 Chaoqiang Zhang1Ruifeng Yang2,3Mengyue Wang2,3Jiayin Zhang4Jingting Shen5Bin Yang1Dongzhi Zhang1Liang Yin6Xiaoming Wang6
Chaoqiang Zhang1Ruifeng Yang2,3Mengyue Wang2,3Jiayin Zhang4Jingting Shen5Bin Yang1Dongzhi Zhang1Liang Yin6Xiaoming Wang6 Chien-Hsun Huang2,3*
Chien-Hsun Huang2,3* Jinglong Li2,3*
Jinglong Li2,3*- 1Key Laboratory of Hexi Corridor Resources Utilization of Gansu, College of Life Sciences and Engineering, Hexi University, Zhangye, Gansu, China
- 2State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, School of Life Sciences, Inner Mongolia University, Hohhot, Inner Mongolia, China
- 3Key Laboratory of Herbage and Endemic Crop Biology, Ministry of Education, School of Life Sciences, Inner Mongolia University, Hohhot, Inner Mongolia, China
- 4Ministry of Education Key Laboratory for Biodiversity Science and Ecological Engineering, Institute of Biodiversity Science, Fudan University, Shanghai, China
- 5Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 6College of Agriculture and Ecological Engineering, Hexi University, Zhangye, Gansu, China
Introduction: Gypsophila vaccaria (Caryophyllaceae) is a medicinal plant with over 2,000 years of documented use in China. Despite its known pharmacological properties and phytochemical profile, no organellar genomic resources are currently available, limiting evolutionary studies and molecular breeding efforts.
Methods: We assembled the complete mitochondrial (361,814 bp) and quadripartite chloroplast (150,050 bp) genomes of G. vaccaria using HiFi sequencing. Codon usage, RNA editing, and selection pressure were analyzed, and phylogenomic relationships were inferred. Species-specific SSR markers were identified for potential molecular applications.
Results: HiFi-based assembly revealed exceptional mitochondrial genome plasticity, with 15.6% (56.7 Kb) derived from chloroplast DNA transfers—the highest reported in Caryophyllaceae—including 12 functional genes (e.g., rps7, ndhB, rrn16S). Both organellar genomes show A/U-biased codon usage (mitochondrial RSCU: 29/44 codons) and divergent RNA editing (257 mitochondrial vs. 105 chloroplast C-to-U sites). Positive selection (Ka/Ks > 1) was detected in cytochrome c maturation genes (ccmFN, ccmB, ccmFC), contrasting with overall purifying selection (median ω = 0.32). Phylogenomic analyses robustly resolved Caryophyllaceae–Amaranthaceae sisterhood (BS = 100%).
Discussion: As the first organellar genomes from Gypsophila, this study provides insights into lineage-specific adaptations through chloroplast-mitochondrial co-evolution. The 56.7 Kb MTPTs and positively selected cytochrome c genes serve as targets for adaptive evolution research, while 81 species-specific SSRs facilitate molecular marker development in Caryophyllaceae.
1 Introduction
Gypsophila vaccaria Sm. (Caryophyllaceae) is an herbaceous plant native to temperate regions of Asia, Europe and other parts of the world. In China, it is widely distributed except in the southern regions (Tian et al., 2021). The dried, mature seeds of G. vaccaria, commonly known as “Wang Bu Liu Xing”, have long been used in traditional Chinese medicine (TCM) to treat amenorrhea, dysmenorrhea, mastitis, and urinary diseases (Sang et al., 2003; Committee, 2020; Tian et al., 2021), as well as to promote diuresis and milk secretion and to relieve carbuncles (Sang et al., 2003). As documented in the oldest materia medica, Shen Nong Ben Cao Jing, G. vaccaria has been used in China for 2000 years (Tian et al., 2021). In recent years, its clinical applications have expanded, with new uses identified, such as the treatment of gallstones (You, 1989), shingles (Tan, 2006), rhinitis (Xing and Jin, 2013), benign prostatic hyperplasia (Qi et al., 2013; Zhang et al., 2013), and hypertension (Liu, 2018). Phytochemical studies have revealed that flavonoids, triterpenoid saponins, cyclic peptides, and polysaccharides are the main bioactive components of G. vaccaria seeds (Zhou et al., 2016; Tian et al., 2021). These compounds exhibit vasodilatory, anticoagulant, anti-inflammatory, antitumor, antiangiogenic, and antioxidant activities, as well as estrogen-like effects (Li and Liang, 2007; Hu et al., 2014; Tian et al., 2021; Bing et al., 2024). Additionally, they have been shown to alleviate osteoporosis and promote vasodilation (Hu et al., 2014). Overall, G. vaccaria has attracted increasing attention in recent years.
To date, studies on this species have primarily focused on its chemical constituents (Zhou et al., 2016) and pharmacological properties (Tian et al., 2021; Li N et al., 2022). However, no studies have reported its mitochondrial or chloroplast genomes, significantly limiting further research. In angiosperms, the nuclear genome is biparentally inherited, whereas chloroplasts and mitochondrial DNA are maternally inherited (Cheng et al., 2021; Tian et al., 2021). This uniparental inheritance pattern, which excludes paternal genetic contributions, serves as a powerful tool for investigating species origin, genetic diversity, classification, and phylogenetic relationships (Birky, 2001; Sibbald et al., 2021; Fan et al., 2022). The mitogenomes of Angiosperm display substantial size variation, spanning from 22 Kilobases (Kb) in Avicennia marina (Friis et al., 2021) to 11.7 Megabases (Mb) in Larix sibirica (Putintseva et al., 2020), although most species harboring genomes between 200 and 750 Kb. Most mitogenomes contain 19 to 64 known genes (excluding duplicate genes and ORFs), 5 to 25 introns, and highly variable intergenic regions (Burger et al., 2003; Bi et al., 2022). The in vivo conformation of plant mitogenomes displays greater diversity. While most assembled plant mitogenomes are circular, polycyclic structures have been reported in maize (Fauron et al., 1995) and kiwifruit (Wang et al., 2019), linear structures in Thuja sutchuenensis (Xia et al., 2023), and multi-branched structures in Punica granatum (Feng et al., 2023; Lu et al., 2023). This structural complexity makes mitogenome assembly challenging, resulting in significantly fewer studies compared to chloroplast and plastid genomes. As of May 2025, the NCBI Organelle Genome Database (https://www.ncbi.nlm.nih.gov/genome/browse#!/organelles/) identified 688 mitogenomes, 15,396 chloroplast genomes, and 1,718 plastid genomes, highlighting the urgent need for expanded mitogenomic investigations. Recent work has demonstrated that mitochondrial genomes can provide unique insights for medicinal species. In Dendrobium and Salvia, structural variation and recombinogenic repeats have yielded diagnostic markers useful for authentication (Yang et al., 2022; Tong et al., 2024). RNA editing in plant mitochondrial genome plays a role in energy metabolism and stress responses, potentially influencing the accumulation of pharmacologically active compounds (Li et al., 2024). These findings highlight the dual importance of mitogenomes for both basic and applied research in medicinal plants. Given its long history of medicinal use and growing clinical applications, G. vaccaria represents a valuable yet genomically neglected taxon. The absence of organellar genomic data, particularly the mitochondrial genome, presents a major knowledge gap. Therefore, assembling and annotating the complete mitogenome of G. vaccaria is critical.
In this study, we assembled and annotated the complete G.vaccaria mitogenome for the first time based on PacBio HiFi long-read sequencing data. We also assembled its chloroplast genome from the same dataset. Characteristics of the G.vaccaria mitochondrial genome and chloroplast genome were analyzed, including codon preference, RNA editing events, repetitive sequence analysis, Ka/Ks analysis, phylogenetic analysis, and sequence migration analysis. These results will provide a foundation for understanding the evolution, molecular breeding, structural characteristics and organelles inheritance in Gypsophila species.
2 Results
2.1 Characteristics of the complete organelle genomes of G. vaccaria
HiFi reads unambiguously resolved the chloroplast genome as a circular quadripartite molecule (150,050 bp; Figure 1A), with IR regions (25,167 bp each), a large single copy (LSC) region (82,717 bp), and a small single copy (SSC) region (16,999 bp) (Figure 1A). A total of 121 functional genes were annotated, including 77 protein-coding genes, 37 tRNAs, and 8 rRNAs (Table 1). Intron analysis identified 16 genes (e.g., trnK-UUU、rps16、trnS-CGA、atpF、rpoC1、trnL-UAA、trnV-UAC、rpl16、ndhB、trnE-UUC、trnA-UGC、ycf1、ndhA、trnA-UGC、trnE-UUC and ndhB) containing single intron, while two genes (ycf3 and ycf1) possessed two introns each (Supplementary Table S1).
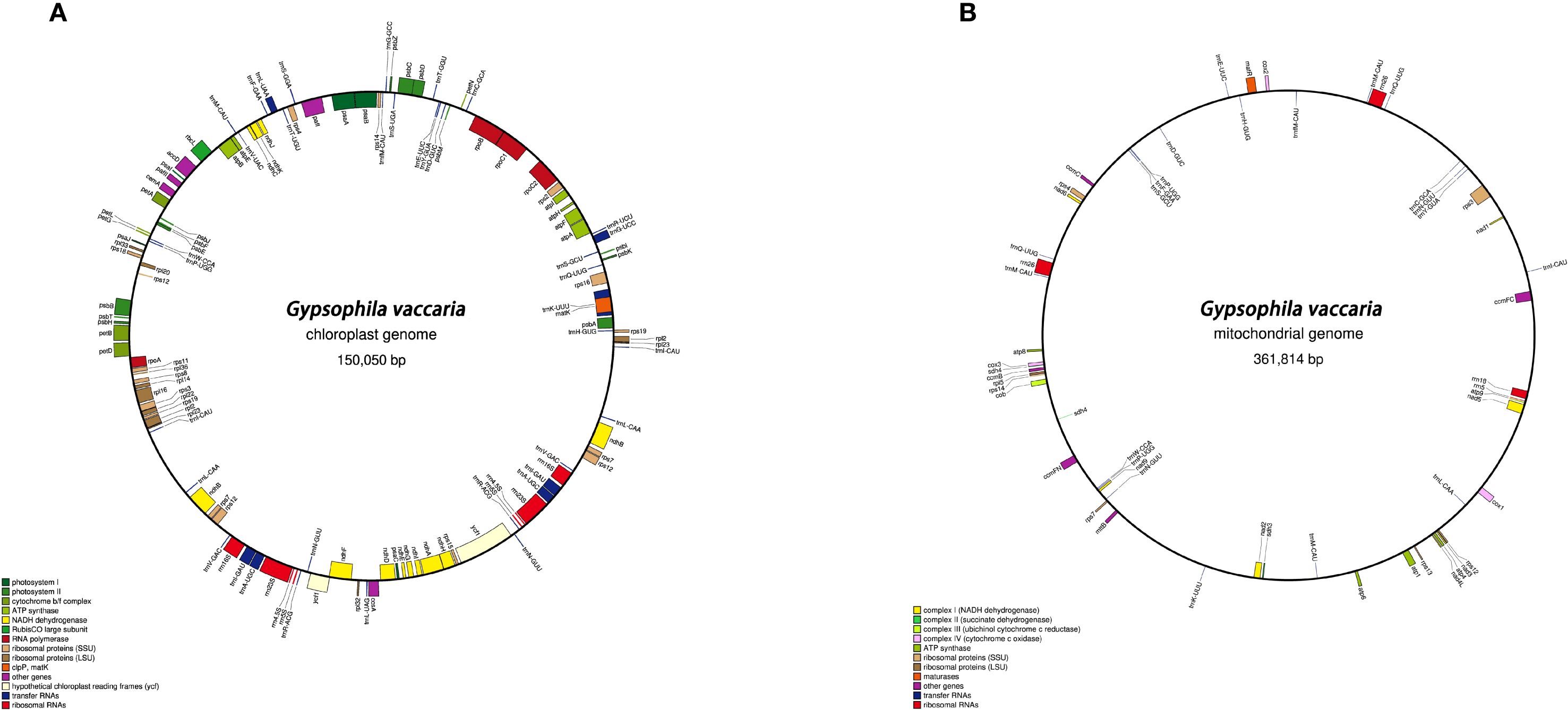
Figure 1. (A) Chloroplast genome of G.vaccaria. (B) Mitochondrial genome of of G.vaccaria. Genes are color-coded based on their functional groups. GC content is represented on the inner circle by the dark gray plot.
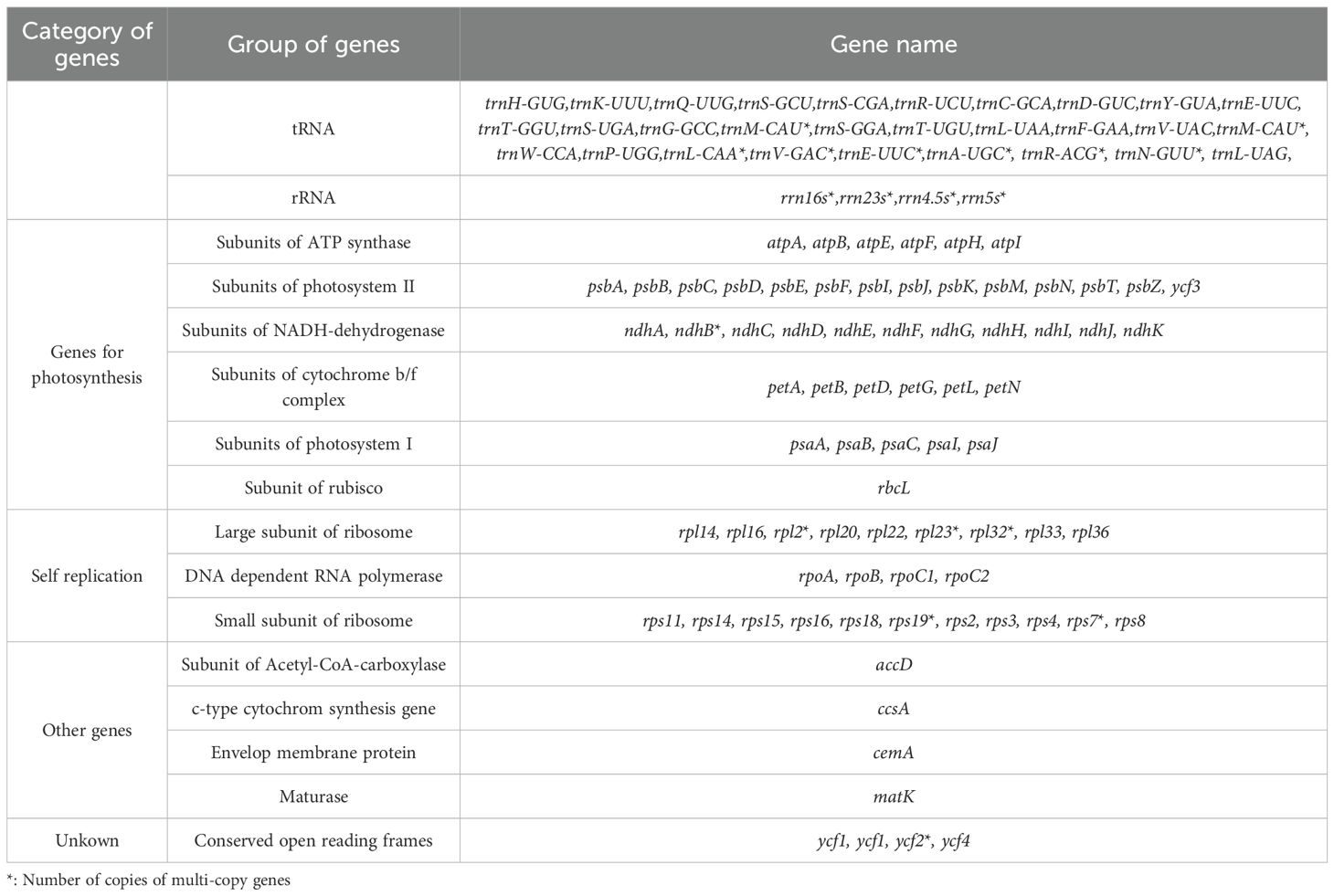
Table 1. Gene composition in the chloroplast genome of Gypsophila vaccaria plants.
The complete mitochondrial genome of G.vaccaria is a circular DNA molecule with a total length of 361,814 bp. (Figure 1B). Nucleotide composition analysis revealed a total GC content of 43.82%, comprising 28.17% (A), 28.00% (T), 21.93% (C), and 21.88% (G). A total of 60 genes were annotated in the G. vaccaria mitogenome (Figure 1B; Table 2), including 35 protein-coding genes (PCGs: 28 core genes and 7 variable genes), 21 tRNA genes (4 multicopy), and 4 rRNA genes (1 multicopy) (Table 2). Interestingly, the nad7 and sdh4 genes were present in two copies, while one rRNA gene (rrn26) and four tRNA genes (trnM-CAU, trnN-GUU, trnP-UUG trnQ-UUG) exhibited either two or three copies. Among the annotated genes in G. vaccaria, seven contained introns: ccmFC, nad1, nad2, nad4, nad5, nad7, and rps3. (Table 2; Supplementary Table S2).
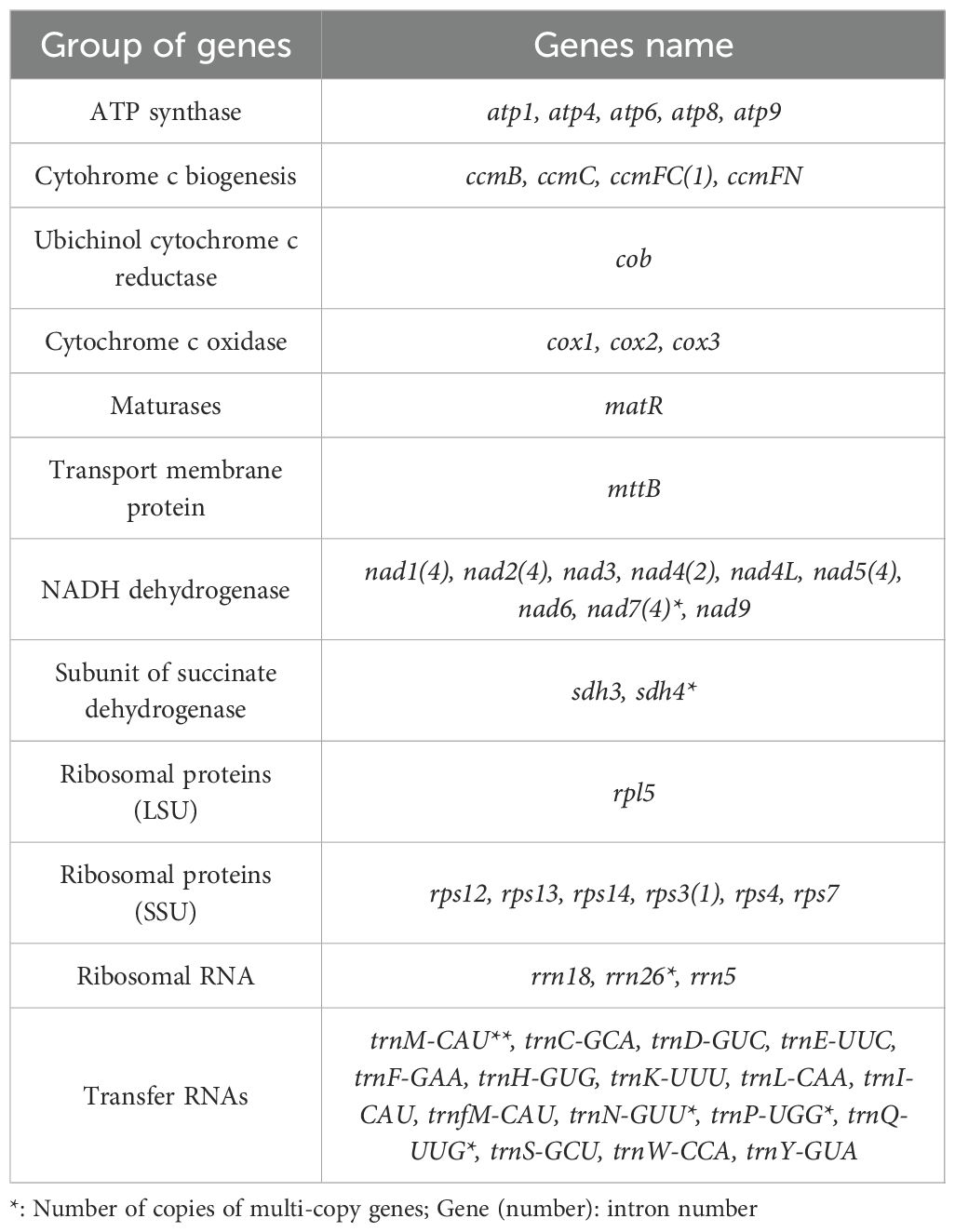
Table 2. Genetic composition of mitochondrial genome of the G. vaccaria.
2.2 Repeat sequences analysis in the G. vaccaria organelle genomes
In this study, 47 SSRs were detected in the chloroplast genome, whereas 34 in the mitochondrial genome (Figure 2; Supplementary Tables S3, Supplementary Tables S4). In the chloroplast genome, mononucleotide repeats—particularly A/T units—were the most abundant SSR type, representing 85.1% of the total. In the mitochondrial genome, they were similarly predominant (88.2%), closely matching the chloroplast pattern. Notably, the mitochondrial genome contained a trinucleotide repeat (TTC) that was undetected in the chloroplast genome. this TTC repeat could serve as a potential marker for identifying G. vaccaria.
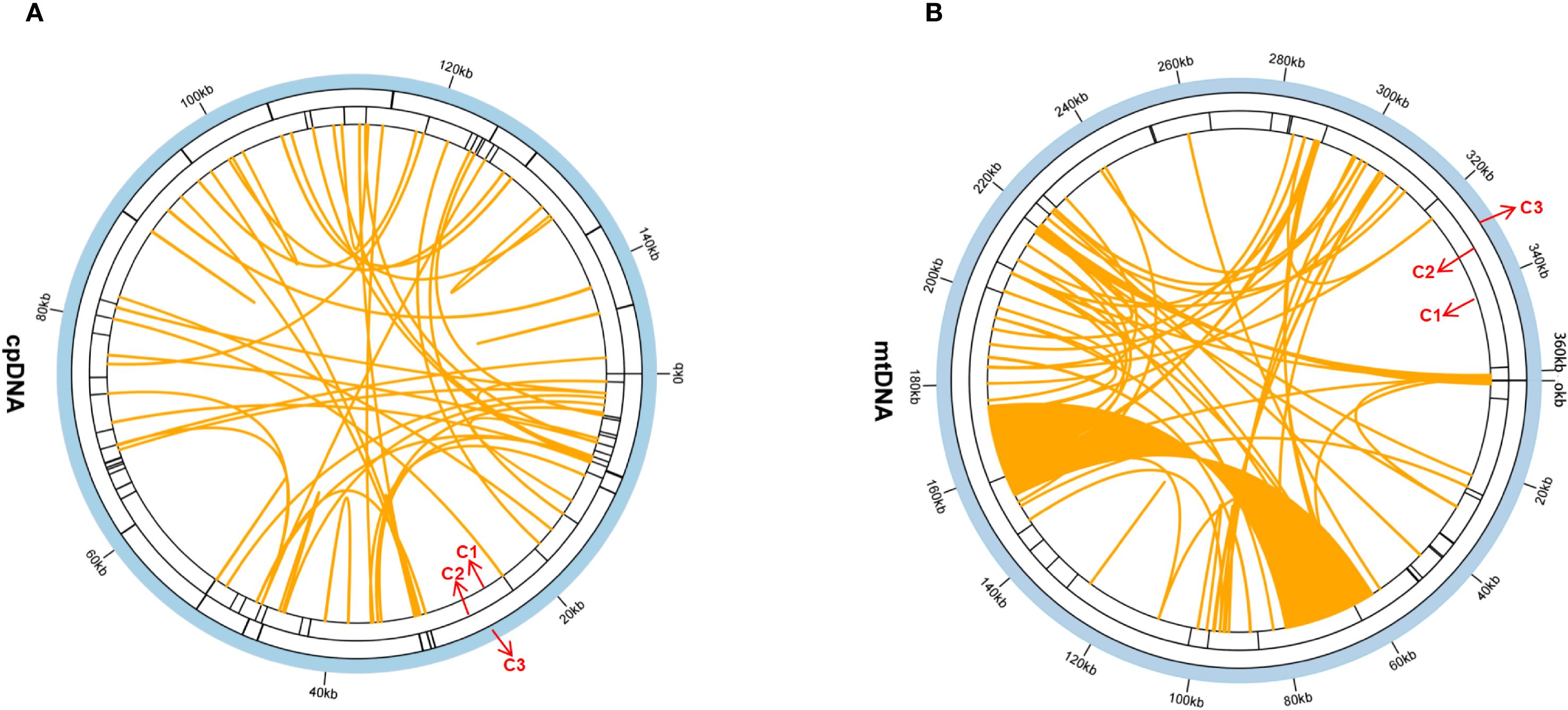
Figure 2. The repeat analysis of the G.vaccaria chloroplast genomes. (A) The repeat sequences identified in the chloroplast genome. (B) The repeat sequences identified in the mitochondrial genome. The C1 circle shows the dispersed repeats connected with yellow, blue, purple, and pink arcs from the center going outward. The C2 circle shows the tandem repeats as short bars. The C3 circle shows the microsatellite sequences identified using MISA. The scale is shown on the C3 circle, with intervals of 20 Kb.
Twenty-two tandem repeats (TRs) were detected in the chloroplast genome and 15 in the mitochondrial genome. Most TRs are located in intergenic regions (chloroplast: 18/22; mitochondrial: 12/15), with minor occurrences in coding regions (chloroplast: 3/22; mitochondrial: 2/15) and pseudogenes (chloroplast: 1/22) (Figure 2; Supplementary Tables S5, Supplementary Tables S6).
Surprisingly, only forward repeats were detected in both chloroplast and mitochondrial genomes (Figure 2, Supplementary Tables S7, Supplementary Tables S8). The absence of other repeat types suggests distinct evolutionary constraints on repeat-driven genomic plasticity in organellar compared to nuclear genomes.
2.3 Sequence similarity between mitochondrial and chloroplast genomes
The mitochondrial genome of G. vaccaria spans 361,814 bp, representing a 2.41-fold size expansion compared to its chloroplast counterpart (150,050 bp). Comparative analysis identified 20 chloroplast-derived DNA (MTPT) fragments in the mitogenome through BLASTn alignment, collectively spanning 56,732 bp and constituting 15.6% of the mitochondrial genome (Figure 3; Supplementary Table S9). Notably, 12 chloroplast-derived genes retained intact ORFs in the mitogenome, including five protein-coding genes (rps7, ndhB, psaB, petG, ycf15), 6 tRNA genes (trnL-CAA, trnW-CCA, trnP-UGG, trnN-GUU, trnD-GUC, trnM-CAU), and 1 rRNA gene (rrn16S). Among these, ndhB gene-encoding a NADH dehydrogenase subunit critical for the electron transport chain—represented the longest intact MTPT fragment (757 bp), suggesting potential functional retention in the mitochondrial genome (Figure 3; Supplementary Table S9), while trnM-CAU was the shortest at 77 bp. The presence of these homologous fragments suggests possible genomic recombination or transfer of genetic material between the chloroplast and mitochondrial genomes.
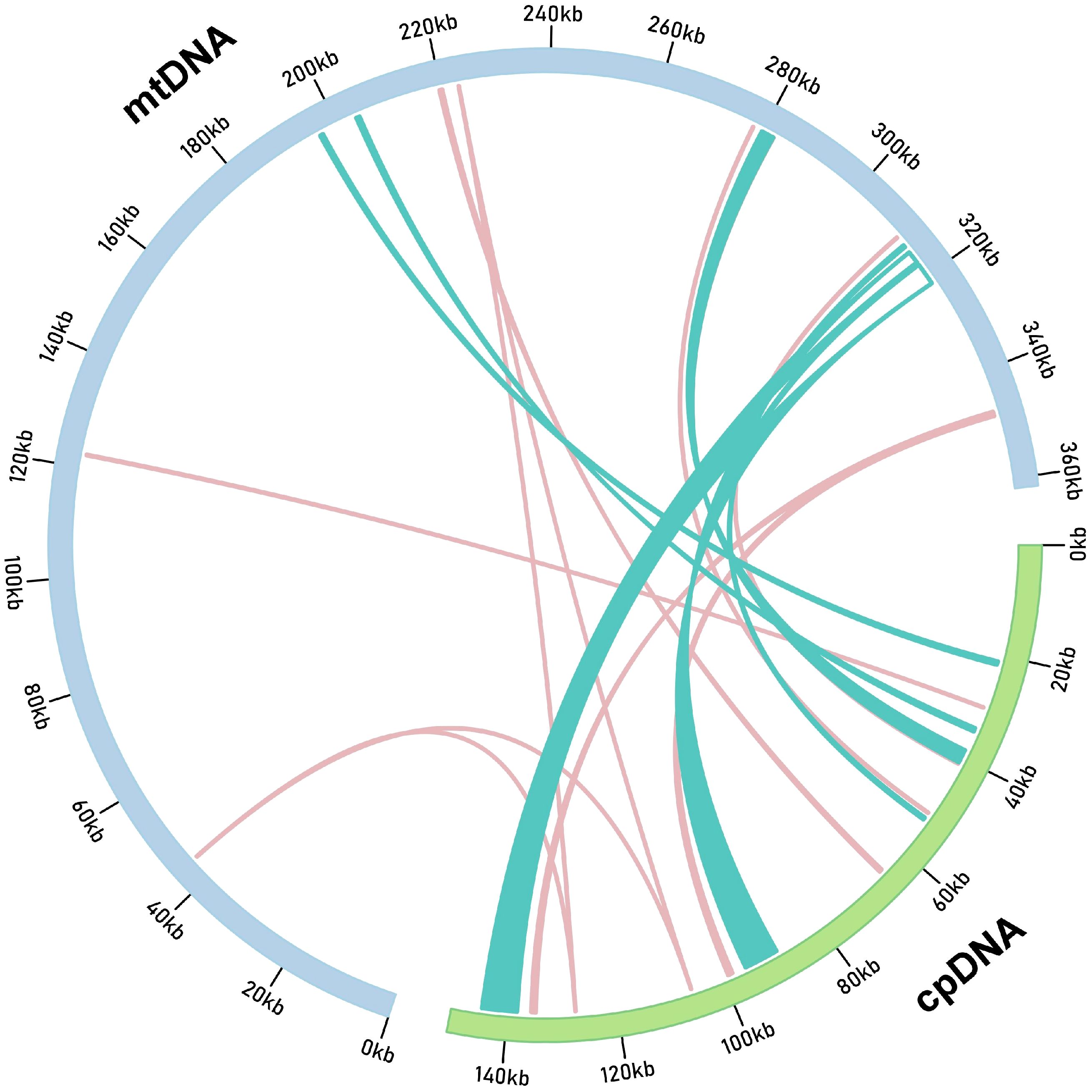
Figure 3. G. vaccaria of the transfer of chloroplast genes to the mitochondrial genome. The orange and teal blue lines within the circles represent the distribution of homologous genes on the positive and negative strands, respectively. (cpDNA: chloroplast genome; mtDNA: mitochondrial genome).
2.4 Comparative analysis of codon usage bias between organellar genomes
A total of 9,600 codons were identified in the G. vaccaria mitogenome. Among these codons, Phenylalanine (Phe, TTT codon) was the most frequently used amino acid, with a total of 412 codons, accounting for 4.29%, followed by isoleucine (Ile), with a total of 334 codons, accounting for 3.48%, and the lowest usage rate was termination codon, with a total of 22 codons, accounting for 0.23%. The AUG codon of methionine showed a high degree of preference in the G. vaccaria mt genome. Forty-five codons had RSCU values higher than one, indicating a preference for these codons. Among the 45 preferred codons, the third base of 29 codons ended with A or U, nine codons ended with G, and eight codons ended with C, which fully demonstrated the A/U preference of the third base of the codon (Figure 4A; Supplementary Table S10).
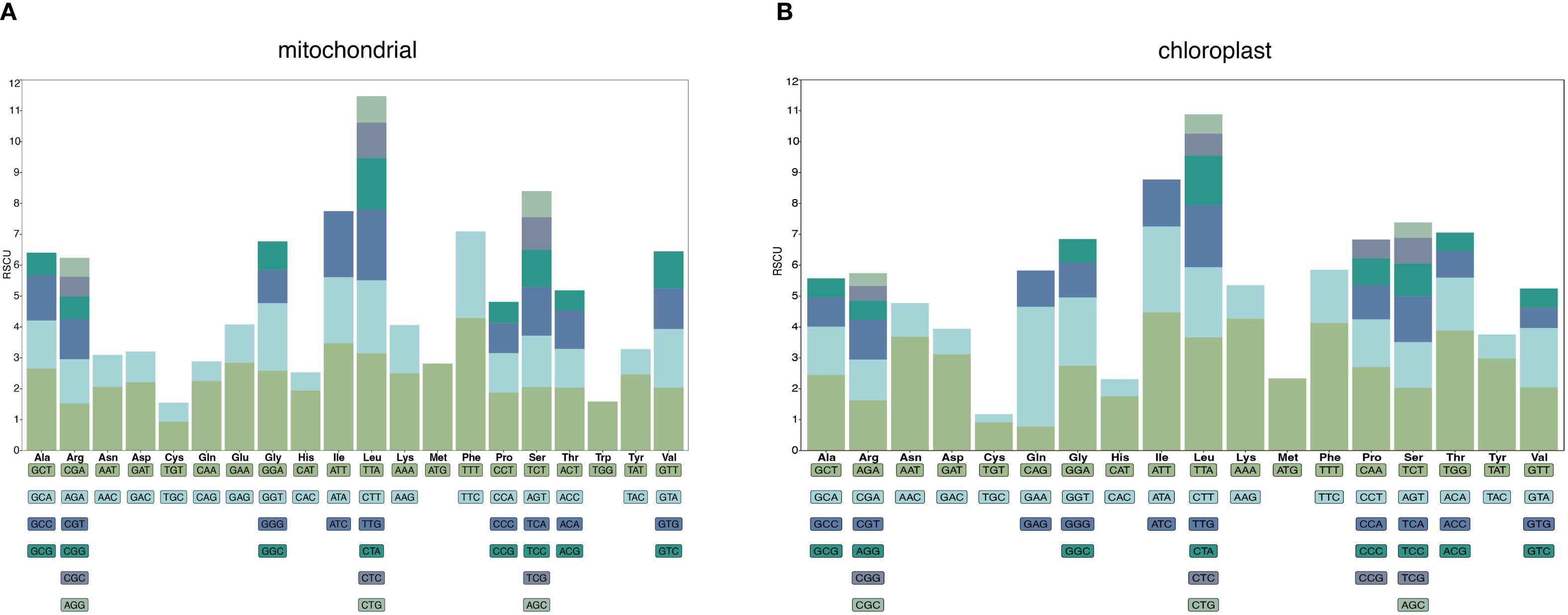
Figure 4. Analysis of relative synonymous codon usage (RSCU) in the G vaccaria mitochondrial (A) and chloroplast (B) genome. The following blocks represent all codons encoding each amino acid, and the height of the upper column represents the sum of RSCU values of all codons.
In contrast, the complete coding sequences of the G. vaccaria chloroplast genome comprised 23,984 codons. Among these codons, isoleucine (Ile, 1073 codons, 4.47%) was the most frequently encoded amino acid, followed by Lysine (Lys, 1024 codons, 4.27%), glutamine (Gln, 928 codons, 3.87%) and leucine (Leu, 879, 3.66%), while stop codons showed the lowest usage (82 codons, 0.34%). 39 codons had RSCU values higher than one, indicating a preference for these codons. Among the 39 preferred codons, the third base of 29 codons ended with A or U, six codons ended with G, and four codons ended with C, which fully demonstrated the A/U preference of the third base of the codon (Figure 4B; Supplementary Table S10).
2.5 Prediction of RNA editing sites in organellar genomes
We employed online prediction tools to analyze RNA editing patterns in 35 mitochondrial and 77 chloroplast protein-coding genes (PCGs). Analysis of the mitochondrial genome revealed 257 predicted RNA editing sites across all PCGs, exclusively involving C-to-U conversions (Figure 5A). The nad7 (32 sites) genes exhibited the highest editing frequency, followed nad5 (25 sites). Notably, certain genes, such as cox1, cox3, rps7, rpsl3, and rpsl4, exhibit only a single RNA editing event. In the chloroplast genome, 105 potential RNA editing sites were identified (Figure 5B). The ndhB (20 sites each) showed the highest editing activity, representing a 40.85% decrease compared to their mitochondrial counterparts. The ycf2 genes ranked second with 12 editing sites. Among them, more than half of the genes (23/43) undergo only one RNA editing event. These findings demonstrate conserved RNA editing distribution patterns between mitochondrial and chloroplast genomes in G. vaccaria, while revealing organelle-specific differences in editing site abundance for homologous genes.
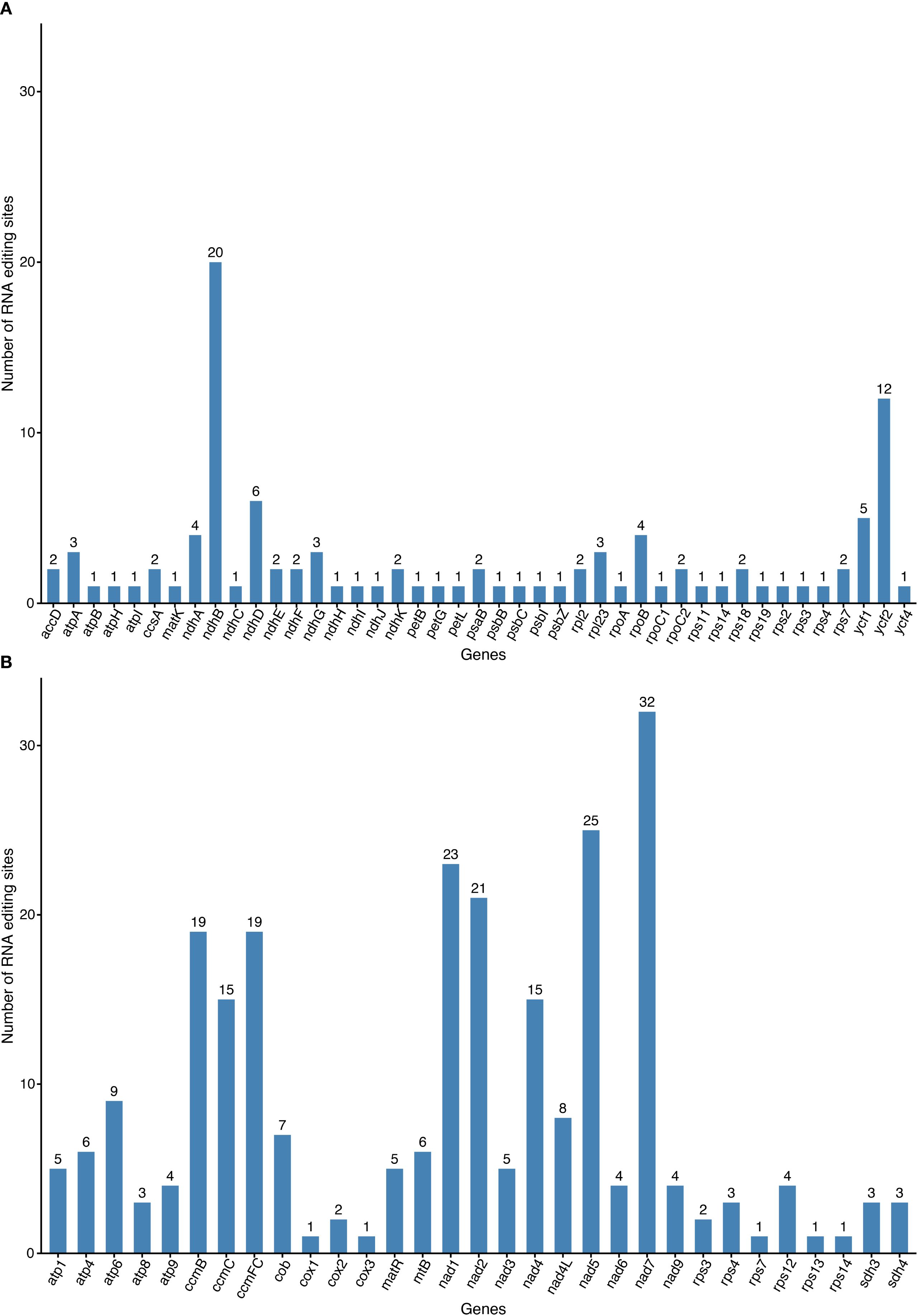
Figure 5. RNA editing events in the (A) mitochondrial and (B) chloroplast genomes of G vaccaria. The x-axis represents gene names, and the y-axis represents the number of RNA editing events.
2.6 Ka/Ks analysis and phylogenetic analysis
Evolutionary selection patterns were assessed through Ka/Ks (ω) ratio analysis of 20 conserved mitochondrial genes across nine Caryophyllales species. The median ω value across all genes was 0.32 (IQR:0.18-0.47), indicating predominant purifying selection (Figure 6). Three cytochrome c maturation genes showed signatures of positive selection (ω>1) in Caryophyllaceae.
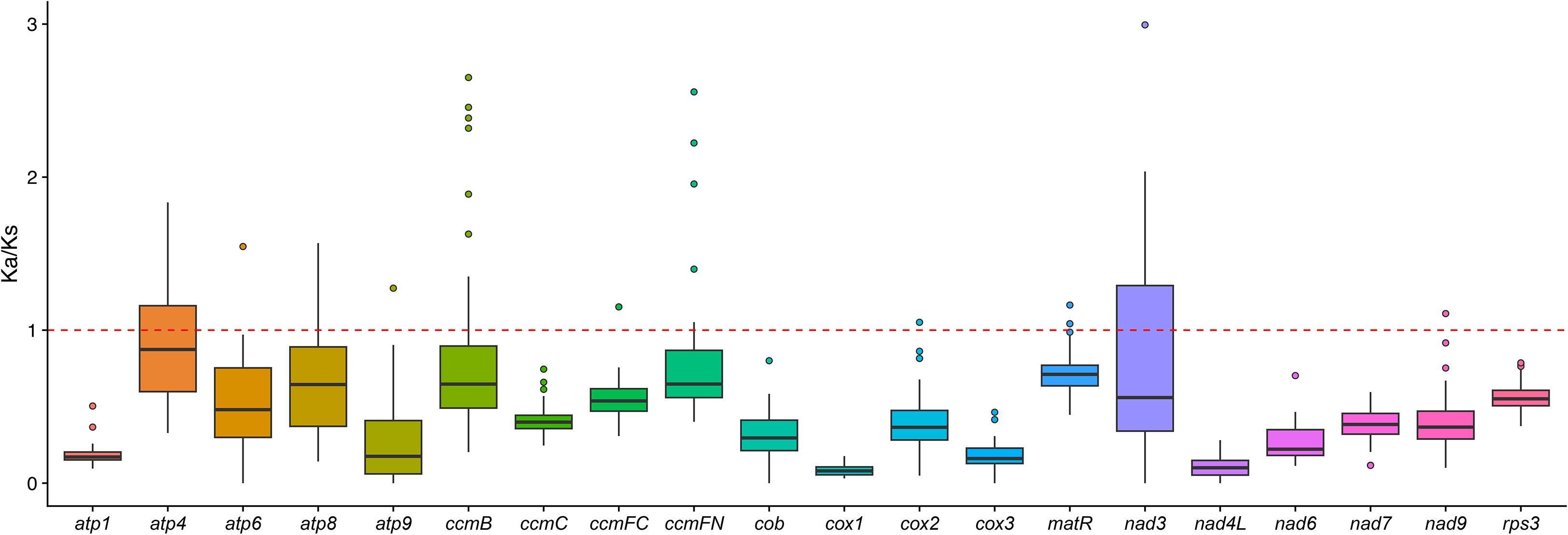
Figure 6. Ka/Ks ratio of G. vaccaria and eight other Caryophyllales plant species.
In our study, we calculated the Ka/Ks ratios for the shared genes in the mitochondrial genomes of nine Caryophyllales species. The median Ka/Ks value for all 20 shared genes was found to be less than 1, indicating that these genes have undergone purifying selection during the evolutionary process of Caryophyllales species. This suggests that these genes are evolutionarily conserved.
Further comparisons of Ka/Ks ratios between G. vaccaria (a Caryophyllaceae species) and two other Caryophyllales species revealed that the Ka/Ks ratio of the ccmFN gene was greater than 1 in G. vaccaria, indicating that this gene underwent positive selection within the Caryophyllaceae family. Similarly, the ccmB and ccmFC genes also showed Ka/Ks ratios greater than 1 in the comparison between G. vaccaria and Agrostemma githago, suggesting that these two genes experienced positive selection during the evolution of the two species. Additionally, the Ka/Ks ratios of the atp4, matR, and nad3 genes in G. vaccaria were greater than 1 when compared to Rheum palmatum, indicating that these three genes underwent positive selection in both species.
A phylogenetic analysis was conducted on the mitochondrial and chloroplast genomes of two species from the Caryophyllaceae family, along with six species from six different families in the order Caryophyllales, including Dianthus (carnation). The species Myricaria laxiflora from the Tamaricaceae family was used as the outgroup (Figure 7). The phylogenetic trees constructed from both mitochondrial and chloroplast genomes showed that the three Caryophyllaceae species clustered together as a single clade. In addition, the Caryophyllaceae family initially grouped with Amaranthaceae, followed by a further clustering with Nyctaginaceae and Cactaceae. In the mitochondrial tree, Nepenthaceae and Polygonaceae formed a distinct clade, although in the chloroplast tree, these two families did not cluster together. Overall, our phylogenetic analysis of Caryophyllales species with publicly available mitochondrial and plastid genomes from NCBI supports a close relationship between the Caryophyllaceae family and the Amaranthaceae family, which is consistent with previous studies.
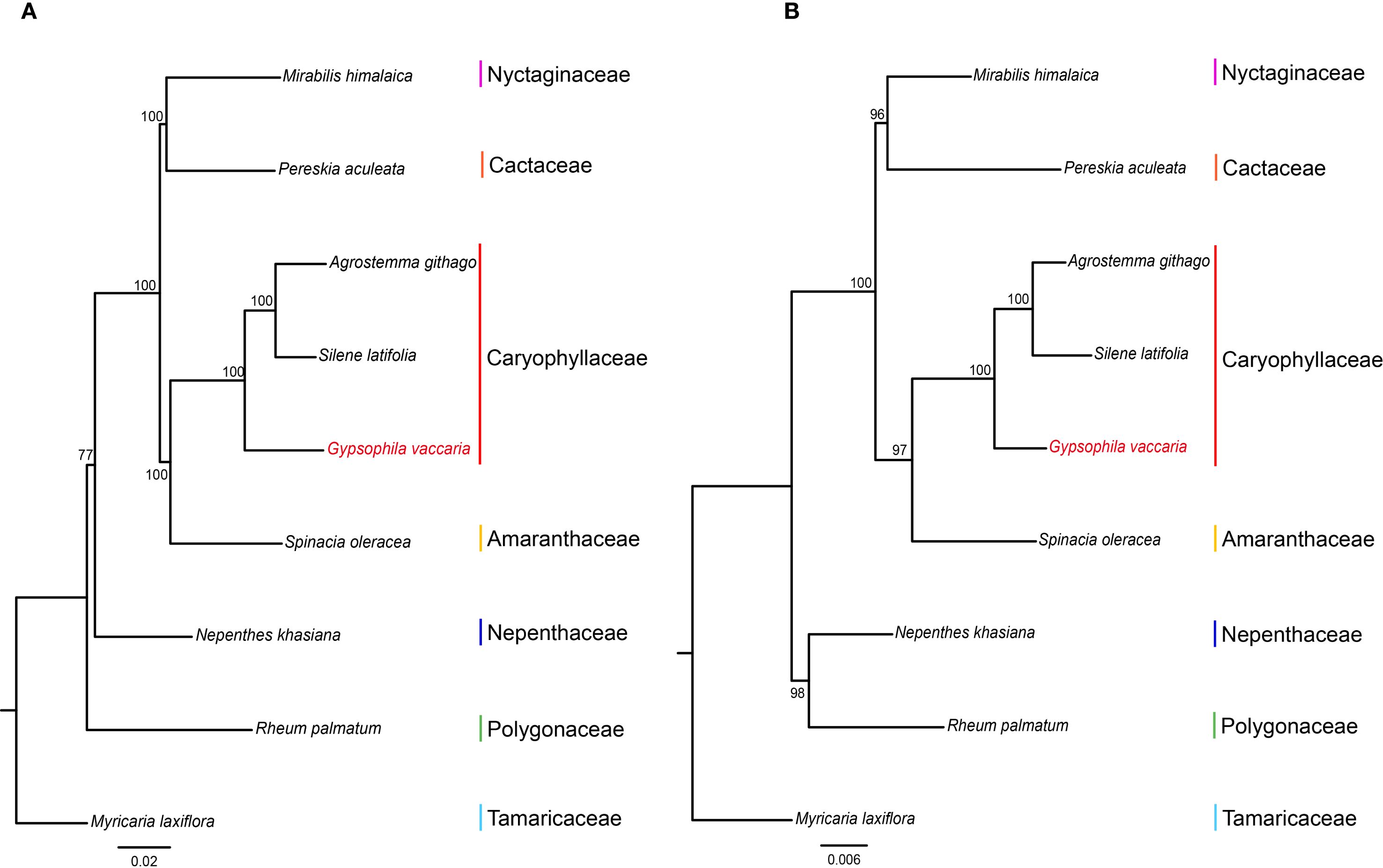
Figure 7. Phylogenetic relationships of G vaccaria and eight Caryophyllales species based on shared genes of mitochondrial genome and chloroplast genome. (A) A phylogenetic tree constructed by Maximum likelihood (ML) based on shared genes of 9 chloroplast genomes. (B) A phylogenetic tree constructed by Maximum likelihood (ML) based on shared genes of 9 mitochondrial genomes.
3 Discussion
Mitochondria drive ATP synthesis in plant cells, critically regulating growth and development (Mackenzie and McIntosh, 1999). Recent comparative studies revealed that the evolutionary trajectories of mitogenomes exhibit striking divergence, leading to extensive structural variations among plant lineages. For example, while Cinnamomum chekiangense, and Salix truncatum maintain a canonical circular mitochondrial genome (Han et al., 2022; Bi et al., 2024), other species exhibit structural diversity—Abelmoschus esculentus harbors two distinct configurations (Li et al., 2022), and Sorghum bicolor and Populus simonii demonstrate three or more structural isoforms (Bi et al., 2022; Zhang et al., 2023a). In this study, using high-fidelity PacBio HiFi long-read sequencing data, we assembled and annotated the complete mitochondrial (361,814 bp) and chloroplast (150,050 bp) genomes of G. vaccaria (Caryophyllaceae). The chloroplast genome exhibits a canonical quadripartite structure with conserved gene content (122 genes). Comparative analysis revealed that the chloroplast genome size, GC content, and gene number of G. vaccaria are similar to those of other reported Caryophyllales species (e.g., Agrostemma and Silene), where the inverted repeat (IR) regions may stabilize the genomic structure by preventing recombination-induced gene loss. In contrast, the mitochondrial genome exhibits a closed circular structure with 2.4-fold size expansion relative to chloroplast genomes. This expansion aligns with a characteristic trend observed in Caryophyllaceae (e.g., Silene), primarily driven by proliferated repetitive elements (e.g., SSRs, TRs; accounting for 88.2% of the mitogenome) and large-scale chloroplast-to- mitochondrion DNA transfers (MTPTs; spanning 56,732 Kb, 15.6% of the mitogenome), as resolved unambiguously by HiFi reads.
To place these findings into a broader evolutionary context, we compared the G. vaccaria mitogenome with those of related taxa within Caryophyllales. Within the order Caryophyllales, substantial variation in mitogenome size and structure (circular/multipartite) has been reported (Gao et al., 2023). While angiosperm mitogenomes typically range between 200–750 Kb (Burger et al., 2003; Gualberto and Newton, 2017), extreme cases include highly reduced (<100 Kb; Viscum scurruloideum) (Gao et al., 2023) or expanded (>11 Mb; Silene conica) (Gualberto and Newton, 2017) genomes. Within the Caryophyllaceae family, the mitogenome of G. vaccaria (361,814 bp) is notably larger than those of its nonfamilial relatives Agrostemma githago (262,903 bp) (Gao et al., 2023) and Silene latifolia (253,413 bp) (Sloan et al., 2010). However, when compared to other families within Caryophyllales, its size aligns with Mirabilis himalaica (Nyctaginaceae;346,363 bp) (Yuan et al., 2020), Spinacia oleracea (Amaranthaceae; 329,613 bp) (Cai et al., 2017), Rheum palmatum (Polygonaceae; 302,993 bp) (Gao et al., 2023), and Myricaria laxiflora (Tamaricaceae; 389,949 bp) (Wang et al., 2024). In contrast, it remains smaller than the mitogenomes of Pereskia aculeata (Cactaceae; 515,187 bp) (Zhang et al., 2023b) and Nepenthes khasiana (Nepenthaceae; 900,031 bp) (Konhar et al., 2025). Notably, extreme expansions occur in some Caryophyllaceae species, such as Silene noctiflora (6.7–7.1 Mb) (Wu et al., 2015) and Silene conica (11.3 Mb) (Smith, 2020), highlighting family-specific genomic plasticity (Supplementary Table S11). Despite these expansions, 99% of the increased genome size in large mitogenomes consists of intergenic sequences, with only ~1% derived from chloroplast or nuclear DNA (Sloan et al., 2012a; Gualberto and Newton, 2017). The origins of the remaining intergenic regions, potentially involving de novo repeats or horizontal transfers, remain unresolved. While recent studies suggested that genome-size circular molecules are rare in mitogenomes, with multi-isoform structures being more prevalent (Smith and Keeling, 2015), our analysis of 27 Caryophyllales mitogenomes revealed that 41.46% (17/41) were assembled as single circular maps. However, 41.18% (7/17) of these single circular assemblies relied on Illumina short-read sequencing (<300 bp), which has limited power to resolve complex structural variants such as alternative conformations or recombination-derived isoforms (Kozik et al., 2019) (Supplementary Table S11). During the assembly of the G. vaccaria mitochondrial genome, we carefully considered the possibility that the observed circular conformation might represent an assembly artifact. Read-mapping depth and HiFi coverage analyses revealed consistent support across the genome, suggesting that the circular form is likely genuine. Although PCR validation of the circular junctions was not performed in this study, future work could experimentally confirm genome circularity and reconcile potential discrepancies with earlier studies that may have overestimated single circular mitogenomes.
In addition, the GC content of the G. vaccaria mitogenome (43.82%) falls within the range of other Caryophyllales species (42.6–45.23%, Supplementary Table S11), Specifically, values for Caryophyllaceae species are 44.7% (Agrostemma githago) and 42.6% (Silene latifolia), while non-Caryophyllaceae members exhibit 43.4-45.23 (e.g., Spinacia oleracea, Rheum palmatum), suggesting family-level GC homogeneity. This study represents the first mitogenomic characterization of the genus Gypsophila, providing a foundation for exploring evolutionary mechanisms in Caryophyllaceae.
Plant mitogenomes frequently harbor plastid-derived sequences (MTPTs), with the extent of plastid-to-mitochondrion DNA transfer exhibiting significant interspecific variation (Wang et al., 2018). Comparative analyses have demonstrated that the total length of MTPTs can range from <1 Kb in Vigna angularis to >130 Kb in Amborella trichopoda (Sloan and Wu, 2014), suggesting lineage-specific dynamics of inter-organellar gene transfer. In this study, the total length of MTPTs in G. vaccaria was 56,732 bp, accounting for 15.69% of the entire mitogenome. This proportion markedly exceeds the 1-12% range reported in most angiosperms (Kubo and Mikami, 2007; Alverson et al., 2010), suggesting exceptional frequency of plastid-to-mitochondrion DNA transfer in G. vaccaria. Comparative analysis revealed divergent patterns of chloroplast-to-mitochondrion DNA transfer, with 12 out of 20 chloroplast-derived homologous sequences retaining intact functional genes—five protein-coding genes (e.g., rps7, ndhB), six tRNAs (e.g., trnL-CAA, trnW-CCA), and one rRNA (rrn16S). These MTPTs likely originated via retrotransposition or double-strand break repair, consistent with unidirectional transfer patterns observed in Silene. Notably, the ndhB (757 bp) and rrn16S genes retained full-length ORFs, raising questions about their functionality in the mitochondrial context. The exclusive presence of forward repeats (0 inverted repeats) in MTPTs implies unidirectional integration via non-homologous end joining (NHEJ), contrasting with recombination-driven plastid DNA incorporation in Silene (Smith, 2020). This Caryophyllaceae-specific mechanism may explain the exceptional MTPT accumulation (15.6% vs. 1-12% in most angiosperms).
Beyond protein-coding genes, tRNA gene sequences showing high similarity between the mitogenome and chloroplast genome were considered to be plastid-derived sequences. The trnH-GUG, trnM-CAU, trnN-GUU, trnW-CCA, trnP-UGG, and trnS-GGA are common plastid-derived genes in angiosperm, trnD-GUC is common only in dicots (Sugiyama et al., 2005). In G. vaccaria, we annotated five plastid-derived tRNAs (trnH-GUG, trnW-CCA, trnN-GUU, trnM-CAU, and trnP-UGG) within MTPTs, but trnS-GGA was absent. tRNA transfer events provide additional insights into MTPT dynamics, revealing both conserved transfer pathways and lineage-specific gene loss patterns in Caryophyllaceae. Although some MTPTs retain homologies to PVGs (plastid-derived viral genes) and functional tRNAs, empirical studies demonstrate their progressive degradation post-transfer (Notsu et al., 2002; Ogihara et al., 2005; Wang et al., 2007), rendering them non-functional in mitochondria. Our identification of MTPTs in G. vaccaria will facilitate the accurate assembly of chloroplast and mitochondrial genomes, and enhance our understanding of organelle genome evolution.
Codon usage bias refers to the phenomenon in which certain codons are used more frequently than others in the DNA or RNA sequences of specific organisms, often associated with gene expression efficiency and protein assembly processes (Sloan et al., 2012a; Smith and Keeling, 2015). Significant differences in codon usage preferences are observed between different species, and even between different organelles within the same species. To investigate the codon usage bias and its differences between the chloroplast and mitochondrial genomes of G. vaccaria, we calculated the relative synonymous codon usage (RSCU) values for each. Codons with RSCU values greater than 1 indicate a preference for the corresponding amino acid. Mitochondrial genes exhibited pronounced A/U-ending codon preference (29/44 high-RSCU codons), correlating with low GC content (43.8%) and likely reflecting mutational bias. Phenylalanine (TTT) and isoleucine were the most frequent amino acids, possibly linked to elevated demand for respiratory chain proteins. Chloroplast genes showed similar but weaker A/U bias, implying convergent translational optimization across organelles. HiFi sequencing resolved codon-level patterns with high accuracy, eliminating ambiguities from homopolymer errors common in short-read data. RNA editing, a ubiquitous post-transcriptional modification in plant organelles, fine-tunes mitochondrial gene expression by introducing nucleotide substitutions (e.g., C-to-U conversions), thereby regulating crucial physiological processes such as energy metabolism and stress responses (Small et al., 2020). We predicted 257 C-to-U RNA editing sites in mitochondrial PCGs—far exceeding the 105 sites in chloroplasts—with nad7 (32 sites) and nad5 (25 sites) being hotspots. This divergence may reflect stricter functional constraints on mitochondrial electron transport chain genes. Chloroplast ndhB (20 sites) and ycf2 (12 sites) showed reduced editing activity (-40.8% vs. mitochondrial), possibly due to relaxed selection or alternative regulatory mechanisms. HiFi data enabled precise mapping of editing sites, avoiding false positives from misaligned short reads. The strong positive selection on cytochrome c maturation genes (ccmFN/B/FC; ω=1.2-1.5) may reflect adaptations to arid environments, as ccm complexes regulate heme biosynthesis critical for stress-responsive hemoproteins (Stevens et al., 2011).
Beyond genomic characterization, our study provides high-quality mitochondrial and chloroplast genome assemblies and 81 species-specific SSR markers for G. vaccaria, offering reliable molecular tools for cultivar identification, parent selection, and marker-assisted breeding. Organelle-derived SSRs have proven effective in species discrimination and genetic diversity analyses in other medicinal and crop plants (Li et al., 2025), highlighting their potential to accelerate breeding and track desirable traits. The integration of plastid DNA into the mitogenome in G. vaccaria, likely mediated by unidirectional non-homologous end joining (NHEJ), appears to differ from the recombination-driven incorporation reported in Silene (Sloan et al., 2012b; Wu et al., 2015), implying that Caryophyllaceae may exhibit distinct mechanisms influencing organelle genome evolution.
Additionally, the expansion of the mitochondrial genome may be partly influenced by nuclear-integrated mitochondrial sequences (NUMTs). Although a systematic analysis of NUMTs was beyond the scope of this study, previous research suggests that their insertions can contribute to increased genome size and structural complexity (Kleine et al., 2009). In the present study, we did not perform a systematic analysis of NUMTs in G. vaccaria, and therefore the potential contribution of NUMTs to mitochondrial–nuclear genome interactions remain unexplored. Future research could employ genome-wide comparative approaches to identify NUMTs, assess their abundance and insertional patterns, and evaluate their potential functional or evolutionary implications in G. vaccaria. Such investigations would provide deeper insights into the dynamics of intracellular DNA transfer and their role in shaping genome architecture in this species, thereby enhancing our understanding of plant genome complexity and evolution.
Collectively, these findings not only advance our understanding of Caryophyllaceae mitogenome evolution but also establish a foundation for molecular breeding, functional genomics, and stress adaptation studies in this medicinally important species.
4 Materials and methods
4.1 Plant materials, DNA extraction, and sequencing
Fresh leaves from the same Gypsophila vaccaria plant were collected at the Medicinal Plant Germplasm College of Hexi University (100.441871°E, 38.948448° N; 1,256 m). The leaves were carefully cleaned with DEPC water and stored at -80 °C in the key laboratory of Hexi Corridor Resources Utilization of Gansu, Hexi University. High-quality genomic DNA was extracted using a DNA plant extraction kit (Tiangen, China). The DNA quality was evaluated with agarose gel electrophoresis, and its concentration was measured using a Nanodrop instrument (Thermo Fisher Scientific, Waltham, MA, USA). The qualified DNA samples were subsequently sent to Beijing BerryGenomics Co., Ltd. for Illumina sequencing and high fidelity (HiFi) sequencing.
4.2 Organelle genome assembly and annotation
The chloroplast and mitochondrial genomes of G. vaccaria were de novo assembled using Oatk v1.0 with default parameters based on HiFi reads (Zhou et al., 2025). The assembly results were visualized and manually curated in Bandage v0.8.1 with default parameters to generate fasta format files (Wick et al., 2015). To offer a more accurate view of the genome assembly, these two organelle genomes were mapped back HiFi reads to calculate the sequencing coverage depth using minimap2 v2.26 and samtools v1.17 with default parameters (Li and Liang, 2007; Li, 2018).
The chloroplast genome of G. vaccaria was annotated using CPGAVAS2 (Shi et al., 2019), GeSeq (Tillich et al., 2017) and CPGview (Liu et al., 2023), while the mitochondrial genome was annotated using IPMGA (http://www.1kmpg.cn/ipmga/), GeSeq (Tillich et al., 2017) and CPGview with default parameters (Liu et al., 2023). The tRNA genes were annotated using tRNAscan-SE v2.0.7 (Chan et al., 2021), and the open reading frames (ORFs) were annotated using the Open Reading Frame Finder (Rombel et al., 2002). During the annotation process, sequences overlapped with known genes were excluded, and sequences longer than 300 bp were further aligned against the NR database for additional functional annotation. To ensure the accuracy of the annotation results, all data were manually inspected and corrected. Additionally, the visualization of the chloroplast and mitochondrial genomes was automatically generated by CPGAVAS2 (Shi et al., 2019) and IPMGA, respectively.
4.3 Analysis of repeated sequences
The repeat sequence analysis includes microsatellite repeats (also known as simple sequence repeats, SSRs), tandem sequence repeats (TSRs), and dispersed sequence repeats (DSRs). In this study, microsatellite repeats were identified using the Misa-web tool (Beier et al., 2017). Tandem repeats (with lengths >6 bp) were detected using Tandem Repeats Finder with default parameters (Benson, 1999). Dispersed repeats were analyzed through the BiBiserv2 platform(https://bibiserv.cebitec.uni-bielefeld.de/). Finally, the identified repeat sequences were visualized using the Circos package in TBtools v2.056 (Chen et al., 2020).
4.4 DNA transfer between the chloroplast and the mitochondrion
To detect potential DNA transfers between the chloroplast genome and the mitochondrial genome of G. vaccaria, BLASTN was used for sequence similarity analysis with an e-value threshold of 1e-5 (Camacho et al., 2009). This analysis identified potential regions of DNA fragment transfer. Finally, the results were visualized using the Circos package in TBtools v2.056 (Chen et al., 2020).
4.5 Codon usage and RNA editing analysis
To explore the combined effects of natural selection, mutation, and genetic drift on codon usage, this study analyzed the relative synonymous codon usage (RSCU). A custom Perl script was used to perform a detailed analysis of the codon composition in the chloroplast and mitochondrial genomes of G. vaccaria. The analysis included the identification of unique coding sequences (CDS), determination of the codon counts for each gene, calculation of the effective number of codons (Nc), and further comparison of the RSCU values of synonymous codons between the chloroplast and mitochondrial genomes.
In this study, plant mitochondrial protein-coding genes were used as reference sequences, and the PREPACT v3.12.0 with default parameters was employed to identify RNA editing sites in the chloroplast and mitochondrial RNAs of G. vaccaria (Lenz et al., 2018).
4.6 Phylogenetic and Ka/Ks analysis
To determine the phylogenetic relationship of G. vaccaria, eight sequenced chloroplast and mitochondrial genomes of Caryophyllales species, including Myricaria laxiflora, Rheum palmatum, Nepenthes khasiana, Spinacia oleracea, Silene latifolia, Agrostemma githago, Mirabilis himalaica, Pereskia aculeata were downloaded from NCBI database (Supplementary Table S12). Phylogenetic trees were conducted based on shared chloroplast and mitochondrial genes among these 9 species including G. vaccaria. The shared chloroplast and mitochondrial gene CDS sequences were aligned using MAFFT (Lenz et al., 2018). Phylogenetic trees were constructed by RAxML with GTRGAMMA model and 1000 bootstraps (Stamatakis, 2014). Synonymous and nonsynonymous substitution ratios (Ka/Ks) were calculated by KaKs_Calculator v 3.0 (Zhang, 2022).
5 Conclusions
We present the first complete chloroplast and mitochondrial genomes of Gypsophila vaccaria, revealing key evolutionary features. The chloroplast genome (quadripartite structure) contains 77 PCGs, 37 tRNAs, and 8 rRNAs, while the mitochondrial genome (circular, 361,814 bp) harbors 35 PCGs, 21 tRNAs, and 3 rRNAs. Both organelles exhibit strong A/U codon bias (mitochondrial GC 44.2% vs. chloroplast 36.8%). Phylogenomic analyses resolve Caryophyllaceae-Amaranthaceae divergence (BS = 100%) and identify 81 species-specific SSRs. Positively selected sites in ccmFN, ccmB, ccmFC (ω=1.2-1.5, P<0.05) contrast with genome-wide purifying selection (median ω=0.32), suggesting adaptive evolution in cytochrome c maturation. Mitochondrial RNA editing (257 sites, e.g., 32 in nad7) exceeds chloroplast activity (105 sites), indicating divergent post-transcriptional regulation. Strikingly, 56.7 Kb of plastid-derived DNA (15.7% of mitogenome), including intact rps7 and ndhB, underscores mitochondrial genome plasticity. These high-quality resources facilitate future studies on Caryophyllaceae evolution and biotechnology applications.
Data availability statement
The complete mitochondrial and chloroplast genome sequences were deposited in the NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank) under accession numbers PV700085 and PV700086. All supporting data are included in Supplementary Table 1.
Author contributions
CZ: Funding acquisition, Project administration, Supervision, Validation, Writing – review & editing. RY: Formal Analysis, Investigation, Methodology, Writing – original draft. MW: Methodology, Visualization, Writing – original draft. JZ: Formal Analysis, Methodology, Visualization, Writing – review & editing. JS: Formal Analysis, Investigation, Methodology, Writing – review & editing. BY: Data curation, Funding acquisition, Methodology, Writing – original draft. DZ: Methodology, Writing – original draft. LY: Funding acquisition, Methodology, Writing – original draft. XW: Data curation, Methodology, Writing – original draft. CH: Project administration, Supervision, Writing – review & editing. JL: Investigation, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Doctoral Research Initiation fund Project of Hexi University (KYQD2022013; KYQD2020018); President’s Fund Project of Hexi University: Innovative Research on Improving the Quality of Wine in the Characteristic Industry of Hexi Corridor (CXTD001). None of these funding bodies have any relationship with the publication of this manuscript.
Acknowledgments
We would like to thank the College of Life Sciences and Engineering, Key Laboratory of Hexi Corridor Resources Utilization of Gansu, Hexi University, Zhangye, Gansu 734000, China; College of Agriculture and Ecological Engineering, Hexi University, Zhangye, Gansu 734000, China; Ministry of Education Key Laboratory for Biodiversity Science and Ecological Engineering, Institute of Biodiversity Science, Fudan University, Shanghai 200438, China; State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, Key Laboratory of Herbage and Endemic Crop Biology, Ministry of Education, School of Life Sciences, Inner Mongolia University, Hohhot, Inner Mongolia, 010070, China; and also thank the peer reviewers for their valuable and constructive comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1684062/full#supplementary-material
References
Alverson, A. J., Wei, X., Rice, D. W., Stern, D. B., Barry, K., and Palmer, J. D. (2010). Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 27, 1436–1448. doi: 10.1093/molbev/msq029
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., and Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of Populus simonii. Front. Plant Sci 13, 914635. doi: 10.3389/fpls.2022.914635
Bi, C., Sun, N., Han, F., Xu, K., Yang, Y., and Ferguson, D. K. (2024). The first mitogenome of Lauraceae (Cinnamomum chekiangense). Plant Diversity 46, 144–148. doi: 10.1016/j.pld.2023.11.001
Bing, Y., Sun, Z., Wu, S., Zheng, Y., Xi, Y., Li, W., et al. (2024). Discovery and verification of Q-markers for promoting blood circulation and removing stasis of raw and wine-steamed Vaccaria segetalis based on pharmacological evaluation combined with chemometrics. J. Ethnopharmacology 319, 117120. doi: 10.1016/j.jep.2023.117120
Birky, C. W., Jr. (2001). The inheritance of genes in mitochondria and chloroplasts: laws, mechanisms, and models. Annu. Rev. Genet. 35, 125–148. doi: 10.1146/annurev.genet.35.102401.090231
Burger, G., Gray, M. W., and Lang, B. F. (2003). Mitochondrial genomes: anything goes. Trends Genet. 19, 709–716. doi: 10.1016/j.tig.2003.10.012
Cai, X., Jiao, C., Sun, H., Wang, X., Xu, C., Fei, Z., et al. (2017). The complete mitochondrial genome sequence of spinach, Spinacia oleracea L. Mitochondrial DNA Part B 2, 339–340. doi: 10.1080/23802359.2017.1334518
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Chan, P. P., Lin, B. Y., Mak, A. J., and Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cheng, Y., He, X., Priyadarshani, S., Wang, Y., Ye, L., Shi, C., et al. (2021). Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genomics 22, 167. doi: 10.1186/s12864-021-07490-9
Committee, N. (2020). Pharmacopoeia of the people’s republic of China (Beijing: China Pharmaceutical Science and Technology Press).
Fan, W., Liu, F., Jia, Q., Du, H., Chen, W., Ruan, J., et al. (2022). Fragaria mitogenomes evolve rapidly in structure but slowly in sequence and incur frequent multinucleotide mutations mediated by microinversions. New Phytol. 236, 745–759. doi: 10.1111/nph.18334
Fauron, C., Casper, M., Gao, Y., and Moore, B. (1995). The maize mitochondrial genome: dynamic, yet functional. Trends Genet. 11, 228–235. doi: 10.1016/S0168-9525(00)89056-3
Feng, L., Wang, Z., Wang, C., Yang, X., An, M., and Yin, Y. (2023). Multichromosomal mitochondrial genome of Punica granatum: comparative evolutionary analysis and gene transformation from chloroplast genomes. BMC Plant Biol. 23, 512. doi: 10.1186/s12870-023-04538-8
Friis, G., Vizueta, J., Smith, E. G., Nelson, D. R., Khraiwesh, B., Qudeimat, E., et al. (2021). A high-quality genome assembly and annotation of the gray mangrove. Avicennia marina. G3 11, jkaa025. doi: 10.1093/g3journal/jkaa025
Gao, L., Hao, L., Xu, W., Xin, T., Song, C., Lin, Y., et al. (2023). Characterization of Rheum palmatum mitochondrial genome and comparative analysis among Caryophyllales species. Acupuncture Herbal Med. 3, 323–332. doi: 10.1097/HM9.0000000000000078
Gualberto, J. M. and Newton, K. J. (2017). Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 68, 225–252. doi: 10.1146/annurev-arplant-043015-112232
Han, F., Qu, Y., and Chen, Y. (2022). Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. Plant Sci 13, 1031769. doi: 10.3389/fpls.2022.1031769
Hu, J., Hu, H., and Yang, L. (2014). Studies on the chemical constituents from the seeds of Vaccaria segetalis. J. Pharm. Res. 33 (2), 71–72. Krupanidhi College of Pharmacy, Jinan, China.
Kleine, T., Maier, U. G., and Leister, D. (2009). DNA transfer from organelles to the nucleus: the idiosyncratic genetics of endosymbiosis. Annu. Rev. Plant Biol. 60, 115–138. doi: 10.1146/annurev.arplant.043008.092119
Konhar, R., Dash, D., and Biswal, D. K. (2025). First report of the chloroplast and mitochondrial genomes of the Indian pitcher plant, Nepenthes khasiana Hook. f. BMC Res. Notes 18, 45. doi: 10.1186/s13104-025-07082-w
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PloS Genet. 15, e1008373. doi: 10.1371/journal.pgen.1008373
Kubo, T. and Mikami, T. (2007). Organization and variation of angiosperm mitochondrial genome. Physiologia Plantarum 129, 6–13. doi: 10.1111/j.1399-3054.2006.00768.x
Lenz, H., Hein, A., and Knoop, V. (2018). Plant organelle RNA editing and its specificity factors: enhancements of analyses and new database features in PREPACT 3.0. BMC Bioinf. 19, 255. doi: 10.1186/s12859-018-2244-9
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, J., Li, J., Ma, Y., Kou, L., Wei, J., and Wang, W. (2022). The complete mitochondrial genome of okra (Abelmoschus esculentus): using nanopore long reads to investigate gene transfer from chloroplast genomes and rearrangements of mitochondrial DNA molecules. BMC Genomics 23, 481. doi: 10.1186/s12864-022-08706-2
Li, C., Liu, H., Qin, M., Tan, Y. J., Ou, X. L., Chen, X. Y., et al. (2024). RNA editing events and expression profiles of mitochondrial protein-coding genes in the endemic and endangered medicinal plant, Corydalis saxicola. Front. Plant Sci 15, 1332460. doi: 10.3389/fpls.2024.1332460
Li, J., Quan, C., Wei, R., Wei, F., Ma, Q., Huang, Y., et al. (2025). Genome-wide identification and development of SSR molecular markers for genetic diversity studies in Ilex asprella. Front. Plant Sci 16, 1582154. doi: 10.3389/fpls.2025.1582154
Li, N., Zhou, S., Li, W., Zhang, J., and Zhang, J. (2022). Effects of flavonoids from the seeds of Vaccaria segetalis on aortic vascular remodeling in hypertension and diabetes mellitus. Mol. Plant Breed. 20, 7.
Liu, Y. (2018). Auricular pressing therapy in the treatment of hypertension for 30 cases. Chin. Med. Mod. Dist. Educ. China 16, 127–128.
Liu, S., Ni, Y., Li, J., Zhang, X., Yang, H., Chen, H., et al. (2023). CPGView: a package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 23, 694–704. doi: 10.1111/1755-0998.13729
Lu, G., Zhang, K., Que, Y., and Li, Y. (2023). Assembly and analysis of the first complete mitochondrial genome of Punica granatum and the gene transfer from chloroplast genome. Front. Plant Sci 14, 1132551. doi: 10.3389/fpls.2023.1132551
Mackenzie, S. and McIntosh, L. (1999). Higher plant mitochondria. Plant Cell 11, 571–585. doi: 10.1105/tpc.11.4.571
Notsu, Y., Masood, S., Nishikawa, T., Kubo, N., Akiduki, G., Nakazono, M., et al. (2002). The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genomics 268, 434–445. doi: 10.1007/s00438-002-0767-1
Ogihara, Y., Yamazaki, Y., Murai, K., Kanno, A., Terachi, T., Shiina, T., et al. (2005). Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33, 6235–6250. doi: 10.1093/nar/gki925
Putintseva, Y. A., Bondar, E. I., Simonov, E. P., Sharov, V. V., Oreshkova, N. V., Kuzmin, D. A., et al. (2020). Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genomics 21, 654. doi: 10.1186/s12864-020-07061-4
Qi, P., Li, Z., Chen, M., Sun, Z., and Huang, C. (2013). Metabolism and tissue distribution study of Vaccaria seeds (Wang-Bu-Liu-Xing) in benign prostatic hyperplasia model rat: Toward an in-depth study for its bioactive components. J. Pharm. Biomed. Anal. 85, 218–230. doi: 10.1016/j.jpba.2013.07.037
Rombel, I. T., Sykes, K. F., Rayner, S., and Johnston, S. A. (2002). ORF-FINDER: a vector for high-throughput gene identification. Gene 282, 33–41. doi: 10.1016/S0378-1119(01)00819-8
Sang, S., Lao, A., Chen, Z., Uzawa, J., and Fujimoto, Y. (2003). Chemistry and bioactivity of the seeds of Vaccaria segetalis. ACS Symposium Series. 859, 279–291.
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi: 10.1093/nar/gkz345
Sibbald, S. J., Lawton, M., and Archibald, J. M. (2021). Mitochondrial genome evolution in pelagophyte algae. Genome Biol. Evol. 13, evab018. doi: 10.1093/gbe/evab018
Sloan, D. B., Alverson, A. J., Chuckalovcak, J. P., Wu, M., McCauley, D. E., Palmer, J. D., et al. (2012a). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PloS Biol. 10, e1001241. doi: 10.1371/journal.pbio.1001241
Sloan, D. B., Alverson, A. J., Štorchová, H., Palmer, J. D., and Taylor, D. R. (2010). Extensive loss of translational genes in the structurally dynamic mitochondrial genome of the angiosperm Silene latifolia. BMC Evolutionary Biol. 10, 274. doi: 10.1186/1471-2148-10-274
Sloan, D. B., Alverson, A. J., Wu, M., Palmer, J. D., and Taylor, D. R. (2012b). Recent acceleration of plastid sequence and structural evolution coincides with extreme mitochondrial divergence in the angiosperm genus Silene. Genome Biol. Evol. 4, 294–306. doi: 10.1093/gbe/evs006
Sloan, D. B. and Wu, Z. (2014). History of plastid DNA insertions reveals weak deletion and at mutation biases in angiosperm mitochondrial genomes. Genome Biol. Evol. 6, 3210–3221. doi: 10.1093/gbe/evu253
Small, I. D., Schallenberg-Rüdinger, M., Takenaka, M., Mireau, H., and Ostersetzer-Biran, O. (2020). Plant organellar RNA editing: what 30 years of research has revealed. Plant J. 101, 1040–1056. doi: 10.1111/tpj.14578
Smith, D. R. (2020). Revisiting ceriantharian (Anthozoa) mitochondrial genomes: casting doubts about their structure and size. Genome Biol. Evol. 12, 1440–1443. doi: 10.1093/gbe/evaa130
Smith, D. R. and Keeling, P. J. (2015). Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proc. Natl. Acad. Sci. 112, 10177–10184. doi: 10.1073/pnas.1422049112
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stevens, J. M., Mavridou, D. A., Hamer, R., Kritsiligkou, P., Goddard, A. D., and Ferguson, S. J. (2011). Cytochrome c biogenesis System I. FEBS J. 278, 4170–4178. doi: 10.1111/j.1742-4658.2011.08376.x
Sugiyama, Y., Watase, Y., Nagase, M., Makita, N., Yagura, S., Hirai, A., et al. (2005). The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: comparative analysis of mitochondrial genomes in higher plants. Mol. Genet. Genomics 272, 603–615. doi: 10.1007/s00438-004-1075-8
Tan, M. (2006). 59 cases of herpes zoster treated with Vaccaria segetalis. Chin. J. Rural Med. Pharm. 13, 51–51.
Tian, M., Huang, Y., Wang, X., Cao, M., Zhao, Z., Chen, T., et al. (2021). Vaccaria segetalis: A review of ethnomedicinal, phytochemical, pharmacological, and toxicological findings. Front. Chem. Volume 9. doi: 10.3389/fchem.2021.666280
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Tong, W., Yu, D., Zhu, X., Le, Z., Chen, H., Hu, F., et al. (2024). The whole mitochondrial genome sequence of dendrobium loddigesii rolfe, an endangered orchid species in China, reveals a complex multi-chromosome structure. Genes 15, 834. doi: 10.3390/genes15070834
Wang, X.-C., Chen, H., Yang, D., and Liu, C. (2018). Diversity of mitochondrial plastid DNAs (MTPTs) in seed plants. Mitochondrial DNA Part A 29, 635–642. doi: 10.1080/24701394.2017.1334772
Wang, S., Li, D., Yao, X., Song, Q., Wang, Z., Zhang, Q., et al. (2019). Evolution and diversification of kiwifruit mitogenomes through extensive whole-genome rearrangement and mosaic loss of intergenic sequences in a highly variable region. Genome Biol. Evol. 11, 1192–1206. doi: 10.1093/gbe/evz063
Wang, C., Ma, L., Xie, Y., Ding, B., Huang, J., and Wang, Q. (2024). Mitochondrial genome analysis of myricaria laxiflora, a protected endangered plant. Available online at SSRN 4689867.doi: 10.32604/phyton.2024.050099
Wang, D., Wu, Y.-W., Shih, A. C.-C., Wu, C.-S., Wang, Y.-N., and Chaw, S.-M. (2007). Transfer of chloroplast genomic DNA to mitochondrial genome occurred at least 300 MYA. Mol. Biol. Evol. 24, 2040–2048. doi: 10.1093/molbev/msm133
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Z., Cuthbert, J. M., Taylor, D. R., and Sloan, D. B. (2015). The massive mitochondrial genome of the angiosperm Silene noctiflora is evolving by gain or loss of entire chromosomes. Proc. Natl. Acad. Sci. 112, 10185–10191. doi: 10.1073/pnas.1421397112
Xia, C., Li, J., Zuo, Y., He, P., Zhang, H., Zhang, X., et al. (2023). Complete mitochondrial genome of Thuja sutchuenensis and its implications on evolutionary analysis of complex mitogenome architecture in Cupressaceae. BMC Plant Biol. 23, 84. doi: 10.1186/s12870-023-04054-9
Xing, L. and Jin, X. (2013). Observation on the therapeutic effect of auricular point pressing bean on 30 cases of allergic rhinitis. J. North Chin. Med. 45, 181–182.
Yang, H., Chen, H., Ni, Y., Li, J., Cai, Y., Ma, B., et al. (2022). De novo hybrid assembly of the salvia miltiorrhiza mitochondrial genome provides the first evidence of the multi-chromosomal mitochondrial DNA structure of salvia species. Int. J. Mol. Sci. 23, 14267. doi: 10.3390/ijms232214267
You, Z. (1989). The effect of auricular plaster with Gypsophila segetalis on gallbladder contraction-a report of 70 cases observed by ultrasound. J. Gansu Univ Chin. Med. 1), 31–32.
Yuan, F., Xu, Y., and Lan, X. (2020). The complete mitochondrial genome sequence of Mirabilis himalaica, an endemic plant to Tibet. Mitochondrial DNA Part B 5, 2802–2804. doi: 10.1080/23802359.2020.1788451
Zhang, Z. (2022). KaKs_Calculator 3.0: calculating selective pressure on coding and non-coding sequences. Genomics Proteomics Bioinf. 20, 536–540. doi: 10.1016/j.gpb.2021.12.002
Zhang, H., Jing, Y., and Wu, G. (2013). Inhibitory effects of crude polysaccharides from Semen vaccariae on benign prostatic hyperplasia in mice. J. Ethnopharmacology 145, 667–669. doi: 10.1016/j.jep.2012.11.027
Zhang, X., Shan, Y., Li, J., Qin, Q., Yu, J., and Deng, H. (2023b). Assembly of the complete mitochondrial genome of Pereskia aculeata revealed that two pairs of repetitive elements mediated the recombination of the genome. Int. J. Mol. Sci. 24, 8366. doi: 10.3390/ijms24098366
Zhang, S., Wang, J., He, W., Kan, S., Liao, X., Jordan, D. R., et al. (2023a). Variation in mitogenome structural conformation in wild and cultivated lineages of sorghum corresponds with domestication history and plastome evolution. BMC Plant Biol. 23, 91. doi: 10.1186/s12870-023-04104-2
Zhou, C., Brown, M., Blaxter, M., Consortium, D. T., McCarthy, S. A., and Durbin, R. (2025). Oatk: a de novo assembly tool for complex plant organelle genomes. BioRxiv. 26, 235. doi: 10.1186/s13059-025-03676-6
Keywords: Gypsophila vaccaria, mitochondrial genome, chloroplast genome, phylogenetic analysis, RNA editing events
Citation: Zhang C, Yang R, Wang M, Zhang J, Shen J, Yang B, Zhang D, Yin L, Wang X, Huang C-H and Li J (2025) Unveiling the complete organelle genomes of Gypsophila vaccaria: de novo assembly and evolutionary insights into a medicinally important species. Front. Plant Sci. 16:1684062. doi: 10.3389/fpls.2025.1684062
Received: 12 August 2025; Accepted: 18 September 2025;
Published: 01 October 2025.
Edited by:
Ahmad A. Omar, University of Florida, United StatesReviewed by:
Azza H. Mohamed, University of Florida, United StatesRongpeng Liu, Beijing University of Chinese Medicine, China
Copyright © 2025 Zhang, Yang, Wang, Zhang, Shen, Yang, Zhang, Yin, Wang, Huang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chien-Hsun Huang, Y2hodWFuZzE5ODFAMTM5LmNvbQ==; Jinglong Li, bGpsNjUyOEAxNjMuY29t