 Aniket Sengupta
Aniket Sengupta Lena C. Hileman
Lena C. Hileman- The Hileman Lab, Department of Ecology and Evolutionary Biology, The University of Kansas, Lawrence, KS, United States
A common feature in developmental networks is the autoregulation of transcription factors which, in turn, positively or negatively regulate additional genes critical for developmental patterning. When a transcription factor regulates its own expression by binding to cis-regulatory sites in its gene, the regulation is direct transcriptional autoregulation (DTA). Indirect transcriptional autoregulation (ITA) involves regulation by proteins expressed downstream of the target transcription factor. We review evidence for a hypothesized role of DTA in the evolution and development of novel flowering plant phenotypes. We additionally provide new bioinformatic and experimental analyses that support a role for transcriptional autoregulation in the evolution of flower symmetry. We find that 5′ upstream non-coding regions are significantly enriched for predicted autoregulatory sites in Lamiales CYCLOIDEA genes—an upstream regulator of flower monosymmetry. This suggests a possible correlation between autoregulation of CYCLOIDEA and the origin of monosymmetric flowers near the base of Lamiales, a pattern that may be correlated with independently derived monosymmetry across eudicot lineages. We find additional evidence for transcriptional autoregulation in the flower symmetry program, and report that Antirrhinum DRIF2 may undergo ITA. In light of existing data and new data presented here, we hypothesize how cis-acting autoregulatory sites originate, and find evidence that such sites (and DTA) can arise subsequent to the evolution of a novel phenotype.
Introduction
A common feature in developmental networks is the autoregulation of transcription factors which, in turn, positively or negatively regulate additional genes critical for developmental patterning. A trans-acting protein is considered transcriptionally autoregulated when the protein itself, or downstream factors, modulate its expression. Transcriptional autoregulation can be either direct, or indirect. In direct transcriptional autoregulation (DTA), a protein binds to cis-regulatory sites in its gene and modulates expression. Indirect transcriptional autoregulation (ITA) involves regulation by proteins expressed downstream of the target transcription factor (Figure 1). Both DTA and ITA have the potential to enter run-away positive feedback processes. Expression of such genes is likely reduced or stabilized by additional regulatory factors. Transcription factor autoregulation is widespread. For example, at least 40% of transcription factors in Escherichia coli are autoregulated (Rosenfeld et al., 2002), and similar direct and indirect autoregulation has been reported across the tree of life—in viruses, prokaryotes, and eukaryotes (for example, Hochschild, 2002; Martínez-Antonio and Collado-Vides, 2003; Holloway et al., 2011; Tao et al., 2012; Gallo-Ebert et al., 2013; and reviewed in Bateman, 1998; Crews and Pearson, 2009), including those with complex development (for example, Cripps et al., 2004; Holloway et al., 2011; Ye et al., 2016). DTA has been demonstrated in processes as diverse, and crucial as the origin of certain cancers (Pasqualucci et al., 2003), and the onset of flowering (Tao et al., 2012).
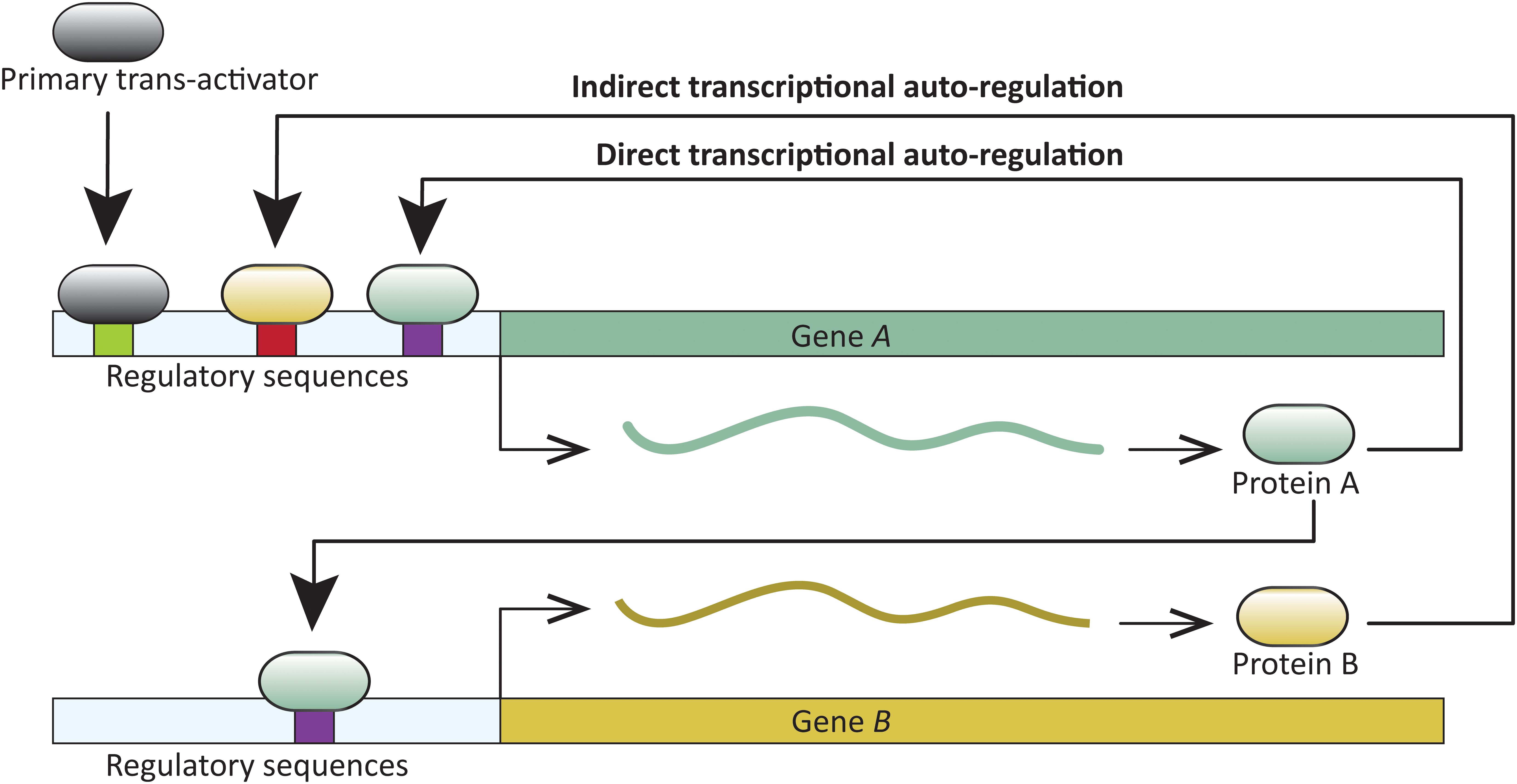
FIGURE 1. Schematic representation of direct and indirect transcriptional autoregulation (DTA and ITA, respectively) involving two transcription factors. Gene A undergoes DTA and also regulates transcription of gene B. In turn, Gene A undergoes ITA when B regulates its transcription.
The widespread occurrence of transcription factor autoregulation suggests a beneficial role in the function and evolution of genetic programs. Here, we provide a review of evidence for DTA in key flowering plant developmental programs. We provide new data supporting the hypothesis that DTA facilitated the evolution of flower monosymmetry in Lamiales. Together these data provide compelling evidence for the hypothesis that DTA plays a role in facilitating the evolution of novelty.
Advantages of Autoregulation
Several models suggest that autoregulation, especially DTA, can maintain a steady level of expression independent of other factors. If so, genes that are more likely to be autoregulated should be those that experience fleeting regulatory signals, or are positioned upstream in genetic regulatory networks with crucial developmental functions (Crews and Pearson, 2009; Singh and Hespanha, 2009). For example, several transcription factors involved in antibiotic resistance are reported to be autoregulated, resistance being a crucial phenotype (Hoot et al., 2010; Hervay et al., 2011). Similarly, entering or exiting lytic and lysogenic stages is a key developmental decision in lambda bacteriophages, and this decision is partly controlled by the autoregulation of a transcription factor, CI (Hochschild, 2002). The prediction that transcription factors upstream in regulatory networks are more likely to undergo autoregulation has been tested in the model eukaryote yeast, Saccharomyces cerevisiae. In yeasts, where all possible transcription factor interactions have either been tested or predicted, master regulatory genes are significantly more likely to experience autoregulation than are other regulators (Odom et al., 2006). Similarly, five out of six master regulatory genes in human hepatocytes bind to their own promoters, i.e., undergo DTA (Odom et al., 2006).
How regulatory networks define stable phenotypes is an important question in evolution and development. Simulations of developmental network evolution suggest that autoregulated genes are more robust when faced with random mutations and environmental perturbation (Pinho et al., 2014). The model that DTA stabilizes expression by reducing system noise has been tested in the gene hunchback in Drosophila melanogaster. Models where the HOX transcription factor Hunchback binds to the hunchback promoter (i.e., hunchback undergoes DTA) predict less promoter binding-unbinding noise, making the system more robust (Holloway et al., 2011). Experimental work in hunchback mutants whose protein cannot bind to DNA (hence, cannot undergo DTA) supports this prediction (Holloway et al., 2011).
In addition to enhancing system robustness, autoregulation provides a mechanism for maintaining expression through key stages of development (reviewed below) that are potentially critical for patterning phenotype. However, the developmental role of DTA has only been tested by mutational studies in a handful of cases. To determine the role of transcription factor DTA, the direct binding between the protein product of a gene and that gene’s cis-regulatory DNA can be either intensified or weakened through direct DNA manipulation. For example, addition or deletion of cis-regulatory self-binding sites can be used to test for the specific developmental role of DTA within a given species (Espley et al., 2009; Tao et al., 2012; Gallo-Ebert et al., 2013). A complementary, but more difficult approach is to alter transcription factor peptide sequence by mutagenesis in order to modify affinity toward the self-binding sites, e.g., in the hunchback exampled discussed above (Holloway et al., 2011). In some model systems, it is possible to repress activity of a transcription factor by overexpressing a dominant chimeric version of the peptide with a repressor domain added to the carboxy-terminus. The chimeric protein can repress the function of the native transcription factor by competitive inhibition (for example, Hiratsu et al., 2003; Koyama et al., 2010). Recent advances in CRISPR/Cas9 gene-editing technologies (Ma et al., 2016) will certainly facilitate exploration of DTA function, at least in model species.
Review of DTA in Flowering Plant Developmental Evolution
Once an initial signal for activation of gene expression has been received, a transcription factor capable of DTA can contribute to swift developmental decisions. A clear example comes from work on the developmental transition to flowering (Figure 2A). Flowering time is a key life-history transition in plant development, intimately tied to environmental cues and aging in order to ensure reproductive success (reviewed in Ó’Maoiléidigh et al., 2014). In Arabidopsis thaliana, the transition from vegetative to reproductive development is regulated in part by a MADS-box transcription factor, SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1). SOC1 undergoes DTA through the binding of SOC1 protein to four cis-regulatory CArG-box self-binding sites close to the SOC1 transcription start site (Tao et al., 2012). The flowering transition is significantly delayed in the insertional mutant soc1-2 which carries a loss-of-function mutation in the coding sequence of SOC1. The delayed flowering phenotype is largely rescued when soc1-2 lines are transformed with a wild type SOC1 allele (including the wild type promoter). This mutant-rescue system with known self-binding sites in the SOC1 promoter creates an elegant system for testing the specific role of SOC1 DTA in establishing tight control of the flowering time phenotype. In heterozygous rescue lines where the self-binding sites in the transgenic allele have been mutated by substituting nucleotides at the first two and last two positions of the CArG-box binding site, flowering is delayed (Tao et al., 2012). This suggests that the DTA of SOC1 has a key role in transition to flowering. Tao et al. (2012) provide further evidence of SOC1 autoregulation using an estradiol-inducible expression system. Estradiol-induction allows tight control over transgenic protein entering the nucleus and functioning as a transcription factor. Within 2 h of estradiol-induction of transgenic SOC1, expression of endogenous SOC1 tripled in comparison to a control. This rapid increase in SOC1 expression after releasing transgenic SOC1 protein to the nucleus suggests SOC1 plays a direct role in its own upregulation. Together, these SOC1 experiments in Arabidopsis provide clear evidence that once induced, a transcription factor undergoing DTA can rapidly increase its expression level to swiftly respond to a signal and affect developmental outcomes.
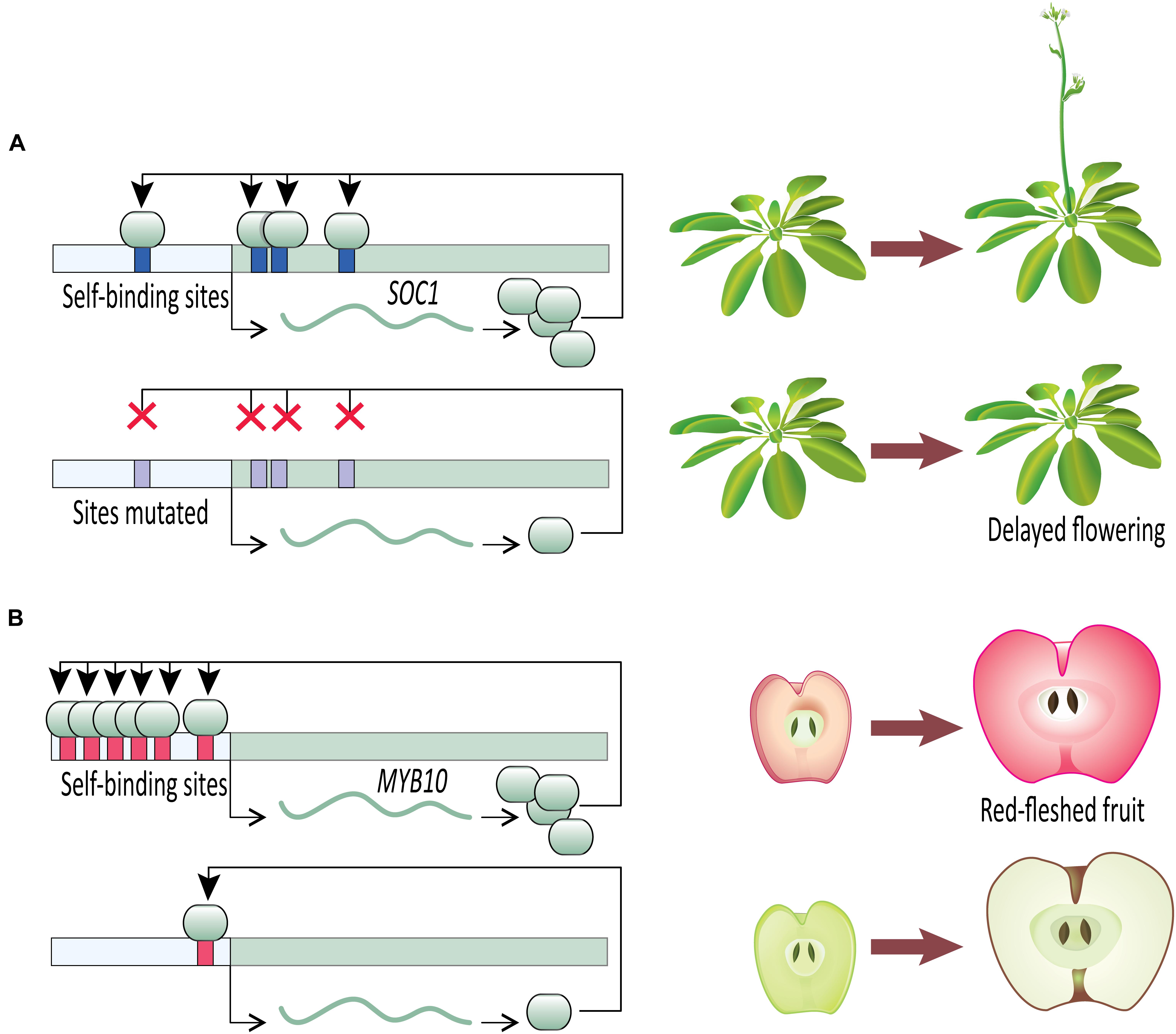
FIGURE 2. Direct transcriptional autoregulation (DTA) in novel plant phenotypes. (A) Disrupting DTA by removal of cis-acting autoregulatory sites in Arabidopsis SOC1 delays onset of flowering. (B) The number of autoregulatory sites in apple MYB10 cis-regulatory sequence is correlated with fruit flesh color.
Sustained, stable, and high expression is likely key to defining complex phenotypes. Other than increasing the expression level at a certain point during development (as described in SOC1 above), DTA would provide selective advantage if it could sustain the expression for an extended time through consecutive developmental events. A way to test this would be to determine how expression changes when homologous autoregulatory and non-autoregulatory sites between a pair of recently diverged paralogs are swapped. Arabidopsis APETALA1 (AtAP1) and CAULIFLOWER (AtCAL) are two recently duplicated paralogs (Wang et al., 2012) and this system was employed by Ye et al. (2016) to test the role of DTA for sustaining expression in developmental patterning. AtAP1 defines sepal development, and Ye et al. (2016) found that strong expression of AtAP1 is initiated in floral meristems, and that the expression continues to near-mature flower stages (stage-12). AtAP1 also undergoes DTA wherein it binds to a CArG-box located in its cis-regulatory region and activates AtAP1 transcription. On the other hand, AtCAL does not undergo DTA, is expressed at a low level in early stage flowers, with the expression vanishing soon after stage-4 (Ye et al., 2016). In an elegant system, Ye et al. (2016) generated β-glucuronidase (GUS) reporter-constructs driven by AtAP1 and AtCAL promoter regions. When the CArG-box in the GUS reporter construct with the AtAP1 promoter was replaced with the homologous non-autoregulatory nucleotides from the AtCAL promoter, two changes occurred. First, the overall expression level of GUS dropped, and second, the expression duration was shortened, approximating that of AtCAL in wild type plants. On the contrary, when GUS was placed under the control of an AtCAL promoter whose non-autoregulatory nucleotides had been replaced with the homologous CArG-box from the AtAP1 promoter, GUS expression level increased and extended to near-mature stage flowers. This suggests that DTA of AtAP1 not only has a role in maintaining high expression levels compared to the non-autoregulated paralog, but has a critical role in sustaining the expression for an extended period. This study did not directly test the role of AtAP1 DTA, its loss or acquisition, in defining phenotype. However, direct evidence for acquisition or loss of DTA on the evolution of a novel phenotype comes from domesticated apples.
Malus domestica (domesticated apple) provides compelling evidence for the importance of DTA on phenotypic outcomes (Figure 2B). The color of fruit flesh in many domesticated apple varieties ranges from white to red. Variation in fruit color is regulated by the transcription factor MYB10, which upregulates anthocyanin expression, especially cyanidin-3-galactoside (Espley et al., 2007, 2009, 2013). Anthocyanin-regulating MYBs have been reported from a wide variety of angiosperm species (reviewed in Lin-Wang et al., 2010), including Malus (Espley et al., 2009), Prunus (Starkeviè et al., 2015), Myrica (Niu et al., 2010), Arabidopsis (Gonzalez et al., 2008), and Ipomoea (Mano et al., 2007). Malus domestica has two alleles of MYB10 that are identical in their coding sequences but differ in their promoter sequences. Allele R1 promoter contains one MBY10 autoregulatory binding site, whereas allele R6 promoter contains six repeats of the autoregulatory site (Espley et al., 2009). The white-fleshed domestic apple varieties are homozygous for the one-repeat R1 allele, whereas the red-fleshed varieties are R1/R6 heterozygotes or R6/R6 homozygotes, which leads to increased anthocyanin production via DTA (Espley et al., 2009). It is not clear which allele is ancestral in domesticated apples. Of the four Malus species that contributed to the domesticated apple genome (Cornille et al., 2014), M. sieversii can be either R6/R6 (Espley et al., 2009; Lin-Wang et al., 2010) or R1/R6 (Espley et al., 2009; van Nocker et al., 2012), and M. baccata is R1/R1 (van Nocker et al., 2012). Of the other species in the genus Malus tested for MYB10 promoter sequence, all but one have the R1/R1 genotype (van Nocker et al., 2012).
Though it is not clear whether R1/R1 (white flesh) or R6/R6 (red flesh) is ancestral in the genus Malus, it is clear from studies in domesticated apple that changes to fruit flesh color are regulated by addition or loss of autoregulatory sites in the MYB10 promoter. The evidence from flesh coloration in apples suggests an interesting possibility. Self-activating loops of DTA can serve as easy modules for evolving elevated or reduced gene expression levels. Such evolutionary shifts in gene expression have potentially adaptive developmental consequences accompanied by minimal pleiotropy. Genes, including transcription factors, are often regulated by trans-activators that bind to the cis-acting elements in the regulatory region of the target gene. Theoretically, these target genes can be upregulated in three ways: adding more cis-regulatory sites recognized by either the existing or novel trans-activators, upregulating the expression of the existing trans-activators, or acquiring new (or additional) self-binding sites in the promoter region. Addition of cis-regulatory sites recognized by trans-activators can be ineffective if the expression level of the trans-activator is limiting. Additionally, increasing the expression level of the trans-activator can have pleiotropic consequences. However, acquiring new (or additional) cis-regulatory self-binding sites can lead to increased expression of the target gene while bypassing the limitations associated with trans-activation. Similarly, reduced expression levels can evolve with minimal pleiotropic consequences through the loss of existing autoregulatory sites.
The evidence from SOC1, AtAP1, and MYB10 provide insight into why genes involved in defining novel phenotypes are likely to undergo DTA. Autoregulatory loops can serve as a quick developmental switch that can rapidly respond to an inbound signal, they can provide high expression levels, and extend that expression through consecutive developmental events. Lastly, DTA can act as a module that can be used to evolve increased or decreased expression with minimal pleiotropic effect, allowing the evolution of novel phenotypes that require such directional changes in protein levels. Quick evolutionary shifts in developmental function of paralogs and divergent alleles can therefore occur through gain or loss of DTA, most likely through gain or amplification of self-binding sites in cis-regulatory sequences of focal genes.
Evidence for DTA in Flower Symmetry Evolution
An emerging system for studying the role of DTA in both development and evolution is flower symmetry. DTA has been implicated in the control of monosymmetry (bilateral symmetry; zygomorphy) (Yang et al., 2012), and may represent a critical step for the evolution of this floral novelty. Monosymmetric flowers are considered a key innovation defining flower form in many species-rich flowering plant lineages including Lamiales, asterids, legumes, and orchids (Sargent, 2004; Vamosi and Vamosi, 2010). Therefore, assessing the role of DTA in the development of flower monosymmetry may provide critical insights into patterns of gene network modification that facilitate novel trait evolution. Below, we review the genetic control of monosymmetry in Lamiales alongside the evidence for DTA. We test for previously unreported regulatory interactions in the Antirrhinum majus flower symmetry program, as well as the potential for DTA in a major radiation of taxa with primarily monosymmetric flowers, the Lamiales. Lastly, we comment on possible wide-spread DTA in repeated origins of monosymmetry across flowering plants.
Flowering plants are ancestrally polysymmetric (radially symmetric; actinomorphic; Figure 3) (Sauquet et al., 2017). Evolutionary shifts away from polysymmetry include asymmetry (no axis of flower symmetry) and disymmetry (two non-equivalent axes of flower symmetry), but monosymmetry (a single axis of flower mirror-image symmetry; Figure 3) is by far the most common form of non-radial symmetry in flowering plants. Monosymmetric flowers have evolved at least 130 times independently during flowering plant diversification (Reyes et al., 2016). The role of floral symmetry in pollination was recognized as early as 1793 by Sprengel in his monumental German work Das entdeckte Geheimniss der Natur im Bau und in der Befruchtung der Blumen (reviewed in Neal et al., 1998; Endress, 1999; Fenster et al., 2004, 2009). Monosymmetric flowers are often associated with specialized pollination by animals (Kampny, 1995; reviewed in Neal et al., 1998), rarely in wind pollinated species (Yuan et al., 2009), and transitions to monosymmetry are strongly associated with increased speciation rates (Sargent, 2004; O’Meara et al., 2016).
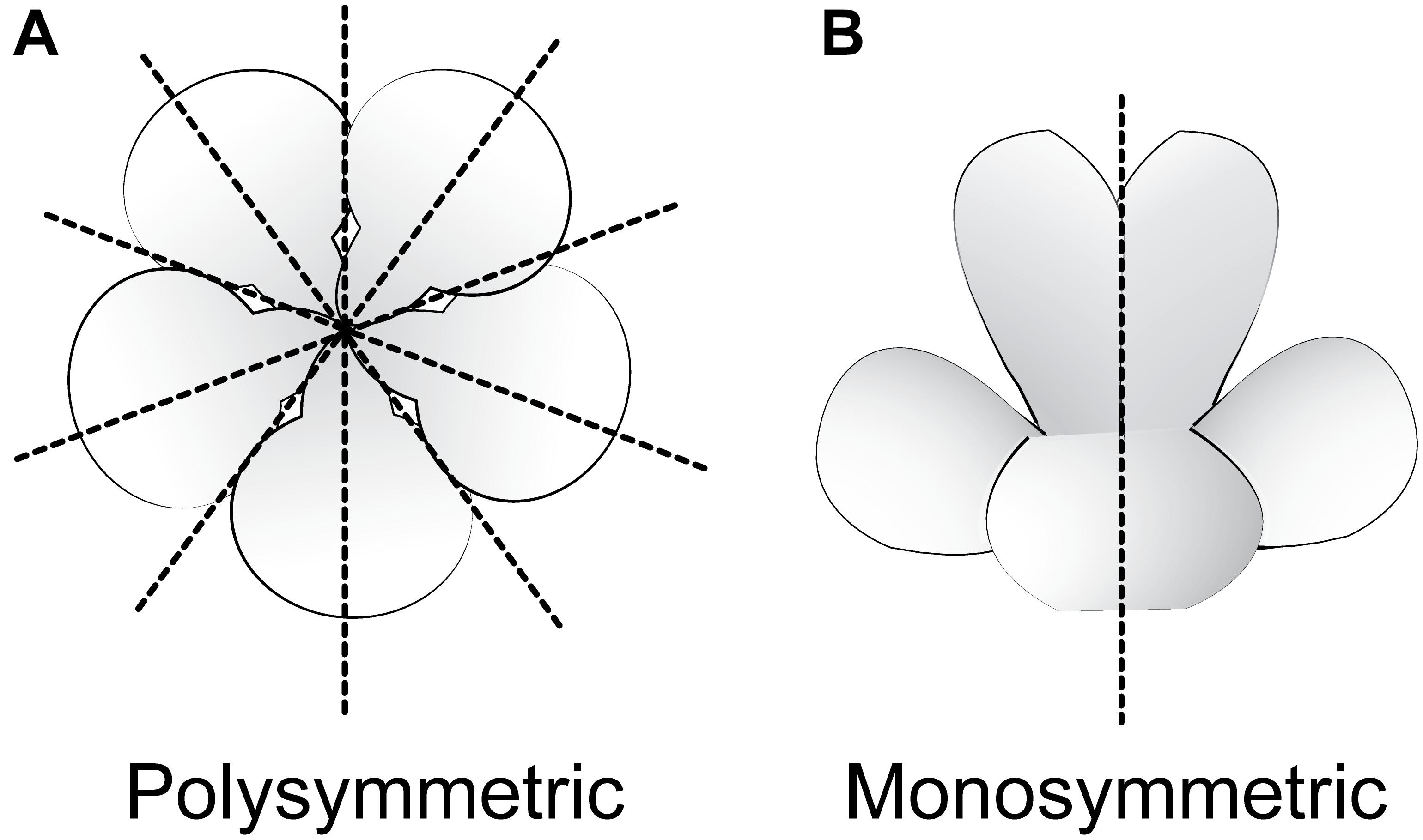
FIGURE 3. Major types of flower symmetry shown with hypothetical flowers. (A) Polysymmetric (radially symmetric, actinomorphic) flower. (B) Monosymmetric (bilaterally symmetric, zygomorphic) flower.
The genetics of monosymmetry is best understood in the model species A. majus (snapdragon, Lamiales). The flowers of A. majus have two distinct morphological regions—the dorsal (top; adaxial) side, and the ventral (bottom; abaxial) side (Figure 4). Monosymmetry of A. majus flowers along the dorso-ventral axis is defined by a competitive interaction involving TCP and MYB transcription factors. TCP (TEOSINTE BRANCHED1, CYCLOIDEA, and PROLIFERATING CELL FACTORS) and MYB (first described from avian myeloblastosis virus) proteins are found as large gene families in flowering plants (Yanhui et al., 2006; Martín-Trillo and Cubas, 2010) and play diverse roles in aspects of vegetative and reproductive developmental patterning (Martín-Trillo and Cubas, 2010; Ambawat et al., 2013; Parapunova et al., 2014). The dorsal side of A. majus flowers is defined by the combined action of two recently duplicated TCP paralogs, CYCLOIDEA (AmCYC) and DICHOTOMA (AmDICH) (Luo et al., 1996, 1999; Hileman and Baum, 2003; Corley et al., 2005). These two transcription factors define dorsal flower morphology partly by activating the transcription of a downstream MYB protein, RADIALIS (AmRAD; Figure 4) (Corley et al., 2005). AmRAD post-translationally negatively regulates another MYB protein, DIVARICATA (AmDIV), which defines ventral flower morphology. Through this negative interaction, AmRAD excludes the ventral flower identity specified by AmDIV from the dorsal side of the developing A. majus flower (Figure 4). Specifically, AmRAD and AmDIV compete for interaction with two MYB-family protein partners called DIV and RAD Interacting Factors 1 and 2 (AmDRIF1 and AmDRIF2) (Almeida et al., 1997; Galego and Almeida, 2002; Corley et al., 2005; Raimundo et al., 2013). AmDIV requires protein-protein interaction with AmDRIF1 or 2 to function as a transcription factor and upregulate its own transcription, as well as to regulate downstream targets (Figure 4) (Perez-Rodriguez et al., 2005; Raimundo et al., 2013). In the dorsal flower domain, AmRAD outcompetes AmDIV for interaction with AmDRIF1/2, preventing accumulation of AmDIV protein (Raimundo et al., 2013).
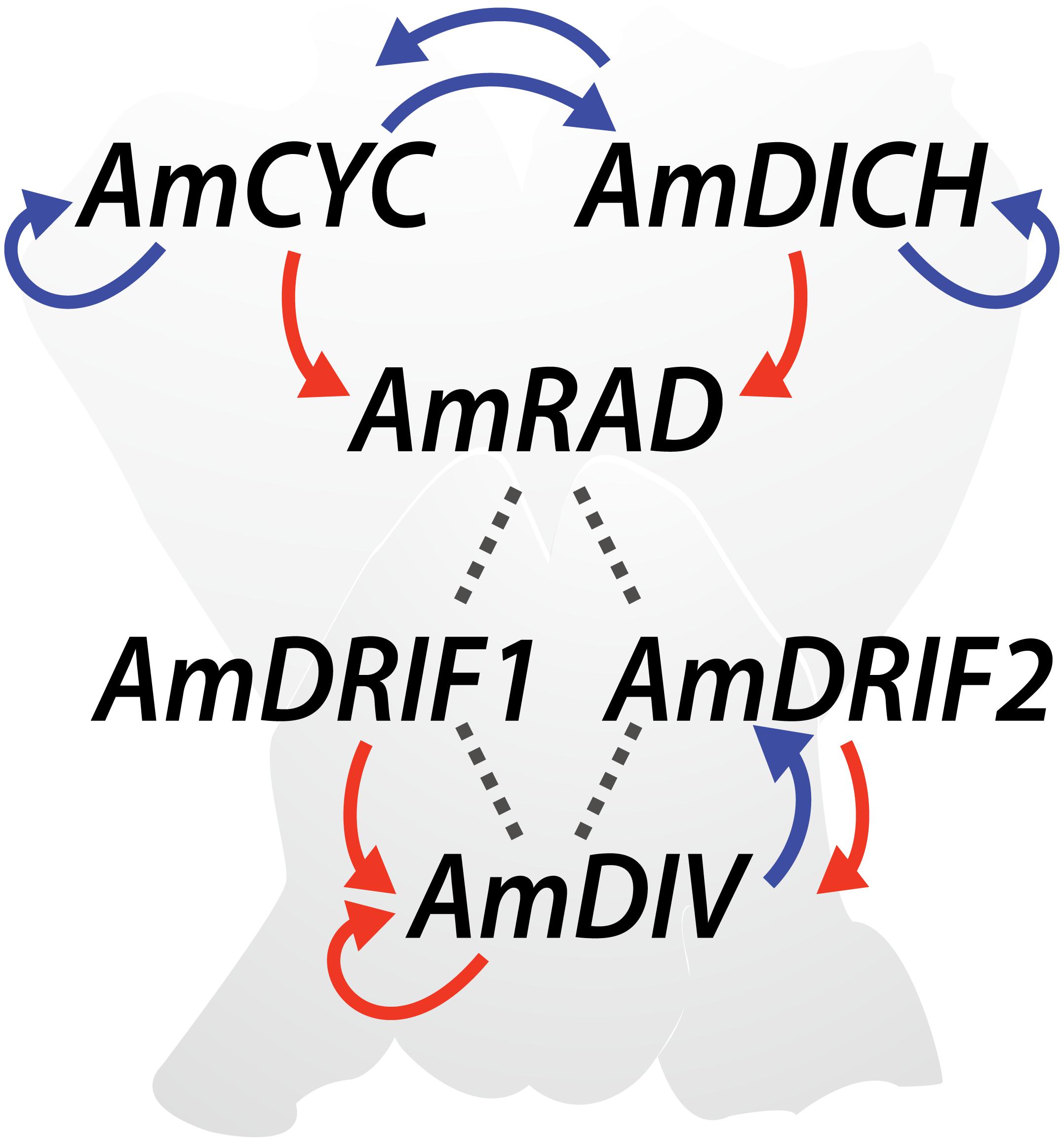
FIGURE 4. Regulatory mechanisms involved in Antirrhinum majus flower symmetry. Previously reported transcriptional regulation (red arrows), transcriptional regulation predicted in this study (blue arrows), and previously reported protein-protein interactions (dotted lines) are shown.
Because flower monosymmetry has evolved multiple times, a considerable amount of effort has gone into testing whether elements of the A. majus symmetry program function to specify dorso-ventral differentiation in other flowering plant lineages. Interestingly, all monosymmetric species tested at a molecular level so far show evidence that a TCP-based regulatory network is likely involved in differentiation along the dorso-ventral flower axis. These studies span eudicot and monocot lineages and primarily, but not exclusively, show a pattern of dorsal-specific floral expression of TCP homologs (for example, Citerne et al., 2003, 2010; Busch and Zachgo, 2007; Wang et al., 2008; Yuan et al., 2009; Bartlett and Specht, 2011; Howarth et al., 2011; Chapman et al., 2012; Preston and Hileman, 2012; Damerval et al., 2013; and reviewed in Hileman, 2014). In core eudicots, there are three lineages of CYCLOIDEA (CYC)-like TCP genes resulting from two rounds of duplication near the origin of core eudicots: the CYC1-, CYC2-, and CYC3-lineages (Howarth and Donoghue, 2006; Citerne et al., 2013). AmCYC and AmDICH belong to the CYC2-lineage, and in an interesting pattern, all TCP genes implicated in floral monosymmetry in core eudicots belong to the same CYC2-lineage (Howarth and Donoghue, 2006; Citerne et al., 2010; and reviewed in Hileman, 2014). How these orthologous genes were recruited convergently during the multiple evolutionary origins of floral monosymmetry, from an as yet unclear function in species with ancestral polysymmetry, remains an open question.
Detailed developmental studies in A. majus have provided key insights into the regulatory interactions that shape flower monosymmetry, and A. majus as a model represents a species-rich lineage of flowering plants, Lamiales. Monosymmetry evolved early in Lamiales diversification (Zhong and Kellogg, 2015; Reyes et al., 2016), and developmental genetic studies in additional Lamiales species provide further insight into the regulatory network that shapes bilateral flower symmetry across the entire lineage. Notably, detailed expression and functional studies of CYC, RAD and DIV orthologs in Gesneriaceae, a sister lineage to the bulk of Lamiales species diversity, have contributed to a fuller understanding of regulatory interactions that shape Lamiales flower monosymmetry (Citerne et al., 2000; Smith et al., 2004; Gao et al., 2008; Zhou et al., 2008; Yang et al., 2010, 2012; Liu et al., 2014a). From studies in A. majus (Plantaginaceae) and Primulina heterotricha (syn. Chirita heterotricha; Gesneriaceae), there is strong evidence that at least two components of the flower symmetry network undergo DTA–DIV and CYC (Figure 4).
As mentioned above, AmDIV forms heterodimers with AmDRIF1 and 2 to specify ventral flower identity in A. majus (Raimundo et al., 2013). AmDIV-AmDRIF dimers bind to a consensus sequence that includes the conserved I-box motif, 5′-GATAAG-3′ located 2596 bp upstream of the AmDIV transcription start site (Raimundo et al., 2013), providing compelling evidence that AmDIV is involved in an autoregulatory loop. Autoregulation of DIV orthologs has not been tested outside of A. majus. In P. heterotricha, peloric (radialized) forms due to flower ventralization have reduced expression levels of CYC orthologs, PhCYC1C and PhCYC1D (Yang et al., 2012), presenting strong evidence that these two genes define dorsal identity of monosymmetric P. heterotricha flowers. Experimental evidence suggests that PhCYC1 and PhCYC2 undergo DTA; PhCYC1 and PhCYC2 proteins bind to the consensus TCP-binding sequence 5′-GGNCCC-3′ in the putative promoter regions of both PhCYC1 and PhCYC2 (Yang et al., 2010, 2012). Autoregulation of CYC orthologs has not been tested outside of P. heterotricha.
These initial insights from A. majus and P. heterotricha lead to a set of important evolutionary questions. Is autoregulation of CYC orthologs conserved across Lamiales? And has a pattern of autoregulation repeatedly evolved in CYC2-lineage orthologs from lineages with independently derived monosymmetric flowers? This second question is especially compelling given that CYC2-lineage ortholog expression is expected to persist from early through later stages of flower development in order to specify asymmetric morphological differentiation along the dorso-ventral floral axis in lineages with flower monosymmetry.
Methods: Evidence for DTA in Flower Symmetry Evolution
Homolog Predictions and Phylogenetic Analyses
AmCYC, AmDICH, AmRAD, and AmDIV orthologs were identified from published sources and online databases by tBLASTx (Altschul et al., 1990). The gene names/identifiers and sources are listed in Supplementary Tables 1, 2. Gene identifiers are also included with terminal genes on the phylogenies (Supplementary Figures 1, 2). A subset of included genes were available as full-length coding sequences from public databases. A subset of included genes were available as partial coding sequences from public databases. For partial coding sequences from species with available genome data, we predicted the full-length coding sequences either manually by aligning to previously reported homologs, or by prediction with AUGUSTUS (Stanke et al., 2004). A subset of included genes were identified by BLAST (Altschul et al., 1990) from annotated genomes. We predicted the coding sequences either manually or with AUGUSTUS when our BLAST searches hit a region in a genome where no or partial genes were predicted. For Mimulus lewisii DIV and RAD homologs, we first BLAST searched the available transcriptome and subsequently mapped the hits to the genome. Two sets of sequences used here were not publicly available, the genes from Ipomoea lacunosa whose genome sequence was generously shared by Dr. Mark Rausher (Duke University), and Mimulus guttatus RADlike1, which was shared by Dr. Jinshun Zhong (University of Vermont; the sequence was reported in Zhong et al., 2017).
We translationally aligned the coding sequences (omitting the stop codon) of CYC-like genes using MAFFT v7.388 (Katoh et al., 2002) in Geneious 10.2.3 (Kearse et al., 2012) with the following parameters: algorithm–auto, scoring matrix–BLOSUM62, gap opening penalty–1.1, offset value–0.124. The entire alignment was used for downstream phylogenetic analyses. The CYC-like gene tree was estimated using a Bayesian approach (Metropolis-coupled Markov chain Monte Carlo) in MrBayes 3.2.6 (Ronquist et al., 2012) with uninformative priors for 10 million generations on the online CIPRES portal at https://www.phylo.org (Miller et al., 2010). The core-eudicot CYC-like tree was rooted with Rananculales CYC-like genes in FigTree1.
DIV- and RAD-like genes were translationally aligned using an approach similar to CYC-like genes except for the following: gap opening penalty–1.53, and offset value–0.123. We removed the columns with 70% or more gaps from the alignment, and from the subsequent file used only the conserved first MYBI domain and nucleotides immediately 3′ to this domain. DIV- and RAD-like gene trees were estimated using the same approach as for CYC-like genes. Resulting DIV- and RAD-like trees were mid-point rooted in FigTree1. For all sequences included in our phylogenetic analyses, nexus format nucleotide alignment along with the Bayesian parameter block, and the unaligned coding sequences in fasta format available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.tv54037.
Consensus TCP and DIV-Binding Site Predictions
We downloaded up to 3 kb non-coding sequence upstream of the transcription start sites of target Lamiales CYC, RAD, and DIV homologs from corresponding genomes. We downloaded up to 3 kb non-coding sequence upstream of the transcription start sites of representative core eudicot CYC homologs from corresponding genomes. All genomic sources are listed in Supplementary Table 1. Within these sequences, we searched for the consensus TCP-binding site 5′-GGNCCC-3′ (Kosugi and Ohashi, 2002; Costa et al., 2005; Yang et al., 2012; Gao et al., 2015) on both strands using Geneious 10.2.3 (Kearse et al., 2012). In A. majus only, we searched for the consensus DIV-binding site, 5′-[AGC]GATA[AC][GC][GAC]-3′ (Raimundo et al., 2013) in 3 kb upstream non-coding sequences of the six genes known to be involved in A. majus flower symmetry (Figure 4) using Geneious 10.2.3 (Kearse et al., 2012). To determine whether the consensus TCP-binding sites found in the A. majus and M. lewisii upstream CYC homolog sequences were derived from other genomic locations, we used the predicted TCP-binding sites, plus 100 bp on either side, as BLAST queries against the available genomes in Geneious 10.2.3 (Kearse et al., 2012).
Analysis of Motif Enrichment
We tested for consensus TCP-binding site enrichment using Analysis of Motif Enrichment (AME2; McLeay and Bailey, 2010). AME can identify known or user-provided motifs that are relatively enriched in a given set of sequences compared with shuffled versions of those sequences or with user-provided control sequences. AME does not discriminate among motifs based on their locations within the sequences. The following options were selected: sequence scoring method—average odds score, motif enrichment test—rank sum test, and background model—uniform model. We defined the consensus TCP-binding site as 5′-GGNCCC-3′ (Kosugi and Ohashi, 2002; Costa et al., 2005; Yang et al., 2012; Gao et al., 2015), and query sequences as 3 kb upstream of transcription start sites of focal genes, and used shuffled sequences as the control. The upstream non-coding sequences are available in fasta format from the Dryad Digital Repository: https://doi.org/10.5061/dryad.tv54037.
Quantitative Reverse-Transcriptase PCR (rt-PCR)
Antirrhinum majus wild type (genotype JI 7) and divaricata mutants (genotype JI 13) were acquired from John Innes Centre, United Kingdom, under USDA Permit No. P37-16-01034. Five flower buds of the same developmental stage (stage-11, flower bud ca. 4.0 mm in length, corolla equal in length to calyx, petal tips white in wild type; Vincent and Coen, 2004) were sampled from each genotype. RNA was extracted using RNeasy plant minikit (Qiagen, Germantown, MD, United States), followed by DNase treatment (TURBOTM DNase, Thermo Fisher Scientific, Waltham, MA, United States), and cDNA synthesis (iScript cDNA Synthesis Kit, Bio-Rad, Hercules, CA, United States). Quantitative rt-PCR was performed on a StepOnePlusTM Real-Time PCR System (Thermo Fisher Scientific) using SYBRTM Select Master Mix (Thermo Fisher Scientific). Quantitative rt-PCR was carried out for three technical replicates for each of five biological replicates per genotype. Expression was normalized against UBIQUITIN5. This gene has been reported to have little transcriptional variation across tissue types and developmental stages (Preston and Hileman, 2010). Expression was analyzed by the ΔΔCt method. Significant differences in relative expression between genotypes were determined using two sample t-test assuming equal variances in Minitab. The quantitative rt-PCR primers were as follows: AmDRIF1_RT_F4: GCCTTGGATCAAATTTCGGC; AmDRIF1_RT_R4: AGGAAGAATGGAGCTGGCAA; AmDRIF2_RT_F1a: AATGGTCATGGAGAGTGGGG; AmDRIF2_RT_R1:TATAGCTTGCTCCTCTGGGG; AmUBQ5_qPCR_F1: GCGCAAGAAGAAGACCTACAC; AmUBQ5_qPCR_R1: CTTCCTGAGCCTCTGCACTT. Efficiency of PCR was determined using DART (Peirson et al., 2003).
Results: Evidence for DTA in Flower Symmetry Evolution
Predicted TCP- and DIV-Binding Sites in A. majus Are Consistent With Known and Hypothesized Transcriptional Regulation
In A. majus, we found consensus TCP-binding sites in four of the six genes known to be involved in A. majus flower symmetry (Figure 4 and Table 1). AmCYC and AmDICH had eight and four predicted TCP-binding sites in their upstream non-coding sequences, respectively, and likely regulate their own and each other’s expression. Notably, AmCYC DTA has been hypothesized previously (Costa et al., 2005), and the presence of predicted autoregulatory sites in AmCYC and AmDICH is consistent with the putative auto and cross-regulation of P. heterotricha PhCYC1C and PhCYC1D (Yang et al., 2012). AmRAD, known to be positively regulated by AmCYC and AmDICH (Corley et al., 2005; Costa et al., 2005), had two predicted consensus TCP-binding sites in its upstream non-coding sequence. AmDIV and AmDRIF2 did not have predicted TCP-binding sites in their upstream non-coding sequences, consistent with evidence that they are unlikely to be under direct transcriptional regulation by AmCYC, AmDICH, or any other more distantly related TCP transcription factors.
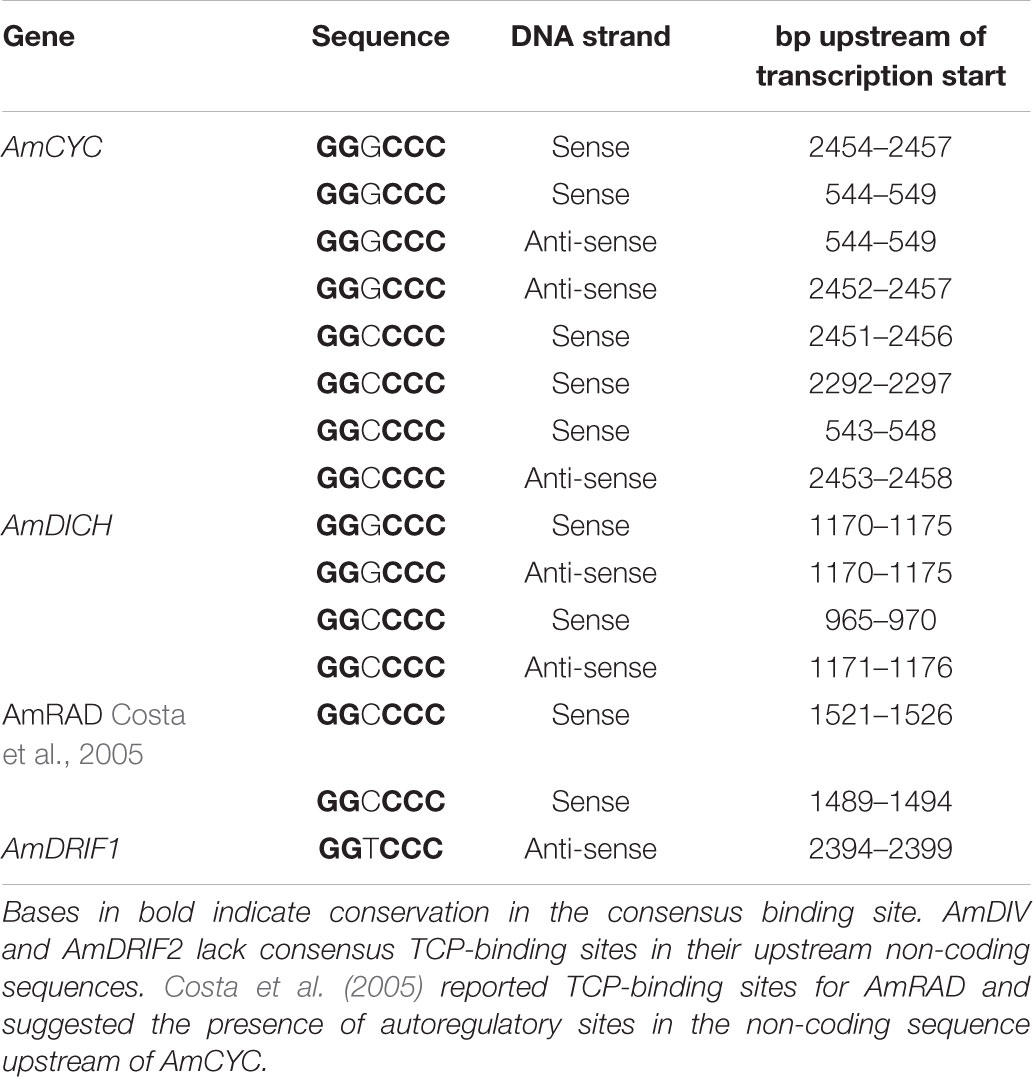
TABLE 1. Predicted consensus TCP-binding sites in the upstream non-coding sequences of A. majus flower symmetry genes.
Consensus TCP-binding sites (plus 100 bp flanking sequence from either side) initially identified in the upstream non-coding sequences of AmCYC and AmDICH were used to search for similar sites elsewhere in the A. majus genome. These searches resulted in only self-hits to AmCYC and AmDICH upstream non-coding sequences or cross-paralog matches between AmCYC and AmDICH. This result suggests that these sites evolved de novo and not through translocation of existing sites from elsewhere in the genome. Similarly, our search for consensus TCP-binding sites from M. lewisii CYC2-lineage genes in the M. lewisii genome resulted in only self-hits.
We identified two consensus DIV-binding sites in the AmDIV upstream non-coding sequence (Table 2), one of which was previously implicated by Raimundo et al. (2013) in AmDIV DTA. AmCYC, AmRAD, AmDRIF1 and AmDRIF2, but not AmDICH, also had predicted DIV-binding sites in their upstream non-coding sequences (Table 2). It is unlikely that the predicted DIV-binding sites in the upstream non-coding sequences of AmCYC or AmRAD function for AmDIV binding. This is because AmDIV function is impaired in the presence of AmRAD proteins through competitive inhibition.
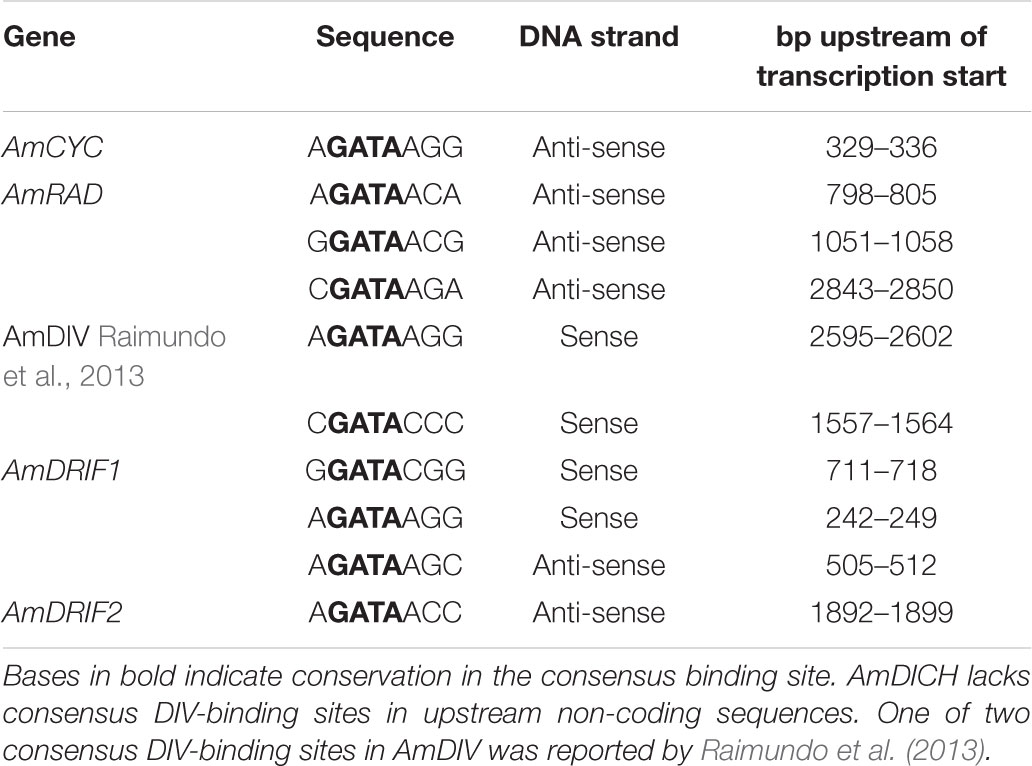
TABLE 2. Predicted consensus DIV-binding sites in the upstream non-coding sequences of A. majus flower symmetry genes.
Expression Analyses Suggest Additional Autoregulation of DIV in A. majus
Given the presence of predicted DIV-binding sites in AmDRIF1 and AmDRIF2 upstream non-coding sequences (Table 2), we tested whether AmDRIF1 and/or AmDRIF2 expression is significantly altered in the A. majus div mutant background compared to wild type. We found that AmDRIF1, despite having multiple DIV consensus binding sites in its upstream region, was not under either direct or indirect regulation by AmDIV (p = 0.453; Figure 5A). AmDRIF1 may be regulated by a non-DIV MYB transcription factor(s) that binds to the consensus DIV-binding motif. On the other hand, we found significantly lower levels of AmDRIF2 expression in div mutant flower buds compared to wild type (p = 0.031; Figure 5B). This suggests that AmDRIF2 is either directly or indirectly positively regulated by AmDIV. In turn, AmDIV is positively regulated by AmDRIF2-AmDIV heterodimers (Raimundo et al., 2013). Therefore, AmDIV appears to experience both direct and ITA through interaction of AmDIV cis-regulatory sequences with AmDRIF2-AmDIV heterodimers.
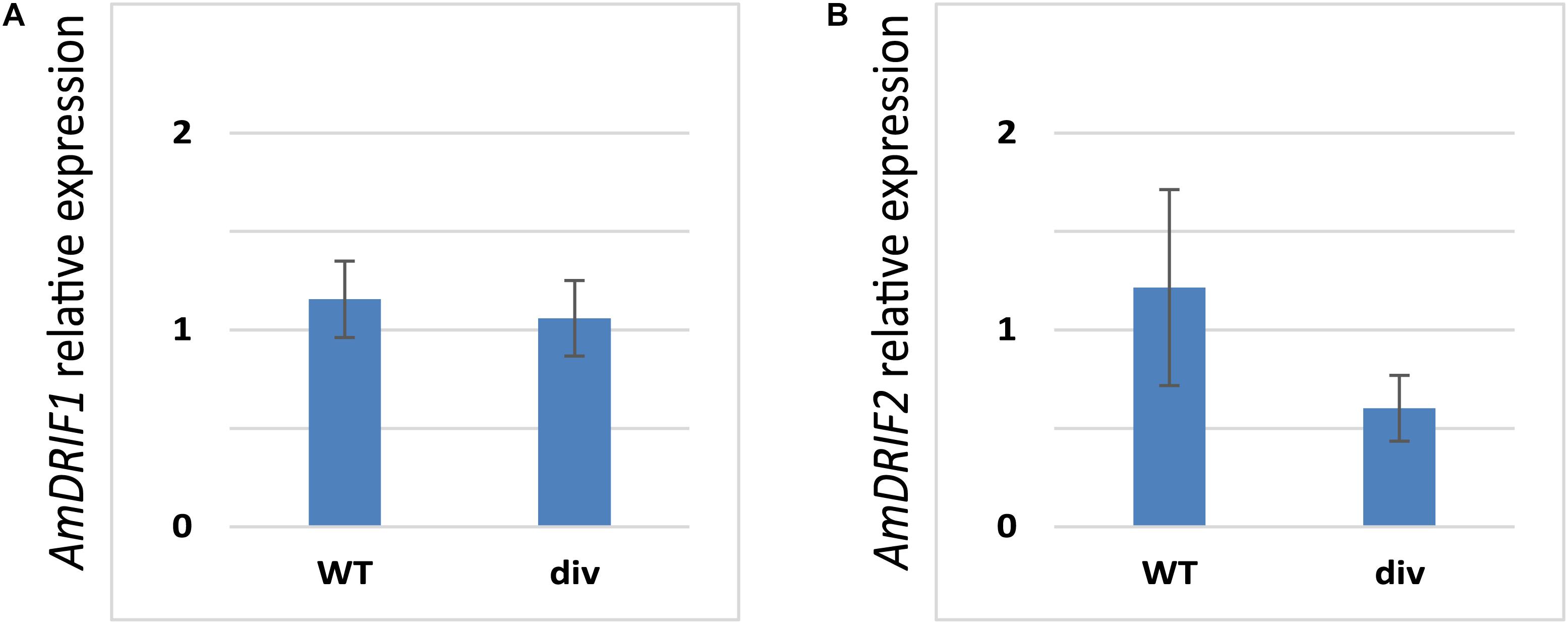
FIGURE 5. Relative expression of AmDRIF1 (A) and AmDRIF2 (B) in wild type and divaricata mutant lines. The expression level of AmDRIF2 is significantly lower in the div background suggesting that AmDIV positively regulates AmDRIF2 transcription. The values are mean ± standard deviation.
Putative TCP-Binding Sites Are Enriched in Upstream Non-coding Sequences of Lamiales CYC2-Lineage Genes
While no CYC2-lineage gene outside P. heterotricha has been experimentally tested for DTA, it is possible to infer the potential for DTA by screening for the consensus TCP-binding site, 5′-GGNCCC-3′ (Kosugi and Ohashi, 2002; Yang et al., 2012; Gao et al., 2015), in putative cis-regulatory regions of Lamiales CYC2-lineage genes. Given that flower monosymmetry is homologous in P. heterotricha and A. majus, evolving early in the diversification of Lamiales (Zhong and Kellogg, 2015; Reyes et al., 2016), a straight-forward hypothesis is that CYC2-lineage DTA evolved early in Lamiales and has been retained in Lamiales lineages with monosymmetric flowers. Under this hypothesis, Lamiales with flower monosymmetry will retain consensus TCP-binding site(s) in putative CYC2-lineage cis-regulatory sequences. The availability of multiple Lamiales genomes (Supplementary Table 1) allowed us to begin testing the hypothesis that autoregulation is potentially conserved across Lamiales CYC orthologs.
We identified orthologs of AmCYC/AmDICH (CYC2-lineage genes) from genome-sequenced Lamiales plus representative core eudicots (Supplementary Figure 1 and Table 1). We identified orthologs of AmRAD and AmDIV from genome-sequenced Lamiales plus representative orthologs from sister lineages to Lamiales, Gentianales, and Solanales (Supplementary Figure 2 and Table 2). As with P. heterotricha and A. majus, recent duplication events lead to paralog complexity for CYC2-lineage genes (Supplementary Figure 1). We found that at least one CYC2-lineage gene from each core eudicot species had consensus TCP-binding sites(s) in the upstream non-coding sequence (Supplementary Tables 3, 4), with two exceptions. The only CYC2-lineage genes in Vitis vinifera (Vitales), CYCLOIDEA-like 2a, and Gossypium raimondii (Malvales), TCP1, had no consensus TCP-binding sites in their upstream non-coding sequences.
We found consensus TCP-binding sites in the upstream non-coding sequences of CYC2-lineage genes in a wide variety of core eudicots with flowers with mono-, poly-, and dissymmetry (Supplementary Tables 3, 4). However, prima facia, the CYC2-lineage orthologs from Lamiales appeared to be enriched for consensus TCP-binding sites. We tested for enrichment of consensus TCP-binding sites in the non-coding sequences upstream of Lamiales CYC2-lineage genes. Additionally, we tested the upstream non-coding sequences of non-Lamiales core-eudicot CYC2-lineage genes, and Lamiales RAD and DIV orthologs for enrichment in consensus TCP-binding sites. We predict that RAD orthologs may show enrichment of the consensus TCP-binding site due to conserved regulation of RAD by CYC-like transcription factors across Lamiales, but that Lamiales DIV orthologs are not likely to be enriched for the consensus TCP-binding site given that there is no previous data indicating regulation of DIV orthologs by CYC-like transcription factors or other TCP proteins.
As expected, we found that the upstream non-coding sequences of Lamiales DIV orthologs were not significantly enriched for the consensus TCP-binding sites (p = 0.517; Table 3), and that the upstream non-coding sequences of Lamiales RAD orthologs were significantly enriched for the consensus TCP-binding site (p = 0.0406; Table 3). This result is consistent with CYC-like transcription factors acting as regulators of RAD, but not DIV across Lamiales. Strikingly, we found that the upstream non-coding sequences of CYC2-lineage genes in Lamiales were significantly enriched in consensus TCP-binding sites (p = 0.0169; Table 3) in-line with the hypothesis that CYC autoregulation evolved early in Lamiales, coincident with the evolution of monosymmetric flower, and has been maintained during Lamiales diversification. Notably, this pattern of enrichment appears specific to Lamiales. We tested for similar enrichment of the consensus TCP-binding site in non-Lamiales core eudicot CYC2-lineage orthologs and found no evidence for a similar pattern of binding site enrichment (p = 0.352; Table 3).

TABLE 3. Results from analysis of motif enrichment (AME) tests for consensus TCP-binding sites in the upstream non-coding sequences of symmetry gene orthologs.
Discussion
Binding Site Enrichment Supports the Hypothesis That DTA of CYC Is Associated With the Origin of Flower Monosymmetry in Lamiales
Positive regulation of RAD by CYC2-lineage genes for specifying flower monosymmetry is conserved across much of Lamiales (Corley et al., 2005; Zhou et al., 2008; Su et al., 2017). That we find significant enrichment of consensus TCP-binding sites in Lamiales RAD upstream non-coding sequences is in-line with conservation of this CYC-RAD regulatory module. Strikingly, our data demonstrate that Lamiales CYC2-lineage genes are also significantly enriched for consensus TCP-binding sites in upstream non-coding sequences. This supports the hypothesis that the origin of Lamiales flower monosymmetry coincides with the evolution of CYC2-lineage DTA. Further empirical studies in emerging Lamiales models (e.g., Liu et al., 2014b; Su et al., 2017) will allow this hypothesis to be tested, as well as the alternative, that CYC2-lineage genes undergo transcriptional regulation by other TCP family proteins. As additional eudicot genomes become available, tests for TCP-binding site enrichment can be carried out in other lineages with bilaterally symmetrical flowers for which a role of CYC2-lineage genes has been implicated, for example, Fabaceae (Wang et al., 2008; Xu et al., 2013) and Malpighiaceae (Zhang et al., 2010).
Evaluating the Pan-Eudicot Model for Monosymmetry Involving DTA of CYC2-Lineage Genes
A model hypothesizing the role of DTA for the parallel origin of monosymmetric flowers across eudicots was put forward by Yang et al. (2012; Figure 6) based on two primary lines of evidence. First, the observed differences in duration of flower specific expression of CYC2-lineage genes between species with monosymmetric vs. non-monosymmetric flowers. Second, the reported absence of consensus TCP-binding sites in the upstream non-coding sequences of CYC2-lineage genes from non-monosymmetric flowers. Specifically, Arabidopsis thaliana, Brassica rapa, Vitis vinifera, and Solanum lycopersicum do not have monosymmetric flowers and were reported to lack consensus TCP-binding sites in their CYC2-lineage genes compared to Glycine max, Medicago trunculata, Mimulus guttatus, Primulina heterotricha, Oryza sativa, and Zea mays (representing three independent origins of monosymmetry) that have consensus TCP-binding sites (Yang et al., 2012).
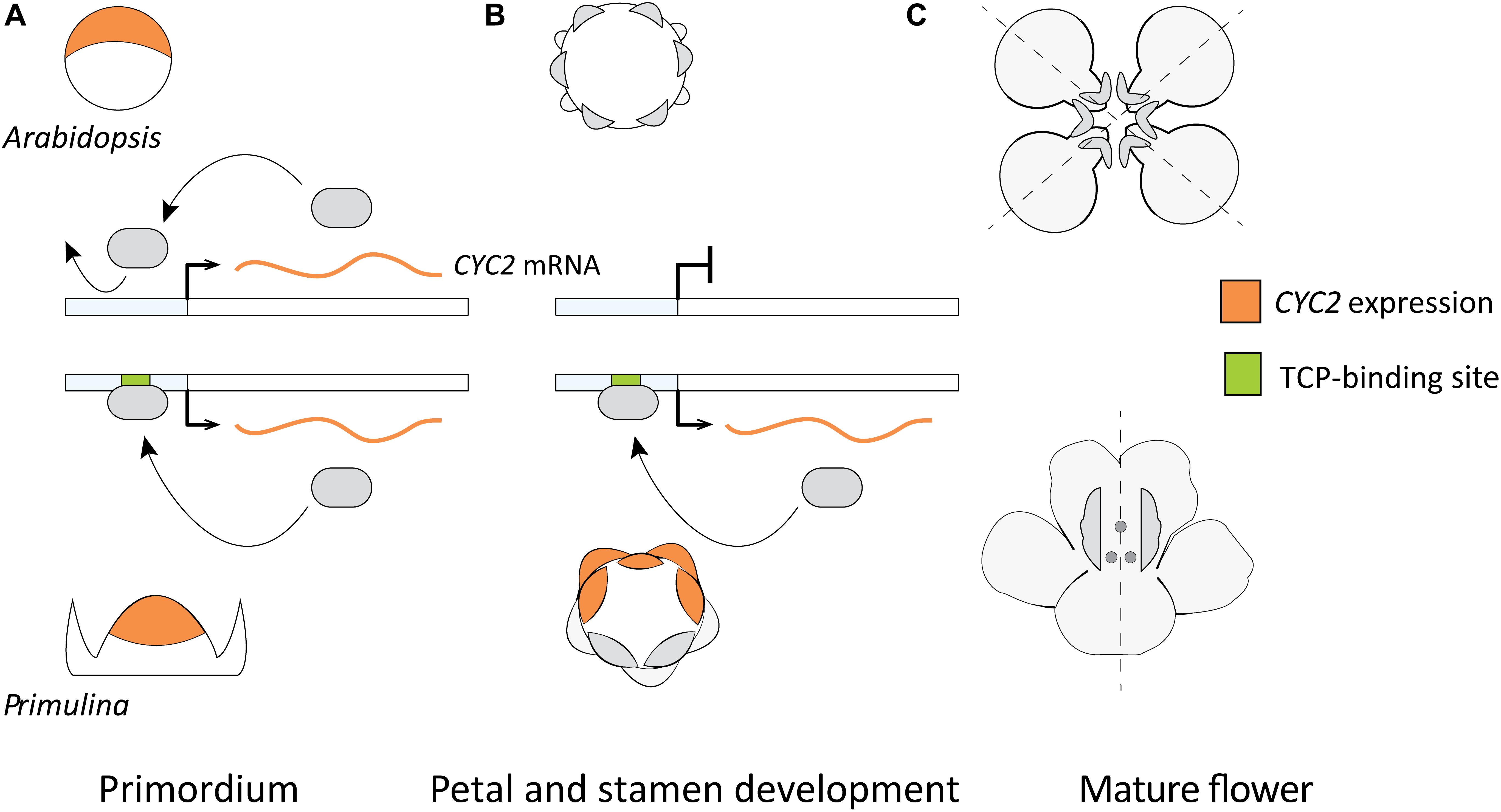
FIGURE 6. A previously proposed model explaining flower symmetry in Primulina heterotricha and Arabidopsis. (A) CYC2-lineage genes are expressed in early stage flower primordia of both Arabidopsis (dorsal part) and P. heterotricha (apical part). (B) Expression in P. heterotricha continues by DTA to later stages crucial for defining flower monosymmetry, this is not the case in Arabidopsis. (C) At anthesis, P. heterotricha is monosymmetric, Arabidopsis is not.
This model relies heavily on observations from Arabidopsis flowers where the expression of the sole CYC2-lineage gene (AtTCP1) is transiently dorsal-specific and the flowers are non-monosymmetric (Cubas et al., 2001). It is clear that AtTCP1 does not play a critical role in floral organ differentiation in Arabidopsis, given no floral-specific DTA or other means by which expression can persist to later stages of flower differentiation. However, the pattern in Arabidopsis may not be universal for non-monosymmetric flowers. Closely related monosymmetric and non-monosymmetric Brassicaceae flowers do not exhibit a consistent pattern of early dorsal-specific expression (Busch et al., 2012). Evidence from Brassicaceae suggests that Arabidopsis-like dorsal-restricted expression early in flower development is not a pre-requisite for the evolution of flower monosymmetry via DTA. Beyond Brassicaceae, there are examples of ancestrally non-monosymmetric flowers in core-eudicots where expression of CYC2-lineage genes is not localized spatially and/or restricted to an early developmental stage. These examples include Bergia texana (Elatinaceae) (Zhang et al., 2010), Viburnum plicatum (Adoxaceae) (Howarth et al., 2011), and Solanum lycoperscicum (Solanaceae, ancestral state ambiguous) (Parapunova et al., 2014), as well as an early-diverging eudicot, Eschscholzia californica (Papaveraceae) (Kölsch and Gleissberg, 2006).
Yang et al. (2012) reported a correlation between flower monosymmetry vs. non-monosymmetry and the presence vs. absence of consensus TCP-binding sites in corresponding upstream non-coding sequences of CYC2-lineage genes. This contributed to the model for the origin of flower monosymmetry facilitated by the evolution of CYC2-lineage DTA. In our expanded sampling we find that consensus TCP-binding sites are present in the upstream non-coding sequences of many CYC2-lineage genes across eudicots irrespective of flower symmetry. Yet, in an interesting pattern, all species with independently derived monosymmetric flowers that we investigated (Fabales, Lamiales, Brassicales, Asterales) have at least one CYC2-lineage ortholog with a consensus TCP-binding sequence in the upstream non-coding sequences (Supplementary Tables 3, 4). On the other hand, many species with non-monosymmetric flowers also have at least one CYC2-lineage ortholog with a consensus TCP-binding sequence in their upstream non-coding sequences (Supplementary Tables 3, 4). Notably, we find that the sole CYC2-lineage gene in Arabidopsis (AtTCP1), and a second CYC2-lineage gene in tomato that was not included in Yang et al. (2012), Solanum lycopersicum TCP26 (Solyc03g045030.1), have consensus TCP-binding sites in their upstream non-coding sequences (Supplementary Table 4).
AtTCP1 binds to all combinations of the consensus sequence 5′-GGNCCC-3′ in vitro, and flanking regions have limited significance in this interaction (Gao et al., 2015). In vivo, AtTCP1 can directly bind to the two TCP-binding sites located in the regulatory region of the a downstream gene DWARF4 (Gao et al., 2015). This suggests that the Arabidopsis TCP1 transcription factor can likely bind to the predicted TCP-binding site in its own upstream non-coding sequence, and hence possibly undergoes DTA. AtTCP1 is expressed and is functional across the shoot organs throughout development, from seedlings to inflorescences (Koyama et al., 2010). This persistent expression is consistent with it having a predicted autoregulatory site. Expression surveys employing in situ mRNA hybridization (Cubas et al., 2001) and AtTCP1 promoter fused to a β-glucuronidase (GUS) construct (Koyama et al., 2010) did not detect AtTCP1 expression in later stages of flower development. It is interesting that the expression of a gene that is widely expressed in and controls development of many different organs is specifically downregulated in flowers. It is possible that AtTCP1 is negatively regulated during late stages of Arabidopsis flower development, or continues to be expressed in flowers but a level that can only be detected by more sensitive methods, like quantitative rt-PCR.
Predicted CYC2-lineage autoregulatory sites are strongly associated with monosymmetry supporting the potential importance for DTA in establishing high and continuous asymmetric expression through later stages of flower organ differentiation (Figure 6). However, this pattern is not exclusive: CYC2-lineage orthologs from many species lacking monosymmetry also have predicted TCP-binding sites. This may be autoregulation for alternative developmental pathways, or regulation of CYC2-lineage genes by upstream TCP activators. At this point, experimental tests of TCP gene autoregulation are too sparse to draw solid conclusions regarding the role of DTA in independent origins of flower monosymmetry across core eudicots.
Origin and Evolution of Autoregulatory Sites in DTA
Any cis-regulatory site can evolve by two primary processes, de novo by mutation and/or recombination in ancestral non-regulatory sequences, or by duplication of existing regulatory sites from a different location in the genome. Both have been reported in the origin of cis-regulatory sites involved in DTA. For example, the CArG-box sites involved in Arabidopsis AP1 autoregulation discussed earlier evolved by substitutions in the ancestral sequence that likely had a weak affinity for AP1 (Ye et al., 2016). Once evolved, these sites can undergo duplications, as reported in the apple MYB10 gene that controls fruit flesh color (Espley et al., 2009; van Nocker et al., 2012).
How did the predicted autoregulatory sites in CYC2-lineage genes originate? We did not detect consensus TCP-binding sites with accompanying flanking sequences elsewhere in the A. majus or M. lewisii genomes. This suggests that these predicted autoregulatory sites evolved in situ and are not a result of duplication from a different part of the genome, i.e., similar to the origin of the autoregulatory sites in Arabidopsis AP1 (Ye et al., 2016). However, multiple consensus TCP-binding sites are present within single A. majus and M. lewisii CYC2-lineage genes. To further test whether these multiple TCP-binding sites within a single putative regulatory region evolved by local, intra-genic duplication, as in the case of MYB10 promoter in apples (Espley et al., 2009; van Nocker et al., 2012), we aligned all A. majus and M. lewisii consensus TCP-binding sites, along with 100 bp flanking on either side, from within single upstream non-coding regions. We found no evidence that any of the predicted TCP-binding sites are derived from tandem duplication within CYC regulatory regions, again suggesting that multiple binding sites evolved de novo.
Chicken or Egg: Novel Function or DTA First?
We have discussed potential roles of DTA in development, but how does DTA itself evolve? Autoregulation is common among genes positioned upstream in genetic regulatory networks with crucial developmental functions (discussed in Crews and Pearson, 2009; Hoot et al., 2010; specifically tested in yeasts and hepatocytes by Pasqualucci et al., 2003; Odom et al., 2006; Hervay et al., 2011; Tao et al., 2012). This observed pattern leads to an interesting chicken or egg conundrum. Which evolves first in genes recruited to new developmental functions: the novel function, or the autoregulation? Two scenarios can explain the observed pattern that crucial genes are often autoregulated. (1) DTA evolves first, and such genes are recruited for new functions that require extended stable expression. Or, (2) New function evolves first, and such genes, under selective pressure to provide extended stable expression, evolve DTA.
Evidence supporting scenario 2 is found in the Arabidopsis AtAP1 example. This A-class floral homeotic gene in Brassicaceae underwent a duplication that generated the paralogs AP1 and CAL gene lineages (Wang et al., 2012). AtAP1 defines sepals in Arabidopsis thaliana, but this function has not been reported elsewhere, and is likely an innovation in the genus Arabidopsis (Huijser et al., 1992; Lowman and Purugganan, 1999; Shepard and Purugganan, 2002; Litt, 2007; Ruokolainen et al., 2010). Except for the AP1 paralog in Arabidopsis species, no Brassicaceae AP1/CAL gene tested to date undergoes DTA (Ye et al., 2016). And, as described above, DTA is an integral component of AtAP1 A-class function in flower development. Further, while the AP1 orthologs of two Arabidopsis species have CArG-box in their cis-regulatory region that allows them to undergo DTA, other Brassicaceae species have CArG-box-like sequences with mismatches in the homologous gene region. In one such homolog, Capsella rubella AP1, the binding affinity of the mismatched CArG-box-like sequence was tested and can only weakly bind to AP1 protein. Hence, Capsella rubella AP1 is likely not autoregulated (Ye et al., 2016). This suggests that the autoregulation of Arabidopsis AP1 evolved either after or during, but not before, its recruitment to A-class function.
A major unanswered question that will clarify the origin of DTA in Arabidopsis AP1 is whether its orthologs have similar functions in other Brassicaceae species. It is challenging to identify the ancestral state of autoregulation for any gene primarily for two reasons: there has been little functional work outside the model species, and predictive surveys are limited because genomes sequencing has been biased toward lineages with those model species. As plant sciences expands away from models systems (Poaceae, Brassicaceae, and Solanaceae), a wider phylogenetic sampling will facilitate reconstruction of ancestral molecular interactions.
Conclusion
The origins and evolution of autoregulation will likely remain elusive until extensive experimental evidence emerges from multiple plant (and animal) lineages that inform ancestral and derived roles for autoregulation in development. It is, however, not surprising that a large number of transcription factors involved in defining crucial or novel phenotypes undergo DTA, as this form of regulation is expected to both enhance and stabilize gene expression patterns critical for developmental patterning. We find evidence for enrichment of self-binding sites in Lamiales CYC2-lineages genes. This enrichment may reflect evolution of a novel pattern of DTA early in Lamiales diversification, coincident with the origin of a key morphological innovation, floral monosymmetry. It is likely that the putative autoregulatory binding sites associated with Lamiales CYC2-lineages genes evolved via de novo mutations. Whether DTA is conserved across Lamiales awaits further experimental evidence, as does the hypothesis that independent origins of flower monosymmetry may be associated with the evolution of positive transcriptional autoregulation.
Author Contributions
LH co-conceived of this project, oversaw analyses, and contributed to writing the manuscript. AS co-conceived of this project, carried out analyses, and contributed to writing the manuscript.
Funding
This work was supported by Ecology and Evolutionary Biology General Research Fund and The Botany Endowment at The University of Kansas.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank members of the Hileman lab for insightful discussions and Dr. Mark Rausher for sharing an early draft of the Ipomoea lacunosa genome.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.01561/full#supplementary-material
Footnotes
References
Almeida, J., Rocheta, M., and Galego, L. (1997). Genetic control of flower shape in Antirrhinum majus. Development 124, 1387–1392.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Ambawat, S., Sharma, P., Yadav, N. R., and Yadav, R. C. (2013). MYB transcription factor genes as regulators for plant responses: an overview. Physiol. Mol. Biol. Plants 19, 307–321. doi: 10.1007/s12298-013-0179-1
Bartlett, M. E., and Specht, C. D. (2011). Changes in expression pattern of the TEOSINTE BRANCHED1-like genes in the Zingiberales provide a mechanism for evolutionary shifts in symmetry across the order. Am. J. Bot. 98, 227–243. doi: 10.3732/ajb.1000246
Bateman, E. (1998). Autoregulation of eukaryotic transcription factors. Prog. Nucleic Acid Res. Mol. Biol. 60, 133–168. doi: 10.1016/S0079-6603(08)60892-2
Busch, A., Horn, S., Mühlhausen, A., Mummenhoff, K., and Zachgo, S. (2012). Corolla monosymmetry: evolution of a morphological novelty in the brassicaceae family. Mol. Biol. Evol. 29, 1241–1254. doi: 10.1093/molbev/msr297
Busch, A., and Zachgo, S. (2007). Control of corolla monosymmetry in the Brassicaceae Iberis amara. Proc. Natl. Acad. Sci. U.S.A. 104, 16714–16719. doi: 10.1073/pnas.0705338104
Chapman, M. A., Tang, S., Draeger, D., Nambeesan, S., Shaffer, H., Barb, J. G., et al. (2012). Genetic analysis of floral symmetry in Van Gogh’s sunflowers reveals independent recruitment of CYCLOIDEA genes in the Asteraceae. Public Libr. Sci. Genet. 8:e1002628. doi: 10.1371/journal.pgen.1002628
Citerne, H., Jabbour, F., Nadot, S., and Damerval, C. (2010). The evolution of floral symmetry. Adv. Bot. Res. 54, 85–137.
Citerne, H. L., Le Guilloux, M., Sannier, J., Nadot, S., and Damerval, C. (2013). Combining phylogenetic and syntenic analyses for understanding the evolution of TCP ECE genes in Eudicots. PLoS One 8:e74803. doi: 10.1371/journal.pone.0074803
Citerne, H. L., Luo, D., Pennington, R. T., Coen, E., and Cronk, Q. C. B. (2003). A phylogenomic investigation of CYCLOIDEA-Like TCP genes in the Leguminosae. Plant Physiol. 131, 1042–1053. doi: 10.1104/pp.102.016311
Citerne, H. L., Möller, M., and Cronk, Q. C. B. (2000). Diversity of cycloidea-like Genes in gesneriaceae in relation to floral symmetry. Ann. Bot. 86, 167–176. doi: 10.1006/anbo.2000.1178
Corley, S. B., Carpenter, R., Copsey, L., and Coen, E. (2005). Floral asymmetry involves an interplay between TCP and MYB transcription factors in Antirrhinum. Proc. Natl. Acad. Sci. U.S.A. 102, 5068–5073. doi: 10.1073/pnas.0501340102
Cornille, A., Giraud, T., Smulders, M. J. M., Roldán-Ruiz, I., and Gladieux, P. (2014). The domestication and evolutionary ecology of apples. Trends Genet. 30, 57–65. doi: 10.1016/j.tig.2013.10.002
Costa, M. M. R., Fox, S., Hanna, A. I., Baxter, C., and Coen, E. (2005). Evolution of regulatory interactions controlling floral asymmetry. Development 132, 5093–5101. doi: 10.1242/dev.02085
Crews, S. T., and Pearson, J. C. (2009). Transcriptional autoregulation in development. Curr. Biol. 19, R241–R246. doi: 10.1016/j.cub.2009.01.015
Cripps, R. M., Lovato, T. L., and Olson, E. N. (2004). Positive autoregulation of the Myocyte enhancer factor-2 myogenic control gene during somatic muscle development in Drosophila. Dev. Biol. 267, 536–547. doi: 10.1016/j.ydbio.2003.12.004
Cubas, P., Coen, E., and Zapater, J. M. M. (2001). Ancient asymmetries in the evolution of flowers. Curr. Biol. 11, 1050–1052. doi: 10.1016/S0960-9822(01)00295-0
Damerval, C., Citerne, H., Guilloux, M. L., Domenichini, S., Dutheil, J., Ronse de Craene, L., et al. (2013). Asymmetric morphogenetic cues along the transverse plane: shift from disymmetry to zygomorphy in the flower of Fumarioideae. Am. J. Bot. 100, 391–402. doi: 10.3732/ajb.1200376
Endress, P. K. (1999). Symmetry in flowers: diversity and evolution. Int. J. Plant Sci. 160, S3–S23.
Espley, R. V., Bovy, A., Bava, C., Jaeger, S. R., Tomes, S., Norling, C., et al. (2013). Analysis of genetically modified red-fleshed apples reveals effects on growth and consumer attributes. Plant Biotechnol. J. 11, 408–419. doi: 10.1111/pbi.12017
Espley, R. V., Brendolise, C., Chagné, D., Kutty-Amma, S., Green, S., Volz, R., et al. (2009). Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell 21, 168–183. doi: 10.1105/tpc.108.059329
Espley, R. V., Hellens, R. P., Putterill, J., Stevenson, D. E., Kutty-Amma, S., and Allan, A. C. (2007). Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 49, 414–427. doi: 10.1111/j.1365-313X.2006.02964.x
Fenster, C. B., Armbruster, W. S., and Dudash, M. R. (2009). Specialization of flowers: is floral orientation an overlooked first step? New Phytol. 183, 502–506. doi: 10.1111/j.1469-8137.2009.02852.x
Fenster, C. B., Armbruster, W. S., Wilson, P., Dudash, M. R., and Thomson, J. D. (2004). Pollination syndromes and floral specialization. Annu. Rev. Ecol. Evol. Syst. 35, 375–403. doi: 10.1146/annurev.ecolsys.34.011802.132347
Galego, L., and Almeida, J. (2002). Role of DIVARICATA in the control of dorsoventral asymmetry in Antirrhinum flowers. Genes Dev. 16, 880–891. doi: 10.1101/gad.221002
Gallo-Ebert, C., Donigan, M., Liu, H.-Y., Pascual, F., Manners, M., Pandya, D., et al. (2013). The yeast anaerobic response element AR1b regulates aerobic antifungal drug-dependent sterol gene expression. J. Biol. Chem. 288, 35466–35477. doi: 10.1074/jbc.M113.526087
Gao, Q., Tao, J.-H., Yan, D., Wang, Y.-Z., and Li, Z.-Y. (2008). Expression differentiation of CYC-like floral symmetry genes correlated with their protein sequence divergence in Chirita heterotricha (Gesneriaceae). Dev. Genes Evol. 218, 341–351. doi: 10.1007/s00427-008-0227-y
Gao, Y., Zhang, D., and Li, J. (2015). TCP1 modulates DWF4 expression via directly interacting with the GGNCCC Motifs in the promoter region of DWF4 in Arabidopsis thaliana. J. Genet. Genomics 42, 383–392. doi: 10.1016/j.jgg.2015.04.009
Gonzalez, A., Zhao, M., Leavitt, J. M., and Lloyd, A. M. (2008). Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. Cell Mol. Biol. 53, 814–827. doi: 10.1111/j.1365-313X.2007.03373.x
Hervay, N. T., Hodurova, Z., Balazfyova, Z., and Gbelska, Y. (2011). Autoactivated KlPDR1 gene in the control of multidrug resistance in Kluyveromyces lactis. Can. J. Microbiol. 57, 844–849. doi: 10.1139/w11-071
Hileman, L. C. (2014). Bilateral flower symmetry — how, when and why? Curr. Opin. Plant Biol. 17, 146–152. doi: 10.1016/j.pbi.2013.12.002
Hileman, L. C., and Baum, D. A. (2003). Why do paralogs persist? Molecular evolution of CYCLOIDEA and related floral symmetry genes in Antirrhineae (Veronicaceae). Mol. Biol. Evol. 20, 591–600. doi: 10.1093/molbev/msg063
Hiratsu, K., Matsui, K., Koyama, T., and Ohme-Takagi, M. (2003). Dominant repression of target genes by chimeric repressors that include the EAR motif, a repression domain, in Arabidopsis. Plant J. 34, 733–739. doi: 10.1046/j.1365-313X.2003.01759.x
Hochschild, A. (2002). The λ switch: cI closes the gap in autoregulation. Curr. Biol. 12, R87–R89. doi: 10.1016/S0960-9822(02)00667-X
Holloway, D. M., Lopes, F. J., da Fontoura Costa, F., Travençolo, B. A., Golyandina, N., Usevich, K., et al. (2011). Gene expression noise in spatial patterning: hunchback promoter structure affects noise amplitude and distribution in Drosophila segmentation. PLoS Comput. Biol. 7:e1001069. doi: 10.1371/journal.pcbi.1001069
Hoot, S. J., Brown, R. P., Oliver, B. G., and White, T. C. (2010). The UPC2 promoter in Candida albicans contains two cis-acting elements that bind directly to Upc2p, resulting in transcriptional autoregulation. Eukaryot. Cell 9, 1354–1362. doi: 10.1128/EC.00130-10
Howarth, D. G., and Donoghue, M. J. (2006). Phylogenetic analysis of the “ECE” (CYC/TB1) clade reveals duplications predating the core eudicots. Proc. Natl. Acad. Sci. U.S.A. 103, 9101–9106. doi: 10.1073/pnas.0602827103
Howarth, D. G., Martins, T., Chimney, E., and Donoghue, M. J. (2011). Diversification of CYCLOIDEA expression in the evolution of bilateral flower symmetry in Caprifoliaceae and Lonicera (Dipsacales). Ann. Bot. 107, 1521–1532. doi: 10.1093/aob/mcr049
Huijser, P., Klein, J., Lönnig, W. E., Meijer, H., Saedler, H., and Sommer, H. (1992). Bracteomania, an inflorescence anomaly, is caused by the loss of function of the MADS-box gene squamosa in Antirrhinum majus. EMBO J. 11, 1239–1249.
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kölsch, A., and Gleissberg, S. (2006). Diversification of CYCLOIDEA-like TCP Genes in the basal Eudicot families Fumariaceae and Papaveraceae s.str. Plant Biol. 8, 680–687. doi: 10.1055/s-2006-924286
Kosugi, S., and Ohashi, Y. (2002). DNA binding and dimerization specificity and potential targets for the TCP protein family. Plant J. 30, 337–348. doi: 10.1046/j.1365-313X.2002.01294.x
Koyama, T., Sato, F., and Ohme-Takagi, M. (2010). A role of TCP1 in the longitudinal elongation of leaves in Arabidopsis. Biosci. Biotechnol. Biochem. 74, 2145–2147. doi: 10.1271/bbb.100442
Lin-Wang, K., Bolitho, K., Grafton, K., Kortstee, A., Karunairetnam, S., McGhie, T. K., et al. (2010). An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biol. 10:50. doi: 10.1186/1471-2229-10-50
Litt, A. (2007). An evaluation of A-function: evidence from the APETALA1 and APETALA2 gene lineages. Int. J. Plant Sci. 168, 73–91. doi: 10.1086/509662
Liu, B.-L., Pang, H.-B., Yang, X., and Wang, Y.-Z. (2014a). Functional and evolutionary analyses of Primulina heterotricha CYC1C gene in tobacco and Arabidopsis transformation systems. J. Syst. Evol. 52, 112–123. doi: 10.1111/jse.12067
Liu, B.-L., Yang, X., Liu, J., Dong, Y., and Wang, Y.-Z. (2014b). Characterization, efficient transformation and regeneration of Chirita pumila (Gesneriaceae), a potential evo-devo model plant. Plant Cell Tissue Organ Cult. 118, 357–371. doi: 10.1007/s11240-014-0488-2
Lowman, A. C., and Purugganan, M. D. (1999). Duplication of the Brassica oleracea APETALA1 floral homeotic gene and the evolution of domesticated cauliflower. J. Hered. 90, 514–520.
Luo, D., Carpenter, R., Copsey, L., Vincent, C., Clark, J., and Coen, E. (1999). Control of organ asymmetry in flowers of Antirrhinum. Cell 99, 367–376. doi: 10.1016/S0092-8674(00)81523-8
Luo, D., Carpenter, R., Vincent, C., Copsey, L., and Coen, E. (1996). Origin of floral asymmetry in Antirrhinum. Nature 383, 794–799. doi: 10.1038/383794a0
Ma, X., Zhu, Q., Chen, Y., and Liu, Y.-G. (2016). CRISPR/Cas9 platforms for genome editing in plants: developments and applications. Mol. Plant 9, 961–974. doi: 10.1016/j.molp.2016.04.009
Mano, H., Ogasawara, F., Sato, K., Higo, H., and Minobe, Y. (2007). Isolation of a regulatory gene of anthocyanin biosynthesis in tuberous roots of purple-fleshed sweet potato. Plant Physiol. 143, 1252–1268. doi: 10.1104/pp.106.094425
Martínez-Antonio, A., and Collado-Vides, J. (2003). Identifying global regulators in transcriptional regulatory networks in bacteria. Curr. Opin. Microbiol. 6, 482–489. doi: 10.1016/j.mib.2003.09.002
Martín-Trillo, M., and Cubas, P. (2010). TCP genes: a family snapshot ten years later. Trends Plant Sci. 15, 31–39. doi: 10.1016/j.tplants.2009.11.003
McLeay, R. C., and Bailey, T. L. (2010). Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11:165. doi: 10.1186/1471-2105-11-165
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in Proceedings of the 2010 Gateway Computing Environments Workshop (GCE) (New Orleans, LA: IEEE), 1–8. doi: 10.1109/GCE.2010.5676129
Neal, P. R., Dafni, A., and Giurfa, M. (1998). Floral symmetry and its role in plant-pollinator sysTEMS: terminology, distribution, and hypotheses. Annu. Rev. Ecol. Syst. 29, 345–373. doi: 10.1146/annurev.ecolsys.29.1.345
Niu, S.-S., Xu, C.-J., Zhang, W.-S., Zhang, B., Li, X., Lin-Wang, K., et al. (2010). Coordinated regulation of anthocyanin biosynthesis in Chinese bayberry (Myrica rubra) fruit by a R2R3 MYB transcription factor. Planta 231, 887–899. doi: 10.1007/s00425-009-1095-z
Odom, D. T., Dowell, R. D., Jacobsen, E. S., Nekludova, L., Rolfe, P. A., Danford, T. W., et al. (2006). Core transcriptional regulatory circuitry in human hepatocytes. Mol. Syst. Biol. 2:2006.0017. doi: 10.1038/msb4100059
Ó’Maoiléidigh, D. S., Graciet, E., and Wellmer, F. (2014). Gene networks controlling Arabidopsis thaliana flower development. New Phytol. 201, 16–30. doi: 10.1111/nph.12444
O’Meara, B. C., Smith, S. D., Armbruster, W. S., Harder, L. D., Hardy, C. R., Hileman, L. C., et al. (2016). Non-equilibrium dynamics and floral trait interactions shape extant angiosperm diversity. Proc. R. Soc. B Biol. Sci. 283:20152304. doi: 10.1098/rspb.2015.2304
Parapunova, V., Busscher, M., Busscher-Lange, J., Lammers, M., Karlova, R., Bovy, A. G., et al. (2014). Identification, cloning and characterization of the tomato TCP transcription factor family. BMC Plant Biol. 14:157. doi: 10.1186/1471-2229-14-157
Pasqualucci, L., Migliazza, A., Basso, K., Houldsworth, J., Chaganti, R. S. K., and Dalla-Favera, R. (2003). Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood 101, 2914–2923. doi: 10.1182/blood-2002-11-3387
Peirson, S. N., Butler, J. N., and Foster, R. G. (2003). Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 31:e73. doi: 10.1093/nar/gng073
Perez-Rodriguez, M., Jaffe, F. W., Butelli, E., Glover, B. J., and Martin, C. (2005). Development of three different cell types is associated with the activity of a specific MYB transcription factor in the ventral petal of Antirrhinum majus flowers. Development 132, 359–370. doi: 10.1242/dev.01584
Pinho, R., Garcia, V., Irimia, M., and Feldman, M. W. (2014). Stability depends on positive autoregulation in boolean gene regulatory networks. PLoS Comput. Biol. 10:e1003916. doi: 10.1371/journal.pcbi.1003916
Preston, J. C., and Hileman, L. C. (2010). SQUAMOSA-PROMOTER BINDING PROTEIN 1 initiates flowering in Antirrhinum majus through the activation of meristem identity genes. Plant J. Cell Mol. Biol. 62, 704–712. doi: 10.1111/j.1365-313X.2010.04184.x
Preston, J. C., and Hileman, L. C. (2012). Parallel evolution of TCP and B-class genes in Commelinaceae flower bilateral symmetry. EvoDevo 3:6. doi: 10.1186/2041-9139-3-6
Raimundo, J., Sobral, R., Bailey, P., Azevedo, H., Galego, L., Almeida, J., et al. (2013). A subcellular tug of war involving three MYB-like proteins underlies a molecular antagonism in Antirrhinum flower asymmetry. Plant J. 75, 527–538. doi: 10.1111/tpj.12225
Reyes, E., Sauquet, H., and Nadot, S. (2016). Perianth symmetry changed at least 199 times in angiosperm evolution. Taxon 65, 945–964. doi: 10.12705/655.1
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rosenfeld, N., Elowitz, M. B., and Alon, U. (2002). Negative autoregulation speeds the response times of transcription networks. J. Mol. Biol. 323, 785–793. doi: 10.1016/S0022-2836(02)00994-4
Ruokolainen, S., Ng, Y. P., Broholm, S. K., Albert, V. A., Elomaa, P., and Teeri, T. H. (2010). Characterization of SQUAMOSA-like genes in Gerbera hybrida, including one involved in reproductive transition. BMC Plant Biol. 10:128. doi: 10.1186/1471-2229-10-128
Sargent, R. D. (2004). Floral symmetry affects speciation rates in angiosperms. Proc. R. Soc. Lond. B Biol. Sci. 271, 603–608. doi: 10.1098/rspb.2003.2644
Sauquet, H., von Balthazar, M., Magallón, S., Doyle, J. A., Endress, P. K., Bailes, E. J., et al. (2017). The ancestral flower of angiosperms and its early diversification. Nat. Commun. 8:16047. doi: 10.1038/ncomms16047
Shepard, K. A., and Purugganan, M. D. (2002). The genetics of plant morphological evolution. Curr. Opin. Plant Biol. 5, 49–55. doi: 10.1016/S1369-5266(01)00227-8
Singh, A., and Hespanha, J. P. (2009). Evolution of gene auto-regulation in the presence of noise. IET Syst. Biol. 3, 368–378. doi: 10.1049/iet-syb.2009.0002
Smith, J. F., Hileman, L. C., Powell, M. P., and Baum, D. A. (2004). Evolution of GCYC, a Gesneriaceae homolog of CYCLOIDEA, within Gesnerioideae (Gesneriaceae). Mol. Phylogenet. Evol. 31, 765–779. doi: 10.1016/j.ympev.2003.09.012
Stanke, M., Steinkamp, R., Waack, S., and Morgenstern, B. (2004). AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312. doi: 10.1093/nar/gkh379
Starkeviè, P., Paukštytë, J., Kazanavièiûtë, V., Denkovskienë, E., Stanys, V., Bendokas, V., et al. (2015). Expression and anthocyanin biosynthesis-modulating potential of sweet cherry (Prunus avium L.) MYB10 and bHLH Genes. PLoS One 10:e0126991. doi: 10.1371/journal.pone.0126991
Su, S., Xiao, W., Guo, W., Yao, X., Xiao, J., Ye, Z., et al. (2017). The CYCLOIDEA–RADIALIS module regulates petal shape and pigmentation, leading to bilateral corolla symmetry in Torenia fournieri (Linderniaceae). New Phytol. 215, 1582–1593. doi: 10.1111/nph.14673
Tao, Z., Shen, L., Liu, C., Liu, L., Yan, Y., and Yu, H. (2012). Genome-wide identification of SOC1 and SVP targets during the floral transition in Arabidopsis. Plant J. 70, 549–561. doi: 10.1111/j.1365-313X.2012.04919.x
Vamosi, J. C., and Vamosi, S. M. (2010). Key innovations within a geographical context in flowering plants: towards resolving Darwin’s abominable mystery. Ecol. Lett. 13, 1270–1279. doi: 10.1111/j.1461-0248.2010.01521.x
van Nocker, S., Berry, G., Najdowski, J., Michelutti, R., Luffman, M., Forsline, P., et al. (2012). Genetic diversity of red-fleshed apples (Malus). Euphytica 185, 281–293. doi: 10.1007/s10681-011-0579-7
Vincent, C. A., and Coen, E. S. (2004). A temporal and morphological framework for flower development in Antirrhinum majus. Can. J. Bot. 82, 681–690. doi: 10.1139/b04-042
Wang, B., Zhang, N., Guo, C.-C., Xu, G.-X., Kong, H.-Z., and Shan, H.-Y. (2012). Evolutionary divergence of the APETALA1 and CAULIFLOWER proteins. J. Syst. Evol. 50, 502–511. doi: 10.1111/j.1759-6831.2012.00211.x
Wang, Z., Luo, Y., Li, X., Wang, L., Xu, S., Yang, J., et al. (2008). Genetic control of floral zygomorphy in pea (Pisum sativum L.). Proc. Natl. Acad. Sci. 105, 10414–10419. doi: 10.1073/pnas.0803291105
Xu, S., Luo, Y., Cai, Z., Cao, X., Hu, X., Yang, J., et al. (2013). Functional Diversity of CYCLOIDEA-like TCP genes in the control of zygomorphic flower development in Lotus japonicus. J. Integr. Plant Biol. 55, 221–231. doi: 10.1111/j.1744-7909.2012.01169.x
Yang, X., Cui, H., Yuan, Z.-L., and Wang, Y.-Z. (2010). Significance of consensus CYC-binding sites found in the promoters of both ChCYC and ChRAD genes in Chirita heterotricha (Gesneriaceae). J. Syst. Evol. 48, 249–256. doi: 10.1111/j.1759-6831.2010.00086.x
Yang, X., Pang, H.-B., Liu, B.-L., Qiu, Z.-J., Gao, Q., Wei, L., et al. (2012). Evolution of double positive autoregulatory feedback loops in CYCLOIDEA2 clade genes is associated with the origin of floral zygomorphy. Plant Cell 24, 1834–1847. doi: 10.1105/tpc.112.099457
Yanhui, C., Xiaoyuan, Y., Kun, H., Meihua, L., Jigang, L., Zhaofeng, G., et al. (2006). The MYB transcription factor superfamily of Arabidopsis: expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol. Biol. 60, 107–124. doi: 10.1007/s11103-005-2910-y
Ye, L., Wang, B., Zhang, W., Shan, H., and Kong, H. (2016). Gains and Losses of Cis-regulatory elements led to divergence of the Arabidopsis APETALA1 and CAULIFLOWER duplicate genes in the time, space, and level of expression and regulation of one paralog by the other. Plant Physiol. 171, 1055–1069. doi: 10.1104/pp.16.00320
Yuan, Z., Gao, S., Xue, D.-W., Luo, D., Li, L.-T., Ding, S.-Y., et al. (2009). RETARDED PALEA1 controls palea development and floral zygomorphy in rice. Plant Physiol. 149, 235–244. doi: 10.1104/pp.108.128231
Zhang, W., Kramer, E. M., and Davis, C. C. (2010). Floral symmetry genes and the origin and maintenance of zygomorphy in a plant-pollinator mutualism. Proc. Natl. Acad. Sci. U.S.A. 107, 6388–6393. doi: 10.1073/pnas.0910155107
Zhong, J., and Kellogg, E. A. (2015). Stepwise evolution of corolla symmetry in CYCLOIDEA2-like and RADIALIS-like gene expression patterns in Lamiales. Am. J. Bot. 102, 1260–1267. doi: 10.3732/ajb.1500191
Zhong, J., Preston, J. C., Hileman, L. C., and Kellogg, E. A. (2017). Repeated and diverse losses of corolla bilateral symmetry in the Lamiaceae. Ann. Bot. 119, 1211–1223. doi: 10.1093/aob/mcx012
Keywords: CYCLOIDEA, evolution, flower development, symmetry, transcriptional autoregulation
Citation: Sengupta A and Hileman LC (2018) Novel Traits, Flower Symmetry, and Transcriptional Autoregulation: New Hypotheses From Bioinformatic and Experimental Data. Front. Plant Sci. 9:1561. doi: 10.3389/fpls.2018.01561
Received: 11 May 2018; Accepted: 05 October 2018;
Published: 26 October 2018.
Edited by:
Verónica S. Di Stilio, University of Washington, United StatesReviewed by:
Lydia Gramzow, Friedrich-Schiller-Universität Jena, GermanyAna Maria Rocha De Almeida, California State University, East Bay, United States
Copyright © 2018 Sengupta and Hileman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lena C. Hileman, bGhpbGVtYW5Aa3UuZWR1