 Iago Hale1*†
Iago Hale1*† Xiao Ma2,3Arthur T. O. Melo1Francis Kwame Padi4
Xiao Ma2,3Arthur T. O. Melo1Francis Kwame Padi4 Prasad S. Hendre5Sarah B. Kingan6
Prasad S. Hendre5Sarah B. Kingan6 Shawn T. Sullivan7
Shawn T. Sullivan7 Shiyu Chen8Jean-Marc Boffa5Alice Muchugi5,9
Shiyu Chen8Jean-Marc Boffa5Alice Muchugi5,9 Agyemang Danquah10Michael Teye Barnor4Ramni Jamnadass5Yves Van de Peer2,3,11,12†
Agyemang Danquah10Michael Teye Barnor4Ramni Jamnadass5Yves Van de Peer2,3,11,12† Allen Van Deynze5,8†
Allen Van Deynze5,8†- 1Department of Agriculture, Nutrition, and Food Systems, University of New Hampshire, Durham, NH, United States
- 2Department of Plant Biotechnology and Bioinformatics, Ghent University, Ghent, Belgium
- 3Center for Plant Systems Biology, VIB, Ghent, Belgium
- 4Plant Breeding Division, Cocoa Research Institute of Ghana, Ghana Cocoa Board, New Tafo, Ghana
- 5AOCC Genomics Laboratory and Tree Genebank Research Unit, World Agroforestry (CIFOR-ICRAF), Nairobi, Kenya
- 6Pacific Biosciences, Menlo Park, CA, United States
- 7Phase Genomics, Seattle, WA, United States
- 8Seed Biotechnology Center, University of California, Davis, Davis, CA, United States
- 9The Forage Genebank, Feed and Forage Development Program, International Livestock Research Institute, Addis Ababa, Ethiopia
- 10West Africa Centre for Crop Improvement, College of Basic and Applied Sciences, University of Ghana, Accra, Ghana
- 11College of Horticulture, Academy for Advanced Interdisciplinary Studies, Nanjing Agricultural University, Nanjing, China
- 12Centre for Microbial Ecology and Genomics, Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria, South Africa
A defining component of agroforestry parklands across Sahelo-Sudanian Africa (SSA), the shea tree (Vitellaria paradoxa) is central to sustaining local livelihoods and the farming environments of rural communities. Despite its economic and cultural value, however, not to mention the ecological roles it plays as a dominant parkland species, shea remains semi-domesticated with virtually no history of systematic genetic improvement. In truth, shea’s extended juvenile period makes traditional breeding approaches untenable; but the opportunity for genome-assisted breeding is immense, provided the foundational resources are available. Here we report the development and public release of such resources. Using the FALCON-Phase workflow, 162.6 Gb of long-read PacBio sequence data were assembled into a 658.7 Mbp, chromosome-scale reference genome annotated with 38,505 coding genes. Whole genome duplication (WGD) analysis based on this gene space revealed clear signatures of two ancient WGD events in shea’s evolutionary past, one prior to the Astrid-Rosid divergence (116–126 Mya) and the other at the root of the order Ericales (65–90 Mya). In a first genome-wide look at the suite of fatty acid (FA) biosynthesis genes that likely govern stearin content, the primary determinant of shea butter quality, relatively high copy numbers of six key enzymes were found (KASI, KASIII, FATB, FAD2, FAD3, and FAX2), some likely originating in shea’s more recent WGD event. To help translate these findings into practical tools for characterization, selection, and genome-wide association studies (GWAS), resequencing data from a shea diversity panel was used to develop a database of more than 3.5 million functionally annotated, physically anchored SNPs. Two smaller, more curated sets of suggested SNPs, one for GWAS (104,211 SNPs) and the other targeting FA biosynthesis genes (90 SNPs), are also presented. With these resources, the hope is to support national programs across the shea belt in the strategic, genome-enabled conservation and long-term improvement of the shea tree for SSA.
Introduction
Shea tree (Vitellaria paradoxa) is a unique agroforestry tree species central to sustaining local livelihoods and the farming environments of rural communities across Africa’s Sudano-Sahelian agroclimactic belt. Cited as the second most important oil crop in Africa after oil palm, shea tree is likely among the most economically and culturally important indigenous tree species in the Sudano-Sahelian region of Africa where oil palm does not grow (Hall and Tomlinson, 1996). Rural families in hundreds of thousands of villages across the so-called “shea belt,” a 500–750 km wide semi-arid area stretching 6,000 km and spanning 21 countries from Senegal to South Sudan, use shea in their daily lives as an edible butter/oil, soap, cosmetic, and medicine. Shea is also a multimillion dollar export commodity as an ingredient in luxury cosmetic and personal care (e.g., moisturizing creams, sun lotions, and soaps) and pharmaceutical (e.g., cholesterol-lowering and anti-arthritic remedies) products. The largest export demand (∼90%) for shea is, however, linked to the extraction of edible stearin used in the formulation of cocoa butter equivalents (CBE) for chocolate confectionary (Rousseau et al., 2015).
As an indigenous tree species, V. paradoxa is traditionally not planted because of its extensive juvenile period (commonly 10–25 years) and local abundance, not to mention the tenurial challenges associated with being unable to differentiate naturally regenerated from planted trees. Instead, shea agroforestry parklands, comprised of annual crops and scattered shea trees that can reach densities of 20–50 trees/ha in areas of strong shea culture, result from self-sown propagation and systematic management (selection and protection, as opposed to planting) by farmers through successive fallow and cultivation cycles. Farmer selection of preferred individuals and removal of inferior trees for charcoal production or building materials serve to increase the levels of locally valued traits, resulting in what has been called a semi-domestication of the species (Lovett and Haq, 2000; Maranz and Wiesman, 2003).
Based on population and daily shea butter consumption data, the number of women taking part in shea nut collection across the shea belt is estimated at more than 18 million (Naughton et al., 2015). Indeed, shea has been called “women’s gold” because it is one of only a few resources that female members of rural households in the region have control over, from harvesting to commercialization (Elias and Carney, 2007). The sale of shea products allows women to secure additional food for themselves and their children once cereal harvests are exhausted (Pouliot, 2012) and to generate cash for household expenses including clothing, medicine, and school fees. The seasonality of shea availability is also critical to farming households’ nutrition. Shea fruits are the only widely available, energy-rich food source at the time when land is tilled and crops are planted at the end of the dry season. The fruits supply significant amounts of protein, sugar, calcium, potassium, and essential fatty acids (Honfo et al., 2014) during this annual ‘hungry season’ when cereal stocks in granaries are lowest and labor requirements for field preparations with the coming of the rains are highest (Maranz et al., 2004a).
Using a land suitability model parameterized with conservative tree density estimates, Naughton et al. (2015) estimated that there are 1.84 billion shea trees over its distribution range, making it one of the largest populations of an economic tree species in the region, not to mention a source of considerable environmental benefits. In high production areas, such as those found in northern Ghana and Burkina Faso’s Central Plateau, shea can account for up to 80% of tree populations and a large portion of the standing biomass of the farmed parklands (Boffa, 1999). These parkland systems, edified through human management, contain substantial carbon stores with enormous potential to mitigate climate change via future carbon sequestration (Takimoto et al., 2009; Luedeling and Neufeldt, 2012). Currently, the shea value chain is estimated to fix about 1.5 million tons of CO2 equivalent (tCO2-e) yearly, or a negative carbon footprint of 1.04 tCO2-e per ton of shea kernels produced (Bockel et al., 2020).
Despite shea’s widespread importance to rural livelihoods (especially those of women), its growing export market, and its ecological role as a key species in these agroforestry parkland systems, the sustainability of V. paradoxa populations is threatened by increasing demographic pressure and the quickening spread of mechanized farming practices. This raises concerns for possible future production gaps. Given increasing saturation of arable land, cultivation periods are extended and fallow intervals, which were traditionally responsible for the recruitment of young shea trees (Ræbild et al., 2012), are reduced or altogether abandoned. As a result, shea tree densities have been declining for decades; trees have been aging and regeneration is low (Gijsbers et al., 1994; Boffa, 1999). Farmers collecting shea nuts need to travel longer distances, increasing the labor required for nut collection; and when monetary returns to collectors drop, farmers begin to convert shea parklands to plantations of alternative tree crops, such as cashew and mango. Also, the relatively recent emphasis on large-scale land investment and agricultural development projects aiming to intensify maize, cotton, and biofuel production with mechanized farming are an additional threat to the retention of shea trees in farmers’ fields (Poudyal, 2011). Elsewhere in areas of high energy demand, competing uses of shea in parklands or uncultivated woodlands can be favored, such as tree cutting for firewood and charcoal making, leading to the degradation of shea stands.
Notwithstanding these pressures, shea consistently features at the top of farmers’ lists of priority species; and surveys indicate that farmers would plant shea trees if improved varieties were available and visually distinguishable (for purposes of clarifying ownership) from naturally regenerated trees (Poudyal, 2011). Like all high-value fruit trees planted in these systems, deliberately planted shea stands could be established in closer proximity to household compounds, thus reducing walking and collection times. Furthermore, the introduction of improved shea varieties has the potential to ease harvesting, boost and reduce tree-to-tree variation in yield and quality, and improve land and labor use efficiency, especially if combined with improved cultural practices. Various recent efforts to distribute shea seedlings top-worked with scions of identified “plus” (i.e., superior) in situ accessions signify a felt need to improve upon the passive semi-domestication process, even if it requires a fundamental shift in the traditional planting culture.
Stearin buyers on the international market also clamor for improved genetics. With stearic and oleic acids constituting nearly 90% of kernel fat content (Maranz et al., 2004b; Ugese et al., 2010), shea is one of the few economically viable natural sources of stearic–oleic–stearic triacylglycerols, a valued vegetable stearin due to its comparable functional properties to cocoa butter (Allal et al., 2013). But while it is the preferred stearin source for CBE production, the quality, quantity and price of the gathered shea crop are criticized by stearin buyers as erratic (LMC International, 2014). This comes as no surprise, as the out-crossing reproductive physiology of the species results in naturally regenerating heterozygous populations with highly variable biochemical profiles. A genetic improvement program for shea is therefore viewed as a promising means of achieving higher resource supply consistency and quality, with the positive effect of reducing price fluctuations and satisfying industry demands.
In short, the systematic genetic improvement of the shea tree is strongly justified to address both the threats to and rising demand for the resource, as well as development opportunities in the sector; yet there has been virtually no history of such work to date. The body of research relevant to the task, however, is extensive and indicates great promise for progress. In the mid-20th century, pioneering cytogenetic and oil/butter compositional studies on V. paradoxa (then Butyrospermum parkii) were conducted, describing the species as a diploid (2n = 24) (Miège, 1954) and providing the first insights into the fatty acid composition of shea butter (Hilditch and Saletore, 1931), already well-recognized for its cultural importance and economic potential (Chevalier, 1948). But it is within the last 30 years that a surge in shea research has occurred, particularly in the form of distribution, parkland regeneration, and diversity studies. Lovett and Haq (2000) published the results of an isozyme-based diversity analysis of shea in Ghana, signaling a shift from methods based on morphological traits to those based on molecular markers. A suite of similar studies followed, some of impressive pan-African scope (e.g., Bouvet et al., 2004; Allal et al., 2011), making use of various low-density sets of markers, including SSRs (8–14), RAPDs (67), chloroplastic SNPs (4), or combinations thereof (see e.g., Fontaine et al., 2004; Kelly et al., 2004; Cardi et al., 2005; Sanou et al., 2005; Allal et al., 2008; Logossa et al., 2011; Gwali et al., 2015; Abdulai et al., 2017). The conclusions of such studies were consistent. Shea is a weakly selfing (low fixation index) species characterized by moderate to high heterozygosity and extensive gene flow. With the Dahomey Gap serving as a dividing boundary, there is strong genetic differentiation between stearic-rich subspecies paradoxa in the West and oleic-rich nilotica in the East, with relatively higher allelic richness observed in western populations; yet essentially no phylogeographic signal is observed within regions. Such weak intraregional differentiation, manifested by the majority (85–95%) of genetic variation existing within populations, is expected in outcrossing, long-lived trees of widespread occurrence over a continuous range and further suggests little impact of domestication on genetic diversity.
In addition to their shared insights, there is a notable limitation common among these studies as well, namely the inability to associate low-resolution genetic data with factors relevant to shea improvement and cultivation, whether it be ethnovarietal attributes, fatty acid profiles, or basic growth parameters. To this end, such genetic investigations echo the patterns of diversity established by the numerous phenotypic studies of the species. Starting with Chevalier’s first observations in the 1940’s, shea diversity studies based on morphological and chemical profile data consistently reported high trait variability, with most (60–90%) of that variation existing within populations (see e.g., Sanou et al., 2005; Akihisa et al., 2011; Allal et al., 2013). Productivity exhibits a similarly high variation within populations. Desmarest (1958), for example, observed of a natural population in Burkina Faso that over half of the trees had essentially no economic value. The work of Boffa et al. (1996) supports that basic conclusion, noting that 24% of the trees they observed accounted for 55% of total stand production. In sum, phenotypic studies to date indicate the existence of substantial genetic variation in economically relevant traits within highly diverse populations, the same populations which defy dissection via low-density molecular markers. In comparing methods for estimating heritability of various foliar traits in shea, Bouvet et al. (2008) observed that a method relying on marker-based estimates of relatedness (12 SSRs) failed to detect the significant heritabilities revealed by a traditional pedigree-based method. Their conclusion was that new approaches are needed for marker-based methods in an agroforestry species like shea. Fortunately, in the intervening years, tremendous strides have been made in the development of such methods. Indeed, genome-wide association studies (GWAS) in unstructured populations have become common in many species (Cortes et al., 2021), provided the supporting genomic resources, including a well-annotated reference genome and sufficiently dense marker sets, are available.
Conversion through genetic improvement of this semi-domesticated agroforestry species into an early fruiting, highly productive tree displaying desired characteristics (e.g., consistent fruit and nut quality, tree architecture, yield stability, etc.) is required to trigger the shift to a more deliberate and active conservation and deployment of shea genetic diversity. Shea’s extended juvenile period makes traditional breeding approaches untenable for this task, but the opportunity for genome-assisted improvement is immense. The resources developed in this study are intended to support such a new era of research, one requiring high genomic resolution commensurate with shea’s well established patterns of diversity.
Materials and Methods
Plant Genetic Resources
At its research substation in the town of Bole in northern Ghana, the Cocoa Research Institute of Ghana (CRIG) has developed and maintains a national shea germplasm repository consisting of an in situ population of approximately 600 trees that have been evaluated for yield and basic quality traits annually since 1998. Accession ‘KA01’ stands out as the highest yielding genotype in that collection, with a 10-year average annual nut yield of 22 kg and the highest annual yield (60 kg) recorded for any tree among CRIG’s in situ and ex situ germplasm collections. A monumental tree (19.5 m tall, 1.3 m diameter at breast height) estimated to be 100–150 years old, ‘KA01’ has a spherical canopy shape (14.2 m mean diameter) and produces generally ovoid fruits with an average weight of 31.0 g (Figure 1). On average, the fruit’s fleshy pulp (mesocarp) comprises 58% of the total fruit weight and has a high sugar content (26.3°Brix). The average dry nut weight for ‘KA01’ is 7.1 g, with a kernel weight ranging from 4.6–5.3 g (mean shelling percentage of 75%) and an average butter yield of 45%.
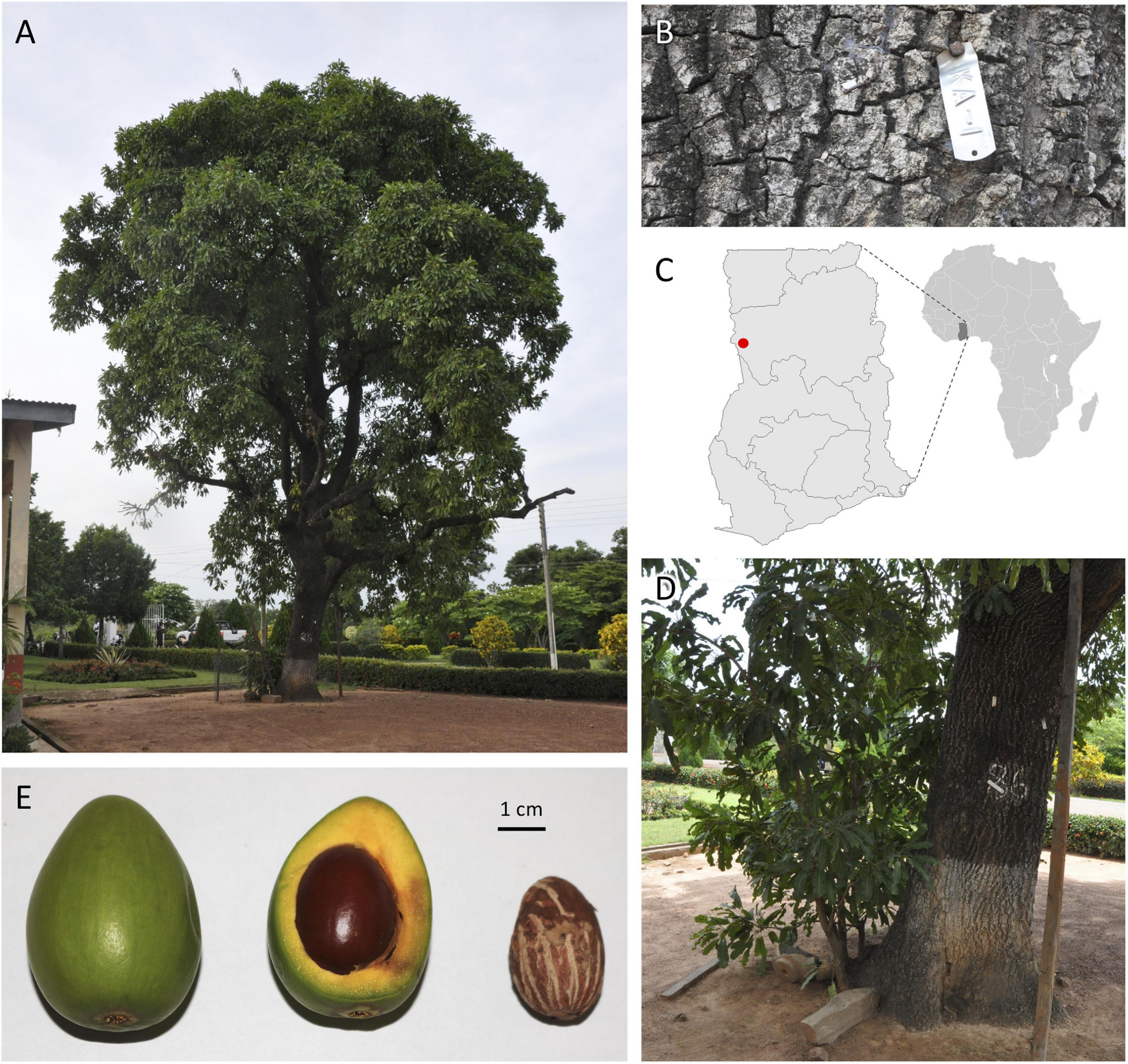
Figure 1. Vitellaria paradoxa reference accession ‘KA01’ is a giant-type shea tree (A,B) found on the CRIG research substation in Bole in northwestern Ghana (C, red dot). For such an aged tree, aggressive root-pruning (D) was required to stimulate the production of leaf tissue conducive to high-quality gDNA extraction appropriate for long-read sequencing. (E) A typical whole fruit (left), fresh cross-section (middle), and dried kernel (right) from this accession.
Fully expanded leaves of ‘KA01’ have undulate margins and an average blade length of 17.9 mm, width of 5.1 mm, and petiole length of 9.4 mm. The tree is an early season bearing genotype, meaning it initiates flowers in the month of October and begins dropping its fruits in April, earlier than most trees in and around Bole which are observed to initiate fruit drop in June. Unlike many accessions in CRIG’s collection, ‘KA01’ has been found to be highly amenable to vegetative propagation through both stem cuttings and grafting. Although the accession is a giant type tree and is susceptible to pestalotia leaf spot, a major disease of shea which attacks both seedlings and mature trees (Akrofi and Amoah, 2009), its relatively stable yield and desirable butter characteristics recommend it as a promising breeding parent; thus it has been used extensively in CRIG’s crossing programs to date, including as the male line in a recent nested association mapping (NAM) population. For these reasons, ‘KA01’ was selected as the V. paradoxa accession for developing an annotated reference genome for this outcrossing, diploid (2n = 24) species (Miège, 1954; Löve, 1980; Johnson, 1991) within the Sapotaceae.
To enable broadly relevant SNP discovery and genome-assisted breeding in shea, a concerted effort was made to collect foliar tissue from a diverse collection of V. paradoxa accessions from across the shea belt. In total, 150 silica-dried leaf samples were received at the genomics laboratory of the African Orphan Crops Consortium (AOCC) at World Agroforestry (CIFOR-ICRAF) in Nairobi, Kenya, for DNA extraction. Following extraction, a subset of 100 accessions was selected to represent a geographically diverse resequencing panel, distributed as follows (west to east): Senegal (5 lines), Côte d’Ivoire (15), Ghana (70), Benin (18), Nigeria (5), Cameroon (4), Chad (5), and Uganda (27 – V. paradoxa ssp. nilotica). Of these 100 lines, however, skim sequencing library preparation was ultimately successful for only 31 due to the challenges of tissue collection in remote areas and the extraction of high-quality genomic DNA (gDNA) from this species (see Methods), thus reducing the geographical representation of the panel to four countries [Ghana (23), Benin (2), Nigeria (4), and Cameroon (2)] (for details, see Additional File 2: Supplementary Table 1). To supplement the shallow resequencing data of the above panel, gDNA was also extracted from fresh leaf tissue of a shea seedling growing in the Nairobi, Kenya, nursery of World Agroforestry (CIFOR-ICRAF genebank accession ‘ICRAFF 11537’) for deeper Illumina shotgun sequencing (see Variant Analysis). According to genebank records, the origin of the seedling is CIFOR-ICRAF’s shea germplasm collection in Bamako, Mali.
Consistent with the Nagoya Protocol on Access and Benefit Sharing, an instrument of the Convention on Biological Diversity (CBD), shea tissues were in all cases collected by collaborating national program scientists (see section “Acknowledgments”) who were responsible for any necessary collection consent in their respective countries. The tissue samples shipped out of the respective countries were used only for DNA/RNA extraction, in accordance with MTA’s. All DNA and RNA samples were then sent to the United States and South Africa, respectively, for analysis for research purposes only; and this research has no direct commercial application.
Reference Genome Sequencing and Assembly
Extraction of DNA and Sequencing
To induce new shoot and leaf growth conducive to high molecular weight gDNA extraction from the mature reference accession ‘KA01,’ the tree was root pruned in the summer of 2016 (Figure 1D). Three different consignments of young leaves were then collected and shipped to the AOCC Genomics Laboratory for sequential rounds of gDNA extraction, in August 2016 (250 leaves), February 2017 (500 leaves), and February 2018 (500 leaves).
Extracting high-quality gDNA from the mucilage-rich foliar tissue of the shea tree proved to be a serious challenge, so it is worth detailing the successful protocol here. Using a modified CTAB procedure developed at the AOCC Laboratory, 4–6 young leaves (3–5 g) at a time were ground in liquid nitrogen, to which 25 mL of preheated (650C) high salt CTAB buffer (100 mM Tris-Cl, pH 8.0; 20 mM EDTA, pH 8.0; 3 M NaCl; freshly added with 3% PVP, β-mercaptoethanol, and 3% CTAB) were added. The solution was incubated for 45 min in a 650C water bath, then cooled in an ice bath. At this point, half the volume (12.5 ml) of 5M NaCl was added, followed by the addition of one-tenth the volume (2.5 ml) of chilled isopropanol, and the solution gently mixed via swirling. Leaf debris was removed via refrigerated centrifugation (3,500 × g at 4°C for 20 min), resulting in the separation of a clear supernatant that was then subjected to two rounds of purification and precipitation.
In the first round, the supernatant was purified by adding an equal volume of dichloromethane, centrifuging as before, then adding the same volume of chloroform:isoamyl alcohol (24:1) and centrifuging again. The purified DNA was precipitated by adding an equal volume of isopropanol and storing at 4°C for 1–3 h. The precipitated DNA was pelletized via centrifugation, washed with 70% ethanol (5–10 mL), dried at room temperature, and then dissolved in 500 μL of TE (10 mM Tris-Cl, pH 8.0;1 mM EDTA, pH 8.0). Following suspension, 10 μL of RNase (10 mg/mL) was added and the solution incubated at room temperature for 1 h. In the second round, an equal volume of chloroform:isoamyl alcohol (24:1) was again added, the solution centrifuged (13,000 × g at 4°C for 15 min), and the supernatant separated. The re-purified DNA, now free of residual RNase, was precipitated by adding double the volume of chilled absolute ethanol and storing at –20°C for 1–2 h. The precipitated DNA was pelletized, washed, and dried as before and finally dissolved in a minimum volume of TE (100–150 μL).
The integrity of the suspended DNA was inspected on a 0.8% agarose gel, its optical density ratios were checked using a Nanodrop spectrophotometer (ND-2000, Thermo Fisher Scientific, Waltham, United States), and final quality checking was performed using a Qubit 2.0 (Thermo Fisher Scientific). As they became available, batches of extracted gDNA were sent to the UC Davis Genome Center, where a 20-kb BluePippin kit (PacBio) was used for Single Molecule Real Time (SMRT) library preparation. Ultimately, three separate libraries were prepared and sequenced on a total of 25 SMRT cells on a PacBio Sequel system, using V2 chemistry.
Assembly, Phasing, and Scaffolding
The FALCON and FALCON-Unzip toolkits (FALCON-integrate v1.8.2) (Chin et al., 2016) were used for whole genome assembly and phasing. FALCON is a Hierarchical Genome Assembly Process (HGAP) pipeline that generates a genome assembly from long PacBio reads through the following basic steps: (1) Raw read error correction via alignment of subreads; (2) Pre-assembly of long, error-corrected reads; (3) Overlap detection among pre-assembled reads; (4) Overlap filtering; (5) Overlap graph construction; and (6) Graph-based contig construction. After the initial assembly, FALCON-Unzip is used in highly heterozygous species like shea to resolve the distinct haplomes (i.e., unzip the genome) based on patterns of structural variants and associated SNPs (i.e., haplotype blocks). This unzipping process gives rise to a set of so-called primary contigs (the primary assembly) and a set of associated haplotigs (phased variants of primary contig segments spanning regions of high heterozygosity). Complete details of the FALCON assembly parameters used in this study are provided in Additional File 3: Supplementary Text 1. Finally, the Arrow algorithm from the ‘GenomicConsensus’ PacBio package1 was used to polish the primary contigs and their associated haplotigs.
High levels of heterozygosity in some genomic regions can lead to the incorrect assignment of haplotigs as distinct primary contigs, thus requiring further polishing and curation of the assembly via the Purge Haplotigs pipeline (Roach et al., 2018). To identify such errors and correctly assign homologous contigs to the haplotig pool, the Purge Haplotigs pipeline first performs a read-depth analysis using BEDTools (Quinlan and Hall, 2010) to flag abnormally low or high coverage contigs as potential chimeras and then performs a BLAST (Camacho et al., 2009) against the entire assembly to identify putative primary contigs exhibiting high homology to one another. During this process, alignment dotplots are produced, and these are manually screened to break likely chimeras, define the final set of primary contigs as the reference sequence, and assign residual syntenic contigs as haplotigs. Complete details of the Purge Haplotigs process are provided in Additional File 3: Supplementary Text 2.
To extend haplotype phasing to chromosome scale, the Purge Haplotigs output was integrated with chromosome conformation capture (Hi-C) data via FALCON-Phase (Kronenberg et al., 2021). The final result of the FALCON-Phase workflow are pairs of scaffolds (phase 0 and phase 1), such that all phase blocks along the scaffolds originate from the same parental haplotype. As part of this workflow, Proximity-Guided Assembly was performed using Phase Genomics’ ProximoTM Hi-C analysis (Burton et al., 2013). Tissue processing, chromatin isolation, library preparation, sequencing, and Hi-C analysis were performed by Phase Genomics (Seattle, WA, United States), including a final stage of manual curation of the Hi-C assembly using JuiceBox (Dudchenko et al., 2018). Due to their slightly superior assembly statistics (total length, length N50, etc.), the phase 0 scaffolds were selected as the reference for all downstream analysis.
Assessment of Assembly Quality
Quality of the final curated assembly was assessed using QUAST (Gurevich et al., 2013), and assembly completeness was evaluated using the set of 1,614 core Embryophta plant genes in BUSCO v4 (Simão et al., 2015). To identify and purge contaminant contigs, the final assembly was aligned using BLAST to the following databases of possible contaminants: plasmid DNA (cpDNA and mtDNA) from angiosperms, the human genome (GRCh38.p7), the Escherichia coli genome (CP017100.1), and 16S and 18S rRNAs. The rRNA database was created using the SILVA project (Quast et al., 2013), and the others were created via sampling from Genbank.
Genome Annotation
Transcriptome Assembly
Total RNA was extracted from nine different tissues: flowers (buds and mature), fruits (skin and pulp), and dried seeds collected from mature reference accession ‘KA01’; and roots, stems, and leaves (buds and mature) collected from juvenile accession ‘ICRAFF 11537.’ All samples were finely chopped, stored, and transported in RNAlater (Thermo Fisher Scientific, Catalog #AM7021) to the CIFOR-ICRAF AOCC Genomics Laboratory, where total RNA was extracted using the Zymo Quick-RNATM Plant Miniprep kit (Catalog #R2024). In addition to the quantification and quality assessment described previously for the gDNA, RIN values of the purified RNA were estimated using BioAnalyzer 2100 (Agilent, Santa Clara, CA, United States). A pooled RNA-seq library, equimolar by tissue type, was prepared using Illumina TruSeq® RNA Library Prep Kits and sequenced via 100 bp paired-end (PE) reads on an Illumina HiSeq 2500 at the Agricultural Research Council Lab in Pretoria, South Africa. The same pool was used to generate a complementary set of long-read RNAseq data via a single-end run of Oxford Nanopore Technology (ONT) at the University of Dundee, United Kingdom.
For the Illumina reads, CASAVA-processed raw sequences were error-corrected using the software BFC v1.0 (Li, 2015), following recommendations from the Oyster River Protocol For Transcriptome Assembly (MacManes, 2016). Error-corrected reads were then processed to remove Illumina adapters and trimmed to remove low quality reads (Phred ≤ 5) using Trimmomatic v.0.33 (Bolger et al., 2014). All post-processed reads from the nine tissues were pooled in silico, and a pure Illumina transcriptome was assembled using Trinity (reference-guided de novo assembly) (Haas et al., 2013). The ONT data set was corrected using proovreads (Hackl et al., 2014), generating a set of corrected ONT reads. Because the ONT platform generates long sequences from single molecules, the sequencing products are considered to be the transcripts themselves. By merging the corrected ONT data set with the pure Illumina assembly and removing redundancy using Vsearch (Rognes et al., 2016), a third ‘hybrid’ assembly was also generated. The relative qualities of the three assemblies were evaluated via basic summary statistics and quality metrics, and the completeness of each was assessed using the set of 1,614 core Embryophyta plant genes in BUSCO v4 (Simão et al., 2015). Finally, the results of the three assemblies were concatenated and the quality and completeness of the integrated RNA assembly was re-evaluated.
Gene Prediction
RepeatModeler v2.0 (Flynn et al., 2020) was used to identify the repeat families in the genome assembly, and RepeatMasker v4.1 (Smit and Hubley, 2013) was used to discover and classify repeats based on the custom repeat libraries from RepeatModeler v2.0. Gene models were predicted via a combination of ab initio prediction, homology search, and RNA-aided annotation. For ab initio gene prediction, model training based on the alignment of short RNAseq data was accomplished using BRAKER2 (Brůna et al., 2021). For homology prediction, protein sequences from three related species within the order Ericales [Actinidia chinensis (kiwifruit), Rhododendron simsii (indoor azalea), and Camellia sinensis (tea plant)] were used as query sequences to search the reference genome using TBLASTN (e-value < 1e-5); and the regions mapped by these query sequences were compared using Exonerate (Slater and Birney, 2005). For transcriptome-guided annotation, the integrated RNA assembly was used as input to the Program to Assemble Spliced Alignments (PASA) pipeline. Finally, EVidenceModeler v1.1.1 (Haas et al., 2008) was used to integrate the results of the three prediction strategies above, based on different evidentiary weights. TransDecoder, a companion software of the Trinity platform, was used to predict open reading frames; and putative gene function was identified using InterProScan in conjunction with different databases, including PFAM, Gene3D, PANTHER, CDD, SUPERFAMILY, ProSite, and GO. Further functional annotation of the predicted genes was obtained by aligning the protein sequences against those in public protein databases and the UniProt database using BLASTP (e-value < 1e−5).
Phylogenetic and Whole Genome Duplication Analyses
Phylogenetic Analysis
To retrieve the evolutionary history of the Ericales, we chose five species [A. chinensis, R. simsii, C. sinensis, Primula vulgaris (primrose), and V. paradoxa], representing five Ericales families, as well as five other species representing extended diversity within the Asterid [Daucus carota (wild carrot), Solanum lycopersicum (tomato), and Camptotheca acuminata (tree of life)] and out-group Rosid [Vitis vinifera (grape) and Arabidopsis thaliana] clades. The amino acid sequences of all proteins were downloaded from PLAZA (Van Bel et al., 2018) and NCBI, and OrthoFinder v1.1.4 (Emms and Kelly, 2019) was used to identify orthologous groups (all-versus-all BLASTP with an e-value < 1e−05). Single-copy orthologs were extracted from the clustering results; and pre-alignment homology filtering was done using PREQUAL (Whelan et al., 2018), followed by multiple sequence alignment of the masked amino acid sequences using MAFFT (Katoh and Standley, 2013) with default parameters. Finally, protein sequence alignments were transformed into codon alignments. The resulting codon alignments from all single-copy orthologs were then concatenated into one representative supergene for species phylogenetic analysis. A maximum likelihood phylogenetic tree of single-copy codon alignments from shea and the nine other angiosperms was constructed using IQ-TREE with GTR + G model and 1,000 bootstrap replicates. Divergence times between the eight Asterid species and the two Rosid outgroups were estimated using MCMCtree from the PAML package (Yang, 2007) under the GTR with Gamma (GTR + G) model, calibrating the results with (1) the crown node of Ericales (89.8 Mya), (2) the Asterid – Rosid divergence (116–126 Mya), and (3) the S. lycopersicum – D. carota divergence (95–106 Mya). MCMCTree was used to obtain 10,000 samples from the posterior, sampling every 150 iterations after a burn in of 500,000 iterations; and convergence was verified via independent runs.
Whole Genome Duplication Analysis
KS distribution analysis was performed using the wgd package (Zwaenepoel and Van de Peer, 2019), with the paranome (entire collection of duplicated genes) obtained via ‘wgd mcl’ using all-against-all BlastP and MCL clustering. The KS distribution of shea was then constructed using ‘wgd ksd’ with default settings [i.e., using MAFFT for multiple sequence alignment, codeml for maximum likelihood estimation of pairwise synonymous distances, and FastTree (Price et al., 2009) for inferring phylogenetic trees used in the node weighting procedure]. So-called “anchors” or “anchor pairs” (i.e., paralogous genes located within broader regions of collinearity/synteny in the genome) were obtained using i-ADHoRe (Simillion et al., 2008; Proost et al., 2012), employing the default settings in ‘wgd syn.’ Within the shea genome, the MCScanX toolkit (Wang et al., 2012) was used to identify collinear blocks; and TBtools (Chen et al., 2020) was used to visualize the results via a circos plot. MCScanX was also used to perform comparative pair-wise genomic analyses among V. paradoxa, C. sinensis, and V. vinifera (Tang et al., 2008).
Analysis of Fatty Acid (FA) Biosynthesis Homologs
To identify genes related to pathways that govern FA biosynthesis in shea, the lipid metabolic pathway was downloaded from the A. thaliana acyl-lipid metabolism database2 and full-length protein sequences of the implicated lipid metabolic genes were downloaded from the TAIR database3. Additional relevant protein sequences from A. thaliana and Theobroma cacao (cocoa tree) were downloaded from PLAZA4. FA biosynthesis homologs in shea and T. cacao were identified via BLASTP searches against the protein sequences of fetched A. thaliana lipid metabolic genes. Candidate FA biosynthesis genes in A. thaliana, T. cacao, and V. paradoxa were aligned using MAFFT (Katoh and Standley, 2013) and concatenated for phylogenetic analysis. Maximum likelihood gene trees based on protein sequences were constructed using IQ-TREE with the GTR + G model and 1,000 bootstrap replicates (Nguyen et al., 2015). Finally, microsynteny plots were generated using MCScan-Jcvi (Tang et al., 2008) as a means of probing the origins of those FA biosynthesis genes exhibiting relatively high copy number in shea.
Variant Analysis
Three sequencing datasets were used to call SNPs independently. The first consists of a total of 92 Gb of 150-bp reads of the reference accession ‘KA01,’ generated by 10x Genomics, with an average 137x coverage of the genome. Long Ranger v2.2.2 was used, in conjunction with GATK v4.0.3.0 (McKenna et al., 2010), to map the 10x reads and call SNPs, which were then phased and screened by 10x specific filters. The second sequencing dataset contains 22 Gb of 150-bp Illumina shotgun reads of shea accession ‘ICRAFF 11537,’ with average 34x coverage of the shea genome. The third data set consists of skim resequencing of 31 individual genotypes using Illumina, with an average of 1.8x coverage of mapped reads across the population. Both Illumina data sets were mapped using BWA v0.7.16a and called for SNPs using GATK v4.1.6.0. Details of all applied filters for each dataset are provided in Additional File 2: Supplementary Table 2. Of all the SNPs called, only those that were shared across at least two datasets (10x data of ‘KA01,’ Illumina shotgun data of ‘ICRAFF 11537,’ and the resequencing panel) were advanced as high-confidence SNPs for functional analysis using the SnpEff pipeline (Cingolani et al., 2012).
SNP Selection and Diversity Analysis
To facilitate the translation of this work into ready-to-use tools of practical value for shea improvement programs, two different SNP panels were developed from the full set of high-confidence SNPs described above (see Variant analysis). The first panel, intended for use in genome-wide association studies (GWAS), genomic selection (GS) programs, germplasm characterization, linkage map development, trait mapping, and the like, requires a set of broadly applicable SNPs evenly distributed across the genome. To accomplish this, the genome was divided into 5 kb contiguous bins and the SNP with highest MAF chosen to represent each bin. The second panel, intended specifically for the targeted characterization of FA biosynthesis genes, requires a set of broadly applicable SNPs within (or in tight linkage with) those genes. To accomplish this, two SNPs were selected per identified FA biosynthesis homolog, with priority given to SNPs of high MAF and secondarily to those with a putative effect on gene function, based on the SnpEff analysis. For both the GWAS and FA panels, selected SNPs were required to be bordered on either side by at least 50 bp of conserved sequence (i.e., no known nearby sequence variation) to facilitate targeted primer and/or bait design. To maximize informativeness and the chance of broad applicability of the panels to other collections of V. paradoxa germplasm, all selected SNPs were required to be present in all genotypic states (homozygous major allele, heterozygous, and homozygous minor allele) within the resequencing panel.
Results
An Annotated, Chromosome-Scale Reference Genome for Shea Tree
Approximately 162.6 Gb of sequence data from reference accession ‘KA01’ was generated from 25 PacBio Single Molecule Real Time (SMRT) cells (V2 chemistry on Sequel), with an average read length of 8,142 bp and a read length N50 of 13,514 bp (Additional File 1: Supplementary Table 3). The FALCON-Unzip pipeline resulted in a 784.3 Mb assembly consisting of 1,215 putative primary contigs with a contig length N50 of 2.03 Mbp (Table 1). Further curation of the primary contigs, in the form of chimera breaking and cryptic haplotig identification (see section “Materials and Methods”), resulted in a final 667.3 Mb assembly consisting of 594 primary contigs with a contig length N50 of 2.44 Mbp (Table 1). Associated with these primary contigs is a corresponding set of 2,517 phased haplotigs (contig length N50 = 376.0 kb), spanning 42.3% of the primary contig space.
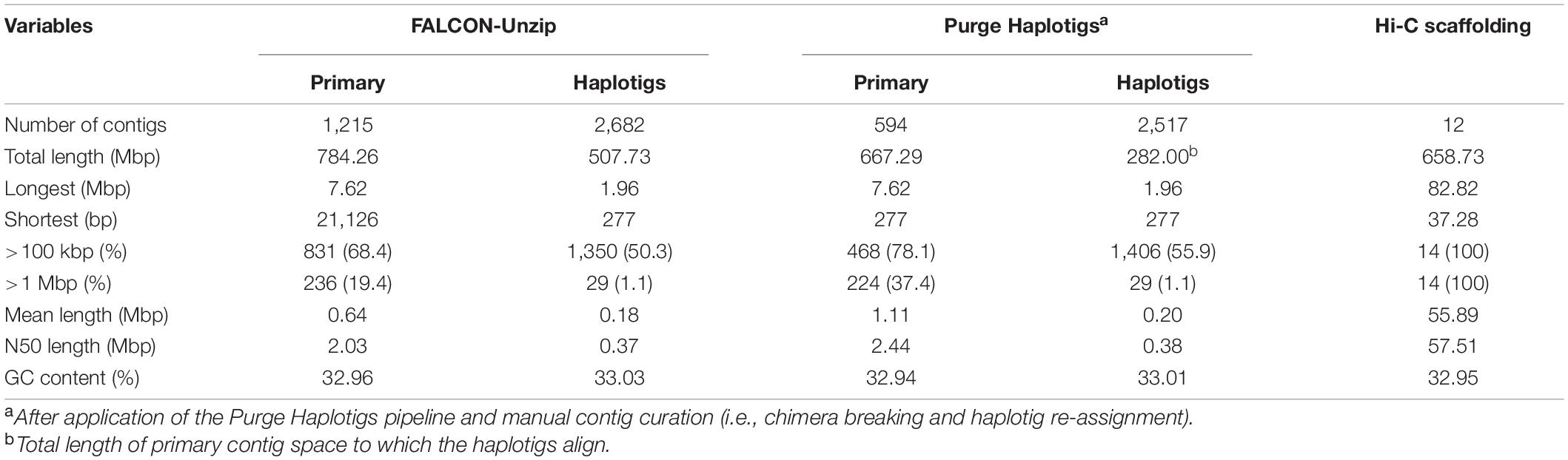
Table 1. Summary statistics of the V. paradoxa accession ‘KA01’ genome assembly, by stage.
The primary contigs from the final assembly were placed into chromosome-level scaffolds (i.e., pseudo-molecules) on the basis of three-dimensional proximity information obtained via chromosome conformation capture (Hi-C) analysis (Burton et al., 2013; Bickhart et al., 2017). Of the 594 primary contigs, 495 (combined length of 658.7 Mb; contig N50 of 2.50 Mbp) successfully assembled into 12 pseudo-molecules, as shown in the Hi-C heatmap (Additional File 1: Supplementary Figure 1). The emergence of 12 distinct Hi-C guided pseudo-molecules lends support to previous cytological studies that placed the chromosome number of V. paradoxa at 2n = 24 (Miège, 1954; Löve, 1980; Johnson, 1991). The remaining 99 contigs (combined length of 8.56 Mbp, contig N50 of 221.0 kbp) were designated as unscaffolded contigs. Detailed summary statistics of the 12 pseudo-molecules and 99 unscaffolded contigs comprising the 667.2 Mb V. paradoxa accession ‘KA01’ reference assembly are reported in Table 2.
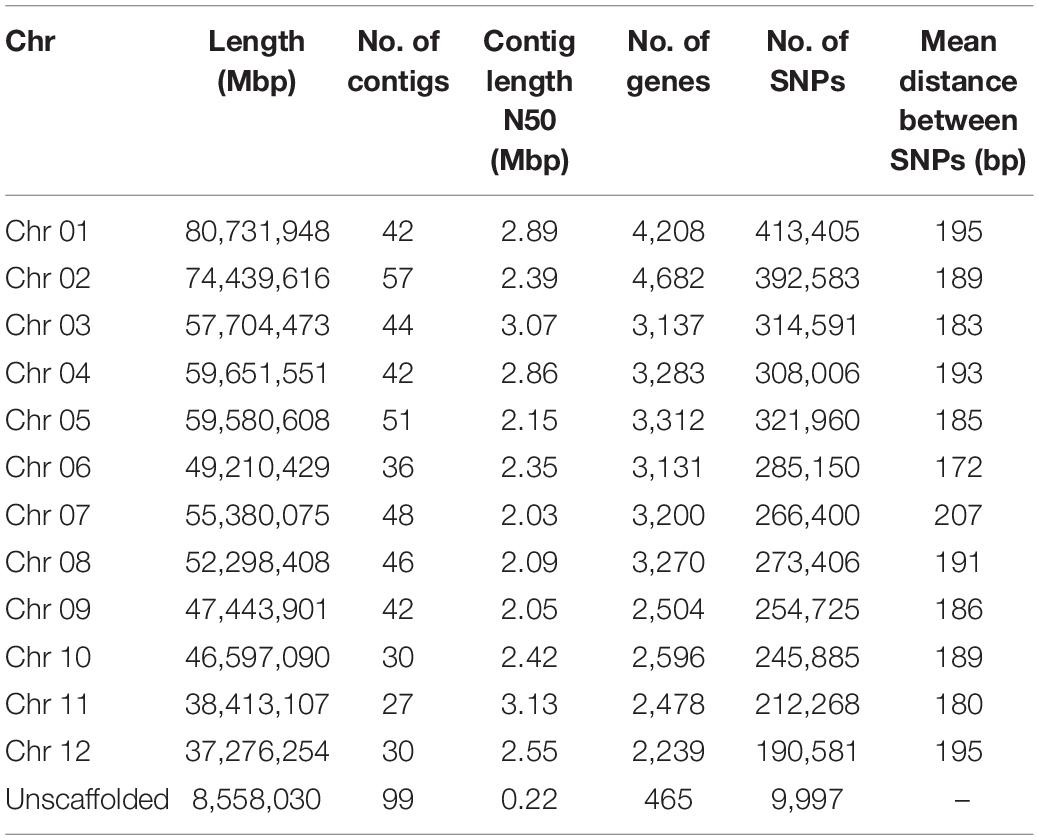
Table 2. Summary statistics of the ‘KA01’ reference genome, by chromosome.
The RNAseq libraries generated 47.02 Gb of Illumina data (∼376 million PE reads) and 0.56 Gb of ONT reads (778,913 reads of variable lengths; length N50 = 886 bp). The three RNA assemblies, namely the pure Illumina Trinity assembly, the pure Oxford Nanopore (ONT) assembly, and the Vsearch-mediated hybrid of the two, were ultimately integrated, providing 118,065 transcripts to support RNA prediction, of which GMAP was able to align 99.98% to the reference genome. In total, 46,868 gene models (including splice variants) were obtained with the PASA pipeline, of which 42,368 had open reading frames predicted by TransDecoder (for details, see Additional File 2: Supplementary Table 4).
In terms of structural annotation, transposable elements (TEs) were found to account for approximately 60% of the 658.7 Mbp ‘KA01’ reference genome, with roughly 17% of those TEs being long terminal repeat (LTR) elements (Additional File 2: Supplementary Table 5). As summarized in Table 3, after masking repeat elements, 38,505 coding gene models were found. On average, protein-coding genes in shea are 4,831 bp long and contain 4.4 exons. BUSCO analysis of the predicted protein sequences against the Embryophyta database10 showed that 87.8% of the BUSCO genes were found as complete genes, with an additional 8.2% of genes represented in fragmented form. In summary, 80.4% of the BUSCO genes were present as single copy genes and 7.5% were found in multiple copies. Only 4.0% of the BUSCO gene set was found missing (Additional File 2: Supplementary Table 4). A visualization of the final V. paradoxa genome assembly is provided in Figure 2, along with genome-wide trends in GC content, TE density, haplotig coverage, and gene density.
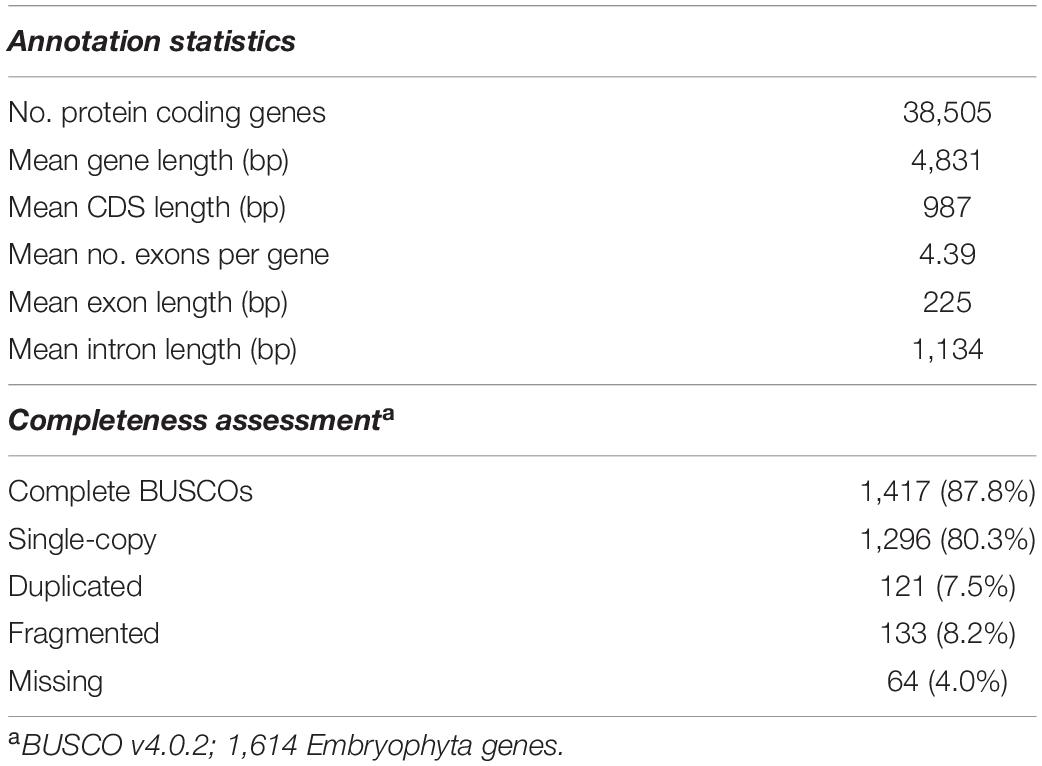
Table 3. Annotation statistics and completeness assessment of the ‘KA01’ reference genome.
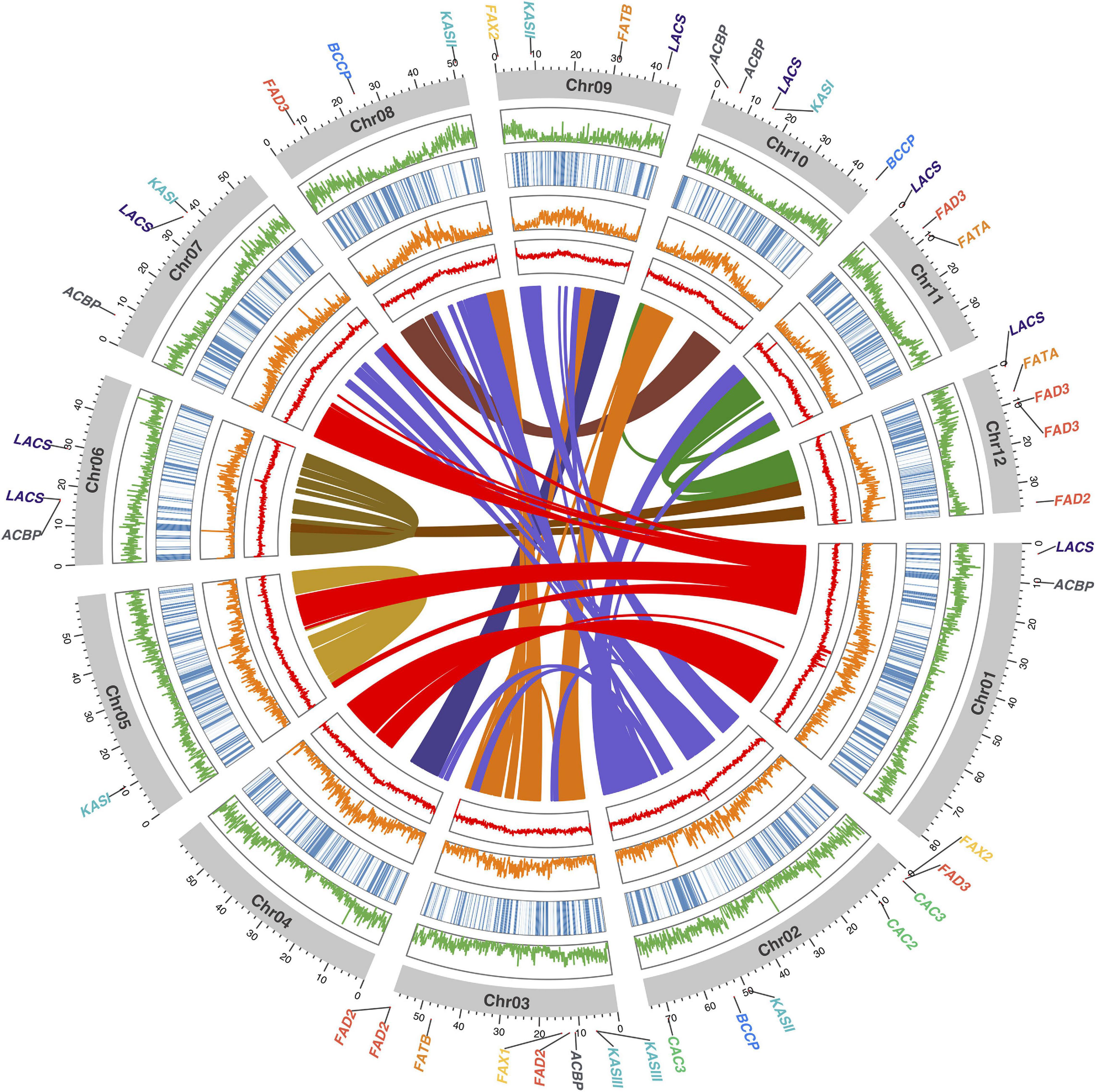
Figure 2. Circos plot of the shea genome, with links between syntenic regions indicated (center). Moving from the center outward, the four circumferential plots per chromosome report mean %GC content (red), number of transposable elements (orange), haplotig coverage (blue heatmap), and number of coding gene models (orange), per 100 kb bin. The coordinate axes track the physical lengths of the chromosomes (Mbp), to which the locations of the 45 fatty acid (FA) biosynthesis homologs identified in this study are anchored.
A Shared Whole Genome Duplication Event in the Ericales
Within the shea genome, a total of 9,800 anchors (i.e., pairs of paralagous genes in duplicated, collinear regions) were found (Figure 2, center). Nearly all collinear blocks on chromosomes 5 and 6 exhibit intrachromosomal linkages and little collinearity with other linkage groups, implying that these two chromosomes may be fusions of the original subgenomes following an ancient whole-genome duplication (WGD) event.
Distributions of synonymous substitutions per synonymous sites (KS) for anchor pairs retained in collinear regions reveal signature peaks at KS ≈ 0.6 for a whole-genome duplication (WGD) event in the shea genome, KS ≈ 0.45 for a WGD event in C. sinensis, and KS ≈ 0.73 for a WGD event in R. simsii (Figure 3). Apart from these three WGD events, KS distributions also provide evidence for two WGDs in A. chinensis, a lineage reported to have gone through three distinct WGD events (Ad-α,17.7–26.5 Mya; Ad-β, 61.9–73.7 Mya; and At-γ, 150.4–159.3 Mya) (Wu et al., 2019).
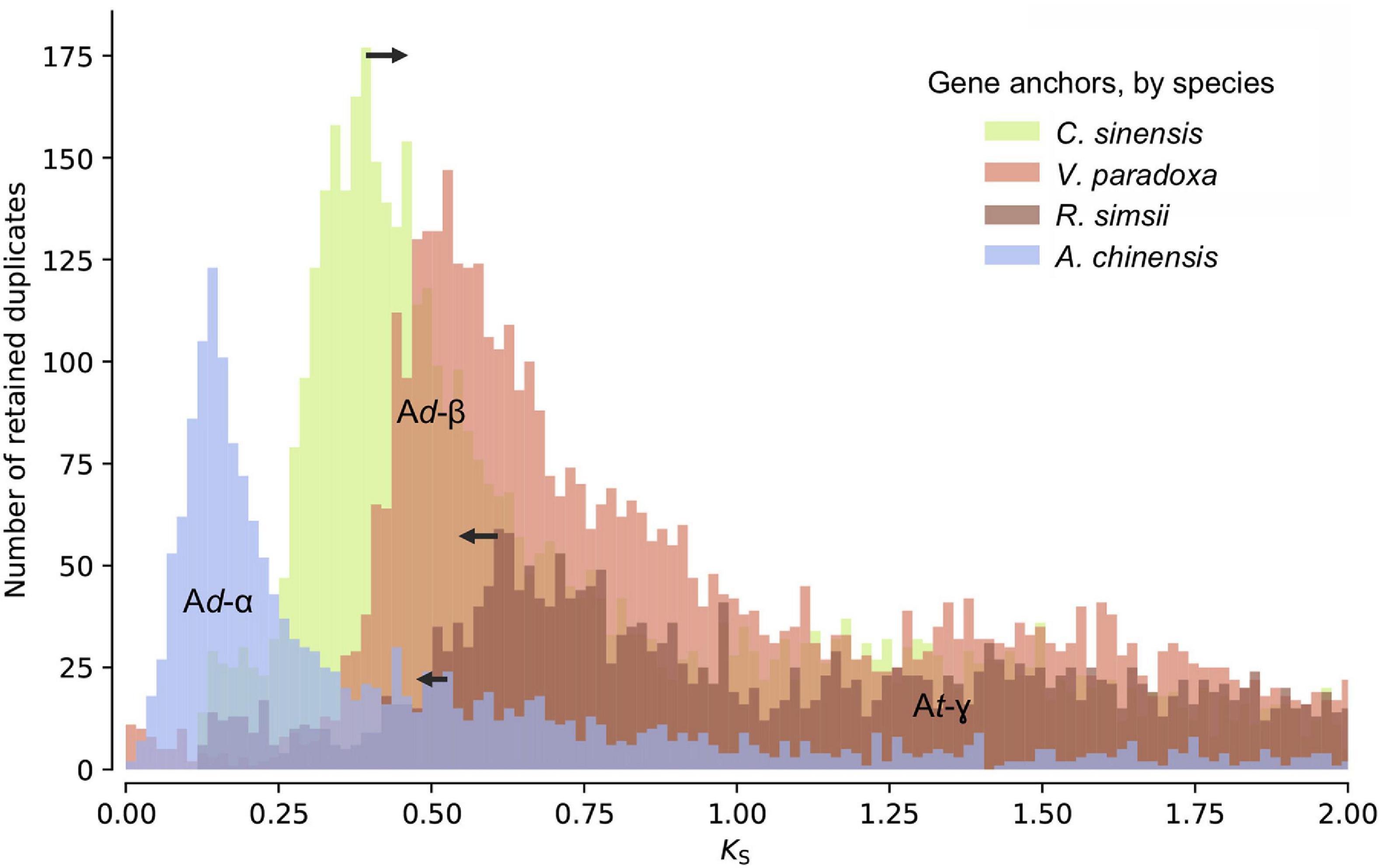
Figure 3. KS age distributions for paralogs of four species within the order Ericales: shea, Camellia sinensis (tea plant), Rhododendron simsii, and Actinidia chinensis (kiwifruit). The blue peak represents the recent WGD event in kiwifruit (Ad-α). The arrows represent adjustments to synonymous substitution rates based on relative rate tests (see text for details), suggesting a convergence of peaks at a likely shared WGD event in the Ericales (Ad-β). The well-established ancient WGD event (At-γ) that pre-dates the Asterid-Rosid divergence is also indicated.
Because no additional WGD events have been reported for well-studied V. vinifera since the ancient At-γ WGD shared by most, if not all, eudicots, we undertook pairwise syntenic analyses between grape and shea, as well as between grape and tea plant. These analyses revealed widespread 2:1 syntenic relationships between shea and grape as well as between tea plant and grape (Figure 4), providing additional evidence for a WGD event in both shea and tea plant since At-γ. Similar results were found by Yang et al. (2020) for R. simsii, a related species in the Ericales. Such evidence, particularly in combination with the clear pattern of synteny between putative homoeologous scaffolds (Figure 2), strongly suggest that all three species experienced second WGD events after the common At-γ event.
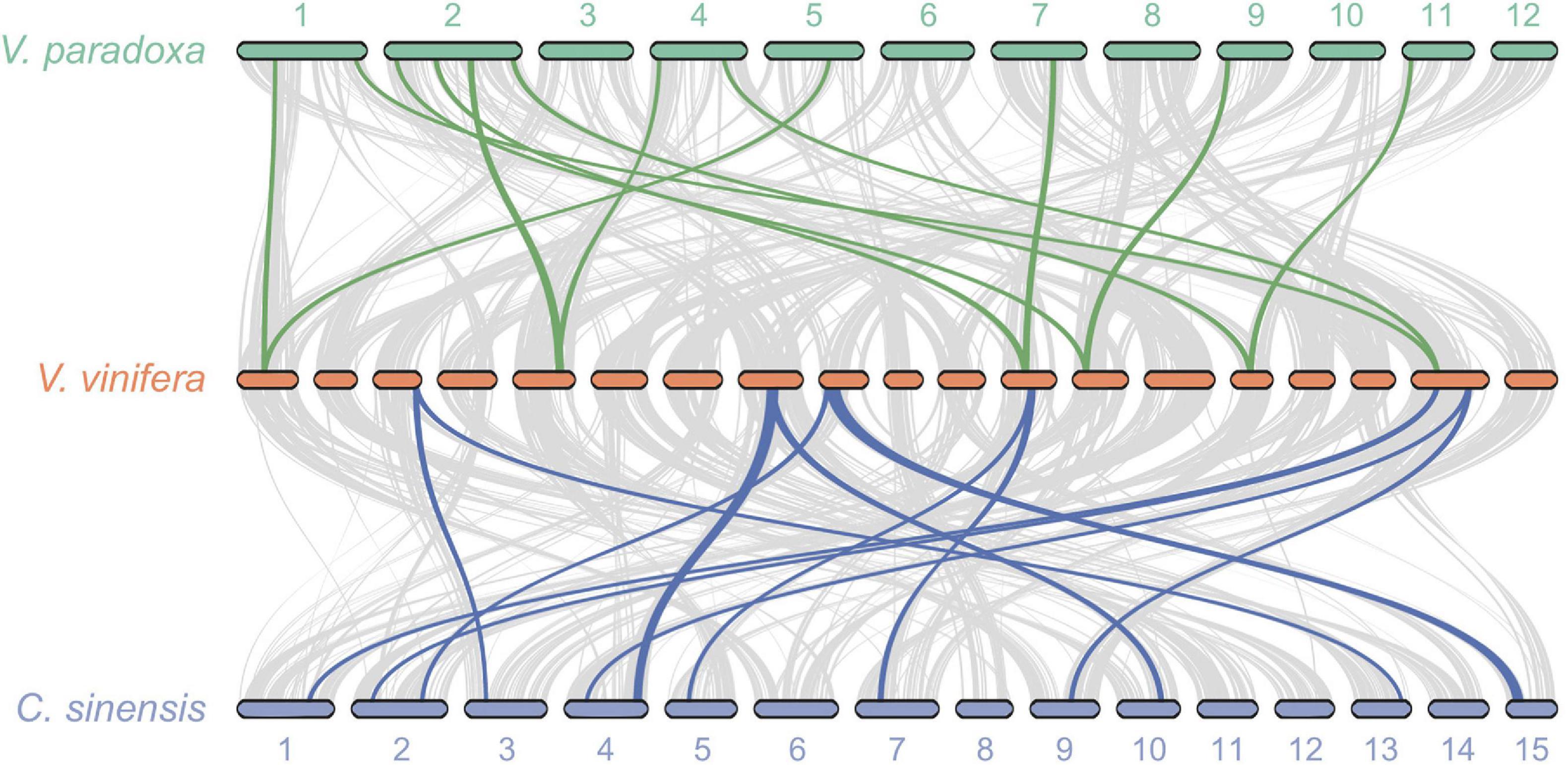
Figure 4. Syntenic relationships between Vitis vinifera (grape) and both shea (top) and the tea plant Camellia sinensis (bottom) reveal widespread patterns of gene duplication, providing further evidence of WGD events in the two latter species subsequent to their divergence from the Rosids. The gray lines connect all regions of synteny between species (i.e., the background collinearity). Between shea and grape, a total of 15 2:1 syntenic blocks are detected, containing 1,035 genes. Of those, an illustrative subset of six blocks are highlighted (green lines). Similarly, of the 21 2:1 syntenic blocks (1,309 genes) between tea plant and grape, six are highlighted (blue lines).
To determine whether these second WGD events were independent or instead shared by shea, tea plant, and R. simsii, and what their relationship might be to those observed in kiwifruit (Ad-α and Ad-β), additional evidence was brought to bear. First, as indicated in the KS distributions (Figure 3), the most recent WGD event (Ad-α) in the kiwifruit genome occurred well after its divergence from shea, supporting the hypothesis that Ad-α occurred independently in Actinidia. Second, a recent study by Wu et al. (2019) indicates a shared Ad-β event in the Ericales; and related work on the tea plant genome concluded that the Ad-β event occurred prior to the divergence between tea plant and kiwifruit (Xia et al., 2017). To determine whether the WGD event in the shea genome is lineage-specific or shared with those reported in kiwifruit, tea plant, and R. simsii (i.e., Ad-β) (Xia et al., 2017; Wu et al., 2019; Yang et al., 2020), we compared one-to-one orthologous KS distributions between shea and each of those species (see Additional File 1: Supplementary Figure 2). The KS peak value representing the divergence between shea and C. sinensis is smaller (i.e., ‘younger’) than those representing the divergences between shea and either R. simsii or kiwifruit, each exhibiting almost equal KS values. In other words, the same speciation event is found to correspond to slightly different KS peak values, likely due to different substitution rates for different species.
When comparing the one-to-one orthologous KS distribution between grape and shea with those between grape and four other species within the order Ericales [tea plant, kiwifruit, R. simsii, and primrose], we similarly observe different KS peak values for the same speciation event (i.e., the Asterid-Rosid divergence) (see Additional File 1: Supplementary Figure 3). These different peaks again suggest different synonymous substitution rates among the species, with primrose (not included in the anchor pair analysis due to the lower quality of its assembly) as the fastest, R. simsii and kiwifruit as relatively slower, and tea plant as the slowest, likely resulting in an overestimation of the divergence time between shea and R. simsii/kiwifruit while an underestimation for the divergence time between shea and tea plant. In light of this, relative rate tests were employed to adjust the synonymous substitution rates (see arrows in Figure 3), providing further support of the idea that the WGD event in the shea genome occurred at the same time as the one in tea plant, R. simsii, and kiwifruit, prior to their divergence.
In conclusion, upon reconstructing the phylogeny of the Ericales and other clades with branch lengths in KS units (i.e., a KS tree) using the codeml package from the PAML software, it can be seen that the KS peak values for the WGD events in shea, tea plant, and R. simsii reconcile at the same node (Figure 5), corresponding to a shared WGD event (Ad-β). Application of the Whale method (Zwaenepoel and Van de Peer, 2019) for gene tree/species tree reconciliation also resulted in additional support for this conclusion of a shared Ad-β event among shea, tea plant, R. simsii, and kiwifruit (and likely P. vulgaris) within the Ericales clade (see Additional File 2: Supplementary Table 6). Considering previous estimates, this shared WGD likely occurred 65 – 90 Mya (Xia et al., 2017; Wu et al., 2019; Yang et al., 2020).
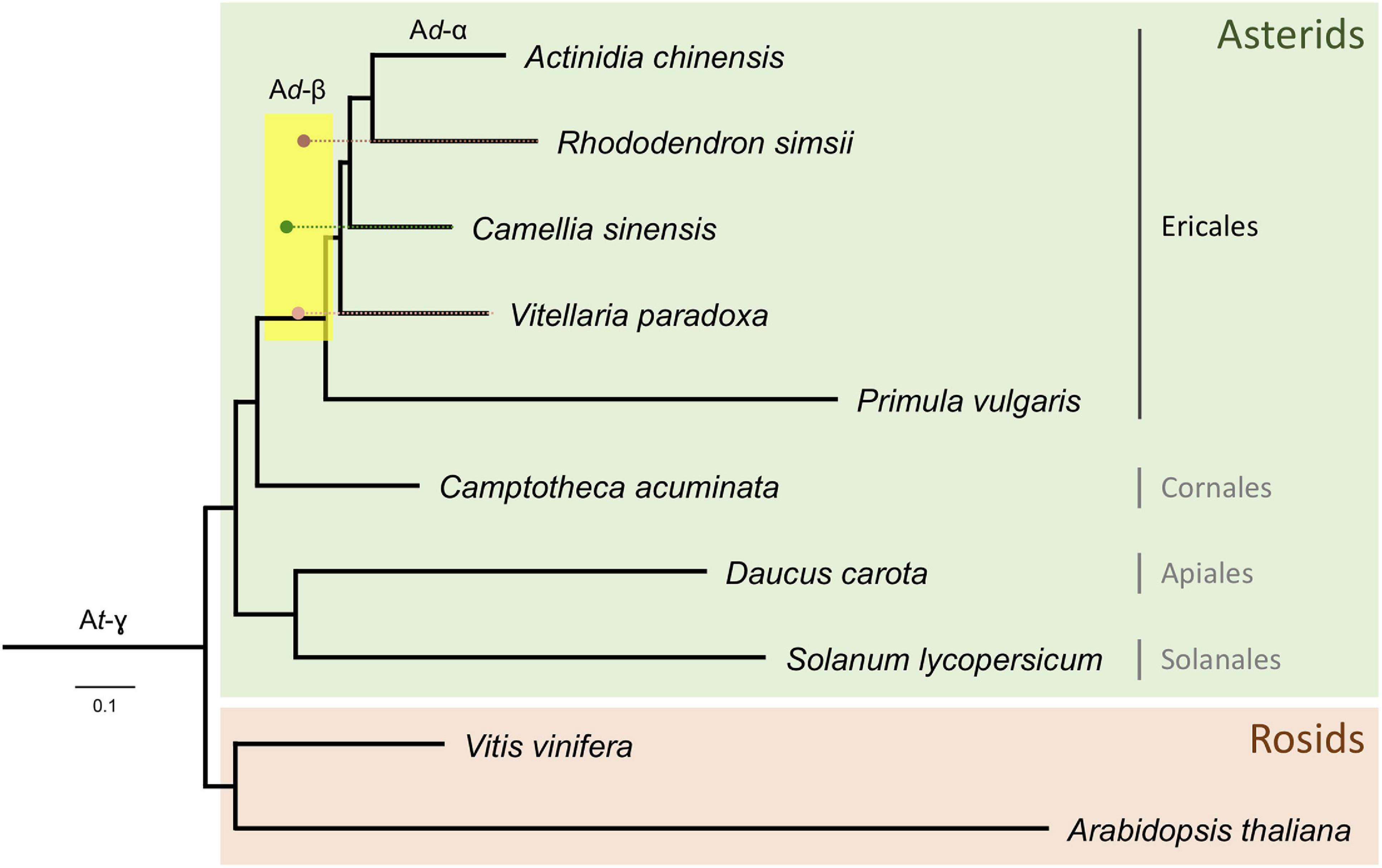
Figure 5. Phylogenetic tree of the Ericales, rooted with Rosids, with branch lengths in KS units. Corrected KS ages (see text for details) of WGD events identified from anchor pairs in R. simsii, C. sinensis, and shea are indicated by brown, green, and pink dots, respectively. A likely shared WGD event in the Ericales (Ad-β) is spanned by the yellow rectangle.
Fatty Acid Biosynthesis Genes in Shea
Shea butter is becoming an increasingly valuable ingredient in cosmetics, but its primary commercial value resides in its serving as a high quality cocoa butter equivalent (CBE) in the international chocolate industry. The fact that shea butter contains higher levels of C18 fatty acids than cocoa butter (Di Vincenzo et al., 2005; Jahurul et al., 2013), particularly in the form of stearic acid, suggests that V. paradoxa may harbor either a greater number and/or more efficient homologs of FA biosynthesis genes than those found in T. cacao. Recent work based on transcriptome data revealed that the shea tree genome indeed encodes a relatively high number of lipid biosynthesis genes (Wei et al., 2019). In this study, the ‘KA01’ reference genome allowed the first whole-genome investigation of the suite of FA biosynthesis genes in shea.
Great strides have been made in characterizing the network of FA biosynthesis enzymes and the underlying genes that encode them. The activity of acetyl-CoA carboxylase (ACC), the key enzyme that catalyzes the necessary and rate-limiting step of de novo FA synthesis, is composed of biotin carboxyl carrier protein (BCCP), biotin carboxylase (BC), and the α- and β-subunits of carboxyltransferase (CT) (Li-Beisson et al., 2013). Previous studies in A. thaliana showed that BCCP is encoded by two genes (CAC1A/BCCP1 and CAC1B/BCCP2), while BC, CTα, and CTβ are encoded by single genes (CAC2, CAC3 and accD, respectively). Of the genes mentioned, accD is the only FA-related gene residing in the plastome; so it was not considered in this analysis. In addition to the ACC enzyme, other key catalytic components of FA biosynthesis belong to the 3-ketoacyl-ACP synthase (KAS) family, members of which include KASIII for the initial combination of acetyl-CoA and malonyl-ACP, KASI for sequential elongation to 16:0-ACP, and KASII (or FAB1) for final extension to 18:0-ACP (see He et al., 2020). KASII activity is therefore a major determinant of the ratio of C18 to C16 FAs in plant cells (Pidkowich et al., 2007). Finally, there are the two sets of fatty acid desaturases (FADs) that catalyze subsequent desaturation steps in the endoplasmic reticulum (ER), namely FAD2 and FAD3. In addition to the genes directly involved in FA biosynthesis, there are others involved in the transmembrane transport of FAs from the plastid to the ER, including acyl-CoA binding proteins (ACBPs), the FATTY ACID EXPORT (FAX) gene, and various Long Chain Acyl-CoA Synthetases (LACS) (Li et al., 2016).
Using the 30 known FA biosynthesis genes in A. thaliana as queries (see Additional File 2: Supplementary Table 7), a search for their homologs in both T. cacao and shea tree resulted in the identification of 35 and 45 genes, respectively, representing 8 gene families. The copy numbers, by gene, are shown in Table 4; and their locations within the shea genome are indicated in Figure 2. Based on copy number, there indeed appears to be more genes associated with FA biosynthesis in shea tree (45) than in either A. thaliana (30) or T. cacao (35). Ketoacyl-ACP synthase (KAS) gene copies are particularly more numerous in shea (9) compared to T. cacao (6), which may contribute to the generally higher lipid content of shea versus cocoa butter. Interestingly, the copy numbers of the FAD2, FAD3, and LACS genes are higher than those found by Wei et al. (2019) based on transcriptome data (4, 5, and 8 vs. 2, 2, and 5, respectively).
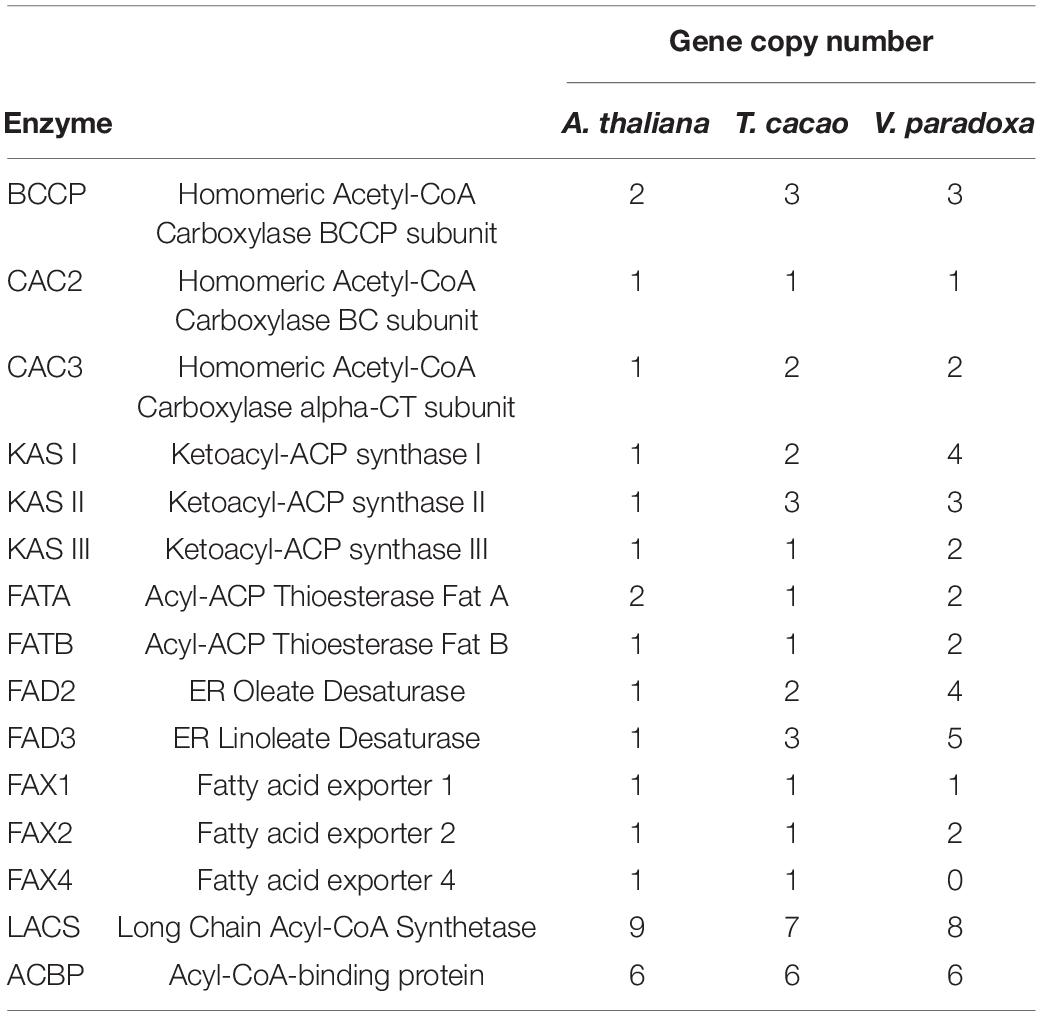
Table 4. Summary table of gene copy numbers of enzymes involved in fatty acid biosynthesis in Arabidopsis thaliana, Theobroma cacao, and Vitellaria paradoxa, using 30 A. thaliana genes as queries.
Based on the genomewide assessment of synteny presented in Figure 2, four of the FA biosynthesis genes exhibiting higher copy number in shea than in either A. thaliana or T. cacao appeared to reside within larger syntenic blocks. For each of those genes (FATB, FAD2, FAD3, and FAX2), subsequent microsynteny analysis confirmed this conclusion (Figure 6; Additional File 1: Supplementary Figures 4–6), suggesting that these copies likely originated in a large-scale event like WGD. Such a signature of large-scale, dispersed duplication for these four genes stands in stark contrast to the clear tandem/local duplication exhibited, for example, by KASIII (Additional File 1: Supplementary Figure 7).
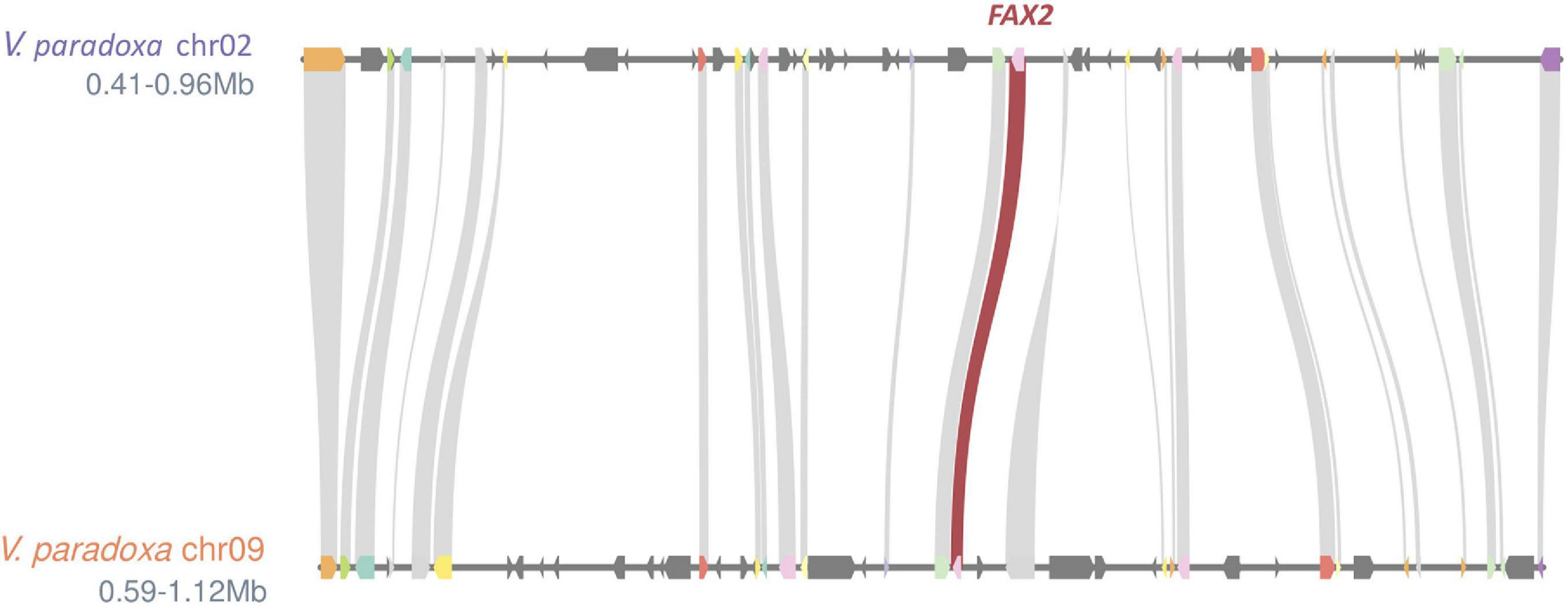
Figure 6. Microsynteny plot showing ∼0.5 Mb regions exhibiting strong synteny (26 gene models) between shea Chromosomes 2 and 9. The existence the two copies of FAX2 within these larger syntenic neighborhoods suggests that shea’s relatively high copy number of this FA biosynthesis gene likely can be traced to a large-scale duplication event like WGD. Microsynteny plots for FATB, FAD2, FAD3, and (as counterpoint) KASIII can be found in Additional File 1: Supplementary Figures 4–7.
Analyses of MEME motifs for each gene family (Additional File 1: Supplementary Figures 8–16) reveal expected similarities within subfamilies. In the FAX family, the FAX2 genes in shea and T. cacao possess an additional motif absent from the A. thaliana genome (Figure 7). Both FAX2 and FAX4 are important for seed oil accumulation. Although no FAX4 homologs were found in the shea genome, two FAX2 genes were identified, compared to the single FAX2 gene found in both A. thaliana and T. cacao.
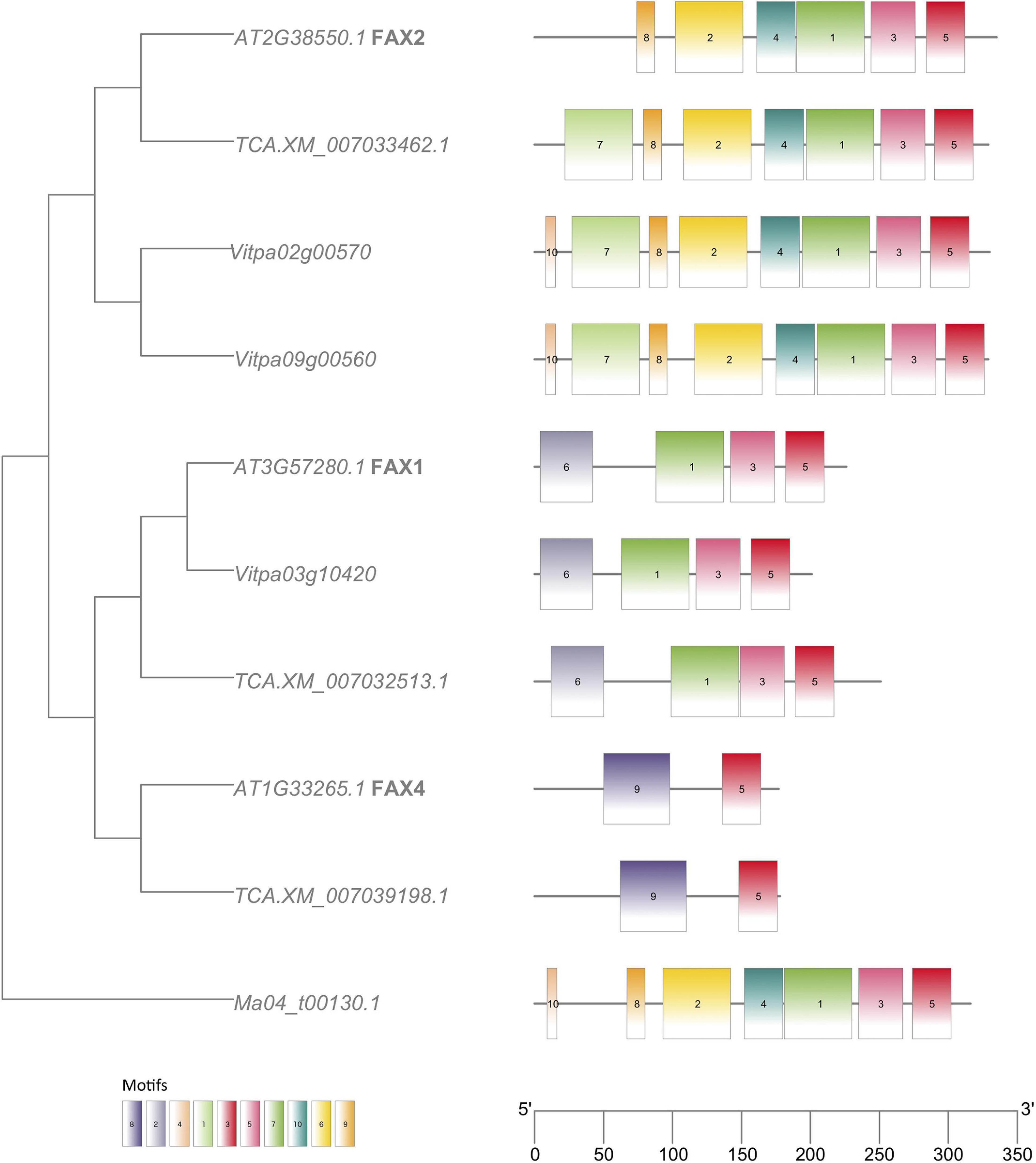
Figure 7. FAX (Fatty acid exporter) gene tree with MEME motifs, comparing orthologs from Arabidopsis thaliana (“AT…”), Theobroma cacao (“TCA.XM…”) and Vitellaria paradoxa (“Vitpa…”). An ortholog from Musa acuminata (“Ma…”) of clade Monocots serves as outgroup. Trees for the other FA biosynthesis genes identified in this study can be found in Additional File 1: Supplementary Figures 8–16.
A SNP Database for the Shea Community
SNP calling under stringent criteria (see Additional File 2: Supplementary Table 2) resulted in the identification of 7.08, 4.45, and 4.47 million bi-allelic SNPs in the resequencing panel of 31 accessions, within reference accession ‘KA01’ (10x data), and within accession ‘ICRAFF 11537’ (Illumina shotgun data), respectively. Of those, a total of 3,488,957 SNPs were found to be shared by at least two of those datasets (see Additional File 1: Supplementary Figure 17). Considering only those ∼3.5 million high-confidence SNPs, SNP density is observed to be similar across shea’s 12 chromosomes, with an average of 1 SNP every 189 bp (Table 2). SnpEff functional annotation of the set of high-confidence SNPs generated 5.6 million predicted effects, more than 97% of which were assigned to the MODIFIER impact class (i.e., no evidence of impact on function; see Table 5). The remaining effects, associated with 152,676 SNPs, are predicted to have HIGH, MODERATE, or LOW impacts on the functions 26,290 implicated genes, as summarized in Table 5.
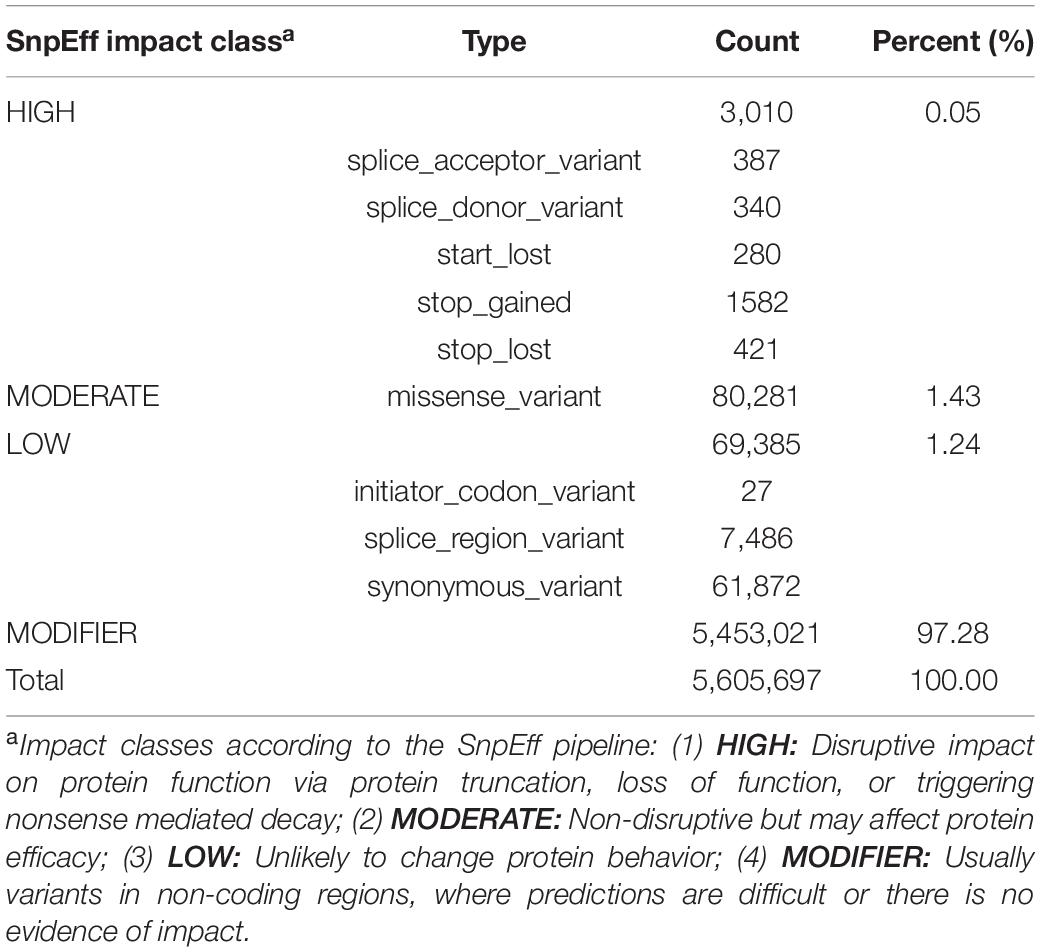
Table 5. Summary of SnpEff functional analysis.
Following the variant selection strategies described above (see section “Materials and Methods”), two different SNP panels were developed for ready use by shea improvement programs. The first, intended to support genome-wide association studies, general germplasm characterization work, linkage map development, genomic selection protocols, trait dissection, and the like, consists of 104,211 SNPs distributed relatively evenly across the reference genome (average spacing of 6,350 bp). The second panel, intended to enable the much more specific task of characterizing the diversity of FA biosynthesis genes within shea, consists of 90 SNPs, two per putative FA biosynthesis homolog identified in this study. Detailed information about the SNPs comprising these two panels can be found in Additional Files 4, 5. For programs interested in developing other targeted SNP panels, details of all ∼3.5 million high-confidence SNPs are also provided (see section “Data Availability Statement”).
Discussion
Despite the shea tree’s long recognized value as a dominant agroecological species fulfilling vital sociocultural, economic, environmental, and nutritional functions for millions of households across rural SSA, it remains semi-domesticated with virtually no history of systematic genetic improvement. The central contribution of this research is an annotated 656.7 Mb reference genome assembly of Vitellaria paradoxa, a necessary resource for future genome-enabled conservation and breeding efforts. Hi-C guided contig assembly resulted in 12 distinct pseudo-molecules, backing previous cytological investigations that placed the chromosome number of V. paradoxa at 2n = 24; and reference annotation revealed 38,505 coding gene models with only 4% missing BUSCO genes, suggesting an acceptable level of completeness for a first assembly.
The annotated reference enabled a whole genome duplication (WGD) analysis which revealed not only that shea underwent a WGD event but that this event (Ad-β) was shared with other important species within the cosmopolitan order Ericales, including C. sinensis, R. simsii, and A. chinensis (and likely P. vulgaris). Evidence of the even earlier WGD event prior to the Asterid-Rosid divergence (At-γ; 150.4–159.3 Mya and shared with V. vinifera) was also detected. Given that gene duplication in general, regardless of mechanism, presents new opportunities for evolution in plants (see e.g., Dubcovsky and Dvorak, 2007; Magadum et al., 2013), this pair of ancient WGD events in shea bodes well in terms of expanding the allelic space relevant to artificial selection targets.
As a first exploration of this idea, the annotated reference was used to provide a first glimpse into the suite of putative fatty acid (FA) biosynthesis genes in shea. Analysis of 30 well-characterized FA biosynthesis genes from A. thaliana resulted in the identification of 45 orthologs in shea, representing 8 gene families. Based on copy numbers, there appears to be more genes associated with FA biosynthesis in shea tree than in T. cacao (35). As a family, Ketoacyl-ACP synthase (KAS) genes in particular were found at higher copy number in shea, which may contribute to the generally higher lipid content of shea versus cocoa butter. Also worth noting are shea’s higher copy numbers of ER desaturase genes FAD2 and FAD3, key enzymes for Omega-3 FA biosynthesis and thus of potential relevance not only to shea’s nutritional value but also its quality for use in cosmetics. Microsynteny analysis implicated a large-scale duplication event, perhaps the Ad-β WGD event described above, in the relatively high copy numbers not only of these ER desaturase genes, but of the FATB and FAX2 genes as well. With the loci governing FA biosynthesis in shea now identified, targeted characterizations of germplasm collections are now possible, a first step toward understanding the role of extant allelic diversity on FA composition. In a long-generation species like shea, such an understanding is required for developing rational strategies for selection, whether within naturally regenerating parkland stands or among breeding lines. Of course, the value of the genomic resources presented here is not limited to FA traits, however important they may be. Combined with accurate phenotyping data from proper germplasm collections (e.g., NAM populations, association mapping panels, etc.), this chromosome-scale annotated genome can facilitate efficient progress on a multitude of traits of importance, including Pestalotia resistance, fruit morphology, flowering time, precocity, tocopherol content, tree architecture, amenability to propagation, and tolerances to any number of abiotic/environmental stresses.
In addition to the raw sequence data, assembled transcriptome, and annotated reference genome (see section “Data Availability Statement”), a database of ∼3.5 million physically anchored and functionally annotated SNPs has also been generated and made publicly available as part of this work. Based not only on variation within reference accession ‘KA01,’ including those regions with sufficient heterozygosity for phased assembly (haplotigs), but also on variation detected within a larger diversity panel, the SNP database should prove to be a widely useful resource for shea improvement programs across SSA. To facilitate the adoption and practical use of this resource, two pre-selected panels of high-confidence annotated SNPs have been developed, one (∼100k SNPs) in support of genome-wide genotyping and the second (90 SNPs) specifically targeting putative FA biosynthesis genes. Effectively serving as first generation in silico SNP chips, these panels provide a ready starting point for programs seeking to undertake the work of genome-assisted germplasm curation, conservation, trait association mapping, and breeding in shea. For fine-mapping or for strategic targeting of other genes of interest, the full SNP database is also available.
Traditionally, the processes of plant domestication and breeding, not to mention deployment of improved genetics and production systems at scale, have been extremely long-term undertakings, even for the most cooperative and fast-cycling of model, annual species. To suggest such painstaking work for a slow-growing tree species like shea, with generation times of 10–25 years and relatively limited global importance, seems wishful at best. And yet, the recent convergence of a suite of biotechnologies (e.g., affordable dense genotyping, gene editing, etc.), breeding strategies [e.g., tricot analysis (Etten et al., 2019) and breeding seedling orchards (Barnes, 1995)], statistical methods [e.g., genomic selection (Heffner et al., 2009) and landscape GWAS (Lasky et al., 2018)], and engagement tools (e.g., smartphone-enabled citizen science) bring the domestication and breeding of even the shea tree into the realm of possibility. Alone, the high-quality reference genome developed here is by no means sufficient for such work; but it is an important foundational resource capable of serving the shea improvement community for years to come. As it has in so many other crops, a well-annotated genome presents new possibilities and is thus likely to stimulate further research and investment.
Data Availability Statement
All raw sequence data generated for this study are publicly available as NCBI Bioproject PRJNA729422, including: (1) Raw PacBio (SMRT) gDNA sequence data of reference accession ‘KA01’ (Biosamples SAMN19116134 and SAMN21031866); and (2) Raw RNAseq data of accessions ‘KA01’ and ‘ICRAFF 11537’ (Biosamples SAMN19488939 and SAMN19488940). All analytical results are publicly available from the ORCAE database (https://bioinformatics.psb.ugent.be/orcae), including: (1) The final Hi-C guided genome assembly of Vitellaria paradoxa reference accession ‘KA01’; (2) A supplemental file of final ‘KA01’ haplotigs; (3) The final transcriptome assembly; (4) A complete set of gene descriptor files; and (5) All genome annotation files; and 6) A vcf file of all ∼3.5 million high-confidence SNPs.
Author Contributions
IH, FP, AD, and RJ conceived and initiated the study. FP, J-MB, AM, and MB acquired, managed, and facilitated the movement of all relevant germplasm. PH and AV coordinated all sample processing and library preparation. IH, ATOM, SK, and SS built the reference assembly. IH and ATOM assembled the transcriptome. XM and YV annotated the assembly and performed the analyses for whole genome duplication and FA biosynthesis genes. SC and AV did the variant calling. IH oversaw all aspects of the study and led the manuscript preparation. All the authors revised the manuscript and approved the final version.
Funding
IH, FP, AD, and RJ acknowledge the Basic Research to Enhance Agricultural Development (BREAD) program (award #1543942), jointly funded by the National Science Foundation (NSF) and the Bill & Melinda Gates Foundation (BMGF), as the primary source of funding for this project. RJ, PH, J-MB, and AM acknowledge that this work was undertaken as part of, and partially funded by, the CGIAR Research Program (CRP) on Forests, Trees, and Agroforestry (FTA) and the CGIAR (ICRAF) Genebank Platform. YV acknowledges funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program (grant agreement no. 833522) and from Ghent University (Methusalem funding, BOF.MET.2021.0005.01). AV, RJ, and PH acknowledge funding from the AOCC and the UC Davis Seed Biotechnology Center.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are indebted to the following colleagues across the shea belt who collected tissue on behalf of the diversity panel for this project: Christophe Djekota and Oumar Dany (Chad); Nafan Diarrassouba (Côte d’Ivoire); Koutchoukalo Faustine Aleza, Césaire Gnanglè, and Yokoue Kiatti (Benin); Alain Tsobeng and Denis Nna (Cameroon); Felix Ugese (Nigeria); Adja Madjiguène Diallo Diop, Ismaïla Diallo, and Marcel Badji (Senegal); Samson Gwali, Clement Okia, and Moses Okao (Uganda); and Wisdom Edem Anyomi (Ghana). For their essential technical contributions to this work, additional acknowledgment is due to Robert Kariba and Samuel Muthemba (AOCC Genomics Laboratory); Jonathan Featherston (Agricultural Research Council); Gordon Simpson and Matthew Parker (University of Dundee); and Julius Yeboah (CRIG). We also gratefully acknowledge Eric Y. Danquah for his support of this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.720670/full#supplementary-material
Additional File 1: Supplementary Figure 1 | Hi-C post-scaffolding heatmap of the primary contigs of V. paradoxa reference accession ‘KA01.’
Additional File 1: Supplementary Figure 2 | One-to-one orthologous KS distributions between shea and three other species within the order Ericales: tea plant (Camellia sinensis), Rhododendron simsii, and kiwifruit (Actinidia chinensis).
Additional File 1: Supplementary Figure 3 | One-to-one orthologous KS distribution between grape (Vitis vinifera) and shea, compared with those between grape and four other species within the order Ericales [tea plant, kiwifruit, Rhododendron simsii, and Primula vulgaris (primrose)].
Additional File 1: Supplementary Figures 4–7 | Microsynteny plots of the regions containing copies of fatty acid biosynthesis genes FATB, FAD2, FAD3, and KASIII genes in KA01.
Additional File 1: Supplementary Figures 8–16 | Gene trees with MEME motifs, comparing fatty acid biosynthesis orthologs from Arabidopsis thaliana, Theobroma cacao, and Vitellaria paradoxa. Orthologs from Musa acuminata of clade Monocots serve as outgroups.
Additional File 1: Supplementary Figure 17 | Venn diagram of SNPs called using the three different data sets in this study.
Additional File 2: Supplementary Table 1 | Geographical provenances of the 31 shea tree accessions used in the skim resequencing panel to facilitate variant calling.
Additional File 2: Supplementary Table 2 | Filters used for SNP calling within the three datasets in this study.
Additional File 2: Supplementary Table 3 | Summary of raw PacBio data obtained for V. paradoxa reference accession ‘KA01’ via 25 SMRT cells.
Additional File 2: Supplementary Table 4 | Descriptive statistics of the various transcriptome assemblies, including the final “Integrated” assembly used for annotation.
Additional File 2: Supplementary Table 5 | Structural annotation summary statistics.
Additional File 2: Supplementary Table 6 | KS peak values of whole-genome duplication events detected in the Ericales.
Additional File 2: Supplementary Table 7 | Summary table of the 30 fatty acid biosynthesis genes from A. thaliana.
Additional File 3: Supplementary Text 1 | Complete FALCON command log for this study, including all specified assembly parameters.
Additional File 3: Supplementary Text 2 | Complete Purge Haplotigs command log for this study, including all specified parameters.
Additional File 4 | Details of the 90 SNPs comprising the FA biosynthesis gene SNP panel for V. paradoxa.
Additional File 5 | Details of the 104,211 SNPs comprising the GWAS SNP panel for V. paradoxa.
Footnotes
- ^ https://github.com/PacificBiosciences/GenomicConsensus
- ^ http://aralip.plantbiology.msu.edu/pathways
- ^ https://www.arabidopsis.org/
- ^ https://bioinformatics.psb.ugent.be/plaza/
References
Abdulai, I., Krutovsky, K. V., and Finkeldey, R. (2017). Morphological and genetic diversity of shea tree (Vitellaria paradoxa) in the savannah regions of Ghana. Genet. Resour. Crop. Evol. 64, 1253–1268. doi: 10.1007/s10722-016-0434-8
Akihisa, T., Kojima, N., Katoh, N., Kikuchi, T., Fukatsu, M., Shimizu, N., et al. (2011). Triacylglycerol and triterpene ester composition of shea nuts from seven African countries. J. Oleo Sci. 60, 385–391.
Akrofi, A. Y., and Amoah, F. M. (2009). Pestalotia spp. causes leaf spot of Vitellaria paradoxa in Ghana. Afr. J. Agric. Res. 4, 330–333.
Allal, F., Piombo, G., Kelly, B. A., Okullo, J. B. L., Thiam, M., Diallo, O. B., et al. (2013). Fatty acid and tocopherol patterns of variation within the natural range of the shea tree (Vitellaria paradoxa). Agroforest. Syst. 5, 1065–1082. doi: 10.1007/s10457-013-9621-1
Allal, F., Sanou, H., Millet, L., Vaillant, A., Camus-Kulandaivelu, L., Logossa, Z. A., et al. (2011). Past climate changes explain the phylogeography of Vitellaria paradoxa over Africa. Heredity 107, 174–186. doi: 10.1038/hdy.2011.5
Allal, F., Vaillant, A., Sanou, H., Kelly, B., and Bouvet, J.-M. (2008). Isolation and characterization of new microsatellite markers in shea tree (Vitellaria paradoxa C. F. Gaertn). Mol. Ecol. Resour. 8, 822–824. doi: 10.1111/j.1755-0998.2007.02079.x
Barnes, R. (1995). The Breeding Seedling Orchard in Multiple Population Breeding Strategy. Italy: Food and Agriculture Organization.
Bickhart, D. M., Rosen, B. D., Koren, S., Sayre, B. L., Hastie, A. R., Chan, S., et al. (2017). Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 49, 643–650. doi: 10.1038/ng.3802
Bockel, L., Veyrier, M., Gopal, P., Adu, A., and Ouedraogo, A. (2020). Shea Value Chain As a Key Pro-Poor Carbon-Fixing Engine in West Africa. Accra: FAO and Global Shea Alliance. doi: 10.4060/ca7406en
Boffa, J.-M. (1999). Agroforestry Parklands in Sub-Saharan Africa. FAO Conservation Guide (FAO). Italy: Food and Agriculture Organization.
Boffa, J.-M., Knudson, D. M., Yameogo, G., and Nikiema, P. (1996). Shea Nut (Vitellaria Paradoxa) Production and Collection in Agroforestry Parklands of Burkina Faso. Non-Wood Forest Products (FAO). Available online at: https://agris.fao.org/agris-search/search.do?recordID=XF1997071761 (accessed August 11, 2021).
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bouvet, J.-M., Fontaine, C., Sanou, H., and Cardi, C. (2004). An analysis of the pattern of genetic variation in Vitellaria paradoxa using RAPD markers. Agrofor. Syst. 60, 61–69. doi: 10.1023/B:AGFO.0000009405.74331.74
Bouvet, J.-M., Kelly, B., Sanou, H., and Allal, F. (2008). Comparison of marker- and pedigree-based methods for estimating heritability in an agroforestry population of Vitellaria paradoxa C.F. Gaertn. (shea tree). Genet. Resour. Crop. Evol. 55:1291. doi: 10.1007/s10722-008-9328-8
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M., and Borodovsky, M. (2021). BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinform. 3:lqaa108. doi: 10.1093/nargab/lqaa108
Burton, J. N., Adey, A., Patwardhan, R. P., Qiu, R., Kitzman, J. O., and Shendure, J. (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. doi: 10.1038/nbt.2727
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2105-10-421
Cardi, C., Vaillant, A., Sanou, H., Kelly, B. A., and Bouvet, J.-M. (2005). Characterization of microsatellite markers in the shea tree (Vitellaria paradoxa C. F Gaertn) in Mali. Mol. Ecol. Notes 5, 524–526. doi: 10.1111/j.1471-8286.2005.00980.x
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chevalier, A. (1948). Recent research on the Shea Butter Tree. Rev. Int. Bot. Appl. Agric. Trop. 28, 241–256.
Chin, C.-S., Peluso, P., Sedlazeck, F. J., Nattestad, M., Concepcion, G. T., Clum, A., et al. (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054. doi: 10.1038/nmeth.4035
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: sNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Cortes, L. T., Zhang, Z., and Yu, J. (2021). Status and prospects of genome-wide association studies in plants. Plant Genome 14:e20077. doi: 10.1002/tpg2.20077
Desmarest, J. (1958). Observations sur la population de karités de Niangoloko de 1953 à 1957. Oléagineux 5, 449–455.
Di Vincenzo, D., Maranz, S., Serraiocco, A., Vito, R., Wiesman, Z., and Bianchi, G. (2005). Regional variation in shea butter lipid and triterpene composition in four African countries. J. Agric. Food Chem. 53, 7473–7479. doi: 10.1021/jf0509759
Dubcovsky, J., and Dvorak, J. (2007). Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 316, 1862–1866. doi: 10.1126/science.1143986
Dudchenko, O., Shamim, M. S., Batra, S. S., Durand, N. C., Musial, N. T., Mostofa, R., et al. (2018). The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. Biorxiv 254797. doi: 10.1101/254797 Preprint.
Elias, M., and Carney, J. (2007). African Shea Butter: a Feminized Subsidy from Nature. Afr. J. Int. Afr. Inst. 77, 37–62. doi: 10.3366/afr.2007.77.1.37
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Etten, J. V., Beza, E., Calderer, L., Duijvendijk, K. V., Fadda, C., Fantahun, B., et al. (2019). First experiences with a novel farmer citizen science approach: crowdsourcing participatory variety selection through on-farm triadic comparisons of technologies (tricot). Exp. Agric. 55, 275–296. doi: 10.1017/S0014479716000739
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. PNAS 117, 9451–9457. doi: 10.1073/pnas.1921046117
Fontaine, C., Lovett, P. N., Sanou, H., Maley, J., and Bouvet, J.-M. (2004). Genetic diversity of the shea tree (Vitellaria paradoxa C.F. Gaertn), detected by RAPD and chloroplast microsatellite markers. Heredity 93, 639–648. doi: 10.1038/sj.hdy.6800591
Gijsbers, H. J. M., Kessler, J. J., and Knevel, M. K. (1994). Dynamics and natural regeneration of woody species in farmed parklands in the Sahel region (Province of Passore. Burkina Faso). For. Ecol. Manag. 64, 1–12. doi: 10.1016/0378-1127(94)90122-8
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Gwali, S., Vaillant, A., Nakabonge, G., Okullo, J. B. L., Eilu, G., Muchugi, A., et al. (2015). Genetic diversity in shea tree (Vitellaria paradoxa subspecies nilotica) ethno-varieties in Uganda assessed with microsatellite markers. For. Trees Livelih. 24, 163–175. doi: 10.1080/14728028.2014.956808
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
Hackl, T., Hedrich, R., Schultz, J., and Förster, F. (2014). proovread : large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30, 3004–3011. doi: 10.1093/bioinformatics/btu392
He, M., Qin, C.-X., Wang, X., and Ding, N.-Z. (2020). Plant Unsaturated Fatty Acids: biosynthesis and Regulation. Front. Plant Sci. 11:390. doi: 10.3389/fpls.2020.00390
Heffner, E. L., Sorrells, M. E., and Jannink, J.-L. (2009). Genomic Selection for Crop Improvement. Crop Science 49, 1–12. doi: 10.2135/cropsci2008.08.0512
Hilditch, T. P., and Saletore, S. A. (1931). fatty acids and glycerides of solid seed fats. 1. composition of the seed fats of allanblackia stuhlmannii, pentadesma butyracea, butyrospermum parkii (shea) and valeria indica (dhupa). J. Soc. Chem. Ind. 50, 468T–472T.
Honfo, F. G., Akissoe, N., Linnemann, A. R., Soumanou, M., and Boekel, M. A. J. S. V. (2014). Nutritional Composition of Shea Products and Chemical Properties of Shea Butter: a Review. Crit. Rev. Food Sci. Nutr. 54, 673–686. doi: 10.1080/10408398.2011.604142
LMC International (2014). Alternatives to Cocoa Butter: the outlook for CBEs, CBSs and exotic fats. United Kingdom: LMC International.
Jahurul, M. H. A., Zaidul, I. S. M., Norulaini, N. A. N., Sahena, F., Jinap, S., Azmir, J., et al. (2013). Cocoa butter fats and possibilities of substitution in food products concerning cocoa varieties, alternative sources, extraction methods, composition, and characteristics. J. Food Eng. 117, 467–476. doi: 10.1016/j.jfoodeng.2012.09.024
Johnson, M. (1991). “Cytology” in The Genera of Sapotaceae. ed. T. D. Pennington (Gran Bretaña: Kew Publishing, Surrey). 15–22.
Katoh, K., and Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kelly, B. A., Hardy, O., and Bouvet, J.-M. (2004). Temporal and spatial genetic structure in Vitellaria paradoxa (shea tree) in an agroforestry system in southern Mali. Mol. Ecol. 13, 1231–1240. doi: 10.1111/j.1365-294X.2004.02144.x
Kronenberg, Z. N., Rhie, A., Koren, S., Concepcion, G. T., Peluso, P., Munson, K. M., et al. (2021). Extended haplotype-phasing of long-read de novo genome assemblies using Hi-C. Nat. Commun. 12:1935. doi: 10.1038/s41467-020-20536-y
Lasky, J. R., Forester, B. R., and Reimherr, M. (2018). Coherent synthesis of genomic associations with phenotypes and home environments. Mol. Ecol. Resour. 18, 91–106. doi: 10.1111/1755-0998.12714
Li, H. (2015). BFC: correcting Illumina sequencing errors. Bioinformatics 31, 2885–2887. doi: 10.1093/bioinformatics/btv290
Li, N., Xu, C., Li-Beisson, Y., and Philippar, K. (2016). Fatty Acid and Lipid Transport in Plant Cells. Trends Plant Sci. 21, 145–158. doi: 10.1016/j.tplants.2015.10.011
Li-Beisson, Y., Shorrosh, B., Beisson, F., Andersson, M., Arondel, V., Bates, P., et al. (2013). Acyl-lipid metabolism. Arabidopsis Book 11:e0161.
Logossa, Z. A., Camus-Kulandaivelu, L., Allal, F., Vaillant, A., Sanou, H., Kokou, K., et al. (2011). Molecular data reveal isolation by distance and past population expansion for the shea tree (Vitellaria paradoxa C.F. Gaertn) in West Africa. Mol. Ecol. 20, 4009–4027. doi: 10.1111/j.1365-294X.2011.05249.x
Lovett, P. N., and Haq, N. (2000). Diversity of the Sheanut tree (Vitellaria paradoxa C.F. Gaertn.) in Ghana. Genet. Resour. Crop Evol. 47, 293–304. doi: 10.1023/A:1008710331325
Luedeling, E., and Neufeldt, H. (2012). Carbon sequestration potential of parkland agroforestry in the Sahel. Clim. Change 115, 443–461. doi: 10.1007/s10584-012-0438-0
MacManes, M. D. (2016). Establishing evidenced-based best practice for the de novo assembly and evaluation of transcriptomes from non-model organisms. BioRxiv 035642. doi: 10.1101/035642
Magadum, S., Banerjee, U., Murugan, P., Gangapur, D., and Ravikesavan, R. (2013). Gene duplication as a major force in evolution. J. Genet. 92, 155–161. doi: 10.1007/s12041-013-0212-8
Maranz, S., Kpikpi, W., Wiesman, Z., De Saint Sauveur, A., and Chapagain, B. (2004a). Nutritional values and indigenous preferences for Shea Fruits(vitellaria paradoxa C.F. Gaertn. F.) in African Agroforestry Parklands. Econ. Bot. 58, 588–600. doi: 10.1663/0013-0001(2004)058[0588:nvaipf]2.0.co;2
Maranz, S., Wiesman, Z., Bisgaard, J., and Bianchi, G. (2004b). Germplasm resources of Vitellaria paradoxa based on variations in fat composition across the species distribution range. Agrofor. Syst. 60, 71–76. doi: 10.1023/B:AGFO.0000009406.19593.90
Maranz, S., and Wiesman, Z. (2003). Evidence for indigenous selection and distribution of the shea tree, Vitellaria paradoxa, and its potential significance to prevailing parkland savanna tree patterns in sub-Saharan Africa north of the equator. J. Biogeogr. 30, 1505–1516. doi: 10.1046/j.1365-2699.2003.00892.x
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Miège, J. (1954). Nombres chromosomiques et répartition géographique de quelques plantes tropicales et équatoriales. Rev. Cytol. Biol. Vég. 15, 312–348.
Naughton, C. C., Lovett, P. N., and Mihelcic, J. R. (2015). Land suitability modeling of shea (Vitellaria paradoxa) distribution across sub-Saharan Africa. Appl. Geogr. 58, 217–227. doi: 10.1016/j.apgeog.2015.02.007
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Pidkowich, M. S., Nguyen, H. T., Heilmann, I., Ischebeck, T., and Shanklin, J. (2007). Modulating seed β-ketoacyl-acyl carrier protein synthase II level converts the composition of a temperate seed oil to that of a palm-like tropical oil. PNAS 104, 4742–4747. doi: 10.1073/pnas.0611141104
Poudyal, M. (2011). Chiefs and Trees: tenures and Incentives in the Management and Use of Two Multipurpose Tree Species in Agroforestry Parklands in Northern Ghana. Soc. Nat. Resour. 24, 1063–1077. doi: 10.1080/08941920.2010.523059
Pouliot, M. (2012). Contribution of “Women’s Gold” to West African Livelihoods: the Case of Shea (Vitellaria paradoxa) in Burkina Faso. Econ. Bot. 66, 237–248. doi: 10.1007/s12231-012-9203-6
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Proost, S., Fostier, J., De Witte, D., Dhoedt, B., Demeester, P., Van de Peer, Y., et al. (2012). i-ADHoRe 3.0—fast and sensitive detection of genomic homology in extremely large data sets. Nucleic Acids Res. 40:e11. doi: 10.1093/nar/gkr955
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Roach, M. J., Schmidt, S. A., and Borneman, A. R. (2018). Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 19:460. doi: 10.1186/s12859-018-2485-7
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ. 4:e2584. doi: 10.7717/peerj.2584
Rousseau, K., Gautier, D., and Wardell, D. A. (2015). Coping with the Upheavals of Globalization in the Shea Value Chain: the Maintenance and Relevance of Upstream Shea Nut Supply Chain Organization in Western Burkina Faso. World Dev. 66, 413–427. doi: 10.1016/j.worlddev.2014.09.004
Ræbild, A., Hansen, U. B., and Kambou, S. (2012). Regeneration of Vitellaria paradoxa and Parkia biglobosa in a parkland in southern Burkina Faso. Agrofor. Syst. 85, 443–453. doi: 10.1007/s10457-011-9397-0
Sanou, H., Lovett, P. N., and Bouvet, J.-M. (2005). Comparison of quantitative and molecular variation in agroforestry populations of the shea tree (Vitellaria paradoxa C.F. Gaertn) in Mali. Mol. Ecol. 14, 2601–2610. doi: 10.1111/j.1365-294X.2005.02601.x
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Simillion, C., Janssens, K., Sterck, L., and Van de Peer, Y. (2008). i-ADHoRe 2.0: an improved tool to detect degenerated genomic homology using genomic profiles. Bioinformatics 24, 127–128. doi: 10.1093/bioinformatics/btm449
Slater, G. S. C., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 6:31. doi: 10.1186/1471-2105-6-31
Smit, A. F. A., and Hubley, R. (2013). 2015 RepeatMasker Open-4.0. RepeatMasker. Available online at: http://www.repeatmasker.org
Takimoto, A., Nair, V. D., and Nair, P. K. R. (2009). Contribution of trees to soil carbon sequestration under agroforestry systems in the West African Sahel. Agrofor. Syst. 76, 11–25. doi: 10.1007/978-90-481-3323-9_2
Tang, H., Bowers, J. E., Wang, X., Ming, R., Alam, M., and Paterson, A. H. (2008). Synteny and Collinearity in Plant Genomes. Science 320, 486–488. doi: 10.1126/science.1153917
Ugese, F. D., Baiyeri, P. K., and Mbah, B. N. (2010). Fatty acid profile of shea tree (Vitellaria paradoxa C. F. Gaertn.) Seeds from the Savanna of Nigeria. For. Trees Livelih. 19, 393–398. doi: 10.1080/14728028.2010.9752680
Van Bel, M., Diels, T., Vancaester, E., Kreft, L., Botzki, A., Van de Peer, Y., et al. (2018). PLAZA 4.0: an integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 46, D1190–D1196. doi: 10.1093/nar/gkx1002
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wei, Y., Ji, B., Siewers, V., Xu, D., Halkier, B. A., and Nielsen, J. (2019). Identification of genes involved in shea butter biosynthesis from Vitellaria paradoxa fruits through transcriptomics and functional heterologous expression. Appl. Microbiol. Biotechnol. 103, 3727–3736. doi: 10.1007/s00253-019-09720-3
Whelan, S., Irisarri, I., and Burki, F. (2018). PREQUAL: detecting non-homologous characters in sets of unaligned homologous sequences. Bioinformatics 34, 3929–3930. doi: 10.1093/bioinformatics/bty448
Wu, H., Ma, T., Kang, M., Ai, F., Zhang, J., Dong, G., et al. (2019). A high-quality Actinidia chinensis (kiwifruit) genome. Hortic. Res. 6, 1–9. doi: 10.1038/s41438-019-0202-y
Xia, E.-H., Zhang, H.-B., Sheng, J., Li, K., Zhang, Q.-J., Kim, C., et al. (2017). The Tea Tree Genome Provides Insights into Tea Flavor and Independent Evolution of Caffeine Biosynthesis. Mol. Plant 10, 866–877. doi: 10.1016/j.molp.2017.04.002
Yang, F.-S., Nie, S., Liu, H., Shi, T.-L., Tian, X.-C., Zhou, S.-S., et al. (2020). Chromosome-level genome assembly of a parent species of widely cultivated azaleas. Nat. Commun. 11:5269. doi: 10.1038/s41467-020-18771-4
Yang, Z. (2007). PAML 4: phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Keywords: shea tree, Vitellaria paradoxa, reference genome, fatty acids, SNPs, whole genome duplication, plant breeding
Citation: Hale I, Ma X, Melo ATO, Padi FK, Hendre PS, Kingan SB, Sullivan ST, Chen S, Boffa J-M, Muchugi A, Danquah A, Barnor MT, Jamnadass R, Van de Peer Y and Van Deynze A (2021) Genomic Resources to Guide Improvement of the Shea Tree. Front. Plant Sci. 12:720670. doi: 10.3389/fpls.2021.720670
Received: 04 June 2021; Accepted: 04 August 2021;
Published: 09 September 2021.
Edited by:
Simon E. Bull, ETH Zürich, SwitzerlandReviewed by:
Chinedu Charles Nwafor, University of Nebraska-Lincoln, United StatesWeiwei Wen, Huazhong Agricultural University, China
Copyright © 2021 Hale, Ma, Melo, Padi, Hendre, Kingan, Sullivan, Chen, Boffa, Muchugi, Danquah, Barnor, Jamnadass, Van de Peer and Van Deynze. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iago Hale, aWFnby5oYWxlQHVuaC5lZHU=
†These authors share senior authorship