mitochondrial length. Additionally, the nad1 and sdh3 genes show strong signals of positive selection in Assam tea (Li et al., 2024). The widespread phenomenon of multichromosomal coexistence in cultivated varieties may enhance ecological adaptability by regulating the activity of key enzymes in the tricarboxylic acid cycle (Zhao and Ma, 2021; Li et al., 2023b, 2024).
RNA editing, a key post-transcriptional regulatory mechanism in mitochondria, has identified 139 C-to-U editing sites in tea trees, with 32% of these events significantly altering the secondary structure of proteins. These include functional remodeling of key components of the electron transport chain, such as nad4 and cox1 (Zhang et al., 2022). It is noteworthy that, compared to the evergreen variety Longjing 43, the albino variety Huabai No. 1 shows a 90% reduction in intron splicing efficiency of the ndhA gene. Additionally, the editing level at the matK-701 site in the yellowing variety is only 30% of that in the normal green-leaf variety (Gao, 2022; Zhang et al., 2022). Interestingly, abnormalities in chloroplast RNA editing have been significantly associated with tea tree albino phenotypes (e.g., the lack of editing at the matK-701 site leads to chloroplast developmental defects) (Zhang et al., 2022), suggesting that mitochondrial editing may be involved in environmental adaptation by regulating reactive oxygen species metabolism. Adaptive evolution analysis shows that the nad1 and sdh3 genes have undergone strong positive selection in the tea plant lineage (Non-synonymous substitution rate (Ka)/Synonymous substitution rate (Ks) > 1). The non-synonymous mutation rate of the sdh3 gene in the Assam variety of large-leaf tea (C. sinensis var. assamica) is 1.8 times higher than that in the Chinese small-leaf variety (C. sinensis var. sinensis), indicating significant differentiation in energy metabolism optimization strategies between the varieties (Li et al., 2024).Their non-synonymous mutation sites may enhance energy metabolism efficiency by optimizing the proton pump functions of complex I and II (Li et al., 2024). However, the cultivar-specific evolutionary trajectories (e.g., the 91 kb structural rearrangement in the mitochondrial genomes of CSSDP and CSSRG varieties (Li et al., 2024)) and their molecular associations with agronomic traits have yet to be systematically analyzed.
This study overcomes the traditional circular genome annotation paradigm by integrating short-read sequencing from Illumina NovaSeq 6000 and long-read sequencing from Nanopore PromethION (Wang et al., 2023). For the first time, it constructs a detailed multichromosomal map of the mitochondrial genome of the Camellia sinensis cv. ‘Zhuyeqi.’ Using comparative genomic methods, this study systematically analyzes the spatiotemporal expression patterns of RNA editing sites, combines positive selection site screening with population genetics data, and reveals the co-evolutionary mechanisms underlying multichromosomal dynamics and ecological adaptation. This research not only provides a new paradigm for the structural biology of the tea tree mitochondrial genome but also lays a molecular foundation for identifying targets for crop stress resistance breeding (Zhao and Ma, 2021; Bi et al., 2022; Li et al., 2023b).
2 Materials and methods
2.1 Preparation of mitochondrial genome DNA
In this study, young shoots with one bud and one leaf from the Camellia sinensis cv. ‘Zhuyeqi’, cultivated at the standardized tea planting demonstration base of Taohuayuan Agricultural Development Co., Ltd., Xinhua County, Hunan Province, China (27°47′N, 110°51′E), were used as experimental materials (Figure 1). Genomic deoxyribonucleic acid (DNA) was extracted using the TIANGEN Plant Genomic DNA Extraction Kit (DP305), following the manufacturer’s standard protocol. DNA purity was assessed using a NanoDrop spectrophotometer (Thermo Scientific), with A260/A280 ratios between 1.8 and 2.0 and A260/A230 ratios ≥2.0. DNA integrity was verified using 1% agarose gel electrophoresis (5 V/cm, 30 min), ensuring DNA fragment lengths >30 kb to meet the high molecular weight DNA requirements for long-read sequencing (Xu et al., 2018; Zhang et al., 2019; Chen et al., 2023).

Figure 1. The planted field (A) and single tea plant of Camellia sinensis cv.’Zhuyeqi’ (B).
2.2 Sequencing data generation and quality control
A combined strategy using second-generation (Illumina NovaSeq 6000) and third-generation (Oxford Nanopore PromethION) sequencing technologies was employed to balance read coverage and long-fragment assembly accuracy (Zhang et al., 2019; Wang et al., 2023). Second-generation sequencing library preparation: Genomic DNA was fragmented into 300–500 bp fragments using a Covaris system, followed by end repair, 3’ poly-A tailing, and Illumina adapter ligation. The library quality was assessed using a Qubit 3.0 fluorometer and an Agilent 2100 Bioanalyzer. Paired-end sequencing (2×150 bp) was performed on the NovaSeq 6000 platform (Zhang et al., 2019). Raw sequencing data were filtered using fastp (v0.23.4) to remove low-quality reads based on the following criteria:1) Reads containing adapter or primer sequences. 2) Reads with an average Phred quality score<Q5. 3) Reads with an N base proportion >5% (An et al., 2021; Li et al., 2024). Third-generation sequencing and quality control: DNA fragments >10 kb were enriched using a magnetic bead-based selection method, followed by end repair and adapter ligation using the SQK-LSK109 library preparation kit. Third-generation sequencing data were filtered using filtlong (v0.2.1) with parameters: –min_length 1000 and –min_mean_q 7. Perl scripts were used to analyze the read N50, average quality scores, and guanine-cytosine (GC) content to ensure the data met the requirements for high-accuracy assembly (Li et al., 2023b; Wang et al., 2023).
2.3 Mitochondrial genome assembly
A hierarchical iterative assembly framework was constructed based on the high conservation of core plant mitochondrial genes (e.g., nad5, atp9, cox2) (Li et al., 2024). Third-generation sequencing data (PacBio/Nanopore) were initially aligned using minimap2 (v2.1). High-confidence seed sequences were selected based on alignment lengths >50 bp and coverage of at least three core genes to avoid interference from repetitive sequences (Li et al., 2024). Seed sequences were extended through multiple rounds of iterative alignment (overlap >1 kb) and integrated into an initial assembly dataset. To address the unique subcircular and non-circular topologies of tea tree mitochondria (Zhang et al., 2019; Bi et al., 2022; Li et al., 2024), third-generation sequencing data were corrected using Canu, and a hybrid assembly strategy was implemented by combining second-generation data (mapped using Bowtie2 v2.3.5.1) with Unicycler (v0.4.8). This approach significantly improved haplotype coverage depth and assembly continuity (Wang et al., 2024b). Further analysis using the visualization tool Bandage (v0.8.1) was performed to examine contig branching patterns. Manual corrections were applied to refine multichromosomal structure boundaries, based on differences in coverage depth and the distribution of repeat sequences. This process enabled the precise reconstruction of the complex multichromosomal configuration (Bi et al., 2022; Li et al., 2024). This strategy effectively overcame the fragmentation issues caused by high repeat sequences, providing a solid structural foundation for subsequent adaptive evolution analyses (Li et al., 2024; Wang et al., 2024b).
2.4 Functional annotation of mitochondrial genes
A multidimensional annotation strategy was applied: (1) Core gene annotation: The mitochondrial genomes of closely related species were used as a reference database. Homologous sequences were identified using BLAST (E-value<1e-5). Exon-intron boundaries were optimized using Geneious Prime, and chloroplast homologous fragments were manually excluded (threshold: sequence similarity ≥80% (Lu et al., 2022; Li et al., 2023b). (2) Transfer RNA (tRNA) identification: tRNAs were predicted using tRNAscan-SE (v2.0) under the default eukaryotic mode, with secondary validation by ARWEN to ensure stability of tRNA secondary structures (Zhang et al., 2019; Li et al., 2024). (3) Open reading frame (ORF) screening and functional annotation: Non-redundant sequences ≥102 bp were identified using ORF Finder (National Center for Biotechnology Information, NCBI). Overlapping regions with known genes and low-complexity regions (analyzed with RepeatMasker) were excluded. ORFs ≥300 bp were annotated using BLASTX against the nr database, focusing on gene clusters related to adaptive evolution Complete chloroplast genome sequence of Camellia sinensis genome structure, adaptive evolution, and phylogenetic relationships (Li et al., 2024).
2.5 Functional analysis of RNA editing sites
To systematically analyze the functional characteristics of mitochondrial RNA editing sites in the Camellia sinensis cv. ‘Zhuyeqi,’ this study employed an integrated multi-omics strategy. First, RNA-seq data generated from the Illumina platform were aligned to mitochondrial genome coding regions using Bowtie2 (v2.3.5.1) with high precision. The alignment results in BAM format were then sorted and filtered for quality using SAMtools (v1.9) (Li et al., 2009; Danecek et al., 2021). Next, single nucleotide polymorphism (SNP) sites between genomic DNA and transcriptome data were identified using BCFtools (v1.9-170). These results were integrated with predictions from the Predictive RNA Editor for Plant Mitochondria to construct a high-confidence set of RNA editing sites (Mower, 2009; Danecek et al., 2021). To further validate the biological significance of editing events, the Kyte-Doolittle hydropathy index was used to quantify changes in the physicochemical properties of amino acids induced by editing (Zhang et al., 2022). Finally, a generalized linear model was applied to analyze the association between RNA editing sites and signals of positive selection (Ka/Ks >1), revealing the regulatory role of RNA editing in the adaptive evolution of tea tree mitochondria (Zhang et al., 2022; Li et al., 2023b).
2.6 Relative synonymous codon usage analysis
To investigate the codon usage bias and the evolutionary driving forces in the mitochondrial genome of the tea plant variety ‘Zhuyeqi’, this study used the Relative Synonymous Codon Usage (RSCU) index for quantitative analysis. The RSCU is calculated using Equation 1:
Here, represents the observed frequency of a synonymous codon, and represents the expected frequency under a neutral evolutionary model. An RSCU value of 1 indicates neutral selection, while RSCU > 1 or< 1 suggests positive or negative selection pressure, respectively (Zhang et al., 2022; Li et al., 2023b). To eliminate redundancy from homologous sequences, unique mitochondrial coding sequences were filtered using CD-HIT (identity = 100%). The RSCU values were then calculated and statistically tested in batches using a custom Perl script, available at https://github.com/ari-dasci/S-TSFE-DL. The expected frequencies were based on a uniform distribution model (Fu et al., 2012).
2.7 Analysis of repetitive sequences in the mitochondrial genome
To systematically analyze the characteristics of repetitive sequences in the mitochondrial genome, this study utilized a multi-dimensional strategy for repetitive sequence detection: (1) Simple Sequence Repeats (SSR): Whole-genome scanning for SSRs was performed using the MISA software (v1.0). The parameters were set based on plant mitochondrial genome research standards. The minimum repeat numbers for mono- to hexanucleotide repeat units were set as 10, 5, 4, 3, 3, and 3, respectively, to balance detection sensitivity and specificity (Sahu et al., 2012; Zhang et al., 2019). (2) Tandem Repeats: Tandem repeats were analyzed using the TRF algorithm (v4.0.9). The parameters were set as follows: match score = 2, mismatch penalty = 7, indel penalty = 7, and minimum alignment score = 80. This ensured the effective identification of periodic repeat units ≥50 bp in length (Benson, 1999; Wynn and Christensen, 2019). (3) Dispersed Repeats: Dispersed repeats were detected by performing a whole-genome self-comparison using BLASTn (v2.10.1), with optimized settings for word size (word_size = 7) and significance threshold (evalue = 1e-5). The REPuter algorithm was then used to remove redundant signals and to exclude tandem repeat interference (Wynn and Christensen, 2019; Liu et al., 2024). Finally, a multi-level interaction network map of repetitive sequences was constructed using Circos (v0.69-5), revealing the spatial distribution patterns of repetitive elements within the genome (Zhang et al., 2019).
2.8 Adaptive evolution (Ka/Ks) analysis
Based on the homologous gene clusters of the following reference species: Zea mays subsp. mays (NC_007982.1), Brassica oleracea (NC_016118.1), Gossypium barbadense (NC_028254.1), Gossypium arboreum (NC_035073.1), Styphnolobium japonicum (NC_039596.1), Chrysanthemum boreale (NC_039757.1), and Camellia sinensis (NC_043914.1), an evolutionary pressure model was constructed. First, global multiple sequence alignment was performed using MAFFT v7.427 (Katoh and Standley, 2013), and low-conserved regions were removed with Gblocks. Then, Ka and Ks values were calculated using the MLWL algorithm in KaKs_Calculator v2.0 (Li et al., 2024), with the significance threshold set at p< 0.05. Particular focus was given to genes under positive selection (Ka/Ks > 1) and genes under strong purifying selection (Ka/Ks< 0.5), which were further analyzed for functional associations with previously reported core functional genes in tea plant mitochondria (Li et al., 2024). Boxplots of Ka/Ks values across evolutionary branches were constructed using the ggpubr package in R, and an evolutionary model of selection pressure over time and across lineages was integrated with PhyloTree (Wang et al., 2024b).
2.9 Nucleotide diversity (Pi Value) analysis
Based on the MAFFT multiple sequence alignment (parameters: –retree 1 –maxiterate 1000) of homologous gene clusters from the following reference species: Zea mays subsp. mays (NC_007982.1), Brassica oleracea (NC_016118.1), Gossypium barbadense (NC_028254.1), Gossypium arboreum (NC_035073.1), Styphnolobium japonicum (NC_039596.1), Chrysanthemum boreale (NC_039757.1), and Camellia sinensis (NC_043914.1), nucleotide polymorphism analysis was performed using the sliding window method (window length: 500 bp, step size: 100 bp) (Li et al., 2024). The average Pi value of single nucleotide polymorphism sites was calculated using DnaSP v5.0, and confidence intervals were assessed through 1,000 bootstrap resampling iterations to quantify the level of genetic differentiation among species (Li et al., 2024; Wang et al., 2024b). To eliminate interference from low-quality alignment regions, highly variable sites were filtered prior to alignment using Gblocks v0.91b (allowing gaps with the parameter: -b5=h) (Wang et al., 2024b).
2.10 Phylogenetic tree construction
Based on the multiple sequence alignment of whole-genome coding sequences generated by MAFFT, conserved regions were selected using trimAl v1.4 (parameters: -gt 0.7 -cons 60) (Li et al., 2024) to retain high-confidence sites. The optimal evolutionary model was determined using PartitionFinder v2.1.1, supporting the GTR+GAMMA+I model (Lanfear et al., 2016). A maximum likelihood tree was constructed with RAxML v8.2.10 (parameters: -m GTRGAMMAI -f a -x 12345), and Bayesian inference was performed using MrBayes v3.2.6 (number of chains = 4, sampling frequency = 1,000, convergence criterion: average standard deviation< 0.01). Finally, 1,000 bootstrap support values and posterior probabilities were integrated to enhance the reliability of the tree topology (Li et al., 2024; Wang et al., 2024b).
2.11 Mitochondrial genome synteny and plastid homologous sequence analysis
Whole-genome synteny alignment was performed using nucmer v4.0 (parameters: –maxmatch -c 65 -l 40), and syntenic blocks and rearrangement events were identified with SyRI v1.6 (Li et al., 2023b). High-resolution dot plots were generated using ggplot2 (Wickham, 2017). For mitochondrial-to-chloroplast horizontal transfer sequences, BLASTN (threshold: E-value ≤ 1e-10, coverage ≥ 80%) (Altschul et al., 1997) was combined with GeneWise v2.4.1 to verify open reading frame integrity (Zhang et al., 2019) and exclude false-positive transfer events. Cross-genome interaction networks were visualized with CIRCOS v0.69-5, and the dynamics of genome structure were revealed based on SSRs and long terminal repeats identified by REPuter v2.74 (Kurtz et al., 2001; Li et al., 2023b; Liu et al., 2024).
3 Results
3.1 Multichromosomal structure of the mitochondrial genome in Camellia sinensis cv. ‘Zhuyeqi’
This study is the first to elucidate the complex multichromosomal configuration of the mitochondrial genome in the Camellia sinensis cv. ‘Zhuyeqi’ (GenBank accession numbers: PQ763484–PQ763490). The genome exhibits a unique structure composed of one circular chromosome and six linear chromosomes (Figure 2; Supplementary Table 1), with a total length of 911,255 bp (individual chromosome lengths ranging from 133 to 311,104 bp) and a GC content of 46% (Supplementary Table 2). The genome size and GC content are highly conserved compared to previously reported Camellia species (701,719–1,081,966 bp, GC 44–46%), supporting the evolutionary stability of mitochondrial genomes in the Theaceae family (Li et al., 2023b). Gene annotation identified 77 functional genes, including 38 protein-coding genes (PCGs), 33 tRNAs, 3 ribosomal RNAs (rRNAs), and 3 pseudogenes (Table 1; Supplementary Table 3). The core functional modules include adenosine triphosphate (ATP) synthase (atp1/4/6/8/9), nicotinamide adenine dinucleotide (NADH) dehydrogenase (nad1–7/9), and cytochrome c oxidase (cox1–3), consistent with the conserved characteristics of mitochondrial genomes in terrestrial plants (Lu et al., 2022; Chen et al., 2023). Notably, the GC content of PCGs (43%) is significantly lower than that of tRNAs (50%) and rRNAs (48%) (Supplementary Table 2). This nucleotide composition heterogeneity may be associated with functional differentiation or post-transcriptional regulatory mechanisms (Chen et al., 2023; Li et al., 2024).
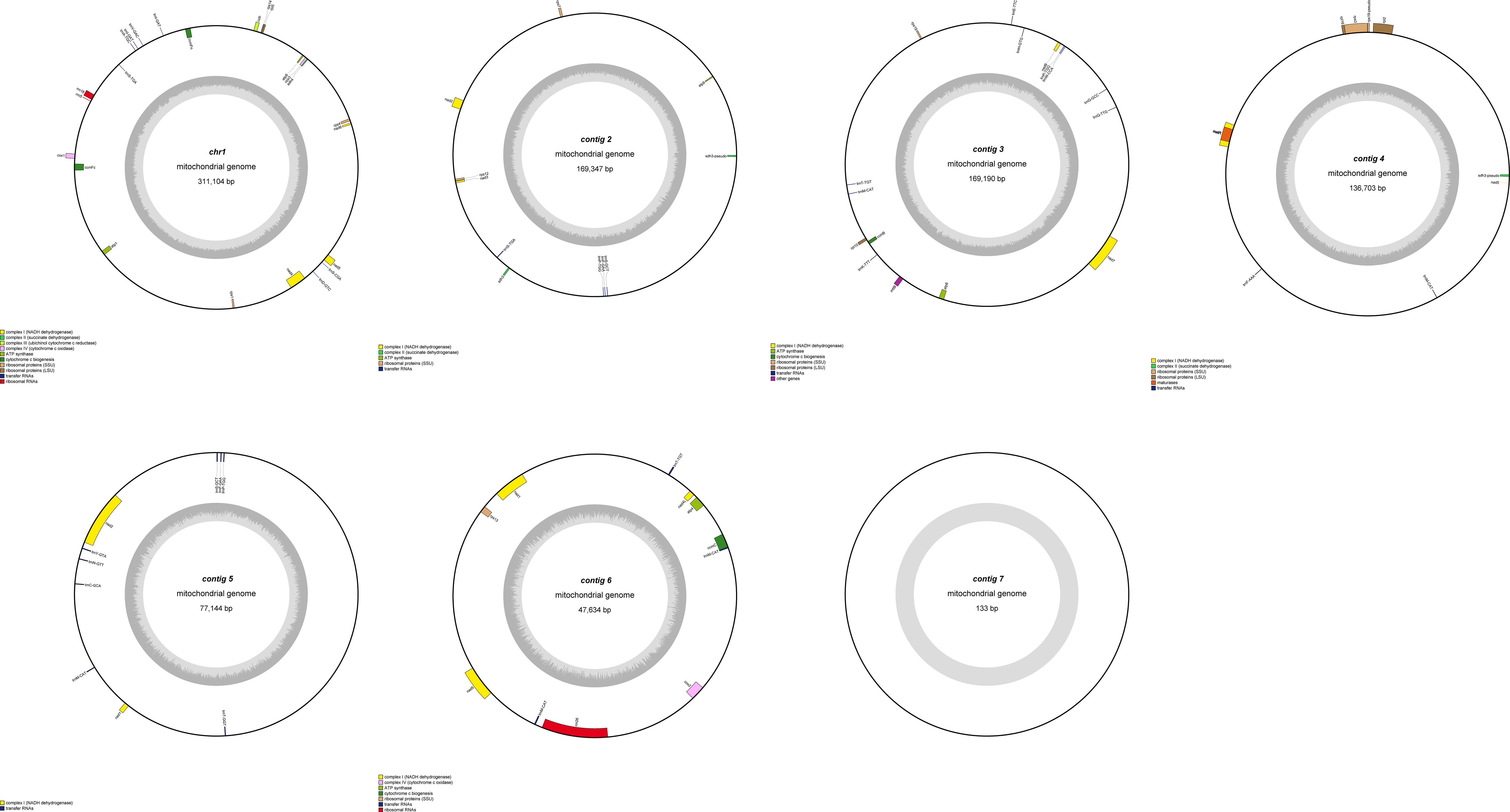
Figure 2. Mitochondrial genome map of Camellia sinensis cv. ‘Zhuyeqi’. The mitochondrial genome consists of seven chromosomes. Genes encoded on the forward strand are located on the outer side of the circle, while genes encoded on the reverse strand are on the inner side. The inner gray circle represents GC content.
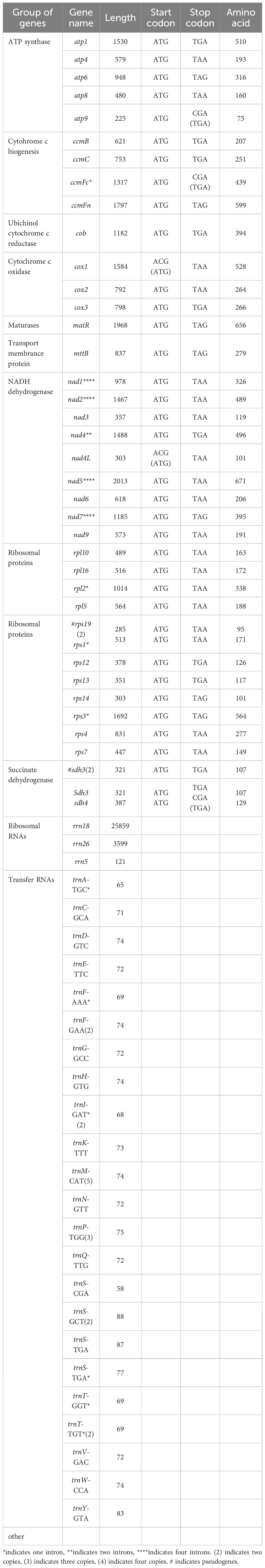
Table 1. Functional classification of genes in the mitochondrial genome of Camellia sinensis cv. ‘Zhuyeqi’.
Structural analysis of the genome revealed multiple copy numbers for some genes, such as trnM-CAT (5 copies), trnP-TGG (3 copies), and trnF-GAA, trnI-GAT, and four other genes with two copies each. These duplications suggest that recombination events mediated by repeat sequences or horizontal gene transfer might drive genome structural variation (Li et al., 2024; Liu et al., 2024). Additionally, the presence of the sdh3 pseudogene may reflect functional redundancy and differentiation of the mitochondrial genome during evolution, aligning with the adaptive evolutionary strategies of the Camellia genus (Li et al., 2023b, 2024). Moreover, the distribution of introns exhibits high heterogeneity: nine genes, including ccmFc and rpl2, contain a single intron; nad4 contains two introns, while nad1/2/5/7 each contain four introns. This complex splicing pattern may contribute to the fine regulation of gene expression through alternative splicing, closely related to the dynamic plasticity of the mitochondrial transcriptome in tea plants (Zhang et al., 2019, 2022). Similar intron structures have also been reported in Camellia sinensis var. assamica, suggesting that these may represent conserved regulatory features of mitochondrial genomes in the Theaceae family (Zhang et al., 2019).
3.2 Prediction and analysis of RNA editing sites
This study systematically analyzed 38 PCGs in the mitochondrial genome of the Camellia sinensis cv. ‘Zhuyeqi’ and identified a total of 556 RNA editing sites (Figure 3), with C-to-U editing being the predominant type. This result is consistent with the dominant C-to-U editing pattern observed in mitochondrial genomes of angiosperms (Li et al., 2021; Zhang et al., 2022). The editing sites were unevenly distributed among the genes: the ccmFn gene contained the most editing sites (38), followed by ccmB (34), mttB (33), and nad2 (30), while sdh3 had only 2 sites. Notably, the high density of editing sites in the ccmB gene (34 sites) aligns with reports from other tea cultivars (Zhang et al., 2022), and its features under positive selection may reflect its critical role in mitochondrial energy metabolism and its adaptive evolutionary demands (Li et al., 2024; Zhang et al., 2024). Additionally, the high number of editing sites in ccmFn is similar to the editing hotspots observed in mitochondrial genes of plants like Populus, suggesting that strong selective pressure due to functional conservation may act on this gene during cross-species evolution (Zhang et al., 2022, 2024).
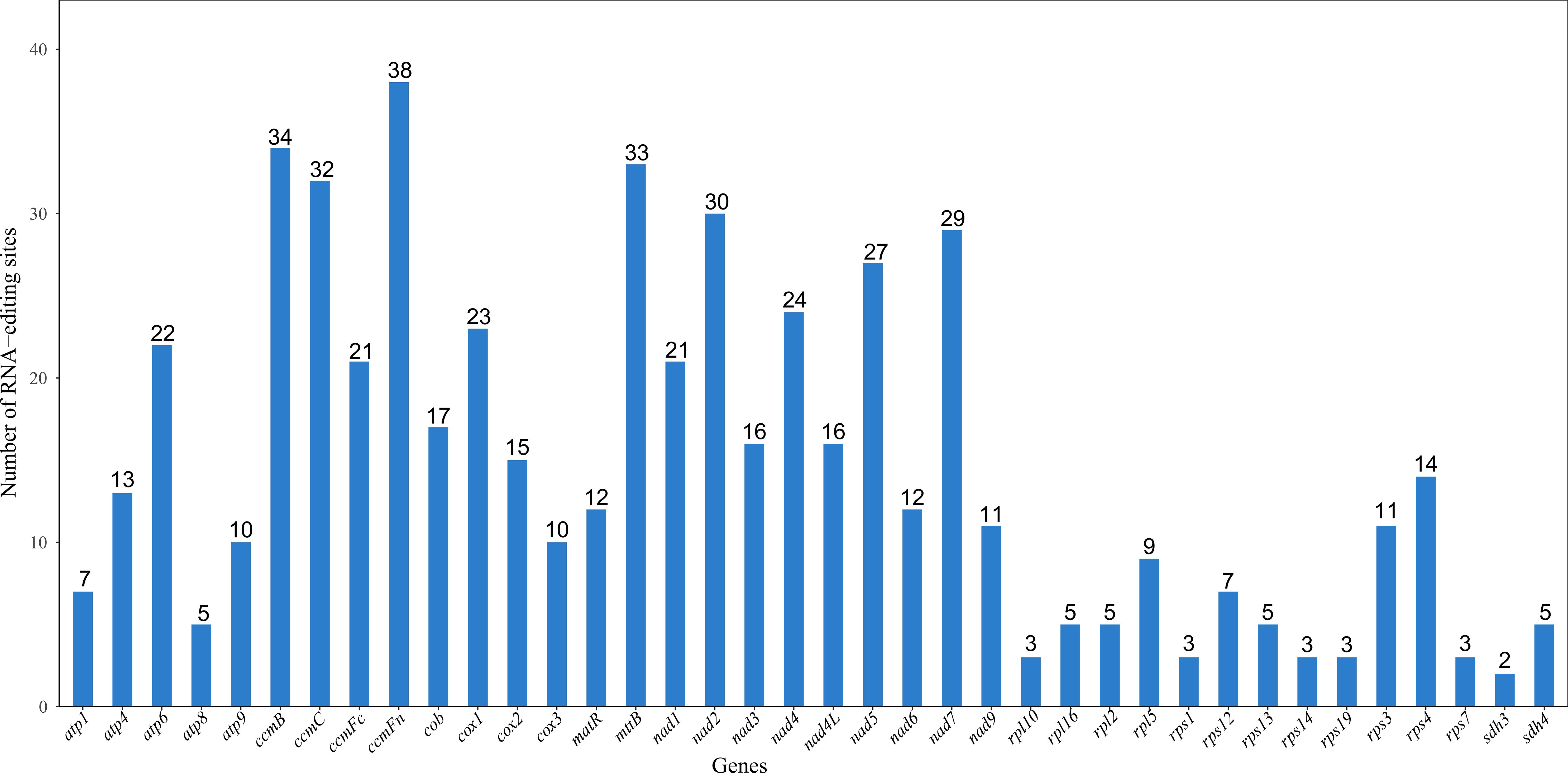
Figure 3. Statistics of RNA editing sites for each gene. The horizontal axis represents genes, and the vertical axis represents the number of RNA editing sites.
The amino acid substitutions caused by RNA editing were classified into five categories: hydrophilic-to-hydrophilic, hydrophilic-to-hydrophobic, hydrophilic-to-stop codon, hydrophobic-to-hydrophilic, and hydrophobic-to-hydrophobic (Table 2). Among these, hydrophilic-to-hydrophobic substitutions were the most frequent (48%), with typical examples including TCA (S, serine) → TTA (L, leucine), TCG (S) → TTG (L), and CGG (R, arginine) → TGG (W, tryptophan). These substitutions significantly enhanced the local hydrophobicity of proteins, which may contribute to remodeling transmembrane domains or the hydrophobic cores of enzyme active sites, thereby regulating the assembly efficiency and functionality of mitochondrial oxidative phosphorylation complexes (Li et al., 2021; Zhang et al., 2022). Hydrophobic-to-hydrophobic substitutions accounted for 31% (e.g., CCA → CTA causing P → L, GCC → GTC causing A → V). Although these substitutions did not change amino acid polarity, differences in side-chain volume and hydrophobicity may lead to subtle conformational adjustments, such as altering the packing density of ATP synthase transmembrane helices or the spatial constraints of substrate-binding pockets (Zhang et al., 2019; Duan and Lai, 2020). Hydrophilic-to-stop codon substitutions were the least frequent (1%), such as CAG (Q) → TAG (X) and CGA (R) → TGA (X).
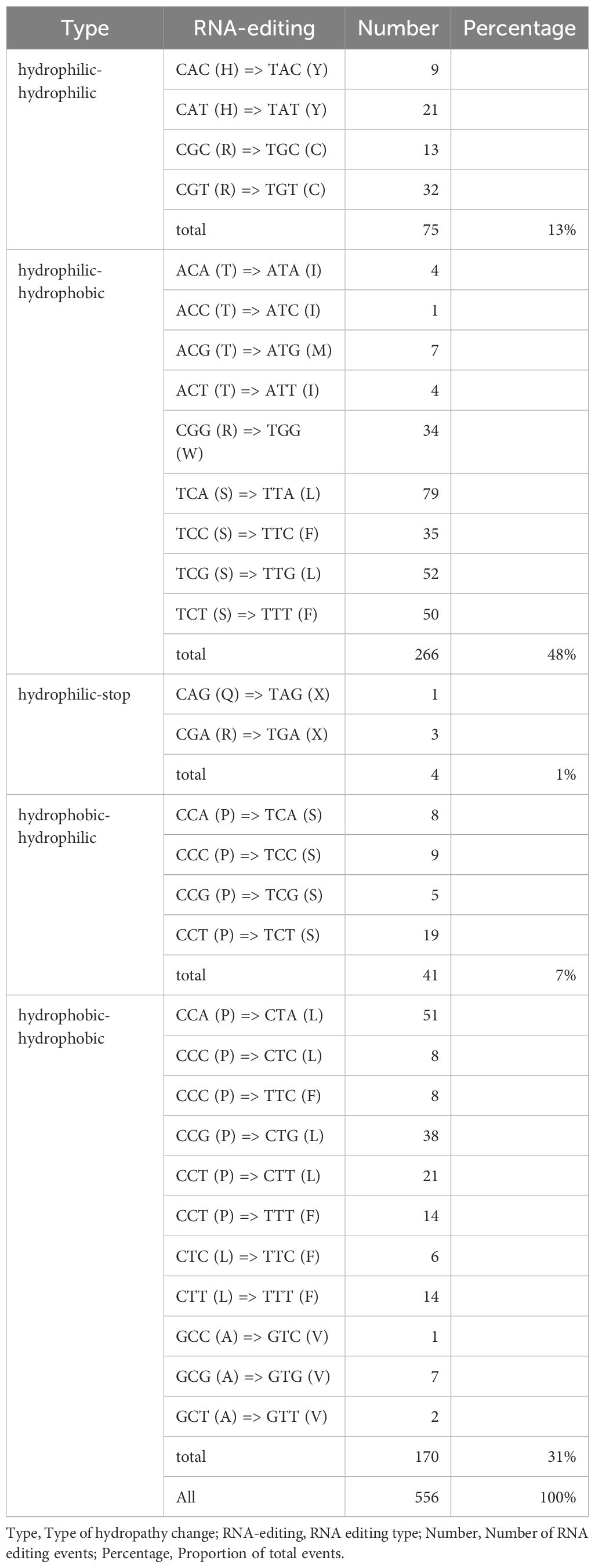
Table 2. Changes in amino acid hydropathy properties caused by RNA editing.
3.3 RSCU analysis
This study conducted a codon usage analysis on 10,828 codons from 38 PCGs in the mitochondrial genome of the Camellia sinensis cv. ‘Zhuyeqi’. The results revealed a significant codon usage bias pattern. Among aliphatic amino acids, leucine (Leu) had the highest expression frequency (1,113 occurrences, accounting for 10%), followed by serine (Ser, 9%) and arginine (Arg, 7%), whereas tryptophan (Trp) showed a significantly lower usage frequency (1%) compared to other amino acids (Figure 4; Supplementary Table 4). This distribution pattern is highly consistent with the conserved evolutionary trends of mitochondrial genomes in Camellia species (Mebeaselassie and Zhu, 2022; Li et al., 2024). Notably, among aromatic amino acids, the phenylalanine (Phe) codon UUU exhibited extreme preference (379 occurrences, 4%), with a 23% higher usage frequency than its synonymous codon UUC (3%). This observation aligns with the preferential selection of A/T-ending codons in the chloroplast genomes of Theaceae plants (Wang et al., 2022). Similarly, the isoleucine (Ile) codon AUU (3%) and the glutamic acid (Glu) codon GAA (3%) also displayed high expression frequencies. In contrast, no expression activity was detected for the methionine (Met) codons CUG/UUG, suggesting a potential functional deficit in the mitochondrial translation system (Mebeaselassie and Zhu, 2022; Chen et al., 2023).
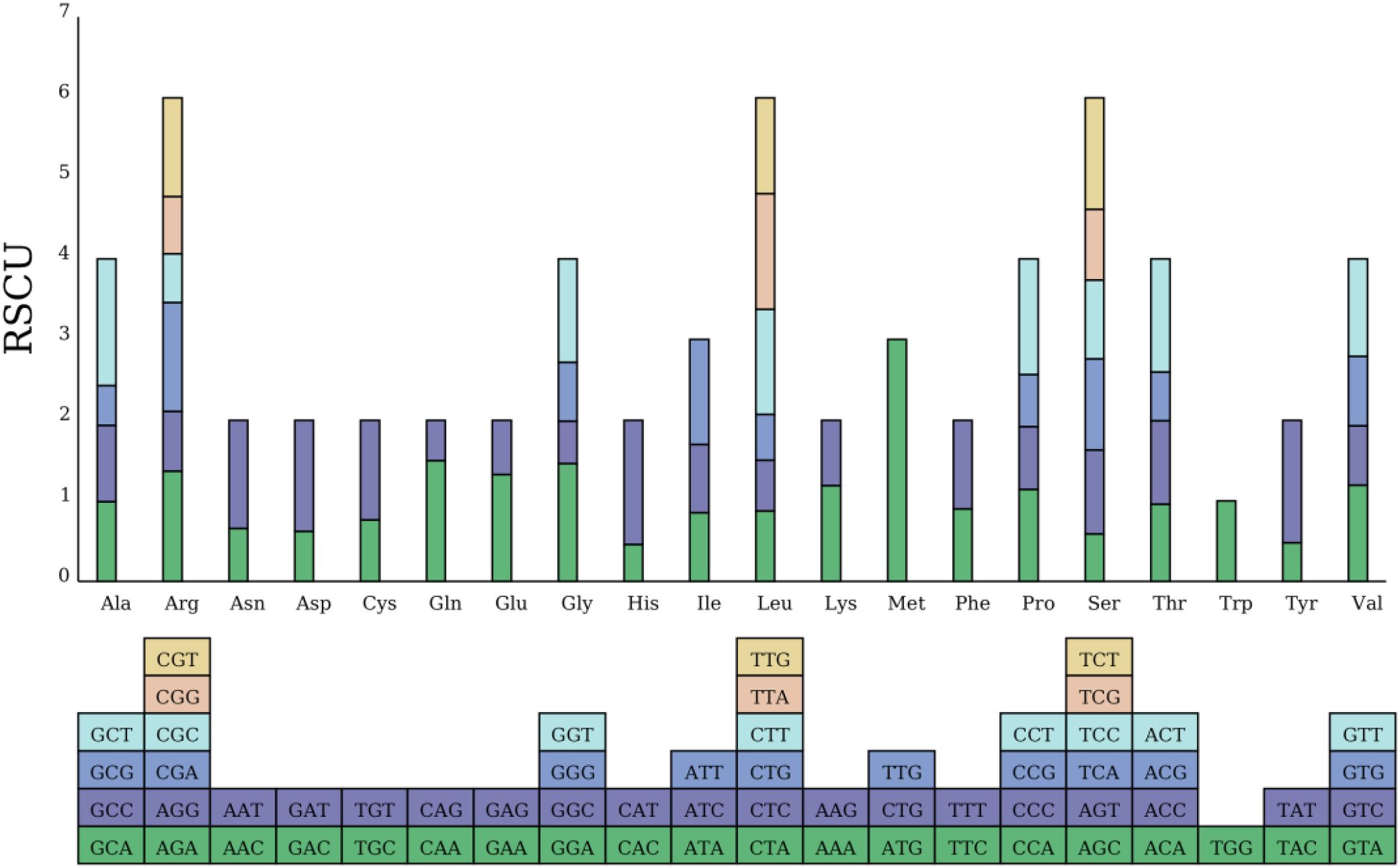
Figure 4. RSCU in the Camellia sinensis cv.’Zhuyeqi’mitogenome. The blocks below represent all codons encoding each amino acid. The height of the columns above represents the total RSCU values of all codons.
Across the genome, 31 codons with relative synonymous codon usage (RSCU) values >1 were identified, accounting for 47% of all codons. Among these, the methionine start codon AUG had the highest RSCU value (3.0) in the genome. This result is consistent with the conserved functional characteristics of start codons reported by Li et al. (2024) in a comparative mitochondrial genomics study of tea variants (Li et al., 2024). Analysis of effective number of codons (ENC) and GC3s data (Supplementary Table 5) showed that high-RSCU codons were predominantly found in AU-rich regions with low GC content (e.g., AUU, UUU). This is highly related to the adaptive evolutionary strategy of terrestrial plant mitochondrial genomes, which reduce translation energy costs through codon deoptimization (Wang et al., 2022; Li et al., 2024).
3.4 Multidimensional analysis of repetitive sequence features
The mitochondrial genome of the Camellia sinensis cv. ‘Zhuyeqi’ exhibits a typical multilayered repetitive sequence structure, including SSRs, tandem repeats, and dispersed repeats. Quantitative analysis identified a total of 1,078 repetitive sequences, with SSRs (269, 25%) and tandem repeats (29, 3%) as minor components, while dispersed repeats (780, 72%) were the predominant type (Figure 5; Supplementary Table 6). This distribution pattern aligns with the conserved characteristics of tea mitochondrial genomes, suggesting that dispersed repeats play a central role in driving the dynamic evolution of the mitochondrial genome (Li et al., 2024).
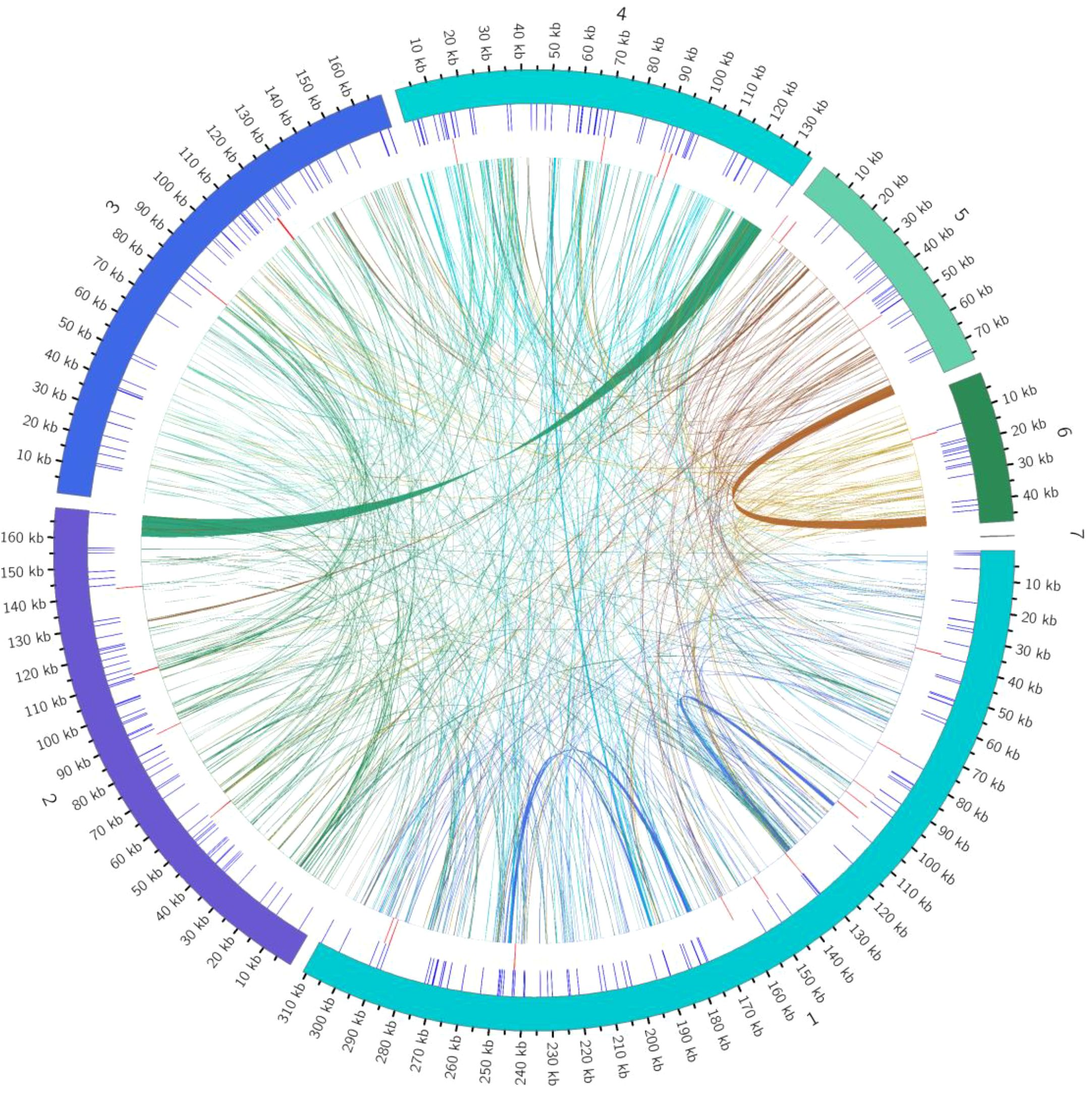
Figure 5. Distribution of repetitive sequences in the genome. The outermost circle represents the mitochondrial genome sequence. Moving inward, the circles represent simple sequence repeats, tandem repeats, and dispersed repeats, respectively.
The total length of dispersed repeats was 68,976 bp, accounting for 8% of the genome, which is significantly higher than reported for other tea cultivars (e.g., C. sinensis var. assamica) (Huang et al., 2014; Li et al., 2023b). The distribution showed marked heterogeneity among chromosomes, with chr4 (8,082 bp), chr6 (3,900 bp), and chr1 (1,281/1,277 bp) having the largest alignment lengths. This may be associated with localized recombination suppression or active transposon activity (Huang et al., 2014). Subtype analysis revealed that forward repeats (354, 45%) and palindromic repeats (426, 55%) were the major types (Supplementary Table 7). Notably, chr4 contained the largest forward repeat (8,082 bp), while chr1 carried the largest palindromic repeat (1,281 bp). These findings support the hypothesis that mitochondrial genomes maintain structural stability through homologous recombination (Zhang et al., 2019; Li et al., 2023b). Short dispersed repeats (<100 bp) accounted for 86% (668) of all dispersed repeats. Their high abundance suggests that DNA slippage replication may drive genome microevolution, a phenomenon widely observed in the adaptive evolution of plant mitochondrial genomes (Li et al., 2023a, 2024).
Satellite DNA analysis identified 29 tandem repeat units with lengths ranging from 6–39 bp, showing significant asymmetry in chromosome distribution: chr1 was enriched with 11 sites, chr3 had 5, chr6 only 1, and chr7–11 lacked tandem repeats entirely (Supplementary Table 8). This uneven distribution is associated with the compartmentalized multichromosomal structure of mitochondria and may reflect the regulation of mitochondrial genome 3D conformation by nuclear-mitochondrial interactions (Huang et al., 2014; Liu et al., 2024).
3.5 Ka/Ks analysis
In molecular evolution studies, the ratio of Ka to Ks is a key indicator for assessing the selective pressure on protein-coding genes. This study systematically compared the evolutionary dynamics of 37 core functional genes in the mitochondrial genome of the Camellia sinensis cv.’Zhuyeqi’with those of representative species, including Camellia sinensis (NC_043914.1), Brassica oleracea (NC_016118.1), Chrysanthemum boreale (NC_039757.1), Gossypium arboreum (NC_035073.1), G. barbadense (NC_028254.1), Styphnolobium japonicum (NC_039596.1), and Zea mays subsp. mays (NC_007982.1) (Figure 6; Supplementary Table 9). The analysis revealed cross-lineage adaptive evolutionary characteristics.
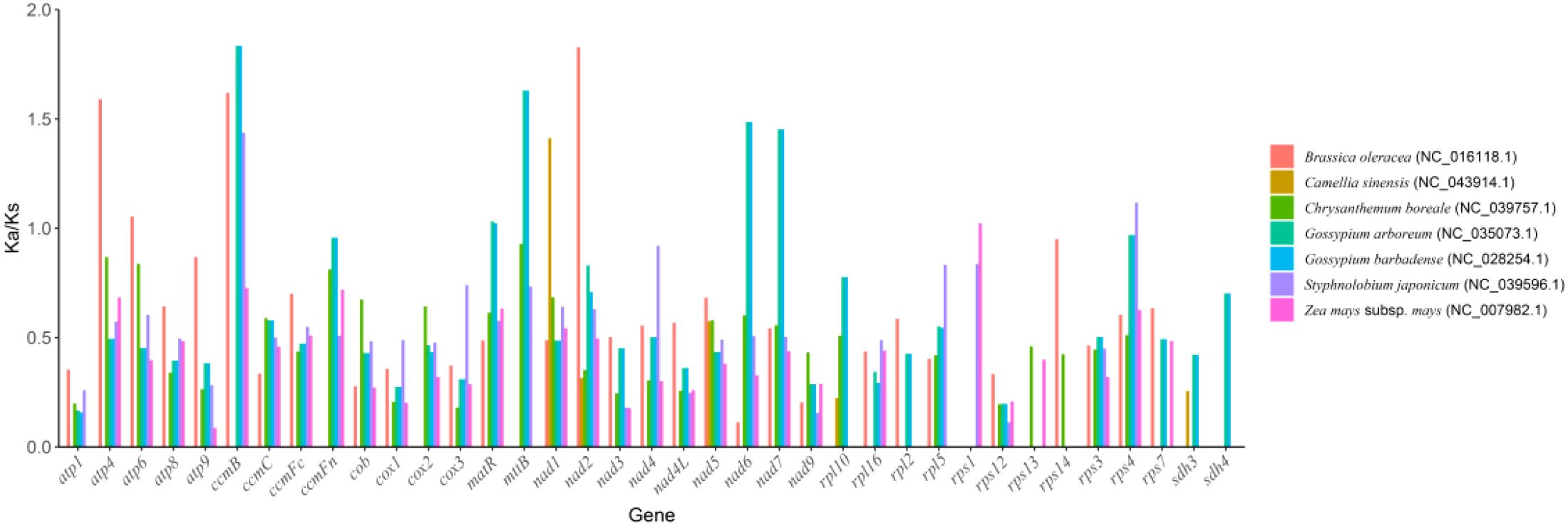
Figure 6. Boxplot of Ka/Ks values.
The Ka/Ks values for the 37 PCGs ranged from 0.09 to 2.70 (Supplementary Table 10), consistent with the Ka/Ks distribution patterns observed in the mitochondrial genome of Camellia species [2]. Notably, the atp9 gene of Zea mays subsp. mays showed the lowest Ka/Ks value (0.09), indicating strong purifying selection, likely associated with the functional conservation of the ATP synthase complex in energy metabolism (Xia et al., 2014; Huang et al., 2022).
In contrast, the sdh4 gene of Chrysanthemum boreale exhibited the highest Ka/Ks value (2.70), followed by the ccmB gene (2.22), suggesting that these genes may have undergone positive selection. This finding aligns with the positive selection observed for Nad1 and Sdh3 genes in the mitochondrial genome of Camellia species, highlighting the convergent evolution of key metabolic genes (e.g., succinate dehydrogenase complex and cytochrome c maturation-related genes) in plant adaptive evolution (Li et al., 2024). Notably, the average Ka/Ks value for the ccmB gene was 1.38, significantly exceeding the neutral evolution threshold (Ka/Ks = 1). Its positive selection characteristics appear conserved across multiple plant lineages (Li et al., 2023b, 2024).
3.6 Nucleotide diversity analysis
This study conducted a phylogenetic analysis of nucleotide diversity (Pi) for 40 core functional genes in the mitochondrial genome of the Camellia sinensis cv. ‘Zhuyeqi’ (Figure 7; Supplementary Table 11). The results revealed multidimensional heterogeneity in genetic variation. The Pi values of functional genes showed a wide range (0–0.09), with the ribosomal protein genes rps4 (Pi = 0.09), ATP synthase subunit gene atp8 (0.09), and rps1 (0.08) exhibiting exceptionally high levels of variation. In contrast, rps19 (Pi = 0) was completely conserved, suggesting it is under strong functional constraint or purifying selection (Ka/Ks< 1) (Li et al., 2024). Notably, the succinate dehydrogenase gene sdh3 (Pi > 0.07) and rps14 (Pi > 0.07) displayed high levels of variation, which is consistent with previous studies reporting positive selection (Ka/Ks > 1) in the nad1 and sdh3 genes (Li et al., 2024), further supporting the critical role of mitochondrial genomes in the adaptive evolution of tea plants (Li et al., 2024).
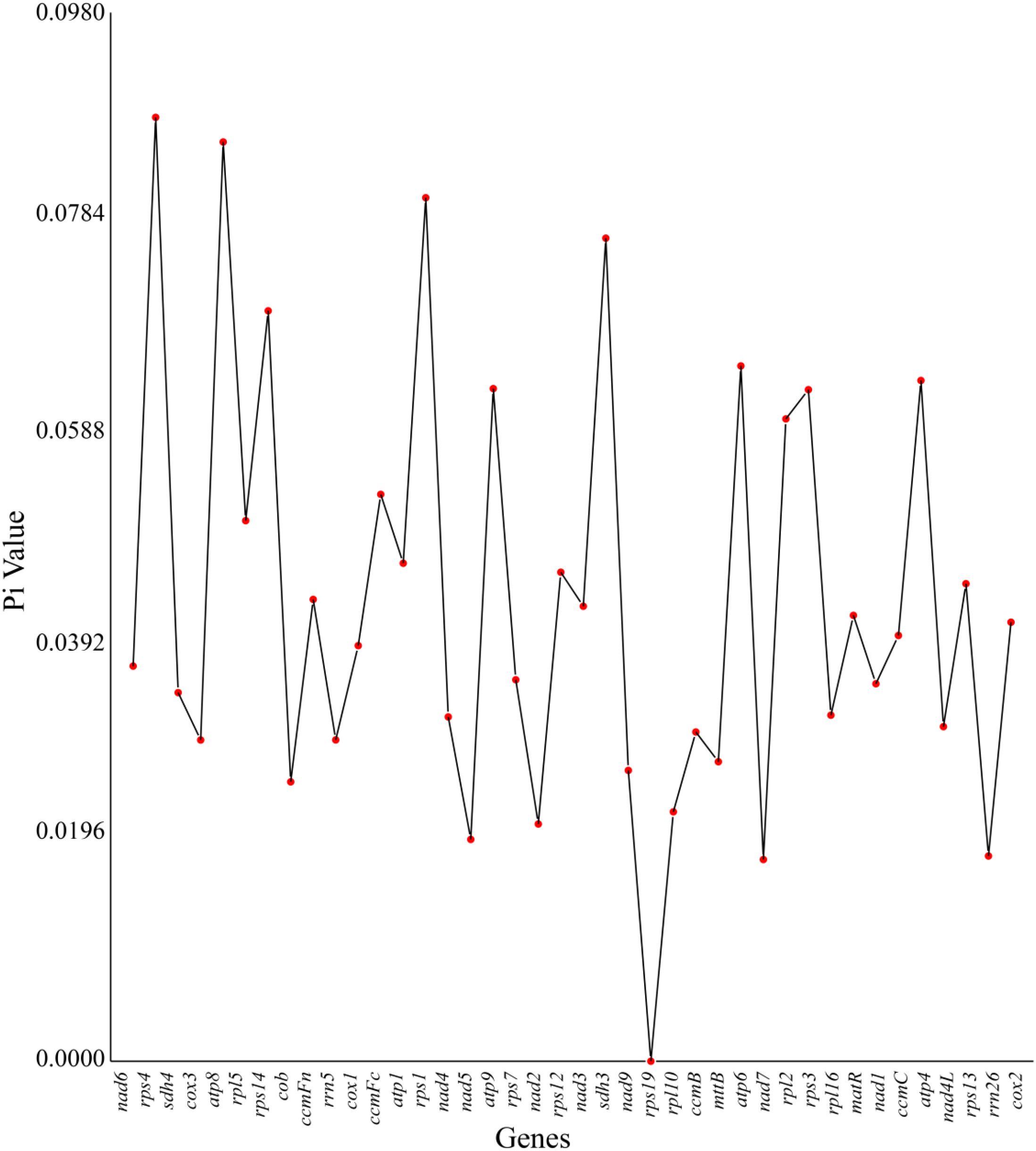
Figure 7. Nucleotide diversity in the mitochondrial genome of Camellia sinensis cv. ‘Zhuyeqi’.
Among rRNA genes, rrn5 (Pi = 0.03) showed significantly higher variation compared to rrn26 (Pi = 0.02), while the absence of Pi values for rrn18 may be attributed to its highly conserved secondary structure and RNA editing mechanisms. Studies have shown that mitochondrial RNA editing is predominantly C-to-U, with a strong preference for T bases in the surrounding sequences (Li et al., 2024). This editing pattern likely contributes to maintaining functional stability by regulating translation efficiency (Zhang et al., 2019). Comparative analysis of genomic structures revealed significant differences in variation patterns between mitochondrial and chloroplast genomes. The chloroplast genome exhibited stronger conservation in inverted repeats (IRs) (Pi< 0.005) (Peng et al., 2021), whereas the mitochondrial genome showed increased structural plasticity due to frequent recombination and horizontal gene transfer events (Li et al., 2024).
3.7 Homologous sequence analysis between mitochondrial and chloroplast genomes
This study systematically analyzed the homologous sequences between the mitochondrial and chloroplast genomes of the Camellia sinensis cv. ‘Zhuyeqi’. The mitochondrial genome, with a total length of 911,255 bp, is approximately 5.8 times larger than the chloroplast genome (157,072 bp) (Figure 8; Supplementary Table 12), consistent with the reported size variation of tea mitochondrial genomes (701,719–1,081,966 bp) (Li et al., 2023b). Whole-genome alignment identified 66 homologous fragments between the chloroplast and mitochondrial genomes, ranging from 32 to 7,721 bp, with a total length of 25,656 bp (Supplementary Table 13). These homologous sequences account for 15% of the chloroplast genome and 2% of the mitochondrial genome. The proportion of mitochondrial homologous sequences (2%) falls within the typical range (1.91%-2.45%) reported for other tea cultivars (Li et al., 2024). This proportion differs significantly from legumes, such as Dalbergia odorifera, where chloroplast-derived sequences account for approximately 4% of the mitochondrial genome (Hong et al., 2021), but both systems highlight the widespread occurrence of inter-organellar DNA transfer (Wang et al., 2012; Li et al., 2023b). It is important to note that the evolutionary rates and recombination mechanisms of plant mitochondrial and chloroplast genomes differ significantly (Sloan et al., 2012; Tyszka et al., 2023), and no direct quantitative relationship has been established between the proportion of homologous sequences and the activity of DNA transfer (Li et al., 2024; Wang et al., 2024a). Therefore, the observed proportion of homologous sequences may represent a baseline level of gene flow between organelles in tea plants, while the activity of the specific transfer mechanisms requires further analysis, including recombination frequency and insertion site characteristics (Hong et al., 2021; Li et al., 2024).
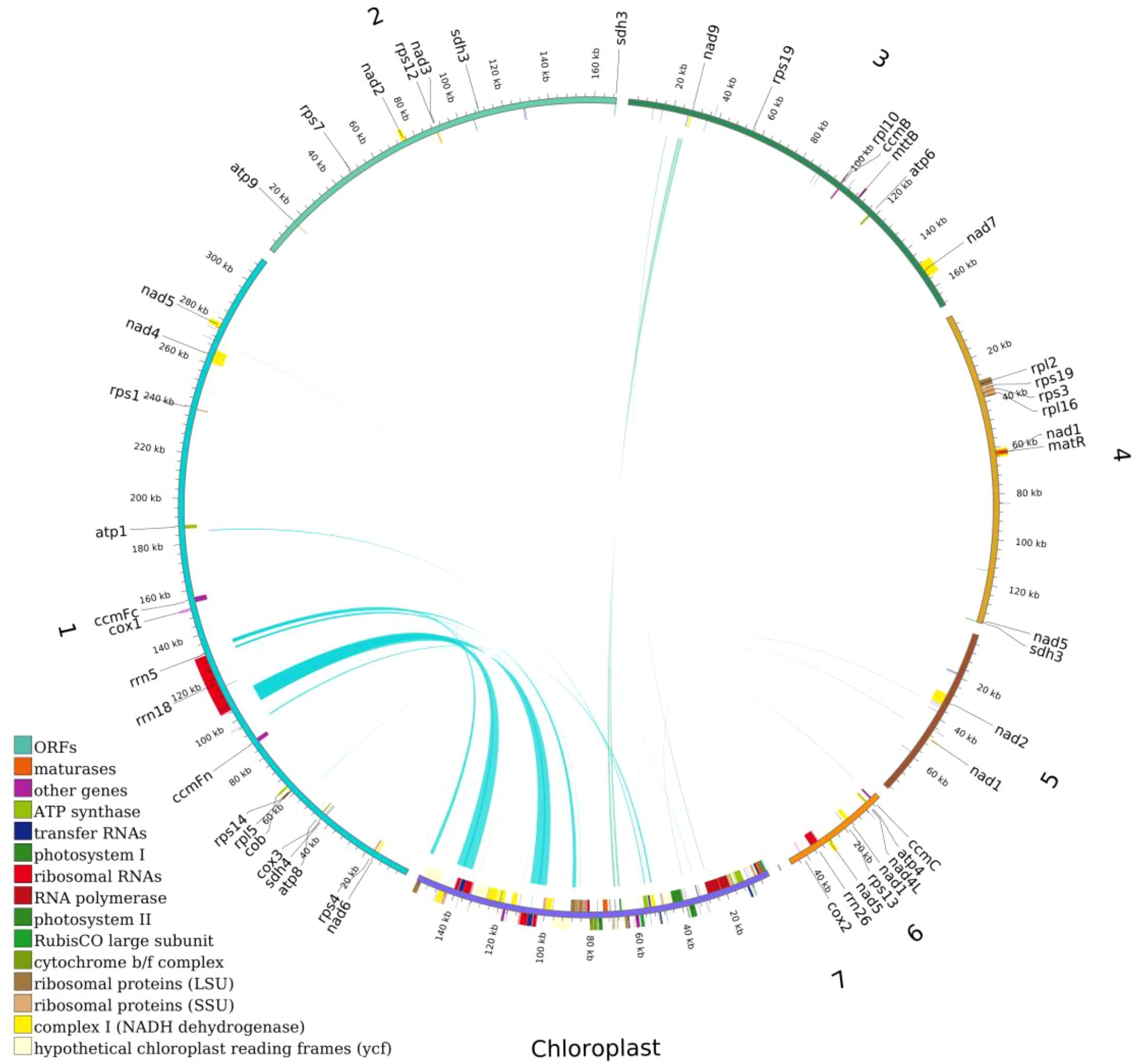
Figure 8. Distribution of homologous sequences between the mitochondrial and chloroplast genomes of Camellia sinensis cv. ‘Zhuyeqi’. “Chloroplast” represents chloroplast sequences, while other labels represent mitochondrial sequences. Genes from the same complex are represented by blocks of the same color. Blocks on the outer and inner circles indicate genes on the positive and negative strands, respectively. The connecting lines in the middle indicate homologous sequences.
In the chloroplast genome, homologous sequences (23,747 bp, 15%) contain 27 complete genes, including key functional genes such as trnA-UGC, rrn16, and rpl2. Additionally, the homologous regions of rps12, atpE, and rrn16 covered 75%, 91%, and 58% of their original gene lengths, respectively (Supplementary Table 13). This suggests that chloroplast DNA transferred to the mitochondria may preferentially retain functional domains (Li et al., 2023b). In contrast, the mitochondrial homologous sequences (14,776 bp, 2%) included only 14 complete genes (e.g., trnV-GAC and trnW-CCA), with some homologous genes showing coverage below 20% (Supplementary Table 13). This indicates that the mitochondrial genome may impose stricter selection or degradation mechanisms on transferred sequences (Li et al., 2024). Such differences may be driven by the strong purifying selection acting on the mitochondrial genome (Li et al., 2024).
3.8 Phylogenetic and evolutionary analysis
A maximum likelihood phylogenetic tree constructed based on 38 mitochondrial protein-coding genes (Figure 9) revealed five monophyletic groups among 26 representative species. The study selected three Poaceae species, Triticum timopheevii (NC_022714.1), Chrysopogon zizanioides (NC_0656367.1), and Zea mays (NC_007982.1), as outgroups (Yang et al., 2024a). The remaining species clustered into branches corresponding to Theaceae, Asteraceae, Brassicaceae, Malvaceae, and Fabaceae. Notably, the Camellia sinensis cv. ‘Zhuyeqi’ clustered wit1h 100% bootstrap support within the Camellia branch, forming a monophyletic group with C. sinensis (OM809792.1) and the Assam variety C. sinensis var. assamica (OL989850.1). This result aligns with the monophyly of the Camellia tribe observed in phylogenetic analyses based on the matR gene (Yang et al., 2006). Genetic differentiation distance analysis showed that Camellia sinensis cv. ‘Zhuyeqi’ shares a closer relationship with three major cultivated tea varieties—C. sinensis var. pubilimba (ON782577.1), C. sinensis (NC_043914.1), and C. sinensis (MH376284.1)—than the inter-family differentiation threshold. This finding suggests that these varieties may have originated from a shared ancestral population that underwent rapid radiation evolution (Xu, 2019; Li et al., 2024).
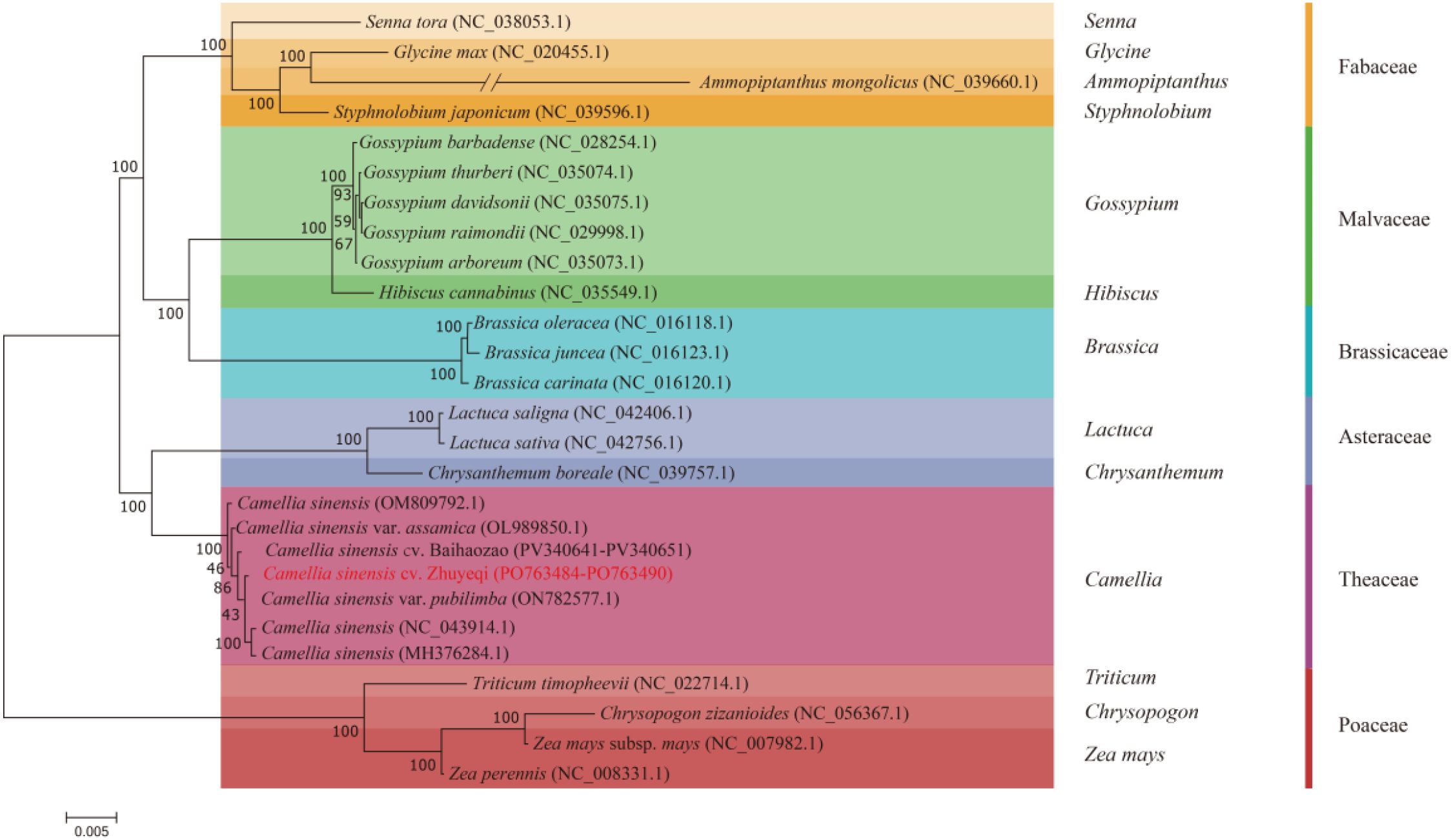
Figure 9. Phylogenetic relationships between Camellia sinensis cv. ‘Zhuyeqi’ and 25 other plant species.
3.9 Synteny analysis of the mitochondrial genome
This study conducted a synteny analysis of the mitochondrial genome of the Camellia sinensis cv. ‘Zhuyeqi’ using whole-genome alignment strategies (Figure 10; Supplementary Table 14). The results revealed significant structural conservation between Camellia sinensis cv. ‘Zhuyeqi’ and other Camellia sinensis species, with homologous regions covering 64% (579,821 bp) of the Camellia sinensis cv. ‘Zhuyeqi’ genome and 88% (620,962 bp) of the other Camellia genome. This observation contrasts sharply with the frequent gene rearrangements and heterogeneous repeat sequences commonly found in Camellia mitochondrial genomes (Lu et al., 2022; Li et al., 2024). Notably, the structural conservation in the Camellia sinensis cv. ‘Zhuyeqi’ may be attributed to artificial selection pressures during domestication, which preserved core functional modules. This aligns closely with the “domestication bottleneck effect” theory proposed by Li et al. (2024) (Li et al., 2024). Specifically, although the mitochondrial genome of Camellia species can contain up to 44% SSRs and exhibit numerous re6combination hotspots (Li et al., 2023b), the observed conservation in this study may result from the selective stabilization of key functional units such as respiratory chain complexes (Wei et al., 2018). Cross-species comparisons showed that syntenic regions between Camellia sinensis cv. ‘Zhuyeqi’ and the monocot Zea mays subsp. mays accounted for only 9% (77,430 bp) of the genome. This difference is likely due to genome remodeling events triggered by early divergences among angiosperms (Wei et al., 2018; Yang et al., 2024a).
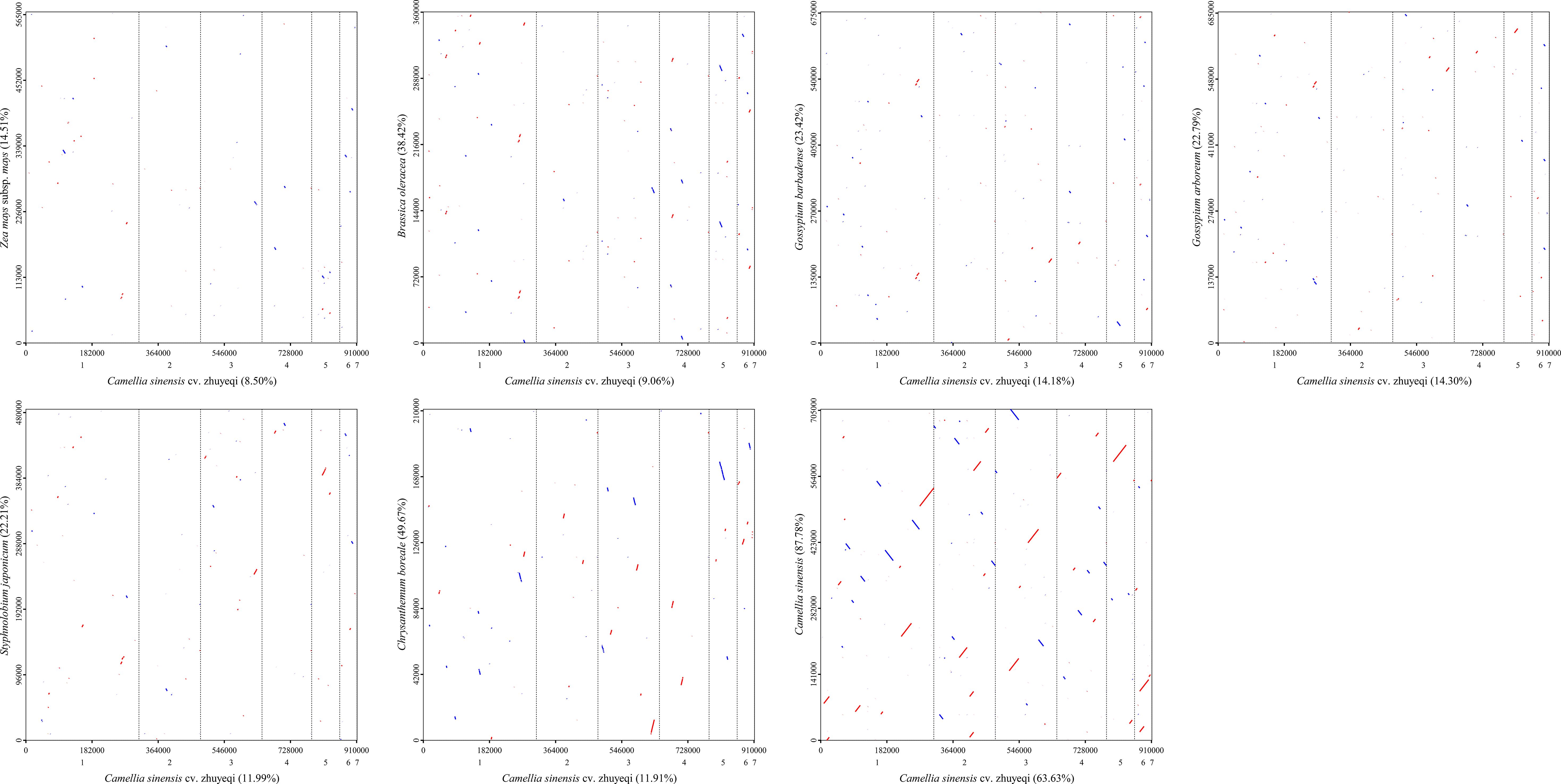
Figure 10. Homologous sequence alignment between the mitochondrial genome of Camellia sinensis cv. ‘Zhuyeqi’ and seven other species. The horizontal axis in each box represents the assembled sequence, while the vertical axis represents sequences from other species. The values in parentheses indicate the proportion of homologous sequences in the total genome. Red lines in the boxes represent forward alignments, while blue lines indicate reverse complementary alignments.
4 Discussion
This study, for the first time, reveals that the mitochondrial genome of the tea plant variety ‘Zhuyeqi’ exhibits a “1 circular + 6 linear” composite chromosome configuration (with a total length of 911,255 bp), which forms a significant contrast to the typical quadripartite circular structure of the ‘Zhuyeqi’ chloroplast. The chloroplast genome of ‘Zhuyeqi’ has a total length of 157,072 bp, encoding 52 protein-coding genes (CDS), 8 rRNA genes, and 37 tRNA genes, and maintains genomic stability through inverted repeat (IR) regions (Zeng et al., 2025). In contrast, the mitochondrial genome relies on dispersed repeat sequences (accounting for 72%) to mediate chromosome breakage/fusion events. The mitochondrial genome of ‘Baihaozao’ consists of 11 linear chromosomes (with a total length of 909,843 bp and a GC content of 45.62%). This structural difference leads to significantly higher recombination activity in the mitochondria (with forward repeats accounting for 45%) compared to the chloroplast (SSR density of only 5.3/kb) (Chen et al., 2025).This structural dynamic balance may be maintained by homologous recombination mediated by direct repeats (45%) and palindromic repeats (55%), similar to the recombination mechanisms of the multi-chromosomal mitochondrial structures in vascular plants such as Quercus acutissima, which rely on intramolecular recombination mediated by repetitive sequences to maintain genome plasticity (Wynn and Christensen, 2019; Liu et al., 2022). Notably, the modular recombination characteristics of linear chromosomes may optimize mitochondrial translation efficiency through the dosage effects of multi-copy genes such as trnM-CAT (5 copies) and trnP-TGG (3 copies). This optimization likely supports the metabolic differentiation between young shoots and mature leaves of tea plants. Compared to the conserved intron distribution in the mitochondrial genome of sweet tea (Rosa rubus) from the Rosaceae family (4 introns in nad1/2/5/7), the dynamic loss of introns in nad4 (only 2 introns) in Camellia sinensis cv. ‘Zhuyeqi’ reveals a unique intron degradation pathway in the Camellia genus (Chen et al., 2023; Li et al., 2024).
A total of 556 C→U RNA editing sites were identified in the mitochondrial PCGs of Camellia sinensis cv. ‘Zhuyeqi’, with the highest editing densities observed in ccmFn (38 sites) and nad2 (30 sites). This indicates precise regulation of the assembly of electron transport chain complexes (Zhang et al., 2022). Comparative analysis shows that the number of editing sites in Camellia sinensis cv. ‘Zhuyeqi’ is significantly higher than the 478 sites in large-leaf tea (Camellia sinensis var. assamica) but lower than the 635 sites in Camellia gigantocarpa, suggesting a positive correlation between RNA editing intensity and mitochondrial genome complexity within the Camellia genus (Zhang et al., 2019; Lu et al., 2022). While the editing pattern is conserved with C. nitidissima and C. taliensis, the number of editing sites is significantly higher than that in C. taliensis (492 sites), suggesting functional divergence in RNA editing among tea species (Li et al., 2024). The editing event at position 701 of the chloroplast matK gene in ‘Huabai No. 1’ is directly associated with leaf color variation, while the C→U conversion at position 15 among the 30 editing sites of the mitochondrial nad2 gene significantly affects the activity of the respiratory chain complex (Yang et al., 2024b). Notably, the downregulation of the CsMORF9.2 factor in ‘Baiye No. 1’ leads to reduced RNA editing efficiency across multiple organelles, highlighting the synergistic role of editing factors between organelles (Yang et al., 2024b).
The high usage frequency of aliphatic amino acids (Leu/Ser/Arg, 27%) corresponds to the high nitrogen metabolism characteristics of tea plants, which may improve the hydrophobicity of respiratory chain proteins and enhance electron transport chain efficiency (Li et al., 2023b; Wang et al., 2024b). This feature is conserved in closely related species of the Camellia genus. For example, the mitochondrial genome of Camellia gigantocarpa shows a leucine (Leu) content as high as 18.5%, significantly higher than that of Arabidopsis thaliana (12.3%) and Asteraceae plants (14.1%), suggesting a unique nitrogen metabolism adaptation strategy in the Camellia genus (Lu et al., 2022). The extreme preference for the phenylalanine codon UUU (4%) may regulate the biosynthesis of phenylpropanoids, a feature also observed in the chloroplast genome of C. taliensis (Chen et al., 2023). The high-frequency usage of aliphatic amino acids (Leu/Ser/Arg accounting for 27%) in the mitochondrial genome complements the A/U-ending codon preference in the chloroplast, forming a functional synergy. The optimal codons in the chloroplast genome of ‘Damianbai’ include 14 codons such as AAU (asparagine) and GAU (aspartic acid), with an ENC value of 46.1. In contrast, high-frequency codons in the mitochondria (e.g., UUU/AUU) account for 47% of the RSCU value, suggesting that mitochondria are under stronger positive selection pressure for synthesizing energy metabolism-related proteins (Yin et al., 2024). Notably, in the chloroplast genome of ‘Xilian No. 1,’ the RSCU value of AUC (isoleucine) reaches 1.98, which synergistically optimizes translational efficiency with the mitochondrial codon AUU (isoleucine) at an RSCU value of 2.83 (LIU et al., 2023). Notably, the complete absence of CUG/UUG Met codons may safeguard translation fidelity by avoiding conflicts with start codons, a feature highly conserved in cultivated tea plants (Chen et al., 2020; Li et al., 2024). This phenomenon contrasts sharply with the low retention rate of the CUG codon (<3%) in Asteraceae, suggesting that mitochondrial genomes in different groups maintain translation accuracy through distinct mechanisms (Zhang et al., 2019).
Dispersed repeats accounted for 72% of the mitochondrial genome, with the largest forward repeat measuring 8,082 bp. This abundance is comparable to that in Camellia nitidissima (72%) but significantly higher than in Camellia taliensis, suggesting that the non-homologous end-joining mechanism plays a central role in chromosome breakage and fusion (Wynn and Christensen, 2019; Li et al., 2024). In contrast to the Rosaceae mitochondrial genome, where tandem repeats account for 35%, tea plants appear to rely more on chromosome breakage/fusion events mediated by the non-homologous end-joining mechanism, rather than the typical microhomology recombination mechanism of Rosaceae (Ponty et al., 2006). The scarcity of tandem repeats (only 29) aligns with the genomic features of Rosaceae plants, indicating that tea mitochondria rely more on dispersed repeats to maintain genetic diversity rather than satellite DNA amplification (Wynn and Christensen, 2019; Li et al., 2024). Notably, in the mitochondrial genome of Asteraceae, 8–10 kb repeat sequences form sub-genomic circular structures through homologous recombination. The 8.1 kb repeat identified in this study may use a similar mechanism, suggesting that this evolutionary strategy is conserved across families in higher plants (Zhang et al., 2019). Notably, the presence of the 8.1 kb large repeat sequence may mediate sub-genomic circularization through recombination, forming a dynamic balance of multi-chromosomal structures. This mechanism is similarly validated in the mitochondrial genome of Stewartia gemmata (an early-branching species of Theaceae), where a 15.7 kb repeat sequence also drives the formation of multi-circular sub-genomes (Liu et al., 2024).
Phylogenetic analysis showed that Camellia sinensis cv. ‘Zhuyeqi’, C. sinensis (OM809792.1), and C. sinensis var. assamica (OL989850.1) formed a monophyletic group (Bootstrap = 100%), 1supporting Camellia sinensis cv. ‘Zhuyeqi’ as a core genotype of Chinese small-leaf tea plants (Chen et al., 2023). Its extremely low p-distance (<0.002) with the cultivated variety pubilimba reflects the influence of multi-generational artificial selection on its genome, providing molecular evidence for the “multi-origin hypothesis” of tea domestication (Chen et al., 2020; Li et al., 2024). Notably, compared to the widespread RNA editing events in 44 protein-coding genes of the Camellia gigantocarpa mitochondrial genome (Lu et al., 2022), the high Pi value (0.09) of key nucleo-mitochondrial interaction genes rps4 and atp8 in Camellia sinensis cv. ‘Zhuyeqi’ suggests their involvement in stress responses by regulating ribosome assembly efficiency and ATP synthesis. It also reflects convergent evolution with the positive selection signals of the mitochondrial atp8 in sweet tea, and similar adaptive selection patterns have been observed in Solanaceae plants (e.g., potato) (Zhang et al., 2019).
The retention of 66 chloroplast homologous fragments (25,656 bp) in the mitochondria is significantly higher than that in sweet tea (55 fragments) and Camellia nitidissima (19 fragments), and far exceeds the cultivated variety pubilimba (40–46 fragments) and the Assam variety (OL989850.1, about 45 fragments) (Chen et al., 2023; Li et al., 2024). This indicates the active role of horizontal gene transfer in the evolution of tea plant organelles (Chen et al., 2023; Li et al., 2023b). The transfer of key genes such as trnA-UGC and rpl2 may compensate for mitochondrial ribosome assembly defects, a mechanism showing molecular homology to the functional replacement of chloroplast rps12 in Arabidopsis (Li et al., 2023b, 2024). Similarly, the transfer of trnM-CAU in the mitochondrial genome of Camellia oleifera exhibits comparable compensatory evolution (Lu et al., 2022). Notably, the retention of trnA-UGC may enhance translation initiation efficiency, facilitating the rapid growth of young shoots. This hypothesis aligns well with the high metabolic activity characteristic of tender leaves in cultivated tea plants (Chen et al., 2023; Li et al., 2024). A comparative analysis shows that the proportion of quadruplex repeat sequences in the mitochondrial genome of Camellia sinensis cv. ‘Zhuyeqi’ (43.9%) is similar to that of the Assam variety ‘Duntsa’ (43.90%) (Li et al., 2023b), but lower than that of Camellia oleifera (47.2%) (Lu et al., 2022). This suggests that the expansion of repeat sequences may be related to species-specific environmental adaptation.
The Ka/Ks ratios of the 37 PCGs analyzed in this study showed a wide range from 0.09 to 2.70. According to molecular evolution theory, Ka/Ks< 1 indicates purifying selection driving conservative evolution, Ka/Ks ≈ 1 reflects neutral evolution, and Ka/Ks > 1 suggests positive selection (Hurst, 2002; Lan et al., 2024). Notably, the average Ka/Ks value of the ccmB gene reached 1.38, significantly deviating from the neutral evolution threshold (p< 0.01), suggesting that this gene may have undergone adaptive evolutionary pressure. This phenomenon is similar to the positive selection pattern of the Nad1 and Sdh3 genes in the tea variety (C. sinensis var. assamica) (Li et al., 2024), but significantly differs from the overall Ka/Ks ratio (0.39) of the mitochondrial genome in C. oleifera (Xia et al., 2014). This indicates significant differences in evolutionary pressures on mitochondrial genes among different Camellia species. The high evolutionary rate of ccmB may be related to its functional characteristics. Similar phenomena have been reported for other metabolism-related genes, such as atp8 (Ka/Ks = 0.81–1.65), which may indicate adaptive adjustments in energy metabolism pathways (Ye et al., 2021; Lu et al., 2023). Comparative analysis shows that the ccmB gene in the mitochondrial genome of Arabidopsis thaliana also exhibits a high Ka/Ks value (1.25-1.89) (Lu et al., 2022), suggesting that the functional plasticity of this gene in the assembly of the mitochondrial respiratory chain complex may be conserved across species. In this study, the strong conservation of cox1 (Ka/Ks = 0.09-0.17) and cytB (0.21-0.47) is consistent with the conserved trend of the mitochondrial genome in Stewartia gemmata (Theaceae) (Liu et al., 2024), and aligns closely with the purifying selection pattern (Ka/Ks< 0.5) of complex I core genes (e.g., nad5) in the closely related C. gigantocarpa (Lu et al., 2022). This conservation may result from the functional constraints of these genes in the mitochondrial electron transport chain. However, compared to the high variability of rps19 and ycf1 genes in the chloroplast genome of tea plants (with a structural rearrangement frequency of 32%) (Chen et al., 2023), the structural stability of mitochondrial genes is more pronounced, indicating coordinated differences in evolutionary rates between organelle genomes. In terms of genome structure dynamics, the multi-chromosomal configuration of Camellia sinensis cv. ‘Zhuyeqi’ mitochondria contrasts sharply with the single circular genome (701-1,082 kb) of the tea variety Duntsa (C. sinensis var. assamica cv. Duntsa) (Li et al., 2023b). However, it is partially similar to the genome fragmentation mediated by multiple repeat sequences observed in C. oleifera, where the SSR density reaches 5.3/kb (Liu et al., 2024). Genome plasticity mediated by repetitive sequences, combined with functional optimization driven by natural selection, constitutes the core mechanism of organellar evolution in tea plants. However, this study has not yet addressed the co-evolutionary network between the nuclear and mitochondrial genomes. Future research should integrate high-throughput chromatin conformation capture data to reveal how nuclear-mitochondrial interactions regulate energy metabolism (Chen et al., 2020; Kong et al., 2023). At the functional evolution level, the positive selection of ccmB in this study complements the expansion of the acyltransferase gene family in Camellia sinensis var. sinensis (driven by whole-genome duplication events) (Wei et al., 2018), suggesting potential co-evolution between mitochondrial respiration and nuclear genome secondary metabolic pathways. In the future, a comparative analysis of the pan-mitochondrial genome map of the Camellia genus, combined with the purifying selection patterns (Ka/Ks< 1) of disease resistance-related genes (e.g., the Camellia sinensis Pectin Methylesterases family) (Huang et al., 2022), could help clarify the molecular trade-off mechanism between disease resistance and quality formation.
5 Conclusions
This study provides a systematic analysis of the mitochondrial genome of the tea cultivar Camellia sinensis cv. ‘Zhuyeqi’, revealing its multi-chromosomal structure and adaptive evolution mechanisms. The mitochondrial genome consists of seven chromosomes (total length: 911,255 bp; GC content: 45%), and its structural features are highly conserved compared to previously reported tea cultivars (GC content: 45%-46%). A total of 38 PCGs were annotated, with core functional gene clusters (e.g., atp, nad, cox) highly conserved among terrestrial plant mitochondria. However, the number of PCGs is significantly lower than that of the large-leaf tea cultivar Duntsa (47 PCGs), suggesting interspecies gene loss or annotation differences. RNA editing analysis identified 556 editing sites, with the ccmFn gene being a hotspot containing 38 sites, far exceeding the mitochondrial average of 139 sites in large-leaf tea cultivars. This suggests that ccmFn may play a unique role in mitochondrial function regulation in Camellia sinensis cv. ‘Zhuyeqi’. C-to-U editing dominated (98%), resulting in hydrophilicity or hydrophobicity changes in 48% of amino acids, which may mediate adaptive regulation of mitochondrial energy metabolism by altering transmembrane domain conformations.
Codon usage bias analysis showed that leucine (Leu, 10%) and tryptophan (Trp, 1%) had the highest and lowest usage frequencies, respectively. Codons with RSCU > 1 preferentially ended with A/T (AT content: 69%), reflecting co-adaptation to the high AT content of the mitochondrial genome. Repetitive sequence analysis revealed that dispersed repeats (72%) played a dominant role in genome evolution, significantly higher than that in cultivars like Duntsa (44%). Short repeats (<100 bp) accounted for 86%, potentially driving structural variations through homologous recombination. The phylogenetic tree clustered Camellia sinensis cv. ‘Zhuyeqi’ within the genus Camellia, showing the closest genetic distance to Camellia sinensis (bootstrap value: 98%). The mitochondrial genome exhibited species-specific single nucleotide polymorphisms in marker genes such as cox1 and nad5, providing molecular evidence for cultivar identification. This study is the first to reveal the high density of RNA editing sites and unique repetitive sequence composition in the mitochondrial genome of Camellia sinensis cv. ‘Zhuyeqi’, offering critical molecular evidence for understanding the adaptive evolution of tea mitochondria. It also provides a theoretical foundation for assessing tea germplasm resources and advancing molecular breeding.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
ZhC: Project administration, Conceptualization, Methodology, Writing – review & editing, Resources, Funding acquisition, Data curation. WZ: Data curation, Conceptualization, Validation, Writing – original draft, Methodology. ZW: Conceptualization, Writing – original draft, Data curation, Validation, Formal analysis, Methodology. ZiC: Methodology, Writing – review & editing, Supervision, Validation. XY: Writing – review & editing, Methodology. YG: Investigation, Resources, Supervision, Conceptualization, Writing – review & editing, Methodology, Visualization, Data curation.
Ethics statement
This study did not involve human subjects or animal experiments; therefore, ethical approval or informed consent from participants was not required. All relevant laws and regulations were strictly followed during the research process to ensure data security and participant privacy protection.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the following projects: Hunan Provincial Natural Science Foundation (awarded to Zhiyin Chen, project number 2023JJ50465); Hunan Provincial Talent Project (awarded to Zhiyin Chen, project number 2024RC8289); Loudi Science and Technology Innovation Program (awarded to Zhiyin Chen, project number 2023RC3501).
Acknowledgments
We express our sincere gratitude to the Taohuayuan Agricultural Development Co., Ltd. of Xinhua County, Hunan Province, for providing a standardized tea plantation as the experimental base and assisting in the standardized collection of research samples.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1644130/full#supplementary-material
Abbreviations
bp, Base pair; IR, Inverted repeat; RNA, Ribonucleic acid; Ka, Non-synonymous substitution rate; Ks, Synonymous substitution rate; DNA, Deoxyribonucleic acid; GC, Guanine-Cytosine; tRNA, Transfer ribonucleic acid; ORF, Open Reading Frame; NCBI, National Center for Biotechnology Information; RSCU, Relative synonymous codon usage; SSR, Simple sequence repeat; Pi, Nucleotide diversity; rRNA, Ribosomal ribonucleic acid; ATP, Adenosine triphosphate; NADH, Nicotinamide adenine dinucleotide, reduced form; PCG, Protein-coding genes.
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
An, Y. L., Chen, L. B., Tao, L. L., Liu, S. R., and Wei, C. L. (2021). QTL mapping for leaf area of tea plants (Camellia sinensis) based on a high-quality genetic map constructed by whole genome resequencing. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.705285
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, C. W., Qu, Y. S., Hou, J., Wu, K., Ye, N., and Yin, T. M. (2022). Deciphering the multi-chromosomal mitochondrial genome of populus simonii. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.914635
Chen, Z. Y., Liu, Q., Xiao, Y., Zhou, G. H., Yu, P. H., Bai, J., et al. (2023). Complete chloroplast genome sequence of Camellia sinensis genome structure, adaptive evolution, and phylogenetic relationships. J. Appl. Genet. 64, 419–429. doi: 10.1007/s13353-023-00767-7
Chen, Z.Y., Wang, Z. X., Zhou, W., Liu, S. J., Xiao, Y. X., Gong, et al. (2025). Complete sequencing of the mitochondrial genome of tea plant Camellia sinensis cv.’Baihaozao’: multichromosomal structure, phylogenetic relationships, and adaptive evolutionary analysis. Front. Plant Sci. 16. doi: 10.3389/fpls.2025.1604404
Chen, J. D., Zheng, C., Ma, J. Q., Jiang, C. K., Ercisli, S., Yao, M. Z., et al. (2020). The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidization event in tea plant. Horticulture Res. 7, 63. doi: 10.1038/s41438-020-0288-2
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10, giab008. doi: 10.1093/gigascience/giab008
Duan, C. and Lai, W. (2020). Systematic analysis reveals cis and trans determinants affecting C-to-U RNA editing in Arabidopsis thaliana. BMC Genet. 21, 98. doi: 10.1186/s12863-020-00907-6
Fu, L. M., Niu, B. F., Zhu, Z. W., Wu, S. T., and Li, W. Z. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gao, R. S. (2022). Study on Post-transcriptional Processing of Chloroplast Genes and RNA Editing Factors in Albino of Tea Plant (Nanjing: Nanjing Agricultural University).
Hong, Z., Liao, X. Z., Ye, Y. J., Zhang, N. N., Yang, Z. J., Zhu, W. D., et al. (2021). A complete mitochondrial genome for fragrant Chinese rosewood (Dalbergia odorifera, Fabaceae) with comparative analyses of genome structure and intergenomic sequence transfers. BMC Genomics 22, 672. doi: 10.1186/s12864-021-07967-7
Huang, D. J., Mao, Y. X., Guo, G. Y., Ni, D. J., and Chen, L. (2022). Genome-wide identification of PME gene family and expression of candidate genes associated with aluminum tolerance in tea plant (Camellia sinensis). BMC Plant Biol. 22, 306. doi: 10.1186/s12870-022-03686-7
Huang, H., Shi, C., Liu, Y., Mao, S. Y., and Gao, L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing genome structure and phylogenetic relationships. BMC Evolutionary Biol. 14, 151. doi: 10.1186/1471-2148-14-151
Hurst, L. D. (2002). The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18, 486–487. doi: 10.1016/s0168-9525(02)02722-1
Katoh, K. and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kong, W. L., Yu, J. X., Yang, J. J., Zhang, Y. B., and Zhang, X. T. (2023). The high-resolution three-dimensional (3D) chromatin map of the tea plant (Camellia sinensis). Horticulture Res. 10, uhad179. doi: 10.1093/hr/uhad179
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lan, G. W., Yu, J. J., Liu, J., Zhang, Y., Ma, R., Zhou, Y. S., et al. (2024). Complete mitochondrial genome and phylogenetic analysis of tarsiger indicus (Aves passeriformes muscicapidae). Genes 15, 90. doi: 10.3390/genes15010090
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2016). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. doi: 10.1093/molbev/msw260
Li, S. C., Chang, L., and Zhang, J. (2021). Advancing organelle genome transformation and editing for crop improvement. Plant Commun. 2, 100141. doi: 10.1016/j.xplc.2021.100141
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignmentMap format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, L., Li, X. R., Liu, Y., Li, J. D., Zhen, X. Y., Huang, Y., et al. (2024). Comparative analysis of the complete mitogenomes of Camellia sinensis var. sinensis and C. sinensis var. assamica provide insights into evolution and phylogeny relationship. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1396389
Li, H. Z., Song, K. K., Li, B., Zhang, X. H., Wang, D., Dong, S. L., et al. (2023a). CRISPR/cas9 editing sites identification and multi-elements association analysis in Camellia sinensis. Int. J. Mol. Sci. 24, 15317. doi: 10.3390/ijms242015317
Li, J., Tang, H., Luo, H., Tang, J., Zhong, N., and Xiao, L. Z. (2023b). Complete mitochondrial genome assembly and comparison of Camellia sinensis var. Assamica cv. Duntsa. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1117002
Liu, D., Guo, H. L., Zhu, J. L., Qu, K., Che, Y., Guo, Y. T., et al. (2022). Complex physical structure of complete mitochondrial genome of quercus acutissima (Fagaceae) A significant energy plant. Genes 13, 1321. doi: 10.3390/genes13081321
Liu, D. L., Zhang, Z. H., Hao, Y. L., Li, M. G., Yu, H. L., Zhang, X. R., et al. (2024). Decoding the complete organelle genomic architecture of Stewartia gemmata: an early-diverging species in Theaceae. BMC Genomics 25, 114–132. doi: 10.1186/s12864-024-10016-8
LIU, Z., ZHAO, Y., YANG, P. D., CHENG, Y., TANG, H. T., and YANG, Y. (2023). Characterization and phylogenetic analysis of the complete chloroplast genome of triploid tea plant Xilian 1. J. Tea Communication 50, 166–175.
Lu, C., Gao, L. Z., and Zhang, Q. J. (2022). A high-quality genome assembly of the mitochondrial genome of the oil-tea tree Camellia gigantocarpa (Theaceae). Diversity 14, 850. doi: 10.3390/d14100850
Lu, C. C., Huang, X. L., and Deng, J. (2023). Mitochondrial genomes of soft scales (Hemiptera Coccidae) features, structures and significance. BMC Genomics 24, 37. doi: 10.1186/s12864-023-09131-9
Mebeaselassie, A. and Zhu, C. Y. (2022). Genome-wide analysis of codon usage in sesame (Sesamum indicum L.). Heliyon 8, e08687. doi: 10.1016/j.heliyon.2021.e08687
Mower, J. P. (2009). The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 37, W253–W259. doi: 10.1093/nar/gkp337
Peng, J., Zhao, Y. L., Dong, M., Liu, S. Q., Hu, Z. Y., Zhong, X. F., et al. (2021). Exploring the evolutionary characteristics between cultivated tea and its wild relatives using complete chloroplast genomes. BMC Ecol. Evol. 21, 71–88. doi: 10.1186/s12862-021-01800-1
Ponty, Y., Termier, M., and Denise, A. (2006). GenRGenS: software for generating random genomic sequences and structures. Bioinformatics 22, 1534–1535. doi: 10.1093/bioinformatics/btl113
Sahu, J., Sarmah, R., Dehury, B., Sarma, K., Sahoo, S., Sahu, M., et al. (2012). Mining for SSRs and FDMs from expressed sequence tags of Camellia sinensis. bioinformation 8, 260–266. doi: 10.6026/97320630008260
Sloan, D. B., Alverson, A. J., Wu, M., Palmer, J. D., and Taylor, D. R. (2012). Recent acceleration of plastid sequence and structural evolution coincides with extreme mitochondrial divergence in the angiosperm genus Silene. Genome Biol. Evol. 4, 294–306. doi: 10.1093/gbe/evs006
Tyszka, A. S., Bretz, E. C., Robertson, H. M., Woodcock-Girard, M. D., Ramanauskas, K., Larson, D. A., et al. (2023). Characterizing conflict and congruence of molecular evolution across organellar genome sequences for phylogenetics in land plants. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1125107
Wang, Z. J., Cai, Q. W., Wang, Y., Li, M. H., Wang, C. C., Wang, Z. X., et al. (2022). Comparative analysis of codon bias in the chloroplast genomes of theaceae species. Front. Genet. 13. doi: 10.3389/fgene.2022.824610
Wang, Y., Chen, S. J., Chen, J. J., Chen, C. J., Lin, X. J., Peng, H., et al. (2023). Characterization and phylogenetic analysis of the complete mitochondrial genome sequence of Photinia serratifolia. Sci. Rep. 13, 770. doi: 10.1038/s41598-022-24327-x
Wang, L., Liu, X., Wang, Y. D., Ming, X. J., Qi, J. S., and Zhou, Y. Q. (2024a). Comparative analysis of the mitochondrial genomes of four Dendrobium species (Orchidaceae) reveals heterogeneity in structure, synteny, intercellular gene transfer, and RNA editing. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1429545
Wang, R., Luo, Y. J., Lan, Z., and Qiu, D. S. (2024b). Insights into structure, codon usage, repeats, and RNA editing of the complete mitochondrial genome of Perilla frutescens (Lamiaceae). Sci. Rep. 14, 13940. doi: 10.1038/s41598-024-64509-3
Wang, D., Rousseau-Gueutin, M., and Timmis, J. N. (2012). Plastid sequences contribute to some plant mitochondrial genes. Mol. Biol. Evol. 29, 1707–1711. doi: 10.1093/molbev/mss016
Wei, C. L., Yang, H., Wang, S. B., Zhao, J., Liu, C., Gao, L. P., et al. (2018). Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. PNAS 115, E4151–E4158. doi: 10.1073/pnas.1719622115
Wickham, H. (2017). ggplot2 – elegant Graphics for Data Analysis. 2nd Edition (New York: Springer-Verlag).
Wynn, E. L. and Christensen, A. C. (2019). Repeats of unusual size in plant mitochondrial genomes identification, incidence and evolution. G3 (Bethesda) 9, 549–559. doi: 10.1534/g3.118.200948
Xia, E. H., Jiang, J. J., Huang, H., Zhang, L. P., Bin, Z. H., and Gao, L. Z. (2014). Transcriptome analysis of the oil-rich tea plant, Camellia oleifera, reveals candidate genes related to lipid metabolism. PloS One 9, e104150. doi: 10.1371/journal.pone.0104150
Xu, L. Y. (2019). Construction of high-density snp genetic map and identification of candidate genes for quantitative traits in camellia sinensis. (Wuhan: Hua zhong agricultural university).
Xu, L. Y., Wang, L. Y., Wei, K., Tan, L. Q., Su, J. J., and Cheng, H. (2018). High-density SNP linkage map construction and QTL mapping for flavonoid-related traits in a tea plant (Camellia sinensis) using 2b-RAD sequencing. BMC Genomics 19, 955. doi: 10.1186/s12864-018-5291-8
Yang, S. Q., Liu, J. F., Jiang, S. C., Guo, B. Q., Guo, J., Cao, L. L., et al. (2024a). Mitochondrial genomic characteristics and phylogenetic analysis of 21 species of poaceae. Genomics Appl. Biol. 43, 1533–1547. doi: 10.13417/j.gab.043.001533
Yang, Y. Q., Tan, J., Wang, F., Peng, S. L., Chen, J., Tan, M. Y., et al. (2024b). Research and application progress in chloroplast genome of tea plant (Camellia sinensis). Biotechnol. Bull. 40, 20–30. doi: 10.13560/j.cnki.biotech.bull.1985.2023-0451
Yang, J. B., Yang, S. X., Li, D. Z., Lei, L. G., Ikeda, T., and Yoshno, H. (2006). Phylogenetic relationships of theaceae inferred from mitochondrial matR gene sequence data. Acta Botanica Yunnanica 28, 29–36.
Ye, F., Li, H., and Xie, Q. (2021). Mitochondrial genomes from two specialized subfamilies of reduviidae (Insecta hemiptera) reveal novel gene rearrangements of true bugs. Genes 12, 1134. doi: 10.3390/genes12081134
Yin, M. H., Zhang, J. X., Le, Y., He, F. F., Huang, T. H., and And Zhang, M. T. (2024). Genomic Characteristics, Codon Preference, and Phylogenetic Analysis of Chloroplasts of Camellia sinensis cv.’Damianbai’. J. Tea Sci. 44, 411–430. doi: 10.13305/j.cnki.jts.2024.03.009
Zeng, W. J., Zhu, Y. P., Chen, J. X., Li, H. Y., Wang, S. H., Gong, Y. H., et al. (2025). Analysis of Codon Usage Bias in Chloroplast and Mitochondrial Genomes of Camellia sinensis cv.’Zhuyeqi’. J. Tea Sci. 45, 201–218. doi: 10.13305/j.cnki.jts.2025.02.008
Zhang, F., Li, W., Gao, C. W., Zhang, D., and Gao, L. Z. (2019). Deciphering tea tree chloroplast and mitochondrial genomes of Camellia sinensis var. assamica. Sci. Data 6, 209–219. doi: 10.1038/s41597-019-0201-8
Zhang, M. Y., Li, Z., Wang, Z. J., Xiao, Y., Bao, L., Wang, M., et al. (2022). Exploring the RNA editing events and their potential regulatory roles in tea plant (Camellia sinensis L.). Int. J. Mol. Sci. 23, 13640. doi: 10.3390/ijms232113640
Zhang, M., Zhang, X. H., Huang, Y. L., Chen, Z. X., and Chen, B. H. (2024). Comparative mitochondrial genomics of Terniopsis yongtaiensis in Malpighiales structural, sequential, and phylogenetic perspectives. BMC Genomics 25, 853. doi: 10.1186/s12864-024-10765-6
Keywords: Camellia sinensis, mitochondrial genome, multichromosomal structure, RNA editing, phylogenetic relationships
Citation: Chen Z, Zhou W, Wang Z, Chen Z, You X and Gong Y (2025) Analysis of the mitochondrial genome of the Camellia sinensis cv. ‘Zhuyeqi’: multichromosomal structure, RNA editing sites, and evolutionary characterization. Front. Plant Sci. 16:1644130. doi: 10.3389/fpls.2025.1644130
Received: 09 June 2025; Accepted: 10 September 2025;
Published: 02 October 2025.
Edited by:
Eirini Sarrou, Hellenic Agricultural Organization – ELGO, GreeceReviewed by:
Zinian Wu, Chinese Academy of Agricultural Sciences, ChinaDahe Qiao, Guizhou Academy of Agricultural Sciences (CAAS), China
Copyright © 2025 Chen, Zhou, Wang, Chen, You and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiyin Chen, Mjk0MEBodWhzdC5lZHUuY24=; Yihui Gong, Z3loemdoQDE2My5jb20=