 Weichao Ren1†
Weichao Ren1† Zhehui Jiang2†Meiqi Zhang2†
Zhehui Jiang2†Meiqi Zhang2† Lingyang Kong1Houliang Zhang3Yunwei Liu3Qifeng Fu4*
Lingyang Kong1Houliang Zhang3Yunwei Liu3Qifeng Fu4* Wei Ma1,4*
Wei Ma1,4*- 1School of Pharmacy, Heilongjiang University of Chinese Medicine, Harbin, China
- 2School of Forestry, Northeast Forestry University, Harbin, China
- 3Yichun Branch of Heilongjiang Academy of Forestry, Yichun, China
- 4Experimental Teaching and Training Center, Heilongjiang University of Chinese Medicine, Harbin, China
Salix floderusii is a rare alpine tree species in the Salix genus. Unfortunately, no extensive germplasm identification, molecular phylogeny, and chloroplast genomics of this plant have been conducted. We sequenced the chloroplast (cp) genome of S. floderusii for the first time using second-generation sequencing technology. The cp genome was 155,540 bp long, including a large single-copy region (LSC, 84,401 bp), a small single-copy region (SSC, 16,221 bp), and inverted repeat regions (IR, 54,918 bp). A total of 131 genes were identified, including 86 protein genes, 37 tRNA genes, and 8 rRNA genes. The S. floderusii cp genome contains 1 complement repeat, 24 forward repeats, 17 palindromic repeats, and 7 reverse repeats. Analysis of the IR borders showed that the IRa and IRb regions of S. floderusii and Salix caprea were shorter than those of Salix cinerea, which may affect plastome evolution. Furthermore, four highly variable regions were found, including the rpl22 coding region, psbM/trnD-GUC non-coding region, petA/psbJ non-coding region, and ycf1 coding region. These high variable regions can be used as candidate molecular markers and as a reference for identifying future Salix species. In addition, phylogenetic analysis indicated that the cp genome of S. floderusii is sister to Salix cupularis and belongs to the Subgenus Vetrix. Genes (Sf-trnI, Sf-PpsbA, aadA, Sf-TpsbA, Sf-trnA) obtained via cloning were inserted into the pBluescript II SK (+) to yield the cp expression vectors, which harbored the selectable marker gene aadA. The results of a spectinomycin resistance test indicated that the cp expression vector had been successfully constructed. Moreover, the aadA gene was efficiently expressed under the regulation of predicted regulatory elements. The present study provides a solid foundation for establishing subsequent S. floderusii cp transformation systems and developing strategies for the genetic improvement of S. floderusii.
Introduction
Salix floderusii (Nakai) is a perennial shrub and member of the genus of Salix in the family Salicaceae. S. floderusii can reach a height of 4–6 m and grow in rocky areas near the top of mountains, 1,000–1,500 m above sea level. S. floderusii is mainly distributed in northeast China, Korea, and the Russian Far East. As an alpine tree species, the unique living environment of S. floderusii may influence its morphological characteristics and species evolution. The flora reflects the composition, distribution, origin and evolutionary history of all plant species in a particular area (Zhengyi et al., 2003). The distribution of S. floderusii indicates that this area belongs to Asian-North American-Arctic vegetation type (Dong et al., 2017). S. floderusii also has a wide range of applications and can be used in painting, baking, and as raw material (Pan et al., 2021).
The chloroplast (cp) genome consists of genetic material unique to plant species. It carries unique genetic information and has its own genetic system composed of a closed circular DNA molecule. The cp genome has a quadripartite structure comprising small single-copy (SSC) and large single-copy (LSC) regions separated by two inverted repeats (Wicke et al., 2011). In recent years, cp genomes have become a better and more popular alternative approach for species identification and phylogenetic analysis of plant relatives as they are more conserved than nuclear genomes (Jansen and Ruhlman, 2012; Yagi and Shiina, 2014; Kim et al., 2015; Park et al., 2018). With the development of high-throughput sequencing technology and low sequencing cost, increasing amounts of cp genomic data have been accumulated (Hatmaker et al., 2020; Shidhi et al., 2021; Wei et al., 2021; Zhang et al., 2021). To date, the cp genome of Salix has been reported (Chen et al., 2019; Guo et al., 2021; Li et al., 2021; Gulyaev et al., 2022; Wang et al., 2022). The published cp genomic data of these species offer important references for related research (Lauron-Moreau et al., 2015; Zong et al., 2019; Meucci et al., 2021).
The cp is an important site for photosynthesis in plants and a potential tool for plant genetic transformation. Plastid gene engineering has attracted wide attention because of its advantages, including high expression levels of foreign genes (De Cosa et al., 2001), the absence of position effect and gene silencing (Lee et al., 2003; Dhingra et al., 2004), and the avoidance of environmental safety problems caused by pollen escape in nuclear transformation systems (Svab and Maliga, 2007). Plastid genetic transformation technology has been used to introduce herbicide resistance (Daniell et al., 1998), disease resistance (Ruhlman et al., 2014), insect resistance (De Cosa et al., 2001), and other related exogenous genes into many herbaceous plants (Okuzaki et al., 2013; Chen et al., 2014). However, studies on cp transformation systems in woody plants remain limited. Therefore, the in-depth study of cp transformation in woody plants would explore endogenous regulatory elements further, establish a more efficient cp transformation system, and provide a theoretical basis for improving and breeding new varieties of woody plants.
Currently, the taxonomic status of S. floderusii in the genus Salix is only at the morphological level. Therefore, it is necessary to clarify the taxonomic status of S. floderusii by cp genome at the molecular level and determine the phylogenetic relationship and evolutionary relatedness with related species. In the present study, we sequenced the cp genome of S. floderusii and analyzed its structural characteristics. In addition, we constructed an evolutionary tree from the cp genome to explore the evolutionary relationship between S. floderusii and other Salix plants. Next, a series of cp regulatory elements were cloned from S. floderusii. A cp expression vector was constructed, and the biological functions of the regulatory elements were verified via a prokaryotic expression system. Our findings provide a basis for establishing subsequent cp genetic transformation systems and for the study of biology and evolution.
Materials and methods
Sample collection and DNA extraction
Mature fresh leaves of S. floderusii were obtained from Fenghuang Mountain National Forest Park in the Heilongjiang Province of China (44.107° N, 128.049° E; 1,730 m above sea level). Fresh samples were stored at −80°C for long-term. The voucher specimen was stored at the Institute of Traditional Chinese Medicine, Heilongjiang University of Chinese Medicine. Total genomic DNA was extracted using the Plant DNA Extraction Kit (SIMGEN, Hangzhou, China) according to the manufacturer’s instructions. DNA purity and integrity were verified via 1% agarose gel electrophoresis. Total plant DNA that met the quality requirements was sent to Novogene Bioinformatics Technology Co., Ltd. (Beijing, China) for sequencing using the Illumina high-throughput sequencing platform NovaSeq 6000 via the double terminal sequencing method (150 pair-ends).
Data filter connected and annotation
Raw data were filtered using the FastQC version 0.11.9 software under the following parameters: (1) remove sequences with N content exceeding 10% of the read length base number, (2) the number of low-quality bases exceeds 50% of the number of bases in the sequence, and (3) retain sequences with unremoved joints to obtain high-quality clean data. The complete cp genome was assembled using the GetOrganelle (Jin et al., 2020) version 1.7.5 software1. The GeSeq2 (Tillich et al., 2017) software was used to check the S. floderusii cp genome sequences after splicing. The online tool CPGAVAS23 (Shi et al., 2019) was used to annotate the cp genomes of S. floderusii and Salix suchowensis (MT551163) as reference sequences with default parameters. The annotated results were submitted to GenBank (GenBank accession number: OK375876). The file containing the sequence annotations was uploaded to the online drawing software OGDRAW4 (Wyman et al., 2004) to generate the cp genome map.
Cp genome analyses
The CDS sequences were extracted S. floderusii cp genome sequence. The relative synonymous codon usage (RSCU) values and codon usage were performed using CodonW version 1.4.2 with default settings (Wong et al., 2010). The MISA (Beier et al., 2017) on-line analysis software5 was used to analyze simple sequence repeat (SSR) of the cp genome. The thresholds used to detect the SSRs were 8,4,4,3,3,3 for mono-nucleotide, di- nucleotide, tri-nucleotide, tetra- nucleotide, penta- nucleotide, and hexa-nucleotides, respectively. The shortest size between any two SSR loci was more than 100 bp. The online software REPuter6 (Kurtz et al., 2001) was carried out on the S. floderusii of cp genome repeat sequence analysis. The analysis included forward, reverse, complement, and palindromic repeats under the following parameters: maximum computed repeats 50; hamming short 3; minimal repeat size of 20 bp; default values for other parameters.
Comparative sequence analysis
Salix sect. Vetrix species were analyzed for genome expansion and shrinkage using the online software IRscope7 (Amiryousefi et al., 2018). Cp genome of three species collinearity analysis was performed using the MAUVE version 2.4.0 (Darling et al., 2004) software. Additionally, the mVISTA8 (Frazer et al., 2004) online analysis software was used under the shuffle-Lagan analysis mode, with p = 0.5 and S. caprea used as a reference. The cp genome sequence diversity of the three species of Salix Sect. Vetrix was analyzed using the DnaSP version 6.12.03 (Rozas et al., 2017) software. The π value of plastids was calculated through the sliding window. The parameters under the sliding window were set as follows: window length = 600 bp; step size = 200 bp.
Phylogenetic analysis
Phylogenetic analysis was conducted on cp genomes of 45 Salix species, using Populus davidiana as an outgroup. Details of the species, downloaded from NCBI, are shown in Supplementary Table 1. First, the cp genomes of the 46 species were aligned using the Mafft version 7.313 software, based on default parameters. Next, a maximum-likelihood (ML) phylogenetic tree was constructed via IQ-TREE version 1.6.8 by selecting the K3Pu + F + G4 computational model with 1,000 bootstraps by PhyloSuite version 1.2.2 (Zhang et al., 2018) analysis software. Bayesian inference (BI) analysis was performed using the Markov Chain Monte Carlo (MCMC) algorithm in MrBayes version 3.1.27 (Ronquist and Huelsenbeck, 2003). The MCMC analysis was run for 1,000,000 generations independently, using a sampling frequency of 100. The initial 25% of trees were discarded as burn-in, and the remaining data were used to construct a majority-rule consensus tree and default values for other parameters. Finally, the evolutionary tree was visualized and refined using MEGA X (Kumar et al., 2018).
Gene cloning of Cp regulatory elements
The Sf-PpsbA fragment contained the S. floderusii psbA promoter and the S. floderusii psbA leader. Sf-TpsbA was the terminator of the psbA gene. The Sf-trnI or Sf-trnA fragment contained the trnI or trnA gene, respectively, as well as part of the flanking sequence of this gene. The target genes (Sf-trnI, Sf-PpsbA, Sf-TpsbA, and Sf-trnA) and a selectable marker gene aadA were cloned using high-fidelity DNA polymerase KOD-Plus (Toyobo, Osaka, Japan). Primer sequences are shown in Supplementary Table 2. Sequence amplification procedures were as follows: pre-denaturation at 98°C for 30 s, followed by 35 cycles of denaturation at 98°C for 10 s, annealing at 58°C for 30 s, and extension at 72°C for 1 min. The target fragment was recycled at 72°C for 7 min and used to construct the cp expression vector.
Construction of recombinant plasmid
We constructed a cp transformation operon structure based on the pBluescript II SK (+) no-load plasmid using the Hieff Clone® Universal One Step Cloning Kit (YEASEN, China). In the process of primer involvement, we followed the homologous arm consisting of a 15–25 bp overlap region at both ends of the target gene and the linearized vector for recombination. All the target segments required to construct the cp expression vector were assembled into synthetic operons, as described below.
During the first round of target gene construction, XhoI and Hind III restriction endonucleases were selected for the enzymatic digestion of pBluescript II SK (+) no-load plasmids to obtain linearized vectors. The Sf-PpsbA, aadA, and Sf-TpsbA gene fragments were assembled into the linearized vector to synthesize pB + aadA using seamless connection kit.
During the second round of target gene construction, the KpnI restriction endonuclease was selected to digest the recombinant plasmid (pB + aadA) —constructed in the previous step—to obtain the linearized vector. pB + Sf-trnI + aadA was synthesized from the trnI gene fragment and assembled into the linearized vector using a seamless connection kit.
During the third round of target gene construction, the recombinant plasmid (pB + Sf-trnI + aadA) constructed in the previous step was digested with the BamHI restriction endonuclease to obtain the linearized vector. pB + Sf-trnI + aadA + Sf-trnA was synthesized by assembling trnA gene fragments with linearized vectors using a seamless connection kit. The steps involved in constructing the cp expression vector are depicted in Supplementary Figure 1.
Expression analysis of the prokaryotic aadA gene
The recombinant plasmid was transformed into E. coli strain DH5α. The transformed Escherichia coli was incubated in LB liquid medium containing ampicillin (50 ng/μL) and spectinomycin (50 ng/μL) at 37°C and 200 rpm/min for 48 h.
Results
Characterization of the Cp genome
Similar to most land angiosperms, the cp genome of S. floderusii was found to be a closed-loop double-stranded DNA molecule with a typical quadripartite structure. It was 155,540 bp long, including a large single copy (LSC) region of 84,401 bp, a small single copy (SSC) region of 16,221 bp, and a pair of inverted repeats (IRa and IRb) 27,459 bp in length (Figure 1). The GC content in the whole genome was 36.7%, including 41.86% in the inverted repeat (IR) region, 34.45% in the LSC region, and 30.97% in the SSC region. A total of 131 genes were identified, including 86 protein genes, 37 tRNA genes, and 8 rRNA genes. There were 16 genes containing introns in the cp genome of S. floderusii, including six tRNA genes (trnA-UGC, trnG-GCC, trnI-GAU, trnK-UUU, trnL-UAA, and trnV-UAC). Ten protein-coding genes (atpF, clpP, ndhA, ndhB, petB, petD, rpl16, rpl2, rpoC1, and ycf3) were identified, among which ycf3 and clpP contained two introns (Table 1).
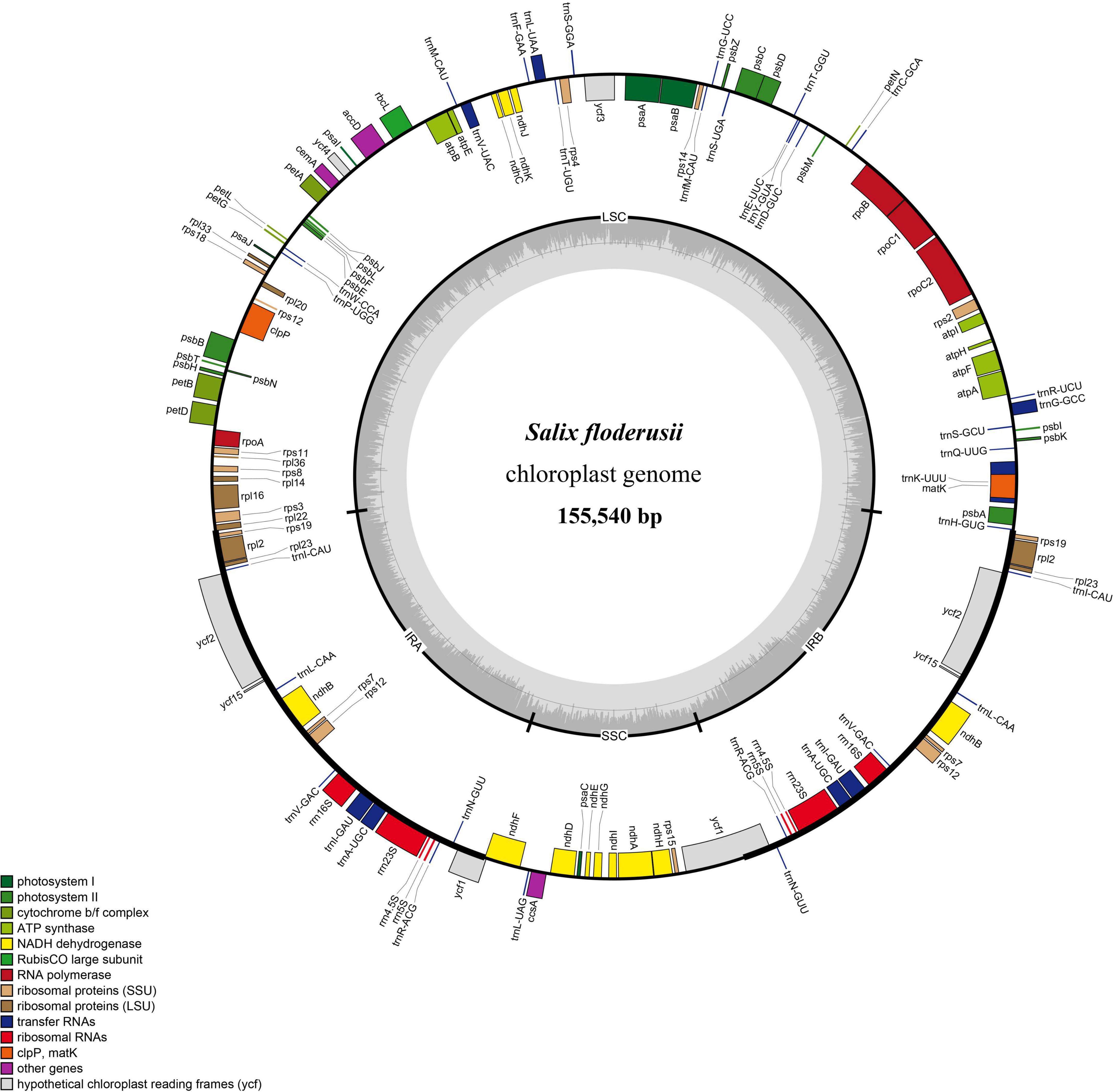
Figure 1. Gene map of the complete Salix floderusii cp genome. Genes drawn within the circle are transcribed in a clockwise direction, and genes drawn out are transcribed in a counterclockwise direction.
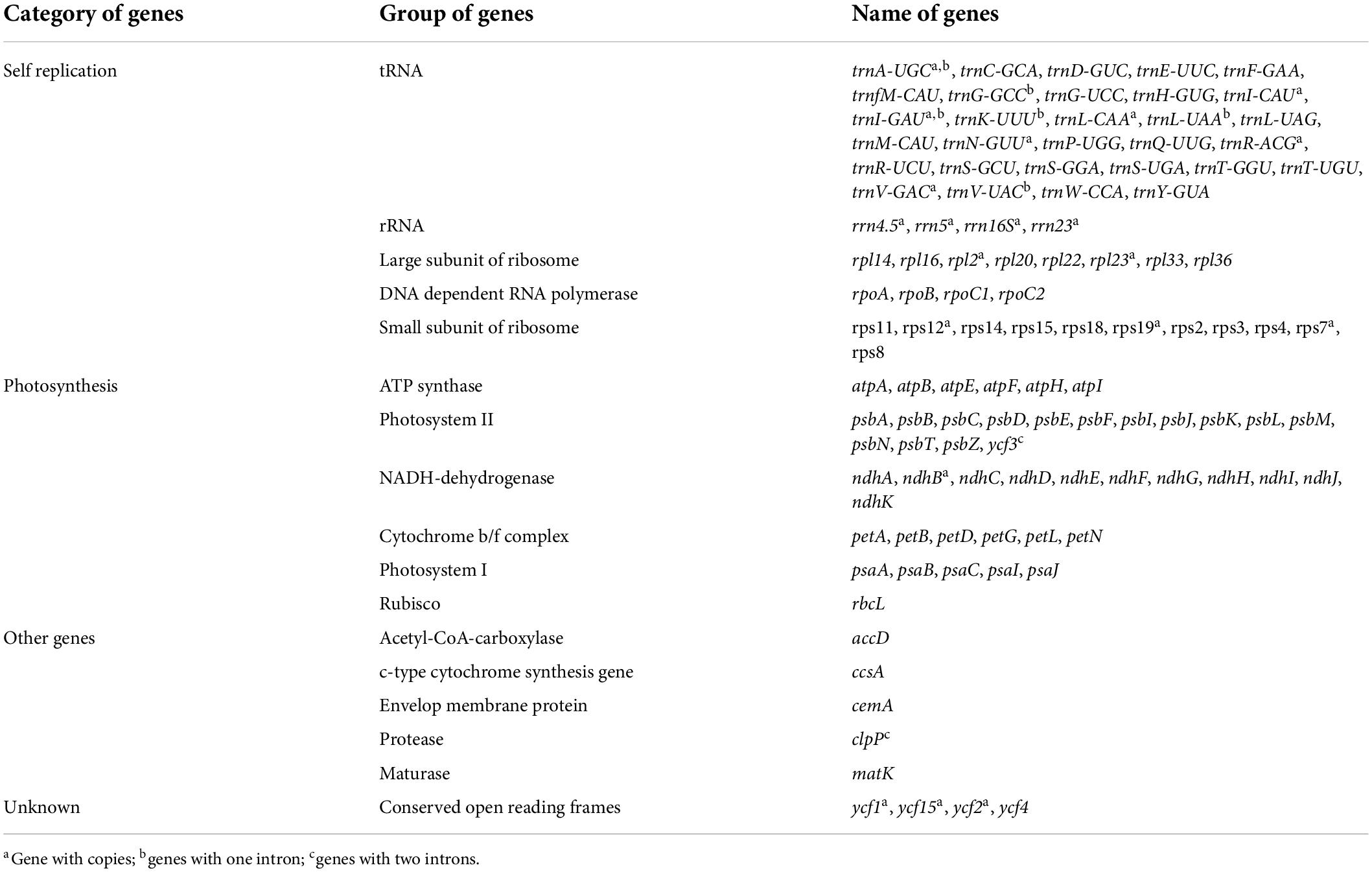
Table 1. List of genes in the Salix floderusii cp genome.
Codon usage
The codon usage frequency and RSCU were analyzed based on 86 protein-coding genes in the S. floderusii cp genome (Figure 2). A total of 20 amino acids and 26,654 codons were identified. Specifically, AUU (1,149) of isoleucine was the most abundant codon, whereas the least abundant codon was UGC (92) of cysteine. Methionine and tryptophan were encoded by only one codon, and the number of amino acids encoding six codons, four codons, three codons, and two codons was 3, 5, 1, and 9. The highest RSCU value (1.9) corresponded to the UUA codon of Leucine (Leu). The lowest RSCU value was 0.36, corresponding to the UAC codon of Tyrosine (Tyr). Furthermore, 29 codons had RSCU values greater than 1, 30 codons had RSCU values less than 1, and the RSCU values of AUG and UGG were equal to 1.
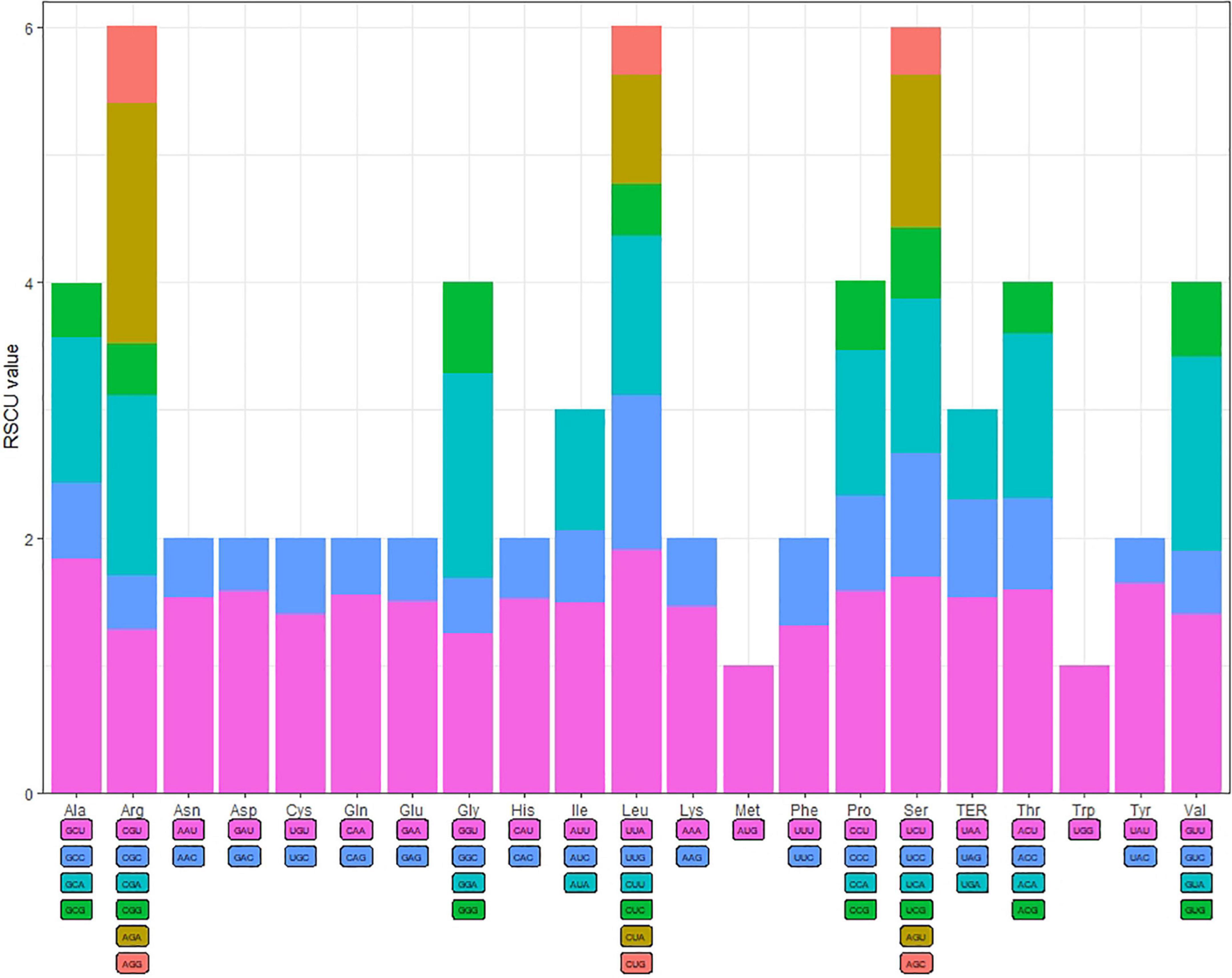
Figure 2. Codon content and codon usage of 20 amino acids and stop codons in the protein-coding genes of the S. floderusii cp genome.
Repeat sequence and simple sequence repeat analysis
We performed repeated sequence analysis on the cp genomes of the three Salix species. The number of repeat sequences for each of the three species is 49 (Figure 3A). S. caprea contains 25 forward repeats, 17 palindromic repeats, and 7 reverse repeats. S. cinerea contains complement repeat one, forward repeat 21, palindromic repeat 19, reverse repeat eight; S. floderusii contains 1 complement repeat, 24 forward repeats, 17 palindromic repeats, and 7 reverse repeats. Although the total number of repeat sequences in the three species was the same, the types of repeat sequences were different. S. cinerea and S. floderusii both contain a complement repeat, unlike S. caprea.
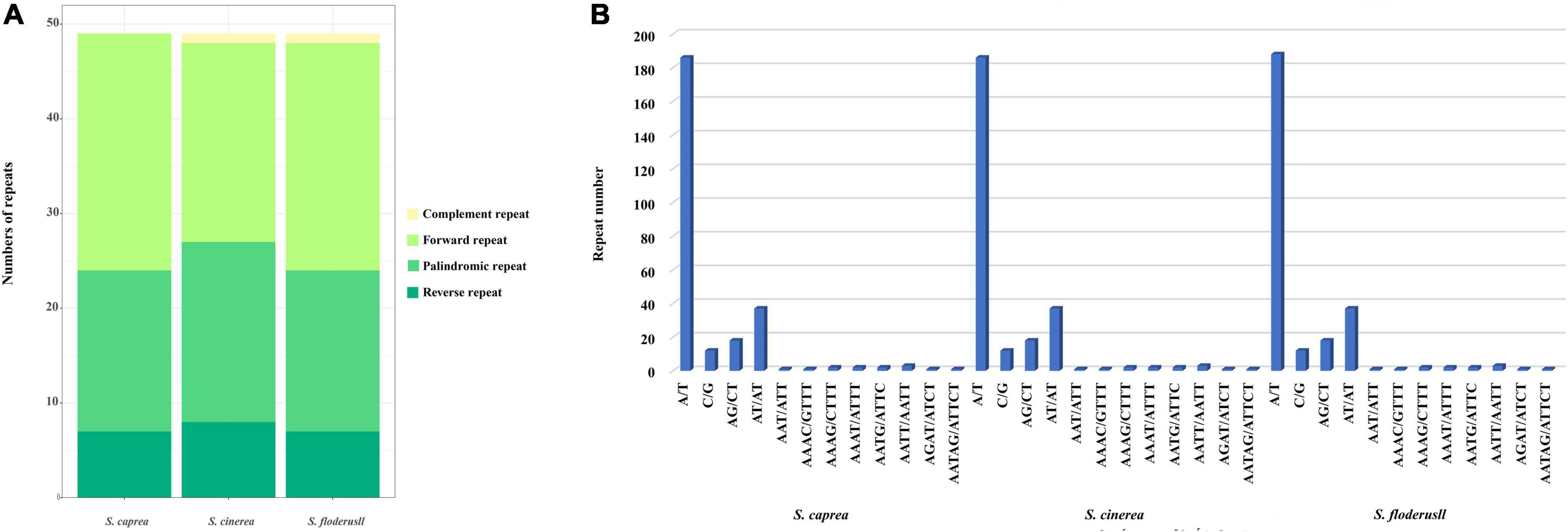
Figure 3. Statistics of simple sequence repeats of the cp genome of S. floderusii. (A) Number of different repeat types. (B) Number of each identified SSRs motif.
Simple sequence repeat analysis was performed on the three cp genomes to identify mononucleotide, dinucleotide, trinucleotide, tetranucleotide, and hexanucleotides repeat motifs, as detailed in Figure 3B. SSRs can be divided into 12 types. A total of 268, 266, and 266 SSRs were detected in S. floderusii, S. caprea, and S. cinerea, respectively. The numbers of SSRs in the three species differed only in A/T type, with 188 for S. floderusii and 186 for S. caprea and S. cinerea. The number of other SSR types in the three species was consistent (Supplementary Table 3). S. floderusii contains polythymine (poly T) repeat at 43,872 bp compared with S. cinerea and S. caprea and S. cinerea was deletion polyadenine (polyA) repeat at 117,925 bp. In addition, S. floderusii (124,782 bp) and S. cinerea (124,813 bp) contain poly T repeat, whereas it was not found in S. caprea (Supplementary Table 4).
Inverted repeat contraction and expansion
The contraction and expansion of the IR region can reflect the evolutionary relationship of species. The cp genome sequences of the three species were very similar in length (155,540–155,572 bp) when compared, and the gene composition and arrangement patterns in each junction region of the three species were roughly equal (Figure 4). The IRa and IRb lengths of S. floderusii and S. caprea were equal; however, LSC and SSC lengths were slightly varied. The LSC of S. floderusii was 23 bp shorter than S. caprea and the SSC of S. floderusii was 1 bp larger than S. caprea. The IRa and IRb regions of S. floderusii and S. caprea contracted compared with those of S. cinerea.
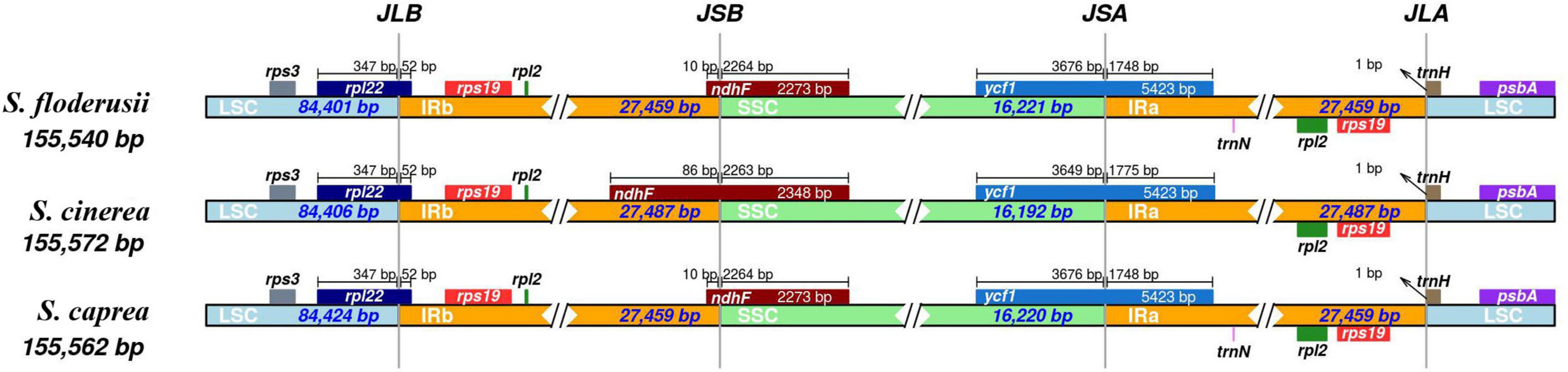
Figure 4. Comparison of the border pattern of large single-copy (LSC) region, small single-copy (SSC) region, and an inverted repeat (IR) region among the S. floderusii, S. cinerea, and S. caprea cp genomes.
In addition, the sequence of genes at the junction of the LSC and IRb regions was rps3, rpl22, rps19, and rpl2. rpl22 extended by 52 bp to the IRb region. The genes ndhF and ycf1 were located at the junction region of IRb/SSC and SSC/IRa, respectively. The length of the ndhF gene and its location at the IRb/SSC junction region were the same in S. floderusii and S. caprea; however, the ndhF gene of S. cinerea had the largest extension (86 pb) into the IRb/SSC border. Only ycf1 was found at the junction of the SSC and IRa. The sequence of genes at the junction of IRa and LSC was rpl2, rps19, trnH, and psbA.
Nucleic acid polymorphism analysis
Nucleotide polymorphism analysis can be used for species identification by comparing the highly variable sequences in related species. Nucleotide polymorphisms of the cp genomes of three Salix species were analyzed, and the results indicated that the Pi value ranged from 0.02111 to 0, with an average value of 0.000262 (Figure 5). Compared with LSC and SSC, the polymorphism of nucleic acids in the IR region was low, indicating that the IR region was more conserved. Pi was greater than 0.005, and we detected two gene spacers, petA/psbJ and trnS-GCU/trnT-CGU. These nucleotide variation sites can be used as molecular markers for species identification and provide an alternative strategy for species identification.
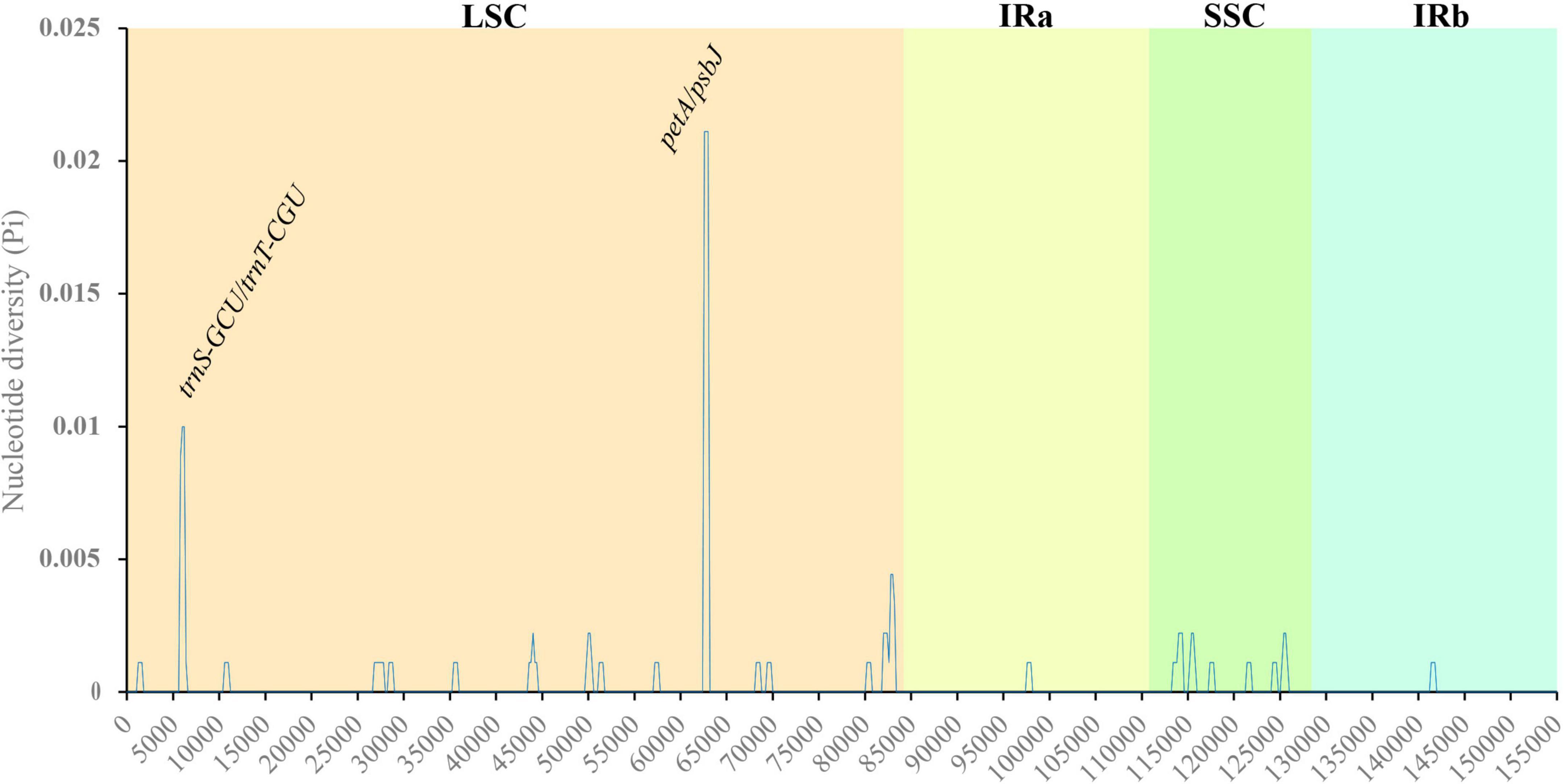
Figure 5. Variation in nucleotide diversity across the cp genomes of the three studied Salix species. X-axis: position of the midpoint of a window; Y-axis: nucleotide diversity of each window.
Comparative genomic analysis
Comparative genome analysis can identify differences in the gene structure and composition of closely related species more intuitively, as well as distinguish among them. In the present study, we performed MAUVE analysis on S. cinerea, S. caprea, and S. floderusii (Figure 6). The results showed that the entire genome sequence was a homologous region with no big indels. The cp genomes of the three Salix species connected with only one locally collinear block, suggesting a high level of similarity, as well as no rearrangement in gene organization.
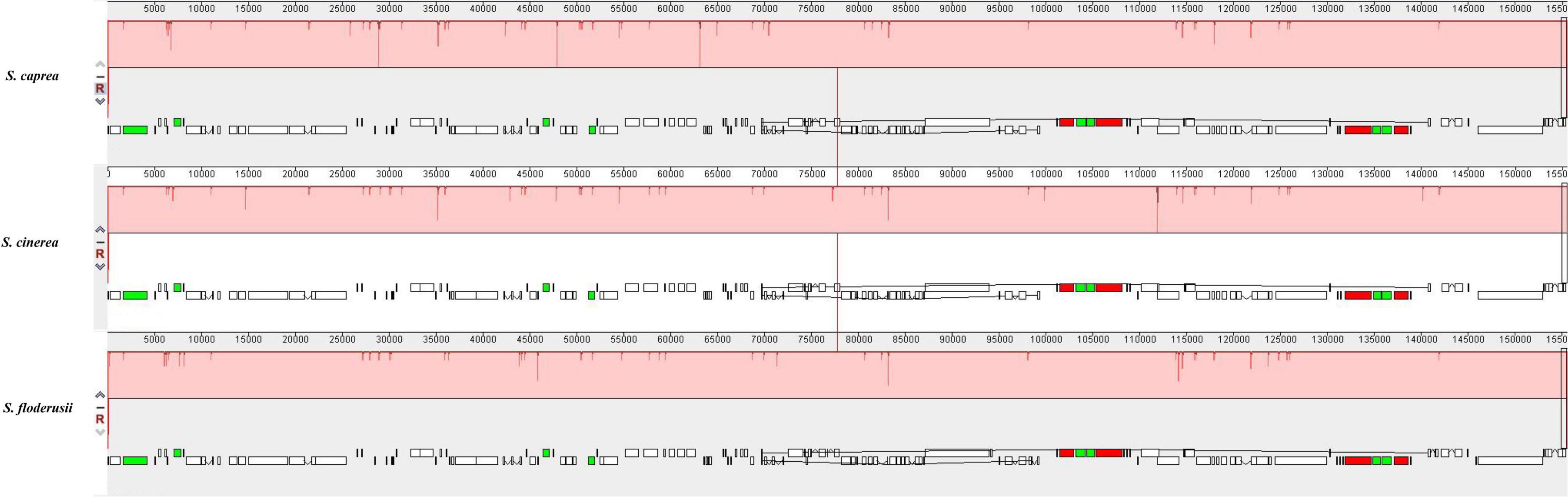
Figure 6. Comparison of the genome structure of three species using the MAUVE program. The DNA sequences above the line are presented in a clockwise direction, and those below the line in a counterclockwise direction.
Analysis using mVISTA showed that the cp genomes of the three Salix species were also highly conservative (Figure 7). In addition, the coding region was more conservative than the non-coding region, and the mutation loci mainly appeared in the non-coding region. Comparative analysis of the LSC region, SSC, and IR regions, showed that the LSC region contained more hypervariable regions than the SSC region. By contrast, the IR region had no hypervariable region. The high variable regions include the rpl22 coding region, psbM/trnD-GUC non-coding region, petA/psbJ non-coding region, and ycf1 coding region.
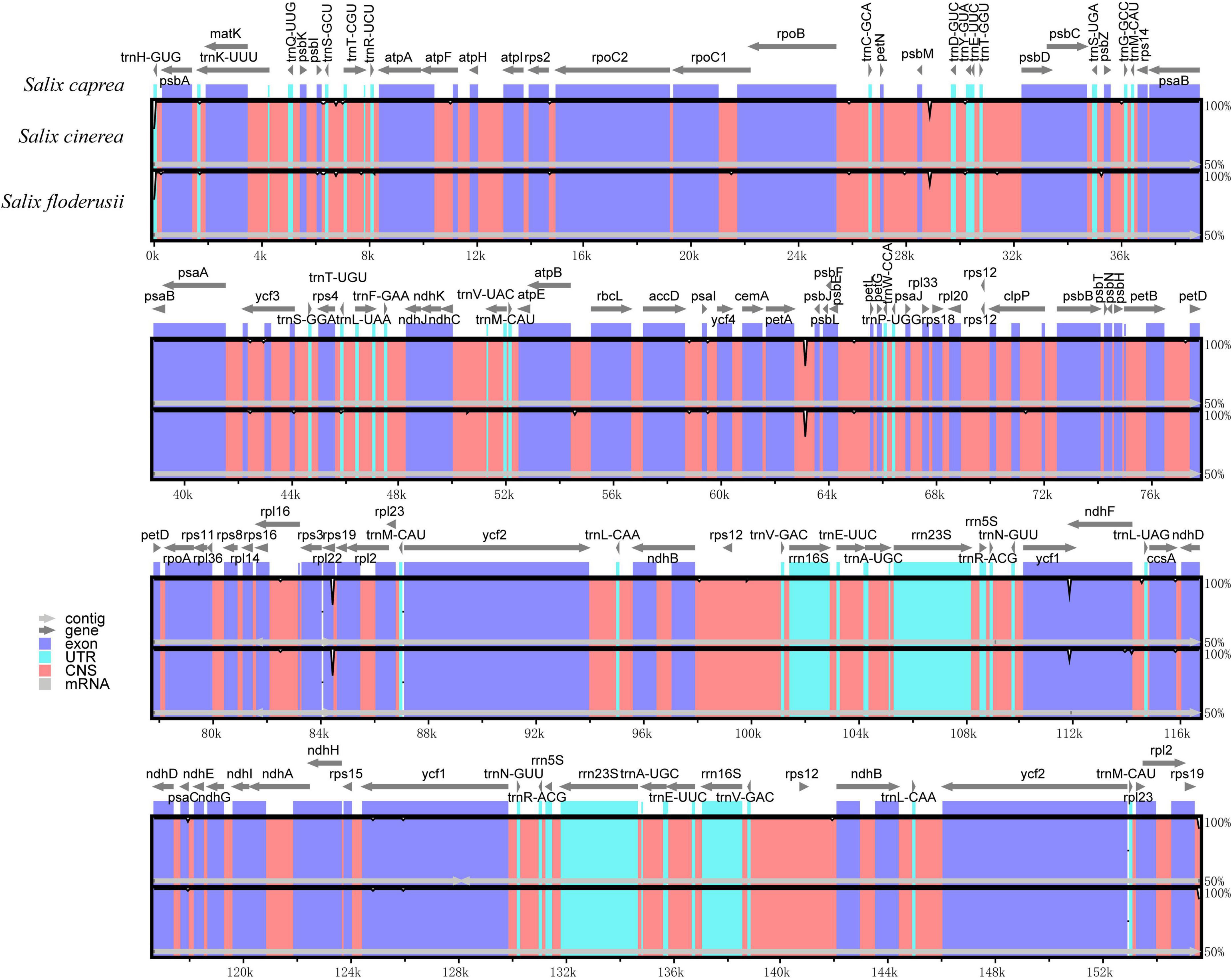
Figure 7. Comparison of three cp genomes using the mVISTA alignment program. Genome regions are color-coded as protein-coding, rRNA coding, tRNA coding, or conserved non-coding sequences. The vertical scale shows the percentage of identity, varying from 50 to 100%.
Phylogenetic analysis
Phylogenetic trees can accurately describe the evolutionary status of species in their genus. Our findings show that the evolutionary tree of the Salix cp genome can be divided into two clades and that all branches have high confidence values (Figure 8). Clade I include Subgenus Salix and a few subgenus Vetrix species. Subgenus Salix includes Sect. Wilsoniana, Sect. Salix, Sect. Pentandrae, and Sect. Tetraspermae. The main body of Clade II is Subgenus Vetrix, and also includes Sect. Amygdalinae of Subgenera Salix and Chamaetia. S. floderusii belongs to Clade II and is closely related to S. cupularis.
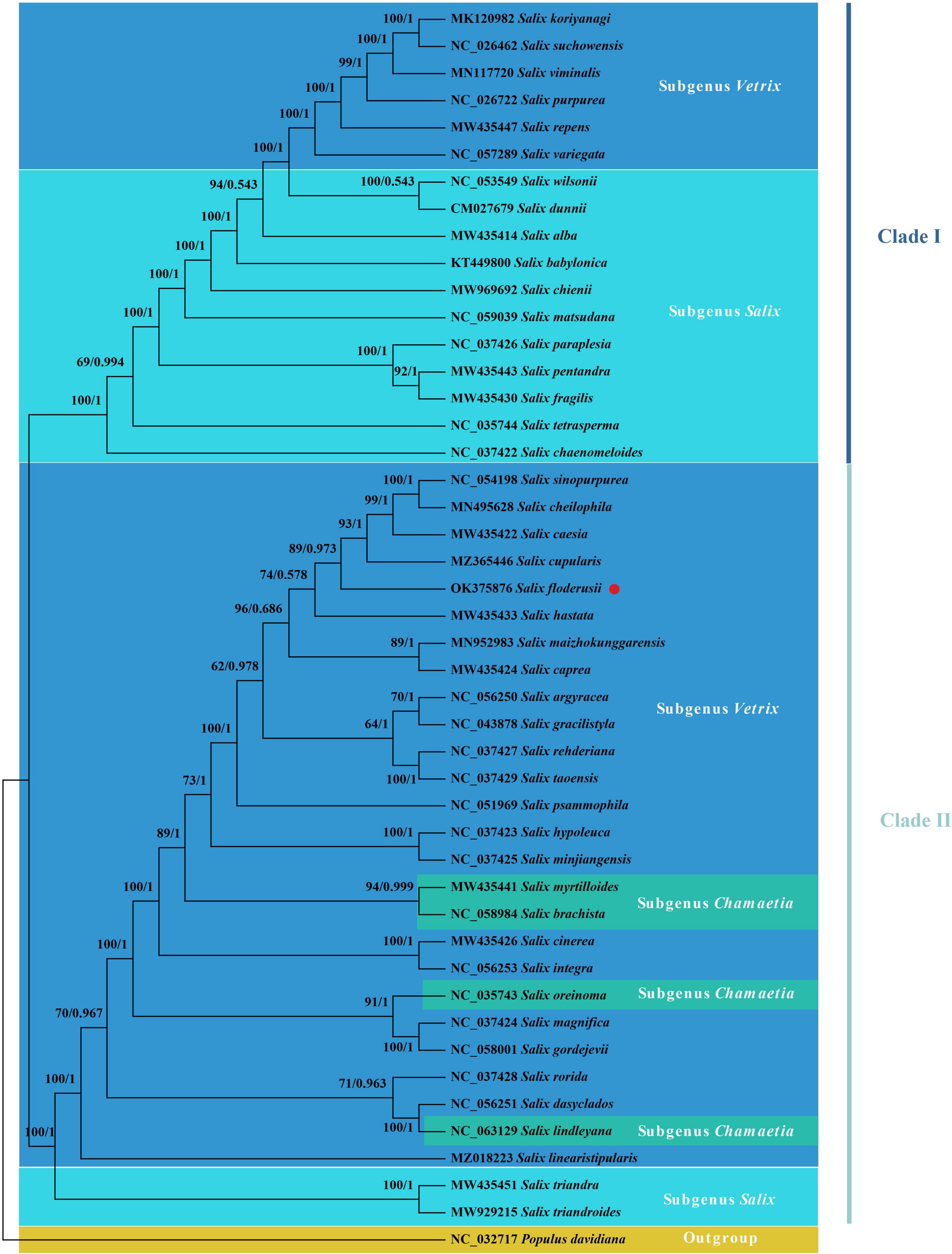
Figure 8. Phylogenetic analysis. The phylogenetic tree inferred from maximum likelihood (ML) and Bayesian inference (BI) of 46 cp genomes from 45 species from the Salix genus and Populus davidiana as an outgroup. The numbers above the branches are the posterior probability and likelihood values, respectively. The species studied in the present study was marked using red circles.
Cp regulatory elements in S. floderusii
The cloned cp regulatory element sequence can be used for subsequent vector construction. The band length of all amplified sequences was equal to that of the target fragment, as shown in Supplementary Figure 2.
Analysis of aadA gene expression
The verified vector was transformed into E. coli strain DH5α for expression. E. coli containing no load could not grow in LB solid medium containing ampicillin and spectinomycin (Figure 9A). However, recombinant plasmids containing repeat sequences (Sf-trnI and Sf-trnA), initiating Sf-PpsbA and terminating Sf-TpsbA and aadA genes were able to grow in LB solid media containing ampicillin and spectinomycin (Figure 9B). The results showed that the selected regulatory elements had prokaryotic characteristics and could regulate the correct expression of exogenous genes.
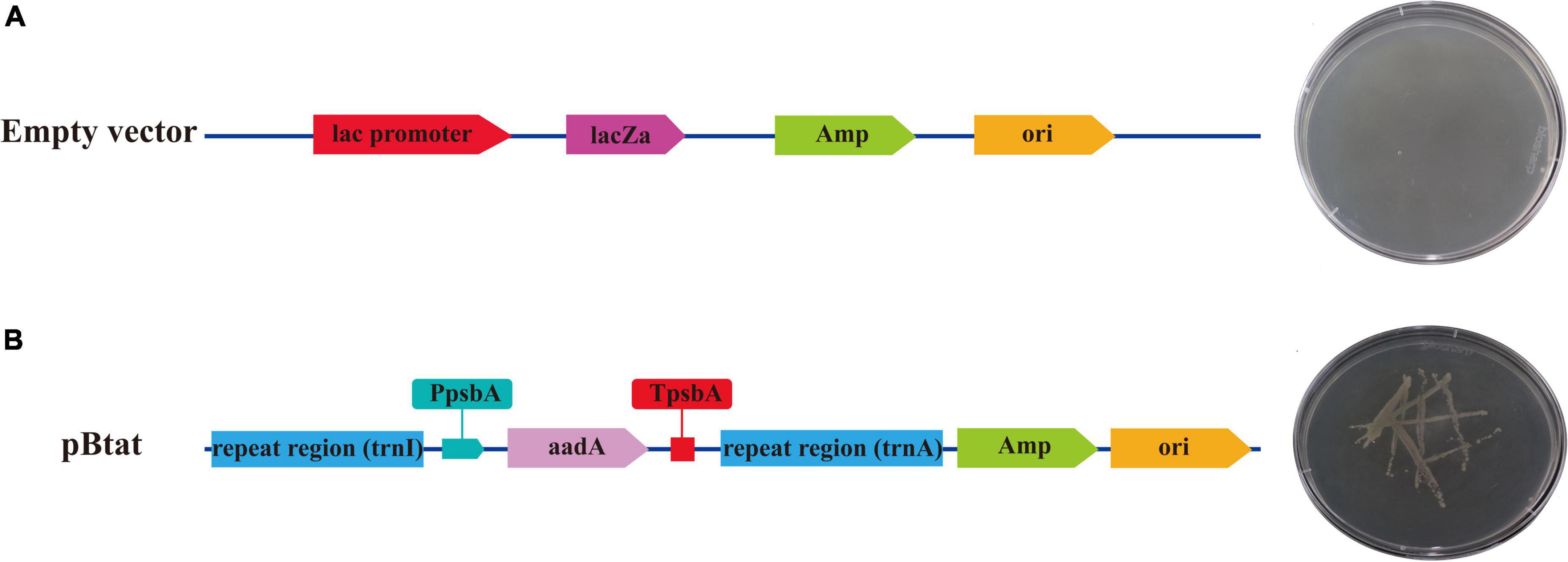
Figure 9. The expression of aadA in Escherichia coli. (A) Escherichia coli containing empty vector. (B) Escherichia coli containing the recombinant vector.
Discussion
Highly conserved Cp genomes of Salix
Chloroplast genomes have low evolution rate and highly conserved and have thus been found to be ideal in studies of plant phylogeography and molecular evolution, as well as phylogenetic analysis (Burke et al., 2012; Huang et al., 2014). Whereas, inversions, gene or intron losses also occur occasionally (Bell et al., 2010). Although introns are not directly expressed and involved in physiological activities, they can enhance the gene expression level, on the special position, in the specific time (Yi et al., 2012). Ycf3 is required for the stable accumulation of the photosystem I (PS I) and PSII. OTP51 is required for the optimal splicing of several plastid introns, especially ycf3 intron 2, and affect the assembly of PS I and PSII (de Longevialle et al., 2008). The previous studies show that the ClpP gene contains two introns make the low selection effect ratio (ka/ks = 0) of the ClpP gene (Liang and Chen, 2021; Liang et al., 2022). The evolution rate of the ClpP gene is related with species that is clearly evident. The rapid evolution of species will lead to the loss of introns, while it will also be subject to the functional constraints of species lineage (Dugas et al., 2015; Williams et al., 2019).
The genetic information carried on DNA is transmitted as triplet codons during the transfer from RNA to protein. Each amino acid corresponds to an unequal number of codons. In the case of natural selection or mutation preference, the use of codons is biased during protein synthesis. All codons, Ile with a frequency of 8.67% and Cysteine (cys) was the least common one with a proportion of 1.14% (Supplementary Table 5). An interesting phenomenon was found after the codon usage analysis of S. floderusii. When the RSCU value was greater than 1, most codons ended in A and U, and only A few codons ended in G. However, when the RSCU value was less than 1, most codons ended with G and C, and only a few codons ended with A. In addition, codon species with RSCU values greater than 1 and less than 1 were very close in number. This phenomenon was also observed in the cp genomes of land plants, such as Ailanthus altissima (Saina et al., 2018), Lycium chinense (Yang et al., 2019), Symplocarpus renifolius (Kim et al., 2019), Alpinia katsumadai (Li et al., 2020b). The codon usage frequency was different in cp genome, which might be related to the hydrophilicity, synonymy substitution rate, gene length (Huang et al., 2014), and expression level of the codon, with highly expressed genes displaying higher codon bias (Zhang et al., 2012). The result is closely correlated with the evolutionary pattern of the species (Liu and Xue, 2005).
The SSRs are tandem repeats distributed across the entire genome. In the cp genome, SSRs exhibit high polymorphism, as well as parthenogenetic patterns, and have been widely used in studies investigating population genetics, phylogenetic relationships, gene flow, and pedigree geography (Khadivi-Khub et al., 2013; Li et al., 2020a). In the present study, SSR sequences in the three cp genomes were mainly composed of poly-A and poly-T repeats, contributing to the AT abundance of these genomes. The mononucleotide motif A/T was the most abundant, accounting for 86.19% of the SSRs, and the SSRs were mostly located in non-coding regions. This trend has been demonstrated in several previous studies (Gao et al., 2019; Yan et al., 2019). These SSR markers can be used to develop specific markers and are the key to systematic research.
Contraction and expansion of inverted repeat regions
The IR region is the most conserved region of the cp genome. The expansion and contraction of IR, LSC, and SSC regions are common phenomena during evolution and the main reason for variations in cp genome length (Daniell et al., 2008; Xue et al., 2019). In the present study, the cp genomes of the three Salix Sect. Vetrix species were highly conserved at the LSC/IRb, SSC/IRa, and IRa/LSC junction sites, which showed slight differences in the length of partial genes, their location at the junction, and the deletion of the gene. The results of the present study, showed that the ndhF gene length and located at the IRb/SSC junction site differed among S. cinerea, S. floderusii, and S. caprea. The length variation of ndhF gene of the three Salix species is similar to those identified in previous studies reported that the ndhF also shows the highest rate of base substitution in the Asteraceae (Olmstead et al., 2000) and Berberidaceae (Kim et al., 2004). By contrast, the ycf1 gene located at the SSC/IRa junction site was conserved among the closely related species of the family. Therefore, we inferred that the junction between the LSC and IR regions was relatively conserved, while the junction between the SSC and IR regions was highly variable. Similar results were obtained in most high plants, as reported in previous studies (Tian et al., 2002; Wang et al., 2013). In addition, the trnN gene, one of the important genes in the cp, was absent from the IRa region of S. cinerea. Studies have shown that the deletion of trnN leads to a significant decrease in the abundance of photosystem I core complex, cytochrome b6f complex, photosystem II core complex, and ATP synthase subunit, which damages cp translation, consequently affecting the accumulation of plastid coding proteins (Legen et al., 2007). Thus, trnN is essential to the mechanism underlying plastid translation.
Comparative genomic analysis
Comparative analysis of the cp genomes of three Salix species revealed four highly variable regions, namely the rpl22 coding region, psbM/trnD-GUC non-coding region, petA/psbJ non-coding region, and ycf1 coding region. Previous research has showed that rpl22-rps19-rpl2, psbM-trnD, and two other highly variable regions can be considered the most promising variable factors for species division and in population genetics studies (Wang et al., 2021). It has been reported that six highly variable loci, namely trnH-GUG/psbA, ndhF, ndhF/rpl32, trl32/trnL-UAG, ndhD, and ycf1, can be used as candidate loci for plasmid barcoding for species identification in the phylogenetic analysis of Lindera species (Tian et al., 2019). Among medicinal plants, 95 ginseng samples have been screened for DNA barcoding. Moreover, significantly different Panax bipinnatifidus species were identified using matK, psbK-I, psbM-trnD, rps16, and nadI (Zuo et al., 2010). The ndhf-rpl32, psbM-trnD, and trnS-trnG regions are the most rapidly evolving regions in ornamental plants (Lu et al., 2018), with psbM-trnD playing an important role in species identification. petA-psbJ may be used as a divergent region to develop potential DNA barcodes to identify Pyrrosia species (Yang et al., 2020). Species diagnosis for most of the Kaempferia genus was conducted using the loci of psbA-trnH and petA-psbJ as DNA barcodes (Techaprasan et al., 2010). petA-psbJ can be used as a candidate fragment for DNA barcoding in phylogeny and species identification of other species, including those of the Rosa (Yin et al., 2020), Bupleurum (Xie et al., 2021), and Zingiber (Li et al., 2020c) genera, as well as the Papaveraceae (Liu et al., 2020) and Lauraceae (Jo et al., 2019) families. These hypervariable regions are suitable candidates for species identification and provide evolutionary information about Salix species.
Comparative analysis of the LSC, SSC, and IR regions, showed that hypervariable regions were mainly distributed in the LSC and SSC regions, with the LSC region containing the highest number of variable regions. By contrast, the IR region contained no highly variable region. The main explanation is that LSC and SSC regions are not conserved, whereas the IR region is more conserved, indicating that the evolution rate of non-coding sequences in Salix is faster than that of protein-coding gene sequences.
Phylogenetic analysis
Phylogenetic analysis of the cp genomes of 45 Salix species showed that all species were monophyletic. Several studies have been published on the phylogeny of the genus Salix and on phylogenetic analyses that used the protein-coding region of the cp genome, as well as the whole cp genome (Azuma et al., 2000; Lauron-Moreau et al., 2015; Huang et al., 2017). Given the limited number of Salix species involved in a previous study (Wu et al., 2015), we compared a higher number of Salix species for phylogenetic analysis and aimed to display the phylogenetic status of Salix more comprehensively. The two main clades formed within the Salix genus were generally consistent, similar to the result obtained by Chen et al. (2019). In the Subgenus Vetrix, each species section is not grouped into one but mixed, consistent with Skvortsov’s morphological description of Sect. Helix and Sect. Incubaceae species. According to Skvortsov, these two species sections have a spiral leaf arrangement, typically seen in the willows (where the divergence angle of 2/5 is retained) (Skvortsov, 1999). The phylogenetic tree showed that most species from the same Section could not cluster into a branch. The main reasons for this phenomenon are as follows: (1) due to the selection of different datasets, the interspecific relationships presented by the phylogenetic tree constructed in this study are slightly inconsistent compared with the results of previous studies; (2) other studies have also shown that Salix is not monophyletic, mainly due to its widespread hybridization, which leading to multiple evolving networks and an unstable morphological classification based on the difference of reproductive organs (Wu et al., 2015); (3) warmer climates were shown to alter the increasing relative abundance of Salix, which showed higher genetic diversity in arctic ecosystems, highlighting the significant role of environmental factors in the evolution of species (Meucci et al., 2021). Therefore, further studies are needed to clarify the classification and phylogeny of Salix.
Analysis of aadA gene expression in the prokaryotic expression vector
Selecting appropriate active promoters with 5′ UTR and 3′ UTR is the first condition to construct prokaryotic expression vectors and establish efficient transformation systems. Generally, using endogenous host genes can markedly facilitate the integration and expression of foreign genes in the host genome (Radakovits et al., 2010). In the present study, two homologous recombinant fragments, one high-efficiency promoter and one 3′ UTR sequence, were identified from the S. floderusii. The 3′ UTR sequence usually contains a stable stem-loop structure—only thus can it ensure the stability of mRNA and facilitate the accumulation of proteins in the prokaryotic expression system (Monde et al., 2000). RNA secondary structure prediction analysis of TpsbA showed that the gene has a stable stem-loop structure.
The aminoglycoside 3-adenylate transferase encoded by aadA can inactivate spectinomycin and streptomycin by adenylate acylation, preventing them from binding to ribosomes. Plants transformed with aadA are thus resistant to spectinomycin and streptomycin—a trait that can be used to screen green transformed cells and white non-transformed cells. Svab and Maliga (1993) successfully transformed tobacco cp (Chen et al., 2014; Yarbakht et al., 2015) using the aadA gene as a selective marker. To date, this transformation system has been applied in Arabidopsis thaliana (Sikdar et al., 1998), potato (Sidorov et al., 1999), soybean (Dufourmantel et al., 2004), rice (Lee et al., 2006), and other herbaceous plants (Liu et al., 2007; Cheng et al., 2010; Wei et al., 2011; Harada et al., 2014; Lelivelt et al., 2014; Muralikrishna et al., 2016). In the present study, the aadA gene was selected as an effective selective marker to ensure that the selected regulatory elements can perform their biological functions normally. Because of the high similarity in the transcription and translation systems between E. coli and chloroplasts, a combined strategy using both the E. coli and chloroplast systems could potentially be applied to develop subsequent chloroplast expression vectors for plant transformation.
Conclusion
In the present study, we sequenced, assembled, and annotated the cp genome of S. floderusii using high-throughput technology and comprehensively analyzed its gene structure. Comparative analysis of the S. floderusii cp genome with those of related species showed that the three species in sect. Vetrix were conserved in terms of gene structure. We identified the phylogenetic position of S. floderusii in the genus Salix by constructing a phylogenetic tree. Furthermore, we cloned and identified a series of cp expression elements with characteristics of prokaryotic expression in S. floderusii. These findings provide a basis for establishing a cp genome transformation system and are essential for conserving and utilizing Salix germplasm resources.
Data availability statement
The original contributions presented in this study are publicly available. The annotated complete chloroplast genome sequences were submitted to the NCBI database with accession number OK375876, and the original source data can be found here: https://www.jianguoyun.com/p/Dap53uUQ3PDeChj5zcsEIAA.
Author contributions
YL, QF, and WM designed the experiments. WR, HZ, and ZJ carried out the experiments. WR, MZ, and LK analyzed the experimental results. WR, ZJ, and MZ wrote the manuscript. All of the authors have approved the final manuscript.
Funding
This work was supported by grants from National Key Research and development Project, research and demonstration of collection, screening and breeding technology of ginseng and other genuine medicinal materials (2021YFD1600901), Heilongjiang Touyan Innovation Team Program (Grant Number: [2019] No. 5), and Study on resource status and protection technology of Endangered Phellodendron amurense (Y0002).
Acknowledgments
Sincere thanks to Yang Ni for providing helpful suggestions and support related to the analysis performed.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.987443/full#supplementary-material
Footnotes
- ^ https://github.com/kinggerm/getorganelle/blob/master/get_organelle_from_assembly.py
- ^ https://chlorobox.mpimp-golm.mpg.de/geseq.html
- ^ http://47.96.249.172:16019/analyzer/home
- ^ https://chlorobox.mpimp-golm.mpg.de/OGDraw.html
- ^ http://pgrc.ipk-gatersleben.de/misa/misa.html
- ^ https://bibiserv.cebitec.uni-bielefeld.de/reputer
- ^ https://irscope.shinyapps.io/irapp/
- ^ https://genome.lbl.gov/vista/mvista/submit.shtml
References
Amiryousefi, A., Hyvonen, J., and Poczai, P. (2018). IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Azuma, T., Kajita, T., Yokoyama, J., and Ohashi, H. (2000). Phylogenetic relationships of Salix Salicaceae based on rbcL sequence data. Am. J. Bot. 87, 67–75. doi: 10.2307/2656686
Beier, S., Thiel, T., Munch, T., Scholz, U., and Mascher, M. (2017). MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bell, C. D., Soltis, D. E., and Soltis, P. S. (2010). The age and diversification of the angiosperms re-revisited. Am. J. Bot. 97, 1296–1303. doi: 10.3732/ajb.0900346
Burke, S. V., Grennan, C. P., and Duvall, M. R. (2012). Plastome sequences of two New World bamboos–Arundinaria gigantea and Cryptochloa strictiflora (Poaceae)–extend phylogenomic understanding of Bambusoideae. Am. J. Bot. 99, 1951–1961. doi: 10.3732/ajb.1200365
Chen, P. J., Senthilkumar, R., Jane, W. N., He, Y., Tian, Z., and Yeh, K. W. (2014). Transplastomic Nicotiana benthamiana plants expressing multiple defence genes encoding protease inhibitors and chitinase display broad-spectrum resistance against insects, pathogens and abiotic stresses. Plant Biotechnol. J. 12, 503–515. doi: 10.1111/pbi.12157
Chen, Y., Hu, N., and Wu, H. (2019). Analyzing and characterizing the chloroplast genome of Salix wilsonii. Biomed Res. Int. 2019:5190425. doi: 10.1155/2019/5190425
Cheng, L., Li, H.-P., Qu, B., Huang, T., Tu, J.-X., Fu, T.-D., et al. (2010). Chloroplast transformation of rapeseed (Brassica napus) by particle bombardment of cotyledons. Plant Cell Rep. 29, 371–381. doi: 10.1007/s00299-010-0828-6
Daniell, H., Datta, R., Varma, S., Gray, S., and Lee, S. B. (1998). Containment of herbicide resistance through genetic engineering of the chloroplast genome. Nat. Biotechnol. 16, 345–348. doi: 10.1038/nbt0498-345
Daniell, H., Wurdack, K. J., Kanagaraj, A., Lee, S. B., Saski, C., and Jansen, R. K. (2008). The complete nucleotide sequence of the cassava (Manihot esculenta) chloroplast genome and the evolution of atpF in Malpighiales: RNA editing and multiple losses of a group II intron. Theor. Appl. Genet. 116, 723–737. doi: 10.1007/s00122-007-0706-y
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
De Cosa, B., Moar, W., Lee, S. B., Miller, M., and Daniell, H. (2001). Overexpression of the Bt cry2Aa2 operon in chloroplasts leads to formation of insecticidal crystals. Nat. Biotechnol. 19, 71–74. doi: 10.1038/83559
de Longevialle, A. F., Hendrickson, L., Taylor, N. L., Delannoy, E., Lurin, C., Badger, M., et al. (2008). The pentatricopeptide repeat gene OTP51 with two LAGLIDADG motifs is required for the cis-splicing of plastid ycf3 intron 2 in Arabidopsis thaliana. Plant J. 56, 157–168. doi: 10.1111/j.1365-313X.2008.03581.x
Dhingra, A., Archie, J., Portis, R., and Daniell, H. (2004). Enhanced translation of a chloroplast-expressed RbcS gene restores small subunit levels and photosynthesis in nuclear RbcS antisense plants. Proc. Natl. Acad. Sci. U.S.A. 101, 6315–6320. doi: 10.1073/pnas.0400981101
Dong, X., Wang, H., and Zhang, J. (2017). Comparative study on floristic component of maoer mountain national forest park around 30 years. Acta Bot. Boreali 37, 2290–2299. doi: 10.7606/j.issn.1000-4025.2017.11.2290
Dufourmantel, N., Pelissier, B., Garcon, F., Peltier, G., Ferullo, J.-M., and Tissot, G. (2004). Generation of fertile transplastomic soybean. Plant Mol. Biol. 55, 479–489. doi: 10.1007/s11103-004-0192-4
Dugas, D. V., Hernandez, D., Koenen, E. J., Schwarz, E., Straub, S., Hughes, C. E., et al. (2015). Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 5:16958. doi: 10.1038/srep16958
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Gao, B., Yuan, L., Tang, T., Hou, J., Pan, K., and Wei, N. (2019). The complete chloroplast genome sequence of Alpinia oxyphylla Miq. and comparison analysis within the Zingiberaceae family. PLoS One 14:e0218817. doi: 10.1371/journal.pone.0218817
Gulyaev, S., Cai, X. J., Guo, F. Y., Kikuchi, S., Applequist, W. L., Zhang, Z. X., et al. (2022). The phylogeny of Salix revealed by whole genome re-sequencing suggests different sex-determination systems in major groups of the genus. Ann. Bot. 129, 485–498. doi: 10.1093/aob/mcac012
Guo, F., Liu, K., Wang, Y., Li, E., Zhan, Z., and Zhang, Z. (2021). Complete chloroplast genome sequence of Salix sinopurpurea (Salicaceae). Mitochondrial DNA B Resour. 6, 718–719. doi: 10.1080/23802359.2020.1858726
Harada, H., Maoka, T., Osawa, A., Hattan, J.-I., Kanamoto, H., Shindo, K., et al. (2014). Construction of transplastomic lettuce (Lactuca sativa) dominantly producing astaxanthin fatty acid esters and detailed chemical analysis of generated carotenoids. Transgenic Res. 23, 303–315. doi: 10.1007/s11248-013-9750-3
Hatmaker, E. A., Wadl, P. A., Rinehart, T. A., Carroll, J., Lane, T. S., Trigiano, R. N., et al. (2020). Complete chloroplast genome comparisons for Pityopsis (Asteraceae). PLoS One 15:e0241391. doi: 10.1371/journal.pone.0241391
Huang, H., Shi, C., Liu, Y., Mao, S.-Y., and Gao, L.-Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. doi: 10.1186/1471-2148-14-151
Huang, Y., Wang, J., Yang, Y., Fan, C., and Chen, J. (2017). Phylogenomic analysis and dynamic evolution of chloroplast genomes in salicaceae. Front. Plant Sci. 8:1050. doi: 10.3389/fpls.2017.01050
Jansen, R. K., and Ruhlman, T. A. (2012). “Plastid genomes of seed plants,” in Genomics of Chloroplasts and Mitochondria, eds R. Bock and V. Knoop (Dordrecht: Springer), 103–126.
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21:241. doi: 10.1186/s13059-020-02154-5
Jo, S., Kim, Y. K., Cheon, S. H., Fan, Q., and Kim, K. J. (2019). Characterization of 20 complete plastomes from the tribe Laureae (Lauraceae) and distribution of small inversions. PLoS One 14:e0224622. doi: 10.1371/journal.pone.0224622
Khadivi-Khub, A., Zamani, Z., Fattahi, R., and Wünsch, A. (2013). Genetic variation in wild Prunus L. subgen. Cerasus germplasm from Iran characterized by nuclear and chloroplast SSR markers. Trees 28, 471–485. doi: 10.1007/s00468-013-0964-z
Kim, K., Lee, S. C., Lee, J., Yu, Y., Yang, K., Choi, B. S., et al. (2015). Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 5:15655. doi: 10.1038/srep15655
Kim, S. H., Yang, J., Park, J., Yamada, T., Maki, M., and Kim, S. C. (2019). Comparison of whole plastome sequences between thermogenic skunk cabbage Symplocarpus renifolius and nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia. Int. J. Mol. Sci. 20:4678. doi: 10.3390/ijms20194678
Kim, Y.-D., Kim, S.-H., Kim, C. H., and Jansen, R. K. (2004). Phylogeny of berberidaceae based on sequences of the chloroplast gene ndhF. Biochem. Syst. Ecol. 32, 291–301. doi: 10.1016/j.bse.2003.08.002
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter the manifold applications of repeat analysis. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lauron-Moreau, A., Pitre, F. E., Argus, G. W., Labrecque, M., and Brouillet, L. (2015). Phylogenetic relationships of American willows (Salix L., Salicaceae). PLoS One 10:e0121965. doi: 10.1371/journal.pone.0121965
Lee, S.-B., Kwon, H.-B., Kwon, S.-J., Park, S.-C., Jeong, M.-J., Han, S.-E., et al. (2003). Accumulation of trehalose within transgenic chloroplasts confers drought tolerance. Mol. Breed. 11, 1–13. doi: 10.1023/a:1022100404542
Lee, S. M., Kang, K., Chung, H., Yoo, S. H., Xu, X. M., Lee, S.-B., et al. (2006). Plastid transformation in the monocotyledonous cereal crop, rice (Oryza sativa) and transmission of transgenes to their progeny. Mol. Cells 21, 401–410. doi: 10.1016/j.cell.2006.04.043
Legen, J., Wanner, G., Herrmann, R. G., Small, I., and Schmitz-Linneweber, C. (2007). Plastid tRNA genes trnC-GCA and trnN-GUU are essential for plant cell development. Plant J. 51, 751–762. doi: 10.1111/j.1365-313X.2007.03177.x
Lelivelt, C. L., van Dun, K. M., Snoo, C., McCabe, M. S., Hogg, B. V., and Nugent, J. M. (2014). Plastid transformation in lettuce (Lactuca sativa L.) by polyethylene glycol treatment of protoplasts. Methods Mol. Biol. 1132, 317–330. doi: 10.1007/978-1-62703-995-6_20
Li, B., Lin, F., Huang, P., Guo, W., and Zheng, Y. (2020a). Development of nuclear SSR and chloroplast genome markers in diverse Liriodendron chinense germplasm based on low-coverage whole genome sequencing. Biol. Res. 53:21. doi: 10.1186/s40659-020-00289-0
Li, D.-M., Zhu, G.-F., Xu, Y.-C., Ye, Y.-J., and Liu, J.-M. (2020b). Complete chloroplast genomes of three medicinal Alpinia species: Genome organization, comparative analyses and phylogenetic relationships in family Zingiberaceae. Plants 9:286. doi: 10.3390/plants9020286
Li, D. M., Ye, Y. J., Xu, Y. C., Liu, J. M., and Zhu, G. F. (2020c). Complete chloroplast genomes of Zingiber montanum and Zingiber zerumbet: Genome structure, comparative and phylogenetic analyses. PLoS One 15:e0236590. doi: 10.1371/journal.pone.0236590
Li, J., Zhuo, Z., Xu, D., Yang, H., and Zhu, T. (2021). The complete chloroplast genome of Salix cupularis Rehder, a sand binder in alpine hillslope, China. Mitochondrial DNA B Resour. 6, 2519–2520. doi: 10.1080/23802359.2021.1959435
Liang, D., Wang, H., Zhang, J., Zhao, Y., and Wu, F. (2022). Complete chloroplast genome sequence of Fagus longipetiolata Seemen (Fagaceae): Genome structure, adaptive evolution, and phylogenetic relationships. Life 12:92. doi: 10.3390/life12010092
Liang, H., and Chen, J. (2021). Comparison and phylogenetic analyses of nine complete chloroplast genomes of Zingibereae. Forests 12:710. doi: 10.3390/f12060710
Liu, C.-W., Lin, C.-C., Chen, J. J., and Tseng, M.-J. (2007). Stable chloroplast transformation in cabbage (Brassica oleracea L. var. capitata L.) by particle bombardment. Plant Cell Rep. 26, 1733–1744. doi: 10.1007/s00299-007-0374-z
Liu, L., Du, Y., Shen, C., Li, R., Lee, J., and Li, P. (2020). The complete chloroplast genome of Papaver setigerum and comparative analyses in Papaveraceae. Genet. Mol. Biol. 43:e20190272. doi: 10.1590/1678-4685-GMB-2019-0272
Liu, Q., and Xue, Q. (2005). Comparative studies on codon usage pattern of chloroplasts and their host nuclear genes in four plant species. J. Genet. 84, 55–62. doi: 10.1007/bf02715890
Lu, Q., Ye, W., Lu, R., Xu, W., and Qiu, Y. (2018). Phylogenomic and comparative analyses of complete plastomes of Croomia and Stemona (Stemonaceae). Int. J. Mol. Sci. 19:2383. doi: 10.3390/ijms19082383
Meucci, S., Schulte, L., Zimmermann, H. H., Stoof-Leichsenring, K. R., Epp, L., Bronken Eidesen, P., et al. (2021). Holocene chloroplast genetic variation of shrubs (Alnus alnobetula, Betula nana, Salix sp.) at the siberian tundra-taiga ecotone inferred from modern chloroplast genome assembly and sedimentary ancient DNA analyses. Ecol. Evol. 11, 2173–2193. doi: 10.1002/ece3.7183
Monde, R.-A., Greene, J. C., and Stern, D. B. (2000). The sequence and secondary structure of the 3’-UTR affect 3’-end maturation, RNA accumulation, and translation in tobacco chloroplasts. Plant Mol. Biol. 44, 529–542. doi: 10.1023/A:1026540310934
Muralikrishna, N., Srinivas, K., Kumar, K. B., and Sadanandam, A. (2016). Stable plastid transformation in Scoparia dulcis L. Physiol. Mol. Biol. Plants 22, 575–581. doi: 10.1007/s12298-016-0386-7
Okuzaki, A., Kida, S., Watanabe, J., Hirasawa, I., and Tabei, Y. (2013). Efficient plastid transformation in tobacco using small gold particles (0.07-0.3μm). Plant Biotechnol. 30, 65–72. doi: 10.5511/plantbiotechnology.12.1227a
Olmstead, R. G., Kim, K. J., Jansen, R. K., and Wagstaff, S. J. (2000). The phylogeny of the Asteridae sensu lato based on chloroplast ndhF gene sequences. Mol. Phylogenet. Evol. 16, 96–112. doi: 10.1006/mpev.1999.0769
Pan, Y., Jiang, M., Cong, X., Xue, Z., Liu, B., Zhang, W., et al. (2021). Composition and floristic characteristics of wild seed plants in the marshes in Sanjiang national nature reserve, Heilongjiang province. Wetl. Sci. 19, 342–352. doi: 10.13248/j.cnki.wetlandsci.2021.03.009
Park, I., Yang, S., Kim, W. J., Noh, P., Lee, H. O., and Moon, B. C. (2018). The complete chloroplast genomes of six Ipomoea species and indel marker development for the discrimination of authentic pharbitidis semen (seeds of I. nil or I. purpurea). Front. Plant Sci. 9:965. doi: 10.3389/fpls.2018.00965
Radakovits, R., Jinkerson, R. E., Darzins, A., and Posewitz, M. C. (2010). Genetic engineering of algae for enhanced biofuel production. Eukaryot. Cell 9, 486–501. doi: 10.1128/EC.00364-09
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Ruhlman, T. A., Rajasekaran, K., and Cary, J. W. (2014). Expression of chloroperoxidase from Pseudomonas pyrrocinia in tobacco plastids for fungal resistance. Plant Sci. 228, 98–106. doi: 10.1016/j.plantsci.2014.02.008
Saina, J. K., Li, Z. Z., Gichira, A. W., and Liao, Y. Y. (2018). The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima (Mill.) (Sapindales: Simaroubaceae), an important pantropical tree. Int. J. Mol. Sci. 19:929. doi: 10.3390/ijms19040929
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi: 10.1093/nar/gkz345
Shidhi, P. R., Nadiya, F., Biju, V. C., Vijayan, S., Sasi, A., Vipin, C. L., et al. (2021). Complete chloroplast genome of the medicinal plant Evolvulus alsinoides: Comparative analysis, identification of mutational hotspots and evolutionary dynamics with species of Solanales. Physiol. Mol. Biol. Plants 27, 1867–1884. doi: 10.1007/s12298-021-01051-w
Sidorov, V. A., Kasten, D., Pang, S. Z., Hajdukiewicz, P. T., Staub, J. M., and Nehra, N. S. (1999). Stable chloroplast transformation in potato: Use of green fluorescent protein as a plastid marker. Plant J. 19, 209–216. doi: 10.1046/j.1365-313X.1999.00508.x
Sikdar, S., Serino, G., Chaudhuri, S., and Maliga, P. (1998). Plastid transformation in Arabidopsis thaliana. Plant Cell Rep. 18, 20–24.
Skvortsov, A. K. (1999). Willows of Russia and adjacent countries: Taxonomical and geographical revision. Joensuu: University of Joensuu.
Svab, Z., and Maliga, P. (1993). High-frequency plastid transformation in tobacco by selection for a chimeric aadA gene. Proc. Natl. Acad. Sci. U.S.A. 90, 913–917. doi: 10.1073/pnas.90.3.913
Svab, Z., and Maliga, P. (2007). Exceptional transmission of plastids and mitochondria from the transplastomic pollen parent and its impact on transgene containment. Proc. Natl. Acad. Sci. U.S.A. 104, 7003–7008. doi: 10.1073/pnas.0700063104
Techaprasan, J., Klinbunga, S., Ngamriabsakul, C., and Jenjittikul, T. (2010). Genetic variation of Kaempferia (Zingiberaceae) in Thailand based on chloroplast DNA (psbA-trnH and petA-psbJ) sequences. Genet. Mol. Res. 9, 1957–1973. doi: 10.4238/vol9-4gmr873
Tian, X., Guo, Z. H., and De-Zhu, L. I. (2002). Phylogeny of aceraceae based on ITS and trn L-F data sets. Acta Bot. Sin. 44, 714–724. doi: 10.1127/0340-269X/2002/0032-0317
Tian, X., Ye, J., and Song, Y. (2019). Plastome sequences help to improve the systematic position of trinerved Lindera species in the family Lauraceae. PeerJ 7:e7662. doi: 10.7717/peerj.7662
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Wang, J., Yu, Z., Yao, X., Wan, J., Wang, Z., and Li, X. (2022). The complete chloroplast genome sequence of Salix kochiana Trautv. and its phylogenetic analysis. Mitochondrial DNA B Resour. 7, 1123–1125. doi: 10.1080/23802359.2022.2087555
Wang, S., Shi, C., and Gao, L. Z. (2013). Plastid genome sequence of a wild woody oil species, Prinsepia utilis, provides insights into evolutionary and mutational patterns of Rosaceae chloroplast genomes. PLoS One 8:e73946. doi: 10.1371/journal.pone.0073946
Wang, Y., Wang, S., Liu, Y., Yuan, Q., Sun, J., and Guo, L. (2021). Chloroplast genome variation and phylogenetic relationships of Atractylodes species. BMC Genomics 22:103. doi: 10.1186/s12864-021-07394-8
Wei, X., Li, X., Chen, T., Chen, Z., Jin, Y., Malik, K., et al. (2021). Complete chloroplast genomes of Achnatherum inebrians and comparative analyses with related species from Poaceae. FEBS Open Bio 11, 1704–1718. doi: 10.1002/2211-5463.13170
Wei, Z., Liu, Y., Lin, C., Wang, Y., Cai, Q. A., Dong, Y., et al. (2011). Transformation of alfalfa chloroplasts and expression of green fluorescent protein in a forage crop. Biotechnol. Lett. 33, 2487–2494. doi: 10.1007/s10529-011-0709-2
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Muller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Williams, A. M., Friso, G., van Wijk, K. J., and Sloan, D. B. (2019). Extreme variation in rates of evolution in the plastid Clp protease complex. Plant J. 98, 243–259. doi: 10.1111/tpj.14208
Wong, E. H., Smith, D. K., Rabadan, R., Peiris, M., and Poon, L. L. (2010). Codon usage bias and the evolution of influenza A viruses. Codon usage biases of influenza virus. BMC Evol. Biol. 10:253. doi: 10.1186/1471-2148-10-253
Wu, J., Nyman, T., Wang, D. C., Argus, G. W., Yang, Y. P., and Chen, J. H. (2015). Phylogeny of Salix subgenus Salix s.l. (Salicaceae): Delimitation, biogeography, and reticulate evolution. BMC Evol. Biol. 15:31. doi: 10.1186/s12862-015-0311-7
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xie, X., Huang, R., Li, F., Tian, E., Li, C., and Chao, Z. (2021). Phylogenetic position of Bupleurum sikangense inferred from the complete chloroplast genome sequence. Gene 798:145801. doi: 10.1016/j.gene.2021.145801
Xue, S., Shi, T., Luo, W., Ni, X., Iqbal, S., Ni, Z., et al. (2019). Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic. Res. 6:89. doi: 10.1038/s41438-019-0171-1
Yagi, Y., and Shiina, T. (2014). Recent advances in the study of chloroplast gene expression and its evolution. Front. Plant Sci. 5:61. doi: 10.3389/fpls.2014.00061
Yan, C., Du, J., Gao, L., Li, Y., and Hou, X. (2019). The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): Genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 699, 24–36. doi: 10.1016/j.gene.2019.02.075
Yang, C.-H., Liu, X., Cui, Y.-X., Nie, L.-P., Lin, Y.-L., Wei, X.-P., et al. (2020). Molecular structure and phylogenetic analyses of the complete chloroplast genomes of three original species of Pyrrosiae Folium. Chin. J. Nat. Med. 18, 573–581. doi: 10.1016/s1875-5364(20)30069-8
Yang, Z., Huang, Y., An, W., Zheng, X., Huang, S., and Liang, L. (2019). Sequencing and structural analysis of the complete chloroplast genome of the medicinal plant Lycium chinense Mill. Plants 8:87. doi: 10.3390/plants8040087
Yarbakht, M., Jalali-Javaran, M., Nikkhah, M., and Mohebodini, M. (2015). Dicistronic expression of human proinsulin–protein A fusion in tobacco chloroplast. Biotechnol. Appl. Biochem. 62, 55–63. doi: 10.1002/bab.1230
Yi, D. K., Lee, H. L., Sun, B. Y., Chung, M. Y., and Kim, K. J. (2012). The complete chloroplast DNA sequence of Eleutherococcus senticosus (Araliaceae); comparative evolutionary analyses with other three asterids. Mol. Cells 33, 497–508. doi: 10.1007/s10059-012-2281-6
Yin, X., Liao, B., Guo, S., Liang, C., Pei, J., Xu, J., et al. (2020). The chloroplasts genomic analyses of Rosa laevigata, R. rugosa and R. canina. Chin. Med. 15:18. doi: 10.1186/s13020-020-0298-x
Zhang, D., Gao, F., Li, W. X., Jakovlić, I., Zou, H., Zhang, J., et al. (2018). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1101/489088
Zhang, X. F., Landis, J. B., Wang, H. X., Zhu, Z. X., and Wang, H. F. (2021). Comparative analysis of chloroplast genome structure and molecular dating in Myrtales. BMC Plant Biol. 21:219. doi: 10.1186/s12870-021-02985-9
Zhang, Y., Nie, X., Jia, X., Zhao, C., Biradar, S. S., Wang, L., et al. (2012). Analysis of codon usage patterns of the chloroplast genomes in the Poaceae family. Aust. J. Bot. 60, 461–470. doi: 10.1071/bt12073
Zhengyi, W., Zhou, Z., Li, D., Peng, H., and Sun, H. (2003). The areal-types of the world families of seed plants. Acta Bot. Yunnanica 25, 245–257. doi: 10.3969/j.issn.2095-0845.2003.03.001
Zong, D., Gan, P., Zhou, A., Zhang, Y., Zou, X., Duan, A., et al. (2019). Plastome sequences help to resolve deep-level relationships of Populus in the family salicaceae. Front. Plant Sci. 10:5. doi: 10.3389/fpls.2019.00005
Keywords: Salix floderusii, chloroplast genome, phylogenetic relationship, chloroplast regulatory elements, prokaryotic expression
Citation: Ren W, Jiang Z, Zhang M, Kong L, Zhang H, Liu Y, Fu Q and Ma W (2022) The chloroplast genome of Salix floderusii and characterization of chloroplast regulatory elements. Front. Plant Sci. 13:987443. doi: 10.3389/fpls.2022.987443
Received: 06 July 2022; Accepted: 11 August 2022;
Published: 26 August 2022.
Edited by:
Niaz Ahmad, National Institute for Biotechnology and Genetic Engineering, PakistanReviewed by:
Linchun Shi, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaXiwen Li, China Academy of Chinese Medical Sciences, China
Copyright © 2022 Ren, Jiang, Zhang, Kong, Zhang, Liu, Fu and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qifeng Fu, MjY5NzAyNjEzQHFxLmNvbQ==; Wei Ma, bWF3ZWlAaGxqdWNtLm5ldA==
†These authors have contributed equally to this work