 Yaling Wang
Yaling Wang Zhongxiao Zhang
Zhongxiao Zhang Penghui Guo
Penghui Guo- School of Life Sciences and Engineering, Northwest Minzu University, Lanzhou, China
The genus Distylium (Hamamelidaceae) is highly valued for its applications in ornamental horticulture, industry, and construction. Although plastid genomes (plastomes) of multiple Distylium species have been characterized, no mitochondrial genomes (mitogenomes) have been reported for this genus. In this study, we assembled and annotated the complete mitogenome of Distylium racemosum using HiFi sequencing data. The mitogenome comprises a longer circular chromosome and a shorter linear chromosome (904,264 bp in total length), revealing a structurally complex conformation. We annotated 67 genes, including 43 protein-coding genes (PCGs), 21 tRNA genes, and three rRNA genes. Analyses identified exceptionally high repetitive sequence content, with 304 simple sequence repeats, 1,508 dispersed repeats, and 50 tandem repeats, representing the highest repeat content among Saxifragales mitogenomes to date. Additionally, 49 mitochondrial plastid DNA sequences were detected, including only one complete plastid-derived gene (trnC-GCA) transferred to the mitogenome. We predicted 697 RNA editing sites across 42 PCGs, further underscoring the genome’s dynamic evolution. Phylogenetic reconstruction based on mitogenomes and plastomes from 18 species indicated D. racemosum occupied a basal position within Saxifragales, which is consistent with the APG IV classification system. This study provides the first comprehensive mitogenomic resource for the Distylium genus, offering valuable insights for molecular classification, species identification, and germplasm conservation of Distylium plants.
1 Introduction
Distylium racemosum, a flowering plant in the genus Distylium (Hamamelidaceae), is endemic to Asia. This evergreen shrub or small tree inhabits warm-temperate lowland forests, distinguished by dense branching, persistent dark-green foliage, and a compact growth form (Dong et al., 2021). Its ornamental appeal is enhanced by clusters of small red flowers produced in spring. The Distylium species exhibits notable resistance to airborne pollutants, including sulfur dioxide and chlorine, making it particularly suitable for urban greening in industrial and mining zones. Notably, Distylium chinense demonstrates exceptional ecological adaptability, serving as a key species for soil conservation and embankment stabilization (Liu et al., 2014; Xiang et al., 2020). Distylium species also engages in a unique ecological interaction with the aphid Nipponaphis distychii, which induces large gall formation on leaves. These galls accumulate tannins, compounds historically utilized as natural dyes (Ismayati et al., 2024). Furthermore, the wood of Distylium is exceptionally dense and durable, rendering it valuable for construction applications such as housing and vehicle manufacturing (Yagi et al., 2019).
Mitochondria, semi-autonomous organelles possessing independent genetic material and distinct replication and expression systems, are critical for cellular respiration and metabolic regulation (Dyall et al., 2004; Gray et al., 1999; van Loo et al., 2002). Unlike animal mitochondrial genomes (mitogenomes, typically 10–20 kb) and plant plastid genomes (plastomes, generally 100–200 kb), which exhibit size conservation (Liu et al., 2022; Wu et al., 2022), plant mitogenomes display substantial size variation, ranging from 66 kb in Viscum scurruloideum to 18.99 Mb in Cathaya argyrophylla (Huang et al., 2024; Skippington et al., 2015). These genomes are pivotal for elucidating cytoplasmic male sterility mechanisms and developing molecular breeding applications (Han et al., 2024; Xiao et al., 2020). However, plant mitogenome assembly remains technically challenging due to extreme structural plasticity, long repetitive sequences, and frequent horizontal transfer of nuclear mitochondrial DNA (NUMTs) and mitochondrial plastid DNA (MTPTs) (Han et al., 2022; Sun et al., 2024). To date, over 34,000 complete plastomes have been reported in Chloroplast Genome Information Resource (CGIR) (https://ngdc.cncb.ac.cn/cgir), while the NCBI Nucleotide database (https://www.ncbi.nlm.nih.gov/) currently contains fewer than one thousand complete mitogenomes for plants (last accessed: February 28th, 2025) (Huang et al., 2025; Wang J. et al., 2024). This disparity highlights the complexity and technical challenges associated with plant mitogenome assembly and annotation.
Previous plastome sequencing of 12 Distylium species has established a phylogenomic framework for understanding the genus’s evolution (Dong et al., 2021). Despite these advances, comprehensive genomic resources for Distylium (particularly nuclear and mitochondrial genomes) remain critically limited. To date, the mitogenomes of Distylium species remain entirely unexplored, and their phylogenetic relationships based on mitogenomic data are still uncleared. While mitogenomes have been assembled for numerous Saxifragales species, only two unannotated mitogenomes from Hamamelidaceae (Rhodoleia championii and Loropetalum chinense) are available in the NCBI database, with none reported for Distylium. This gap underscores the necessity to prioritize mitogenome sequencing and annotation in Hamamelidaceae. Such initiatives will advance our understanding of evolutionary trajectories within the family and improve phylogenomic resolution across Saxifragales.
In this study, we successfully assembled and annotated the mitogenome of D. racemosum, establishing the inaugural mitochondrial genomic resource for the Distylium genus. We comprehensively characterized the mitogenome’s structural features, repeat content, RNA editing sites and MTPTs, filling a critical gap in genomic data for this genus. Comparative genomic analyses and phylogenies reconstructed from mitochondrial and plastid protein-coding genes (PCGs) further clarified evolutionary relationships. These results provide foundational resources for molecular taxonomy, species discrimination, and germplasm conservation in Distylium, while elucidating broader evolutionary dynamics within Saxifragales.
2 Materials and methods
2.1 DNA extraction and sequencing
Fresh leaves of D. racemosum were collected from Nanjing Forestry University (Nanjing, China; 32°08′ N, 118°82′ E) and immediately frozen at −80°C for downstream analysis. High-quality genomic DNA was extracted using the CTAB method (Arseneau et al., 2017). DNA integrity and purity were verified via 1.0% agarose gel electrophoresis and quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) (Shi et al., 2023). Sequencing libraries were prepared with the SMRTbell Express Template Prep Kit 2.0 (PacBio Biosciences, California, USA) using high-molecular-weight DNA. High-fidelity (HiFi) sequencing data was subsequently performed on the PacBio Revio platform.
2.2 Assembly and annotation of D. racemosum mitogenome
The HiFi sequencing data were processed with PMAT v2.0 (Bi et al., 2024a) to assemble the D. racemosum mitogenome using parameters ‘-t hifi -m -F 0.2 -T 50’ in ‘autoMito’ mode. The raw assembly graph was resolved and visualized with Bandage (Wick et al., 2015). Annotation of the D. racemosum mitogenome was carried out using the online program PMGA (http://47.96.249.172:16084/home) (Li et al., 2024). Then the rRNA genes (rRNAs) and tRNA genes (tRNAs) were verified using BLASTn and tRNAscan-SE v2.0, respectively (Camacho et al., 2009; Chan et al., 2021; Chen et al., 2015). Finally, the genome map of D. racemosum mitogenome was visualized using PMGmap with default parameters (Zhang et al., 2024).
2.3 Detection of repeat sequences and prediction of RNA editing sites
Simple sequence repeats (SSRs) in the D. racemosum mitogenome were identified using the online platform MISA (https://webblast.ipk-gatersleben.de/misa/) (Beier et al., 2017). The thresholds for the minimum number of repetitions were set as follows: 10 for mononucleotides, 5 for dinucleotides, 4 for trinucleotides, and 3 for tetranucleotides, pentanucleotides, and hexanucleotides. Dispersed repeats were detected using the program vmatch-2.3.1with parameters ‘-d -p -h 3 -l 30 -best 5000 -noscore -noidentity -absolute’ (Kurtz et al., 2001). Additionally, tandem repeats in the mitogenome were identified using Tandem Repeats Finder v4.09 (https://tandem.bu.edu/trf/trf.html) with default parameters (Benson, 1999). All repeat elements were manually verified and then visualized using the ggplot2 package in R.
RNA editing, a post-transcriptional process involving the insertion, deletion, or substitution of nucleotides in RNA transcripts, plays a critical role in expanding transcriptional and functional diversity within mitochondrial genes (Fan et al., 2019). To construct reference sequences, we extracted coding sequences for all PCGs from the D. racemosum mitogenome (Yang et al., 2023a). Subsequently, the RNA editing sites in the D. racemosum mitogenome were predicted using the Deepred-Mt software (Edera et al., 2021), with a probability threshold set at 0.9 to ensure high-confidence identification.
2.4 Analysis of MTPTs and collinearity
To identify the MTPTs in D. racemosum, the complete plastome of D. racemosum was obtained from NCBI with accession number of NC_059886. Subsequently, BLASTn was utilized to detect homologous fragments between the mitogenome and plastome of D. racemosum with the following parameters: ‘-word_size 9 -evalue 1e-5 -reward 2 -gapopen 5 -gapextend 2 -penalty -3 -outfmt 6’ (Camacho et al., 2009; Chen et al., 2015). Fragments with a matching rate ≥80% and lengths ≥30 bp were selected for further analysis. The selected fragments were manually annotated to determine their locations within the mitogenome and plastome. The results of MTPTs were finally visualized using Circos (v0.69-5) (Zhang et al., 2013).
To investigate the mitogenome structure of D. racemosum, we downloaded three additional Saxifragales mitogenomes (Paeonia lactiflora, Ribes meyeri, and Rhodiola tangutica) from the NCBI database (Supplementary Table S1). Collinear blocks were identified using BLASTn with stringent criteria applied: only alignments with a minimum length of ≥200 bp and a minimum identity of ≥80% were retained for further analysis. The results were visualized using NGenomeSyn to facilitate structural comparisons (He et al., 2023).
2.5 Phylogenetic analysis
For the construction of phylogenetic trees for both mitochondria and plastids, we obtained the mitogenomes and plastomes of 17 additional species via the NCBI Nucleotide database, with Sorghum bicolor designated as the outgroup. Detailed species information and corresponding accession numbers are provided in Supplementary Table S1. We extracted 18 shared conserved PCGs from mitogenomes and 59 common conserved PCGs from plastomes using a self-written python script. Then we performed multiple sequence alignment utilizing MAFFT v7.4 (Katoh and Standley, 2013). After trimming the results using trimAl v1.4 (Capella-Gutiérrez et al., 2009), the maximum likelihood (ML) trees were reconstructed using IQ-TREE v2.0.3 with 1000 bootstrap replicates (Minh et al., 2020). The evolutionary model ‘GTR+F+I+G4’ was selected as the fittest model for both mitochondrial and plastid ML trees based on Bayesian Information Criterion (BIC) scores. Finally, the ML tree results were visualized using the online tool iTOL v5 (https://itol.embl.de/) (Letunic and Bork, 2021).
3 Results
3.1 Characteristics of the D. racemosum mitogenome
Using the Revio sequencing platform, we generated 865,212 HiFi reads totaling 15.49 Gb, with an N50 value of 14,470 bp (Table 1). The mitogenome structure of D. racemosum exhibited significant complexity upon disentangling the mitogenome graph using Bandage (Figures 1A, B), consisting of a dominant circular molecule (834,429 bp) and a smaller linear fragment (69,835 bp) (Figure 1C). The complete mitogenome spans 904,264 bp with 96.9× average coverage, a GC content of 46.28%, and contains 67 genes occupying 5.01% (45,287 bp) of the sequence (Supplementary Table S2). These include 43 PCGs (37,845 bp, 4.19%), three rRNA genes (5,861 bp, 0.65%), and 21 tRNA genes (1,581 bp, 0.17%). All 24 core mitochondrial PCGs (atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, ccmFC, ccmFN, cob, cox1, cox2, cox3, matR, mttB, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7 and nad9) and 15 variable PCGs (rpl2, rpl5, rpl10, rpl16, rps1, rps3, rps4, rps7, rps10, rps12, rps13, rps14, rps19, sdh3, and sdh4) were identified (Table 2). Notable features include three copies of atp1, two copies of rps19 and sdh3, and nad1 presenting on both the circular and linear molecules.
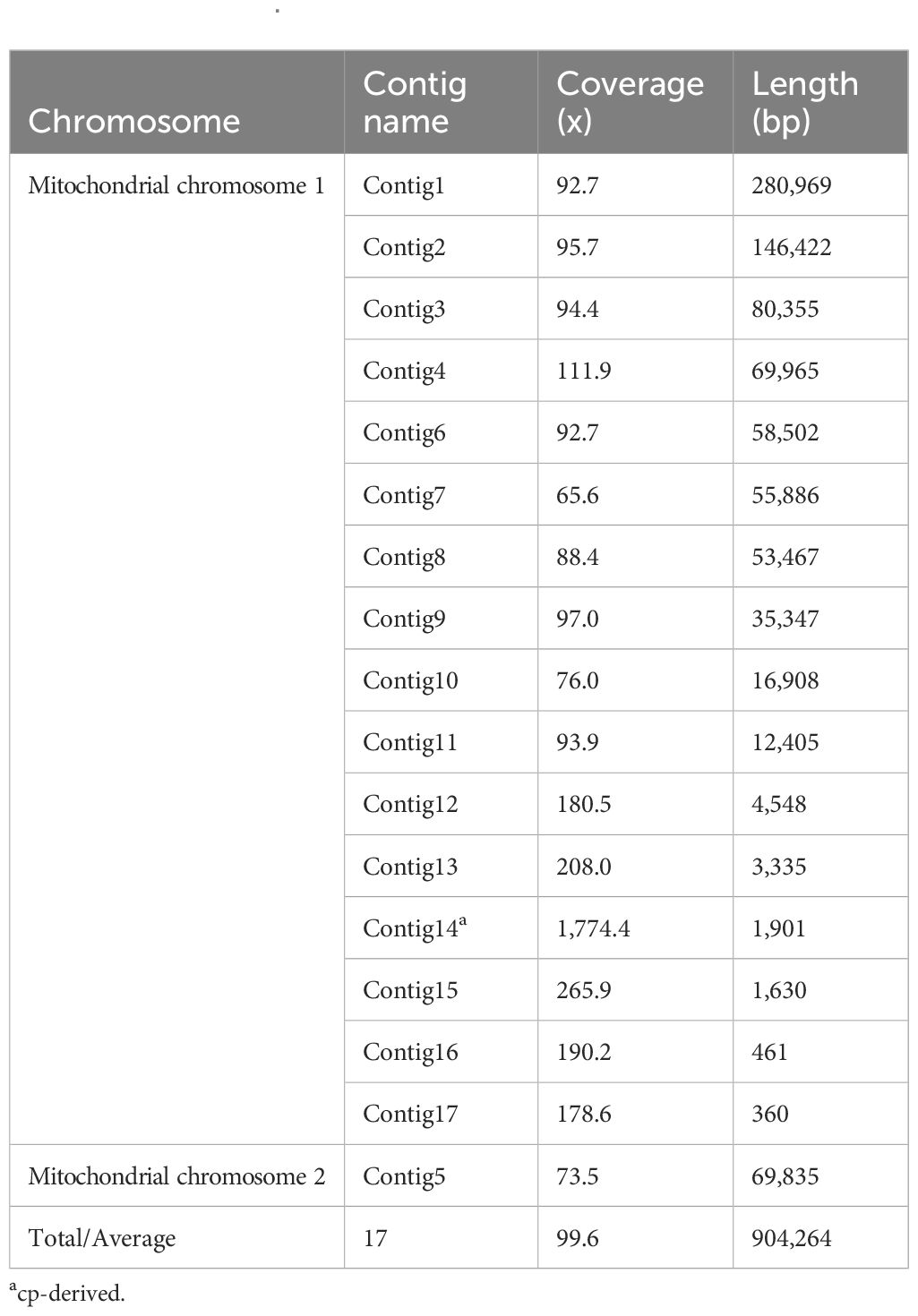
Table 1. The length and depth of each assembled contig in Distylium racemosum.
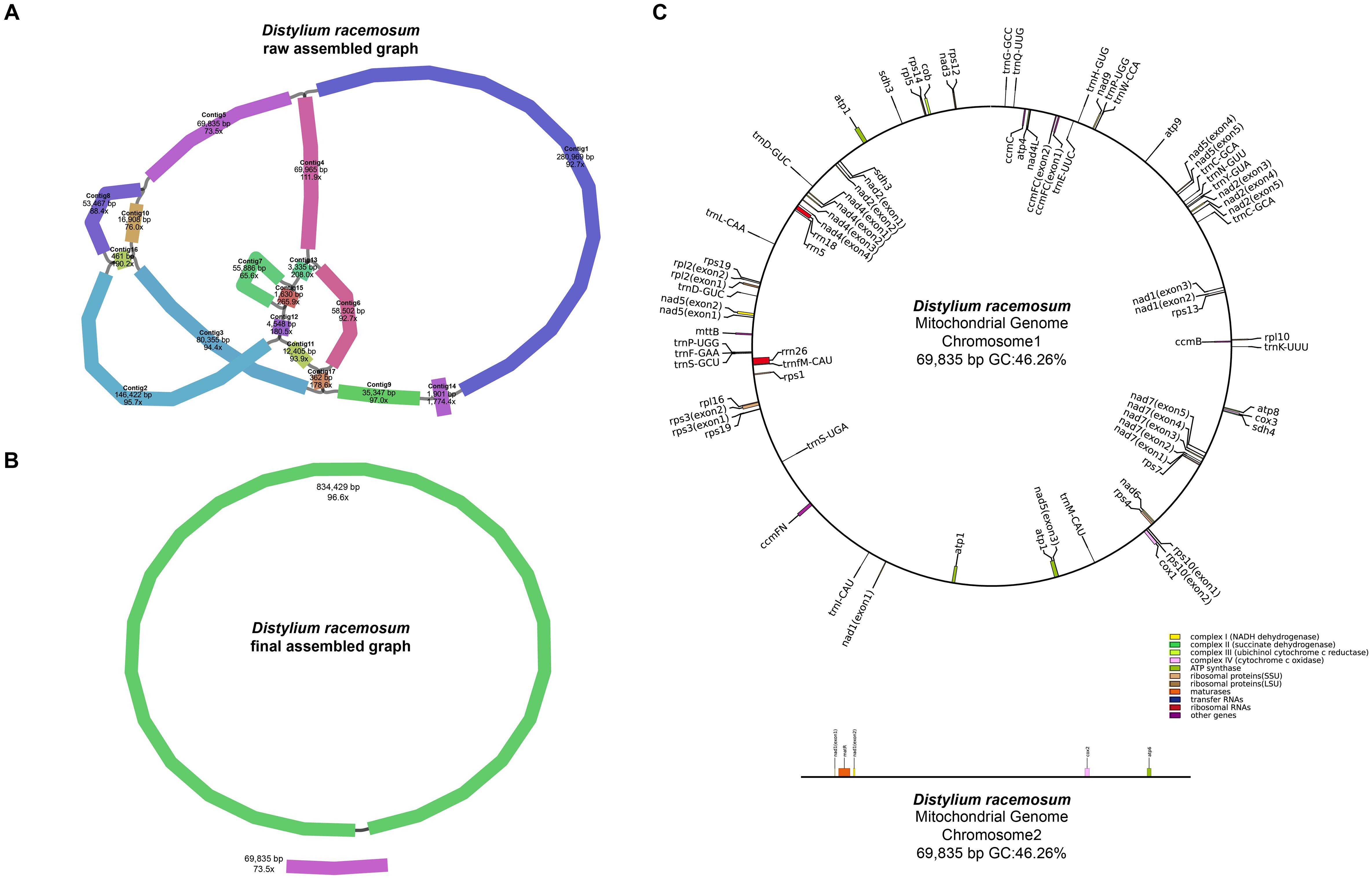
Figure 1. Assembly graphs and genome map of the Distylium racemosum mitogenome. (A) Raw assembly graph. (B) Disentangled assembly graph. (C) The genome map of the Distylium racemosum mitogenome. Different types of genes were represented by distinct color blocks.
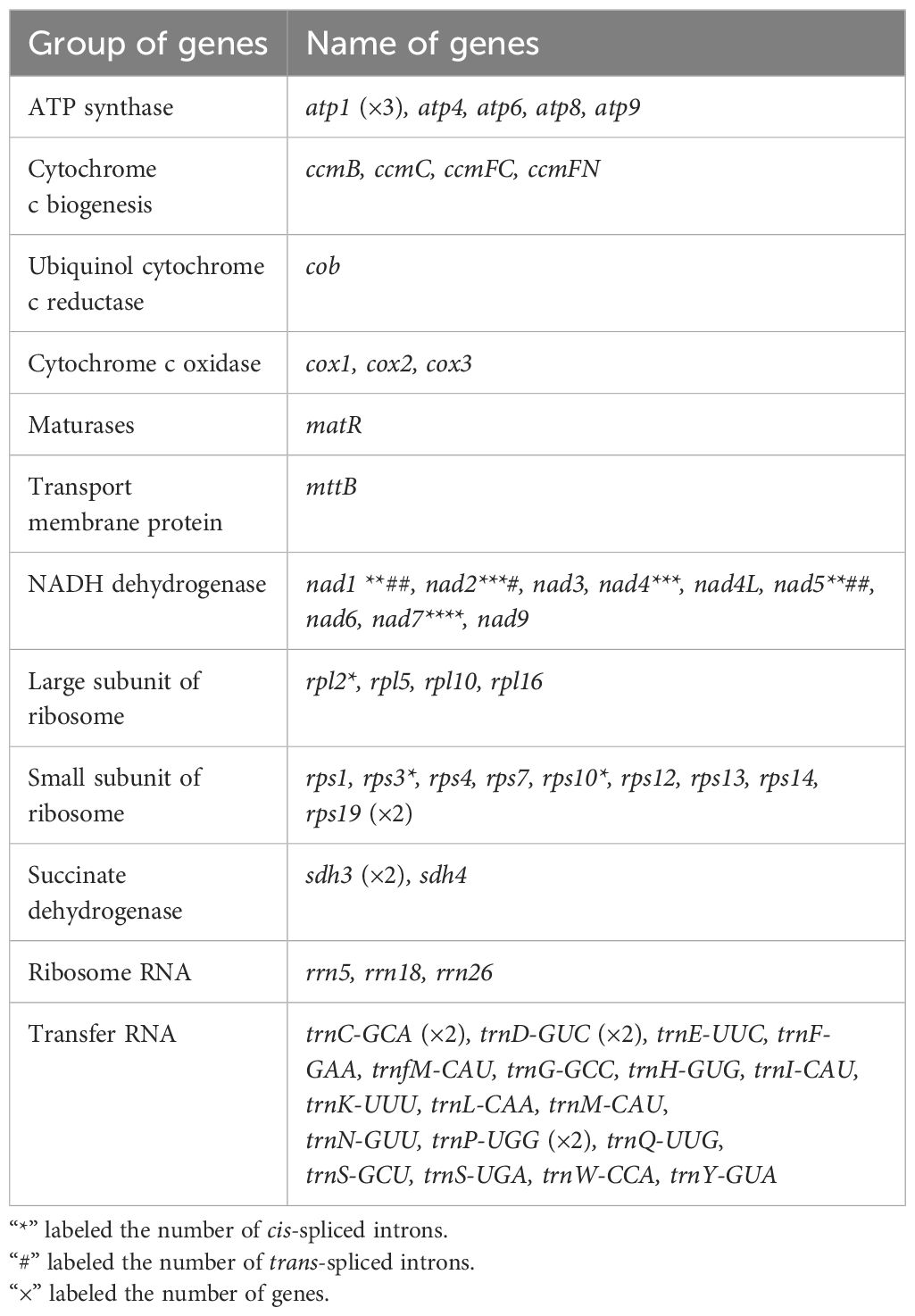
Table 2. Gene composition in the mitogenome of Distylium racemosum.
The mitogenome harbors 23 introns (18 cis-spliced and 5 trans-spliced; Supplementary Figure S1), collectively spanning 30,760 bp (3.40% of the genome; Supplementary Table S2). A total of 37 PCGs start with ATG, while cox1, nad1, and nad4L use ACG as their start codon. The start codons for mttB, rpl16, and rps4 remain unresolved. Stop codons included TAA (21 PCGs), TAG (8), TGA (11), and CGA (3) (Supplementary Table S3). The complete mitogenome of D. racemosum has been deposited in NCBI Nucleotide database under accessions PQ594873 and PQ594874.
3.2 Analysis of repeats
SSRs were widely distributed throughout the mitochondrial genome. Using the online platform MISA, we identified 304 SSRs, including 74 mononucleotide, 53 dinucleotide, 42 trinucleotide, 122 tetranucleotide, 12 pentanucleotide, and one hexanucleotide repeat units (Figure 2A; Supplementary Figure S2; Supplementary Table S4). Analysis of dispersed repeats revealed 1,508 sequences (≥30 bp) spanning 68,859 bp (7.61% of the total genome length), comprising 748 direct and 760 palindromic repeats (Supplementary Table S5). Most of the dispersed repeats (1,481 repeats) were shorter than 100 bp, while four exceeded 500 bp (Figure 2B), with the maximum length reaching 6,180 bp (Supplementary Table S5). Additionally, 50 tandem repeats (8–50 bp in length) with copy identities ≥75% were detected (Figure 2C; Supplementary Table S6).
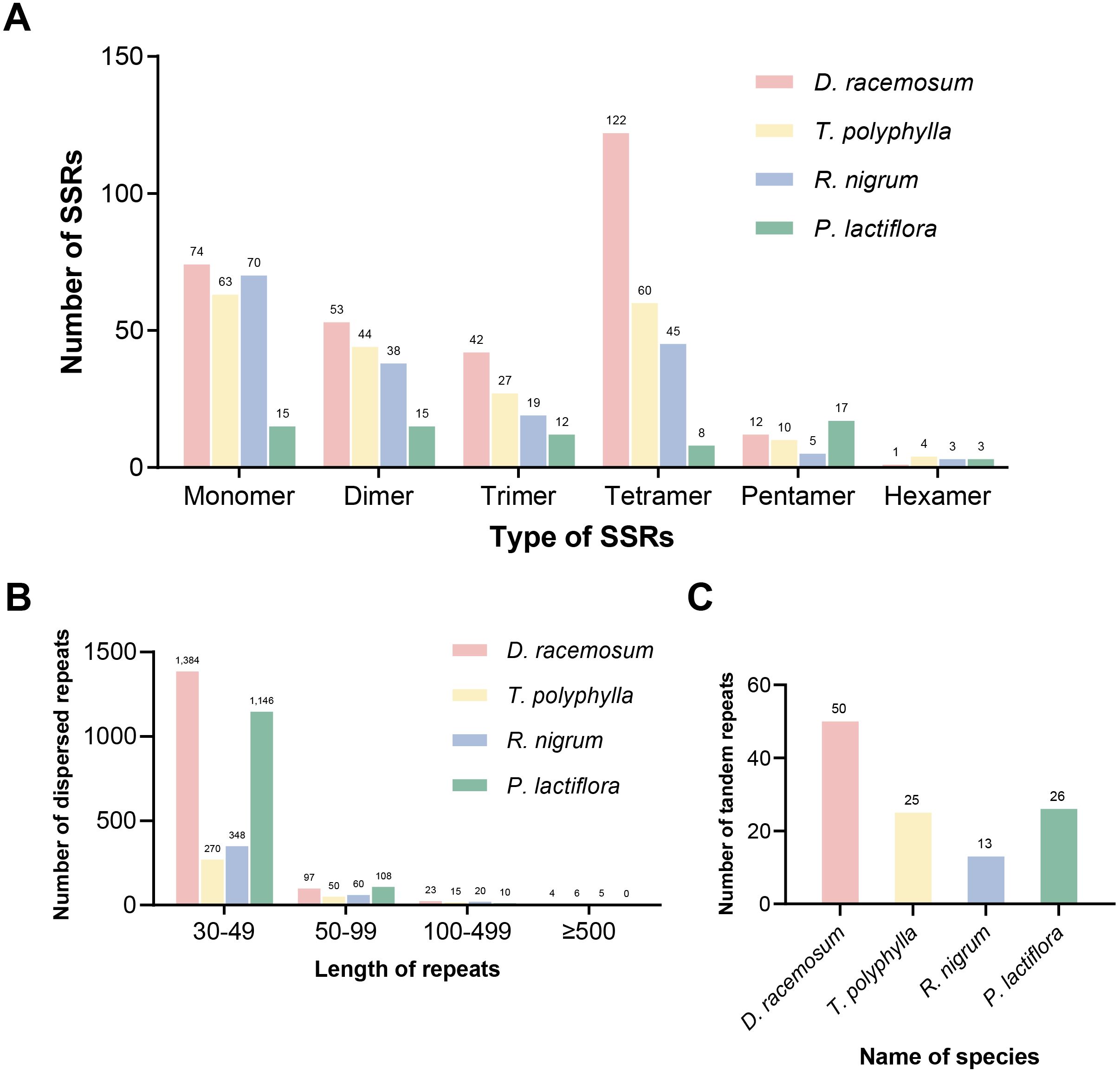
Figure 2. Repeat elements detected in the mitogenomes of four Saxifragales species. (A) Type and number of simple sequence repeats in the mitogenomes of four Saxifragales species. (B) Type and number of dispersed repeats in the mitogenomes of four Saxifragales species. (C) Number of tandem repeats in the mitogenomes of four Saxifragales species.
To further characterize repeat element patterns in Saxifragales mitogenomes, we performed comparative analyses of SSRs, dispersed repeats, and tandem repeats across four species: D. racemosum, Tiarella polyphylla, Ribes nigrum, and P. lactiflora. The D. racemosum mitogenome harbored the highest abundance of all three repeat types. SSR profiling revealed six conserved repeat categories (mono- to hexanucleotide) across all species (Figure 2A). The D. racemosum mitogenome exhibited the highest number of tetranucleotide repeat units, totaling 122. The number of dispersed repeats varied among species, but the length distributions were conserved: 30–49 bp repeats predominated, while only a few exceeded 500 bp (Figure 2B).
3.3 Analysis of MTPTs and prediction of RNA editing events
A total of 49 MTPTs were identified in the D. racemosum mitogenome (Supplementary Table S7), spanning 23,278 bp with ≥80% sequence similarity to the plastome. These MTPTs account for 14.63% of the plastome and 2.57% of the mitogenome, respectively (Figure 3). Most MTPTs ranged from 30 to 500 bp in length, while five exceeded 1,000 bp, including the longest fragment (7,710 bp). Only six plastid-derived genes were retained within these MTPTs: five partial genes (accD, rbcL, ccmC, trnL-CAA, trnY-GUA) and one complete gene (trnC-GCA).
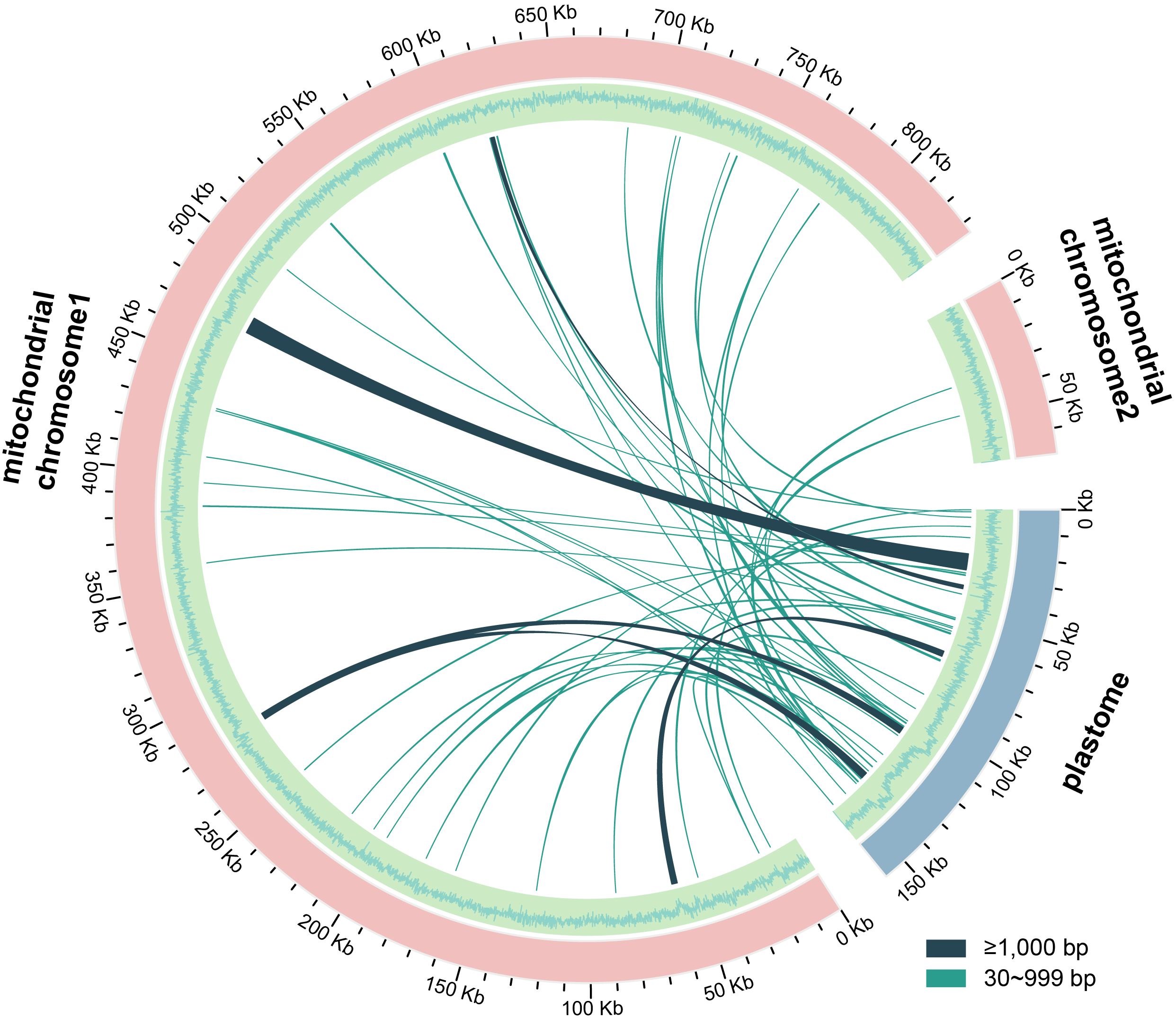
Figure 3. Distribution of mitochondrial plastid DNA sequences (MTPTs) in Distylium racemosum. The plastid and mitochondrial genomes of D. racemosum were illustrated by pink and blue arcs, respectively. The outer circle depicts the GC content of the two mitochondrial chromosomes and the plastid genome, with adjacent bars indicating the length of MTPTs. Connecting lines between the arcs represented the MTPTs. Detailed information of MTPTs was shown in Supplementary Table S7.
A total of 697 RNA editing events were identified in the PCGs of the D. racemosum mitogenome (Supplementary Table S8). The nad4 gene displayed the highest number of RNA editing sites, with 55 events detected, whereas no RNA editing events were observed in the sdh3 gene (Figure 4). RNA editing sites were distributed unevenly across codon positions: 229 (32.85%) occurred at first positions, 434 (62.27%) at second positions, and 34 (4.88%) at third positions.
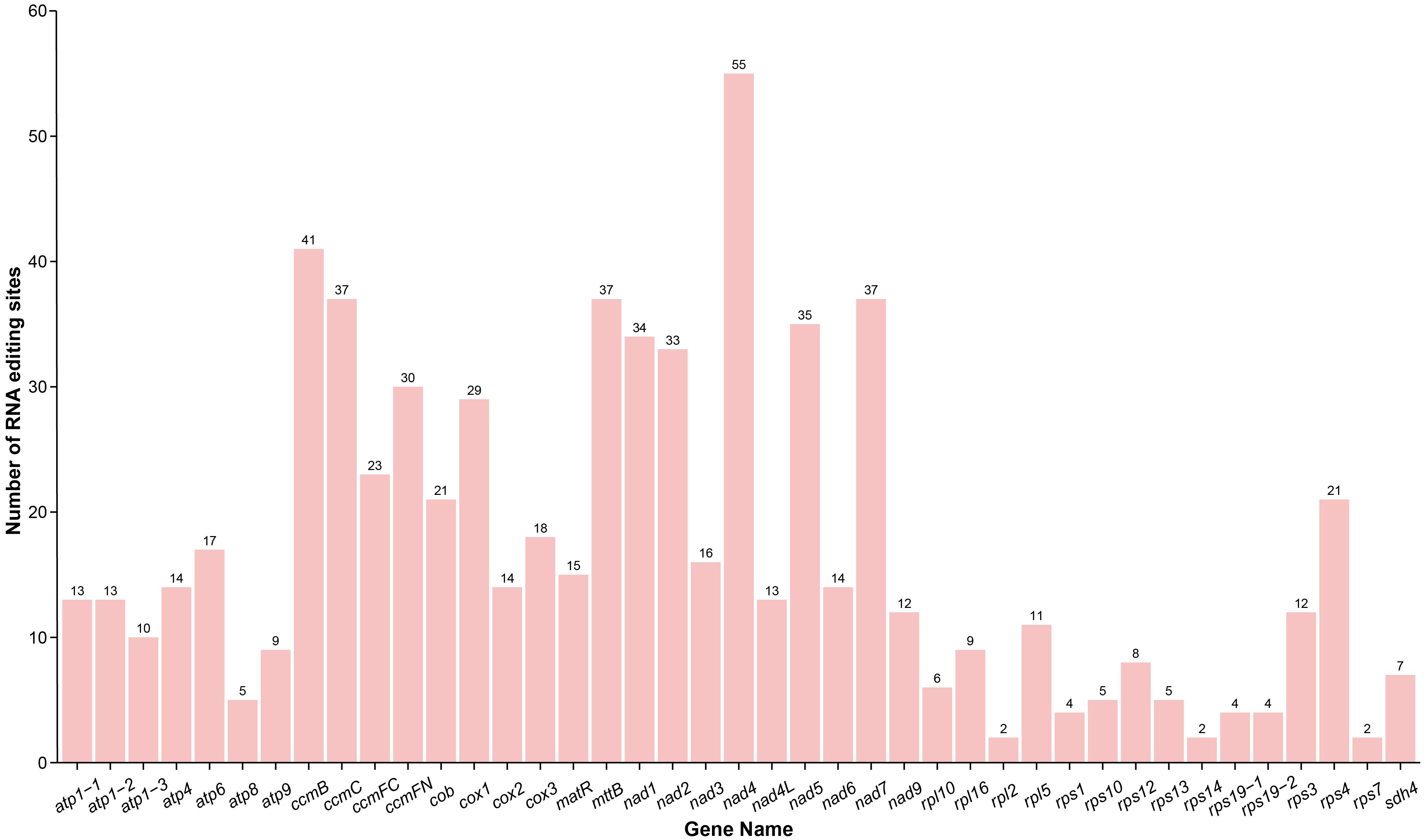
Figure 4. RNA editing events in the Distylium racemosum mitogenome. Characteristics of RNA editing sites across all protein-coding genes in the mitogenome of Distylium racemosum.
3.4 Analysis of whole-genome collinearity
We performed a comparative analysis to investigate the mitogenome collinearity among the four selected Saxifragales species. Extensive sequence rearrangements were evident between the D. racemosum mitogenome and those of the other species analyzed (Figures 5A, B). Between D. racemosum and R. tangutica, we identified 77 locally collinear blocks (LCBs; 81,121 bp), representing 31.52% of the R. tangutica mitogenome (Figure 5B; Supplementary Table S9). Similarly, a comparison of D. racemosum and R. meyeri revealed 129 LCBs, covering 183,570 bp and accounting for 37.92% of the R. meyeri mitogenome (Supplementary Table S9).
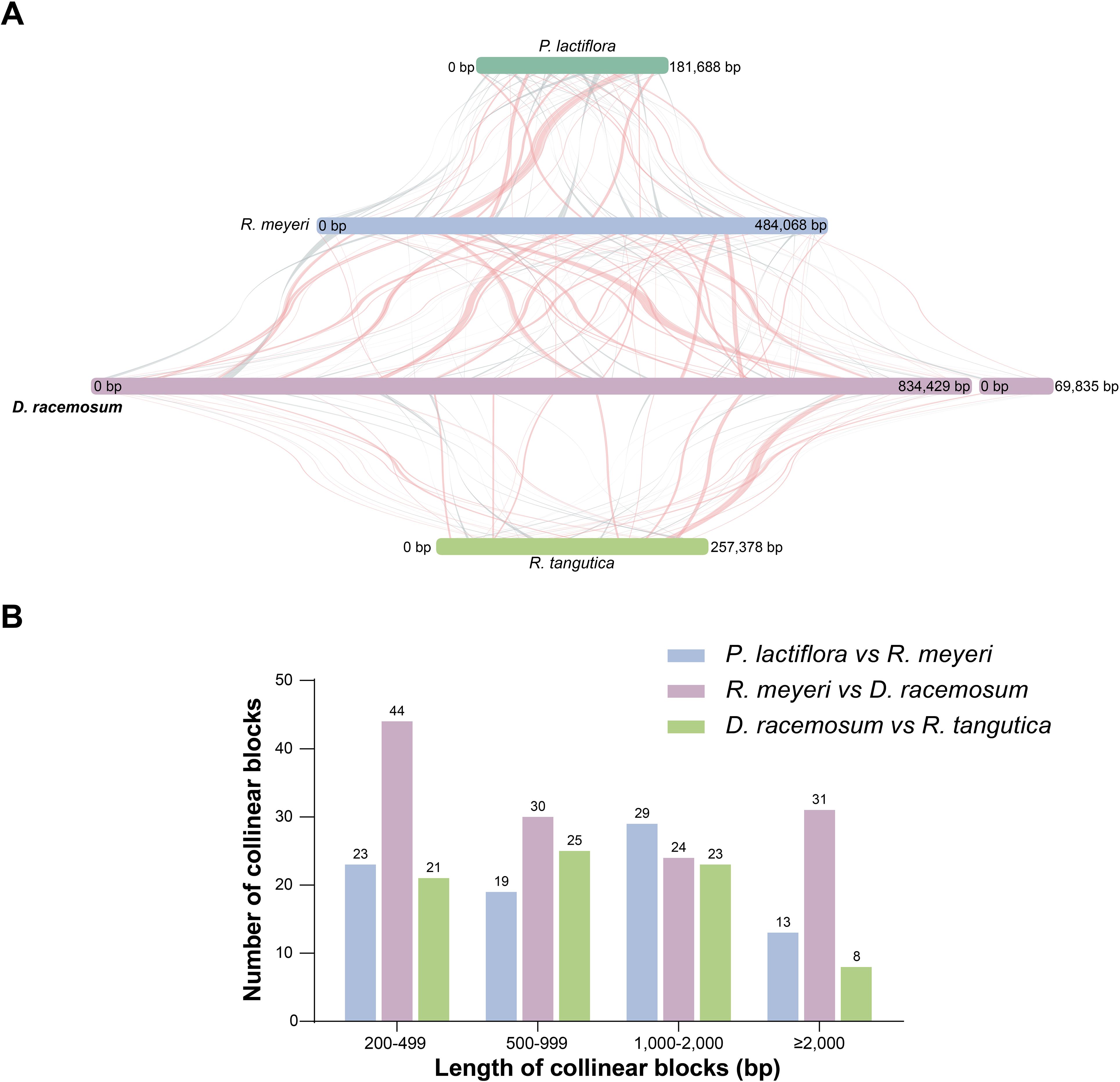
Figure 5. Whole mitogenome collinearity analysis of four Saxifragales species. (A) Collinearity analysis of the four Saxifragales mitogenomes. Strips of varying colors represented distinct mitogenomes, while linear blocks denoted collinear regions. (B) Distribution of collinear block lengths and counts. Color-coded legends indicated homologous fragments among the species. The names and NCBI accession numbers of species used in the collinearity analysis were provided in Supplementary Table S1.
3.5 Phylogenetic analysis
To determine the phylogenetic position of D. racemosum, a ML tree was constructed using 18 conserved mitochondrial PCGs from 17 plant species, with S. bicolor designated as the outgroup. The phylogenetic analysis revealed that D. racemosum clustered with four Saxifragales species (R. meyeri, R. tangutica, P. suffruticosa, and P. lactiflora), and occupied a basal position within the Saxifragales (Figure 6A). The topology received strong nodal support for the Saxifragales phylogeny (bootstrap value = 97). Notably, the five Saxifragales species were positioned at the base of the Superrosids, which is a sister group to the Rosids. To further validate our findings, we reconstructed the phylogenetic tree using plastome sequences. Phylogenetic reconstruction using plastid genomes (plastomes) yielded congruent results (Figure 6B), with topological congruence between mitogenome- and plastome-derived trees confirming the reliability of our phylogenetic inferences.

Figure 6. The ML trees of 18 plant species. (A) The ML tree based on 59 plastid PCGs. (B) The ML tree based on 18 mitochondrial PCGs. The mitogenome of Distylium racemosum was highlighted in bold and marked with an asterisk. Sorghum bicolor was chosen as the outgroup. Bootstrap values were indicated on each branch, and colors represented the respective groups for each species. The plant names and NCBI accession numbers utilized in the phylogenetic analysis were listed in Supplementary Table S1.
4 Discussion
4.1 Variety of structure and size in mitogenomes of Saxifragales species
Plant mitogenomes are organized into diverse structural conformations—including circular, linear, and complex branched molecules due to frequent recombination events (Bi et al., 2024b; Han et al., 2024; Wu et al., 2022). For example, Amborella trichopoda, Rhopalocnemis phalloides, and Panax notoginseng exhibit highly complex mitogenome architectures shaped by recombination (Rice et al., 2013; Yang et al., 2023b; Yu et al., 2022). Within Saxifragales, structural diversity of mitogenome is pronounced. T. polyphylla contains three circular chromosomes (Liu et al., 2024a), while species in Rhodiola genus display contrasting conformations—R. crenulate has a single circular chromosome, whereas R. wallichiana and R. sacra possess two circular chromosomes, reflecting atypical multi-chromosomal organization (Yu et al., 2023). Our study resolved the D. racemosum mitogenome as a bipartite structure comprising one large circular chromosome (834,429 bp) and a smaller linear fragment (69,835 bp). Recent research indicates that species with high GC content possess an enhanced ability to thrive in regions characterized by extremely cold winters or seasonal drought (Lu et al., 2024; Šmarda et al., 2014). The GC content in Saxifragales mitogenomes exhibits limited evolutionary variation, ranging from 44.50% (Sedum plumbizincicola) to 46.28% (D. racemosum) (Ding et al., 2022). The mitogenome size varies significantly across Saxifragales. In the Crassulaceae and Paeoniaceae families, the mitochondrial genome size ranges from approximately 180 bp to 260 kb, while Grossulariaceae and Saxifragaceae families span 400–500 kb, and D. racemosum (Hamamelidaceae) extends to 904,264 bp, which is the longest in Saxifragales to date, suggesting that Hamamelidaceae species may possess the larger mitogenomes among Saxifragales.
4.2 D. racemosum mitogenome exhibiting the highest repeats abundance in Saxifragales
Repeats are primary drivers of dynamic genomic restructuring in plant mitochondrial DNA, facilitating recombination-mediated changes in genome size, gene arrangement, and evolutionary trajectories (Wang et al., 2025; Wynn and Christensen, 2019). These structural modifications may ultimately contribute to phenotypic variation (Bi et al., 2022; Liu et al., 2024b; Tang et al., 2024; Wang Y. et al., 2024). For example, in Zea mays, repeats modulate chloroplast gene expression, directly affecting photosynthetic efficiency and growth (Bedbrook et al., 1977), while in O. sativa, repeat variations correlate with key agronomic traits like plant height and tillering capacity (McCarthy et al., 2002). In the D. racemosum mitogenome, we identified 304 SSRs, 1,508 dispersed repeats (748 direct; 761 palindromic), and 50 tandem repeats, representing the highest repeat density reported in Saxifragales mitogenomes (Liu et al., 2024a; Lu et al., 2024; Tang et al., 2024). This repeat abundance in D. racemosum provides a plausible explanation for its exceptionally large mitochondrial genome size in Saxifragales species (904,264 bp). These findings provide critical insights into Distylium evolution, underscoring repeats as drivers of genomic expansion and diversification.
4.3 Relatively abundant and stable RNA editing events in Saxifragales mitogenomes
RNA editing, a post-transcriptional modification critical for enhancing transcriptome diversity and producing functional mitochondrial proteins, is widespread across plant lineages (Edera et al., 2018; Gommans et al., 2009; Lukeš et al., 2021). Evolutionary trends reveal limited RNA editing in bryophytes (mosses and liverworts) but a marked increase in lycophytes (Rüdinger et al., 2009; Zhang et al., 2020). Among gymnosperms, the frequency of RNA editing demonstrates substantial variation, ranging from 99 editing sites in Welwitschia mirabilis to 1,405 in Ginkgo biloba (Fan et al., 2019; Guo et al., 2016). Angiosperms typically maintain moderate editing activity (400–500 sites). Notably, Cinnamomum chekiangense represents an exceptional case, exhibiting an unprecedented 1,119 RNA editing sites, the highest number recorded in angiosperms to date (Bi et al., 2016, 2024b). Within Saxifragales, studies conducted previously revealed substantial RNA editing activity: 569 sites in P. lactiflora, 653 in Tiarella polyphylla, and 731 in R. nigrum (Liu et al., 2024a; Lu et al., 2024; Tang et al., 2024). Our analysis identified 697 C-to-U editing sites across all PCGs except sdh3, underscoring both the prevalence and evolutionary conservation of RNA editing in this order. However, computational predictions necessitate experimental validation through PCR amplification and Sanger sequencing to confirm site-specific accuracy.
4.4 Low gene transfer level from plastome to mitogenome of D. racemosum
The transfer of plastid DNA sequences to mitogenomes, known as MTPTs, represents a recurrent evolutionary phenomenon in plant mitogenome evolution (Timmis et al., 2004). As a form of horizontal gene transfer (HGT), MTPTs exhibit considerable variation in length and sequence similarity across species (Tang et al., 2024). In this study, we identified 49 MTPTs in the D. racemosum mitogenome, with a cumulative length representing 2.57% of the total mitogenome. This proportion surpasses values reported for P. lactiflora (2.2%), R. nigrum (1.11%), Arabidopsis thaliana (0.8%), Silene conica (0.2%), and Vigna angularis (0.1%) (Lu et al., 2024; Sloan and Wu, 2014; Tang et al., 2024), yet remains lower than those of Michelia figo (5.53%) and Boea hygrometrica (10.5%) (Wang et al., 2025; Zhang et al., 2012). Gene retention within MTPTs also varies significantly. Only three plastid-derived PCGs were retained in D. racemosum—fewer than in P. lactiflora (10 PCGs), R. nigrum (12 PCGs), and R. wallichiana (7 PCGs), but more than in R. crenulata (2 PCGs) (Lu et al., 2024; Tang et al., 2024; Yu et al., 2023). The functional relevance of MTPTs appears minimal due to sequence degradation and lack of RNA editing (Clifton et al., 2004; Notsu et al., 2002), suggesting these genes likely have limited functional roles and represent nonessential genomic remnants (Wang et al., 2007).
4.5 Phylogenetic analysis of D. racemosum and its relatives
Plant mitogenomes demonstrate a remarkable propensity for the incorporation of exogenous or migratory DNA sequences, driving recurrent gains and losses of PCGs (Adams and Palmer, 2003; Garcia et al., 2021; Lu et al., 2024). In this study, we reconstructed two phylogenetic trees using 18 conserved mitochondrial and 59 plastid PCGs from 18 plant species. Both topologies strongly supported the basal divergence of D. racemosum within Saxifragales. Phylogenetic analyses further resolved Saxifragales as a basal lineage of Superrosids, emerging as a sister group to the Rosids, with tree architectures consistent with the Angiosperm Phylogeny Group IV (APG IV) system (Bremer et al., 2016). Nonetheless, limited mitogenome data from Hamamelidaceae species restricts deeper resolution of phylogenetic relationships within this clade.
5 Conclusion
In this study, we de novo assembled the first mitogenome of D. racemosum, comprising a 904,264 bp bipartite structure with a dominant circular chromosome (834,429 bp) and a smaller linear fragment (69,835 bp). The mitogenome encodes 67 genes, including 43PCGs, 3 rRNAs, and 21 tRNAs. Repetitive elements dominate the mitogenome, with 304 SSRs, 1,508 dispersed repeats, and 50 tandem repeats. Our analysis revealed 49 fragments (23,278 bp) that had been transferred from the plastome to the mitogenome of D. racemosum, representing 14.63% of the plastome and 2.57% of the mitogenome. We predicted 697 RNA editing sites across PCGs, predominantly at second codon positions (62.3%). Collinearity analysis revealed pronounced structural divergence between D. racemosum and other Saxifragales mitogenomes. ML trees reconstructed from mitochondrial and plastid genomes congruently resolve D. racemosum as a basal lineage within Saxifragales. As the first annotated mitogenome in Hamamelidaceae, this resource will advance comparative studies of mitochondrial evolution in Hamamelidaceae family and provide a genomic foundation for taxonomic refinement and applied research.
Data availability statement
The datasets presented in this study can be found in online repositories. These data can be found in Genbank repository, accession numbers for the mitogenome of D. racemosum are PQ594873 and PQ594874, respectively.
Author contributions
YW: Formal Analysis, Visualization, Writing – original draft, Writing – review & editing. ZZ: Formal Analysis, Writing – original draft, Writing – review & editing. XC: Formal Analysis, Writing – review & editing. HL: Visualization, Writing – review & editing. CM: Writing – review & editing. PG: Conceptualization, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The work is supported by the Northwest Minzu University Talent Introduction Project (Z2101707), the Science and Technology Department of Gansu Province (23CXNA0045), the 2023 Central University Project (31920230027), and the Lanzhou Science and Technology Bureau (2023-RC-47).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fpls.2025.1632449.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1586341/full#supplementary-material
Supplementary Table 1 | Names and GenBank accession numbers of species used for phylogenetic analysis.
Supplementary Table 2 | Statistical Information of the Distylium racemosum mitogenome.
Supplementary Table 3 | Start and stop codons of the Distylium racemosum mitogenome.
Supplementary Table 4 | Distribution of simple sequence repeats (SSRs) in the Distylium racemosum mitogenome.
Supplementary Table 5 | Distribution of dispersed repeats in the Distylium racemosum mitogenome.
Supplementary Table 6 | Tandem repeats identified in the Distylium racemosum mitogenome.
Supplementary Table 7 | Fragments transferred from plastome to mitogenome in Distylium racemosum.
Supplementary Table 8 | RNA editing events predicted in the Distylium racemosum mitogenome.
Supplementary Table 9 | Synteny results in mitogenomes of four Saxifragales species.
Supplementary Figure 1 | The cis-splicing and trans-splicing introns in the Distylium racemosum mitogenome.
Supplementary Figure 2 | Distribution of simple sequence repeats in the two mitochondrial chromosomes of Distylium racemosum.
References
Adams, K. L. and Palmer, J. D. (2003). Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395. doi: 10.1016/s1055-7903(03)00194-5
Arseneau, J. R., Steeves, R., and Laflamme, M. (2017). Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol. Ecol. Resour. 17, 686–693. doi: 10.1111/1755-0998.12616
Bedbrook, J. R., Kolodner, R., and Bogorad, L. (1977). Zea mays chloroplast ribosomal RNA genes are part of a 22,000 base pair inverted repeat. Cell 11, 739–749. doi: 10.1016/0092-8674(77)90288-4
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, C., Paterson, A. H., Wang, X., Xu, Y., Wu, D., Qu, Y., et al. (2016). Analysis of the complete mitochondrial genome sequence of the diploid cotton gossypium raimondii by comparative genomics approaches. BioMed. Res. Int. 2016, 5040598. doi: 10.1155/2016/5040598
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., and Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of populus simonii. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.914635
Bi, C., Shen, F., Han, F., Qu, Y., Hou, J., Xu, K., et al. (2024a). PMAT: an efficient plant mitogenome assembly toolkit using low-coverage HiFi sequencing data. Hortic. Res. 11, uhae023. doi: 10.1093/hr/uhae023
Bi, C., Sun, N., Han, F., Xu, K., Yang, Y., and Ferguson, D. K. (2024b). The first mitogenome of Lauraceae (Cinnamomum chekiangense). Plant Divers. 46, 144–148. doi: 10.1016/j.pld.2023.11.001
Bremer, K., Chase, M. W., Reveal, J. L., Soltis, D. E., Soltis, P. S., and Stevens, P. F. (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.2016.181.issue-1
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chan, P. P., Lin, B. Y., Mak, A. J., and Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Chen, Y., Ye, W., Zhang, Y., and Xu, Y. (2015). High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 43, 7762–7768. doi: 10.1093/nar/gkv784
Clifton, S. W., Minx, P., Fauron, C. M., Gibson, M., Allen, J. O., Sun, H., et al. (2004). Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiol. 136, 3486–3503. doi: 10.1104/pp.104.044602
Ding, H., Bi, D., Zhang, S., Han, S., Ye, Y., Yi, R., et al. (2022). The Mitogenome of Sedum plumbizincicola (Crassulaceae): Insights into RNA Editing, Lateral Gene Transfer, and Phylogenetic Implications. Biol. (Basel). 11, 1661. doi: 10.3390/biology11111661
Dong, W., Liu, Y., Xu, C., Gao, Y., Yuan, Q., Suo, Z., et al. (2021). Chloroplast phylogenomic insights into the evolution of Distylium (Hamamelidaceae). BMC Genomics 22, 293. doi: 10.1186/s12864-021-07590-6
Dyall, S. D., Brown, M. T., and Johnson, P. J. (2004). Ancient invasions: from endosymbionts to organelles. Science 304, 253–257. doi: 10.1126/science.1094884
Edera, A. A., Gandini, C. L., and Sanchez-Puerta, M. V. (2018). Towards a comprehensive picture of C-to-U RNA editing sites in angiosperm mitochondria. Plant Mol. Biol. 97, 215–231. doi: 10.1007/s11103-018-0734-9
Edera, A. A., Small, I., Milone, D. H., and Sanchez-Puerta, M. V. (2021). Deepred-Mt: Deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 136, 104682. doi: 10.1016/j.compbiomed.2021.104682
Fan, W., Guo, W., Funk, L., Mower, J. P., and Zhu, A. (2019). Complete loss of RNA editing from the plastid genome and most highly expressed mitochondrial genes of Welwitschia mirabilis. Sci. China Life Sci. 62, 498–506. doi: 10.1007/s11427-018-9450-1
Garcia, L. E., Edera, A. A., Palmer, J. D., Sato, H., and Sanchez-Puerta, M. V. (2021). Horizontal gene transfers dominate the functional mitochondrial gene space of a holoparasitic plant. New Phytol. 229, 1701–1714. doi: 10.1111/nph.16926
Gommans, W. M., Mullen, S. P., and Maas, S. (2009). RNA editing: a driving force for adaptive evolution? Bioessays 31, 1137–1145. doi: 10.1002/bies.200900045
Gray, M. W., Burger, G., and Lang, B. F. (1999). Mitochondrial evolution. Science 283, 1476–1481. doi: 10.1126/science.283.5407.1476
Guo, W., Grewe, F., Fan, W., Young, G. J., Knoop, V., Palmer, J. D., et al. (2016). Ginkgo and welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol. Biol. Evol. 33, 1448–1460. doi: 10.1093/molbev/msw024
Han, F., Bi, C., Zhao, Y., Gao, M., Wang, Y., and Chen, Y.-C. (2024). Unraveling the complex evolutionary features of the Cinnamomum camphora mitochondrial genome. Plant Cell Rep. 43, 183. doi: 10.1007/s00299-024-03256-1
Han, F., Qu, Y., Chen, Y., Xu, L., and Bi, C. (2022). Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1031769
He, W., Yang, J., Jing, Y., Xu, L., Yu, K., and Fang, X. (2023). NGenomeSyn: an easy-to-use and flexible tool for publication-ready visualization of syntenic relationships across multiple genomes. Bioinformatics 39, btad121. doi: 10.1093/bioinformatics/btad121
Huang, Y., Xing, Y., Men, W., Xue, H., Hou, W., Huang, Y., et al. (2025). The first complete mitochondrial genome assembly and comparative analysis of the fern Blechnaceae family: Blechnopsis orientalis. Front. Plant Sci. 16. doi: 10.3389/fpls.2025.1534171
Huang, K., Xu, W., Hu, H., Jiang, X., Sun, L., Zhao, W., et al. (2024). The mitochondrial genome of cathaya argyrophylla reaches 18.99 mb: analysis of super-Large mitochondrial genomes in pinaceae. arXiv. e-prints. doi: 10.48550/arXiv.2410.07006.2410.07006
Ismayati, M., Solihat, N. N., Setiawati, F. M., Syafii, W., Tobimatsu, Y., and Zulfiana, D. (2024). Tannins from acacia mangium bark as natural dyes for textiles: characteristics and applications. J. Renewable Mater. 12, 1625–1637. doi: 10.32604/jrm.2024.054739
Katoh, K. and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Letunic, I. and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–w296. doi: 10.1093/nar/gkab301
Li, J., Ni, Y., Lu, Q., Chen, H., and Liu, C. (2024). PMGA: A plant mitochondrial genome annotator. Plant Commun. 6 (3), 101191. doi: 10.1016/j.xplc.2024.101191
Liu, C., Chen, H. H., Tang, L. Z., Khine, P. K., Han, L. H., Song, Y., et al. (2022). Plastid genome evolution of a monophyletic group in the subtribe Lauriineae (Laureae, Lauraceae). Plant Divers. 44, 377–388. doi: 10.1016/j.pld.2021.11.009
Liu, Z., Cheng, R., Xiao, W., Guo, Q., and Wang, N. (2014). Effect of off-season flooding on growth, photosynthesis, carbohydrate partitioning, and nutrient uptake in Distylium chinense. PloS One 9, e107636. doi: 10.1371/journal.pone.0107636
Liu, L., Long, Q., Lv, W., Qian, J., Egan, A. N., Shi, Y., et al. (2024b). Long repeat sequences mediated multiple mitogenome conformations of mulberries (Morus spp.), an important economic plant in China. Genomics Commun. 1, e005. doi: 10.48130/gcomm-0024-0005
Liu, B., Long, Q., Lv, W., Shi, Y., Li, P., and Liu, L. (2024a). Characterization of the complete mitogenome of Tiarella polyphylla, commonly known as Asian foamflower: insights into the multi-chromosomes structure and DNA transfers. BMC Genomics 25, 883. doi: 10.1186/s12864-024-10790-5
Lu, G., Wang, W., Zhang, S., Yang, G., Zhang, K., Que, Y., et al. (2024). The first complete mitochondrial genome of Grossulariaceae: Molecular features, structure recombination, and genetic evolution. BMC Genomics 25, 744. doi: 10.1186/s12864-024-10654-y
Lukeš, J., Kaur, B., and Speijer, D. (2021). RNA editing in mitochondria and plastids: weird and widespread. Trends Genet. 37, 99–102. doi: 10.1016/j.tig.2020.10.004
McCarthy, E. M., Liu, J., Lizhi, G., and McDonald, J. F. (2002). Long terminal repeat retrotransposons of Oryza sativa. Genome Biol. 3, Research0053. doi: 10.1186/gb-2002-3-10-research0053
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Notsu, Y., Masood, S., Nishikawa, T., Kubo, N., Akiduki, G., Nakazono, M., et al. (2002). The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genomics 268, 434–445. doi: 10.1007/s00438-002-0767-1
Rice, D. W., Alverson, A. J., Richardson, A. O., Young, G. J., Sanchez-Puerta, M. V., Munzinger, J., et al. (2013). Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 342, 1468–1473. doi: 10.1126/science.1246275
Rüdinger, M., Funk, H. T., Rensing, S. A., Maier, U. G., and Knoop, V. (2009). RNA editing: only eleven sites are present in the Physcomitrella patens mitochondrial transcriptome and a universal nomenclature proposal. Mol. Genet. Genomics 281, 473–481. doi: 10.1007/s00438-009-0424-z
Shi, Q., Tian, D., Wang, J., Chen, A., Miao, Y., Chen, Y., et al. (2023). Overexpression of miR390b promotes stem elongation and height growth in Populus. Hortic. Res. 10, uhac258. doi: 10.1093/hr/uhac258
Skippington, E., Barkman, T. J., Rice, D. W., and Palmer, J. D. (2015). Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. U. S. A. 112, E3515–E3524. doi: 10.1073/pnas.1504491112
Sloan, D. B. and Wu, Z. (2014). History of plastid DNA insertions reveals weak deletion and at mutation biases in angiosperm mitochondrial genomes. Genome Biol. Evol. 6, 3210–3221. doi: 10.1093/gbe/evu253
Šmarda, P., Bureš, P., Horová, L., Leitch, I. J., Mucina, L., Pacini, E., et al. (2014). Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc. Natl. Acad. Sci. U. S. A. 111, E4096–E4102. doi: 10.1073/pnas.1321152111
Sun, N., Han, F., Wang, S., Shen, F., Liu, W., Fan, W., et al. (2024). Comprehensive analysis of the Lycopodium japonicum mitogenome reveals abundant tRNA genes and cis-spliced introns in Lycopodiaceae species. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1446015
Tang, P., Ni, Y., Li, J., Lu, Q., Liu, C., and Guo, J. (2024). The complete mitochondrial genome of paeonia lactiflora pall. (Saxifragales: paeoniaceae): evidence of gene transfer from chloroplast to mitochondrial genome. Genes (Basel). 15, 239. doi: 10.3390/genes15020239
Timmis, J. N., Ayliffe, M. A., Huang, C. Y., and Martin, W. (2004). Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 5, 123–135. doi: 10.1038/nrg1271
van Loo, G., Saelens, X., van Gurp, M., MacFarlane, M., Martin, S. J., and Vandenabeele, P. (2002). The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 9, 1031–1042. doi: 10.1038/sj.cdd.4401088
Wang, Y., Cui, G., He, K., Xu, K., Wang, Y., Wang, Z., et al. (2024). Assembly and comparative analysis of the complete mitochondrial genome of ilex rotunda thunb. Forests 15, 1117. doi: 10.3390/f15071117
Wang, J., Kan, S., Liao, X., Zhou, J., Tembrock, L. R., Daniell, H., et al. (2024). Plant organellar genomes: much done, much more to do. Trends Plant Sci. 29, 754–769. doi: 10.1016/j.tplants.2023.12.014
Wang, S., Qiu, J., Sun, N., Han, F., Wang, Z., Yang, Y., et al. (2025). Characterization and comparative analysis of the first mitochondrial genome of Michelia (Magnoliaceae). Genomics Commun. 2, e001. doi: 10.48130/gcomm-0025-0001
Wang, D., Wu, Y. W., Shih, A. C., Wu, C. S., Wang, Y. N., and Chaw, S. M. (2007). Transfer of chloroplast genomic DNA to mitochondrial genome occurred at least 300 MYA. Mol. Biol. Evol. 24, 2040–2048. doi: 10.1093/molbev/msm133
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Z.-Q., Liao, X.-Z., Zhang, X.-N., Tembrock, L. R., and Broz, A. (2022). Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Syst. Evol. 60, 160–168. doi: 10.1111/jse.12655
Wynn, E. L. and Christensen, A. C. (2019). Repeats of unusual size in plant mitochondrial genomes: identification, incidence and evolution. G3 (Bethesda). 9, 549–559. doi: 10.1534/g3.118.200948
Xiang, L., Li, X.-L., Wang, X.-S., Yang, J., Lv, K., Xiong, Z.-Q., et al. (2020). Genetic diversity and population structure of Distylium chinense revealed by ISSR and SRAP analysis in the Three Gorges Reservoir Region of the Yangtze River, China. Global Ecol. Conserv. 21, e00805. doi: 10.1016/j.gecco.2019.e00805
Xiao, S., Zang, J., Pei, Y., Liu, J., Liu, J., Song, W., et al. (2020). Activation of mitochondrial orf355 gene expression by a nuclear-encoded DREB transcription factor causes cytoplasmic male sterility in maize. Mol. Plant 13, 1270–1283. doi: 10.1016/j.molp.2020.07.002
Yagi, H., Xu, J., Moriguchi, N., Miyagi, R., Moritsuka, E., Sato, E., et al. (2019). Population genetic analysis of two species of Distylium: D. racemosum growing in East Asian evergreen broad-leaved forests and D. lepidotum endemic to the Ogasawara (Bonin) Islands. Tree Genet. Genomes. 15, 77. doi: 10.1007/s11295-019-1386-x
Yang, H., Chen, H., Ni, Y., Li, J., Cai, Y., Wang, J., et al. (2023a). Mitochondrial Genome Sequence of Salvia officinalis (Lamiales: Lamiaceae) Suggests Diverse Genome Structures in Cogeneric Species and Finds the Stop Gain of Genes through RNA Editing Events. Int. J. Mol. Sci. 24, 5372. doi: 10.3390/ijms24065372
Yang, H., Ni, Y., Zhang, X., Li, J., Chen, H., and Liu, C. (2023b). The mitochondrial genomes of Panax notoginseng reveal recombination mediated by repeats associated with DNA replication. Int. J. Biol. Macromol. 252, 126359. doi: 10.1016/j.ijbiomac.2023.126359
Yu, R., Sun, C., Zhong, Y., Liu, Y., Sanchez-Puerta, M. V., Mower, J. P., et al. (2022). The minicircular and extremely heteroplasmic mitogenome of the holoparasitic plant Rhopalocnemis phalloides. Curr. Biol. 32, 470–479.e5. doi: 10.1016/j.cub.2021.11.053
Yu, X., Wei, P., Chen, Z., Li, X., Zhang, W., Yang, Y., et al. (2023). Comparative analysis of the organelle genomes of three Rhodiola species provide insights into their structural dynamics and sequence divergences. BMC Plant Biol. 23, 156. doi: 10.1186/s12870-023-04159-1
Zhang, X., Chen, H., Ni, Y., Wu, B., Li, J., Burzyński, A., et al. (2024). Plant mitochondrial genome map (PMGmap): A software tool for the comprehensive visualization of coding, noncoding and genome features of plant mitochondrial genomes. Mol. Ecol. Resour. 24, e13952. doi: 10.1111/1755-0998.13952
Zhang, T., Fang, Y., Wang, X., Deng, X., Zhang, X., Hu, S., et al. (2012). The complete chloroplast and mitochondrial genome sequences of Boea hygrometrica: insights into the evolution of plant organellar genomes. PloS One 7, e30531. doi: 10.1371/journal.pone.0030531
Zhang, J., Fu, X. X., Li, R. Q., Zhao, X., Liu, Y., Li, M. H., et al. (2020). The hornwort genome and early land plant evolution. Nat. Plants. 6, 107–118. doi: 10.1038/s41477-019-0588-4
Keywords: Distylium racemosum, mitochondrial genome, phylogenetic analysis, RNA editing events, comparative analysis
Citation: Wang Y, Zhang Z, Chen X, Li H, Ma C and Guo P (2025) De novo assembly and comparative analysis of the first complete mitogenome in Distylium (Distylium racemosum). Front. Plant Sci. 16:1586341. doi: 10.3389/fpls.2025.1586341
Received: 02 March 2025; Accepted: 11 April 2025;
Published: 15 May 2025; Corrected: 06 June 2025.
Edited by:
Changwei Bi, Nanjing Forestry University, ChinaReviewed by:
Qiuyue Ma, Jiangsu Academy of Agricultural Sciences (JAAS), ChinaYang Xuchen, Guangzhou University, China
Copyright © 2025 Wang, Zhang, Chen, Li, Ma and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Penghui Guo, eGJtdXNreUAxNjMuY29t