 Wei-Bing Fan
Wei-Bing Fan Khurram Shahzad
Khurram Shahzad Zhong-Hu Li
Zhong-Hu Li- Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, College of Life Sciences, Northwest University, Xi’an, China
In general, the chloroplast genomes of angiosperms are considered to be highly conserved and affected little by adaptive evolution. In this study, we tested this hypothesis based on sequence differentiation and adaptive variation in the plastid genomes in the order Dipsacales. We sequenced the plastid genomes of one Adoxaceae species and six Caprifoliaceae species, and together with seven previously released Dipsacales chloroplasts, we determined the sequence variations, evolutionary divergence of the plastid genomes, and phylogeny of Dipsacales species. The chloroplast genomes of Adoxaceae species ranged in size from 157,074 bp (Sinadoxa corydalifolia) to 158,305 bp (Sambucus williamsii), and the plastid genomes of Caprifoliaceae varied from 154,732 bp (Lonicera fragrantissima var. lancifolia) to 156,874 bp (Weigela florida). The differences in the number of genes in Caprifoliaceae and Adoxaceae species were largely due to the expansion and contraction of inverted repeat regions. In addition, we found that the number of dispersed repeats (Adoxaceae = 37; Caprifoliaceae = 384) was much higher than that of tandem repeats (Adoxaceae = 34; Caprifoliaceae = 291) in Dipsacales species. Interestingly, we determined 19 genes with positive selection sites, including three genes encoding ATP protein subunits (atpA, atpB, and atpI), four genes for ribosome protein small subunits (rps3, rps7, rps14, and rps15), four genes for photosystem protein subunits (psaA, psaJ, psbC, and pabK), two genes for ribosome protein large subunits (rpl22 and rpl32), and the clpP, infA, matK, rbcL, ycf1, and ycf2 genes. These gene regions may have played key roles in the adaptation of Dipsacales to diverse environments. In addition, phylogenetic analysis based on the plastid genomes strongly supported the division of 14 Dipsacales species into two previously recognized sections. The diversification of Adoxaceae and Caprifoliaceae was dated to the late Cretaceous and Tertiary periods. The availability of these chloroplast genomes provides useful genetic information for studying taxonomy, phylogeny, and species evolution in Dipsacales.
Introduction
Traditionally, the order Dipsacales comprises the families Valerianaceae, Dipsacaceae, Adoxaceae, and Caprifoliaceae sensu lato (including Linnaeaceae, Diervillaceae, and Caprifoliaceae sensu stricto) (Cronquist, 1979; Thorne, 1992). The evolutionary relationships among some Dipsacales species have been revised based on studies of their taxonomy and phylogeny. For example, the families Linnaeaceae, Diervillaceae, and Caprifoliaceae s. s. have been renamed as Linnaeeae, Diervilleae, and Caprifolieae, respectively, and they constitute the new family Caprifoliaceae s. l. (Donoghue et al., 1992; Olmstead et al., 2000; Bell et al., 2001; Zhang et al., 2003). The general consensus is that Dipsacales comprises a monophyletic taxon with two major lineages (Donoghue et al., 1992; Olmstead et al., 1993, 2000; Zhang et al., 2003): (i) the large clade Caprifoliaceae containing Diervilleae, Caprifolieae, Linnaeeae, Morinaceae, Valerianaceae, and Dipsacaceae; and (ii) the smaller clade comprising the family Adoxaceae, which contains the genera Viburnum, and Sambucus, Sinadoxa, Tetradoa, and Adoxa. Within Adoxaceae, analyses of several data sets (e.g., morphological evidence as well as internal transcribed spacer, mitochondrial, and chloroplast (cp) DNA sequences) have suggested that the genera Sambucus and Viburnum (belonging to the traditional Caprifoliaceae) have a close relationship (Eriksson and Donoghue, 1997; Donoghue et al., 2001; Zhang et al., 2002; Winkworth et al., 2008). Within Caprifoliaceae, the genera Lonicera and Triosteum have a very close relationship (Donoghue et al., 2001; Zhang et al., 2002; Winkworth et al., 2008). In addition, other studies based on cp DNA regions trnL-trnF and ndhF, as well as mitochondrial DNA sequence variations have suggested that the genera Dipelta and Kolkwitzia have a very close evolutionary relationship (Donoghue et al., 2001; Zhang et al., 2002; Winkworth et al., 2008). These previous studies have resolved the basic phylogenetic relationships among Dipsacales, but the interspecific divergence and lineage structure of some main genera (e.g., Dipelta, Kolkwitzia, and Weigela) still remain largely controversial.
Many studies have focused on studying the origin and divergence history of Dipsacales as an important angiosperm clade. For example, Backlund (1996) employed DNA sequences and several important fossil points to estimate the origin of Dipsacales species as around 60–70 million years ago (mya) during the late Cretaceous or early Tertiary. Wikström et al. (2001) used the non-parametric rate smoothing method to estimate the origin of Dipsacales as 85–90 mya. In addition, Bell and Donoghue (2005) used the relaxed assumption of rate constancy among lineages to estimate the ages of major lineages and suggested a late Cretaceous origin for Dipsacales. Studies of the divergence times for Dipsacales based on the DNA sequence datasets have progressed greatly, but the divergence dates of the major lineages within Dipsacales remain unclear.
Chloroplasts are multifunctional organelles in plant cells with critical roles in photosynthesis and carbon fixation (Wicke et al., 2011; Daniell et al., 2016). Chloroplasts possess their own genetic material, which exists as a quadripartite circular molecule of double-stranded DNA containing two copies of inverted repeats (IRs) separated by two regions: the large (LSC) and small (SSC) single copy regions (Huotari and Korpelainen, 2012). Most angiosperm cp genomes are remarkably conserved in terms of their structure, gene content, and order (Wicke et al., 2011). The cp genomes of land plants usually encode 110–130 genes with sizes in the range of 120–160 kb (Zhang et al., 2014). In general, plant cp genomes are recombination-free, maternally inherited, and with low rates of nucleotide substitutions, which make them valuable sources of genetic markers for phylogenetic and population genetic analyses (Korpelainen, 2004; Ravi et al., 2008). In recent years, cp genomes have been used widely to study phylogenetic relationships and species evolution in different taxa, as well as for constructing the phylogenetic lineages of angiosperms. For example, studies based on cp genome datasets have shown that Chloranthaceae and Magnoliids are sisters to a clade of monocots, as well as eudicots including Ceratophyllaceae (Moore et al., 2007). In addition, orchids and grasses together form a monophyletic group nested within the remaining angiosperms (Chang et al., 2006). Similarly, important advances have been made based on the complete cp genomes in elucidating the relationships within the larger monocot (Graham et al., 2006) and asterid (Bremer et al., 2002) clades. Recently, several cp genome sequences have been published for Dipsacales species (Wang Y. et al., 2016; Bai et al., 2017; He et al., 2017). However, there has been little research into the phylogenetic evolution and interspecific divergence of Dipsacales species due to the lack of conjoint analyses of large cp genome datasets.
In general, the chloroplast genomes of angiosperms have slow substitution rates and they are affected little by adaptive evolution (Erixon and Oxelman, 2008). Excluding genes that have evolved very rapidly, non-synonymous nucleotide substitutions have occurred less frequently than synonymous substitutions due to the action of purifying selection (Ivanova et al., 2017). Previous studies have suggested that plant cp genes have lower rates of synonymous nucleotide substitutions than nuclear genes (Palmer, 1985; Wolfe et al., 1987). In addition, positive selection is expected to speed up non-synonymous substitution rates whereas synonymous rates are expected to be unaffected. However, until recently, adaptive evolution by positive selection had rarely been determined in Dipsacales cp genes.
In the present study, we investigated adaptive evolution in the cp genomes of Dipsacales. We collected materials from one Adoxaceae species and six Caprifoliaceae species, and we assembled and annotated their complete cp genomes, before comparing these cp genomes and other published cp genomes to explore genome differentiation and sequence divergence in Dipsacales. We also identified the variant hotspot regions in cp genomes and reconstructed the phylogenetic relationships and molecular divergence dates for the major lineages within the order Dipsacales.
Materials and Methods
Sample Collection, Genome Sequencing, and Assembly
Fresh leaves from one Adoxaceae species, Viburnum betulifolium, and six Caprifoliaceae species, Lonicera fragrantissima var. lancifolia, Lonicera stephanocarpa, Lonicera tragophylla, Triosteum pinnatifidum, Weigela florida, and Dipelta floribunda, were collected in Shaanxi Province in 2016 (Supplementary Table S1). Voucher specimens of each sample were deposited in the Key Laboratory of Resource Biology and Biotechnology in Western China (Xi’an, China). Total genomic DNA was isolated from 1 g of each fresh leaf sample using the modified CTAB method (Doyle, 1987). In addition, we downloaded the available complete cp genomes of seven other Dipsacales species from GenBank (Viburnum utile, NC_032296; Sambucus williamsii, NC_033878; Sinadoxa corydalifolia, NC_032040; Lonicera japonica, NC_026839; Kolkwitzia amabilis, NC_029874; Adoxa moschatellina, KX258652; and Trachelium caeruleum, NC_010442). The cp genomes of Helianthus annuus (NC_007977) and Guizotia abyssinica (NC_010601) were also downloaded for subsequent analyses.
After extracting the genomic DNA, approximately 5–10 μg of DNA was sheared, before adapter ligation and library amplification. The fragmented DNA was subjected to library preparation and paired-end read (PE150/PE125) sequencing was then conducted with the Illumina Hiseq 2500 platform. Raw reads were filtered to remove sequences shorter than 50 bp and adapter sequences, using the NGSQCToolkit_v2.3.3 tool (Patel and Jain, 2012). The Dipsacales cp genomes were then reconstructed by de novo assembly combined with reference-based assembly. We aligned the short reads obtained from the Illumina sequencing to the reference chloroplast genomes (L. japonica, K. amabilis, and V. utile) using Bowtie 2.2.6 (Langmead and Salzberg, 2012). Then, the cp reads that mapped to the reference genome were extracted to be used as input for de novo assembly using SPAdes 3.9.0 (Bankevich et al., 2012). For the reference-based assembly, the clean reads for L. fragrantissima var. lancifolia, L. stephanocarpa, L. tragophylla, T. pinnatifidum, W. florida, D. floribunda, and V. betulifolium were first assembled using MIRA 4.0.2 (Chevreux et al., 2004), where the references comprised the cp genomes of the closely related species Lonicera japonica (NC_026839), Kolkwitzia amabilis (NC_029874), and Viburnum utile (NC_032296). Subsequently, some ambiguous regions were selected for extension by using a baiting and iteration method with the MITObim v1.8 program (Hahn et al., 2013). The contigs obtained were used to generate consensus sequences with Geneious R v9.0.5 (Kearse et al., 2012). A small number of gaps and low coverage regions in the assembled cp genomes were validated using the Sanger sequencing method, with primers (Supplementary Table S2) developed using Primer3 (Untergasser et al., 2012).
Genome Annotation
The consensus sequences were imported into the online program Dual Organellar Genome Annotator (DOGMA, Wyman et al., 2004) for gene annotation, guided by the other cp genomes. In addition, all of the tRNA genes were further verified using tRNAscan-SE1.21 (Schattner et al., 2005). Sequences were aligned using the Mauve program to compare the structure and gene contents within the genomes (Darling et al., 2004). We also re-annotated the sequences downloaded from NCBI Genbank before using them in our analyses. The newly obtained chloroplast genomes and the raw reads of Dipsacales species were submitted into the GenBank under accession numbers are MG738664-MG738664 and SRR6898410-SRR6898416, respectively. Finally, circular plastid genome maps were drawn using OGDRAW (Lohse et al., 2013).
Repeat Element Analysis
Repeat motifs are very useful markers with important roles in phylogenetic analysis (Cavalier-Smith, 2002; Nie et al., 2012). In general, large repeated elements comprise dispersed, palindromic, and tandem repeats. Tandem repeat in DNA is the pattern of two or more adjacent, approximate copies of nucleotides. Dispersed repeats are nucleotide sequences present in multiple copies in the genome. Palindromic repeat is an inverted repeat sequence with no intervening nucleotides between the initial sequence and its downstream reverse complements. In order to identify repeat elements, the web-based REPuter program (Kurtz et al., 2001) was used to analyze the dispersed and palindromic repeats based on the following conditions; (1) Hamming distance = 1; (2) sequence identity ≥ 90%; and (3) minimum repeat size = 30 bp. In addition, the tandem repeat sequences (>10 bp in length) were detected using the online Tandem Repeats Finder program (Benson, 1999), where the alignment match, mismatch, and indel parameters were set as two, seven, and seven, respectively. The minimum alignment score and maximum period size were 80 and 500, respectively.
Sequence Divergence Analysis
Alignments of the 14 Dipsacales complete cp genome sequences were visualized using mVISTA (Frazer et al., 2004). We extracted all the coding regions and intergenic spacers to examine regions of divergence within Adoxaceae and Caprifoliaceae for further phylogenetic analysis. The percentage of variable sites was calculated within each homologous region.
Adaptive Evolution Analysis
To analyze the non-synonymous (dN) and synonymous (dS) substitution rates, and their ratio (ω = dN/dS), the same unique functional protein coding sequences for each gene were extracted and aligned separately using Geneious R v9.0.5, and maximum likelihood phylogenetic trees were reconstructed based on the complete cp genomes using RAxML v 7.2.8 (Stamatakis, 2006). The values of dN, dS, and ω for each protein-coding exon were calculated using the site-specific model implemented in the codeml package (seqtype = 1, model = 0, NSsites = 1, 2, 7, 8) in PAML4.7 (Yang et al., 2005). This model allowed the ω ratio to vary among sites with a fixed ω ratio in all branches in order to test for site-specific evolution in the gene phylogeny (Yang and Nielsen, 2002). Two likelihood ratio tests were performed to check for the presence of positively selected sites: M1 (neutral) vs. M2 (positive selection), and M7 (beta) vs. M8 (beta and ω), which were compared using site-specific models (Yang and Nielsen, 2002; Yang et al., 2005). Model M1 distinguished two site classes with ω < 1 and ω = 1, and model M2 allowed for a third site class with ω > 1. Models M7 and M8 both described the distribution of ω as a beta function. The beta null model M7 restricted ω to (0, 1), and the alternative beta and ω model M8 allowed for positively selected extra site classes. Only candidate sites for positive selection with significant support from the posterior probability [p(ω > 1) ≥ 0.99]; Bayes Empirical Bayes approach) identified by M2 and M8 were considered further.
Phylogenetic Analysis
We used the 16 cp genomes to analyze the phylogenetic relationships among Dipsacales species, including six Adoxaceae species, eight Caprifoliaceae species, and two outgroups Guizotia abyssinica (NC_010601) and Helianthus annuus (NC_007977). The phylogenetic analysis were conducted based on the following five data partitions: (1) complete cp genomes; (2) protein-coding sequences; (3) LSC region; (4) IR region; and (5) SSC region. These regions were aligned using Mauve (Darling et al., 2004) and the best-fitting model was determined using MrModeltest 2.3 (Nylander, 2004). Maximum likelihood analysis was conducted using the program RAxML v 7.2.8 (Stamatakis, 2006) with 1,000 bootstrap replicates. Bayesian inference was performed using MrBayes v3.1.2 (Ronquist and Huelsenbeck, 2003) with the following settings: Markov chain Monte Carlo simulations for 1,000,000 generations with four incrementally heated chains, starting from random trees and sampling one out of every 1,000 generations. The first 25% of the trees were regarded as burn-ins (Meng et al., 2008; Ma et al., 2014).
Divergence Time Estimation
The divergence times between lineages were estimated using a Yule process speciation prior and the uncorrelated lognormal model of rate change with a relaxed clock in BEAST v1.8.0 (Drummond et al., 2012). We set the stem of Dipelta with: lognormal mean = 0, SD = 1.0, offset = 36 mya; and the Dipsacales node was constrained to: 79.9 mya, with a normal prior, mean = 79.9 mya, SD = 5. The GTRAGMMA nucleotide substitution model was selected using MrModeltest 2.3 (Nylander, 2004). A normal prior probability distribution was used to consider the uncertainty of prior knowledge. The analyses were run for 20,000,000 generations and the parameters were sampled every 5,000 generations. The effective sample size (>200) was determined using Tracer v 1.6 (Drummond et al., 2012) and the first 10% of the samples were discarded as burn-ins. Tree Annotator v.1.8.0 (Drummond et al., 2012) was used to summarize the set of post burn-in trees and their parameters in order to produce a maximum clade credibility chronogram showing the mean divergence time estimates with 95% highest posterior density (PHD) intervals. FigTree V1.3.1 (Drummond et al., 2012) was used for visualize the resulting divergence times.
Results
Features of cp Genomes of 14 Dipsacales Species
In this study, we determined the structural characteristics and gene contents of the complete cp genomes of six Adoxaceae species (V. betulifolium, MG738665; V. utile, S. williamsii, S. corydalifolia, A. moschatellina, and T. omeiensis) and eight Caprifoliaceae species (L. fragrantissima var. lancifolia, MG738669; L. stephanocarpa, MG738668; L. tragophylla, MG738667; T. pinnatifidum, MG738666; W. florida, MG738664; D. floribunda, MG738670; L. japonica, and K. amabilis) within the order Dipsacales. The cp genomes of the six Adoxaceae species ranged in size from 157,074 bp (S. corydalifolia) to 158,305 bp (S. williamsii), and the eight Caprifoliaceae cp genomes ranged from 154,732 bp (L. fragrantissima var. lancifolia) to 156,874 bp (W. florida) (Figure 1 and Table 1). All of the cp genomes had a typical quadripartite structure and they were similar to those of most land plants. The LSC length in the six Adoxaceae cp genomes ranged from 86,171 bp (S. corydalifolia) to 86,810 bp (S. williamsii), and the SSC and IR lengths ranged from 18,338 bp (V. betulifolium) to 18,993 bp (S. williamsii) and 26,112 bp (A. moschatellina) to 26,462 bp (V. betulifolium), respectively. All eight Caprifoliaceae cp genomes had an LSC region of 88,504 bp (L. fragrantissima var. lancifolia) to 89,964 bp (K. amabilis), an SSC region of 18,672 bp (L. japonica to 20,543 bp (T. pinnatifidum), and an IR of 22,673 bp (T. pinnatifidum) to 23,946 bp (K. amabilis) (Table 1). In addition, the six Adoxaceae cp genomes encoded 129 functional genes, with 84 protein-coding genes, 37 tRNA genes, and eight ribosomal RNA genes. The eight Caprifoliaceae cp genomes encoded 128 genes (Supplementary Table S8), with 82 protein-coding genes, 37 tRNA genes, eight ribosomal RNA genes and one pseudogene. The cp genomes of 14 Dipsacales species had the same average GC contents (mean = 38.23%). The GC contents of the SC regions in the six Adoxaceae (mean = 33.9%) and eight Caprifoliaceae (mean = 34.96%) species were lower than those of the IR regions (mean = 43.9%, 43.7%, respectively) (Table 1). The high GC percentage in the IR regions was possibly due to the presence of four rRNA genes in these regions. These results are similar to a previous report of a high GC percentage in the IR regions (Qian et al., 2013).
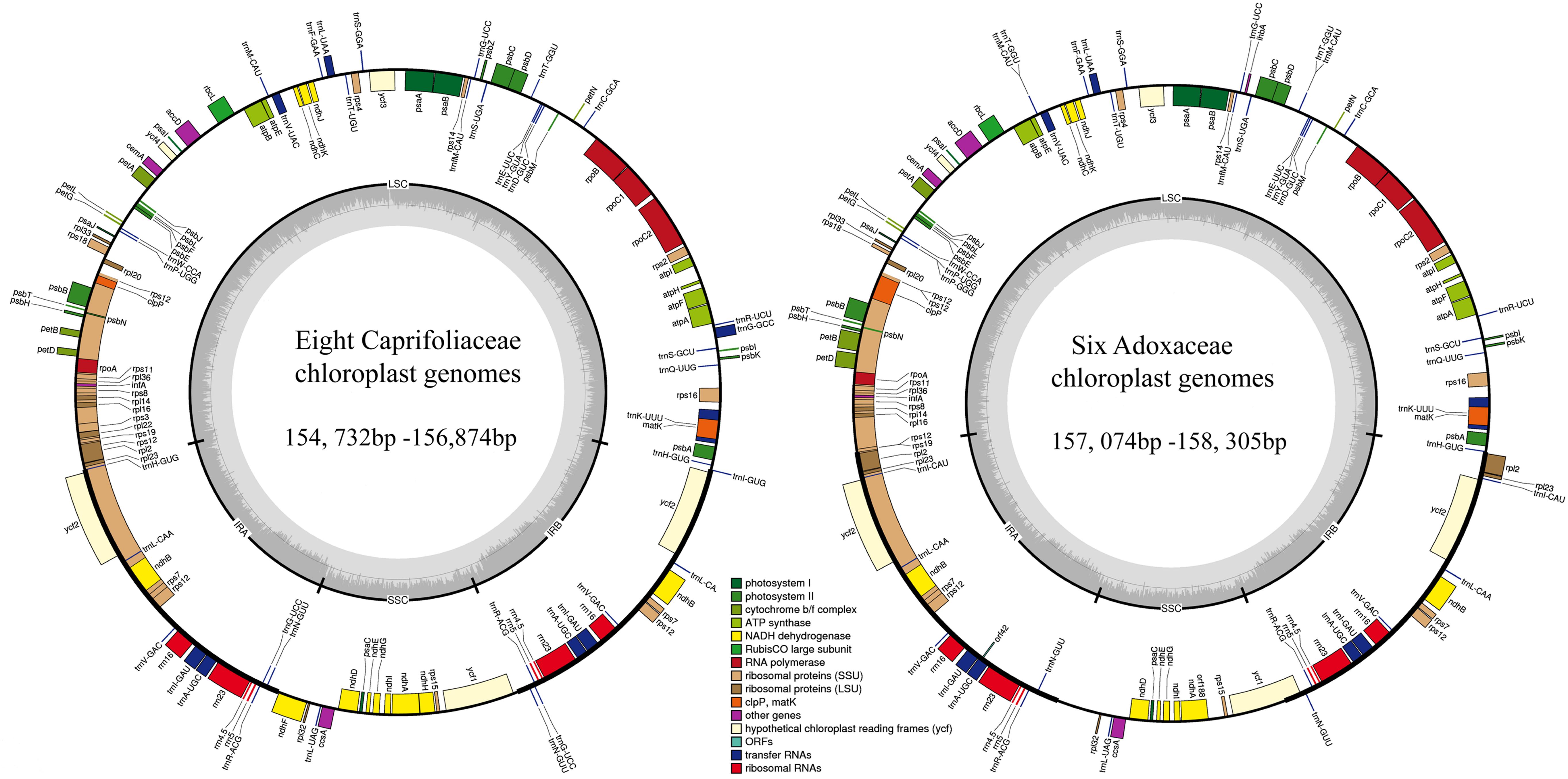
FIGURE 1. Chloroplast genome map for eight Caprifoliaceae species and six Adoxaceae species. Genes located outside the outer rim are transcribed in a counterclockwise direction, whereas genes inside the outer rim are transcribed in a clockwise direction. The colored bars indicate known different functional groups. The dashed gray area in the inner circle shows the percentage GC contents of the corresponding genes. LSC, SSC, and IR denote large single copy, small single copy, and inverted repeat, respectively.
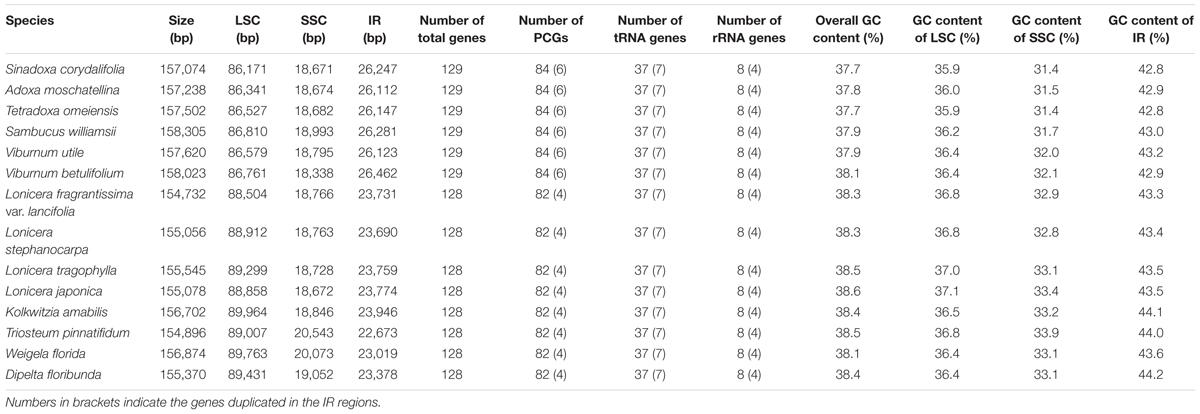
TABLE 1. Features of chloroplast genomes in six Adoxaceae and eight Caprifoliaceae species.
Repeat Element Analysis
We divided the repeats into three categories: tandem, dispersed, and palindromic (Supplementary Tables S3, S4). The number of tandem repeats (92) was higher than that of dispersed repeats (51) and palindromic repeats (35) in the six Adoxaceae species, and the number of dispersed repeats (384) was higher than that of tandem repeats (291) and palindromic repeats (224) in the eight Caprifoliaceae species (Figure 2). In addition, the total number of repeats in the six Adoxaceae species (178) was much lower than that in the eight Caprifoliaceae species (904). In the Adoxaceae family, the number of repeats was highest in A. moschatellina (42) and lowest in V. utile (19). In Caprifoliaceae, the numbers of all repeats, tandem repeats, and palindromic repeats were 155, 58, and 47 in W. florida, respectively, 141, 52, and 40 in K. amabilis, and 124, 38, and 37 in D. floribunda. The repeat units were mainly 21–50 bp regions in Caprifolieae and W. florida. In K. amabilis, most of the repeat units comprised 21–50 bp (48), followed by repeat units measuring 21–30 bp (20), and 0–20 bp (six). Within D. floribunda, most of the repeat units were 31–40 bp (43), followed by repeat units measuring > 80 bp (22), 21–30 bp (22), and 51–60 bp (three). Most of the repeats were distributed in intergenic or intron regions, and only a minority were located in gene regions in the order Dipsacales.

FIGURE 2. Maps obtained by repeat sequence analyses. (A) Histogram showing the number of repeats in the six Adoxaceae chloroplast genomes. (B) Histogram showing the number of repeats in the eight Caprifoliaceae chloroplast genomes. (C) Compositions of the repeats in six Adoxaceae species. (D) Compositions of the repeats in six Adoxaceae species. (E) Pie chart showing the numbers of the three repeat types in Adoxaceae. (F) Pie chart showing the numbers of the three repeat types in Caprifoliaceae.
Contraction and Expansion of IRs
We analyzed the IR/single copy (SC) region border positions and their adjacent genes in the six Adoxaceae and eight Caprifoliaceae cp genomes (Figure 3). The rpl2 and rpl32 genes were detected around the junctions of the IRb/SSC and IRa/LSC regions in six Adoxaceae species, and the ndhF and trnI-CAU genes appeared in these two regions in eight Caprifoliaceae species. In addition, the IR/SC boundary structure was similar in six Adoxaceae species, where the rps19 gene was located in the junction of the LSC/IRb region in S. williamsii, V. betulifolium, V. utile, and T. omeiensis, and the rps19 gene was located in the LSC region in S. corydalifolia and A. moschatellina. There was high variability in the IRb/SSC and SSC/IRa boundaries in the eight Caprifoliaceae species. The junction position between IRb and SSC was located in the ndhF gene, except it was in the IRb region in L. japonica. In addition, the trnN-GUU gene extended into the SSC region and appeared twice in T. pinnatifidum.
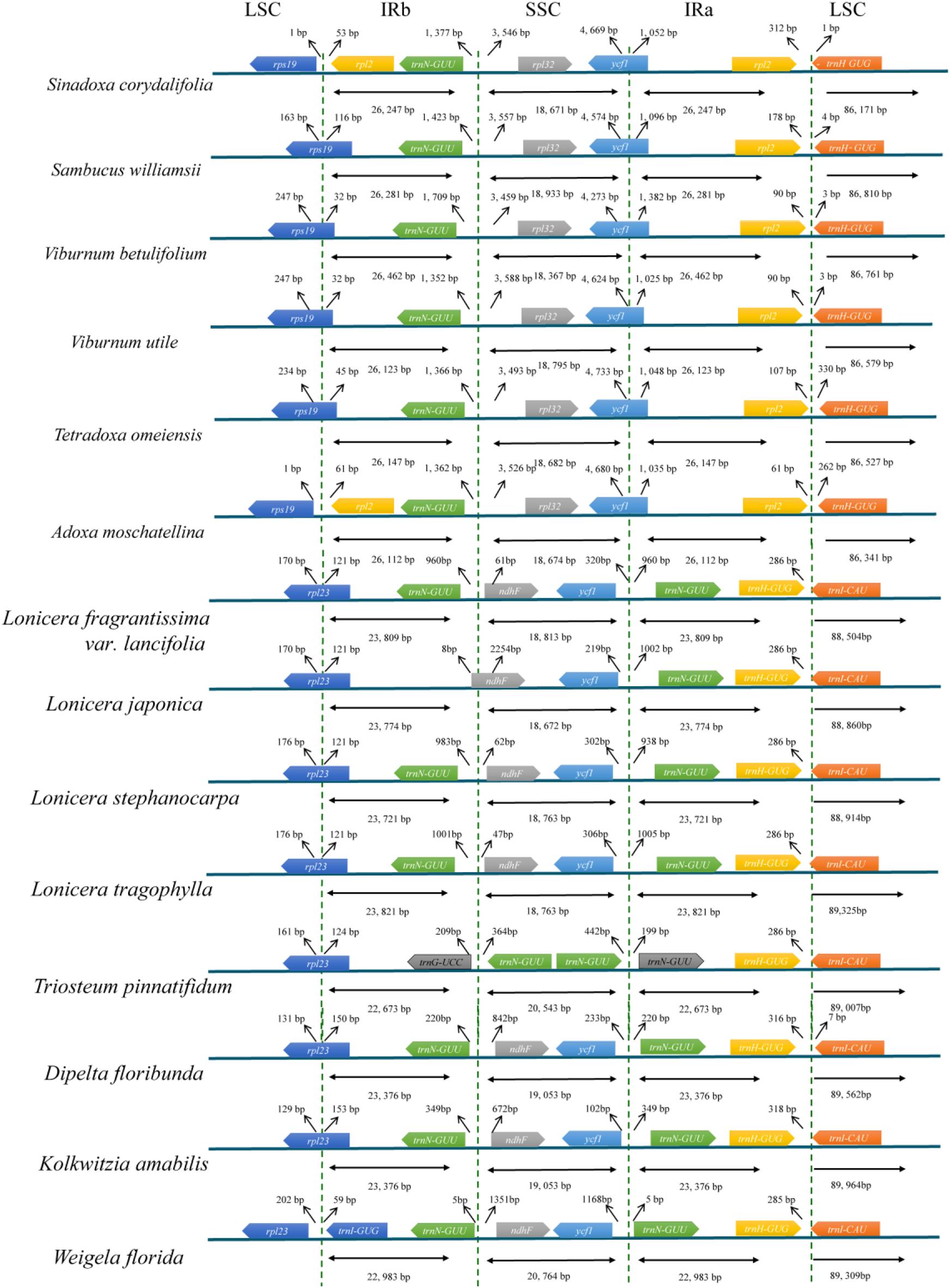
FIGURE 3. Comparison of the border positions of LSC, SSC, and IR regions in the chloroplast genomes in six Adoxaceae species and eight Caprifoliaceae species.
Sequence Divergence Analysis
The multiple complete cp genomes allowed us to estimate sequence variation. The divergence of sequences in the cp genomes of six Adoxaceae species and eight Caprifoliaceae species was plotted using the mVISTA program with annotations for S. corydalifolia as the reference (Supplementary Figure S1). In addition, the percentage variation was calculated in each of the 14 Dipsacales cp genomes (Figure 4 and Supplementary Table S5). The results showed that the mean percentage of variation was 12.8% in the six Adoxaceae cp genomes and 18.77% in the eight Caprifoliaceae species. In addition, the percentage variations in the coding regions (Adoxaceae, mean = 4.56%; Caprifoliaceae, mean = 7.11%) were smaller than those in the non-coding regions (Adoxaceae, mean = 17.95%; Caprifoliaceae, mean = 24.72%). Interestingly, the single copy regions (mean = 12.75%) had a higher average percentage of variation than the inverted repeats (mean = 4.14%) in six Adoxaceae species. However, the variation in single copy regions (mean = 17.61%) was lower than that in inverted repeat regions (mean = 21.25%) in Caprifoliaceae. In the coding regions, the five genes with the greatest variability (>10%) were rpl22, ndhI, ycf1, clpP, and rps16 in Adoxaceae, and the percentage variation in seven genes (rps16, clpP, rps3, ycf2, rps7, rps15, and ycf1) exceeded 15%. In non-coding regions, 11 regions (trnH-GUG-psbA, trnR-UCU-atpA, trnC-GCA-petN, ycf3-trnS-GGA, trnL-UAA-trnF-GAA, accD-psaI, ycf4-cemA, psbH-petB, rpl32-trnL-UAG, trnL-UAG-ccsA, and rps3-rpl22) in Adoxaceae and 16 regions (trnH-GUG-psbA, rpoC2-rpoC1, trnC-GCA-petN, ndhC-trnV-UAC, rbcL-accD, accD-psaI, psbJ-psbL, rps18 intron, infA-rps8, trnI-CAU-ycf2, ycf2-trnL-CAA, rrn5-trnR-ACG, trnR-ACG-trnN-GUU, trnN-GUU-rpl32, rps15-ycf1, and ycf1-trnN-GUU) in Caprifoliaceae had high levels of variation (percentage variation > 40%, 45%, respectively).
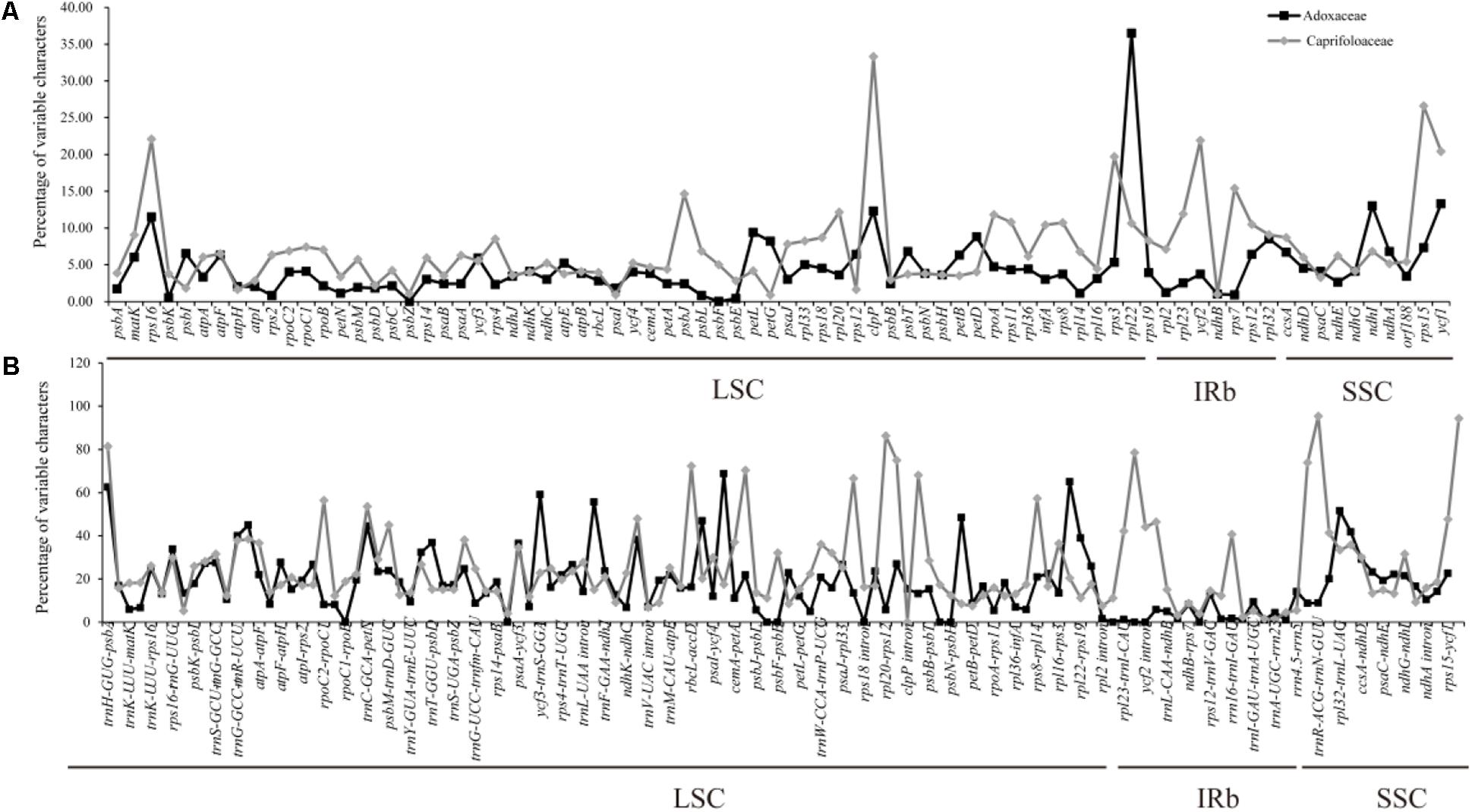
FIGURE 4. Percentages of variable characters in homologous regions among the chloroplast genomes of 14 Dipsacales species. (A) Coding region. (B) Non-coding region. The homologous regions are oriented according to their locations in the chloroplast genome.
Positive Selection Analysis
We identified 19 genes with positively selected sites in the order Dipsacales (Supplementary Tables S6, S7). Interestingly, these genes included three ATP subunit genes (atpA, atpB, and atpI), four ribosome small subunit genes (rps3, rps8, rps14, and rps15), four photosystem subunit genes (psaA, psaJ, psbC, and pabK), two ribosome large subunit genes (rpl22 and rpl32), and the clpP, infA, matK, and rbcL genes. In addition, according to the M2 and M8 models, the ycf2 gene harbored 15 and 18 sites under positive selection, respectively, with five and 16 in ycf1, three and five in rbcL, one and three in atpA, and two and two in clpP, and the other 14 genes each had only one active site. Both likelihood ratio tests (M1 vs. M2; M7 vs. M8) supported the presence of positively selected codon sites (p < 0.01) (Supplementary Table S9).
Phylogenetic Relationships
In this study, five data partitions from the cp genomes of 16 Dipsacales species were used to reconstruct the phylogenetic relationships. The topologies obtained were largely consistent with the different datasets (Figure 5 and Supplementary Figure S2) where two major clades were identified comprising a large clade and a small clade with 100% bootstrap values (except the LSC dataset which had a bootstrap value of 48%). A small clade in Adoxaceae included the genera Viburnum, Sambucus, Adoxa, Tetradoxa, and Tetradoxa. V. utile and V. betulifolium formed a clade with 100% bootstrap support. Viburnum and Sambucus were very closely related. In another clade, D. floribunda was placed as a sister to K. amabilis with high bootstrap values. L. fragrantissima var. lancifolia and L. stephanocarpa had close relationships with L. japonica and L. tragophylla in the genus Lonicera. In addition, W. florida was the earliest diverging lineage in the family Caprifolieae s. s.
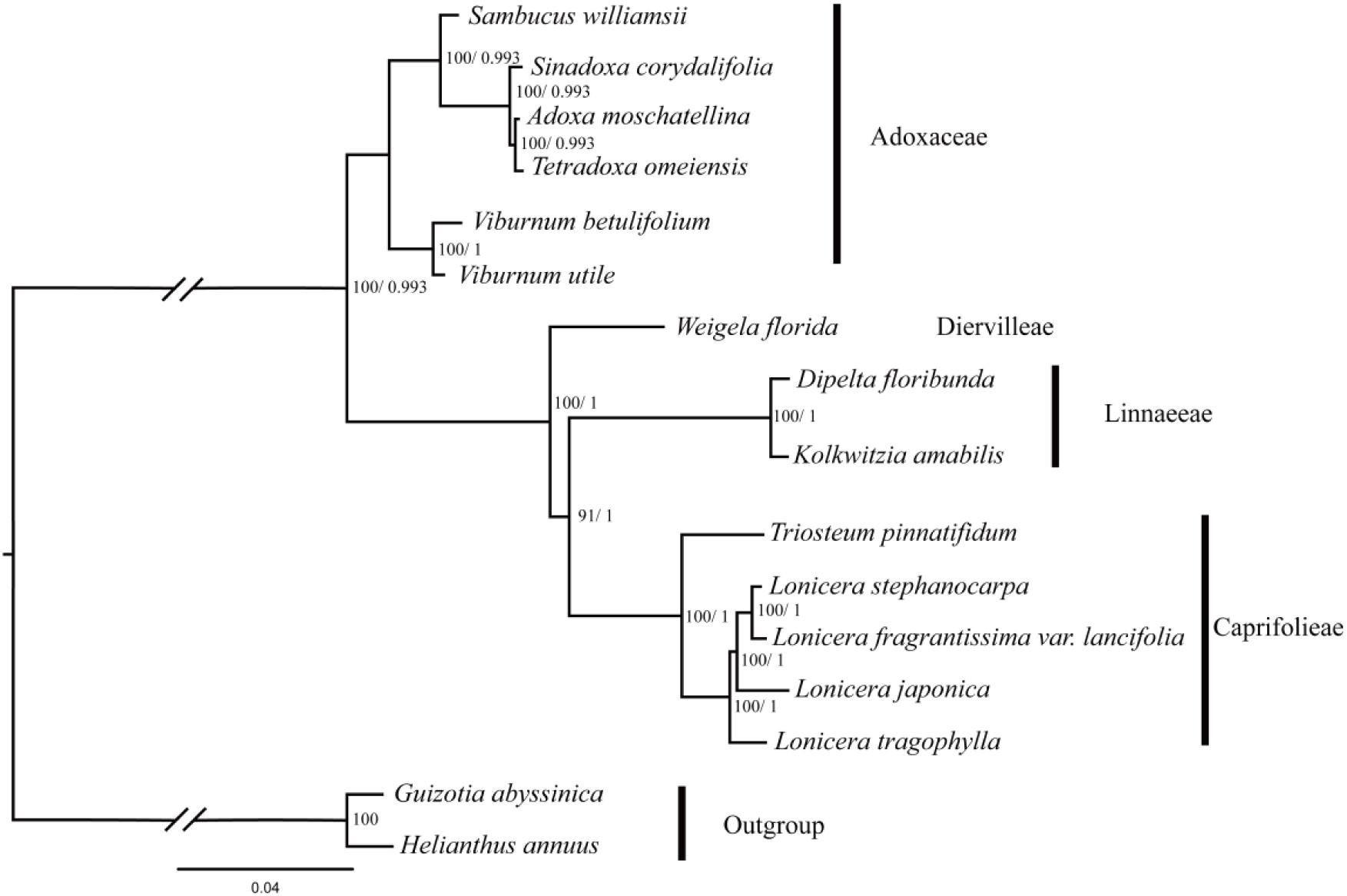
FIGURE 5. Phylogenetic tree obtained for 14 Dipsacales species based on the complete chloroplast genomes. The numbers to the left of the slashes on the braches show the bootstrap values obtained by maximum likelihood analyses, and those to the right show the posterior probabilities according to Bayesian inference.
Divergence Time Estimation
We computed the molecular divergence dates of the 14 Dipsacales species based on the protein-coding sequences (Figure 6). The results showed that the divergence time between Adoxaceae and Caprifoliaceae was about 81.14 mya (95% PHD = 58.28–94.57 mya, calibration point = 79.9 mya), thereby suggesting a late Cretaceous origin for these two families within the order Dipsacales. The split of the Viburnum lineage to yield Adoxa, Tetradoxa and Sinadoxa occurred in the Eocene, with an estimated mean age of 47.16 mya (95% PHD = 42.08–76.45 mya). We estimated that Sambucus split from its ancestor about 34.02 mya (95% PHD = 24.36–44.13 mya). The diversification of Sinadoxa, Tetradoxa, and Adoxa occurred in the Miocene about 15–16 mya. Within Caprifoliaceae, the diversification of Diervilleae occurred in the Palaeocene, with an estimated mean age of 61.71 mya. Linnaeeae diversified from Caprifolieae in the Eocene (95% PHD = 25.75–46.99 mya, mean age of 39.79 mya). The divergence time of L. tragophylla (mean age of 19.05 mya) was much earlier than that of L. japonica (mean age = 3.10 mya), and that of L. stephanocarpa and L. fragrantissima var. lancifolia (mean age = 1.10 mya).
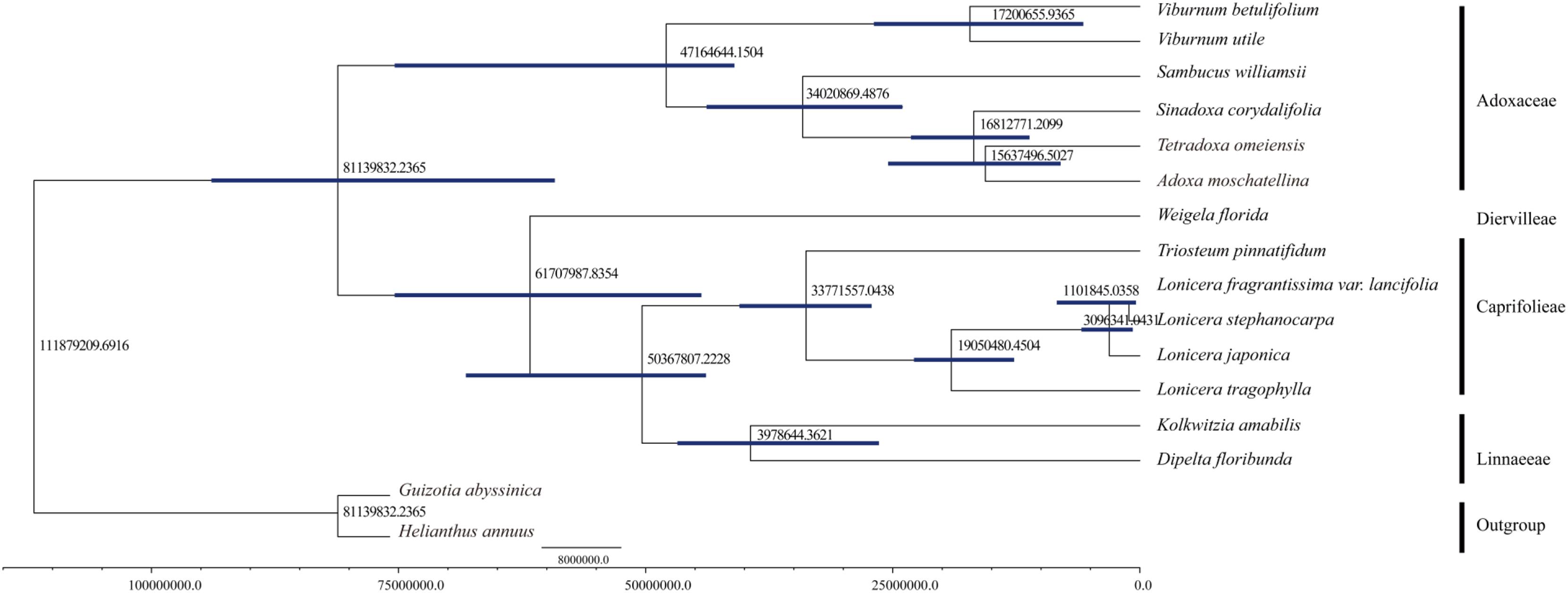
FIGURE 6. Molecular dating of 14 Dipsacales species based on the protein-coding sequences in chloroplast genomes.
Discussion
Sequence Differentiation
In terms of genome size, the six Adoxaceae (mean = 157,755 bp) cp genomes were much larger than those of the eight Caprifoliaceae species (mean = 155,675 bp). Meanwhile, the sizes of inverted repeat regions in six Adoxaceae species (mean = 26,229 bp) were much larger than those of the eight Caprifoliaceae species (mean = 23,496 bp). The difference of these cp genomes size may have been due to the expansion and contraction of the border positions between the IR regions and SC regions (Wang and Messing, 2011). In general, most angiosperms contain 74 protein-coding genes, and evidence of gene loss-and-gain events and rearrangements is present in some species, although the cp genomes of land plants are considered highly conserved (Millen et al., 2001; Kim et al., 2009). In this study, we found that the Adoxaceae and Caprifoliaceae cp genomes encoded 84 and 82 protein-coding genes, respectively. In addition, the members of these two families all had 37 tRNA genes and eight rRNA genes. We detected only single copies of rpl2 and rpl23 in Caprifoliaceae, located at the LSC region, but two copies of these genes in Adoxaceae were located in the IR regions. Meanwhile, rpl2 and rpl23 was located in LSC/IR border positions, which possibly due to the expansion and contraction of the border regions (Wang and Messing, 2011). In addition, the ndhF gene only appeared in Caprifoliaceae and it was located around the junction of the IRb/SSC regions. N. flexilis and P. stellaris lacked the ndh gene, which may be related to their adaptation to a submerged environment (Peredo et al., 2013) and non-photosynthetic lifestyle (Straub et al., 2012). Similarly, ndh gene losses have been frequent in Orchidaceae (Kim et al., 2015), where Chang et al. (2006) found that the ndhA, ndhF, and ndhH genes had transferred to the nuclear genome in Phalaenopsis aphrodite. In addition, the ndhF gene plays a role in IR/SSC junction stability (Kim et al., 2015). We also found that the accD gene encoding a subunit of heteromeric cetyl-CoA carboxylase was present as a pseudogene in eight Caprifoliaceae species. Previous studies have also shown that the accD gene has been lost from some plant species (Downie and Palmer, 1992; Cosner et al., 1997), and that it is present as a pseudogene in Jasminum nudiflorum, Trachelium caeruleum, and Eucommia ulmoides (Wang L. et al., 2016).
Divergence Hotspot Regions
In order to determine the divergence hotspots, we compared the whole cp genome sequences of the Adoxaceae and Caprifoliaceae species using mVISTA, where we computed the percentages of variable characters in coding and non-coding regions. Our results indicated that the proportions and distributions of the variable sites were strikingly different among the cp genomes. Overall, the proportion of variable sites was higher in the non-coding regions than the coding regions, which is generally consistent with most previous studies of the plastid genomes of angiosperms (Wolfe et al., 1987; Clegg et al., 1994; Perry and Wolfe, 2002; Jansen and Ruhlman, 2012; Huang et al., 2014). Interestingly, the SC regions (mean = 12.75%) had a higher average percentage of variation than the IRs (mean = 4.14%) in six Adoxaceae species. However, the variation in single copy regions (mean = 17.61%) was lower than that in inverted repeat regions (mean = 21.25%) in Caprifoliaceae, possibly due to the instability of the boundary of the IR region. Considering the proportion and number of variable sites, we propose 17 (rpl22, ndhI, ycf1, trnH-GUG-psbA, trnR-UCU-atpA, trnC-GCA-petN, ycf3-trnS-GGA, trnL-UAA-trnF-GAA, accD-psaI, ycf4-cemA, psbH-petB, rpl32-trnL-UAG and trnL-UAG-ccsA) and 23 (rps16, clpP, rps3, ycf2, rps7, rps15, ycf1, trnH-GUG-psbA, rpoC2-rpoC1, trnC-GCA-petN, ndhC-trnV-UAC, rbcL-accD, accD-psaI, psbJ-psbL, rps18 intron, infA-rps8, trnI-CAU-ycf2, ycf2-trnL-CAA, rrn5-trnR-ACG, trnR-ACG-trnN-GUU, trnN-GUU-rpl32, rps15-ycf1, and ycf1-trnN-GUU) of the most variable hotspot regions as candidate DNA barcodes for Adoxaceae and Caprifoliaceae species for future studies. These regions may be very useful for assessing the phylogenetic relationships and interspecific divergence in Dipsacales species.
Adaptive Selection
Synonymous and non-synonymous nucleotide substitution patterns are very important markers for gene evolution studies. In most genes, synonymous nucleotide substitutions have occurred more frequently than non-synonymous ones (Ogawa et al., 1999). Thus, a ratio of dN/dS < 1 indicates purifying selection, dN/dS > 1 denotes probable positive selection, and dN/dS values close to one indicate neutral evolution. Our analysis identified 19 genes with positive selection sites. These genes included three ATP subunit genes (atpA, atpB, and atpI), four ribosome small subunit genes (rps3, rps7, rps14, and rps15), four photosystem subunit genes (psaA, psaJ, psbC, and pabK), two ribosome large subunit genes (rpl22 and rpl32), and the clpP, infA, matK, rbcL, ycf1, and ycf2 genes. ATP synthase is essential during photosynthesis and it is usually the product of two genetic systems in plants (Westhoff et al., 1985). Six ATP subunit genes (atpA, atpB, atpE, atpF, atpH, and atpI) are encoded and synthesized in the chloroplasts (Westhoff et al., 1985), and three genes exhibited site-specific selection in this study. In addition, 21 genes were identified that encode ribosome subunits and four of these genes were under positive selection. Two photosystem I subunit genes (psaJ and psaA) and two photosystem II subunit genes (psbC and psbK) were under positive selection. Maturase enzymes catalyze non-autocatalytic intron removal from premature RNAs, such as RNA transcripts for the trnK, trnA, trnI, rps12, rpl2, and atpF genes (Vogel et al., 1999). The clpP gene is a member of a gene family within the cp genome that encodes clpP proteases. In general, the clpP gene is essential for plant cells and the main function of its product is the degradation of polypeptides (Clarke, 1999; Shikanai et al., 2001; Kuroda and Maliga, 2003). We identified positively selected sites in the clpP gene in our study, which might have played key roles in the adaptive evolution of Dipsacales species. In addition, the rbcL gene plays an important role as a modulator of photosynthetic electron transport and it is essential for photosynthesis because it encodes the large subunit of RuBisCO (Allahverdiyeva et al., 2005). A previous study showed that rbcL is often under positive selection in land plants (Kapralov and Filatov, 2007). In particular, the rbcL gene evolved under strong positive selection after the C3–C4 photosynthetic transition (Piot et al., 2017). Similarly, selection analysis in Haberlea rhodopensis showed that 17 genes were under site-specific selection, and the rbcL gene harbored 13 sites under positive selection (Ivanova et al., 2017). We also found that the ycf2 gene had 18 sites under positive selection, with 16 in ycf1, five in rbcL, three in atpA, and two in clpP, but only one site in each of the other 14 genes. These positively selected genes may have played key roles in the adaptation of species in the order Dipsacales to various environments.
Phylogenetic Relationships
The phylogenetic trees based on five different datasets had similar topologies. In the order Dipsacales, two major lineages were clearly defined: Adoxaceae and Caprifoliaceae. Viburnum and Sambucus are the most closely related. In addition, the traditional Caprifolieae, Diervilleae, and Linnaeeae form a larger branch comprising the new family Caprifolieae. These results are similar to previous analyses based on morphological characters and various molecular data sets (Donoghue et al., 2001, 2003; Zhang et al., 2002; Winkworth et al., 2008). Zhang et al. (2002) suggested that Caprifoliaceae s. l. (excluding Sambucus and Viburnum) is a polyphyletic clade based on the sequence variations in trnL and trnF markers. In addition, other studies found that Morinaceae, Valerianaceae, and Dipsacaceae were derived from the polyphyletic Caprifoliaceae s. l., which is divided into three separate lineages, i.e., Caprifoliaceae s. str., Diervillaceae, and Linnaeaceae (Backlund and Pyck, 1998; Pyck et al., 1999). Our results showed that W. florida belongs to Diervilleae, D. floribunda and K. amabilis belong to Linnaeeae, and T. pinnatifidum, L. tragophylla, L. stephanocarpa, L. japonica, and L. fragrantissima var. lancifolia cluster in the Caprifoliaceae clade. Four Lonicera and one Triosteum species form a single clade in Caprifoliaceae with high bootstrap support, and D. floribunda and K. amabilis have a close relationship. These results agree well with previous evidence based on trnL-F, ndhF, and mitochondrial markers (Donoghue et al., 2001; Zhang et al., 2002; Winkworth et al., 2008). The similar topologies obtained based on various analyses, including these obtained in this study, indicate the clear resolution of the phylogenetic relationships in Dipsacales. In future research, it will be necessary to collect more species in order to verify the species relationships and interspecific divergence in Dipsacales.
Molecular Dating
We estimated the divergence times of 14 Dipsacales species based on the protein-coding sequences in the complete cp genomes. The results showed that the diversification into Adoxaceae and Caprifoliaceae occurred about 81.14 mya at the Cretaceous/Tertiary boundary. These results are similar to the molecular divergence dates obtained in previous studies. For example, Bell and Donoghue (2005) used multiple methods to estimate the divergence dates of the major Dipsacales lineages, and suggested that the diversification of Adoxaceae and Caprifoliaceae mainly occurred in the Tertiary, and the major lineages mainly originated during the Eocene. In addition, in the Adoxaceae family, the split into Viburnum and Sambucus occurred in the Eocene. Within Caprifoliaceae, the splits to yield Diervilleae, Caprifolieae, and Linnaeeae also occurred in the Eocene period. Our results are similar to those obtained in previous studies. Smith et al. (2010) estimated the divergence date for Lonicera and suggested that an ancestor of this genus had a widespread distribution across the Northern Hemisphere about 7–17 mya. We found that the spilt to yield L. tragophylla (mean age = 19 mya) occurred earlier than that for other Lonicera species. To the best of our knowledge, the present study is the first to use all of the protein-coding sequences in cp genomes to estimate the divergence dates of Dipsacales, although the results could be improved by larger phylogenetic analyses.
Author Contributions
Z-HL conceived and designed the study. W-BF and JY performed the experiments. Z-HL, W-BF, YW, and KS contributed materials and analysis tools. W-BF and Z-HL wrote the paper. Z-HL and W-BF revised the paper. All authors approved the final manuscript.
Funding
This study was co-supported by the National Natural Science Foundation of China (31470400), the Shaanxi Provincial Key Laboratory Project of Department of Education (Grant No. 17JS135), and the Open Foundation of Key Laboratory of Resource Biology and Biotechnology in Western China (Ministry of Education).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00689/full#supplementary-material
FIGURE S1 | Sequence alignment of chloroplast genomes from 14 Dipsacales species. Sequences of chloroplast genomes were aligned and compared using the mVISTA program. The vertical scale indicates the percentage identity, ranging from 50% to 100%.
FIGURE S2 | Phylogenetic relationships among the 16 species inferred by maximum likelihood and Bayesian analyses based on five datasets. (A) Complete chloroplast genome; (B) Coding region; (C) LSC region; (D) SSC region; and (E) IR region. The numbers to the right of the slashes on the braches show the bootstrap values obtained by maximum likelihood analyses, and those to the left show the posterior probabilities according to Bayesian inference.
TABLE S1 | Sampling and assembly information for the 16 species.
TABLE S2 | Primers for low coverage regions in seven Dipsacales species.
TABLE S3 | List of tandem repeats in the chloroplast genomes of 14 Dipsacales species.
TABLE S4 | List of dispersed repeats and palindromic repeats in the chloroplast genomes of 14 Dipsacales species.
TABLE S5 | Percentages of variable characters in coding and non-coding regions within 14 Dipsacales species.
TABLE S6 | Maximum likelihood parameter estimates for 75 genes in Dipsacales.
TABLE S7 | Maximum likelihood parameter estimates for 19 genes with positive selection sites.
TABLE S8 | List of genes present in the chloroplast genomes of six Adoxaceae species and eight Caprifoliaceae species: (a) two gene copies in Caprifoliaceae; (b) two gene copies in Adoxaceae; (c) pseudogene in the chloroplast genome of Caprifoliaceae; and (d) only present in Caprifoliaceae.
TABLE S9 | Likelihood ratio test (LRT) of the variable ω ratio under different models.
References
Allahverdiyeva, Y., Mamedov, F., Mäenpää, P., Vass, I., and Aro, E. M. (2005). Modulation of photosynthetic electron transport in the absence of terminal electron acceptors: characterization of the rbcL deletion mutant of tobacco. Biochim. Biophys. Acta 1709, 69–83. doi: 10.1016/j.bbabio.2005.06.004
Backlund, A., and Pyck, N. (1998). Diervillaceae and Linnaeaceae, two new families of caprifolioids. Taxon 47, 657–661. doi: 10.2307/1223583
Bai, G. Q., Zhou, T., Zhao, J. X., Li, W. M., Han, G. J., and Li, S. F. (2017). The complete chloroplast genome of kolkwitzia amabilis (caprifoliaceae), an endangered horticultural plant in China. Mitochondrial DNA 28, 296–297. doi: 10.3109/19401736.2015.1118087
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., and Dvorkin, M. (2012). Spades: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bell, C. D., and Donoghue, M. J. (2005). Dating the Dipsacales: comparing models, genes, and evolutionary implications. Am. J. Bot. 92, 284–296. doi: 10.3732/ajb.92.2.284
Bell, C. D., Edwards, E. J., Kim, S. T., and Donoghue, M. J. (2001). Dipsacales phylogeny based on chloroplast DNA sequences. Harvard Papers Bot. 6, 481–499.
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bremer, B., Bremer, K., Heidari, N., Erixon, P., and Olmstead, R. G. (2002). Phylogenetics of asterids based on 3 coding and 3 noncoding chloroplast DNA markers and the utility of non-coding DNA at higher taxonomic levels. Mol. Phylogenet. Evol. 24, 274–301. doi: 10.1016/S1055-7903(02)00240-3
Cavalier-Smith, T. (2002). Chloroplast evolution: secondary symbiogenesis and multiple losses. Curr. Biol. 12, R62–R64. doi: 10.1016/S0960-9822(01)00675-3
Chang, C. C., Lin, H. C., Lin, I. P., Chow, T. Y., and Chen, H. H. (2006). The chloroplast genome of Phalaenopsis aphrodite (Orchidaceae): comparative analysis of evolutionary rate with that of grasses and its phylogenetic implications. Mol. Biol. Evol. 23, 279–291. doi: 10.1093/molbev/msj029
Chevreux, B., Pfisterer, T., Drescher, B., Driesel, A. J., and Müller, W. E. (2004). Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 14, 1147–1159. doi: 10.1101/gr.1917404
Clarke, A. K. (1999). ATP-dependent Clp proteases in photosynthetic organisms—a cut above the rest. Ann. Bot. 83, 593–599. doi: 10.1006/anbo.1999.0878
Clegg, M. T., Gaut, B. S., Learn, G. H., and Morton, B. R. (1994). Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. U.S.A. 91, 6795–6801. doi: 10.1073/pnas.91.15.6795
Cosner, M. E., Jansen, R. K., Palmer, J. D., and Downie, S. R. (1997). The highly rearranged chloroplast genome of Trachelium caeruleum (Campanulaceae): multiple inversions, inverted repeat expansion and contraction, transposition, insertions/deletions, and several repeat families. Curr. Genet. 31, 419–429. doi: 10.1007/s002940050225
Cronquist, A. (1979). The evolution and classification of flowering plants. Brittonia 31, 293–293. doi: 10.1007/BF02806171
Daniell, H., Lin, C. S., Yu, M., and Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Donoghue, M. J., Bell, C. D., and Winkworth, R. C. (2003). The evolution of reproductive characters in Dipsacales. Int. J. Plant Sci. 164, S453–S464. doi: 10.1086/376874
Donoghue, M. J., Eriksson, T., Reeves, P. A., and Olmstead, R. G. (2001). Phylogeny and phylogenetic taxonomy of Dipsacales, with special reference to Sinadoxa and Tetradoxa (Adoxaceae). Harvard Papers Bot. 6, 459–479.
Donoghue, M. J., Olmstead, R. G., Smith, J. F., and Palmer, J. D. (1992). Phylogenetic relationships of Dipsacales based on rbcL sequences. Ann. Mo. Bot. Gard. 79, 333–345. doi: 10.2307/2399772
Downie, S. R., and Palmer, J. D. (1992). “Use of chloroplast DNA rearrangements in reconstructing plant phylogeny,” in Molecular Systematics of Plants, eds P. S. Soltis, D. E. Soltis, and J. J. Doyle (New York, NY: Chapman & Hall), 14–35.
Doyle, J. J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Drummond, A. J., Suchard, M. A., Xie, D., and Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Eriksson, T., and Donoghue, M. J. (1997). Phylogenetic relationships of Sambucus and Adoxa (Adoxoideae, Adoxaceae) based on nuclear ribosomal ITS sequences and preliminary morphological data. Syst. Bot. 22, 555–573. doi: 10.2307/2419828
Erixon, P., and Oxelman, B. (2008). Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the chloroplast, clpp1, gene. PLoS One 3:e1386. doi: 10.1371/journal.pone.0001386
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). mVISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Graham, S. W., Zgurski, J. M., McPherson, M. A., Cherniawsky, D. M., and Saarela, J. M. (2006). Robust inference of monocot deep phylogeny using an expanded multigene plastid data set. Aliso 22, 3–20. doi: 10.5642/aliso.20062201.02
Hahn, C., Bachmann, L., and Chevreux, B. (2013). Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129. doi: 10.1093/nar/gkt371
He, L., Qian, J., Li, X., Sun, Z., Xu, X., and Chen, S. (2017). Complete chloroplast genome of medicinal plant Lonicera japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 22:E249. doi: 10.3390/molecules22020249
Huang, H., Shi, C., Liu, Y., Mao, S. Y., and Gao, L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. doi: 10.1186/1471-2148-14-151
Huotari, T., and Korpelainen, H. (2012). Complete chloroplast genome sequence of Elodea canadensis and comparative analyses with other monocot plastid genomes. Gene 508, 96–105. doi: 10.1016/j.gene.2012.07.020
Ivanova, Z., Sablok, G., Daskalova, E., Zahmanova, G., and Apostolova, E. (2017). Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Front. Plant Sci. 8:204. doi: 10.3389/fpls.2017.00204
Jansen, R. K., and Ruhlman, T. A. (2012). Plastid Genomes of Seed Plants, Genomics of Chloroplasts, and Mitochondria. Dordrecht: Springer, 103–126. doi: 10.1007/978-94-007-2920-9_5
Kapralov, M. V., and Filatov, D. A. (2007). Widespread positive selection in the photosynthetic Rubisco enzyme. BMC Evol. Biol. 7:73. doi: 10.1186/1471-2148-7-73
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, H. T., Kim, J. S., Moore, M. J., Neubig, K. M., and Williams, N. H. (2015). Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across orchids and associated instability of the inverted repeat/small single-copy region boundaries. PLoS One 10:e0142215. doi: 10.1371/journal.pone.0142215
Kim, Y. K., Park, C. W., and Kim, K. J. (2009). Complete chloroplast DNA sequence from a Korean endemic genus, Megaleranthis saniculifolia, and its evolutionary implications. Mol. Cells 27, 365–381. doi: 10.1007/s10059-009-0047-6
Korpelainen, H. (2004). The evolutionary processes of mitochondrial and chloroplast genomes differ from those of nuclear genomes. Naturwissenschaften 91, 505–518. doi: 10.1007/s00114-004-0571-3
Kuroda, H., and Maliga, P. (2003). The plastid clpP1 protease gene is essential for plant development. Nature 425, 86–89. doi: 10.1007/s00114-004-0571-3
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). Organellar Genome DRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Ma, P., Zhang, Y., Zeng, C., Guo, Z., and Li, D. (2014). Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe arundinarieae (Poaceae). Syst. Biol. 63, 933–950. doi: 10.1093/sysbio/syu054
Meng, Y., Wen, J., Nie, Z., Sun, H., and Yang, Y. (2008). Phylogeny and biogeographic diversification of Maianthemum (Ruscaceae: Polygonatae). Mol. Phylogenet. Evol. 49, 424–434. doi: 10.1016/j.ympev.2008.07.017
Millen, R. S., Olmstead, R. G., Adams, K. L., Palmer, J. D., Lao, N. T., Heggie, L., et al. (2001). Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13, 645–658. doi: 10.1105/tpc.13.3.645
Moore, M. J., Bell, C. D., Soltis, P. S., and Soltis, D. E. (2007). Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. U.S.A. 104, 19363–19368. doi: 10.1073/pnas.0708072104
Nie, X., Lv, S., Zhang, Y., Du, X., and Wang, L. (2012). Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS One 7:e36869. doi: 10.1371/journal.pone.0036869
Nylander, J. (2004). MrModeltest V2. Program Distributed by the Author. Uppsala: Uppsala University.
Ogawa, T., Ishii, C., Kagawa, D., Muramoto, K., and Kamiya, H. (1999). Accelerated evolution in the protein-coding region of galectin cDNAs, congerin I and congerin II, from skin mucus of conger eel (Conger myriaster). Biosci. Biotechnol. Biochem. 63, 1203–1208. doi: 10.1271/bbb.63.1203
Olmstead, R. G., Bremer, B., Scott, K. M., and Palmer, J. D. (1993). A parsimony analysis of the Asteridae sensu lato based on rbcL sequences. Ann. Mo. Bot. Gard. 80, 700–722. doi: 10.2307/2399855
Olmstead, R. G., Kim, K.-J., Jansen, R. K., and Wagstaff, S. J. (2000). The Phylogeny of the Asteridae sensu lato based on chloroplast ndhF gene sequences. Mol. Phylogenet. Evol. 16, 96–112. doi: 10.1016/S1055-7903(02)00303-2
Palmer, J. D. (1985). Molecular Evolutionary Genetics, ed. R. J. MacIntyre (New York, NY: Plenum), 131–240. doi: 10.1007/978-1-4684-4988-4_3
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e0030619. doi: 10.1371/journal.pone.0030619
Peredo, E. L., King, U. M., and Les, D. H. (2013). The plastid genome of Najas flexilis: adaptation to submersed environments is accompanied by the complete loss of the NDH complex in an aquatic angiosperm. PLoS One 8:e0068591. doi: 10.1371/journal.pone.0068591
Perry, A. S., and Wolfe, K. H. (2002). Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 55, 501–508. doi: 10.1007/s00239-002-2333-y
Piot, A., Hackel, J., Christin, P. A., and Besnard, G. (2017). One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 247, 255–266. doi: 10.1007/s00425-017-2781-x
Pyck, N., Roels, P., and Smets, E. (1999). Tribal relationships in Caprifoliaceae: evidence from a cladistic analysis using ndhF sequences. Systemat. Geogr. Plants 62, 145–159. doi: 10.2307/3668539
Qian, J., Song, J., Gao, H., Zhu, Y., and Xu, J. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS One 8:e57607. doi: 10.1371/journal.pone.0057607
Ravi, V., Khurana, J. P., Tyagi, A. K., and Khurana, P. (2008). An update on chloroplast genomes. Plant Syst. Evol. 270, 101–122. doi: 10.1007/s00606-007-0608-0
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Shikanai, T., Shimizu, K., Ueda, K., Nishimura, Y., Kuroiwa, T., and Hashimoto, T. (2001). The chloroplast clpP gene, encoding a proteolytic subunit of ATP-dependent protease, is indispensable for chloroplast development in tobacco. Plant Cell Physiol. 42, 264–273. doi: 10.1093/pcp/pce031
Smith, S. A., Beaulieu, J. M., and Donoghue, M. J. (2010). An uncorrelated relaxed-clock analysis suggests an earlier origin for flowering plants. Proc. Natl. Acad. Sci. U.S.A. 107, 5897–5902. doi: 10.1073/pnas.1001225107
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Straub, S. C. K., Parks, M., Weitemier, K., Fishbein, M., Cronn, R. C., and Liston, A. (2012). Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am. J. Bot. 99, 349–364. doi: 10.3732/ajb.1100335
Thorne, R. F. (1992). Classification and geography of flowering plants. Bot. Rev. 58, 225–327. doi: 10.1007/BF02858611
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res. 40:e115. doi: 10.1093/nar/gks596
Vogel, J., Börner, T., and Hess, W. R. (1999). Comparative analysis of splicing of the complete set of chloroplast group II introns in three higher plant mutants. Nucleic Acids Res. 27, 3866–3874. doi: 10.1093/nar/27.19.3866
Wang, L., Wuyun, T. N., Du, H., Wang, D., and Cao, D. (2016). Complete chloroplast genome sequences of Eucommia ulmoides: genome structure and evolution. Tree Genet. Genomes 12:12. doi: 10.1007/s11295-016-0970-6
Wang, W., and Messing, J. (2011). High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS One 6:e24670. doi: 10.1371/journal.pone.0024670
Wang, Y., Guo, X., Hao, G., Wang, T., and Wang, K. (2016). The complete chloroplast genome of Sinadoxa corydalifolia (Adoxaceae). Conserv. Genet. Res. 8, 303–305. doi: 10.1007/s12686-016-0559-2
Westhoff, P., Alt, J., Nelson, N., and Herrmann, R. G. (1985). Genes and transcripts for the ATP synthase CF0 subunits I and II from spinach thylakoid membranes. Mol. Gen. Genet. 199, 290–299. doi: 10.1007/BF00330271
Wicke, S., Schneeweiss, G. M., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wikström, N., Savolainen, V., and Chase, M. W. (2001). Evolution of the angiosperms: calibrating the family tree. Proc. Biol. Sci. 268, 2211–2220. doi: 10.1098/rspb.2001.1782
Winkworth, R. C., Bell, C. D., and Donoghue, M. J. (2008). Mitochondrial sequence data and Dipsacales phylogeny: mixed models, partitioned Bayesian analyses, and model selection. Mol. Phylogenet. Evol. 46, 830–843. doi: 10.1016/j.ympev.2007.11.021
Wolfe, K. H., Li, W. H., and Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. U.S.A. 84, 9054–9058. doi: 10.1073/pnas.84.24.9054
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Yang, Z. H., and Nielsen, R. (2002). Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 19, 908–917. doi: 10.1093/oxfordjournals.molbev.a004148
Yang, Z. H., Wong, W. S., and Nielsen, R. (2005). Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. doi: 10.1093/molbev/msi097
Zhang, W. H., Chen, Z. D., Li, J. H., Chen, H. B., and Tang, Y. C. (2003). Phylogeny of the Dipsacales s. l. based on chloroplast trnL-F and ndhF sequences. Mol. Phylogenet. Evol. 26, 176–189. doi: 10.1016/S1055-7903(02)00303-2
Zhang, Y., Li, L., Yan, T. L., and Liu, Q. (2014). Complete chloroplast genome sequences of Praxelis (Eupatorium catarium Veldkamp), an important invasive species. Gene 549, 58–69. doi: 10.1016/j.gene.2014.07.041
Keywords: Adoxaceae, Caprifoliaceae, chloroplast genome, Dipsacales, phylogenetic relationship, positive selection
Citation: Fan W-B, Wu Y, Yang J, Shahzad K and Li Z-H (2018) Comparative Chloroplast Genomics of Dipsacales Species: Insights Into Sequence Variation, Adaptive Evolution, and Phylogenetic Relationships. Front. Plant Sci. 9:689. doi: 10.3389/fpls.2018.00689
Received: 03 January 2018; Accepted: 04 May 2018;
Published: 23 May 2018.
Edited by:
Genlou Sun, Saint Mary’s University, CanadaReviewed by:
Marcelo R. S. Briones, Federal University of São Paulo, BrazilWei Lun Ng, Sun Yat-sen University, China
Copyright © 2018 Fan, Wu, Yang, Shahzad and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhong-Hu Li, bGl6aG9uZ2h1QG53dS5lZHUuY24=