 Arthur Zwaenepoel1,2,3
Arthur Zwaenepoel1,2,3 Tim Diels1,2,3
Tim Diels1,2,3 David Amar4
David Amar4 Thomas Van Parys1,2,3
Thomas Van Parys1,2,3 Ron Shamir5Yves Van de Peer1,2,3,6*
Ron Shamir5Yves Van de Peer1,2,3,6* Oren Tzfadia1,2,3*
Oren Tzfadia1,2,3*- 1Department of Plant Biotechnology and Bioinformatics, Ghent University, Ghent, Belgium
- 2VIB Center for Plant Systems Biology, Ghent, Belgium
- 3Bioinformatics Institute Ghent, Ghent University, Ghent, Belgium
- 4Stanford Center for Inherited Cardiovascular Disease, Stanford University, Stanford, CA, United States
- 5Blavatnik School of Computer Science, Tel-Aviv University, Tel-Aviv, Israel
- 6Genomics Research Institute, University of Pretoria, Pretoria, South Africa
Recent times have seen an enormous growth of “omics” data, of which high-throughput gene expression data are arguably the most important from a functional perspective. Despite huge improvements in computational techniques for the functional classification of gene sequences, common similarity-based methods often fall short of providing full and reliable functional information. Recently, the combination of comparative genomics with approaches in functional genomics has received considerable interest for gene function analysis, leveraging both gene expression based guilt-by-association methods and annotation efforts in closely related model organisms. Besides the identification of missing genes in pathways, these methods also typically enable the discovery of biological regulators (i.e., transcription factors or signaling genes). A previously built guilt-by-association method is MORPH, which was proven to be an efficient algorithm that performs particularly well in identifying and prioritizing missing genes in plant metabolic pathways. Here, we present MorphDB, a resource where MORPH-based candidate genes for large-scale functional annotations (Gene Ontology, MapMan bins) are integrated across multiple plant species. Besides a gene centric query utility, we present a comparative network approach that enables researchers to efficiently browse MORPH predictions across functional gene sets and species, facilitating efficient gene discovery and candidate gene prioritization. MorphDB is available at http://bioinformatics.psb.ugent.be/webtools/morphdb/morphDB/index/. We also provide a toolkit, named “MORPH bulk” (https://github.com/arzwa/morph-bulk), for running MORPH in bulk mode on novel data sets, enabling researchers to apply MORPH to their own species of interest.
Introduction
Groups of genes involved in a common biological process are often defined as pathways, which are traditionally studied as if they were isolated groups. However, pathway boundaries are inherently fuzzy which greatly compromises their systematic delineation. In plants, the understanding of secondary metabolism and stress regulated pathways is of paramount importance and even though these pathways have been studied extensively, discovering missing genes and understanding the regulatory interrelations among them remains a fundamental challenge. Moreover, despite more than two decades of functional genomics research, the functions of most plant genes remain unknown. These problems are exacerbated in newly sequenced genomes and non-model organisms (Rhee and Mutwil, 2014). Sequence similarity based tools such as Blast2GO (Conesa et al., 2005; Conesa and Götz, 2008), BlastKOALA (Kanehisa et al., 2016), PlantCyc's EP2P (Schläpfer et al., 2017) and InterProScan (Zdobnov and Apweiler, 2001; Jones et al., 2014), are often used to provide a first clue about the function of a gene in a newly sequenced and annotated genome. Other comparative genomics methods aim to leverage annotation efforts in model organisms, typically by utilizing clustering analysis, using e.g., OrthoMCL (Li et al., 2003) or OrthoFinder (Emms and Kelly, 2015). After the delineation of groups of homologous genes, annotations are transferred between orthologs under the assumption that evolutionary conservation implies a conserved function.
A complementary approach for gene function prediction is to use “omics” data (e.g., transcriptomics and proteomics) within an integrative analysis pipeline that builds on the guilt-by-association (GBA) principle. GBA involves inferring putative gene functions for unknown genes from genes with known functions that show similar behavior across different experimental conditions or data sets. For example, co-expression based GBA with genes from known Gene Ontology (GO) terms has been shown to be ubiquitously applicable across the transcriptome of different species (Wolfe et al., 2005). Because of the demonstrated general applicability of the GBA principle and the fact that transcriptomic data is the most straightforward 'omics' data to gather, there is an increasing usage of co-expression networks for candidate gene prioritization in the plant science community (Rhee and Mutwil, 2014; Serin et al., 2016).
A related methodology for in-depth analysis of gene functions is comparative transcriptomics, in which evolutionary relationships between genes are used to integrate expression data across species (Movahedi et al., 2011, 2012; Hansen et al., 2014). Such methods often use integrative network approaches to allow discovery of conserved co-expression modules (Zarrineh et al., 2011) across multiple species, again possibly leveraging knowledge from model to non-model organisms. These networks can often unveil missing pathway genes and regulators, as they naturally cope with the fuzzy nature of pathway boundaries while incorporating evolutionary relationships that can serve as constraints and can discriminate between highly interesting evolutionary conserved candidate genes and potential noise. Indeed, it has been shown that comparative co-expression networks may yield more accurate gene function predictions (Hansen et al., 2014). Some important (comparative) co-expression based tools for gene function analysis are ATTED-II (Aoki et al., 2016), PlaNet (Proost and Mutwil, 2017), CORNET (De Bodt et al., 2010; Van Bel and Coppens, 2017), AraNet (Lee et al., 2015), MORPH (Tzfadia et al., 2012; Amar et al., 2015), and CoExpNetViz (Tzfadia et al., 2016).
While co-expression based methods are extremely relevant for gene function analysis, some important caveats are to be noted. First and foremost, these networks are based on correlation measures which are prone to spurious associations, indirect functional links, and noise (both false positives and false negatives) (Mutwil et al., 2011; Hansen et al., 2014). Therefore, when analyzing large data sets, co-expression networks quickly become dense, limiting their biological interpretability (Usadel et al., 2009; Serin et al., 2016). Second, and associated with these issues, is the problem of reproducibility, as many distinct steps and filtering decisions have to be taken to produce a co-expression network, while a standardized protocol does not exist. Third, these networks are more suitable for inference of biological processes than of molecular functions (Hansen et al., 2014). Fourth, the conditions, tissues and perturbations used in the expression compendium are also of great importance, especially when one is interested in a specific tissue or condition-dependent regulatory processes (Hansen et al., 2014; Serin et al., 2016). Finally, co-expression analysis is expected to be more suitable for genes and processes under strong transcriptional control, whereas they are not well-suited for processes that are mostly controlled at the translational or post-translational level. For example, Kleessen et al. (2013) showed that co-expression based GBA performs much better for primary and secondary metabolism pathways than for hormone and cell wall related biological processes. These reasons also make it desirable to have some estimate of the performance of GBA on a particular process of interest. A distinct and more practical issue is that most available tools (see above) cannot be easily applied to custom data sets or novel species, limiting their usage to a handful of model organisms.
MORPH (MOdule-guided Ranking of candidate PatHway genes) is an algorithm for unveiling missing genes in biological pathways (Tzfadia et al., 2012; Amar et al., 2015) and uses multiple expression datasets and clustering thereof for the prioritization of candidate genes. As with other gene expression based GBA methods, it relies on an input set of 'bait' genes that are associated with the biological process of interest, and uses the expression profiles of these bait genes across conditions to prioritize candidate genes. MORPH uses clustering solutions of the expression data to calculate a module partitioned co-expression metric for each candidate gene with regard to the input bait genes. Based on the input set of bait genes, MORPH selects the optimal expression data—clustering combination to be used for the prioritization of candidate genes. This configuration learning step follows an approach commonly known in machine learning as “model selection” (Guyon et al., 2010). To this end, MORPH uses a leave-one-out cross validation (LOOCV) procedure. For every gene gi in a given bait gene set G, the MORPH algorithm is run with as input bait genes the set G′ defined as the set G with gi left out (i.e., G′ = G \ {gi}). Using this bait gene set, the “self-rank” for gi is determined, defined as the rank assigned to gi by MORPH using the set G′. The self-ranks for every gi are collected and can then be plotted in a self-rank curve, which shows for increasing rank threshold, the proportion of bait genes with a rank higher than the threshold. The area under the self-rank curve (AUSR) can then be used as a model selection metric, as the data set—clustering combination that results in the highest AUSR can be regarded as the one most appropriate to use for GBA based candidate gene prioritization. Interestingly, besides its use for model selection, this AUSR metric can also be used as an estimate of the performance (and relevance) of GBA based methods on a process of interest. While powerful and with proven success, GBA and co-expression based methods in general have not been fully exploited and their real value for plant functional genomics is yet to be explored (Rhee and Mutwil, 2014).
In this paper we extend and improve MORPH. We present a new tool, called MorphDB, which covers more organisms and functional annotations, and provides advanced visualizations that can help researchers in performing genome-wide comparative analyses for a series of model organisms. The new analyses (genome-wide and comparative modes, functional annotation of newly sequenced species) are explored using multiple datasets and functional annotations. Gene-centric and process-centric networks are used for visualization of predicted candidate genes across species and functional categories, which is instrumental in guiding knowledge discovery. Several examples of use cases are shown, illustrating the potential of MorphDB for gene discovery and advancing our understanding of plant gene functions. The tool and the results are accessible via a web interface: http://bioinformatics.psb.ugent.be/webtools/morphdb/morphDB/index/. Besides, we offer a framework for running genome-wide MORPH analyses, called “MORPH bulk” (https://github.com/arzwa/morph-bulk), enabling researchers to perform large scale MORPH analyses on their species and data sets of interest.
Materials and Methods
Expression Data Processing and Functional Annotation Data
The functional annotation data for the model species was retrieved from the PLAZA 3.0 comparative genomics platform (Van Bel et al., 2012). For C. roseus and Z. marina, GO annotations were acquired using InterProScan + InterPro2GO and Blast2GO. The expression data and clustering solutions used for A. thaliana, S. lycopersicum, S. tuberosum, O. sativa, and C. roseus were those already configured for the MORPH web tool (Amar et al., 2015). Expression data for M. truncatula was obtained from the Medicago truncatula gene expression atlas (Benedito et al., 2008). For P. trichocarpa expression data from Shi et al. (2017) was used (GEO accession ID: GSE81077), acquired as count tables. For Z. marina, RNA-Seq data from the original genome project was used (Olsen et al., 2016), obtained as both count and fragments per kilobase of exon per million reads mapped (fpkm) data sets (GEO: GSE67579). All expression data sets were filtered by gene-wise standard deviation, such that ~75% of the genes were retained. All microarray data sets were normalized using quantile normalization (Irizarry et al., 2003), while all RNA-Seq data sets were normalized using the trimmed mean of M-values (TMM) method (Robinson and Oshlack, 2010). Where expression data from previous MORPH releases was used, the original clustering solutions were used as well. Gene expression data sets that were not included in previous MORPH analyses (M. truncatula, P. trichocarpa, and Z. marina) were clustered using CLICK (Sharan and Shamir, 2000). For M. truncatula, a metabolic clustering, with pathways as clusters, was included as well.
MORPH Bulk Runs
To efficiently apply MORPH in a genome wide fashion, a Python3.5 command line interface (CLI) was developed named “morph-bulk.” The morph-bulk CLI uses the highly computationally efficient MORPH C++ implementation (v1.0.6) enabling very fast genome wide MORPH analyses. The morph-bulk CLI enables performing MORPH bulk runs in automatic pipeline mode or step by step, allowing full control over the analysis pipeline. The morph-bulk CLI, including installation instructions and a step-by-step protocol for MORPH bulk analyses, is available at https://github.com/arzwa/morph-bulk. We also provide a Singularity container (Kurtzer et al., 2017) further ensuring portability of the software.
The analysis pipeline proceeds as follows: first, a new species is automatically “added” to MORPH by generating the required configuration files based on the input data (expression matrices and corresponding clustering solutions). The different MORPH jobs are then defined for a given functional annotation (e.g., GO or MapMan) by taking the sets of genes annotated with a specific ontology term for example, and using them as input bait genes. MORPH is then run in bulk on all bait gene sets. Jobs with fewer than 5 genes in all data sets are discarded since co-expression based GBA methods are expected to give unreliable results for few genes, especially in the module partitioned framework used by MORPH. If desired, random MORPH bulk runs can then be performed to perform permutation test based significance assessment. In a random run, for each desired bait set size, n random sets of bait genes are picked from a randomly chosen data set and used in MORPH. The applied range of bait set sizes was from 5 to 30 and the number of random bait sets to analyze for each bait set size was set at 1,000. The corresponding AUSR values are recorded and used to empirically estimate the probability to observe AUSR value for a gene set size. This p-value for a bait gene set of size S with observed AUSR* is then defined as the fraction of occurrences of AUSR scores larger than AUSR* among 1,000 random gene sets of size S. The empirical probability distributions constructed in this fashion are shown in Figure S1. We note that no considerable differences were observed when using random gene sets drawn from the pool of functionally annotated genes vs. random sets drawn from the full genome (Figure S2). For extended annotation purposes, these p-values were corrected for multiple testing using the Benjamini and Hochberg procedure (Benjamini and Hochberg, 1995). After the main analysis, the results are summarized and extended functional annotations are generated. If desired, a Resource Description Framework (RDF) graph for MorphDB can be generated using the same CLI.
The MorphDB Database and Web Tool
MORPH bulk run data was parsed into an RDF graph using the rdflib (v4.2.2) Python package. The main objects (subjects) in the RDF graph are genes, gene sets (GO/MapMan terms), gene families and scores of a gene for a specific gene set term. The full list of predicates and objects is included in the about section of the MorphDB website. The RDF graph as constructed using rdflib was serialized to Turtle format [W3C recommendation (W3C, 2014)] and loaded in a triple store using Apache Jena (The Apache Software Foundation, 2011), queryable with the SPARQL query language (W3C, 2008). For in-browser network construction and visualization, the Cytoscape.js JavaScript library was used (Franz et al., 2015).
Results
MORPH Bulk Mode
The MORPH algorithm for candidate gene prediction uses as input a set of genes known to belong to a specific pathway or to have a common function (this set is referred to as the bait set) and aims to propose and rank additional genes of the same function or pathway. Expression profiles, their clustering solutions, and biological networks are used in the prediction. We applied MOPRH in a genome-wide fashion, hereafter called MORPH bulk mode, on six important model organisms: Arabidopsis thaliana, Medicago truncatula, Solanum lycopersicum, Solanum tuberosum, Oryza sativa, and Populus trichocarpa, and two non-model organisms, the recently sequenced seagrass Zostera marina (Olsen et al., 2016) and the medicinal plant Catharanthus roseus. In genome-wide runs, we provide as input to MORPH a genome-scale functional annotation as acquired from public repositories or popular software tools (e.g., Blast2GO or InterProScan). As bait gene sets, Gene Ontology (GO) annotations (Ashburner et al., 2000) were used, as well as MapMan annotations (Thimm et al., 2004) when available. MORPH uses a machine learning approach for performance estimation based on LOOCV and reports the area under the self-rank curve (AUSR) as a metric for the performance on a specific bait gene set. The AUSR ranges from 0 to 1 (perfect score), but its reliability is strongly dependent on the size of the bait gene set. Smaller sets are more likely to have larger AUSR values by chance. Therefore, for each bait gene set analyzed with MORPH, empirical p-values were computed using a permutation test. For each candidate, MORPH calculates a within-module Pearson correlation co-expression metric and subsequently converts these values to z-scores, which enables a common ranking across different modules. This z-score can then be used to rank and select relevant candidates.
Investigating the overall performance of MORPH illustrates its potential for gene function prioritization. Table 1 shows the number of significant bait sets for different p-value thresholds. MORPH performed best for A. thaliana, with 1,985 (66%) of the 3,005 GO terms and 279 (64%) of the 467 MapMan categories showing significant AUSR scores (p < 0.05). For M. truncatula, O. sativa and P. trichocarpa, a considerably smaller fraction of the analyzed gene sets showed significant AUSR scores (43, 33, and 24% respectively). For the Solanaceae species included (S. lycopersicum and S. tuberosum), fewer bait sets had good scores, probably due to a more limited GO annotation.
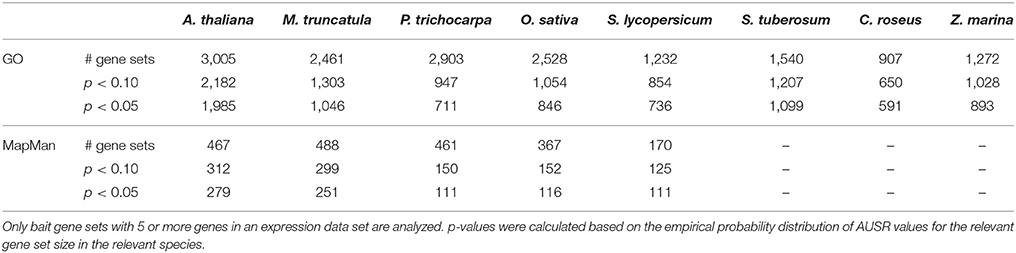
Table 1. Number of bait gene sets (GO/MapMan) successfully analyzed with MORPH in bulk mode.
The performance of MORPH strongly depends on the available functional annotation, the expression data, and the clustering solutions. Interestingly, performance seems not to differ dramatically among different GO sub-ontologies, namely Biological Process (BP), Cellular Compartment (CC) and Molecular Function (MF), as shown in Figure 1, indicating that module-partitioned co-expression is manifest in every sub-ontology. However, a closer look reveals that, with the exception of O. sativa and P. trichocarpa, CC categories seem to systematically have higher fractions of significantly scoring gene sets. This observation may be explained by the fact that in all species the average gene set size is larger for CC GO categories than for BP or MF categories, e.g., 76 (CC) compared to 53 (BP) and 58 (MF) for A. thaliana, or 64 (CC) compared to 25 (BP) and 26 (MF) for M. truncatula. For larger gene sets, the AUSR may be significant even when the underlying co-expression strength is moderate. This can occur when a large set of bait genes that shows moderate overall co-expression contains some strongly co-expressed clusters of genes, which is a scenario that is directly addressed by the MORPH algorithm. While the BP ontology is probably the most directly relevant for candidate gene prediction, the other ontologies are also informative and hence included in MorphDB.
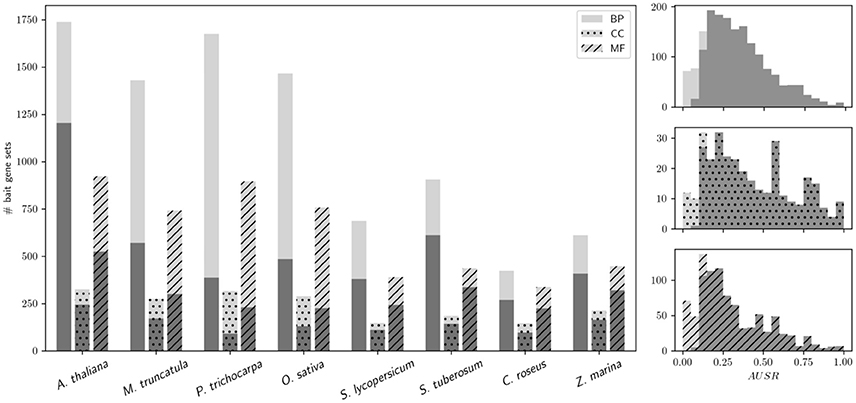
Figure 1. Performance of MORPH in bulk mode for different GO sub-ontologies. (Left) MORPH bulk results partitioned by sub-ontology (BP, CC, and MF) for the different species under study are shown. Light gray: the total number of analyzed bait gene sets with MORPH; dark gray: the number of significantly scoring (p < 0.05) bait gene sets. (Right) Histograms of AUSR values for different sub-ontologies in A. thaliana (using the same colors as in the left plot).
Extending MORPH to Non-model Organisms
We used MORPH in bulk mode to predict putative gene functions for two non-model organisms, namely C. roseus and Z. marina. As in-depth functional annotations are not available for these organisms, we used established sequence-based algorithms to obtain predicted GO terms. This is common practice when analyzing newly sequenced organisms and non-model organisms (Amar et al., 2014). Using the predicted GO terms as bait sets, we applied MORPH using 5% FDR-corrected p-values for determining sets with significant predictions. For significant GO terms, we selected the top genes whose co-expression z-score was larger than the 97.5% percentile of the theoretical null distribution (z > 1.96). Our analysis resulted in 521 GO terms that could be assigned to 18,842 genes for C. roseus. Of these genes, 11,738 are currently unannotated, resulting in a considerable improvement of the primary GO annotation for these specific z-score cut-offs. For Z. marina, 794 GO categories were significant, which could be assigned to 13,343 genes using the same procedure as for C. roseus. Of these, 3,060 genes had no GO terms assigned previously, again showing the potential of this approach for improving automatically generated GO annotations with putative gene functions.
The analysis above may result in a high false positive rate and the resulting functional predictions are to be taken as a set of possible annotations that should be further tested. Nevertheless, we here show some specific examples of how these results can be used for generating biological hypotheses for C. roseus, for which the community is particularly interested in specialized metabolism pathways. C. roseus is an important medicinal plant that serves as a source for the potent indole alkaloid chemotherapeutic compounds vinblastine and vincristine (Almagro et al., 2015). Mining basic functional annotation data will often not suffice for finding interesting unknown regulators and pathway genes, while constructing co-expression networks and analyzing them can become very laborious and complicated. Mining functional annotations extended by MORPH offers an alternative. For example, considering transcription factors that are assigned by MORPH to aromatic compound biosynthetic processes (GO:0019438 and similar categories), several interesting candidates are suggested. Three top-scoring candidates (z > 3.0) are shown in Table 2. These genes also had high scores for other relevant GO terms, such as flavonoid and quercetin (also a flavonoid) metabolism related terms as well as response to wounding and other more general metabolism related terms (O-methyltransferase, NAD binding, Thiamine pyrophosphate (TPP) binding and malate metabolism). Flavonoids, as well as other phenylpropanoid compounds, are well known for their roles in plant defense (Falcone Ferreyra et al., 2012; Tohge et al., 2013). Plant defense responses are well known to correlate with enhanced production of many specialized metabolites, and such responses have been described for C. roseus as well (Menke et al., 1999; Roepke et al., 2010). This simple example demonstrates how extended functional annotations acquired by MORPH can provide a valuable starting point for identifying interesting candidates for pathways of interest in non-model genomes.
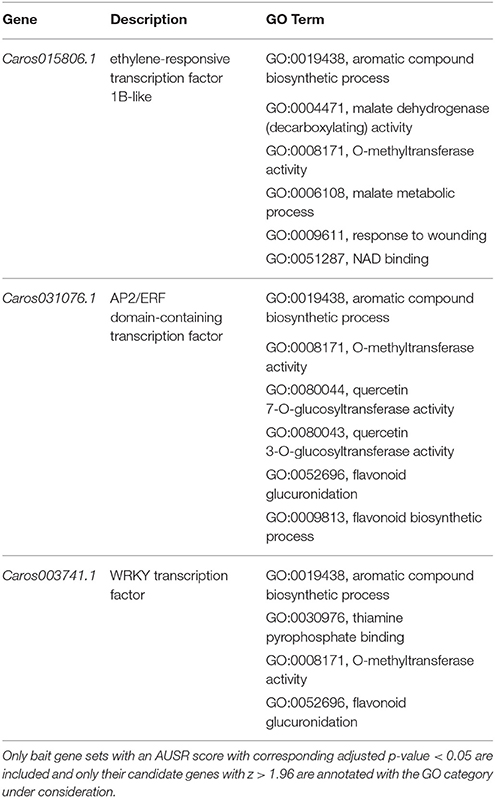
Table 2. Subset of transcription factors in C. roseus associated with GO:0019438 (aromatic compound biosynthetic process) by MORPH and their respective other predicted GO terms.
MorphDB
We created a web-based tool named MorphDB that provides access to MORPH's predictions for the six important model organisms discussed above, as well as for C. roseus and Z. marina. For each species, MorphDB stores the top 100 candidates with z-scores that exceed the 90% percentile of the theoretical null distribution (z > 1.28) for all gene sets with empirical p-value < 0.10. The primary goal of MorphDB is to integrate the MORPH candidate gene predictions across species using orthogroup data as retrieved from the PLAZA comparative genomics platform (Proost et al., 2009; Van Bel et al., 2012). Both candidates and bait genes are linked to their respective homologous candidates and to bait genes in the other species in the MorphDB database. MorphDB allows querying in a gene centric manner, enabling users to provide a set of genes of interest and view the GO categories or MapMan terms that were predicted for it by MORPH. Moreover, gene sets can also be queried and visualized in a comparative network, i.e., across species, which allows identification of candidate genes that manifest conserved signatures across different species (e.g., Figure 2). This analysis can be used for highlighting candidates in less thoroughly studied species based on knowledge in other organisms. Lastly, MorphDB has a SPARQL endpoint, allowing arbitrarily complex queries of the database. We illustrate the use and the potential of the tools in MorphDB in the next section.
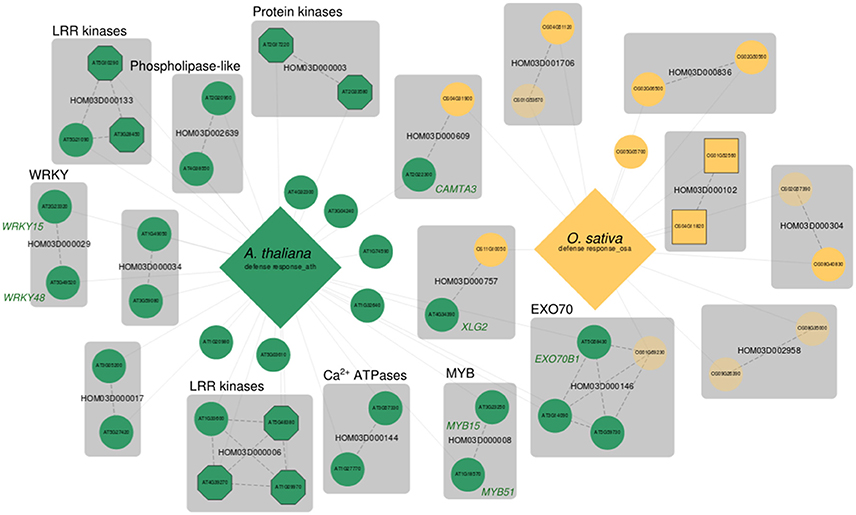
Figure 2. Comparative MorphDB network of GO:0006952 (defense response) for A. thaliana and O. sativa. A. thaliana genes are indicated in green, O. sativa genes indicated in yellow. Only genes among the top 100 candidates for this process with a homologous relation to another candidate in MorphDB for the same GO category are included for clarity. Large diamonds represent the bait gene set and candidate genes are connected to their respective bait set. Bait genes themselves are omitted for clarity as well. Genes with no functional description are depicted transparently. Transporters are shown as squares and kinases and receptors as octagons. All other candidate genes are shown as circles. Gene families are shown as gray compound nodes and orthology relationships as dashed edges.
Prioritizing Regulatory Genes for the Plant Defense Response
In this section, we focus on plant defense responses and related specialized metabolism pathways in A. thaliana, M. truncatula, O. sativa, S. tuberosum, and S. lycopersicum. A case study is shown as an illustration of the potential of MorphDB.
Figure 2 shows a comparative network generated by MorphDB for the GO category “defense response” (GO:0006952) in A. thaliana (AUSR = 0.15, p = 0.02, 269 bait genes), and O. sativa (AUSR = 0.23, p = 0.02, 150 bait genes). The network shows mainly signaling related genes for A. thaliana, with high-scoring gene families such as HOM03D000133 and HOM03D000006 (Leucine-rich receptor (LRR) kinases, all z > 2.42), HOM03D000003 (protein kinases, all z > 2.76), HOM03D002639 (phospolipase-like proteins, both z > 2.50) and HOM03D000144 (autoinhibited Ca2+ ATPases, both z > 2.90). Besides these putatively signaling related genes, putative transcription factors (TFs) are retrieved, such as WRKY TFs (HOM03D000029) and MYB domain TFs (HOM03D000008). Both AT2G23320 (WRKY15, z = 2.71) and AT5G49520 (WRKY48, z = 2.40) have been associated with the response to chitin, an import plant-defense elicitor from fungal origin (Libault et al., 2007). WRKY48 was also shown to be involved in the defense response to bacterial pathogens (Xing et al., 2008). Both TFs have been associated with diverse stress responses in another large scale computational study (Heyndrickx and Vandepoele, 2012). Both AT3G23250 (MYB15, z = 2.44) and AT1G18570 (MYB51, z = 2.42) have been associated with a whole range of hormone metabolism and stress response related processes. MYB51 regulates glucosinolate biosynthesis (Gigolashvili et al., 2007; Frerigmann et al., 2012), specialized metabolites that act as antiherbivore compounds in plants. MYB15 has been associated with the response to chitin (Libault et al., 2007).
In addition, a highly remarkable group of predicted candidates from the HOM03D000146 gene family is retrieved. These genes belong to the EXO70 gene family, which are putative exocyst subunits conserved in land plants (Chong et al., 2010; Wang et al., 2010). EXO70B1 has been associated with autophagy-related transport in A. thaliana (Kulich et al., 2013), a crucial process in diverse plant stress responses. Interestingly, Zhao et al. (2015) reported that exo70B1 mutants showed enhanced defense response through activation of a nucleotide binding domain and leucine-rich repeat-containing (NLR)-like disease resistance protein. Their study provides a link between the plant immune system and the exocyst complex, and they suggest that pathogen effectors may manipulate and interact with the plant secretion machinery. The MORPH results presented here support this hypothesis, as for two species, independently, exocyst related proteins are among the top 100 candidates with acceptable to high scores (AT5G58430 (EXO70B1): z = 2.58, AT3G14090 (EXO70D3): z = 2.57, AT5G59730 (EXO70H7): z = 2.69 and OS01G69230: z = 1.44). Looking at the processes for which EXO70B1 was predicted as a candidate in A. thaliana (Table 3), several defense related GO terms are obtained (e.g., GO:0010337, GO:0031347, GO:0009410, GO:0009816, and GO:0008219), further supporting the hypothesis of exocyst related functions in plant defense responses.
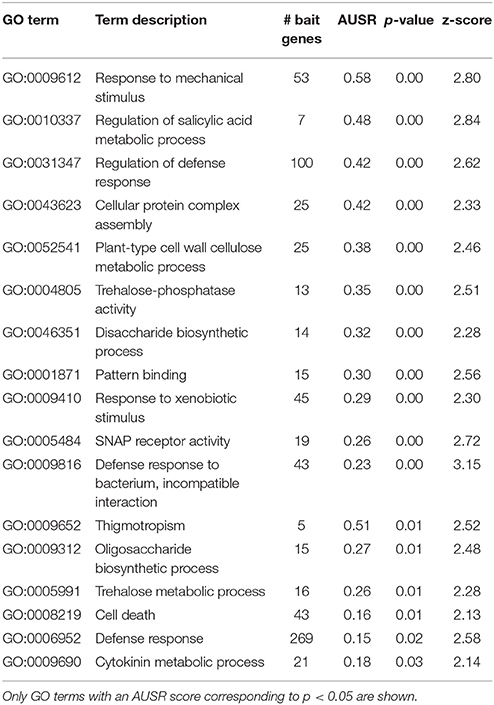
Table 3. GO terms for which AT5G58430 is among the top 100 MORPH-predicted candidates.
Other candidate gene predictions that are consistent over species can be retrieved, such as the candidates found in gene families HOM03D000609 and HOM03D000757. The first family again consists of an already known defense response regulator in A. thaliana, namely AT2G22300 (z = 2.48) encoding CAMTA3 (Calmodulin-binding transcription activator), a putative CAM binding TF. CAMTA3 mutants (camta3-1 and camta3-2) show enhanced defense responses, with a high fraction of defense associated upregulated genes in both the camta3-1 and camta3-2 mutant (Galon et al., 2008). The homolog in O. sativa (OS04G31900) predicted by MORPH for GO:000652 has not been associated with defense responses before. Lastly, HOM03D000757 also has a candidate predicted in both A. thaliana (AT4G34390, z = 2.82) and O. sativa (OS11G10050, z = 1.44). AT4G34390 encodes an extra-large GTP-binding protein (XLG2), which has been shown to be involved in root morphogenesis (Ding et al., 2007) and defense responses to bacteria (Zhu et al., 2009). Again, as expected, the rice homolog predicted by MORPH has not been functionally characterized, and the MORPH prediction supports the hypothesis of a conserved function in defense responses.
A more specific defense response related GO term that is also well represented in MorphDB is GO:0002679 (respiratory burst involved in defense response). The respiratory burst is defined as the biological process in which elevated metabolic activity increases oxygen consumption, and through an NADH dependent system reactive oxygen species are formed (ROS), such as hydrogen peroxide (Kawano, 2003). Again, a MorphDB network was constructed, with a focus on comparative aspects between A. thaliana (AUSR = 0.74, p < 0.005) and M. truncatula (AUSR = 0.54, p < 0.01) (Figure 3). Below, we focus on several interesting observations that can be made from the network.
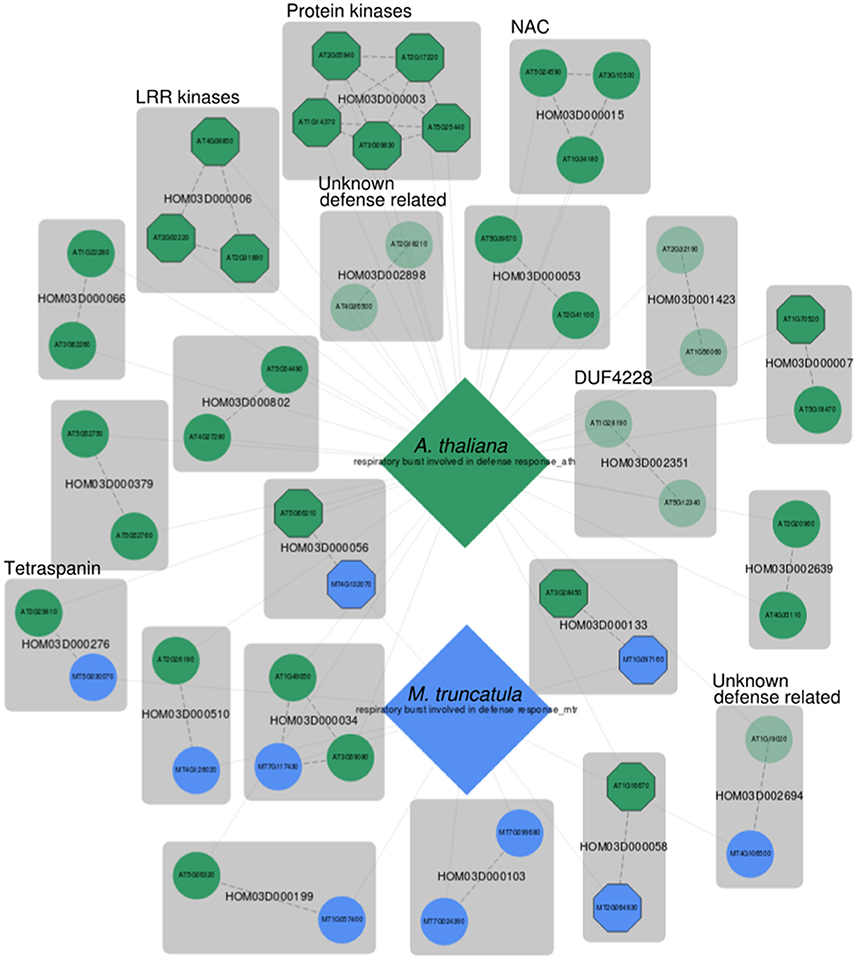
Figure 3. MorphDB network for GO:0002679 (respiratory burst involved in defense response) for A. thaliana (green) and M. truncatula (blue). For more details, see Figure 2.
Interestingly, several unknown Arabidopsis genes are predicted as candidates for this biological process. For HOM03D002351, both Arabidopsis gene family members are among the top 100 MORPH predicted candidates. This gene family consists of proteins with a domain of unknown function (DUF) DUF4228, which is functionally uncharacterized. One of the two Arabidopsis duplicates (AT1G28190) has been linked to defense response related processes (JA and SA signaling and hypersensitive response) in a large-scale systems biology study (Heyndrickx and Vandepoele, 2012). The other Arabidopsis homolog (AT5G12340) has no functional term assigned and was predicted to have a mitochondrial subcellular localization, which is consistent with a putative role in respiratory burst. Interestingly, the unknown gene AT5G12340 is ranked higher (z = 3.01) than the previously associated homolog AT1G28190 (z = 2.71). An inspection of the phylogenetic tree of this gene family on PLAZA shows that the family is angiosperm (Magnoliophyta) specific and that it is conserved across this clade. The tree indicates that the duplication event from which the Arabidopsis homologs are derived precedes the divergence of the angiosperms, as inferred from the position of the Amborella trichopoda homologs in the tree. The ancient origin of this gene family and the conservation across the angiosperm tree indicates a high likelihood of functional importance.
HOM03D002694 is another gene family without functional characterization. The Arabidopsis gene AT1G19020 has been shown to be involved in oxidative stress signaling in a mutant phenotype screen (Luhua et al., 2008), and has been associated with response to wounding, response to insect, SAR, SA mediated signaling and defense response to fungus by Heyndrickx and Vandepoele (2012). A gene centric search in MorphDB shows that AT1G19020 is predicted for a plethora of stress and defense response related GO terms (Table S1). A similar gene centric search in MorphDB reveals that the functionally uncharacterized Medicago homolog MT4G106500 is also predicted to be involved in anthocyanin-containing compound biosynthesis (GO:0009781). Anthocyanin biosynthesis is regulated by JA signaling (Shan et al., 2009) and anthocyanin accumulation is associated with enhanced herbivore resistance in Arabidopsis (Khan et al., 2016).
Interestingly, tetraspanin gene family members (HOM03D000276) are also present in the network for both Arabidopsis and Medicago. This family of membrane proteins has been mainly studied in the context of development (Wang et al., 2012, 2015) and it has been suggested that tetraspanins have a role in cell-cell communication during various developmental stages. However, it has been observed that many tetraspanins remain active also in mature differentiated tissues (Wang et al., 2015), and some tetraspanin promoter regions contain defense and pathogen response elements (Wang et al., 2015). Therefore, it is tempting to suggest a role in defense response through sensing of pathogen related molecules, because of the putative role in developmental cell-cell communication, the presence of extracellular loops and the presence of pathogen response related promoter elements. Also, this gene family has undergone several duplications and has been shown to contain putative functionally divergent clades (Wang et al., 2012), supporting the possibility of tetraspanins involved in defense response.
Lastly, we analyzed jasmonate (JA) and salicylic acid (SA) signaling. SA is one of the major important signaling molecules involved in the plant defense response (Loake and Grant, 2007; Zhang et al., 2013). SA biosynthesis is activated in response to a wide variety of phytopathogens, and SA mediated signaling results in the accumulation of pathogenesis-related (PR) proteins (Loake and Grant, 2007). It is the main molecular signal involved in the establishment of both local and systemic acquired resistance (SAR) (Loake and Grant, 2007). Besides its roles in defense and disease resistance, SA is known to regulate leaf senescence, flowering and thermogenesis (Dempsey et al., 2011; Zhang et al., 2013). Next to salicylic acid (SA) mediated signaling, JA mediated signaling is the main signaling pathway for plant defense responses (Turner et al., 2002; Chini et al., 2007), and JA is thought to be the key regulator for many specialized metabolism pathways that are triggered during biotic and abiotic stresses. Investigation of functional gene sets for SA and JA mediated signaling is therefore highly relevant in the context of this case study. MORPH results for GO:0009753 (response to jasmonic acid stimulus) for A. thaliana (AUSR = 0.27, p < 0.01), and S. tuberosum (AUSR = 0.26, p < 0.01) were based on gene sets of 236 and 74 bait genes respectively. GO:0009863 (SA mediated signaling pathway) scored an AUSR of 0.44 (p < 0.001) for a bait set consisting of 132 genes. The network of the top 50 candidates is shown in Figure 4.
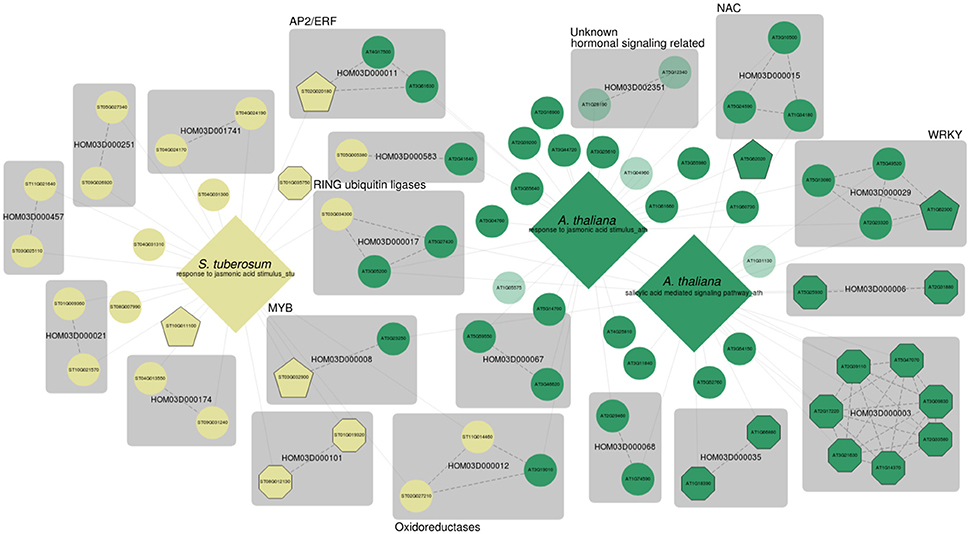
Figure 4. MorphDB network for GO:0009753 (response to jasmonic acid stimulus) and GO:0009863 (SA mediated signaling pathway) for A. thaliana (green) and S. tuberosum (pale yellow). Only the top 50 candidates for each GO category that have a homolog predicted in some other species in MorphDB are shown. For more details, see Figure 2.
Again, the results illustrate the strength of a systematic network-based analysis. Many relevant gene families are identified, some with putative TF genes. For example, the MYB TF family HOM03D00008, which has roles in defense related specialized metabolism (Liu et al., 2015), is represented in the network. The gene family HOM03D000011 consists of AP2/ERF domain containing TFs, and the selected Arabidopsis gene AT4G17500 in this family has been associated with defense response (Fujimoto et al., 2000; Onate-Sanchez and Singh, 2002). HOM03D000029 consists of WRKY domain containing TFs, already discussed above. HOM03D000015 consists of NAC DNA-binding domain containing proteins of which NAC16, NAC53 and TCV-interacting protein are among the top 50 candidates for either GO:0009751 or GO:0009863 in A. thaliana, all with high co-expression scores (z > 2.59). NAC proteins are widely recognized for their roles in hormonally controlled development (Aida et al., 1997; Xie et al., 2000) and biotic and abiotic stress responses (Lee et al., 2012; Nuruzzaman et al., 2013) (NAC53). However, A. thaliana consists of 92 NAC domain containing proteins, of which many have not been functionally characterized in detail. NAC16 has been previously associated with the response to chitin in Arabidopsis (Libault et al., 2007) and TCV-interacting protein has been shown to physically interact with turnip crinkle virus (TCV) viral capsids (Ren et al., 2000; Donze et al., 2014).
Interestingly, a family of RING type ubiquitin ligases is also present in the network (HOM03D000017). Both AT5G27420, which encodes CNI1 (Carbon/Nitrogen Insensitive1, also known as ATL31), and AT3G05200 (ATL6) have been linked previously to both fungal (Libault et al., 2007) and bacterial defense responses (Maekawa et al., 2012). The potato homolog ST03G034300 has not been functionally characterized before, and MORPH strongly suggests a role in defense responses. An interesting family of oxidoreductases is obtained (HOM03D000012) as well, with putative functions in flavonoid biosynthesis. Lastly, this network also shows several totally uncharacterized genes, of which the Arabidopsis genes in the gene family HOM03D002351 seem particularly interesting. AT1G28190 was previously associated with various hormonal signaling pathways, among which JA, SA, abscisic acid and ethylene signaling, by Heyndrickx and Vandepoele (2012) consistent with a putative role in defense. The homolog AT5G12340 could only be associated with a mitochondrial subcellular localization and is further not functionally characterized. A gene centric query for AT5G12340 shows that this gene is predicted as a high scoring candidate for a plethora of stress response related processes, further supporting a role in stress and defense responses. MORPH results as integrated in MorphDB can provide useful hints on gene functions for these enigmatic genes.
Discussion
MORPH is a highly valuable tool that was developed to accelerate gene discovery for plant metabolic pathways (Tzfadia et al., 2012). Here, the usage of MORPH was reconsidered from a genome-wide and comparative viewpoint in the context of functional annotation, gene discovery and candidate gene prioritization. Besides a framework and methodology for performing MORPH bulk runs, a database and web-tool, MorphDB, were developed, providing a friendly interface for consulting MORPH bulk predictions of several important model organisms. An additional key feature of the MORPH bulk framework is the easy usage of MORPH with custom data or novel species, which was not supported previously. This enables researchers to use the MORPH algorithm for candidate gene prioritization in their species of interest and tackle specific research questions.
In this work, we showed how MORPH can be used in bulk mode on non-model species, such as C. roseus and Z. marina, for rendering putative gene functions by analyzing bait gene sets defined by GO categories. MORPH bulk runs were also performed for already well-studied organisms, with gene discovery and candidate gene prioritization as main objectives. The integration of MORPH results with homology information from PLAZA (Van Bel et al., 2012) in MorphDB, as well as the comparative network visualization implemented in the same web tool, were shown to be particularly useful for gene discovery and candidate gene prioritization objectives in a case study concerning the plant defense response in several model organisms. MORPH predictions were shown to be well in accordance with the literature or with previously described functions for homologous genes. Our findings illustrate the relevance and potential of MORPH predictions, which may be particularly interesting for the elucidation and prioritization of regulatory roles for members of large gene families of TFs and signaling genes. Indeed, while sequence similarity and profile based methods can easily assign a TF, kinase or receptor function based on characteristic protein domains, it remains virtually impossible to associate these functions with specific biological processes. For these purposes, gene expression based methods provide a valuable solution, as we have shown in our defense response case study.
In our case study, we showed how the integration of MORPH results with homology data strengthens hypotheses suggested by MORPH and renders highly interesting candidates. A key advantage of using MORPH in a comparative fashion over classical comparative co-expression networks such as in CoExpNetViz (Tzfadia et al., 2016) is that using candidate genes predicted by MORPH instead of the top co-expressed genes based on Pearson correlation values is expected to render a higher fraction of relevant candidates. Even relatively small MORPH networks can therefore render highly relevant candidates, with the additional advantage that these networks are relatively simple and easy to browse using the MorphDB resource. Here also the tight integration with the PLAZA platform accelerates biological discovery. The case study also showed that the visual highlighting of classes of regulatory sets of genes (defined here as: transcription factors, kinases, receptors and transporters) is quite helpful in browsing the networks for interesting candidates efficiently. As functional biology is shifting to multi-omics analyses, interest in network based approaches for visualization and data exploration is growing. Networks enable the easy integration of additional experimental data such as proteomic, protein-protein interaction or genetic interaction data.
We expect that gene expression based analysis will remain central in the future elucidation of gene functions. High performing candidate gene prioritization algorithms such as MORPH enable further in-depth exploration of the functional gene space in both model and non-model organisms, but the results remain largely speculative. This is in contrast with similarity based approaches, which render quite confident gene functions but result in a largely incomplete exploration of the functional landscape of a genome. Applying MORPH in bulk mode enables researchers to generate a large set of putative functional associations, which can be further mined by domain experts to address specific research questions, as we have shown in this work. Moreover, the MorphDB web resource enables efficient querying and interpretation in a comparative setting, further aiding researchers in the prioritization of candidate genes for their particular biological process of interest.
Author Contributions
AZ, OT, and YV designed the research; AZ and TD developed MORPH and MORPH bulk; AZ and TV developed the MorphDB database and web interface; AZ, OT, YV, DA, and RS analyzed data and wrote the manuscript.
Funding
AZ acknowledges financial support from the special research fund (BOF) of Ghent University. YV acknowledges the Multidisciplinary Research Partnership Bioinformatics: from nucleotides to networks Project (no. 01MR0310W) of Ghent University, and funding from the European Union Seventh Framework Programme (FP7/2007-2013) under European Research Council Advanced Grant Agreement 322739 – DOUBLEUP.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00352/full#supplementary-material
References
Aida, M., Ishida, T., Fukaki, H., Fujisawa, H., and Tasaka, M. (1997). Genes involved in organ separation in Arabidopsis: an analysis of the cup-shaped cotyledon mutant. Plant Cell 9, 841–857. doi: 10.1105/tpc.9.6.841
Almagro, L., Fernández-Pérez, F., and Pedreño, M. (2015). Indole alkaloids from Catharanthus roseus: bioproduction and their effect on human health. Molecules 20, 2973–3000. doi: 10.3390/molecules20022973
Amar, D., Frades, I., Danek, A., Goldberg, T., Sharma, S. K., Hedley, P. E., et al. (2014). Evaluation and integration of functional annotation pipelines for newly sequenced organisms: the potato genome as a test case. BMC Plant Biol. 14:329. doi: 10.1186/s12870-014-0329-9
Amar, D., Frades, I., Diels, T., Zaltzman, D., Ghatan, N., Hedley, P. E., et al. (2015). The MORPH-R web server and software tool for predicting missing genes in biological pathways. Physiol Plant. 155, 12–20. doi: 10.1111/ppl.12326
Aoki, Y., Okamura, Y., Tadaka, S., Kinoshita, K., and Obayashi, T. (2016). ATTED-II in 2016: a plant coexpression database towards lineage-specific coexpression. Plant Cell Physiol. 57:e5. doi: 10.1093/pcp/pcv165
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Benedito, V. A., Torres-Jerez, I., Murray, J. D., Andriankaja, A., Allen, S., Kakar, K., et al. (2008). A gene expression atlas of the model legume Medicago truncatula. Plant J. 55, 504–513. doi: 10.1111/j.1365-313X.2008.03519.x
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57, 289–300.
Chini, A., Fonseca, S., Fernández, G., Adie, B., Chico, J. M., Lorenzo, O., et al. (2007). The JAZ family of repressors is the missing link in jasmonate signaling. Nature 448, 666–671. doi: 10.1038/nature06006
Chong, Y. T., Gidda, S. K., Sanford, C., Parkinson, J., Mullen, R. T., and Goring, D. R. (2010). Characterization of the Arabidopsis thaliana exocyst complex gene families by phylogenetic, expression profiling, and subcellular localization studies. New Phytol. 185, 401–419. doi: 10.1111/j.1469-8137.2009.03070.x
Conesa, A., and Götz, S. (2008). Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 1–12. doi: 10.1155/2008/619832
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
De Bodt, S., Carvajal, D., Hollunder, J., Van den Cruyce, J., Movahedi, S., and Inze, D. (2010). CORNET: a user-friendly tool for data mining and integration. Plant Physiol. 152, 1167–1179. doi: 10.1104/pp.109.147215
Dempsey, D. A., Vlot, A. C., Wildermuth, M. C., and Klessig, D. F. (2011). Salicylic acid biosynthesis and metabolism. Arabidopsis Book 9:e0156. doi: 10.1199/tab.0156
Ding, L., Pandey, S., and Assmann, S. M. (2007). Arabidopsis extra-large G proteins (XLGs) regulate root morphogenesis. Plant J. 53, 248–263. doi: 10.1111/j.1365-313X.2007.03335.x
Donze, T., Qu, F., Twigg, P., and Morris, T. J. (2014). Turnip crinkle virus coat protein inhibits the basal immune response to virus invasion in Arabidopsis by binding to the NAC transcription factor TIP. Virology 449, 207–214. doi: 10.1016/j.virol.2013.11.018
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Falcone Ferreyra, M. L., Rius, S. P., and Casati, P. (2012). Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 3:222. doi: 10.3389/fpls.2012.00222
Franz, M., Lopes, C. T., Huck, G., Dong, Y., Sumer, O., and Bader, G. D. (2015). Cytoscape.js: a graph theory library for visualisation and analysis. Bioinformatics 32, 309–311. doi: 10.1093/bioinformatics/btv557
Frerigmann, H., Boettcher, C., Baatout, D., and Gigolashvili, T. (2012). Glucosinolates are produced in trichomes of Arabidopsis thaliana. Front. Plant Sci. 3:242. doi: 10.3389/fpls.2012.00242
Fujimoto, S. Y., Ohta, M., Usui, A., Shinshi, H., and Ohme-Takagi, M. (2000). Arabidopsis ethylene-responsive element binding factors act as transcriptional activators or repressors of GCC box-mediated gene expression. Plant Cell 12, 393–404. doi: 10.1105/tpc.12.3.393
Galon, Y., Nave, R., Boyce, J. M., Nachmias, D., Knight, M. R., and Fromm, H. (2008). Calmodulin-binding transcription activator (CAMTA) 3 mediates biotic defense responses in Arabidopsis. FEBS Lett. 582, 943–948. doi: 10.1016/j.febslet.2008.02.037
Gigolashvili, T., Berger, B., Mock, H.-P., Müller, C., Weisshaar, B., and Flügge, U.-I. (2007). The transcription factor HIG1/MYB51 regulates indolic glucosinolate biosynthesis in Arabidopsis thaliana. Plant J. 50, 886–901. doi: 10.1111/j.1365-313X.2007.03099.x
Guyon, I., Saffari, A., Dror, G., and Cawley, G. (2010). Model selection: beyond the Bayesian/frequentist divide. J. Mach. Learn. Res. 11, 61–87.
Hansen, B. O., Vaid, N., Musialak-Lange, M., Janowski, M., and Mutwil, M. (2014). Elucidating gene function and function evolution through comparison of co-expression networks of plants. Front. Plant Sci. 5:394. doi: 10.3389/fpls.2014.00394
Heyndrickx, K. S., and Vandepoele, K. (2012). Systematic identification of functional plant modules through the integration of complementary data sources. Plant Physiol. 159, 884–901. doi: 10.1104/pp.112.196725
Irizarry, R. A., Hobbs, B., Collin, F., Beazer-Barclay, Y. D., Antonellis, K. J., Scherf, U., et al. (2003). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264. doi: 10.1093/biostatistics/4.2.249
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kawano, T. (2003). Roles of the reactive oxygen species-generating peroxidase reactions in plant defense and growth induction. Plant Cell 21, 829–837. doi: 10.1007/s00299-003-0591-z
Khan, G. A., Vogiatzaki, E., Glauser, G., and Poirier, Y. (2016). Phosphate deficiency induces the jasmonate pathway and enhances resistance to insect herbivory. Plant Physiol. 171, 632–644. doi: 10.1104/pp.16.00278
Kleessen, S., Klie, S., and Nikoloski, Z. (2013). Data integration through proximity-based networks provides biological principles of organization across scales. Plant Cell 25, 1917–1927. doi: 10.1105/tpc.113.111039
Kulich, I., Pečenková, T., Sekereš, J., Smetana, O., Fendrych, M., Foissner, I., et al. (2013). Arabidopsis exocyst subcomplex containing subunit EXO70B1 is involved in the autophagy-related transport to the vacuole. Traffic 14, 1155–1165. doi: 10.1111/tra.12101
Kurtzer, G. M., Sochat, V., and Bauer, M. W. (2017). Singularity: scientific containers for mobility of compute. PLoS ONE 12:e0177459. doi: 10.1371/journal.pone.0177459
Lee, S., Seo, P. J., Lee, H.-J., and Park, C.-M. (2012). A NAC transcription factor NTL4 promotes reactive oxygen species production during drought-induced leaf senescence in Arabidopsis. Plant J. 70, 831–844. doi: 10.1111/j.1365-313X.2012.04932.x
Lee, T., Yang, S., Kim, E., Ko, Y., Hwang, S., Shin, J., et al. (2015). AraNet v2: an improved database of co-functional gene networks for the study of Arabidopsis thaliana and 27 other nonmodel plant species. Nucleic Acids Res. 43, D996–D1002. doi: 10.1093/nar/gku1053
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Libault, M., Wan, J., Czechowski, T., Udvardi, M., and Stacey, G. (2007). Identification of 118 Arabidopsis transcription factor and 30 ubiquitin-ligase genes responding to chitin, a plant-defense elicitor. Mol. Plant Microbe Interact. 20, 900–911. doi: 10.1094/MPMI-20-8-0900
Liu, J., Osbourn, A., and Ma, P. (2015). MYB transcription factors as regulators of phenylpropanoid metabolism in plants. Mol Plant. 8, 689–708. doi: 10.1016/j.molp.2015.03.012
Loake, G., and Grant, M. (2007). Salicylic acid in plant defence—the players and protagonists. Curr. Opin. Plant Biol. 10, 466–472. doi: 10.1016/j.pbi.2007.08.008
Luhua, S., Ciftci-Yilmaz, S., Harper, J., Cushman, J., and Mittler, R. (2008). Enhanced tolerance to oxidative stress in transgenic arabidopsis plants expressing proteins of unknown function. Plant Physiol. 148, 280–292. doi: 10.1104/pp.108.124875
Maekawa, S., Sato, T., Asada, Y., Yasuda, S., Yoshida, M., Chiba, Y., et al. (2012). The Arabidopsis ubiquitin ligases ATL31 and ATL6 control the defense response as well as the carbon-nitrogen response. Plant Mol. Biol. 79, 217–227. doi: 10.1007/s11103-012-9907-0
Menke, F. L., Parchmann, S., Mueller, M. J., Kijne, J. W., and Memelink, J. (1999). Involvement of the octadecanoid pathway and protein phosphorylation in fungal elicitor-induced expression of terpenoid indole alkaloid biosynthetic genes in catharanthus roseus. Plant Physiol. 119, 1289–1296. doi: 10.1104/pp.119.4.1289
Movahedi, S., Van Bel, M., Heyndrickx, K. S., and Vandepoele, K. (2012). Comparative co-expression analysis in plant biology. Plant Cell Environ. 35, 1787–1798. doi: 10.1111/j.1365-3040.2012.02517.x
Movahedi, S., Van de Peer, Y., and Vandepoele, K. (2011). Comparative network analysis reveals that tissue specificity and gene function are important factors influencing the mode of expression evolution in Arabidopsis and rice. Plant Physiol. 156, 1316–1330. doi: 10.1104/pp.111.177865
Mutwil, M., Klie, S., Tohge, T., Giorgi, F. M., Wilkins, O., Campbell, M. M., et al. (2011). PlaNet: combined sequence and expression comparisons across plant networks derived from seven species. Plant Cell 23, 895–910. doi: 10.1105/tpc.111.083667
Nuruzzaman, M., Sharoni, A. M., and Kikuchi, S. (2013). Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 4:248. doi: 10.3389/fmicb.2013.00248
Olsen, J. L., Rouzé, P., Verhelst, B., Lin, Y.-C., Bayer, T., Collen, J., et al. (2016). The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 530, 331–335. doi: 10.1038/nature16548
Oñate-Sánchez, L., and Singh, K. B. (2002). Identification of Arabidopsis ethylene-responsive element binding factors with distinct induction kinetics after pathogen infection. Plant Physiol. 128, 1313–1322. doi: 10.1104/pp.010862
Proost, S., and Mutwil, M. (2017). Planet: comparative co-expression network analyses for plants. Methods Mol. Biol. 1533, 213–227. doi: 10.1007/978-1-4939-6658-5_12
Proost, S., Van Bel, M., Sterck, L., Billiau, K., Van Parys, T., Van de Peer, Y., et al. (2009). PLAZA: a comparative genomics resource to study gene and genome evolution in plants. Plant Cell 21, 3718–3731. doi: 10.1105/tpc.109.071506
Ren, T., Qu, F., and Morris, T. J. (2000). HRT gene function requires interaction between a NAC protein and viral capsid protein to confer resistance to turnip crinkle virus. Plant Cell 12, 1917–1926. doi: 10.1105/tpc.12.10.1917
Rhee, S. Y., and Mutwil, M. (2014). Towards revealing the functions of all genes in plants. Trends Plant Sci. 19, 212–221. doi: 10.1016/j.tplants.2013.10.006
Robinson, M. D., and Oshlack, A. (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11:R25. doi: 10.1186/gb-2010-11-3-r25
Roepke, J., Salim, V., Wu, M., Thamm, A. M. K., Murata, J., Ploss, K., et al. (2010). Vinca drug components accumulate exclusively in leaf exudates of Madagascar periwinkle. Proc. Natl. Acad. Sci. U.S.A. 107, 15287–15292. doi: 10.1073/pnas.0911451107
Schläpfer, P., Zhang, P., Wang, C., Kim, T., Banf, M., Chae, L., et al. (2017). Genome-wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiol. 173, 2041–2059. doi: 10.1104/pp.16.01942
Serin, E. A. R., Nijveen, H., Hilhorst, H. W. M., and Ligterink, W. (2016). Learning from co-expression networks: possibilities and challenges. Front. Plant Sci. 7:444. doi: 10.3389/fpls.2016.00444
Shan, X., Zhang, Y., Peng, W., Wang, Z., and Xie, D. (2009). Molecular mechanism for jasmonate-induction of anthocyanin accumulation in Arabidopsis. J. Exp. Bot. 60, 3849–3860. doi: 10.1093/jxb/erp223
Sharan, R., and Shamir, R. (2000). CLICK: a clustering algorithm with applications to gene expression analysis. Proc. Int. Conf. Intell. Syst. Mol. Biol. 8, 307–316.
Shi, R., Wang, J. P., Lin, Y.-C., Li, Q., Sun, Y.-H., Chen, H., et al. (2017). Tissue and cell-type co-expression networks of transcription factors and wood component genes in Populus trichocarpa. Planta 245, 927–938. doi: 10.1007/s00425-016-2640-1
Thimm, O., Bläsing, O., Gibon, Y., Nagel, A., Meyer, S., Krüger, P., et al. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939. doi: 10.1111/j.1365-313X.2004.02016.x
Tohge, T., Watanabe, M., Hoefgen, R., and Fernie, A. R. (2013). The evolution of phenylpropanoid metabolism in the green lineage. Crit. Rev. Biochem. Mol. Biol. 48, 123–152. doi: 10.3109/10409238.2012.758083
Turner, J. G., Ellis, C., and Devoto, A. (2002). The jasmonate signal pathway. Plant Cell 14(Suppl), S153–S164. doi: 10.1105/tpc.000679
Tzfadia, O., Amar, D., Bradbury, L. M. T., Wurtzel, E. T., and Shamir, R. (2012). The MORPH algorithm: ranking candidate genes for membership in Arabidopsis and tomato pathways. Plant Cell 24, 4389–4406. doi: 10.1105/tpc.112.104513
Tzfadia, O., Diels, T., De Meyer, S., Vandepoele, K., Aharoni, A., and Van de Peer, Y. (2016). CoExpNetViz: comparative co-expression networks construction and visualization tool. Front. Plant Sci. 6:1194. doi: 10.3389/fpls.2015.01194
Usadel, B., Obayashi, T., Mutwil, M., Giorgi, F. M., Bassel, G. W., Tanimoto, M., et al. (2009). Co-expression tools for plant biology: opportunities for hypothesis generation and caveats. Plant Cell Environ. 32, 1633–1651. doi: 10.1111/j.1365-3040.2009.02040.x
Van Bel, M., and Coppens, F. (2017). Exploring plant co-expression and gene-gene interactions with CORNET 3.0. Methods Mol. Biol. 1533, 201–212. doi: 10.1007/978-1-4939-6658-5_11
Van Bel, M., Proost, S., Wischnitzki, E., Movahedi, S., Scheerlinck, C., Van de Peer, Y., et al. (2012). Dissecting plant genomes with the PLAZA comparative genomics platform. Plant Physiol. 158, 590–600. doi: 10.1104/pp.111.189514
Wang, F., Muto, A., Van de Velde, J., Neyt, P., Himanen, K., Vandepoele, K., et al. (2015). Functional analysis of the Arabidopsis TETRASPANIN gene family in plant growth and development. Plant Physiol. 169, 2200–2214. doi: 10.1104/pp.15.01310
Wang, F., Vandepoele, K., and Van Lijsebettens, M. (2012). Tetraspanin genes in plants. Plant Sci. 190, 9–15. doi: 10.1016/j.plantsci.2012.03.005
Wang, J., Ding, Y., Wang, J., Hillmer, S., Miao, Y., Lo, S. W., et al. (2010). EXPO, an exocyst-positive organelle distinct from multivesicular endosomes and Autophagosomes, mediates cytosol to cell wall exocytosis in Arabidopsis and tobacco cells. Plant Cell 22, 4009–4030. doi: 10.1105/tpc.110.080697
Wolfe, C. J., Kohane, I. S., and Butte, A. J. (2005). Systematic survey reveals general applicability of “guilt-by-association” within gene coexpression networks. BMC Bioinformatics 6:227. doi: 10.1186/1471-2105-6-227
Xie, Q., Frugis, G., Colgan, D., and Chua, N. H. (2000). Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 14, 3024–3036. doi: 10.1101/gad.852200
Xing, D.-H., Lai, Z.-B., Zheng, Z.-Y., Vinod, K., Fan, B.-F., and Chen, Z.-X. (2008). Stress- and pathogen-induced Arabidopsis WRKY48 is a transcriptional activator that represses plant basal defense. Mol. Plant. 1, 459–470. doi: 10.1093/mp/ssn020
Zarrineh, P., Fierro, A. C., Sánchez-Rodríguez, A., De Moor, B., Engelen, K., and Marchal, K. (2011). COMODO: an adaptive coclustering strategy to identify conserved coexpression modules between organisms. Nucleic Acids Res. 39:e41. doi: 10.1093/nar/gkq1275
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Zhang, K., Halitschke, R., Yin, C., Liu, C.-J., and Gan, S.-S. (2013). Salicylic acid 3-hydroxylase regulates Arabidopsis leaf longevity by mediating salicylic acid catabolism. Proc. Natl. Acad. Sci. U.S.A. 110, 14807–14812. doi: 10.1073/pnas.1302702110
Zhao, T., Rui, L., Li, J., Nishimura, M. T., Vogel, J. P., Liu, N., et al. (2015). A truncated NLR protein, TIR-NBS2, is required for activated defense responses in the exo70B1 mutant. PLoS Genet. 11:e1004945. doi: 10.1371/journal.pgen.1004945
Keywords: comparative co-expression networks, candidate gene prioritization, functional annotation, MORPH, defense response
Citation: Zwaenepoel A, Diels T, Amar D, Van Parys T, Shamir R, Van de Peer Y and Tzfadia O (2018) MorphDB: Prioritizing Genes for Specialized Metabolism Pathways and Gene Ontology Categories in Plants. Front. Plant Sci. 9:352. doi: 10.3389/fpls.2018.00352
Received: 04 January 2018; Accepted: 02 March 2018;
Published: 19 March 2018.
Edited by:
Thiago Motta Venancio, State University of Norte Fluminense, BrazilReviewed by:
Xiyin Wang, North China University of Science and Technology, ChinaGuilherme Corrêa De Oliveira, Instituto Tecnológico Vale (ITV), Brazil
Copyright © 2018 Zwaenepoel, Diels, Amar, Van Parys, Shamir, Van de Peer and Tzfadia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yves Van de Peer, eXZwZWVAcHNiLnVnZW50LmJl
Oren Tzfadia, b3J0emZAcHNiLnZpYi11Z2VudC5iZQ==