 Wei Chen1†
Wei Chen1† Qiuming Yao2†
Qiuming Yao2† Gunvant B. Patil1†
Gunvant B. Patil1† Gaurav Agarwal1,3†
Gaurav Agarwal1,3† Rupesh K. Deshmukh1
Rupesh K. Deshmukh1 Li Lin1
Li Lin1 Biao Wang1,4
Biao Wang1,4 Yongqin Wang1
Yongqin Wang1 Silvas J. Prince1
Silvas J. Prince1 Li Song1
Li Song1 Dong Xu2
Dong Xu2 Yongqiang C. An5
Yongqiang C. An5 Babu Valliyodan1
Babu Valliyodan1 Rajeev K. Varshney3
Rajeev K. Varshney3 Henry T. Nguyen1*
Henry T. Nguyen1*- 1Division of Plant Sciences, University of Missouri, Columbia, MO, USA
- 2Department of Computer Science and Christopher S. Bond Life Sciences Center, University of Missouri, Columbia, MO, USA
- 3Center of Excellence in Genomics, International Crops Research Institute for the Semi-Arid Tropics, Hyderabad, India
- 4Legume Biotechnology Laboratory, School of Agriculture and Biology, Shanghai Jiao Tong University, Shanghai, China
- 5Plant Genetics Research Unit, Donald Danforth Plant Science Center, US Department of Agriculture, Agricultural Research Service, Midwest Area, St. Louis, MO, USA
Drought and flooding are two major causes of severe yield loss in soybean worldwide. A lack of knowledge of the molecular mechanisms involved in drought and flood stress has been a limiting factor for the effective management of soybeans; therefore, it is imperative to assess the expression of genes involved in response to flood and drought stress. In this study, differentially expressed genes (DEGs) under drought and flooding conditions were investigated using Illumina RNA-Seq transcriptome profiling. A total of 2724 and 3498 DEGs were identified under drought and flooding treatments, respectively. These genes comprise 289 Transcription Factors (TFs) representing Basic Helix-loop Helix (bHLH), Ethylene Response Factors (ERFs), myeloblastosis (MYB), No apical meristem (NAC), and WRKY amino acid motif (WRKY) type major families known to be involved in the mechanism of stress tolerance. The expression of photosynthesis and chlorophyll synthesis related genes were significantly reduced under both types of stresses, which limit the metabolic processes and thus help prolong survival under extreme conditions. However, cell wall synthesis related genes were up-regulated under drought stress and down-regulated under flooding stress. Transcript profiles involved in the starch and sugar metabolism pathways were also affected under both stress conditions. The changes in expression of genes involved in regulating the flux of cell wall precursors and starch/sugar content can serve as an adaptive mechanism for soybean survival under stress conditions. This study has revealed the involvement of TFs, transporters, and photosynthetic genes, and has also given a glimpse of hormonal cross talk under the extreme water regimes, which will aid as an important resource for soybean crop improvement.
Introduction
Soybeans, the most important legume crop worldwide, are an essential source of oil and protein for humans and livestock, and are also considered a potential source of bio-diesel (Koberg et al., 2011). Considering the importance of soybeans for food and nutritional security, there have been extensive efforts toward increasing soybean production. However, due to increases in the global population and its demand, there is a need for enhancing soybean productivity and production. Despite extensive efforts, soybean yield improvement is facing severe challenges and suffers with yield loss due to a range of biotic and abiotic stresses. Global climatic changes also play a vital role in influencing the abiotic and biotic conditions. Among abiotic stresses, extreme water regimes such as drought and flooding cause severe yield losses in all major crops including soybeans, rice, corn, and wheat (Perata et al., 2011). Drought is caused by insufficient water supply either from rainfall or groundwater, and results in the soil drying. In contrast, flooding is caused by heavy rainfall and results in water logging and submergence. In soybeans, both drought and flooding can cause up to a 40–60% yield loss worldwide (Valliyodan and Nguyen, 2006; Komatsu et al., 2009; Ahmed et al., 2013).
The mechanism that overcomes drought stress in plants is facilitated by drought avoidance, drought tolerance, drought escape, and drought recovery (Cruz De Carvalho, 2008). Drought causes a negative impact on all developmental stages, starting from germination to seed maturation (Valliyodan and Nguyen, 2006). Flooding also leads to very sensitive responses that affect both soybean growth and yield (Komatsu et al., 2009). Stress-specific adaptive mechanisms and various molecular, biochemical, and physiological responses in plants help maintain normal growth and survival under drought and flooding conditions (Dat et al., 2004). The perception of the environmental signal and subsequent molecular signaling is the first step in a stress response. In this direction, several signal compounds including abscisic acid (ABA), have been identified and studied for drought stress. The ABA level is up-regulated under drought conditions and the downstream responsive network is induced (Xiong and Zhu, 2002). The initiation of the ABA signaling process involves PYR/PYL/RCAR (Pyrabactin Resistance /PYR1-Like/Regulatory Components of ABA Receptors) ABA receptors, and two enzymes, protein phosphatase 2c family (PP2Cs) and SNF-related kinase (SnRK2), with opposite functions of phosphatase/kinase (Park et al., 2009; Kim, 2014). The binding of ABA to PP2Cs can inhibit PP2C activity, which causes it to lose its ability to inhibit SnRK2s. Then, activated SnRK2s can phosphorylate and activate the downstream ABFs (ABA responsive element binding factor), which can bind to their own targets (or other genes) and activate the downstream response pathways (Park et al., 2009). This causes plant to express a drought tolerance phenotype. The ABA level is also increased in flooding treatments in tomato (Else et al., 1995). However, the ABA level is reduced under flooding treatment in rice seedlings during submergence (Saika et al., 2007). Among different pathways of stress response signaling, most of the ABA dependent and independent responses involve TFs. These TFs belong to very diverse families representing ~10% of the genes in soybean, which are involved in most of the biotic and abiotic stress responses (http://planttfdb_v1.cbi.pku.edu.cn:9010/web/index.php?sp=gm). In rice, SUBMERGENCE1 (SUB1) has been identified as an important TF involved in submergence tolerance (Xu et al., 2006).
Antioxidants, such as ascorbate, glutathione, and tocopherol, are accumulated to protect the plants against reactive oxygen species (ROS), which are over-generated under drought and flooding conditions (Valliyodan and Nguyen, 2006; Chen et al., 2015). Photosynthesis is the major producer of ROS that enables the chloroplasts either to avoid its production or process it in the antioxidant network (Foyer and Shigeoka, 2011). Photosynthetic activity is reduced and carbohydrate metabolism changes under drought stress (Tabaeizadeh, 1998; Krasensky and Jonak, 2012; Chen et al., 2015). Similarly, ROS production increases due to heat, pathogen invasion, wounding, and low oxygen, while it decreases due to low light conditions that may arise during submergence (Steffens et al., 2013).
The complex molecular mechanisms, signaling perception, integrated responses, and molecular cross talk activated in response to different abiotic stress are not well-understood in soybean. In this regard, several efforts have been made to elucidate the molecular mechanisms. Genome-wide transcriptome profiling will be helpful in understanding a global view of gene expression under drought. Recently, drought-responsive candidate genes (GmNAC) have been identified using transcriptome profiling of soybean under drought conditions performed with a 66K Affymetrix microarray platform (Le et al., 2012). It has been reported that transcription profiles of the cold- and dehydration-responsive genes were similar among Arabidopsis, rice, and soybean, which shows representative up-regulated late embryogenesis abundant (dehydrin/LEA) and down-regulated (photosynthesis-related) genes. This suggests that different species have conserved different stress responses (Maruyama et al., 2012). Similarly, several studies using microarray platforms have been conducted and expression data for over 5000 soybean samples have been deposited in a public database (http://www.ncbi.nlm.nih.gov/geo/). However, the microarray have some drawbacks such as cross-hybridization, non-specific hybridization, and a limited sensitivity. Most importantly, microarray platforms provides the information on only those genes that are available on the microarray, which is a problem for soybean whose gene models are not well-characterized. These limitations can be overcome using recently advanced sequencing-based techniques, such as RNA sequencing (RNA-Seq; Varshney et al., 2009; Trapnell et al., 2013). In soybean, RNA-Seq has been deployed to generate an expression atlas for soybean genes using several tissues in seed development stages (Severin et al., 2010). In another study, variability in commercial and developing cultivars under drought conditions was identified using single nucleotide polymorphisms (SNPs) in RNA-Seq data (Vidal et al., 2012). In this study, genome-wide transcriptome profiling of soybean leaf tissue using the RNA-Seq approach was performed to understand the response to drought and flooding stress. The comparative analysis has enabled us to understand molecular responses against the extreme water availability conditions.
Materials and Methods
Plant Material and Stress Conditions
Soybean (William 82 genotype) plants were grown in 26.5 L pots with a dimension of 30 × 27 × 37 cm (top diameter × bottom diameter × height), filled with a mixture of turface and sand (2:1). Osmocote (Scotts Co., Marysville, OH) with a nutrient content of N:P2O5:K2O = 14:14:14 was added as a nutrient source at a rate of 20 g per pot. The plants were grown in a well-watered conditions [up to vegetative (V4) stage] under a 16 h photoperiod (~10,200 lux) at 28°C. Drought stress was imposed by withdrawing water for 7 days, while flooding stress was imposed by placing the pots into a bigger pot with a trashcan liner filled up to a water level of 4 cm above the soil surface for 7 days. After 7 days of treatment, all the leaves (irrespective of their response to stress) were sampled from the drought and flood treatments and control plants for RNA sequencing and real-time PCR analysis. The soil moisture was measured after the stress imposition in the drought experiment using a PR2 moisture probe (Delta T, UK).
RNA Isolation and Library Preparation
The total RNA was extracted using RNeasy Plant Mini Kit (Qiagen, Valencia, CA) and on-column DNase digestion was performed according to manufacturer's protocol. The concentration and quality of RNA were checked by a Nanodrop 1000 (Grand Island, NY) and by running the product on an agarose gel. The RNA-Seq libraries were prepared using a TruSeq Stranded mRNA LT Sample Prep Kit—Set A (Catalog #: RS-122-2101, Illumina, USA) according to the manufacturer's instructions. Three biological replicates were used for RNA-Seq and real-time PCR analysis.
Quality Filtering and Mapping of RNA-Seq Reads
The single end RNA-Seq reads were generated on the Illumina Genome Analyzer (San Diego, CA) platform. The processing of the initial reads was performed using the Illumina analysis pipeline (in the Fastq format) using custom and default parameters. Additional filtering was performed by removing adaptor sequences and low quality bases. To facilitate the read mapping, the Glycine max reference genome (Gmax1.1version) was indexed by Bowtie (http://www.phytozome.net; Langmead and Salzberg, 2012). The read mapping was performed using the Tophat software package (Trapnell et al., 2009; Kim et al., 2013). The reads were first mapped directly to the genome using indexing and then some of the unmapped reads were resolved by identifying novel splicing events. Two mismatched base pairs were allowed and the multiple position matching was reported up to 40 alignments using the Tophat mapping procedure. The transcriptome raw sequencing data from this study have been submitted on the NCBI (http://www.ncbi.nlm.nih.gov/) database as individual BioProjects: PRJNA324522.
Sequence Assembly and Differential Counting
The binary read alignment files were used as input to Cufflinks (Trapnell et al., 2009), which assembled the reads into transfrags (transcripts). The estimated gene abundance was then measured in terms of the fragments per kilobase of transcript per million mapped reads (FPKM). The differentially expressed genes (DEGs) between the two sets of samples were identified using cuffdiff. The significant up-regulated and down-regulated gene lists were obtained for the drought and flood samples, respectively. Only the genes with a log2 fold change ≥+2 and ≤ −2, but without infinite values and a FDR adjusted p ≤ 0.05 after Benjamini-Hochberg correction for multiple-testing with significance level “yes,” were considered as significantly DEGs.
Functional Annotation and Gene Ontology (GO) Enrichment
The DEGs were annotated for gene ontology (GO) terms (Ashburner et al., 2000) and categorized into Molecular Function (MF), Cellular Component (CC), and Biological Process (BP) categories. A gene enrichment test was then performed on each of the gene lists to obtain the significant terms. Fisher's exact test, which is based on the hypergeometric distribution, was used to calculate the p-value. The TreeView (http://jtreeview.sourceforge.net/docs/overview.html) program was used to draw the heatmap of the significant DEGs in response to drought and flood stress.
Pathways and TF Identification and Analysis
The drought and flood responsive pathways were identified and plotted using MapMan (Usadel et al., 2009). Multiple biological or metabolic pathways were plotted together with the mapped gene intensity of the fold change (≥ +2 and ≤ −2) by a blue and red schema. The TFs and transcription related genes were also mapped and plotted by MapMan. A more detailed TF family annotation was obtained from plantTFDB (Jin et al., 2014).
Analysis of Cis-Motif Pattern
Using the collection of the motif sequences from the “Database of Plant Cis-acting Regulatory DNA Elements” (Higo et al., 1999), the 3 kb upstream of the 5′ translation start base was searched for all of the annotated soybean genes (phytozome v9.1). The number of genes was counted for each matched motif (gene count) and the total number of matches to all of the genes by each motif (hit count). Furthermore, counts from the subset of genes in each of the gene sets with all the genes were compared using Fisher's test. Any of the comparisons with a p ≤ 0.05 is presented in Supplementary Tables 3A,B.
cDNA Synthesis and qPCR
Total RNA was extracted from each sample using the Qiagen RNeasy mini kit (Qiagen, CA, USA).
The first strand cDNA from 1 μg of total RNA was synthesized using the EcoDry premix (Clontech, CA, USA), following the manufacturer's instructions. Quantitative PCR (qPCR) was performed using 10-fold diluted cDNA product in a 10 μL reaction volume using the Maxima SYBR Green/ROX qPCR Master Mix (Thermo, Waltham, MA, USA) on ABI7900HT detection system (Foster City, CA, USA). Three biological replicates and two technical replicates were used for analysis. The PCR was performed using two-step cycling protocol as follows: 50°C for 2 min; 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, and 60°C for 1 min (https://www.thermofisher.com/order/catalog/product/K0221). To normalize the gene expression, Actin (Glyma18g52780) was selected as a housekeeping gene. All the primers were designed using QuantPrime software (http://www.quantprime.de) (Supplementary Table 5).
Results
Drought and Flood Treatment
Three biological replications were subjected to well-watered, drought, and flooding conditions at the V4 growth stage. Soil moisture, the major limiting factor used to impose drought stress on growing plants (Else et al., 1995), and was measured before and after the drought treatment. The soil moisture of the well-watered plant was 19% and was reduced to 10% after 7 days of drought treatment; the plants showed symptoms of leaf wilting. The degree of drought and flooding was determined by monitoring the canopy temperature. The canopy temperature (°F) increased from 76.8 ± 1.03 in normal conditions to 85.7 ± 4.42 and 79.4 ± 1.06 under the drought and flooding conditions, respectively.
Mapping and Differential Gene Expression Analysis under Drought and Flooding Stresses
The RNA samples from the soybean leaves under the control/drought/flooding conditions were used for sequencing by the Illumina Genome Analyzer. Approximately 28 million reads were generated from each sample (Table 1). The RNA-Seq analysis workflow is shown in Supplementary Figure 1 and was utilized for the data analysis. Approximately 72% of the reads were mapped to the soybean reference genome and ~2.5 to 4 million reads were mapped to multiple regions (Table 1).
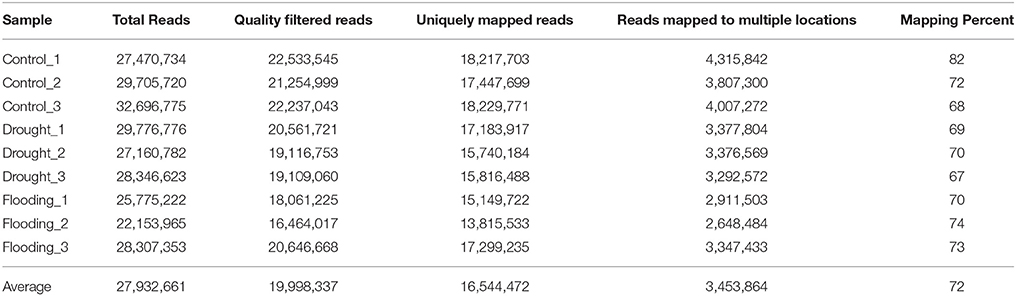
Table 1. Summary of RNA-Seq performed for soybean leaf tissue under drought and flooding stress.
To quantify and identify the genes, a core set of DEGs under drought and flood stress in soybean were examined. High-throughput RNA-sequencing analysis using Cufflinks pipeline (Trapnell et al., 2012) was performed in the following three combinations: (i) control vs. drought, (ii) control vs. flood, and (iii) drought vs. flood. The DEGs specific to and common between the stresses were also identified (Figures 1,2). A total of 36,968 and 37,434 genes with confident expression in drought and flood conditions, respectively, when compared to the control (Supplementary Tables 1A,B). Out of these genes, 36,239 were found to be common between the two conditions. The top 50 genes with >500 FPKM featured in the drought and flooding conditions were identified (Tables 2, 3). Interestingly, there are 15 genes among the top 50 highly expressed ones enriched for photosystem II (PS II) and dehydrin family proteins featured in both stress conditions. Flooding stress specific genes were found to be enriched for lipoxygenase 2, ethylene forming enzyme, matrixin family proteins, and 12-oxophytodienoate reductase 2, whereas drought specific genes were enriched for S-adenosylmethionine synthetase, calmodulin 5, 1-deoxy-D-xylulose-5-phosphate-reductoisomerase, and carbonic anhydrase (Tables 2, 3).
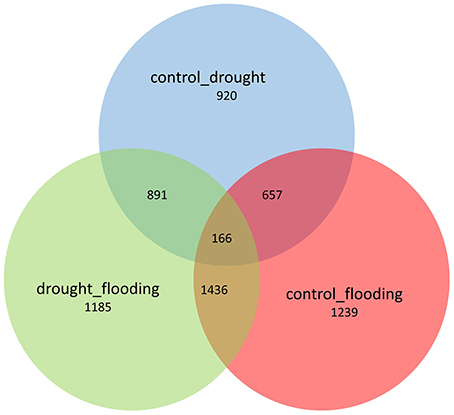
Figure 1. Venn diagram representing the specific and common DEGs across the control vs. drought, control vs. flooding, and drought vs. flooding conditions.
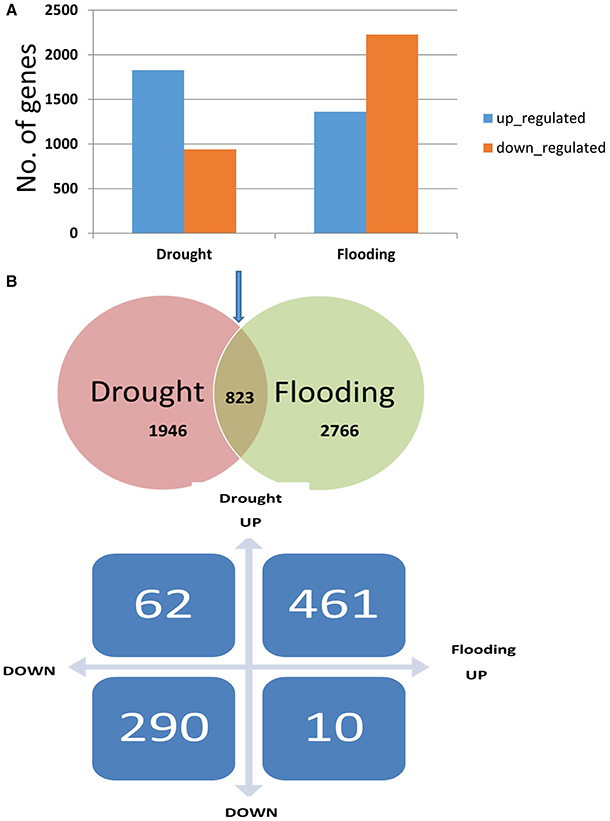
Figure 2. The number of up- and down-regulated genes in drought and flooding conditions. (A) Proportion of significant results (p ≤ 0.05, log2 fold change ≥2 for up-regulated and ≤ −2) for down-regulated genes in drought and flooding conditions. (B) The number of overlapped genes in two conditions.
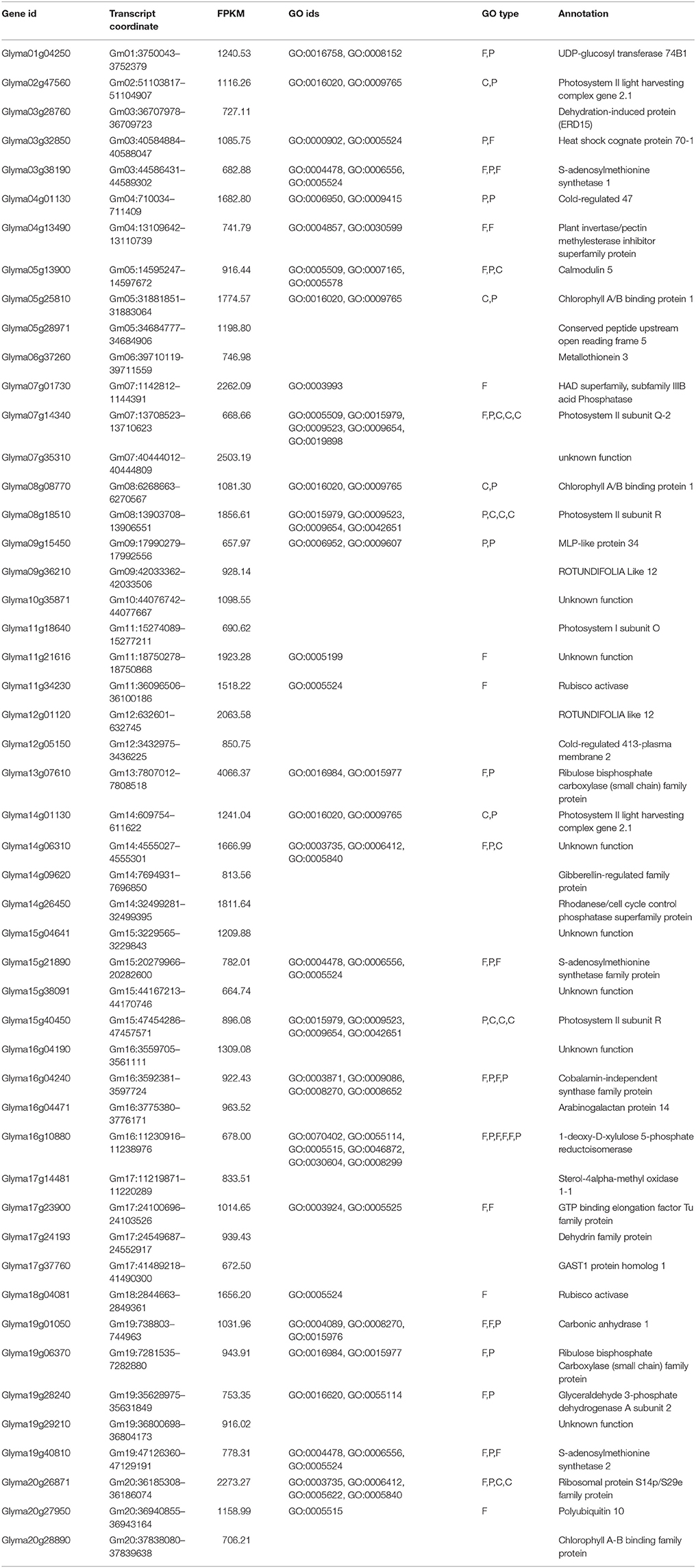
Table 2. List of the 50 most highly expressed transcripts with their ontology and annotations in drought stressed leaf tissue compared to non-stressed control tissues.
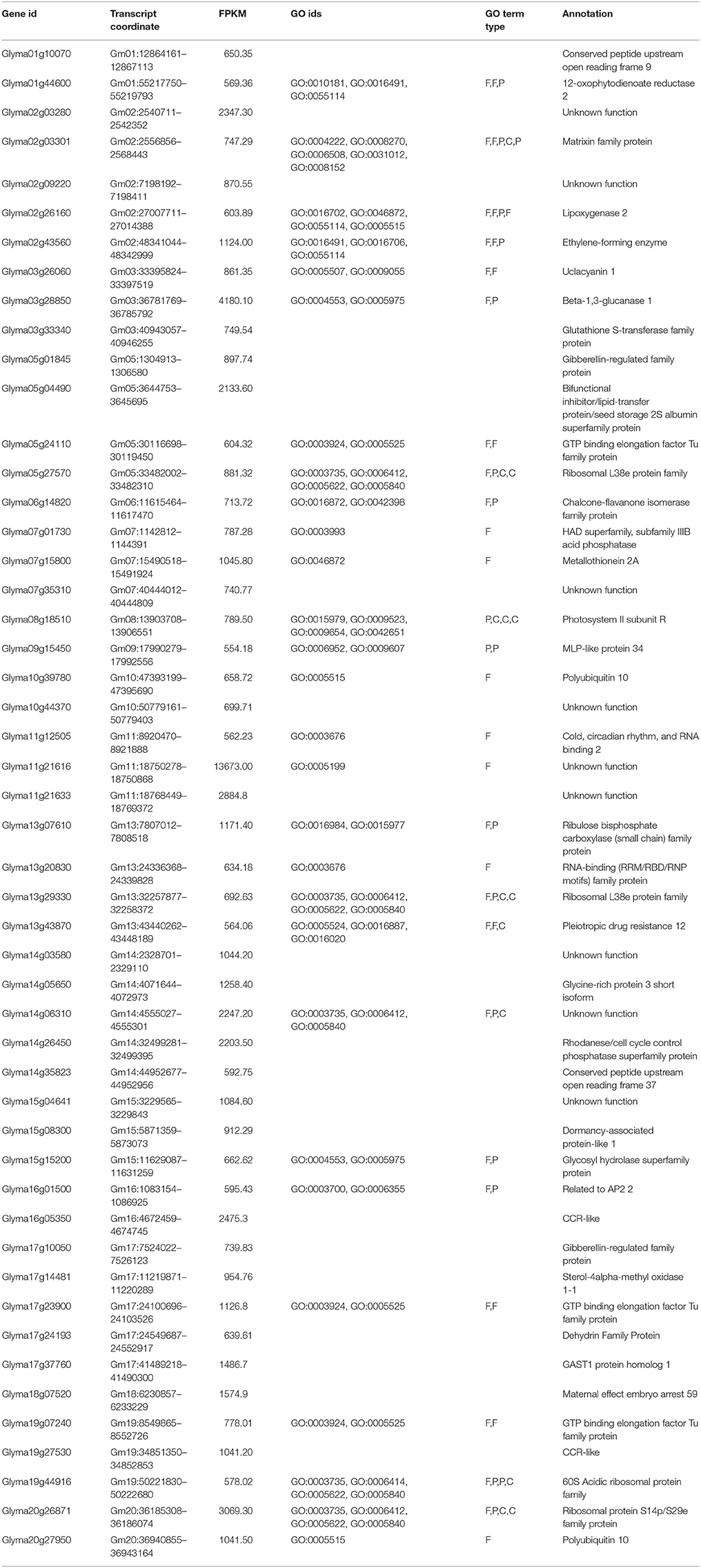
Table 3. List of the 50 most highly expressed transcripts with their ontology and annotations in flooding stressed leaf tissue compared to non-stressed control tissue.
The distribution trends in terms of fold change ranged from ~8- to 9-fold change for DEGs under drought and flood stress (Supplementary Figure 2). A total of 2724 DEGs were identified under the drought conditions when compared to control, and 1802 genes were up-regulated and 922 genes were down-regulated (Supplementary Table 3A). During flood stress, a total of 3498 DEGs were identified when compared to the control, and 1303 genes were up-regulated and 2195 genes were down-regulated (Supplementary Table 2B). The total number of DEGs was greater in the flood condition compared to the drought. However, a greater number of up-regulated genes were observed under the drought condition compared to the flood condition with an overlap of 166 genes across the three comparisons (Figure 1). A total of 3768 genes were found to be differentially expressed when flood and drought conditions were compared to each other (Supplementary Table 2C). However, among the 2724 and 3498 DEGs identified in the drought and flood conditions, respectively, 823 DEGs were found to be in common (Figure 2A). Among these genes, 461 genes were both up-regulated under the drought and flood conditions, 290 genes were both down-regulated under the two conditions, 62 genes were up-regulated in the drought but down-regulated in the flood conditions, and 10 genes were down-regulated in the drought but up-regulated during flood conditions (Figure 2B).
Gene Ontology (GO) Annotation of Differentially Expressed Genes
The profiles of biological processes represented by GO enriched DEGs under drought and flood stresses were studied and represented (Figure 3, Supplementary Table 3). The heat-map revealed a stark difference in biological processes under different GO categories, such as phagocytosis, cell morphogenesis, cell cycle, isoprenoid biosynthesis, and transcriptional regulation represented by the enriched genes in these categories. However, little differences were observed in the profiles of the genes enriched for the GO categories “responsive to stress” and “defense response” under drought and flooding conditions. Biological processes such as “tRNA processing,” “cell wall macromolecule catabolic process,” “ubiquitin-dependent protein catabolic process,” and “carbohydrate transport” were reduced under both conditions (Figure 3). Most of the GO categories showed an opposite and comparable profile under drought and flood conditions, which suggested differential and specific gene regulation in both conditions. The GO categories of “cell redox homeostasis,” “intracellular protein transport,” “phosphatidylinositol phosphorylation,” “phagocytosis,” “embryo development,” and “Golgi vesicle transport” were enriched under flood condition but reduced under the drought conditions. On the contrary, “protein-heme linkage,” “regulation of transcription DNA-dependent,” “nucleoside metabolic process,” and “metabolic process” were enriched under drought but reduced under flooding conditions.

Figure 3. The p-value heatmap of significant biological processes under drought and flooding conditions. Darker color indicates greater significance.
Differentially Expressed Transcription Factors (TFs) under Drought and Flooding Stress
The RNA-Seq expression profiling revealed 289 and 271 differentially expressed TFs under drought and flood stress, respectively. A total of 213 and 199 TFs were up-regulated, whereas 76 and 72 TFs were down-regulated under drought and flood conditions, respectively (Table 4). Interestingly, the majority of differentially expressed TFs, irrespective of a family, were up-regulated under both conditions (Table 4). Genes belonging to the bHLH, ERF, MYB, NAC, and WRKY family represent most of the differentially expressed TFs. The Ethylene Response Factor (ERF) represented the highest number of significantly expressed genes under both drought and flooding conditions followed by bHLB, MYB, NAC, and WRKY (Table 4, Figure 4, Supplementary Figure 5).
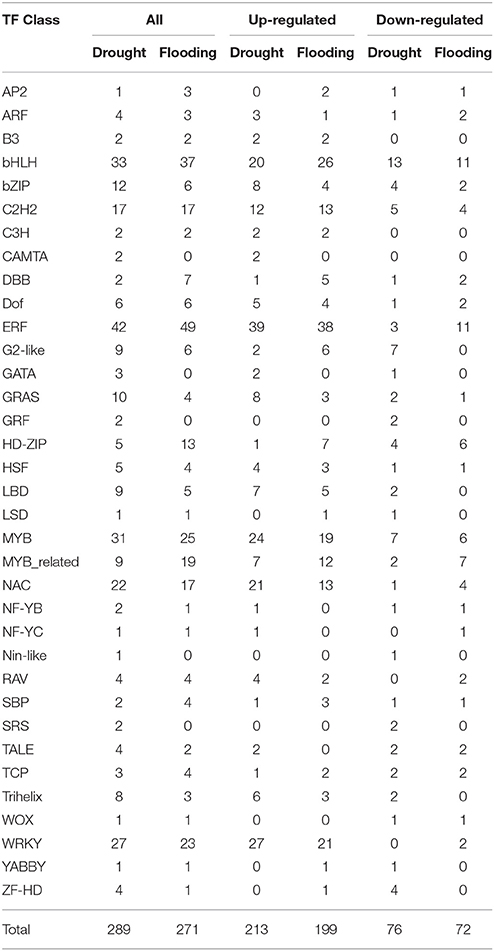
Table 4. Details of differentially expressed TFs identified by RNA-Seq and performed for drought and flooding conditions in soybean.
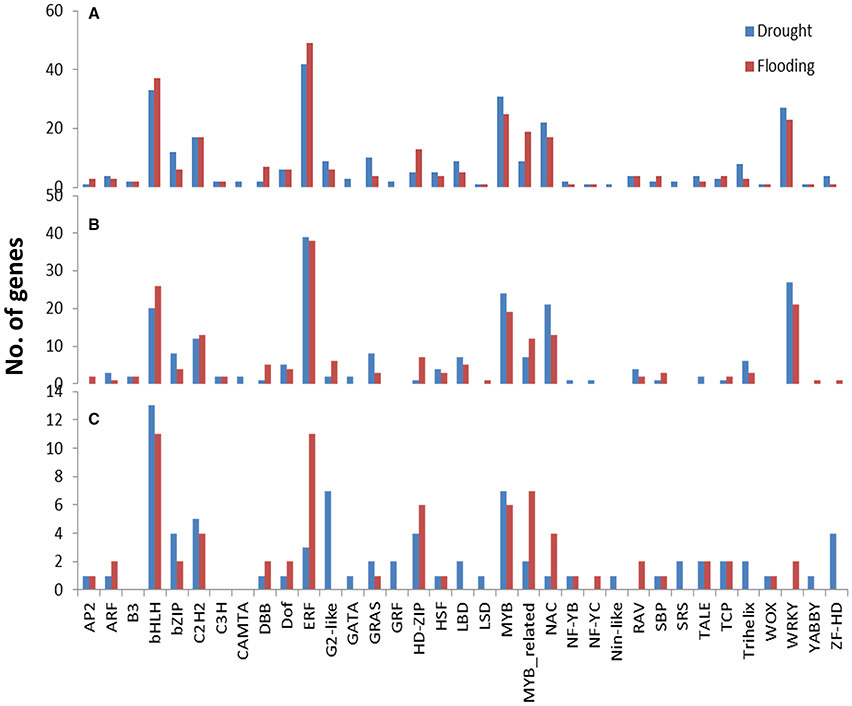
Figure 4. Class-wise counts of transcription factors identified in DEGs under drought and flooding stress. A total of 289 TFs (UP: 213, DOWN: 76) were found in drought and 271 TFs (UP: 199, DOWN: 72) in flooding stress. Identified from (A) all regulated genes, (B) only up-regulated genes, and (C) only down-regulated genes.
Photosynthesis Associated Responses
The photosynthesis related genes were down-regulated under both drought and flooding conditions. The expression level of three PS-II light harvesting complex (LHC) related genes, Glyma16g28030.1, Glyma16g28070.1, and Glyma10g04630.1, were repressed significantly under drought conditions. Similarly, three photosynthesis light reaction related genes, Glyma07g21150.1, Glyma20g24500.1, and Glyma20g24500.1, and one Calvin cycle related gene, Glyma01g34300.1, were also down-regulated under drought conditions. Only one gene from the photosynthetic system encoding an electron carrier was induced under the drought conditions (Supplementary Figure 6A). However, compared to the drought, more photosynthesis related genes were found to be down-regulated under flooding conditions (Supplementary Figure 6B). The relative expression level of 34 genes encoding PS-II LHC and 45 genes encoding PS-II subunits were repressed (Supplementary Figure 6B). In addition, 41 genes related to the Calvin cycle and eight electron carrier related genes were also repressed. In total, seven genes were down- and one was up-regulated under drought stress. However, 128 genes were down-regulated under flood treatment (Supplementary Figure 6B). The dynamic expression of photosynthesis related genes can be correlated with the chlorophyll content under drought and flood conditions, which concurs with the inhibition of photosynthesis (Supplementary Figures 3A,B).
Sugar and Starch Metabolism Pathways
Changes in the expression levels of a large number of sucrose and starch metabolism pathway genes were observed under the drought and flood conditions. Genes encoding sucrose and starch biosynthesis genes, namely, amylose synthase, ADP-Glucose synthase, and starch and maltose synthase, showed a significant decrease in the transcript level in drought and flooding treatments (Supplementary Figure 4). No change in expression was observed for sucrose phosphate synthase genes (Glyma08g42140.1 and Glyma06g48200.1) in the drought condition, in contrast to the significant repression in expression of these genes under the flood conditions. However, the genes encoding beta-fructofuranosidase (Glyma13g42530.1), fructokinase (Glyma16g32530.1), hexokinase (Glyma11g10130.1), and three maltose synthase genes (Glyma13g28630.1, Glyma11g04210.1, and Glyma15g10480.1) were up-regulated under the drought conditions. Overall, transcript abundance for starch biosynthesis encoding genes was reduced more in the flooding stress compared to the drought stress. This could result in the decrease of starch content under flooding conditions (Supplementary Figure 4B). Only one maltose synthesis gene, Glyma11g04210.1, was induced during flooding (Supplementary Figures 4, 6). We also identified six genes of the soybean sugar-effluxer (SWEET) family that was differentially expressed under the two extreme water regimes. Three genes, Glyma12g36300, Glyma14g27610, and Glyma06g13110, were up-regulated under the drought conditions, and the other three genes, Glyma06g17520, Glyma09g04840, and Glyma13g33950, were down-regulated under flooding conditions. Interestingly, one SWEET gene, Glyma14g27610, was up-regulated in both drought and flooding conditions (Supplementary Tables 1A,B).
Cell Wall Associated Responses
The annotated RNA-Seq data was analyzed for the cell wall precursor's related genes. Expression of 11 cell wall precursor's related genes were up-regulated in the drought conditions, whereas not a single gene was observed to be down-regulated. On the contrary, the flooding condition showed a reduced transcript level for 10 cell wall precursor synthase genes and three genes were up-regulated (Supplementary Figure 7). The UDP-Glucosyl Transferase enzyme coding gene under drought stress was found to be highly expressed (FPKM = 1240.53; Table 2). This enzyme is known to be involved in the formation of β-glucans, which are thought to be involved in cell wall formation (Kosegarten et al., 1988).
Analysis of Cis-Motif Pattern
Genes with similar expression patterns are likely co-regulated and have the same cis-regulatory elements (CREs or known as motifs; Priest et al., 2009). The set of DEGs (up- and down-regulated genes) identified under flood and drought treatments in this study was utilized (Supplementary Figure 2). Using the collection of motif sequences from the “Database of Plant Cis-acting Regulatory DNA Elements,” three kb upstream of the 5′ most translation start base for all of the annotated soy genes was searched (Phytozome v9.1). Sixteen and 76 over-represented motifs in the down- and up-regulated drought response genes, respectively, and 48 and 5 over-represented motifs in the down- and up-regulated flood response genes, respectively were identified (Supplementary Table 4A). No overlapping motifs were found among the drought and flood response up-regulated genes; however, eight overlapped motifs were found between the drought and flooding down-regulated genes. Additionally, no overlapped motifs were found between the drought down-regulated genes and flooding up-regulated genes, and 30 overlapped motifs were found between the up- and down-regulated drought and flood genes (Supplementary Table 4B). Among the 30 overlapped motifs, 24 showed the ACGT sequence, which serves as a binding site for the bZIP class of TFs. It has been reported that the flanking sequence of the ACGT core motif is responsible for differential distribution of the stress responsive regulatory elements in the stress responsive promoters (Suzuki et al., 2005). The nucleotides flanking the ACGT motif determine the binding affinity and specificity of bZIP TFs, which in turn leads to distinct physiological functions in stress responses, light regulation, etc. (Izawa et al., 1993; Foster et al., 1994).
Although drought stress up-regulated genes shared several cis-regulatory elements with down-regulated flood stress genes, few genes were found to be common between drought up-regulated and flooding down-regulated genes. For example, there are 324 up-regulated drought responsive genes and 457 down-regulated flood response genes with only one shared motif, ACGTABREMOTIFA2OSEM. Importantly, this motif acts at the core of ACGT of motif A in the rice ABRE gene (Hattori et al., 2002). However, there were only 16 common genes representing 5% of the drought up-regulated genes and 4% of the flood down-regulated genes.
Drought and flood down-regulated genes had ~15% of their genes in common (Supplementary Table 3D). Interestingly, flood down-regulated genes shared most their over-represented motifs (30/48) with drought up-regulated genes. However, they did not share many genes between the down-regulated flooding response and the up-regulated drought responsive genes (Supplementary Tables 3C,D). The molecular function categories were also examined using the GO enrichment analysis for both of the datasets represented by the ACGTABREMOTIFA2OSEM motif. Interestingly, the top three molecular functions for both of the datasets were “catalytic activity,” “sequence-specific DNA binding TF activity,” and “DNA binding.”
Quantitative Real-Time PCR (qRT-PCR) Validation of DEGs from RNA-Seq
To validate RNA-Seq analysis, qRT-PCR was performed. A few key genes were selected based on increased or reduced transcript abundance under drought and flooding conditions. Although the RNA-Seq values showed slight variations compared with the corresponding values from the qRT-PCR analyses, the expression data and pattern from RNA-Seq were largely consistent with those obtained by qRT-PCR with a high correlation coefficient value (R2 = 0.70, 0.76 for drought and flood responsive genes, respectively; Figure 5).
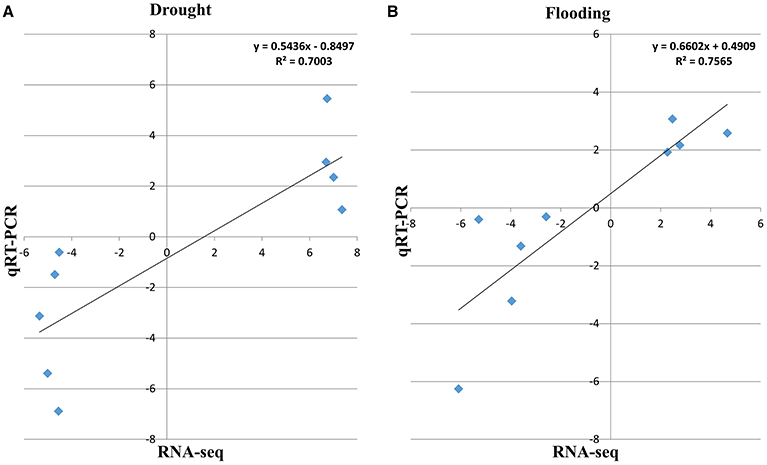
Figure 5. The qRT-PCR validation of differentially expressed genes in soybean under drought and flooding conditions. The correlation of the fold change analyzed by RNA-Seq (x-axis) with data obtained using real-time PCR (y-axis; A) under drought and (B) under flooding stress.
Discussion
The results presented here demonstrate the unique and differential response of soybean leaf tissue under two extreme water regimes, drought and flood. Several 100 genes are known to be differentially expressed under abiotic stresses such as drought, high salinity, low temperature, and flooding (Deshmukh et al., 2014; Patil et al., 2016). In addition to the involvement of various physiological and molecular mechanisms, most of the pathways and genes are found to be common across the different stress responses (Deshmukh et al., 2014). Transcriptome profiling by RNA-Seq has enabled comparison of a transcriptional response under drought and flooding stress in soybeans, and also development of a catalog of DEGs. The direct comparison of genes expressed under different conditions or experiments would require a meta-analysis (Bhargava et al., 2013) to have a better insight into the modus-operandi of genes specifically and commonly involved in various stress responses. Nevertheless, the ratio of up- and down-regulated genes under two extreme water regimes in this study could be compared and ultimately shed some light on the comparative transcriptional response. However, a higher number of DEGs were identified under flood conditions, and the number of up-regulated genes was much higher under drought conditions (Figure 2). Similarly, fewer up-regulated genes under flood conditions in soybean have been reported by Nanjo et al. (2011). In agreement with Nanjo et al. (2011), we observed the lowered metabolic activity under flooding as compared to the drought, which was evident from the global gene expression profiling under the two stresses.
Transcription Factors Regulation under Drought and Flooding Stress in Soybean
The involvement of TFs was examined in the drought and flood stress responses through both the ABA dependent and independent pathways. The ABA and other upstream signals can trigger the downstream regulation pathway through the TF regulatory network in both drought and flood conditions. In this research, ABA related TFs: AP2, bZIP, MYB, and NAC were up-regulated under both drought and flood conditions (Figure 4). These TF families have been extensively studied in many plants under several different stress conditions (Dubos et al., 2010; Hao et al., 2011; Mizoi et al., 2012; Nakashima et al., 2012,). In several abiotic stress regulation studies, the TFs identified are initially involved in tolerance against a specific stress and later observed to be important for other stresses. For instance, the SUB1A TF was initially identified as a key regulator for the flood responsive pathways in rice, and later transcriptional and functional studies confirmed its important role in drought tolerance (Xu et al., 2006; Fukao et al., 2011). However, the flood response in soybean seems to be independent of the SUB1 pathway since no SUB1A homolog was found to be differentially expressed in our study. On the other hand, recent studies have shown the involvement of SUB1 and core-clock related genes mediated through alternatively spliced forms (Syed et al., 2015).
Hormonal Signaling and Transcriptional Regulations under the Extreme Water Regime
Genes including TFs involved in the ABA dependent stress response were found to be differentially expressed under both drought and flood stress conditions (Table 4, Supplementary Table 2). Although ABA is the most specifically studied hormone for its regulatory role in different stress conditions, other hormones such as cytokinins, brassinosteroids, auxins, and ethylene also played an important functional and regulatory role (Peleg and Blumwald, 2011; Patil and Nicander, 2013). The highest number of TFs belonging to the ERF family coincided with the higher number of ethylene related genes observed under drought and flooding conditions (Table 4). This suggests the role of ethylene in both drought and flood responsive pathways. Ethylene biosynthesis has been found to be affected by auxin, particularly with the auxin dependent roles of several members of the 1-aminocyclopropane-1-carboxylate synthase (ACS) family genes (Nakatsuka et al., 1998). S-Adenosylmethionine synthetase (SAM synthetase) coding genes were found to be highly enriched and featured among the top 50 highly expressed genes under drought stress. SAM acts as a precursor for polyamines and ethylene biosynthesis, which indicates a high efflux of ethylene hormone under stress to counteract the adverse conditions. Higher ethylene levels are known to induce the ethylene signaling cascade, which in turn involves the ERF TF activation. That is also found to be highly expressed under the stresses imposed in the current study. Higher ethylene levels are manifested as the ethylene triple response (Knight et al., 1910) to prevent root and shoot elongation, radial swelling of the root and shoot, and horizontal growth of the embryonic leaves and meristem instead of vertical growth (a consequence of auxin imbalance across the stem axis). This triple response prevents the plants from damage. It would also be interesting to see the receptor response against elevated ethylene levels as reviewed by Agarwal et al. (2012) because different plants perceive ethylene in their own unique way and the receptors have a very important role in signal perception and the downstream cascade.
The 1-Deoxy-D-xylulose-5-phosphate reductoisomerase (DXR) is an important enzyme involved in the 2-C-methyl-D-erythritol-4-phosphate (MEP) pathway for isoprenoid biosynthesis and was identified among the highly expressed drought responsive genes (Table 2). Xing et al. (2010) reported that dxr mutants with a dwarf phenotype significantly reduced the number of trichomes and abnormal stomatal closure, which was rescued with exogenous application of ABA and gibberellic acids (GAs). Thus, the high expression of DXR under drought stress implies a plant's response to withstand stress and compensate for compromised photosynthetic capacity and abnormal stomatal conductance. Auxin, ethylene, and ABA were previously identified to be involved in the drought responsive pathway in soybean (Le et al., 2012). Additionally, ethylene, ABA, and GA are known to be involved in a regulatory network under flooding stress (Bailey-Serres et al., 2012). Hormonal cross-talk at different levels of the stress response and the resultant synergetic or antagonist interactions have been found to be important for stress tolerance (Chinnusamy et al., 2004; Peleg and Blumwald, 2011). The biosynthesis of jasmonic acid (JA) through the octadecanoic pathway (Vick and Zimmerman, 1984) with α-linolenic acid (LA) as a precursor, is mediated by 12-oxophytodienoic acid reductase 2 (OPDAR2) (Glyma01g44600), which is found to be up-regulated under both drought and flooding stress (Supplementary Tables 1B, 2A) but highly expressed under flooding stress (Table 3). The high expression of OPDRA2 is correlated with higher JA synthesis and other systemic responses. The enzyme has also been shown to detoxify 2,4,6-trinitro toluene (TNT) in polluted water contaminated by explosives as a result of manufacturing and munitions (Beynon et al., 2009). The upsurge of OPDAR2 under flooding stress suggests plants' adaptive response against stress and this could also help in detoxification of contaminants such as TNT if any are present in the water. DEGs involved in different hormonal pathways identified here under drought and flooding stress could be good candidates to manipulate hormonal cross-talk for the effective regulation of stress tolerance in plants.
Transcriptional Response Related to Photosynthesis Efficiency and Chlorophyll
The genes involved in photosynthesis were found to be preferentially down-regulated under both drought and flood conditions in this study (Supplementary Figure 6). This agrees with the hypothesis that a plant's photosynthetic capacity is affected under stresses such as drought and flooding stresses. Thus, this implies that the reduction in photosynthesis activity is the major effect of drought and flooding in soybean. Similarly, the down-regulation of genes involved in photosynthesis has been previously reported under a water deficit situation in other crops (Wang et al., 2011). Interestingly, Nanjo et al. (2011) reported the up-regulation of photosynthesis related genes under flooding; however, they imposed stress at the 2 day-old seedling stage of soybean and performed transcriptome profiling with hypocotyl tissue. Similarly, during the early stress response in Arabidopsis (low oxygen, 3 h) and gray poplar (flooded 5 h), the up-regulation of photosynthesis related genes was observed (Kreuzwieser et al., 2009). In our study, the down-regulation of photosynthesis related genes was observed after 7 days of flood treatment (Supplementary Figure 6). The down-regulation of photosynthesis related genes has also been reported in the leaves of rape seedlings waterlogged for 3 days (Lee et al., 2014). This suggests that the flooding or low-oxygen stress stimulates the expression of photosynthesis related genes during the initial stress response but it is repressed at later stages. Flooding results in reduced photosynthesis manifested by depleted carbon dioxide (CO2) concentration in leaves mediated by the stomatal constraint to the exchange gases (Bailey-Serres et al., 2012). Reduced CO2 levels lead to a depleted electron sink in chloroplasts, which ultimately results in accumulation of photons that are a potential source of ROS. The shrunken electron sink under flooding conditions is also substantiated in our findings and is demonstrated as the repressed expression of eight electron carriers and PS II related genes (Supplementary Figure 6B). The chlorophyll synthesis related genes were found to be repressed under both drought and flooding conditions in this study (Supplementary Figure 3). The lower availability of oxygen (O2) and CO2 under flooding caused by the low gas exchange ratio (Bailey-Serres et al., 2012) may result in decreased photosynthesis efficiency and carbohydrate content. This was consistent with the reduced expression level of starch synthesis genes in the present study (Supplementary Figure 4).
Reduced photosynthesis during prolonged abiotic stresses such as drought and flooding can be correlated with the production of ROS (Klok et al., 2002). High levels of ROS can damage the cell through peroxidation of the lipids, oxidation of proteins and other pathways, and finally the death of the cell (Clement et al., 2008; Wrzaczek et al., 2011). Glutathione S-transferase, one of the important enzyme in the regulation of ROS flux, is featured as a highly enriched gene among the top 50 highly expressed genes under flooding stress in this study, which indicates another adaptive response under the stress conditions (Table 3). However, the opposite (high GST expression in response to drought compared to flooding stress) was reported by Oh and Komatsu (2015).
Drought also has severe effects on the stomatal conductance and low CO2 assimilation. It is often considered to be one of the most prominent reasons for poor photosynthesis. However, it is not the only reason. Another factor is reduced ATP synthesis leading to depleted ribulose 1,5-bisphosphate content (Tezara et al., 1999), which produces a 6C compound when combined with CO2 in the dark reaction (Calvin cycle) of photosynthesis. The repressed expression levels of genes associated with the Calvin cycle in this study (Supplementary Figure 6A) substantiates the decreased photosynthetic capacity mediated by down-regulated dark reaction genes. However, the observed up-regulated electron carrier related gene in drought stressed plants needs to be justified when the Calvin cycle and LHC related genes are down-regulated. This is because under lesser CO2 assimilation in drought stress, the photosynthetically accumulated energy becomes excess for the dark reaction, and to counteract this condition, over reduction of the electron transport chain occurs (Biehler and Fock, 1996).
Transcriptional Response toward Cell Wall Plasticity under Drought and Flooding
Water stress can change the turgor pressure of the plant cell and it responds accordingly, either by tightening or loosening the wall under the extreme water conditions of drought or flooding conditions (Moore et al., 2008). UDP-glucuronic acid is the major precursor of cell wall polysaccharides and comes from two sources: UDP-glucose and myo-inositol (Siddique et al., 2013). The genes involved in both UDP-glucuronic acid synthesis pathways were up-regulated under drought stress (Supplementary Figure 7A). This can cause the accumulation of cell wall precursors and the increase of cell turgor under drought conditions. However, an adverse result was observed under flooding. The GDP-glucuronic acid synthesis genes were reduced under flooding (Supplementary Figure 4). This implies the loosening of the cell wall and stimulation of shoot elongation under flooding (Voesenek et al., 2006).
Regulation of Sucrose and Starch Synthesis under Drought and Flood Conditions
Most research on the effect of drought and flooding on sugar metabolism and starch biosynthesis has demonstrated that the carbohydrate levels are altered in leaves (Lemoine et al., 2013). Under stress conditions, the increase in hexose amounts is associated with the induction of starch hydrolysis (Pelleschi et al., 1997). In agreement with these findings, we observed an increased expression of the hexokinase (Glyma11g10130.1) gene in the drought treatment. The gradual slowing down of starch metabolism is a general mechanism under flooding (Fukao et al., 2011) and drought (Burke, 2007) conditions for rice and cotton, respectively, which reserves the energy source for a prolonged energy supply to maintain cell survival. It is conceivable that starch metabolism during flooding and drought response may also be controlled/gated by the up- and down-regulation of carbohydrate metabolism genes in soybean. One of the prominent processes under sugar metabolism that gets affected in water stressed plants is the sugar efflux rate, which is carried out by proteins coded by sugar transporter genes. They ensure sugar transport from the source (leaves) to the sink (roots and seeds) to counteract the depleted sugar levels under severe conditions such as drought and flooding. Utilizing the available resource generated by Patil et al. (2015), six SWEET family genes were featured among the DEGs identified in this study under drought and flood conditions. The findings suggest that SWEET genes in general are induced in drought conditions and repressed in flooding conditions, which indicate that sucrose efflux genes are activated under drought conditions and repressed under flooding conditions. However, one gene (Glyma14g276210) was found to be up-regulated under both of the conditions. Syed et al. (2015) also identified the up-regulation of soybean SWEET genes under drought stress. The activated gene expression under drought stress could be accounted for by sugar transportation due to an uninterrupted energy supply under such adversity. The down-regulation of three SWEET family genes under flooding stress in the current study is contrary to the findings of Lemoine et al. (2013), where it was suggested that the up-regulated sugar effluxer genes may contribute to sugar homeostasis in a flood tolerant genotype. In this study, the repressed genes might be playing the same role by controlling the efflux of sugar under flooding stress and thus putting a check on normal efflux of sugar. However, this hypothesis needs additional substantiation.
Glycolytic enzymes along with inducers of heat shock proteins are key factors in the early response to flooding and drought in soybean (Nanjo et al., 2011; Oh and Komatsu, 2015), which suggests the significance of the glycolytic pathway in the adaptation to flooding and drought conditions. Oh and Komatsu (2015) reported increased expression of all glycolysis related proteins under both drought and flooding responses. The up-regulated expression of genes coding for the glycolytic pathway enzymes, fructokinase (Glyma16g32530.1) and hexokinase (Glyma11g10130.1), under drought stress can be considered an adaptive response of soybean under drought stress. On the contrary, the transcript levels of glycolytic pathway enzymes were not significantly affected under water deficit conditions in chickpea (Khanna et al., 2014). This suggests a legume specific adaptive variability under similar stress.
Cis-Regulation during Drought and Flooding Treatments
The analysis of cis regulatory elements showed that although the genes between drought up-regulated and flooding down-regulated DEGs shared a common motif, they might have a similar function with opposite regulation under contrasting stress treatments. It has been reported that proteins with up to 67% amino acid sequence similarity may share similar high-affinity binding sequences, and prefer different low-affinity sites. However, even closely related TFs may have distinct DNA-binding profiles (Badis et al., 2009). The positions of the flanking sequence of motif sites also play an important role in achieving regulatory specificity within the TF families (Ciolkowski et al., 2008; Gordan et al., 2013). Therefore, it is possible that this opposite regulation between drought up-regulated genes and flooding down-regulated genes with the same motif could be due to the different TF binding sites or because of the different site specific cofactors with the same affinity for the same TF binding sites, which leads to activation or repression of the target genes.
Conclusion
Plant responses toward abiotic stress are complex and involve several different mechanisms regulated by crosstalk between genes related to hormonal signaling, photosynthesis, respiration, and transcriptional regulations. Comparative transcriptome profiling performed in this study under drought and flooding has provided an opportunity to evaluate the categorized molecular responses. Besides having contrasting stress conditions, many common molecular mechanisms were observed to be involved under drought and flooding. A notable example is the ABA related TFs that are up-regulated under both drought and flood conditions. ABA is considered as the stress related hormone and seems to be a key regulator under both conditions. Unlike rice and Arabidopsis, the ABA mediated stress response in soybean under flooding stress is independent of the SUB1 pathway. Another important aspect highlighted here is the reduced overall metabolic activity under flooding stress, which supports the quiescence rather than the escape mechanism.
The resource of DEGs and also the validated expression of key genes featured in various metabolic pathways under drought and flooding stress will be helpful in understanding the complex mechanisms of stress tolerance. The study further substantiates and elaborates the affected photosynthetic capacity and sugar metabolism under these stresses in soybean. In addition, an in-depth insight into the TFs associated with drought and flooding responsive genes points toward a more complex and intricate gene regulatory network. In particular, genes encoding for LHC, sugar transporters, and the cell wall composition were identified to play a crucial role in combating stress. These genes could be targeted as potential candidates for functional validation and can also be considered for study in other legumes affected by similar stresses.
Author Contributions
WC, QY, GP, and GA are all equal contributing authors for this manuscript. WC, BV, HTN designed the experiment. QY, GP, GA, RD, LL, DX, YCA, performed bioinformatics analysis. WC, GP, GA, and RD performed data mining, analysis and interpretation. RD, GP, and GA contributed to drafting the manuscript. WC, BW, SP, and LS performed greenhouse experiment, tissue collection, RNA isolation and qRT-PCR analysis. YW constructed RNAseq library. RKV and HTN conceived the study and edited the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge the funding support for this study from United Soybean Board and Missouri Soybean Merchandising Council. The authors acknowledge Mr. Rick Meyer for his help in conducting cis-element analysis. The authors also would like to thank Theresa A. Musket for editing the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01044
Supplementary Figure 1. RNA-Seq analysis workflow for drought and flood stressed soybean leaf samples.
Supplementary Figure 2. Scatter plots of differentially expressed genes under (A) drought (B) flooding stress.
Supplementary Figure 3. Tetrapyrrole under (A) drought and (B) flooding. Under flooding stress, this pathway is down-regulated.
Supplementary Figure 4. The sucrose starch synthesis pathway under (A) drought and (B) flooding.
Supplementary Figure 5. DEGs categorized in different metabolic functions. Up- and down-regulation of genes under (A) drought and (B) flooding are represented by colored squares.
Supplementary Figure 6. DEGs categorized and mapped in primary metabolic pathways under (A) drought and (B) flooding conditions in soybean. The log2 fold change value of a DEG is represented by colored squares.
Supplementary Figure 7. The cell wall precursors synthesis pathway under (A) drought and (B) flooding.
Supplementary Table 1. List of genes with confident expression in drought stressed leaf tissue compared to non-stressed control tissue. (B) List of genes with confident expression in flooding stressed leaf tissue compared to non-stressed control tissue.
Supplementary Table 2. List of DEGs in, (A) drought, (B) flooding stressed leaf tissue compared to non-stressed control tissue. (C) List of DEGs in flooding stressed leaf tissue compared to drought stressed leaf tissue.
Supplementary Table 3. Significant GO terms among (A) up-regulated genes under drought; (B) down-regulated genes under drought; (C) up-regulated genes under flooding, and (D) down-regulated genes under flooding conditions.
Supplementary Table 4. (A) Overrepresented motifs from the significantly changed flooding and drought gene sets. (B) Common motifs shared by flooding and drought response genes.
Supplementary Table 5. Primers used in the qRT-PCR analysis.
References
Agarwal, G., Choudhary, D., Singh, V. P., and Arora, A. (2012). Role of ethylene receptors during senescence and ripening in horticultural crops. Plant Signal. Behav. 7, 827–846. doi: 10.4161/psb.20321
Ahmed, F., Rafii, M. Y., Ismail, M. R., Juraimi, A. S., Rahim, H. A., Asfaliza, R., et al. (2013). Waterlogging tolerance of crops: breeding, mechanism of tolerance, molecular approaches, and future prospects. BioMed Res. Int. 2013:963525. doi: 10.1155/2013/963525
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Badis, G., Berger, M. F., Philippakis, A. A., Talukder, S., Gehrke, A. R., Jaeger, S. A., et al. (2009). Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723. doi: 10.1126/science.1162327
Bailey-Serres, J., Lee, S. C., and Brinton, E. (2012). Waterproofing crops: effective flooding survival strategies. Plant Physiol. 160, 1698–1709. doi: 10.1104/pp.112.208173
Beynon, E. R., Symons, Z. C., Jackson, R. G., Lorenz, A., Rylott, E. L., and Bruce, N. C. (2009). The role of oxophytodienoate reductases in the detoxification of the explosive 2, 4, 6-trinitrotoluene by Arabidopsis. Plant Physiol. 151, 253–261. doi: 10.1104/pp.109.141598
Bhargava, A., Clabaugh, I., To, J. P., Maxwell, B. B., Chiang, Y. H., Schaller, G. E., et al. (2013). Identification of cytokinin-responsive genes using microarray meta-analysis and RNA-Seq in Arabidopsis. Plant Physiol. 162, 272–294. doi: 10.1104/pp.113.217026
Biehler, K., and Fock, H. (1996). Evidence for the contribution of the Mehler-peroxidase reaction in dissipating excess electrons in drought-stressed wheat. Plant Physiol. 112, 265–272.
Burke, J. J. (2007). Evaluation of source leaf responses to water-deficit stresses in cotton using a novel stress bioassay. Plant Physiol. 143, 108–121. doi: 10.1104/pp.106.087783
Chen, W., He, S., Liu, D., Patil, G. B., Zhai, H., Wang, F., et al. (2015). A Sweetpotato geranylgeranyl pyrophosphate synthase gene, IbGGPS, increases carotenoid content and enhances osmotic stress tolerance in Arabidopsis thaliana. PLoS ONE 16:e0137623. doi: 10.1371/journal.pone.0137623
Chinnusamy, V., Schumaker, K., and Zhu, J. K. (2004). Molecular genetic perspectives on cross-talk and specificity in abiotic stress signaling in plants. J. Exp. Bot. 55, 225–236. doi: 10.1093/jxb/erh005
Ciolkowski, I., Wanke, D., Birkenbihl, R. P., and Somssich, I. E. (2008). Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function. Plant Mol. Biol. 68, 81–92. doi: 10.1007/s11103-008-9353-1
Clement, M., Lambert, A., Herouart, D., and Boncompagni, E. (2008). Identification of new up-regulated genes under drought stress in soybean nodules. Gene 426, 15–22. doi: 10.1016/j.gene.2008.08.016
Cruz De Carvalho, M. H. (2008). Drought stress and reactive oxygen species: production, scavenging and signaling. Plant Signal. Behav. 3, 156–165. doi: 10.4161/psb.3.3.5536
Dat, J. F., Capelli, N., Folzer, H., Bourgeade, P., and Badot, P. M. (2004). Sensing and signaling during plant flooding. Plant Physiol. Biochem. 42, 273–282. doi: 10.1016/j.plaphy.2004.02.003
Deshmukh, R., Sonah, H., Patil, G., Chen, W., Prince, S., Mutava, R., et al. (2014). Integrating omic approaches for abiotic stress tolerance in soybean. Front. Plant Sci. 5:244. doi: 10.3389/fpls.2014.00244
Dubos, C., Stracke, R., Grotewold, E., Weisshaar, B., Martin, C., and Lepiniec, L. (2010). MYB transcription factors in Arabidopsis. Trends Plant Sci. 15, 573–581. doi: 10.1016/j.tplants.2010.06.005
Else, M. A., Davies, W. J., Malone, M., and Jackson, M. B. (1995). A negative hydraulic message from oxygen-deficient roots of tomato plants? (influence of soil flooding on leaf water potential, leaf expansion, and synchrony between stomatal conductance and root hydraulic conductivity). Plant Physiol. 109, 1017–1024.
Foster, R., Izawa, T., and Chua, N. H. (1994). Plant basic leucine zipper proteins gather at ACGT elements. EMBO J. 8, 192–200.
Foyer, C. H., and Shigeoka, S. (2011). Understanding oxidative stress and antioxidant functions to enhance photosynthesis. Plant Physiol. 155, 93–100. doi: 10.1104/pp.110.166181
Fukao, T., Yeung, E., and Bailey-Serres, J. (2011). The submergence tolerance regulator SUB1A mediates crosstalk between submergence and drought tolerance in rice. Plant Cell 23, 412–427. doi: 10.1105/tpc.110.080325
Gordan, R., Shen, N., Dror, I., Zhou, T., Horton, J., Rohs, R., et al. (2013). Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 3, 1093–1104. doi: 10.1016/j.celrep.2013.03.014
Hao, Y. J., Wei, W., Song, Q. X., Chen, H. W., Zhang, Y. Q., Wang, F., et al. (2011). Soybean NAC transcription factors promote abiotic stress tolerance and lateral root formation in transgenic plants. Plant J. 68, 302–313. doi: 10.1111/j.1365-313X.2011.04687.x
Hattori, T., Totsuka, M., Hobo, T., Kagaya, Y., and Yamamoto-Toyoda, A. (2002). Experimentally determined sequence requirement of ACGT-containing abscisic acid response element. Plant Cell Physiol. 43, 136–140. doi: 10.1093/pcp/pcf014
Higo, K., Ugawa, Y., Iwamoto, M., and Korenaga, T. (1999). Plant cis-acting regulatory DNA elements (PLACE) database. Nucleic Acids Res. 27, 297–300. doi: 10.1093/nar/27.1.297
Izawa, T., Foster, R., and Chua, N. H. (1993). Plant bZIP protein DNA binding specificity. J. Mol. Biol. 230, 1131–1144. doi: 10.1006/jmbi.1993.1230
Jin, J., Zhang, H., Kong, L., Gao, G., and Luo, J. (2014). PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–1187. doi: 10.1093/nar/gkt1016
Khanna, S. M., Taxak, P. C., Jain, P. K., Saini, R., and Srinivasan, R. (2014). Glycolytic enzyme activities and gene expression in Cicer arietinum exposed to water-deficit stress. Appl. Biochem. Biotechnol. 173, 2241–2253. doi: 10.1007/s12010-014-1028-6
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Kim, T. H. (2014). Mechanism of ABA signal transduction: agricultural highlights for improving drought tolerance. J. Plant Biol. 57, 1. doi: 10.1007/s12374-014-0901-8
Klok, E. J., Wilson, I. W., Wilson, D., Chapman, S. C., Ewing, R. M., Somerville, S. C., et al. (2002). Expression profile analysis of the low-oxygen response in Arabidopsis root cultures. Plant Cell 14, 2481–2494. doi: 10.1105/tpc.004747
Knight, L. I., Rose, R. C., and Crocker, W. (1910). Effect of various gases and vapors upon etiolated seedlings of the sweet pea. Science 31, 635–636.
Koberg, M., Abu-Much, R., and Gedanken, A. (2011). Optimization of bio-diesel production from soybean and wastes of cooked oil: combining dielectric microwave irradiation and a SrO catalyst. Bioresour. Technol. 102, 1073–1078. doi: 10.1016/j.biortech.2010.08.055
Komatsu, S., Yamamoto, R., Nanjo, Y., Mikami, Y., Yunokawa, H., and Sakata, K. (2009). A comprehensive analysis of the soybean genes and proteins expressed under flooding stress using transcriptome and proteome techniques. J Proteom. Res. 8, 4766–4778. doi: 10.1021/pr900460x
Kosegarten, H., Judel, G. K., and Mengel, K. (1988). Activity of UDP-glucose glucosyl transferase and its role in the carbohydrate metabolism of wheat caryopsis. J. Plant Physiol. 133, 126–128. doi: 10.1016/S0176-1617(88)80100-7
Krasensky, J., and Jonak, C. (2012). Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 63, 1593–1608. doi: 10.1093/jxb/err460
Kreuzwieser, J., Hauberg, J., Howell, K. A., Carroll, A., Rennenberg, H., Millar, A. H., et al. (2009). Differential response of gray poplar leaves and roots underpins stress adaptation during hypoxia. Plant Physiol. 149, 461–473. doi: 10.1104/pp.108.125989
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Le, D. T., Nishiyama, R., Watanabe, Y., Tanaka, M., Seki, M., Ham Le, H., et al. (2012). Differential gene expression in soybean leaf tissues at late developmental stages under drought stress revealed by genome-wide transcriptome analysis. PLoS ONE 7:e49522. doi: 10.1371/journal.pone.0049522
Lee, Y. H., Kim, K. S., Jang, Y. S., Hwang, J. H., Lee, D. H., and Choi, I. H. (2014). Global gene expression responses to waterlogging in leaves of rape seedlings. Plant Cell Rep. 33, 289–299. doi: 10.1007/s00299-013-1529-8
Lemoine, R., La Camera, S., Atanassova, R., Dedaldechamp, F., Allario, T., Pourtau, N., et al. (2013). Source-to-sink transport of sugar and regulation by environmental factors. Front. Plant Sci. 4:272. doi: 10.3389/fpls.2013.00272
Maruyama, K., Todaka, D., Mizoi, J., Yoshida, T., Kidokoro, S., Matsukura, S., et al. (2012). Identification of cis-acting promoter elements in cold- and dehydration-induced transcriptional pathways in Arabidopsis, rice, and soybean. DNA Res. 19, 37–49. doi: 10.1093/dnares/dsr040
Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2012). AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 1819, 86–96. doi: 10.1016/j.bbagrm.2011.08.004
Moore, J. P., Vicre-Gibouin, M., Farrant, J. M., and Driouich, A. (2008). Adaptations of higher plant cell walls to water loss: drought vs. desiccation. Physiol. Plant. 134, 237–245. doi: 10.1111/j.1399-3054.2008.01134.x
Nakashima, K., Takasaki, H., Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2012). NAC transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 1819, 97–103. doi: 10.1016/j.bbagrm.2011.10.005
Nakatsuka, A., Murachi, S., Okunishi, H., Shiomi, S., Nakano, R., Kubo, Y., et al. (1998). Differential expression and internal feedback regulation of 1-aminocyclopropane-1-carboxylate synthase, 1-aminocyclopropane-1-carboxylate oxidase, and ethylene receptor genes in tomato fruit during development and ripening. Plant Physiol. 118, 1295–1305. doi: 10.1104/pp.118.4.1295
Nanjo, Y., Maruyama, K., Yasue, H., Yamaguchi-Shinozaki, K., Shinozaki, K., and Komatsu, S. (2011). Transcriptional responses to flooding stress in roots including hypocotyl of soybean seedlings. Plant Mol. Biol. 77, 129–144. doi: 10.1007/s11103-011-9799-4
Oh, M., and Komatsu, S. (2015). Characterization of proteins in soybean roots under flooding and drought stresses. J. Proteomics 114, 161–181. doi: 10.1016/j.jprot.2014.11.008
Park, S. Y., Fung, P., Nishimura, N., Jensen, D. R., Fujii, H., Zhao, Y., et al. (2009). Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 324, 1068–1071. doi: 10.1126/science.1173041
Patil, G., and Nicander, B. (2013). Identification of two additional members of the tRNA isopentenyltransferase family in Physcomitrella patens. Plant Mol. Biol. 82, 417–426. doi: 10.1007/s11103-013-0072-x
Patil, G., Do, T., Vuong, T. D., Valliyodan, B., Lee, J. D., Chaudhary, J., et al. (2016). Genomic-assisted haplotype analysis and the development of high-throughput SNP markers for salinity tolerance in soybean. Sci. Rep. 19:19199. doi: 10.1038/srep19199
Patil, G., Valliyodan, B., Deshmukh, R., Prince, S., Nicander, B., Zhao, M., et al. (2015). Soybean (Glycine max) SWEET gene family: insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis. BMC Genomics 16, 520–535. doi: 10.1186/s12864-015-1730-y
Peleg, Z., and Blumwald, E. (2011). Hormone balance and abiotic stress tolerance in crop plants. Curr. Opin. Plant Biol. 14, 290–295. doi: 10.1016/j.pbi.2011.02.001
Pelleschi, S., Rocher, J. P., and Prioul, J. L. (1997). Effect of water restriction on carbohydrate metabolism and photosynthesis in mature maize leaves. Plant Cell Environ. 20, 493–503. doi: 10.1046/j.1365-3040.1997.d01-89.x
Perata, P., Armstrong, W., and Voesenek, L. A. (2011). Plants and flooding stress. New Phytol. 190, 269–273. doi: 10.1111/j.1469-8137.2011.03702.x
Priest, H. D., Filichkin, S. A., and Mockler, T. C. (2009). Cis-regulatory elements in plant cell signaling. Curr. Opin. Plant Biol. 12, 643–649. doi: 10.1016/j.pbi.2009.07.016
Saika, H., Okamoto, M., Miyoshi, K., Kushiro, T., Shinoda, S., Jikumaru, Y., et al. (2007). Ethylene promotes submergence-induced expression of OsABA8ox1, a gene that encodes ABA 8'-hydroxylase in rice. Plant Cell Physiol. 48, 287–298. doi: 10.1093/pcp/pcm003
Severin, A. J., Woody, J. L., Bolon, Y. T., Joseph, B., Diers, B. W., Farmer, A. D., et al. (2010). RNA-Seq Atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol. 10:160. doi: 10.1186/1471-2229-10-160
Siddique, S., Endres, S., Sobczak, M., Radakovic, Z. S., Fragner, L., Grundler, F. M., et al. (2013). Myo-inositol oxygenase is important for the removal of excess myo-inositol from syncytia induced by Heterodera schachtii in Arabidopsis roots. New Phytol. 201, 476–485. doi: 10.1111/nph.12535
Steffens, B., Steffen-Heins, A., and Sauter, M. (2013). Reactive oxygen species mediate growth and death in submerged plants. Front. Plant Sci. 4:179. doi: 10.3389/fpls.2013.00179
Suzuki, M., Ketterling, M. G., and Mccarty, D. R. (2005). Quantitative statistical analysis of cis-regulatory sequences in ABA/VP1- and CBF/DREB1-regulated genes of Arabidopsis. Plant Physiol. 139, 437–447. doi: 10.1104/pp.104.058412
Syed, N. H., Prince, S. J., Mutava, R. N., Patil, G., Li, S., Chen, W., et al. (2015). Core clock, SUB1, and ABAR genes mediate flooding and drought responses via alternative splicing in soybean. J. Exp. Bot. 66, 7129–7149. doi: 10.1093/jxb/erv407
Tabaeizadeh, Z. (1998). Drought-induced responses in plant cells. Int. Rev. Cytol. 182, 193–247. doi: 10.1016/S0074-7696(08)62170-1
Tezara, W., Mitchell, V. J., Driscoll, S. D., and Lawlor, D. W. (1999). Water stress inhibits plant photosynthesis by decreasing coupling factor and ATP. Nature 401, 914–917. doi: 10.1038/44842
Trapnell, C., Hendrickson, D. G., Sauvageau, M., Goff, L., Rinn, J. L., and Pachter, L. (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31, 46–53. doi: 10.1038/nbt.2450
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Usadel, B., Poree, F., Nagel, A., Lohse, M., Czedik-Eysenberg, A., and Stitt, M. (2009). A guide to using MapMan to visualize and compare Omics data in plants: a case study in the crop species, Maize. Plant Cell Environ. 32, 1211–1229. doi: 10.1111/j.1365-3040.2009.01978.x
Valliyodan, B., and Nguyen, H. T. (2006). Understanding regulatory networks and engineering for enhanced drought tolerance in plants. Curr. Opin. Plant Biol. 9, 189–195. doi: 10.1016/j.pbi.2006.01.019
Varshney, R. K., Nayak, S. N., May, G. D., and Jackson, S. A. (2009). Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 27, 522–530. doi: 10.1016/j.tibtech.2009.05.006
Vick, B. A., and Zimmerman, D. C. (1984). Biosynthesis of jasmonic acid by several plant species. Plant Physiol. 75, 458–461. doi: 10.1104/pp.75.2.458
Vidal, R. O., Do, L. C., Mondego, J. M., Pereira, G. A., and Carazzolle, M. F. (2012). Identification of SNPs in RNA-seq data of two cultivars of Glycine max (soybean) differing in drought resistance. Genet. Mol. Biol. 35, 331–334. doi: 10.1590/S1415-47572012000200014
Voesenek, L. A., Colmer, T. D., Pierik, R., Millenaar, F. F., and Peeters, A. J. (2006). How plants cope with complete submergence. New Phytol. 170, 213–226. doi: 10.1111/j.1469-8137.2006.01692.x
Wang, D., Pan, Y., Zhao, X., Zhu, L., Fu, B., and Li, Z. (2011). Genome-wide temporal-spatial gene expression profiling of drought responsiveness in rice. BMC Genomics 12:149. doi: 10.1186/1471-2164-12-149
Wrzaczek, M., Vainonen, J. P., Gauthier, A., Overmyer, K., and Kangasjärvi, J. (2011). “Reactive oxygen in abiotic stress perception - from genes to proteins,” in Abiotic Stress Response in Plants, eds A. Shanker and B. Venkateswarlu (Rijeka: InTech), 27–55.
Xing, S. F., Miao, J., Li, S. A., Qin, G. J., Tang, S., Li, H. N., et al. (2010). Disruption of the 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR) gene results in albino, dwarf and defects in trichome initiation and stomata closure in Arabidopsis. Cell Res. 20, 688–700. doi: 10.1038/cr.2010.54
Xiong, L., and Zhu, J. K. (2002). Molecular and genetic aspects of plant responses to osmotic stress. Plant Cell Environ. 25, 131–139. doi: 10.1046/j.1365-3040.2002.00782.x
Keywords: RNA-Seq, soybean, drought, flooding, stress, gene expression
Citation: Chen W, Yao Q, Patil GB, Agarwal G, Deshmukh RK, Lin L, Wang B, Wang Y, Prince SJ, Song L, Xu D, An YC, Valliyodan B, Varshney RK and Nguyen HT (2016) Identification and Comparative Analysis of Differential Gene Expression in Soybean Leaf Tissue under Drought and Flooding Stress Revealed by RNA-Seq. Front. Plant Sci. 7:1044. doi: 10.3389/fpls.2016.01044
Received: 11 May 2016; Accepted: 04 July 2016;
Published: 19 July 2016.
Edited by:
Shabir Hussain Wani, Sher-e-Kashmir University of Agricultural Sciences and Technology of Kashmir, IndiaReviewed by:
Biswapriya Biswavas Misra, University of Florida, USATaras P. Pasternak, Institute of Biology II, Germany
Copyright © 2016 Chen, Yao, Patil, Agarwal, Deshmukh, Lin, Wang, Wang, Prince, Song, Xu, An, Valliyodan, Varshney and Nguyen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Henry T. Nguyen, bmd1eWVuaGVucnlAbWlzc291cmkuZWR1
†These authors have contributed equally to this work.