 Shinan Liu
Shinan Liu Yinuo Zhang1
Yinuo Zhang1- 1Guangxi Colleges and Universities Key Laboratory for Cultivation and Utilization of Subtropical Forest Plantation, College of Forestry, Guangxi University, Nanning, China
- 2Guangxi Key Laboratory of Superior Timber Trees Resource Cultivation, Guangxi Forestry Research Institute, Nanning, China
- 3Guangxi Forestry Inventory & Planning Institute, Protected Areas and Wetland Planning & Research Center, Nanning, China
Indocalamus longiauritus, as a dwarf bamboo holds the ecological and economic significance. Although earlier studies have successfully elucidated its chloroplast (cp) genome, the complete mitochondrial (mt) genome still is uncovered. This study undertook the sequencing, assembly, and comprehensive analysis of the complete mt genome of I. longiauritus. Based on the findings, the mt genome contained one circular and two linear contigs with the total length of 491,541bp. Totally, 59 genes were identified, which included 37 protein-coding genes (PCGs), 3 rRNA genes and 19 tRNA genes. In addition, 119 SSRs and 234 dispersed repeats were discovered. We discovered 602 RNA editing sites, with a striking 78.9% of them involving the conversion of hydrophilic amino acid to hydrophobic ones. Furthermore, in the I. longiauritus mt genome, 12 genes included 8 PCGs (petB, psbH, psbN, atpE, ndhJ, rps4, psaB, and ndhI) and 4 tRNA genes (trnM-CAU, trnV-UAC, trnF-GAA, and trnS-GGA) were found to transfer from the cp genome. Phylogenetic analysis showed a close genetic relationship of I. longiauritus with the species Fargesia qinlingensis and I. tessellatus. Collinearity analysis suggested that significant rearrangements existed in the mt genome of I. longiauritus. Selection pressure analysis revealed that more than half of PCGs had Ka/Ks values less than 1. Obviously, certain genes including rpl2, rpl5, rpl10, rpl14, rps2, rps11, rps12, rps14, rps19, and sdh4 were absent in the mt genomes of I. longiauritus and nine relative Poeceae species. Interestingly, the rpl14 gene was uniquely present in the mt genome of I. longiauritus. This study provides a significant genetic resource for the Bambusoideae family, which will facilitate further investigations in the molecular diversity and genetic evolution of bamboos.
1 Introduction
Mitochondria are crucial for the synthesis and conversion of energy, which support a variety of cellular processes, making them vital for the growth of plants (Li et al., 2024; Jacoby et al., 2012). These organelles, through the process of phosphorylation, convert biomass energy into chemical energy and participate in essential cellular functions including division, differentiation, and apoptosis (Braun, 2020; Ghifari et al., 2023). Despite being separate from the nucleus, mitochondria possess their own genomes, which are maternally passed down in a haploid (Cheng et al., 2021). These organelles contain their own distinct genomes, which are vital for various biological functions and research purposes (Liu et al., 2025). In accordance with the endosymbiotic theory, it is considered that mitochondria originate from a symbiotic relationship between alpha-proteobacteria and ancestral archaea host cells, evolving into essential organelles within eukaryotic cells (Roger et al., 2017). Furthermore, mitochondrial (mt) genomes and plastidial genomes exhibit maternal inheritance but with significant differences (Zervas et al., 2019). The size of these mt genomes varies considerably among plant species, ranging from 0.25Mb to 11.7Mb (Wu et al., 2022; Sloan et al., 2012; Putintseva et al., 2020). While it has been commonly observed that the mt genomes in plants typically appear as circular structures, more recent studies have revealed their presence in various forms, including multiple branched, linear, circular, branching, or reticular structures (Wu et al., 2015; Hong et al., 2021; Sloan, 2013). The mutation rates in the mt genomes of plants are generally higher than those found in nuclear genomes, primarily due to inadequate the DNA repair mechanisms, causing both duplications and rearrangements (Cole et al., 2018; Kong et al., 2025). In addition to being directly inherited from ancestral mitochondria, plant mitochondrial tRNAs also originate from the migration of sequences from their own chloroplast (cp) genomes (Xiong et al., 2008; Szandar et al., 2022). Therefore, the mt genomes of higher plants harbors a wealth of genetic variation, making them the excellent molecular marker for investigating species origin, evolution, and population genetic diversity (Yang et al., 2022; Liu et al., 2023a).
The Poaceae family, which encompasses around 11,800 species across 12 subfamilies hold significant economic and ecological value (Soreng et al., 2022; Arthan et al., 2025). To date, complete mt genome of over 20 Poaceae species have been sequenced, with genome sizes ranging from 321kb (Zizania latifolia) to 704kb (Tripsacum dactyloides) (Liu et al., 2023b; Yang et al., 2024; Luo et al., 2024). Moreover, diverse structural forms have been identified within Poaceae, as exemplified by single circular DNA molecules in Oryza minuta and Avena longiglumis (Asaf et al., 2016; Liu et al., 2023b), and dual circular DNA molecules in Z. latifolia and Setaria italica (Luo et al., 2024; Zhang et al., 2024a). In contrast, Fargesia qinlingensis, a Bambusoideae subfamily species, has a linear mt structure (Wu et al., 2024a). Bamboos, despite being classified as grasses, exhibit tree-like traits, which may be contributed to the different structure in mt genome. However, limited high-quality bamboo mt genomes in public databases hinder the exploration (Wang et al., 2024a). Besides, molecular phylogenetic analyses based on cp genomes and nuclear genes have been widely used to resolve the evolutionary questions in Bambusoideae (Wang et al., 2024a). Studies showed that Indocalamus was not monophyletic, which is clustered with Gelidocalamus, Chimonobambusa, Bashania, and Pseudosasa (Zeng et al., 2010; Wang et al., 2024a). This underscores the controversy in Indocalamus phylogenetic relationships. When compared with the cp genome, the plants’ mt genome is variable in structures (Richardson et al., 2013). Therefore, the analysis of mt genome sequence is crucial for comprehending the evolution of various plant species (Wang et al., 2024b). In this study, we assembled and annotated the mt genomes of I. longiauritus from Indocalamus genus (Bambusoideae), and further analyze the key genomic features such as codon usage, repetitive sequences, and mt plastid DNAs (MTPTs), RNA editing sites, and the phylogenetic position. Moreover, this will provide a valuable reference for understanding the structural characteristics and evolutionary diversity of the mt genome in the Bambusoideae family.
2 Materials and methods
2.1 Plant materials sampling, DNA extraction, and sequencing
The young and healthy leaves of I. longiauritus were harvested from the bamboo garden at the Guangxi Forestry Research Institute, located in Yongwu district, Nanning, Guangxi Zhuang Autonomous Region (coordinates: 108°20′51"E, 22°55′38"N). Immediately after collection, the samples were flash-frozen in liquid nitrogen and then stored at -80°C for further processing. To extract genomic DNA, the Plant DNAzol Reagent (Invitrogen) was utilized according to the manufacturer’s guidelines. The agarose gel electrophoresis and a NanoDrop spectrophotometer (Thermo Fisher Scientific) were used to evaluate the quality and concentration of the extracted DNA. A 15-kb library was constructed using a SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, CA, USA). The construction included DNA shearing, AMPure PB bead purification, ssDNA overhang removal, damage repair, end repair, hairpin adapter ligation, and library bead purification. Following quality control, a SMRTbell library was obtained. The library was sequenced on the PacBio Revio platform (Pacific Biosciences, CA, USA) by Shenzhen Huitong Biotechnology Co., Ltd. The CCS algorithm (version 6.0.0) was employed to process the raw data. To generate highly accurate HiFi reads, the parameters were set as follows: -minPasses 3, -minPredictedAccuracy 0.99, and -maxLength 21,000.
2.2 Mitochondrial genome assembly and annotation
The PMAT software (v 1.5.3, -g 3.8G) was used to assemble the PacBio HIFI data that the sequencing data volume and an average fragment length were greater than 7G and 7kb (Bi et al., 2024). Subsequently, the assembly results were visualized and manually refined with Bandage v 0.8.1 software to obtain preliminary results (Wick et al., 2015). Thereafter, the HIFI data were aligned with the assembled sequences using the minimap2 software (Li, 2018). The results were polished through NextPolish v 1.3.1 (https://github.com/Nextomics/NextPolish) and manually adjusted to obtain the final assembly sequences along with the corresponding graph files. The complete mt genome of I. longiauritus was later annotated using MITOFY (Alverson et al., 2010) and MFANNOT (Beck and Lang, 2010), and its map was generated with OGDRAW (Greiner et al., 2019).
2.3 Relative synonymous codon usage analysis
The PCGs from the mt genome were isolated using Perl scripting. To analyze these sequences, CUSP, an online analytical tool, was used in conjunction with Codon W v1.4.4 software (Sharp and Li, 1986) to calculate the essential nucleotide and codon features, including GC content, relative synonymous codon usage (RSCU), and effective number of codons (ENC). To visualize the data, a scatter plot was constructed using the R package ‘ggplot2’, with ENC values being plotted on the vertical axis and GC3 values on the horizontal axis. The expected ENC curve was derived by the formula: ENC = 2 + GC3 + 29/(GC3² + (1 - GC3)²), as established by Romero et al. (2000). In this plot, data points positioned above or close to the standard curve suggested that codon usage bias is primarily shaped by mutational pressures, whereas points lying below indicated that natural selection exerted a dominant role in shaping codon preferences (Sueoka, 1988). Additionally, a GC plot was generated using the ‘ggplot2’ package, with the average GC content of positions 1 and 2 (referred to as GC12) displayed on the vertical axis and GC3 value on the horizontal axis.
2.4 Analysis of repeat sequences
SSRs were identified with the MISA v1.0 tool (Thiel et al., 2003). The motif length of one- to six-nucleotides SSR was set as 10, 5, 4, 3, 3 and 3, respectively. Tandem repeat sequences were detected through TRF v4.09, employing parameters of 2, 7, 7, 80, 10, 50, 2000, -f, -d -m (Benson, 1999). Intersperse repeat sequences were identified with REPuter (Kurtz et al., 2001), with the minimum size and identity thresholds being set at 30bp and 90%, respectively. The results were visualized based on the Circos v0.69-6 (Zhang et al., 2013).
2.5 Gene transfer analysis and identification of RNA editing sites
The mt genome of I. longiauritus was aligned to its cp genome by blastn v2.9.0, with parameters being set as the -evalue 1E-5 and -word size 7 (Altschul et al., 1990). Next, the results were visualized with the Circos v0.69-6 (Zhang et al., 2013). RNA editing sites in the PCGs were analyzed using the PmtREP program with a cutoff value of 0.2 (Mower, 2005).
2.6 Phylogenetic and collinearity analysis
To determine the phylogenetic position of I. longiauritus, mt and cp genome sequences were utilized from 17 related species available in NCBI, with Aconitum kusnezoffii and Anemone maxima being outgroups (Supplementary Table S1). Six single-copy orthologous genes were identified and extracted from mt genomes (ccmFc, ccmFn, matR, mttB, rpl16, and rps1) and 48 genes from cp genomes (e.g., atpA, atpB, atpE, atpF, atpH, atpI, cemA, clpP, and others) using Perl scripts. These sequences were aligned with MAFFT v7.429 (Katoh and Standley, 2013). The resulting alignments were processed in Gblocks 0.91b, applying default parameters with the exception of gap handling. Then, maximum likelihood phylogeny was constructed via IQ-Tree v1.6.12 (Nguyen et al., 2015), employing the GTR+F+R2 model for mt genomes and TVM+F+R2 for cp genomes with a bootstrap of 1000. The comparison of mt genomes across 10 related species, including I. longiauritus and Fargesia qinlingensis, I. tessellatus, Bambusa oldhamii, Paspalum vaginatum, Setaria italica, Eleusine indica, Oryza sativa Indica Group, Lolium perenne, and Triticum aestivum cultivar Chinese Yumai, was performed using the Blastn v2.9.0 with the parameters of -evalue 1E-5 and -word size 7 (Altschul et al., 1990). In addition, the concordance factors including gene concordance factors (gCF) and site concordance factors (sCF) were calculated by adopting IQ-TREE v.2.4.0 (Minh et al., 2020). Syntenic relationships were visualized with TBtools v2.119 (Chen et al., 2020).
2.7 Ka/Ks and PI analysis
To evaluate the substitution rates of nonsynonymous (Ka) and synonymous (Ks), pairs of homologous genes between I. longiauritus and F. qinlingensis, I. tessellatus, B. oldhamii, P. vaginatum, S. italica, E. indica, O. sativa Indica Group, L. perenne, as well as T. aestivum cultivar Chinese Yumai were analyzed. Using ParaAR2.0 with its default settings (Zhang et al., 2012), the homologous sequences were aligned across above species. Subsequently, the Ka and Ks values for each gene were identified through the KaKs_Calculator v2.0 tool with the YN method (Zhang et al., 2006). To conduct a comprehensive comparison of nucleotide variability across the cp genome, MAFFT v7.429 (Katoh and Standley, 2013) was employed to analyze the homologous gene sequences. Finally, DnaSP v 6.0 software was used for calculating the nucleotide diversity (Pi) across the cp genome through a sliding window approach with a step size of 100 and a window length of 200 base pairs (Rozas et al., 2017).
3 Results
3.1 Features of the I. longiauritus mt genome
The complete mt genome of I. longiauritus exhibited a multi-branched conformation (Figure 1A) that was 491,541 bp with a GC content of 42.4%. After de-catenation, it was composed of three contiguous sequences (Figure 1B). Chromosome 1, chromosome 2, and chromosome 3 had length 290,324 bp, 195,199 bp, and 6,018 bp with GC contents of 44.03%, 44.29%, and 38.33%, separately. Their average depths were 87.11X, 61.27X, and 40.36X for long read, respectively. For subsequent analysis, we restructured Chromosome 1 into a circular molecule following the sequence of contigs 1-2-3-4-5-6-7-8-9-10-9-11-12-3-13-14. Chromosome 2 was also arranged into a linear molecule following the sequence of contigs 15-16-17-18. Chromosome 3 consisted solely of linear fragment, namely contig 19 (Figure 1B).
Figure 1. The structural features of I longiauritus (A) Branched conformation of I longiauritus mt genome. (B) Three contiguous sequences of I longiauritus mt genome after de-catenation. (C) The mt genome maps of the I longiauritus contig1 and contig2.
The mt genome of I. longiauritus included 24 core PCGs, 13 variable PCGs, 3 rRNA genes, and 19 tRNA genes (Figure 1C). These PCGs consisted of 9 NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7 and nad9), 5 ATP synthase genes (atp1, atp4, atp6, atp8 and atp9), 4 cytochrome c biogenesis genes (ccmB, ccmC, ccmFc and ccmFn), 3 cytochrome c oxidase genes (cox1, cox2 and cox3), as well as 1 cytochrome c reductase gene (cob), 1 maturation enzyme gene (matR), and 1 transport membrane protein gene (mttB) (Table 1). Moreover, the cob, rpl5, trnM-CAT and trnM-CAT genes each had two copies (Table 1). Furthermore, there were one to four introns for presence in some genes. For instance, the ccmFc, cox2 and rps3 genes each contained one intron, while rpl2 contained two; obviously, only nad7 had four introns, and the remaining genes (nad1, nad2, nad4, and nad5) each possessed three introns (Table 1).
Table 1. Gene composition of the I. longiauritus mt genome.
3.2 Codon usage analysis
In order to perform the codon usage analysis, the RSCU of PCGs in the mt genome of I. longiauritus were calculated (Supplementary Table S2). The 37 PCGs encoded totally 10,064 codons including termination codons. The most frequently occurring amino acid was leucine (Leu) with 1,049 codons, accounting for 10.42% of the total. The second most frequent one was serine (Ser) with 888 codons, which occupied 8.82%. The termination codon was the least frequent, with 30 occurrences, occupying 0.030% of the total. Moreover, 33 codons exhibited an RSCU value ≥1, among them, 27 codons ended with A or U, while 4 codons (UGG, UUG, UAG, and AUG) ended with G, and 2 codons (UCC and ACC) ended with C (Figure 2A, Supplementary Table S2). As illustrated in Supplementary Table S2, glutamine preferentially utilized the CAA codon, which exhibited the highest RSCU value of 1.55 among these PCGs, followed by histidine (His) and alanine (Ala), showing significant codon usage for CAU (1.53) and GCU (1.5), respectively.
Figure 2. The landscape of codon usage bias in the I longiauritus mt genome. (A) Relative synonymous codon usage (RSCU) analysis in the I longiauritus mt genome. (B) The ENC plot of PCGs genes in the I longiauritus mt genome. (C) The analysis of neutrality plot of PCGs genes in the I longiauritus mt genome.
The CUSP analysis indicated that the GC contents at positions GC1, GC2, GC3, and GCall were 29.29% ~ 61.36%, 27.18% ~ 52.91%, 21.57% ~ 54.57%, and 30.46% ~ 56.28%, respectively (Supplementary Table S3). Furthermore, their average values were as follows: GC1 (47.69%) > GCall (42.56%) > GC2(42.03%) > GC3 (37.97%) (Supplementary Table S3). This indicated that the uneven distribution of GC content at different positions of codons and a preference for A-ended or U-ended in the mt genome of I. longiauritus. Moreover, the ENC values of different genes ranged from 42.25 to 61, with an average value of 51.78 (Supplementary Table S3). Among them, only rps13 gene had an ENC value below 45, suggesting that these codons in the mt genome had a relatively weak preference in usage. The ENC-plot analysis revealed that 63% of ENC ratios ranged from 0.05 to 0.15, while the remaining proportion (37%) changed from -0.05 to 0.05 (Figure 2B). The results indicated that the actual ENC values of most genes deviated significantly from the theoretical ones, implying that the codon usage bias was affected by natural selection rather than mutation. Subsequently, we analyzed the distribution of the gene on a scatter plot illustrating GC3 and GC12 (Figure 2C). A low regression line slope (0.14) indicated that the association between GC3 and GC12 was very weak, indicating that mutations exerted a minor role in the formation of codon usage bias in the mt genes of I. longiauritus.
3.3 Repeat sequence analysis
Totally 119 SSRs were observed in the mt genome of I. longiauritus, comprising 32 monomers (26.89%), 24 dimers (20.17%), 15 trimers (12.60%), 44 tetramers (36.97%), 4 pentamers (3.36%), and no hexamers (Figure 3A). Next, 28 of the monomeric repeat sequences were classified as repeat type of A or T, identified as the most abundant SSR type, occupying 23.53% of the total (Figure 3B). In addition, 15 of the dimeric repeat sequences were AT/AT, recognized as the second most abundant type of SSRs, occupying 12.61% of the total (Figure 3B). Moreover, there were 26 tandem repeats in the mt genome with over 75% match and a length of 11 ~ 35bp. In total, 208 pairs of interspersed repeats of at least 30 bp in length were identified, which included 107 pairs of palindromic repeats, 101 pairs of forward repeats, and no pairs of reverse or complement repeats (Figures 3C, D).
Figure 3. The repeats of I longiauritus mt genome. (A) Type and number of SSRs repeats. (B) Type and number of SSRs motifs. (C) Type and number of interspersed repeats. (D) The length distribution of interspersed repeats.
3.4 Prediction of RNA editing sites
Totally 602 RNA editing sites across 37 PCGs in the I. longiauritus mt genome were detected. The genes ccmFn, nad2, mttB, ccmC, and ccmB were identified with 36 to 43 RNA editing sites, each significantly more than the other genes (Figure 4). Moreover, the three different types were found in total RNA editing sites (Table 2). After RNA editing, 122 positions of amino acids hydrophobicity remained unchanged, while 475 positions were changed from hydrophilic to hydrophobic. Furthermore, only 5 positions of amino acids were changed from hydrophilic to stop. All the identified editing events were the C to T type.
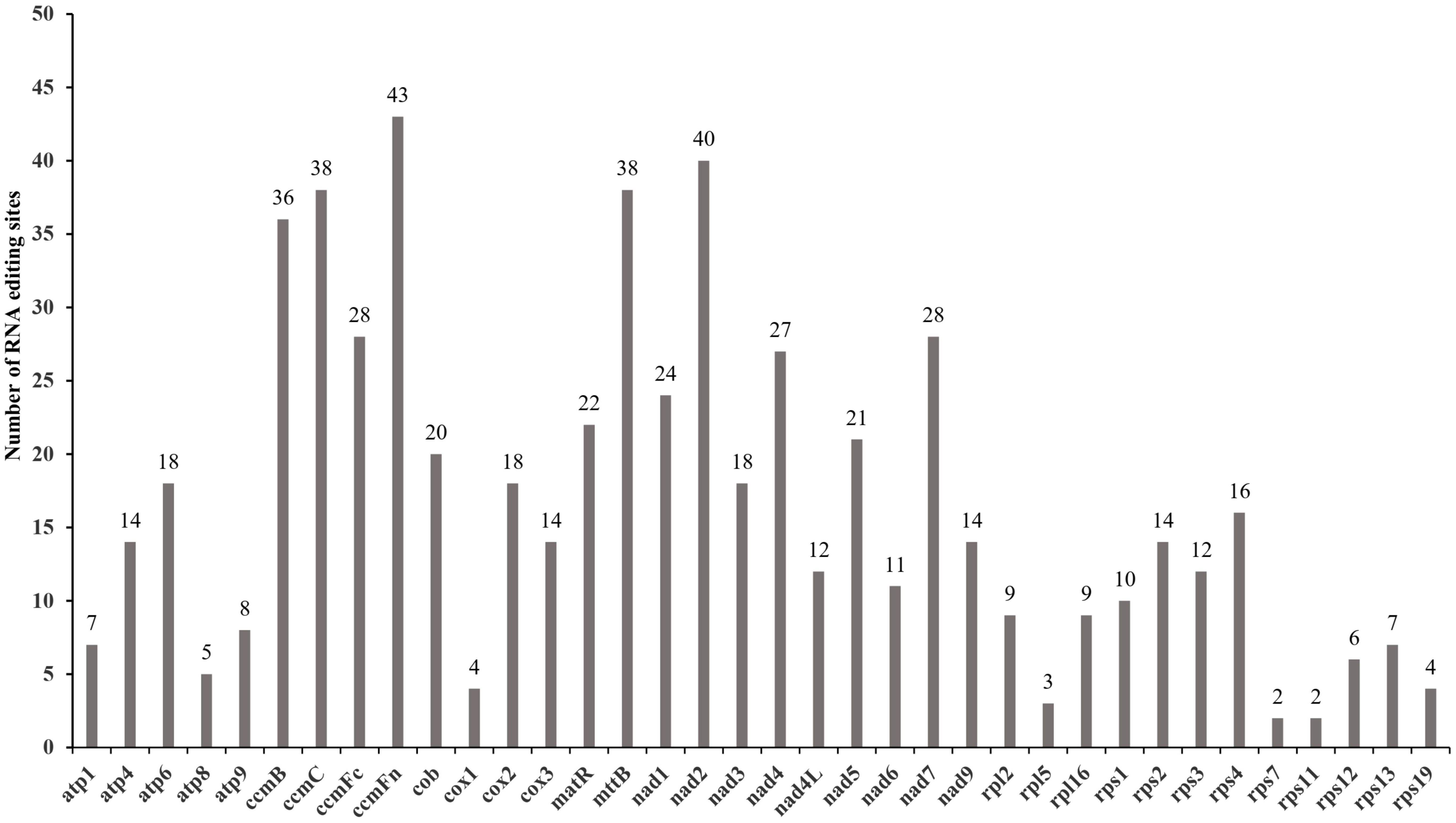
Figure 4. The distribution of RNA editing sites among PCGs in the I. longiauritus mt genome.
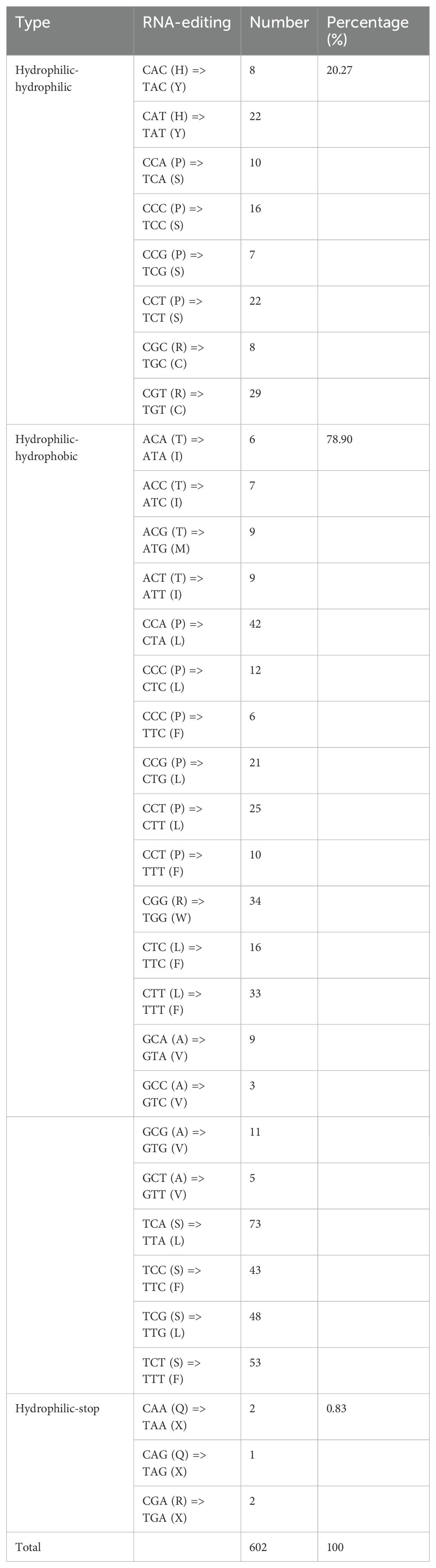
Table 2. Prediction of RNA editing sites in the I. longiauritus mt genome.
3.5 DNA migration from chloroplast to mitochondria
According to sequence similarity analysis, 10 homologous fragments with the more than 1,000 bp in length were screened (Figure 5) between the cp and mt genomes of I. longiauritus. These fragments had a total length of 19,808 bp, while the longest of them was 4,117 bp (Supplementary Table S4). Furthermore, the annotated results demonstrated that 12 complete genes were identified in these homologous sequences, transferring from the cp genome to mt genome. They included 8 protein-coding genes (petB, psbH, psbN, atpE, ndhJ, rps4, psaB, and ndhI) and 4 tRNA genes (trnM-CAU, trnV-UAC, trnF-GAA, and trnS-GGA).
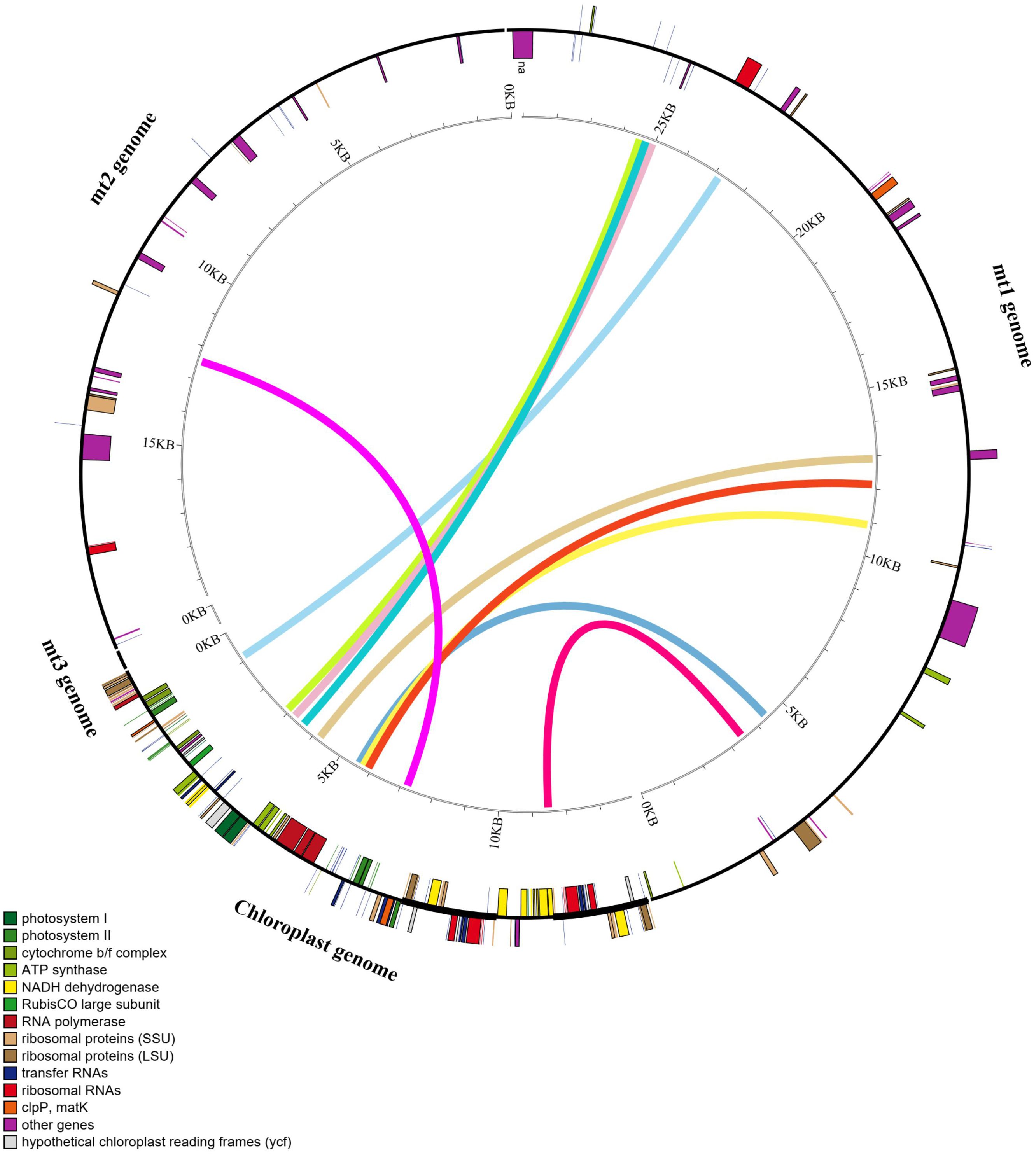
Figure 5. Similar sequences shared between the mt genome and cp genome.
3.6 Phylogenetic and colinear analysis
A phylogenetic analysis was perfomed using the six single-copy orthologous genes from 18 species to better understand the evolutionary position of I. longiauritus. The corresponding information of these species was shown in Supplementary Table S1. Among these selected plants, 16 belong to the grass family, including 3 species from Bambusoideae, 9 species from Panicoideae, 2 species from Pooideae, 1 species from Eragrostoideae, and 1 species from Oryzoideae, while 2 species from Ranunculaceae were used as the outgroup. The tree indicated that I. longiauritus clustered with 3 other species from Bambusoideae, exhibiting a close relationship with F. qinlingensis and I. tessellatus (Figure 6A). In addition, the result was also supported by the phylogenetic analysis based on their cp genomes (Figure 6B).
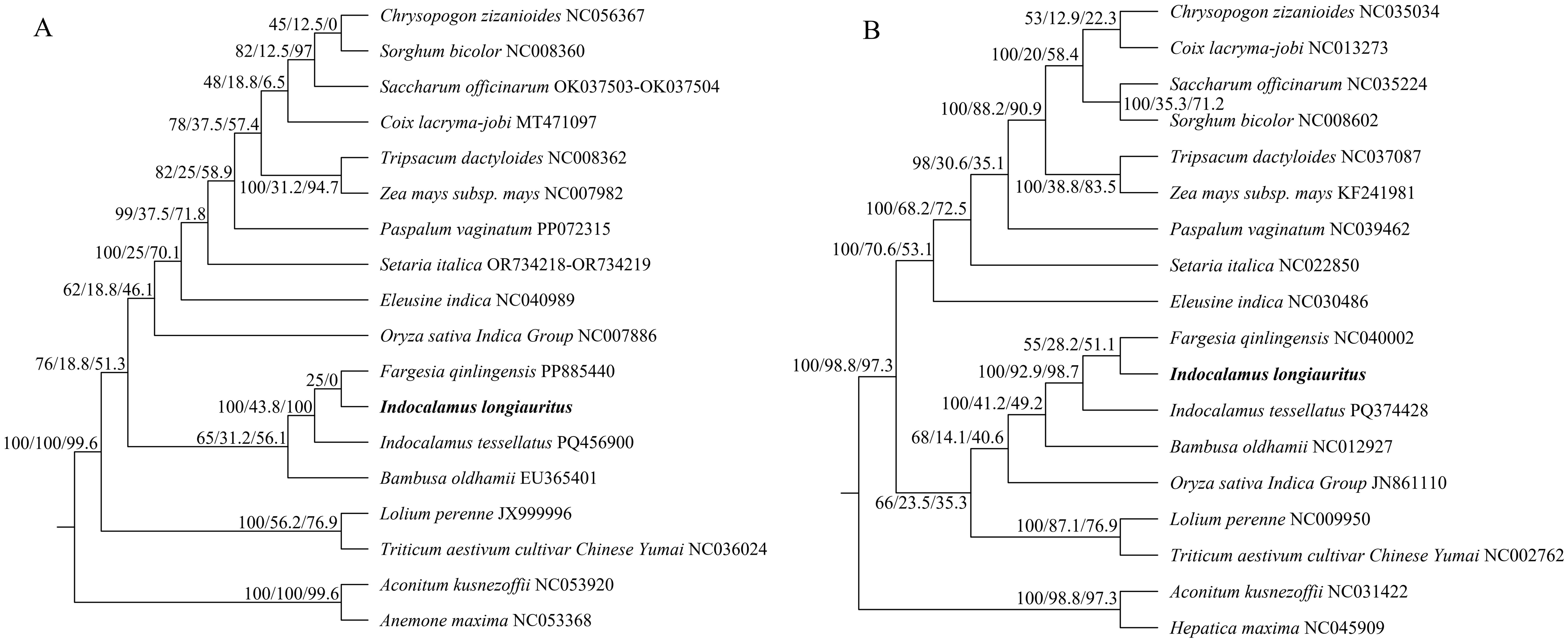
Figure 6. The phylogenetic relationships of I longiauritus with other 17 plant species by Maximum likelihood based on the single-copy orthologous genes from mt genomes (A) and cp genomes (B). Both trees were annotated with supports which were indicated by concordance factors (UFB/gCF/sCF).
To further assess the mt relationship of I. longiauritus with its closely related species, 26 homologous genes and their sequence arrangements were compared using the BLAST program (Figure 7). The homologous collinear blocks among 9 pairs of species ranged from 30 to 95, with lengths exceeding 1000 bp. The total length of collinear blocks ranged from 371,935 to 590,899 bp from four bamboo species, significantly greater than that of other species. Meanwhile, the order of collinear blocks among different mt genomes was found to be inconsistent. These results indicated that the mt genomes of I. longiauritus and its closely related species underwent extensive genome rearrangements, suggesting that the mt genome showed a highly non-conservative structure.

Figure 7. Collinear analysis of the mt genome of I longiauritus, F qinlingensis, I tessellatus, B oldhamii, P. vaginatum, S. italica, E indica, O. sativa Indica Group, L. perenne, and T. aestivum cultivar Chinese Yumai. The red arcs represented homologous regions of these mt genomes.
3.7 Comparison of the I. longiauritus with other nine closely related species (Ka/Ks) and Pi analysis
Based on the result of phylogenetic analysis, nine other Poaceae species were selected for examining of the selective pressure on PCGs. A total of 26 common PCGs which included atp1, atp4, atp6, atp8, ccmB, ccmC, ccmFc, ccmFn, cox1, cox2, matR, mttB, nad1, nad2, nad3, nad4, nad4L, nad5, nad7, nad9, rpl16, rps1, rps3, rps4, rps7, and rps13 were identified and their Ka/Ks values were calculated using I. longiauritus as a reference (Figure 8, Supplementary Table S5). The average Ka/Ks value was shown to be less than one. Among these genes, 12 genes including atp4, ccmB, ccmFc, ccmFn, matR, mttB, nad1, nad2, nad5, rps1, rps3, and rps4 revealed Ka/Ks values greater than one, while the remaining 11 genes had Ka/Ks values less than one. Moreover, this indicated that the proportions of genes undergoing positive and negative selection were similar.
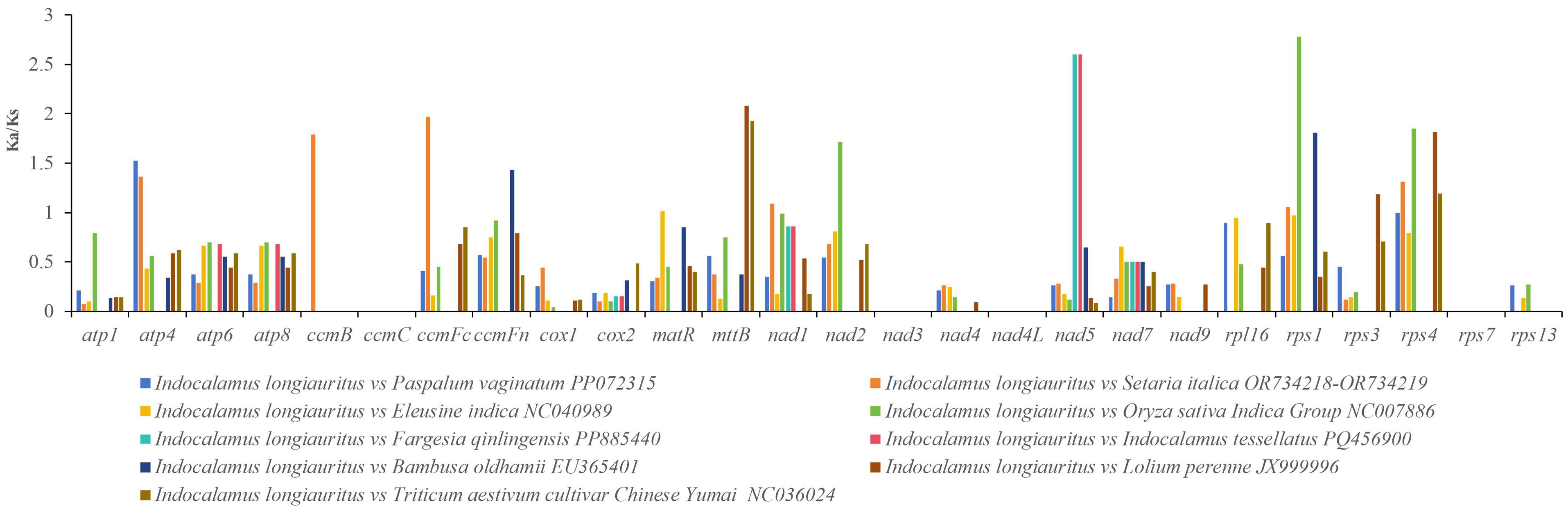
Figure 8. Ka/Ks ratios of 26 PCGs between I. longiauritus and other nine species.
Furthermore, the nucleotide diversity (Pi) of the 26 PCGs within the mt genome was calculated to assess the level of sequence divergence among these 10 species. The Pi values ranged from 0.00175 to 0.04008 (Figure 9). The atp8 gene exhibited the highest Pi value (0.04008), followed by the atp6 gene (0.0347). In contrast, the ccmC and cox2 genes showed the lowest Pi values (0.00175). All Pi values for the 26 PCGs were all less than 0.05, suggesting a high level of conservation in the nucleotide sequences of these mt genes.
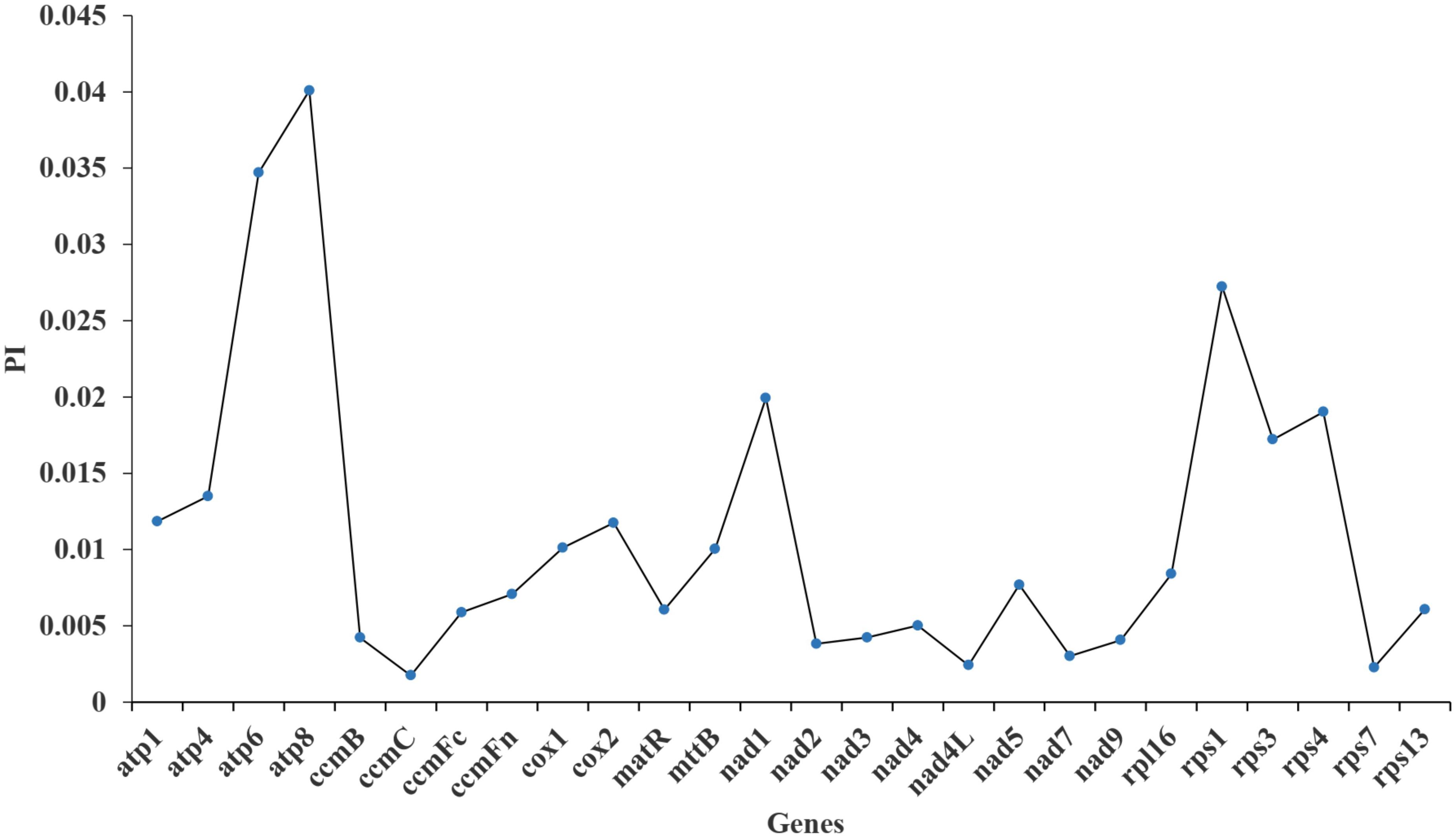
Figure 9. Nucleotide diversity of I. longiauritus mt genome.
3.8 Gene loss and multi-copy gene
Based on the phylogenetic tree, other 9 Poaceae plants owned the closer relationships with I. longiauritus. So, the distribution of PCGs in their mt genomes was compared. Based on Figure 10, most PCGs were conserved, particularly those involved in cytochrome c biogenesis (ccmB, ccmC, ccmFc, and ccmFn), maturation enzymes (matR), and transmembrane protein genes (mttB). On the contrary, the ribosomal protein and succinate dehydrogenase genes exhibited greater variability. Genes including rpl2, rpl5, rpl10, rpl14, rps2, rps11, rps12, rps14, rps19, and sdh4 were absent in some mt genomes. For example, the rps10, rps14, and sdh4 genes were lost in the I. longiauritus mt genome. Furthermore, the rpl14 gene and sdh4 were present only in the mt genome of I. longiauritus and S. italica, respectively. Besides, there were one to nine genes possessing two copies in these mt genomes except for F. qinlingensis. Specifically, the cob and rpl5 genes in I. longiauritus mt genome were demonstrated to contain two copies.
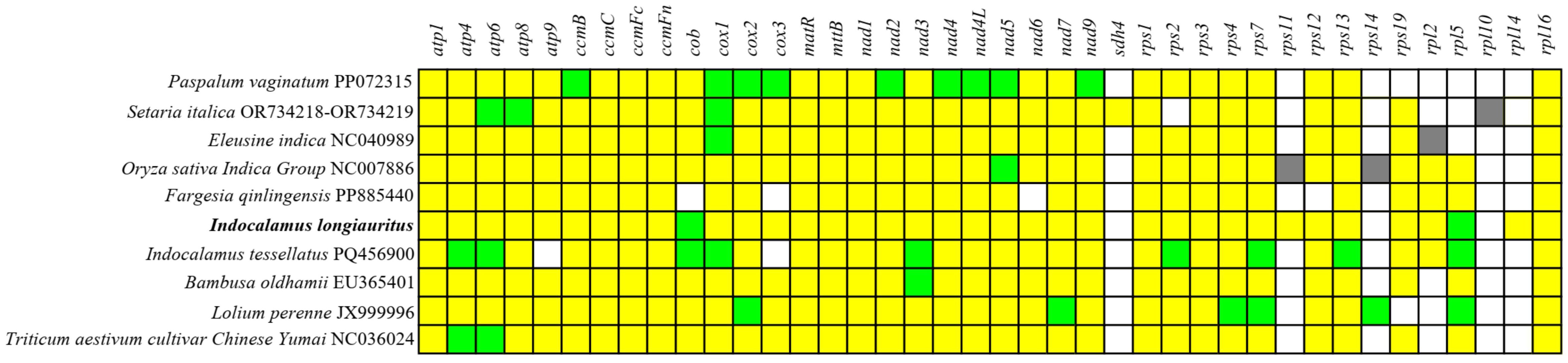
Figure 10. The PCGs distribution of I. longiauritus and other nine species. Yellow and blue boxes indicated that one and two copies in the mt genomes, respectively. White boxes indicated that the gene was absent in the mt genomes. Grey boxes indicated that the gene was pseudogene.
4 Discussion
4.1 Characterization of the I. longiauritus mt genome
In plants, the mt genome is a highly dynamic entity exhibiting significant variation across different species (Petersen et al., 2020). In Poaceae species, relatively few studies have focused on the mt genome structures and their complexities (Evans et al., 2019), so is even Bambusoideae. However, the recent rapid expansion of plant genome sequencing projects has caused a growing number of mt genomes available for comparative analyses. In this study, the sequence, assembly, and analysis of the I. longiauritus mt genome were carried out. We obtained the sequence of 491,541bp in length with one circular and two linear contigs, which was different from F. qinlingensis with three linear contigs (Wu et al., 2024a). Previous studies have demonstrated that the mt genomes of Poaceae species except for Bambusoideae have one or two circular molecules such as Z. latifolia, O. minuta, and S. italica (Zhang et al., 2024a; Luo et al., 2024; Asaf et al., 2016). These results suggest that the mt genome of Bamboo is similar to other species of Poaceae species in sizes, while there is a significant difference in structure. Codon preference can influence amino acid sequences, protein structure and function, influencing an organism’s adaptability and survival (Wu et al., 2024b). It also reflects the evolutionary process and trend of the genome. We identified 33 codons whose RSCU values were greater than 1, indicating that the mt genome displayed a preference in amino acid codon usage, such as CAA in Gln (RSCU=1.55) and CAU in His (RSCU=1.53).
For plants, complete mt genomes were usually consisted of 24 core genes and 17 variable genes. This study compared the distribution of PCGs in the mt genomes of I. longiauritus and nine relative Poeceae species. The same amount of core genes (24) was also observed in these ten mt genomes, while the number of variable genes varied from 8 to 13. The results demonstrated that most PCGs were highly conserved, especially the genes involved in cytochrome c biosynthesis, cytochrome c reductase, maturase, and membrane transport proteins. By contrast, ribosomal protein genes and succinate dehydrogenase genes exhibited greater variability. Genes including rpl2, rpl5, rpl10, rpl14, rps2, rps11, rps12, rps14, rps19, and sdh4 are absent in some mt genomes. This is not surprising, as ribosomal protein and succinate dehydrogenase genes are frequently lost or transferred to the nucleus during the evolution of angiosperm mt genomes (Adams and Palmer, 2003; Ueda and Kadowaki, 2012). Noteworthily, four mt genomes of bamboo all lost sdh4, rps14, and rpl10. Besides, only the mt genome of I. longiauritus had the rpl14 gene while only that of I. tessellatus lost the atp9 gene. Compared with other three bamboos, only F. longiauritus lost cob, nad6, and rps12. These findings showed that the PCGs of mt genomes among four bamboos exhibited diversity, potentially serving as the scientific references for species identification.
The RNA editing phenomenon is one of the essential steps in gene expression for the plant mt genomes, which is widely present in higher plant mitochondria (Small et al., 2023). We acquired 602 RNA editing sites within all the PCGs in the I. longiauritus mt genome, such figure was extremely greater than that reported in the same subfamily such as F. qinlingensis (482) and different subfamilies including S. italica (417), Z. latifolia (93), and other species (0~35) (Yang et al., 2024; Wu et al., 2024a; Zhang et al., 2024a). All the RNA editing sites were the C-T (U) types, and the finding was similar as the identification results from other plant mt genomes (Gong et al., 2024). Number RNA-editing events in the mitogenome result in the conversion of hydrophilic amino acids to hydrophobic ones (Zhang et al., 2024b). Here, 78.9% of amino acids were changed from hydrophilic to hydrophobic in the mt genome of I. longiauritus. The increase in hydrophobic amino acids can improve the overall stability of protein structures of the I. longiauritus mt genome, as hydrophobic interactions exert a vital role in the folding and stability of proteins (Zhang et al., 2024b).
4.2 Repeat sequences and nucleotide diversity analysis
Current studies have indicated that numerous different types of repetitive sequences mediate the high-frequency recombination within or between mt DNA molecules in higher plants, making them the primary cause of mt genome structural diversity (Gualberto and Newton, 2017). Here, 109 SSRs were identified in the mt genome of I. longiauritus. Though the monomeric, dimeric, trimeric, tetrameric, pentameric, hexameric and compound SSRs exist in the Poeceae family, differences are observed among species. For example, E. inadica has mono-, di-, tri-, and compound nucleotide repeats (Hall et al., 2020), while Aegilops speltoides possesses mono- to hexa- types (Li et al., 2023). However, SSRs of I. longiauritus included mono- to penta- types, the same as those of F. qinlingensis (Wu et al., 2024a). Additionally, 234 dispersed repeats were detected in our study, and the number was greater than that obtained in F. qinlingensis (195), while both species were similar in the distribution of different length (Wu et al., 2024a). These findings may explain why the mt genome of I. longiauritus is larger than that of F. qinlingensis. The larger Pi values of genes showed the higher variation of nucleotide sequences, potentially serving as the molecular markers for distinguishing different species (Gong et al., 2024). In addition, our results revealed that the Pi value of the atp8 gene was the largest, suggesting that it evolved more rapidly than other genes. As a component of the oxidative phosphorylation complex on the inner mitochondrial membrane, atp8 is vital for energy production (Weiss et al., 2012). Due to its essential role, this gene may have been subjected to intense natural selection pressure, leading to a high degree of diversity (Zhao et al., 2022). Bamboo, a plant with wide distribution and strong adaptability, may face to different selection pressures in different ecological environments (Li and Sun, 2024), which can contribute to the increased diversity of the atp8 gene. The highly variable atp8 gene can be applied for molecular markers as the mt genome analysis in I. longiauritus.
4.3 Phylogenetic and evolutionary analyses of I. longiauritus
The mt genome exhibits a lower evolutionary rate and fewer repetitive events, which can provide a clear homologous gene relationship, effectively addressing the deep nodal compounds in angiosperm phylogeny, and providing an important example for angiosperm phylogeny and evolution studies (Palmer and Herbon, 1988; Drouin et al., 2008; Lin et al., 2022, 2025). In this study, the phylogenetic tree was also constructed on the basis of the mt genomes of 18 species. I. longiauritus with three bamboos were clustered into a group, conforming to the reported by Wu et al. (2024a). These results indicated that the mt genome data can be effectively applied in distinguishing bamboo from other species from Poaceae. Noteworthily, that I. longiauritus was more closely related to F. qinlingensis rather than I. tessellatus from the same genus. To further confirm this result, their relationships were supported by the constructed tree according to their cp genomes (Figure 6). These findings further corroborated the perspective that Indocalamus was generally clustered into several lineages and other genera based on nuclear or cp genome data (Gao et al., 2025). Collinearity analysis demonstrated that the substantial rearrangements existed in the mt genome of I. longiauritus, leading to the highly variable structure when compared with its close relatives. Furthermore, this phenomenon exerted a crucial role in the evolution and diversification of the mt genome, as explained by Li et al. (2023). Certain regions of the I. longiauritus mt genome display no homology with those of other species, underscoring their distinct presence within this particular mt genome. This remarkable finding has important implications for future studies on the genetics, growth, and development of I. longiauritus. These insights may be beneficial for deepening our understanding of the unique biological characteristics of this species and contribute to broader studies in evolutionary genetics. The transfer of DNA sequences between cp and mt genomes is a common phenomenon in plant mt genomes (Straub et al., 2013).
The Ka/Ks ratios are of great significance for reconstructing the phylogeny and understanding the evolutionary dynamics of protein-coding sequences in closely related species (Fay and Wu, 2003). During the evolution in plants, most of mt genes exhibit negative selections with Ka/Ks ratios <1. In this study, 14 of the shared PCGs had Ka/Ks <1, which were obtained by analyzing the Ka/Ks values between I. longiauritus and nine related species. This was consistent with other Poeceae species such as Z. latifollia (Luo et al., 2024). It was indicated that these PCGs in the mt genome of Poeacea species were highly conserved during evolution. Nevertheless, other genes such as atp4, ccmB, ccmFc, ccmFn, matR, mttB, nad1, nad2, nad5, rps1, rps3, and rps4 were more than 1 with Ka/Ks ratio, suggesting positive selection during the evolution. Particularly, the rps1, nad5, and mttB exhibited a higher Ka/Ks value greater than 2, which might play critical roles in evolutionary history and essential life activities, possibly related to environmental stress (Wang et al., 2024b). These will be advantageous for understanding the adaptability of bamboo species, identifying selection factors, evaluating adaptation potential, and investigating heredity and evolution.
5 Conclusion
I. longiauritus is a bamboo species that holds economic and ecological significance (Bai et al., 2011). In this study, through the integration of multiple sequencing techniques and in-depth bioinformatics analyses, we successfully carried out the assembly and annotation of the mt genome of I. longiauritus. When compared with other species in the Poaceae family, the mt genome of I. longiauritus exhibits both conservation and specificity in its characteristics. Phylogenetic analyses based on mt and cp genomic datasets have revealed discrepancies regarding the evolutionary position of this species within its taxonomic clades. These results provide foundational insights into genetic traits, molecular variations, taxonomic classification of I. longiauritus. Moreover, this research will serve as a reliable genomic resource for future studies on the systematic evolution of the Bambusoideae family.
Data availability statement
The chloroplast genome datasets of I. longiauritus with the accession number PV366311 and its mitochondrial genome datasets with accession number PV366312, PV366313, and PV366314 have been deposited in the GenBank of NCBI. The BioProject, BioSample, and SRA numbers of raw sequencing data were PRJNA1265797, SAMN48631790, and SRR33649504, respectively, were also deposited in the GenBank of NCBI.
Author contributions
SL: Visualization, Methodology, Validation, Writing – original draft. YZ: Writing – original draft, Data curation, Formal analysis. LL: Writing – review & editing, Investigation, Resources. DH: Funding acquisition, Writing – review & editing, Investigation. YQ: Project administration, Writing – review & editing, Validation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work has been supported by the Guangxi Natural Science Foundation of China (2022GXNSFBA035604) and Special Project on the First Comprehensive Survey and Collection of Forest and Grass Genetic Resources in Guangxi (PCZX2022002).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1599464/full#supplementary-material
References
Adams, K. and Palmer, J. D. (2003). Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395. doi: 10.1016/S1055-7903(03)00194-5
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Alverson, A. J., Wei, X., Rice, D. W., Stern, D. B., Barry, K., and Palmer, J. D. (2010). Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 27, 1436–1448. doi: 10.1093/molbev/msq029
Arthan, W., Baker, W. J., Barrett, M. D., Barrett, R. L., Bennetzen, J. L., et al. (2025). A nuclear phylogenomic tree of grasses (Poaceae) recovers current classification despite gene tree incongruence. New Phytol. 1245, 818-834. doi: 10.1111/nph.20263
Asaf, S., Khan, A. L., Khan, A. R., Waqas, M., Kang, S. M., Khan, M. A., et al. (2016). Mitochondrial genome analysis of wild rice (Oryza minuta) and its comparison with other related Species. PloS One 11, e0152937. doi: 10.1371/journal.pone.0152937
Bai, K., Jiang, D., Cao, K., Liao, D., and Wan, X. (2011). The physiological advantage of an ecological filter species, Indocalamus longiauritus, over co-occurring Fagus lucida and Castanopsis lamontii seedlings. Ecol. Res. 26, 15–25. doi: 10.1007/s11284-010-0744-9
Beck, N. and Lang, B. (2010). MFannot, organelle genome annotation webserver (University of Montreal QC, Canada) Available online at: http://megasun.bch.umontreal.ca/RNAweasel/.
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, C. W., Shen, F., Han, F., Qu, Y., Hou, J., Xu, K., et al. (2024). PMAT: an efficient plant mitogenome assembly toolkit using low coverage HiFi sequencing data. Hortic. Res. 11, uhae023. doi: 10.1093/hr/uhae023
Braun, H. P. (2020). The oxidative phosphorylation system of the mitochondria in plants. Mitochondrion 53, 66–75. doi: 10.1016/j.mito.2020.04.007
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cheng, Y., He, X., Priyadarshani, S. V. G. N., Wang, Y., Ye, L., Shi, C., et al. (2021). Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genomics 22, 167. doi: 10.1186/s12864-021-07490-9
Cole, L. W., Guo, W., Mower, J. P., and Palmer, J. D. (2018). High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol. Biol. Evol 35, 2773-2785. doi: 10.10933/molbev/msy176
Drouin, G., Daoud, H., and Xia, J. (2008). Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 49, 827–831. doi: 10.1016/j.ympev.2008.09.009
Evans, D. L., Hlongwane, T. T., Joshi, S. V., and Pachón, D. M. R. (2019). The sugarcane mitochondrial genome: assembly, phylogenetics and transcriptomics. Peer J. 7, e7558. doi: 10.7717/peerj.7558
Fay, J. C. and Wu, C. I. (2003). Sequence divergence, functional constraint, and selection in protein evolution. Annu. Rev. Genomics Hum. Genet. 4, 213–235. doi: 10.1146/annurev.genom.4.020303.162528
Gao, L., Li, Y., Wang, C., Wang, J., Yang, G., and Zhang, W. (2025). New combination and two synonyms of Indocalamus Nakai (Poaceae: Bambusoideae) from China based on morphological characters and phylogenetic evidence. Taxonomy 5, 12. doi: 10.3390/taxonomy5010012
Ghifari, A. S., Saha, S., and Murcha, M. W. (2023). The biogenesis and regulation of the plant oxidative phosphorylation system. Plant Physiol. 192, 728–747. doi: 10.1093/plphys/kiad108
Gong, Y., Xie, X., Zhou, G., Chen, M., Chen, Z., Li, P., et al. (2024). Assembly and comparative analysis of the complete mitochondrial genome of Brassica rapa var. Purpuraria. BMC Genomics 25, 546. doi: 10.1186/s12864-024-10457-1
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Gualberto, J. M. and Newton, K. J. (2017). Plant mitochondrial genomes: dynamics and mechanisms of Mutation. Annu. Rev. Plant Biol. 68, 225–252. doi: 10.1146/annurev-arplant-043015-112232
Hall, N. D., Zhang, H., Mower, J. P., McElroy, J. S., and Goertzen, L. R. (2020). The mitochondrial genome of Eleusine indica and characterization of gene content within Poaceae. Genome Biol. Evol. 12, 3684–3697. doi: 10.1093/gbe/evz229
Hong, Z., Liao, X., Ye, Y., Zhang, N., Yang, Z., Zhu, W., et al. (2021). A complete mitochondrial genome for fragrant Chinese rosewood (Dalbergia odorifera, Fabaceae) with comparative analyses of genome structure and intergenomic sequence transfers. BMC Genomics 22, 672. doi: 10.1186/s12864-021-07967-7
Jacoby, R. P., Li, L., Huang, S., Pong Lee, C., Millar, A. H., and Taylor, N. L. (2012). Mitochondrial composition, function and stress response in plants. J. Integr. Plant Biol. 54, 887–906. doi: 10.1111/j.1744-7909.2012.01177.x
Katoh, K. and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kong, J. L., Wang, J., Nie, L. Y., Tembrock, L. R., Zou, C. S., Kan, S. L., et al. (2025). Evolutionary dynamics of mitochondrial genomes and intracellular transfers among diploid and allopolyploid cotton species. BMC Biol. 23, 9. doi: 10.1186/s12915-025-02115-z
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, J., Chen, Y., Liu, Y., Wang, C., Li, L., and Chao, Y. (2023). Complete mitochondrial genome of Agrostis stolonifera: insights into structure, codon usage, repeats, and RNA editing. BMC Genomics 24, 466. doi: 10.1186/s12864-023-09573-1
Li, X. and Sun, H. (2024). Bamboo breeding strategies in the context of “bamboo as a substitute for plastic initiative. Forests 15, 1180. doi: 10.3390/f15071180
Li, Y., Zhang, Y., He, X., Guo, Z., Yang, N., Bai, G., et al. (2024). The mitochondrial blueprint: unlocking secondary metabolite production. Metabolites 14, 711. doi: 10.3390/metabo14120711
Lin, Q. S., Braukmann, T. W. A., Gomez, M. S., Mayer, J. L. S., Pinheiro, F., Merckx, V. S. F. T., et al. (2022). Mitochondrial genomic data are effective at placing mycoheterotrophic lineages in plant phylogeny. New Phytol. 236, 1908–1921. doi: 10.1111/nph.18335
Lin, D. L., Shao, B. Y., Gao, Z. Y., Li, J. W., Li, Z. H., Li, T. Y., et al. (2025). Phylogenomics of angiosperms based on mitochondrial genes: insights into deep node relationships. BMC Biol. 23, 45. doi: 10.1186/s12915-025-02135-9
Liu, Q., Wu, Z. N., Tian, C. Y., Yang, Y. T., Liu, L. M., Feng, Y. M., et al. (2023a). Complete mitochondrial genome of the endangered Prunus pedunculata (Prunoideae, Rosaceae) in China: characterization and phylogenetic analysis. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1266797
Liu, J., Yu, S., Lu, P., Gong, X., Sun, M., and Tang, M. (2025). De novo assembly and characterization of the complete mitochondrial genome of Phellodendron amurense reveals three repeat-mediated recombination. Gene 935, 149031. doi: 10.1016/j.gene.2024.149031
Liu, Q., Yuan, H. Y., Xu, J. X., Cui, D. L., Xiong, G., Schwarzacher, T., et al. (2023b). The mitochondrial genome of the diploid oat Avena longiglumis. BMC Plant Biol. 23, 218. doi: 10.1186/s12870-023-04217-8
Luo, X., Gu, C., Gao, S., Li, M., Zhang, H., and Zhu, S. (2024). Complete mitochondrial genome assembly of Zizania latifolia and comparative genome analysis. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1381089
Minh, B. Q., Hahn, M. W., and Lanfear, R. (2020). New methods to calculate concordance factors for phylogenomic datasets. Mol. Biol. Evol. 37, 2727–2733. doi: 10.1093/molbev/msaa106
Mower, J. P. (2005). PREP-Mt: predictive RNA editor for plant mitochondrial genes. BMC Bioinf. 6, 96. doi: 10.1186/1471-2105-6-96
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Palmer, J. D. and Herbon, L. A. (1988). Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence. J. Mol. Evol. 28, 87–97. doi: 10.1007/BF02143500
Petersen, G., Anderson, B., Braun, H. P., Meyer, E. H., and Moller, I. M. (2020). Mitochondria in parasitic plants. Mitochondrian 52, 173–182. doi: 10.1016/j.mito.2020.03.008
Putintseva, Y. A., Bondar, E. I., Simonov, E. P., Sharov, V. V., Oreshkova, N. V., Kuzmin, D. A., et al. (2020). Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genomics 21, 654. doi: 10.1186/s12864-020-07061-4
Richardson, A. O., Rice, D. W., Young, G. J., Alverson, A. J., and Palmer, J. D. (2013). The fossilized mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 11, 1–17. doi: 10.1186/1741-7007-11-29
Roger, A. J., Munoz-Gomez, S. A., and Kamikawa, R. (2017). The origin and diversification of mitochondria. Curr. Biol. 27, R1177–R1192. doi: 10.1016/j.cub.2017.09.015
Romero, H., Zavala, A., and Musto, H. (2000). Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic Acids Res. 28, 2084–2090. doi: 10.1093/nar/28.10.2084
Rozas, J., Ferrer-Mata, A., Carlos Sanchez-DelBarrio, J., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sharp, P. M. and Li, W. H. (1986). Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 14, 7737–7749. doi: 10.1093/nar/14.19.7737
Sloan, D. B. (2013). One ring to rule them all? Genome sequencing provides new insights into the “master circle” model of plant mitochondrial DNA structure. New Phytol. 200, 978–985. doi: 10.1111/nph.12395
Sloan, D. B., Alverson, A. J., Chuckalovcak, J. P., Wu, M., McCauley, D. E., Palmer, J. D., et al. (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PloS Biol. 10, e1001241. doi: 10.1371/journal.pbio.1001241
Small, I., Melonek, J., Bohne, A. V., Nickelsen, J., and Schmitz-Linneweber, C. (2023). Plant organellar RNA maturation. Plant Cell 35, 1727–1751. doi: 10.1093/plcell/koad049
Soreng, R. J., Peterson, P. M., Zuloaga, F. O., Romaschenko, K., Clark, L. G., Teisher, J. K., et al. (2022). A worldwide phylogenetic classification of the Poaceae (Gramineae) III: An update. J. S. E. 60, 476–521. doi: 10.1111/jse.12847
Straub, S. C. K., Cronn, R. C., Edwards, C., Fishbein, M., and Liston, A. (2013). Horizontal transfer of DNA from the mitochondrial to the plastid genome and its subsequent evolution in milkweeds (Apocynaceae). Genome Biol. Evol. 5, 1872–1885. doi: 10.1093/gbe/evt140
Sueoka, N. (1988). Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. U. S. A. 85, 2653–2657. doi: 10.1073/pnas.85.8.2653
Szandar, K., Krawczyk, K., Myszczynski, K., Slipiko, M., Sawicki, J., and Szczecinska, M. (2022). Breaking the limits-multichromosomal structure of an early eudicot Pulsatilla patens mitogenome reveals extensive RNA-editing, longest repeats and chloroplast derived regions among sequenced land plant mitogenomes. BMC Plant Biol. 22, 109. doi: 10.1186/s12870-022-03492-1
Thiel, T., Michalek, W., Varshney, R. K., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Ueda, M. and Kadowaki, K. (2012). “Gene Content and Gene Transfer from Mitochondria to the Nucleus During Evolution,” in Mitochondrial Genome Evolution, vol. 21–40 . Ed. Marechal Drouard, L. (Academic Press Ltd-Elsevier Science Ltd, London). doi: 10.1016/B978-0-12-394279-1.00002-8
Wang, C. K., Li, Y. L., Yang, G. Y., Zhang, W. G., and Guo, C. C. (2024a). Comparative analysis of chloroplast genomes and phylogenetic relationships in the endemic Chinese bamboo Gelidocalamus (Bambusoideae). Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1470311
Wang, L., Liu, X., Xu, Y. J., Zhang, Z. W., Wei, Y. S., Hu, Y., et al. (2024b). Assembly and comparative analysis of the first complete mitochondrial genome of a traditional Chinese medicine Angelica Biserrata (Shan Et Yuan) Yuan Et Shan. Int. J. Biol. Macromol. 257, 128571. doi: 10.1016/j.ijbiomac.2023.128571
Weiss, H., Wester-Rosenloef, L., Koch, C., Koch, F., Baltrusch, S., Tiedge, M., et al. (2012). The mitochondrial Atp8 mutation induces mitochondrial ROS generation, secretory dysfunction, and β-cell mass adaptation in conplastic B6-mtFVB mice. Endocrinology 153, 4666–4676. doi: 10.1210/en.2012-1296
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Z., Cuthbert, J. M., Taylor, D. R., and Sloan, D. B. (2015). The massive mitochondrial genome of the angiosperm Silene noctiflora is evolving by gain or loss of entire chromosomes. Proc. Natl. Acad. Sci. U. S. A. 112, 10185–10191. doi: 10.1073/pnas.1421397112
Wu, H., Li, X., Qu, K., Yang, L. L., Su, T., Yong, L. J., et al. (2024a). Integration of Illumina and PacBio HiFi sequencing reveals a three-linear-molecule mitogenome with RNA-editing sites and phylogeny in arrow bamboo (Fargesia qinlingensis). Forests 15, 1267. doi: 10.3390/f15071267
Wu, Z. Q., Liao, X. Z., Zhang, X. N., Tembrock, L. R., and Broz, A. (2022). Genomic architectureal variation of plant mitochondria- a review of multichromosomal structuring. J. Syst. Evol. 60, 160–168. doi: 10.1111/jse.12655
Wu, X., Xu, M., Yang, J. R., and Lu, J. (2024b). Genome-wide impact of codon usage bias on translation optimization in Drosophila melanogaster. Nat. Commun. 15, 8329. doi: 10.1038/s41467-024-52660-4
Xiong, A. S., Peng, R. H., Zhuang, J., Gao, F., Zhu, B., Fu, X. Y., et al. (2008). Gene duplication and transfer events in plant mitochondria genome. Biochem. Biophys. Res. Commun. 376, 1–4. doi: 10.1016/j.bbrc.2008.08.116
Yang, J., Ling, C. C., Zhang, H. M., Hussain, Q., Lyu, S. H., Zheng, G. H., et al. (2022). A comparative genomics approach for analysis of complete mitogenomes of five Actinidiaceae plants. Genes 13, 1827. doi: 10.3390/genes13101827
Yang, S. Q., Liu, J. F., Jiang, S. C., Guo, B. Q., Guo, J., Cao, L. L., et al. (2024). Mitochondrial genomic characteristics and phylogenetic analysis of 21 species of Poaceae. Genomics Appl. Biol. 43, 1533–1547. doi: 10.13417/j.gab.043.001533
Zeng, C. X., Zhang, Y. X., Triplett, J. K., Yang, J. B., and Li, D. Z. (2010). Large multi-locus plastid phylogeny of the tribe Arundinarieae (Poaceae: Bambusoideae) reveals ten major lineages and low rate of molecular divergence. Mol. Phylogenet. Evol. 56, 821–839. doi: 10.1016/j.ympev.2010.03.041
Zervas, A., Petersen, G., and Seberg, O. (2019). Mitochondrial genome evolution in parasitic plants. BMC Evol. Biol. 19, 87. doi: 10.1186/s12862-019-1401-8
Zhang, Z., Li, J., Zhao, X. Q., Wang, J., Wong, G. K. S., and Yu, J. (2006). KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinf. 4, 259–263. doi: 10.1016/S1672-0229(07)60007-2
Zhang, J., Liu, G., and Wei, J. (2024a). Assembly and comparative analysis of the first complete mitochondrial genome of Setaria italica. Planta 260, 23. doi: 10.1007/s00425-024-04386-2
Zhang, H., Meltzer, P., and Davis, S. (2013). RCircos: an R package for Circos 2D track plots. BMC Bioinf. 14, 244. doi: 10.1186/1471-2105-14-244
Zhang, K., Qu, G. Y., Zhang, Y., and Liu, J. X. (2024b). Assembly and comparative analysis of the first complete mitochondrial genome of Astragalus membranaceus (Fisch.) Bunge: an invaluable traditional Chinese medicine. BMC Plant Biol. 24, 1055. doi: 10.1186/s12870-024-05780-4
Zhang, Z., Xiao, J. F., Wu, J. Y., Zhang, H. Y., Liu, G. M., Wang, X. M., et al. (2012). ParaAT: a parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Bioph. Res. Co. 419, 779–781. doi: 10.1016/j.bbrc.2012.02.101
Keywords: Indocalamus longiauritus, mitochondrial genome, repeat sequence, predicted RNA editing sites, phylogenetic analysis
Citation: Liu S, Zhang Y, Li L, Huang D and Qin Y (2025) Assembly and comparative analysis of the complete mitochondrial genome of Indocalamus longiauritus. Front. Plant Sci. 16:1599464. doi: 10.3389/fpls.2025.1599464
Received: 25 March 2025; Accepted: 26 May 2025;
Published: 13 June 2025.
Edited by:
Changwei Bi, Nanjing Forestry University, ChinaReviewed by:
Zinian Wu, Chinese Academy of Agricultural Sciences, ChinaYanpeng Chen, Shenzhen University, China
Copyright © 2025 Liu, Zhang, Li, Huang and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dayong Huang, aGR5MjAyOUAxNjMuY29t; Yonghua Qin, Z3hfcWlueW9uZ2h1YUAxMjYuY29t