



- 1UMR1347 Agroécologie, Institut National de la Recherche Agronomique, Dijon, France
- 2UR1268 Biopolymers, Interactions, Assemblies, Institut National de la Recherche Agronomique, Nantes, France
Understanding the regulatory networks coordinating seed development will help to manipulate seed traits, such as protein content and seed weight, in order to increase yield and seed nutritional value of important food crops, such as legumes. Because of the cardinal role of the nucleus in gene expression, sub-proteome analyses of nuclei from developing seeds were conducted, taking advantage of the sequences available for model species. In this review, we discuss the strategies used to separate and identify the nuclear proteins at a stage when the seed is preparing for reserve accumulation. We present how these data provide an insight into the complexity and distinctive features of the seed nuclear proteome. We discuss the presence of chromatin-modifying enzymes and proteins that have roles in RNA-directed DNA methylation and which may be involved in modifying genome architecture in preparation for seed filling. Specific features of the seed nuclei at the transition between the stage of cell divisions and that of cell expansion and reserve deposition are described here which may help to manipulate seed quality traits, such as seed weight.
Introduction
Because seeds, such as those of legumes and cereals, are a source of nutrients for animal and human nutrition, breeding objectives include improving seed quality and yield and/or stabilizing these traits under fluctuating environmental conditions. To develop an understanding of the genetic factors controlling these traits, omics studies of seed development were performed from the year 2000 onward exploiting the availability of genome sequence for several species, including Arabidopsis, rice, and Medicago truncatula. This last species was adopted in 2001 as a model for legumes because of its small genome size compared to other legumes (Bell et al., 2001). Genomics resources were then developed in this species (Young et al., 2011) and extensively exploited, notably to study seed biology. Proteomics has been used to identify candidate proteins with roles in seed development (Thompson et al., 2009). While only abundant soluble proteins were identified by proteomics targeted to entire seed tissues, transcriptome studies provided information about low-abundance expression of some genes (Thompson et al., 2009). By comparing the timing of appearance of the proteins with that of their corresponding transcripts during seed development, divergent patterns were found for 50% of the proteins detected in the M. truncatula seed proteome (Gallardo et al., 2007). This indication of major post-transcriptional events highlighted the need to choose a proteomics approach to identify the regulatory mechanisms governing seed development. Targeted to the nucleus, proteomics allowed identification of regulatory proteins in leaves, suspension cells, or seedlings from various species, including Arabidopsis, rice (Oryza sativa), maize (Zea mays), and chickpea (Cicer arietinum; Bae et al., 2003; Khan and Komatsu, 2004; Ferreira et al., 2006; Pandey et al., 2006; Tan et al., 2007; Li et al., 2008). To provide a list of nuclear proteins with potential regulatory role(s) in developing M. truncatula seeds, the approach of combining nuclei isolation with proteomics was applied at 12 days after pollination (dap; Repetto et al., 2008). This key stage is characterized by the switch from an embryogenesis-oriented program, with frequent cell divisions, to a filling program associated with embryo cell expansion and reserve accumulation. In a parallel study, a nuclear proteomics approach was applied to the filial tissue of rice seeds (i.e., the endosperm) at 9 dap (Li et al., 2008). At this stage, the embryo is differentiated and the reserves start to accumulate (Luo et al., 2011). Because understanding the processes underlying the embryogenesis/filling transition might help greatly to modulate both seed size and storage capacities, after outlining the strategies used to identify nuclear proteins from developing seeds, we describe the specificities of the seed nuclear proteome, and discuss the proteins that might play key roles in controlling this transition.
Seed Nuclei Purification and Protein Extraction
Nuclear isolation methods based on density gradients were applied to immature seeds or seed tissues in flax (Linum usitatissimum), M. truncatula, rice, and maize (Table 1), with the objective of obtaining nuclei of sufficient yield and quality for protein profiling (Ferreira et al., 2006), proteomics (Li et al., 2008; Repetto et al., 2008), or gel shift experiments (Renouard et al., 2012). Castillo et al. (2000) also succeeded in isolating nuclei from ungerminated pea embryonic axes to purify and sequence a nuclear protein induced by dehydration (Table 1). The isolation of nuclei from developing seeds is challenging due to the presence of storage compounds such as globulins, oils, and carbohydrates (Gallardo et al., 2008). In M. truncatula, we tested several nuclear separation procedures from seeds collected at different developmental stages, including flow cytometry, sucrose or percoll density gradients, before adopting a sucrose-based “semi-pure” nuclear preparation of the CelLytic plant nuclei isolation kit (Sigma-Aldrich) to which we have made some modifications described in Repetto et al. (2008). At the 12 dap stage, the M. truncatula seed possesses nuclei of 5–15 μm diameter with low DNA C-value (0.48 pg; Arumuganathan and Earle, 1991). Observations of nuclei preparations from M. truncatula seeds at later stages reveal few and larger seed nuclei, along with many starch granules probably originating from the seed coats (Abirached-Darmency et al., 2005). Optimizations are necessary to obtain high-purity nuclei at these stages, which differ in the number of contaminants (e.g., protein bodies, starch granules), average nuclear size, and DNA content. Interestingly, a cotton filtration step was set up by Li et al. (2008) for starch grain removal from rice endosperm at 9 dap, and a protocol allowing the removal of mucilage and phenolic compounds from seed coats before nuclei isolation was developed by Renouard et al. (2012); (Table 1).
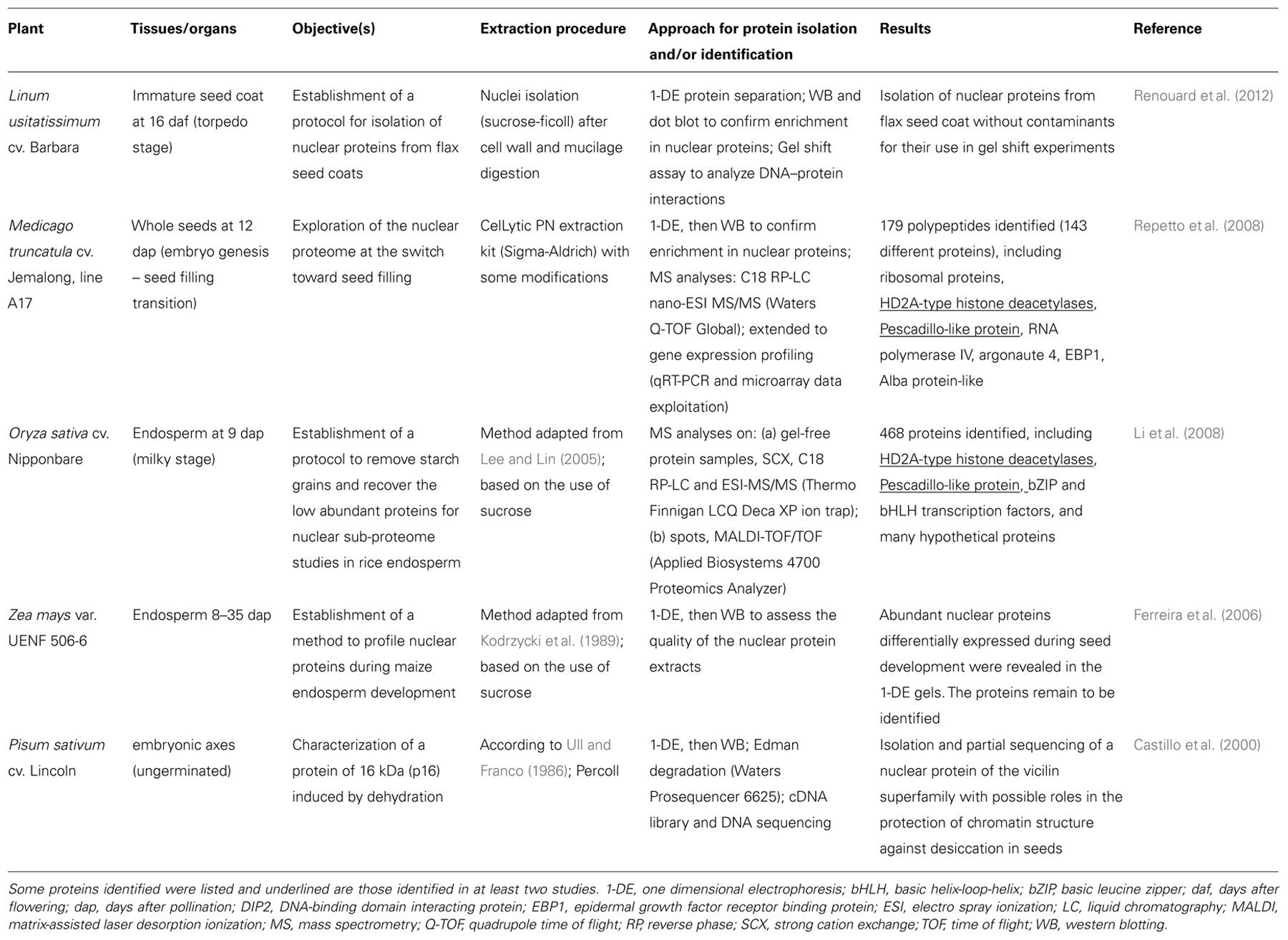
TABLE 1. Isolation of seed nuclei for protein identification or gel shift experiments.
Two of the nuclei isolation methods presented in Table 1 were combined with mass spectrometry (MS) for sub-proteome analyses. In Repetto et al. (2008), the nuclei-containing pellets obtained from 12 dap M. truncatula seeds were directly resuspended in a high salt concentration buffer (1 M NaCl), and then sonicated to destroy the nuclear membranes. After validating the enrichment for nuclear proteins by western blotting with antibodies for histone H1 and for proteins specific for other subcellular compartments, the resulting protein extract was directly separated by mono-dimensional gel electrophoresis (1-DE) and the whole lane was sequentially cut into 36 portions for MS analyses (Figure 1). A different approach was used by Li et al. (2008). They first removed the highly abundant bands corresponding to storage proteins from the 1-DE profile by excision, and then crushed the rest of the gel to extract the low abundance proteins using a phenol extraction buffer. After precipitation, the protein pellet was dissolved in 6 M urea with 100 mM Tris–Cl for MS analyses.
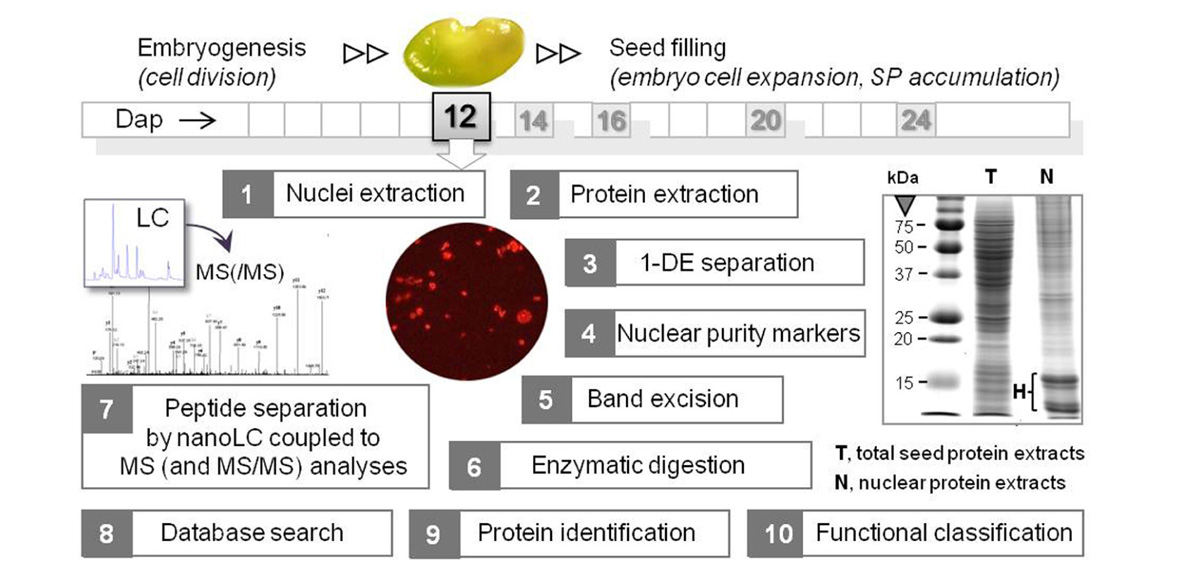
FIGURE 1. Workflow of the nuclear proteomics approach applied to M. truncatula seeds at a key stage between embryogenesis and seed filling. Organelles were purified from 12 dap seeds (1 seed = 1.5 mm length), and the proteins were extracted and separated by mono-dimensional gel electrophoresis (1-DE). A typical 1-DE profile is shown with intense bands at about 10–15 kDa corresponding to histones (H). After assessing the purity of the nuclear protein fraction by Western blotting using antibodies against proteins specific of different cell compartments, the in-gel digested peptides were analyzed by nanoLC coupled to MS and MS/MS analyses. The peptide mass data were subjected to a database search for putative protein identification, and the proteins were functionally classified after a search for nuclear peptide signals. SP, storage proteins. dap, days after pollination.
Identification of Seed Nuclear Proteins
In Repetto et al. (2008), the in-gel trypsin-digested peptides were separated by liquid nano-chromatography (nanoLC) and further measured and fragmented (MS/MS experiments) in a hybrid quadrupole-time-of-flight mass spectrometer. A search in both a wide databank (UniRef100) and a targeted databank made of expressed sequence tags from M. truncatula (the TIGR MtGI release 8 database) was realized from the mass data. The databank search program was MASCOT 2.2 and proteins were identified when at least two of their peptides matched the databank entry with a p-value <0.05. We succeeded in identifying 179 polypeptides, corresponding to 143 distinct proteins, using this approach. Sequence annotations were manually checked or completed by (cross-) BLAST “parameters” searches against the NCBI non-redundant database. The proteins were functionally classified according to the MapMan ontology (Usadel et al., 2005) as well as to a manual assignment not limited to homologs as described in Gallardo et al. (2007). A complete list of proteins is available in Repetto et al. (2008) that remains to date the most comprehensive description of the M. truncatula nuclear proteome.
In a parallel study, a shotgun proteomics approach was used by Li et al. (2008) to characterize the rice nuclear proteome. The complex peptide mixtures derived from trypsin digestion were subjected to 2-D liquid chromatography coupled to an ESI-IT (electro spray ionization-ion trap) mass spectrometer. A search in the rice non-redundant protein database (NCBInrPDB) was done from the mass data, and proteins were identified when at least two of their peptides matched the databank entry with a p-value <0.01. This approach identified 468 proteins from the nuclear enriched fractions of rice endosperm. A nuclear localization was assigned for 47% of these proteins by searching the Gene Ontology (GO) database (http://www.geneontology.org/). It should be noted that prediction of nuclear localization of proteins is far from being easy and entirely reliable. In fact, the nucleo-cytoplasmic protein shuttling through the nuclear pore complex (NPC) is a highly dynamic and complex system (Grünwald and Singer, 2012), and for many proteins (e.g., ribosomal and cytoskeletal) there is a consistent evidence for multiple locations. Moreover, only a fraction of the proteins localized in nuclei possess nuclear localization signals for NPC-mediated transport into the nucleus. Therefore, the prediction of nuclear localization based on the presence of signal peptides (e.g., PSORT; Nakai and Horton, 1999) is usually coupled with homology-based GO annotations, and must ideally be confirmed by further experiments, for example using fluorescent protein fusions or specific antibodies.
Among the proteins identified in the M. truncatula seed nucleus that may be multifunctional and might display different organelle functions and localizations, are certain enzymes of intermediary metabolism. Previous studies also reported the presence of these enzymes in the nucleus although no obvious nuclear localization signal was found in their sequences (Yamamoto et al., 1997; Markova et al., 2006; Li et al., 2008; Lee et al., 2012). As an example, sulfite reductase, a key plastid enzyme involved in sulfur reduction in plants, was identified in the M. truncatula seed nucleus. This enzyme was shown to bind to DNA in the chloroplast, and thus to repress genomic activity (i.e., transcription) through DNA compaction (Sato et al., 2003; Sekine et al., 2007). Although further experiments are needed to confirm their nuclear localization, the presence of such proteins raises the possibility of a regulation of transcriptional activities in seeds through nuclear targeting of metabolic enzymes. They may be able to monitor metabolic status in response to various stimuli by transmitting the changes to the transcriptional apparatus.
Specificities of the Seed Nuclear Proteome
A comparison of nuclear proteomes from different organs and species might help to decipher the level of conservation of nuclear proteins and to identify tissue- or species-specific nuclear functions. With the aim to identify specific nuclear features in seeds, we compared the nucleus proteome of M. truncatula seeds with that of the rice endosperm at a milky stage (Li et al., 2008), and that of chickpea seedlings (Pandey et al., 2006) and Arabidopsis leaves (Bae et al., 2003). Interestingly, two protein classes were particularly enriched in the M. truncatula seed nucleus at a stage preparing for reserve deposition: RNA processing and ribosome biogenesis. In particular, an abundant pool of proteins (22% of the proteins identified) was found that are members of the ribosomal protein families comprising the 40S and 60S subunits synthesized within the nucleolus in eukaryotes. The abundance of their transcripts decreased sharply at the beginning of seed filling (i.e., 14–16 dap; Gallardo et al., 2007). A salient feature of 12 dap M. truncatula seeds is therefore the storage of a large pool of ribosomal proteins within the nucleus, that can presumably be further readily used for storage protein synthesis during seed filling. This may contribute to our understanding of the mechanisms allowing legume seeds to synthesize large amounts of storage proteins while entering into a quiescent state. It also raises an important question of whether the stored ribosomal proteins could be involved in the intricate control of homeostasis of protein amount per seed under challenging environmental conditions. Interestingly, a PESCADILLO-like protein that may play a role in the biogenesis of ribosomal subunits was identified in the nuclear proteome of both the rice endosperm and M. truncatula seeds (Table 1). This protein is not functionally characterized in plants but implicated in rRNA precursor processing and ribosomal subunit assembly in human and mammalian cells (Andersen et al., 2002; Lerch-Gaggl et al., 2002).
In the nuclear proteomes of both the M. truncatula seed and rice endosperm the proportion of functionally annotated proteins belonging to the DNA metabolism class (12% in M. truncatula and 29% in rice) exceeded that found in chickpea seedlings (Pandey et al., 2006) and Arabidopsis leaves (Bae et al., 2003). Some of these proteins are involved in the epigenetic regulation of the genome (Li et al., 2008; Repetto et al., 2008). There is increasing evidence that some components of the chromatin modification machinery play a significant role in developing seeds. Recent surveys demonstrated that genomic imprinting primarily occurs in the endosperm in both rice and Arabidopsis, and that gene-specific imprinting in the embryo also exists in maize (Ikeda, 2012 and references therein). By comparing candidate imprinted genes from rice and Arabidopsis, Luo et al. (2011) found a low degree of conservation, suggesting that imprinting targets have evolved independently in dicots and monocots. In seeds, the epigenetic regulation of the genome, which modulates chromatin structure to limit the expression of genes to a particular tissue at a specific developmental stage, could play a crucial role in the developmental switch of the dicot embryo cells from division to expansion and filling (Figure 1). In legumes, final seed weight is largely determined by the number of cotyledon cells (Munier-Jolain and Ney, 1998). Therefore, identifying the epigenetic components of legume seeds that regulate the timing of the transition between cell division and cell expansion might help to manipulate final seed weight.
Among the epigenetic components detected in the M. truncatula seed nuclei were histone deacetylases HD2A that are good candidates for regulating the transition from an embryonic program to a filling mode. HD2A are plant-specific chromatin-remodeling factors participating in transcriptional repression via the modification of gene accessibility (Li et al., 2002). Interestingly, these proteins were also identified in the filial tissue of rice (Table 1). HD2A are expressed strongly in embryonic tissues and their ectopic expression under the control of the 35S promoter resulted in developmental abnormalities, including aborted seed development (Zhou et al., 2004). Importantly, Tanaka et al. (2008) demonstrated that histone deacetylases are involved in the repression of embryonic properties upon germination via direct or indirect repression of embryo-specific transcription factors. It is therefore possible that HD2A also plays a role in regulating the switch from embryogenesis to seed filling in eudicots and monocots. Although this hypothesis requires experimental confirmation, it holds promise to resolve the presently unclear mechanisms shifting the seed developmental program to reserve deposition (Figure 1).
The histone modifications induced by HD2A may be associated with other chromatin modifications, such as DNA methylation, to silence gene expression in response to developmental stimuli. Interestingly, two proteins needed for RNA-directed DNA methylation (i.e., DNA methylation guided by 24 nucleotide small interfering RNAs) were identified in the M. truncatula seed nucleus: a subunit of the plant-specific RNA polymerase IV, and argonaute 4 (AGO4). These proteins were not identified in nuclei from rice endosperm (Table 1), chickpea seedlings (Pandey et al., 2006), or Arabidopsis leaves (Bae et al., 2003), suggesting a specific role in legume seeds and/or in immature embryos. RNA polymerase IV is required for the biogenesis of a major class of 24-nucleotide small interfering RNAs, which are predominantly expressed in the developing endosperm of Arabidopsis seeds (Lu et al., 2012). Li et al. (2006) showed that the C-terminal domain of a RNA polymerase IV subunit interacts with AGO4 within nucleolus-associated bodies (i.e., Cajal bodies), which have been proposed to be a site for the generation of siRNA/protein complexes acting in RNA-directed DNA methylation. The detection of these proteins in the M. truncatula seed nucleus suggests they may interact in 12 dap seeds in concert with HD2A to repress the expression of genes via chromatin remodeling. To elucidate the mechanism of repression, it will be necessary to identify the target genes, some putative candidates could be described in the following section.
Proteins Implicated in Transcriptional Regulation
When targeted to the nucleus, proteomics offers the opportunity to identify regulatory factors controlling cell development, differentiation, and cell growth by binding to DNA and regulating gene expression. In seeds, there is great interest in identifying such factors to manipulate seed size and weight. A putative transcriptional regulator which was found specifically in the M. truncatula seed nucleus may control cell division but its function in seeds has not yet been characterized. This protein, named EBP1 (epidermal growth factor receptor binding protein), recruits histone deacetylase activity in human cells to mediate the transcriptional repression of E2 promoter binding factors (E2F) controlling cell cycle progression (Zhang et al., 2003). In potato and Arabidopsis, Horváth et al. (2006) demonstrated that EBP1 regulates organ size through cell growth and proliferation: elevating or decreasing EBP1 levels in transgenic plants resulted in a dose-dependent increase or reduction in leaf surface area, respectively. In the same study, they showed that EBP1 is required for expression of cell cycle genes in an auxin-dependent manner. This is likely to occur through the repression of RBR1 (retinoblastoma binding protein-like) that blocks cell cycle progression by inhibiting E2F-dependent transcription, which is required for expression of many genes involved in S-phase and cell cycle progression (Lai et al., 1999). The presence of EBP1 in the M. truncatula seed nucleus suggests this protein could play a key role in the control of cell division during seed development. Various other regulatory proteins were specifically detected in the rice endosperm nucleus (e.g., basic leucine zipper and basic helix-loop-helix transcription factors) or in the M. truncatula seed nucleus (e.g., DNA-binding domain interacting protein DIP2, Alba protein-like; Table 1). In plants, the exact functions of some of these proteins remain to be defined. The Alba protein has been proposed to control chromatin structure through interaction with histone deacetylase in Archaea and could also have a function in RNA metabolism (Bell et al., 2002; Aravind et al., 2003). The DIP2 protein displays similarities with the animal transcriptional coactivator ALY, suggesting it could be involved in transcriptional regulation. In plants, DIP2 interacts with the DNA-binding domain of plant poly(ADP-ribose) polymerases possibly implicated in chromosome dynamics and modifying proteins involved in different signaling pathways from DNA damage to energy metabolism (Babiychuk et al., 2001; Storozhenko et al., 2001).
Conclusion
The availability of data from next-generation technologies, now used for de novo sequencing of genomes in crops, such as pigeonpea (Varshney et al., 2011), will facilitate the identification of seed nuclear proteins in these species. Ethyl methanesulfonate (EMS) and TnT1 insertion mutant populations have been developed in M. truncatula and EMS mutants in pea and rice (Dalmais et al., 2008; Tadege et al., 2008; Le Signor et al., 2009; Wang T. L., 2012; Cooper et al., 2013). Both reverse and forward genetics can be applied to study mutants from these collections. Moreover, a series of EMS mutations could be identified by TILLING in candidate genes for regulating the embryogenesis-filling transition, which in addition to providing mutants for functional studies, could reveal favorable alleles to be used in selection for seed quality improvements.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work on M. truncatula was supported by an INRA post-doctoral fellowship and by grants #B05796 from the Regional Council of Burgundy. We sincerely thank Steven P. C. Groot and Jan Bergervoët (Plant Research International, Wageningen, Netherlands) for advice and help regarding nuclei preparations.
References
Abirached-Darmency, M., Abdel-gawwad, M. R., Conejero, G., Verdeil, J. L., and Thompson, R. (2005). In situ expression of two storage protein genes in relation to histo-differentiation at mid-embryogenesis in Medicago truncatula and Pisum sativum seeds. J. Exp. Bot. 56, 2019–2028.
Andersen, J. S., Lyon, C. E., Fox, A. H., Leung, A. K., Lam, Y. W., Stehen, H., et al. (2002). Directed proteomic analysis of the human nucleolus. Curr. Biol. 12, 1–11.
Aravind, L., Iyer, L. M., and Anantharaman, V. (2003). The two faces of Alba: the evolutionary connection between proteins participating in chromatin structure and RNA metabolism. Genome Biol. 4, R64.
Arumuganathan, K., and Earle, E. D. (1991). Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–218.
Babiychuk, E., Van Montagu, M., and Kushnir, S. (2001). N-terminal domains of plant poly(ADP-ribose) polymerases define their association with mitotic chromosomes. Plant J. 28, 245–255.
Bae, M. S., Cho, E. J., Choi, E., and Prk, O. K. (2003). Analysis of the Arabidopsis nuclear proteome and its response to cold stress. Plant J. 36, 652–663.
Bell, C. J., Dixon, R. A., Farmer, A. D., Flores, R., Inman, J., Gonzales, R. A., et al. (2001). The Medicago Genome Initiative: a model legume database. Nucleic Acids Res. 29, 114–117.
Bell, S. D., Botting, C. H., Wardleworth, B. N., Jackson, S. P., and White, M. F. (2002). The interaction of Alba, a conserved archaeal chromatin protein, with Sir2 and its regulation by acetylation. Science 296, 148–151.
Cooper, J. L., Henikoff, S., Comai, L., and Till, B. J. (2013). TILLING and Ecotilling for rice. Methods Mol. Biol. 956, 39–56.
Castillo, J., Rodrigo, M. I., Márquez, J. A., Zúñiga, A., and Franco, L. (2000). A pea nuclear protein that is induced by dehydration belongs to the vicilin superfamily. Eur. J. Biochem. 267, 2156–2165.
Dalmais, M., Schmidt, J., Le Signor, C., Moussy, F., Burstin, J., Savois, V., et al. (2008). UTILLdb, a Pisum sativum in silico forward and reverse genetics tool. Genome Biol. 9, R43.
Ferreira, B. S., Branco, A. T., de Oliveira, M. A., Pereira, M. G., and de Souza Filho, G. A. (2006). Methodological improvements on extraction of nuclear proteins and its preliminary analysis during the maize (Zea mays L.) endosperm development. Protein Pept. Lett. 13, 981–984.
Gallardo, K., Firnhaber, C., Zuber, H., Héricher, D., Belghazi, M., Henry, C., et al. (2007). A combined proteome and transcriptome analysis of developing Medicago truncatula seeds: evidence for metabolic specialization of maternal and filial tissues. Mol. Cell. Proteomics 6, 2165–2179.
Gallardo, K., Thompson, R., and Burstin, J. (2008). Reserve accumulation in legume seeds. C. R. Biol. 331, 755–762.
Grünwald, D., and Singer, R. H. (2012). Multiscale dynamics in nucleocytoplasmic transport. Curr. Opin. Cell Biol. 24, 100–106.
Horváth, B. M., Magyar, Z., Zhang, Y., Hamburger, A. W., Bakó, L., Visser, R. G., et al. (2006). EBP1 regulates organ size through cell growth and proliferation in plants. EMBO J. 25, 4909–4920.
Ikeda, Y. (2012). Plant imprinted genes identified by genome-wide approaches and their regulatory mechanisms. Plant Cell Physiol. 53, 809–816.
Khan, M. M. K., and Komatsu, S. (2004). Rice proteomics: recent developments and analysis of nuclear proteins. Phytochemistry 65, 1671–1681.
Kodrzycki, R., Boston, R. S., and Larkins, B. A. (1989). The opaque-2 mutation of maize differentially reduces zein gene transcription. Plant Cell 1, 105–114.
Lai, A., Marcellus, R. C., Corbeil, H. B., and Branton, P. E. (1999). RBP1 induces growth arrest by repression of E2F-dependent transcription. Oncogene 18, 2091–2100.
Lee, H., and Lin, T. Y. (2005). Isolation of plant nuclei Suitable for flow cytometry from recalcitrant tissue by use of a filtration column. Plant Mol. Biol. Rep. 23, 53–58.
Lee, S., Doxey, A. C., McConkey, B. J., and Moffatt, B. A. (2012). Nuclear targeting of methyl-recycling enzymes in Arabidopsis thaliana is mediated by specific protein interactions. Mol. Plant 5, 231–248.
Lerch-Gaggl, A., Haque, J., Li, J., Ning, G., Traktman, P., and Duncan, S. A. (2002). Pescadillo is essential for nucleolar assembly, ribosome biogenesis, and mammalian cell proliferation. J. Biol. Chem. 47, 45347–45355.
Le Signor, C., Savois, V., Aubert, G., Verdier, J., Nicolas, M., Pagny, G., et al. (2009). Optimizing TILLING populations for reverse genetics in Medicago truncatula. Plant Biotechnol. J. 7, 430–441.
Li, G., Hall, T. C., and Holmes-Davis, R. (2002). Plant chromatin: development and gene control. Bioessays 24, 234–243.
Li, C. F., Pontes, O., El-Shami, M., Henderson, I. R., Bernatavichute, Y. V., Chan, S. W., et al. (2006). An ARGONAUTE4-containing nuclear processing center colocalized with Cajal bodies in Arabidopsis thaliana. Cell 126, 93–106.
Li, G., Nallamilli, B. R., Tan., F., and Peng, Z. (2008). Removal of high-abundance proteins for nuclear subproteome studies in rice (Oryza sativa) endosperm. Electrophoresis 29, 604–617.
Lu, J., Zhang, C., Baulcombe, D. C., and Chen, Z. J. (2012). Maternal siRNAs as regulators of parental genome imbalance and gene expression in endosperm of Arabidopsis seeds. Proc. Natl. Acad. Sci. U.S.A. 109, 5529–5534.
Luo, M., Taylor, J. M., Spriggs, A., Zhang, H., Wu, X., Russell, S., et al. (2011). A genome-wide survey of imprinted genes in rice seeds reveals imprinting primarily occurs in the endosperm. PLoS Genet. 7:e1002125. doi: 10.1371/journal.pgen.1002125
Markova, N. G., Pinkas-Sarafova, A., and Simon, M. (2006). A metabolic enzyme of the short-chain dehydrogenase/reductase superfamily may moonlight in the nucleus as a repressor of promoter activity. J. Invest. Dermatol. 126, 2019–2031.
Munier-Jolain, N., and Ney, B. (1998). Seed growth rate in grain legumes II. Seed growth rate depends on cotyledon cell number. J. Exp. Bot. 49, 1971–1976.
Nakai, K., and Horton, P. (1999). PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 24, 34–36.
Pandey, A., Choudhary, M. K., Bhushan, D., Chattopadhyay, A., Chakraborty, S., Datta, A., et al. (2006). The nuclear proteome of chickpea (Cicer arietinum L.) reveals predicted and unexpected proteins. J. Proteome Res. 5, 3301–3311.
Renouard, S., Cyrielle, C., Lopez, T., Lamblin, F., Lainé, E., and Hano, C. (2012). Isolation of nuclear proteins from flax (Linum usitatissimum L.) seed coats for gene expression regulation studies. BMC Res. Notes 5:15. doi: 10.1186/1756-0500-5-15
Repetto, O., Rogniaux, H., Firnhaber, C., Zuber, H., Küster, H., Larré, C., et al. (2008). Exploring the nuclear proteome of Medicago truncatula at the switch towards seed filling. Plant J. 56, 398–410.
Sato, N., Terasawa, K., Miyajima, K., and Kabeya, Y. (2003). Organization, developmental dynamics, and evolution of plastid nucleoids. Int. Rev. Cytol. 232, 217–262.
Sekine, K., Fujiwara, M., Nakayama, M., Takao, T., Hase, T., and Sato N. (2007). DNA binding and partial nucleoid localization of the chloroplast stromal enzyme ferredoxin:sulfite reductase. FEBS J. 274, 2054–2069.
Storozhenko, S., Inzé, D., Van Montagu, M., and Kushnir, S. (2001). Arabidopsis coactivator ALY-like proteins, DIP1 and DIP2, interact physically with the DNA-binding domain of the Zn-finger poly(ADP-ribose) polymerase. J. Exp. Bot. 52, 1375–1380.
Tadege, M., Wen, J. Q., He, J., Tu, H. D., Kwak, Y., Eschstruth, A., et al. (2008). Large-scale insertional mutagenesis using the Tnt1 retrotransposon in the model legume M. truncatula. Plant J. 54, 335–347.
Tan, F., Li, G., Chitteti, B. R., and Peng, Z. (2007). Proteome and phosphoproteome analysis of chromatin associated proteins in rice (Oryza sativa). Proteomics 7, 4511–4527.
Tanaka, M., Kikuchi, A., and Kamada, H. (2008). The Arabidopsis histone deacetylases HDA6 and HDA19 contribute to the repression of embryonic properties after germination. Plant Physiol. 146, 149–161.
Thompson, R., Burstin, J., and Gallardo, K. (2009). Post-genomics studies of developmental processes in legume seeds. Plant Physiol. 151, 1023–1029.
Ull, M. A., and Franco, L. (1986). The nucleosomal repeat length of pea (Pisum sativum) chromatin changes during germination. Plant Mol. Biol. 7, 25–31.
Usadel, B., Nagel, A., Thimm, O., Redestig, H., Blaesing, O. E., Palacios-Rojas, N., et al. (2005). Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 138, 1195–1204.
Varshney, R. K., Chen, W., Li, Y., Bharti, A. K., Saxena, R. K., Schlueter, J. A., et al. (2011). Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 30, 83–89.
Wang T. L., Uauy C., Robson F., and Till B. (2012). TILLING in extremis. Plant Biotechnol. J. 10, 761–772.
Yamamoto, Y. T., Zamski, E., Williamson, J. D., Conkling, M. A., and Pharr, D. M. (1997). Subcellular localization of celery mannitol dehydrogenase: a cytosolic metabolic enzyme in nuclei. Plant Physiol. 115, 1397–1403.
Young, N. D., Debellé, F., Oldroyd, G. E., Geurts, R., Cannon, S. B., Udvardi, M. K., et al. (2011). The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524.
Zhang, Y., Woodford, N., Xia X., and Hamburger, A. W. (2003). Repression of E2F1-mediated transcription by the ErbB3 binding protein Ebp1 involves histone deacetylases. Nucleic Acids Res. 15, 2168–2177.
Keywords: seeds, development, nuclei, proteomics, regulation
Citation: Repetto O, Rogniaux H, Larré C, Thompson R and Gallardo K (2012) The seed nuclear proteome. Front. Plant Sci. 3:289. doi: 10.3389/fpls.2012.00289
Received: 22 October 2012; Paper pending published: 11 November 2012;
Accepted: 05 December 2012; Published online: 20 December 2012.
Edited by:
Nicolas L. Taylor, The University of Western Australia, AustraliaReviewed by:
Jozef Samaj, Centre of the Region Hana for Biotechnological and Agricultural Research, Palacky University Olomouc, Czech RepublicTiago S. Balbuena, State University of Campinas, Brazil
Copyright: © 2012 Repetto, Rogniaux, Larré, Thompson and Gallardo. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Karine Gallardo, UMR1347 Agroécologie, Institut National de la Recherche Agronomique, 17 rue de Sully, BP 86510, Dijon, France. e-mail:Z2FsbGFyZG9AZGlqb24uaW5yYS5mcg==
†Present address:Ombretta Repetto, Proteomics Core Facility, Experimental and Clinical Pharmacology, Centro di Riferimento Oncologico - Istituto di Ricovero e Cura a Carattere Scientifico, Aviano, Italy.