 Laura Piñeiro Fernández1,2*
Laura Piñeiro Fernández1,2* Kelsey J. R .P. Byers2,3
Kelsey J. R .P. Byers2,3 Jing Cai2,4
Jing Cai2,4 Khalid E. M. Sedeek2,5,6Roman T. Kellenberger2,7
Khalid E. M. Sedeek2,5,6Roman T. Kellenberger2,7 Alessia Russo1,2,8Weihong Qi9Catharine Aquino Fournier9
Alessia Russo1,2,8Weihong Qi9Catharine Aquino Fournier9 Philipp M. Schlüter1*
Philipp M. Schlüter1*- 1Institute of Botany, University of Hohenheim, Stuttgart, Germany
- 2Department of Systematic and Evolutionary Botany, University of Zurich, Zurich, Switzerland
- 3Department of Zoology, University of Cambridge, Cambridge, United Kingdom
- 4Center for Ecological and Environmental Sciences, Northwestern Polytechnical University, Xi’an, China
- 5Laboratory for Genome Engineering and Synthetic Biology, Division of Biological Sciences, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
- 6Agricultural Genetic Engineering Research Institute (AGERI), Agriculture Research Centre, Giza, Egypt
- 7Department of Plant Sciences, University of Cambridge, Cambridge, United Kingdom
- 8Department of Plant and Microbial Biology, University of Zurich, Zurich, Switzerland
- 9Functional Genomics Centre Zurich, Zurich, Switzerland
The orchids (Orchidaceae) constitute one of the largest and most diverse families of flowering plants. They have evolved a great variety of adaptations to achieve pollination by a diverse group of pollinators. Many orchids reward their pollinators, typically with nectar, but the family is also well-known for employing deceptive pollination strategies in which there is no reward for the pollinator, in the most extreme case by mimicking sexual signals of pollinators. In the European flora, two examples of these different pollination strategies are the sexually deceptive genus Ophrys and the rewarding genus Gymnadenia, which differ in their level of pollinator specialization; Ophrys is typically pollinated by pseudo-copulation of males of a single insect species, whilst Gymnadenia attracts a broad range of floral visitors. Here, we present and describe the annotated floral transcriptome of Ophrys iricolor, an Andrena-pollinated representative of the genus Ophrys that is widespread throughout the Aegean. Furthermore, we present additional floral transcriptomes of both sexually deceptive and rewarding orchids, specifically the deceptive Ophrys insectifera, Ophrys aymoninii, and an updated floral transcriptome of Ophrys sphegodes, as well as the floral transcriptomes of the rewarding orchids Gymnadenia conopsea, Gymnadenia densiflora, Gymnadenia odoratissima, and Gymnadenia rhellicani (syn. Nigritella rhellicani). Comparisons of these novel floral transcriptomes reveal few annotation differences between deceptive and rewarding orchids. Since together, these transcriptomes provide a representative sample of the genus-wide taxonomic diversity within Ophrys and Gymnadenia (Orchidoideae: Orchidinae), we employ a phylogenomic approach to address open questions of phylogenetic relationships within the genera. Specifically, this includes the controversial placement of O. insectifera within the Ophrys phylogeny and the placement of “Nigritella”-type morphologies within the phylogeny of Gymnadenia. Whereas in Gymnadenia, several conflicting topologies are supported by a similar number of gene trees, a majority of Ophrys gene topologies clearly supports a placement of O. insectifera as sister to a clade containing O. sphegodes.
Introduction
Orchidaceae and Asteraceae constitute the largest families of flowering plants. Over 800 orchid genera and 25,000 species have been described, with an average rate of 500 species and 13 genera described per year (Cribb et al., 2003; Chase et al., 2015). Orchids have colonized a great variety of geographical ranges, from Scandinavia to Tierra del Fuego (Antonelli et al., 2009; Domínguez and Bahamonde, 2013), although the vast majority of species occur in tropical and neotropical areas (Dressler, 1993). The key to their success has variously been hypothesized to reside in their epiphytic habitat (for tropical orchids) or in their high level of pollinator specialization (Gravendeel et al., 2004; Cozzolino and Widmer, 2005). About two thirds of orchid species present rewards to their visitors, in most cases, nectar (Dafni and Ivri, 1979; Bell et al., 2009; Johnson et al., 2013). These rewarding species are commonly generalized in their pollination, attracting a wide range of pollinators (Brantjes, 1981; Claessens and Kleynen, 2017). However, the ability to produce nectar is missing in one third of species across the family. Instead, they have developed alternative mechanisms based on deception (Ackerman, 1986; Jersáková et al., 2006; Schiestl and Schlüter, 2009; Johnson and Schiestl, 2016). Some of these mechanisms target generalist pollinators, e.g., food deception, where orchids attract pollinators by advertising floral cues that resemble those from rewarding plants (Salzmann et al., 2007; Braunschmid et al., 2017). On the other hand, orchids have also developed mechanisms such as sexual deception to attract highly specialized pollinators. Sexually deceptive flowers produce chemical signals that mimic the sexual pheromones of pollinators, and thus, lead the pollinators to “pseudo-copulate“ with the flowers (Kullenberg and Bergström, 1976; Paulus and Gack, 1990; Schiestl et al., 1999). Examples of such behaviour occur in the Australian Chiloglottis spp. (Mant et al., 2002; Schiestl et al., 2003), or the recently discovered sexually deceptive Caladenia abbreviata (Phillips and Peakall, 2018).
In the European flora, one can find representatives of the aforementioned pollination strategies in the sexually deceptive genus Ophrys and the rewarding genus Gymnadenia, both within the subtribe Orchidinae (subfamily Orchidoideae) (Inda et al., 2012). Orchids from the Mediterranean genus Ophrys attract male pollinators by means of sexual deception (Paulus and Gack, 1990; Ayasse et al., 2000; Schiestl et al., 2000). Attractiveness to pollinators in the genus is highly species-specific, that is, each Ophrys species normally attracts a single pollinator species (Paulus and Gack, 1990; Paulus, 2018) by releasing chemicals (for solitary bees, mostly alkenes) mimicking the female sex pheromones (Schiestl et al., 2000; Schlüter and Schiestl, 2008; Xu et al., 2012). This high specificity acts as a pre-zygotic barrier and facilitates reproductive isolation between orchid species (Xu et al., 2011; Xu et al., 2012; Paulus, 2018). Ophrys is a recently diverged genus (crown age estimated ca. 5 Ma) with ancestral wasp pollination (Breitkopf et al., 2015), but extant species are commonly pollinated by solitary bees, e.g. Eucera or Andrena (Paulus and Gack, 1990; Gaskett, 2011). Successful floral isolation and species divergence in the genus may easily be achieved by shifts between similar pollinators, where small changes in genes involved in the pheromone profiles can lead to attraction of new, related pollinators (Schlüter et al., 2011; Sedeek et al., 2014; Schlüter, 2018). For instance, after two independent shifts to (mostly) Andrena solitary bee pollination (Breitkopf et al., 2015), two parallel adaptive radiations have taken place simultaneously within the last ca. 1 Ma, yielding two major clades, the Ophrys sphegodes and the Ophrys fusca species complexes. In line with its recent radiation, a large amount of genetic polymorphism is shared across closely related species within the O. sphegodes complex, which has been attributed to common ancestry rather than independent mutations or recent hybridization, although a hybridization event prior to radiation seems distinctly possible (Sedeek et al., 2014; Roma et al., 2018; Cozzolino et al., 2019). Coalescence theory predicts that in the case of a radiation, the time of coalescence of these polymorphic alleles will often predate the split of species (Takahata, 1989). Yet, or maybe because of this, phylogenetic relationships within Ophrys remain controversial, with different markers in the genome potentially painting different pictures of relationships (Cozzolino et al., 2019). Phylogenetically, the ca. 10 main Ophrys lineages are split into three major clades (where clade α includes Ophrys insectifera, β includes the O. fusca s.l. lineage and γ includes the O. sphegodes s.l. lineage) and the relationships among major lineages within these clades are relatively clear, although one major question remains unclear. In particular, the placement of the wasp-pollinated O. insectifera L. (clade α) within the Ophrys phylogeny has been suggested to be either the earliest-branching lineage [topology: (α,(β,γ))] or more closely related to the O. sphegodes lineage [topology: (β,(α,γ))] (cf. e.g. Breitkopf et al., 2015; Bateman et al., 2018b, and references therein).
The Eurasian genus Gymnadenia is characterized by fragrant, purple to white, resupinate flowers that mainly attract diurnal and nocturnal Lepidoptera species offering nectar as a reward. Although they attract a wide range of Lepidoptera, and some species are found in sympatry, pollinator overlap is minimal between most species (Vöth, 2000; Huber et al., 2005; Claessens and Kleynen, 2011) and strong pollinator-mediated reproductive isolation has been reported between the putative sister species G. odoratissima (L.) Richard and Gymnadenia conopsea (L.) Brown (Sun et al., 2015). The latter species is strongly genetically differentiated from the morphologically similar taxon G. densiflora (Wahlenberg) Dietrich (Stark et al., 2011). Finally, the Alpine G. rhellicani (Teppner & E. Klein) Teppner & E. Klein (syn. Nigritella rhelliani) represents a morphologically distinct lineage within the genus, characterized by extremely dense inflorescences, generally dark red and without resupination, i.e. the labellum remains pointing upwards as opposed to rotated downwards as in other Gymnadenia species. The former genus Nigritella was merged into Gymnadenia only following molecular phylogenies (Hedrén et al., 2000). Previous phylogenetic analysis have shown that Gymnadenia odoratissima is sister to Gymnadenia conopsea, and Gymnadenia densiflora forms a clade with Gymnadenia rhellicani (Bateman et al., 2003; Sun et al., 2015). However, these relationships remain contentious, since other studies support a sister-group relationship among Nigritella and the “classical” genus Gymnadenia (Hedrén et al., 2000; Brandrud et al., 2019). Hence, further attention is warranted, especially to clarify the position of Nigritella. The age of the most recent common ancestor shared among all Gymnadenia/Nigritella species is estimated to be around 2.5–3 Ma (Inda et al., 2012).
Due to the high taxonomic complexity of Orchidaceae, reconstructing phylogenetic patterns to understand relationships in the family remains challenging. In the last decades, phylogenetic studies in orchids moved from a morphological (Chittka and Menzel, 1992; Gravendeel et al., 2004) to a molecular approach aiming to provide a better insight into orchid relationships (Cameron et al., 1999; Stark et al., 2011; Inda et al., 2012; Breitkopf et al., 2015; Givnish et al., 2015; Bateman et al., 2018a). Previously, the focus of these analyses was at the level of using few genetic markers, e.g. ITS, to reconstruct phylogenies. However, this approach can be problematic as some markers are chosen by their relevance or suitability in a certain taxonomic group, even though they could present low resolution for certain taxonomic groups (Capella-Gutiérrez et al., 2014). Moreover, this approach generally focuses on estimating one coherent tree (e.g. by concatenating sequences), which ignores the fact that different loci can have different phylogenetic histories. Especially when dealing with recently diverged groups with incomplete lineage sorting (Pamilo and Nei, 1988), a genomic approach focusing on understanding patterns on different gene genealogies, may allow the quantification of the different phylogenetic scenarios and thus, be more informative on the evolutionary history of a group (Pease and Hahn, 2015; Pease et al., 2016). Orthologous genes, described as homologous genes that originated from a common ancestral gene as a result of the speciation process (Fitch, 1970), tend to retain the original function from the common ancestor over evolutionary time (Jensen, 2001). Thus, groups of orthologous genes within gene families, together with a genome-wide approach, are perfect candidates to resolve orchid phylogeny and effectively clarify their relationships in an evolutionary framework (Li et al., 2003; Deng et al., 2015).
Here, we present the novel floral transcriptome of the Mediterranean sexually deceptive orchid Ophrys iricolor Desf., a representative of the genus Ophrys in the Aegean area, which is considered to be a member of the O. fusca group (clade β) and represents the evolutionarily distinct abdomen-pollinated members of the genus (previous section Pseudophrys) (Schlüter et al., 2009). In addition, we present several floral transcriptomes of both rewarding and deceptive orchids of the subtribe Orchidinae, particularly the rewarding orchids G. conopsea, G. densiflora, G. odoratissima, and G. rhellicani, together with the sexually deceptive O. insectifera, Ophrys aymoninii (Breistroffer) Buttler, and finally, an updated transcriptome of O. sphegodes s.l. (Sedeek et al., 2013). Using a set of orthologous genes, we employ a genome-wide approach to phylogenetic analysis of these novel floral transcriptomes together with published orchid transcriptomic/genomic data, to compare the transcriptomes of deceptive and rewarding orchids. Furthermore, as these transcriptomes cover the genus-wide taxonomic diversity within Ophrys and Gymnadenia, our objectives are to elucidate (1) the placement of the O. insectifera complex within the three major clades in the Ophrys phylogeny, (2) the placement of the morphologically distinct G. rhellicani (and presumably other members of subgenus Nigritella) within the phylogeny of Gymnadenia and (3) whether there is evidence of introgression due to shared pollinators in distinct Ophrys lineages.
Material and Methods
Plant Material
The novel Ophrys iricolor s.l. (O. iricolor s.s. and Ophrys mesaritica H.F. Paulus, C. Alibertis & A. Alibertis) cross-species transcriptome is presented here. Data from the putative sister species O. iricolor s.s. and O. mesaritica (Schlüter et al., 2009) were assembled into a single transcriptome due to expected high levels of allele sharing among the group, as seen in the O. sphegodes complex (Sedeek et al., 2013; Sedeek et al., 2014). Sample size (Ophrys iricolor s.l., N = 16 biological replicates; O. sphegodes s.l., N = 37) and provenance are listed in Table 1. The previously published cross-species O. sphegodes s.l. (O. exaltata subsp. archipelagi (Gölz & H.R. Reinhard) Del Prete, O. garganica Nelson ex O. & E. Danesch, and O. sphegodes Miller) transcriptome (Sedeek et al., 2013) is here updated with data from additional samples, including from O. incubacea Bianca (samples from Sedeek et al., 2014) within the same species complex that is characterized by the aforementioned high levels of allele and transcript sharing among species (Sedeek et al., 2013; Sedeek et al., 2014) and is hence covered in a single cross-species transcriptome assembly. Additionally, O. insectifera and O. aymoninii transcriptomes are also presented here. Data from O. insectifera and O. aymoninii (collected in Gervasi et al., 2017), were assembled into separate transcriptomes because these species are pollinated by different types of pollinators (O. insectifera is wasp-pollinated, while O. aymoninii is Andrena-pollinated) and the assumption of high levels of within-group allele sharing cannot be made. Finally, sampled flowers from the clearly distinct species G. conopsea, G. densiflora, G. odoratissima and G. rhellicani (from Kellenberger et al., 2019) were used to create individual transcriptome assemblies for these species to complement the published cross-species Gymnadenia transcriptome assembly (N = 10, Table 1) (Kellenberger et al., 2019). As far as it was possible to ascertain pollination status (not always possible for Gymnadenia flowers), all samples used in this study were from unpollinated flowers of diploid individuals. Flowers were flash-frozen and stored at ‑80°C until RNA extraction was conducted as detailed by Kellenberger et al. (2019). Since polyploids are known from Gymnadenia and (occasionally) Ophrys and to ensure that all samples sequenced were diploid, ploidy levels of O. iricolor and O. mesaritica were checked via flow cytometry of pollinia as described by Xu et al. (2011) using a Cell Lab Quanta™ SC-MPL flow cytometer (Beckman Coulter, Fullerton, Canada). Phaseolus coccineus “Scarlett Emperor” (sativa Rheinau SG, Switzerland) leaf material was used as internal standard. Ploidy levels were previously described for O. sphegodes s.l. (Sedeek et al., 2014), O. insectifera and O. aymoninii (Gervasi et al., 2017) and the four Gymnadenia species (Kellenberger et al., 2019) used in this study, including all sequenced individuals.
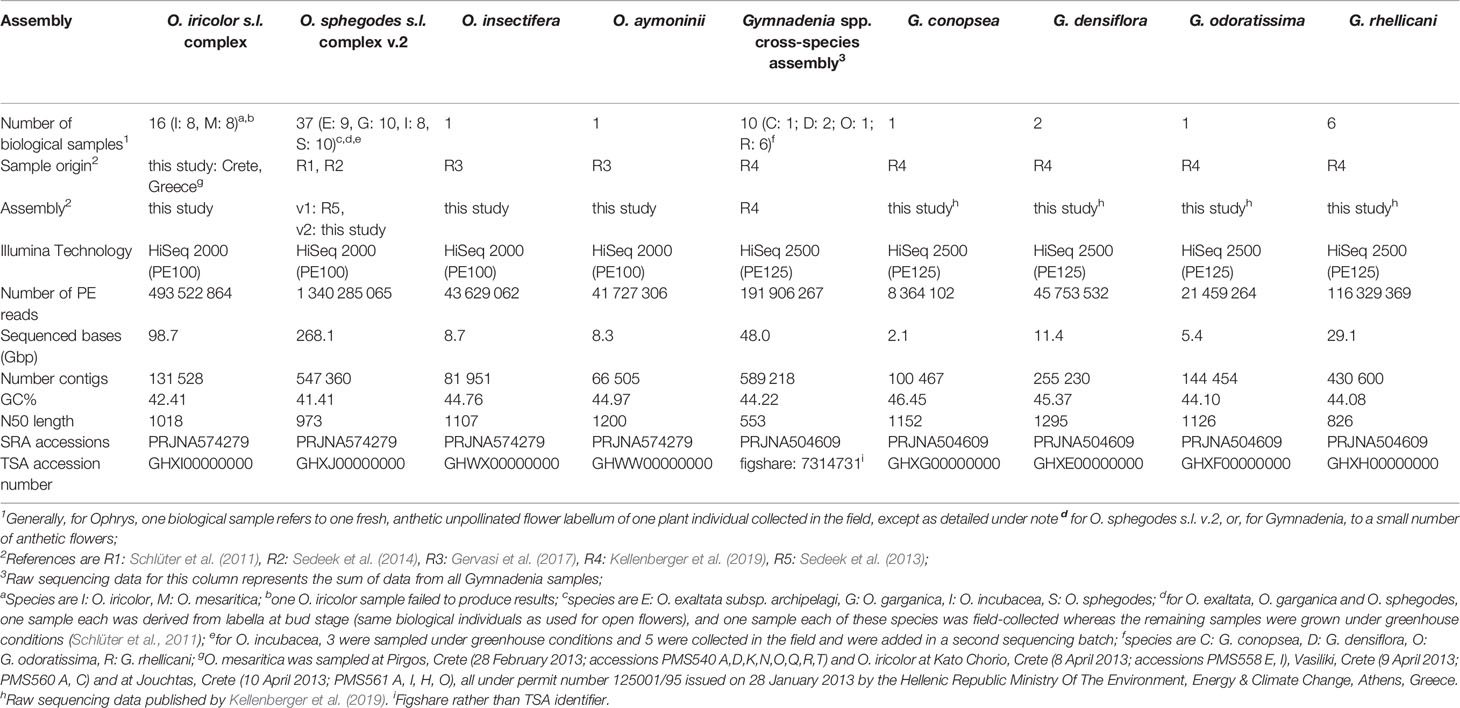
Table 1 Statistics of transcriptomic data for each species/assembly.
RNA Extraction, Library Preparation and Sequencing
Total RNA was extracted separately for each biological individual and tissue with TRIzol reagent (Thermo Fisher Scientific, Massachusetts) according to the manufacturer's protocol followed by a purification step using Qiagen RNeasy MinElute Cleanup Kit (Qiagen, Netherlands). Quality of the isolated RNA was determined with a Qubit® (1.0) Fluorometer (Life Technologies, California, USA) and a Bioanalyzer 2100 (Agilent, Waldbronn, Germany). Paired-end sequencing was performed on the Illumina HiSeq 2000 or 2500 platforms (Illumina, Inc, California, USA) for Ophrys and Gymnadenia samples (Table 1), generating separate files for each biological sample.
Transcriptome Assemblies and Functional Annotation
Individual reads were first aligned to PhiX Control library (Illumina) sequences using bowtie2 v2.2.4 (Langmead and Salzberg, 2012) to remove sequencing control reads. Filtered reads were trimmed using Trimmomatic v. 0.36 (Bolger et al., 2014) to remove any Illumina adapters. Surviving reads were then de-novo assembled to transcripts using Trinity r20140717/v. 2.0.618 (Grabherr et al., 2011). In the case of O. sphegodes, where a previous assembly based on 454, Solexa and Sanger data was available (Sedeek et al., 2013), additional Illumina HiSeq reads were assembled with Trinity as described above and then merged with the published assembly using cd-hit-est (Li and Godzik, 2006; Fu et al., 2012) (95% sequence identity threshold with full length alignment coverage for the shorter sequence). Protein coding regions were analysed using TransDecoder r20140704 (http://transdecoder.github.io) (Haas et al., 2013). The assembled contigs were annotated with the standard Trinotate annotation pipeline (https://trinotate.github.io/) (Grabherr et al., 2011) against Swissprot (Boeckmann et al., 2003), Pfam (Finn et al., 2014), TmHMM (Krogh et al., 2001), Gene Ontology (Ashburner et al., 2000) and SignalP (Petersen et al., 2011). Due to high levels of overlap among the four single-species Gymnadenia transcriptomes (Figure S1B), we annotated only the cross-species Gymnadenia transcriptome from all four species. For purposes of comparison, we also updated the annotation of the previously published, updated (v.2) transcriptome of O. sphegodes (Sedeek et al., 2013) with Trinotate. Finally, to estimate the completeness of the transcriptomes, we performed a BUSCO v3.1.0 assessment (Simão et al., 2015) with the lineage databases embryophyta_odb10 and liliopsida_odb10 (Figures 1A, B).
Phylogenomic Analysis
OrthoMCL v2.0.9 (Li et al., 2003) was used under the MySQL v14.14 server to identify orthologous groups based on annotated coding sequences (CDS) (where no annotated CDS were available, they were derived by TransDecoder as above) of 15 members of the Orchidaceae family including the above described Ophrys and the four Gymnadenia single-species transcriptome assemblies together with the transcriptomes/genomes of Apostasia shenzhenica and Phalaenopsis equesteris (Zhang et al., 2017), Dactylorhiza fuchsii (Balao et al., 2017), Chiloglottis trapeziformis (Wong et al., 2017), Dendrobium catenatum (Zhang et al., 2016), and Platanthera clavellata and Goodyera pubescens (retrieved from the 1KP project; http://www.onekp.com/). Following the TranslatorX pipeline (Abascal et al., 2010), sequences were aligned using Mafft v7.407 (Katoh and Standley, 2013). To construct phylogenetic trees, a pipeline as described in Xu et al. (2017) was followed. In brief, poorly aligned sequences were removed using trimal v1.2 (Capella-Gutiérrez et al., 2009). Selection of the best-fit models of nucleotide substitution was performed with jModelTest 2.1.10 (Santorum et al., 2014), with parameters: -f -i -g 4 -a -AIC -s 3. This allowed the inclusion of models with unequal base frequencies, a proportion invariable sites, rate variation among sites and set 4 categories, model-averaged phylogeny for each active criterion. Moreover, it used AIC (Akaike Information Criterion) for model selection and accounted for 3 substitution schemes. Maximum likelihood trees of the best-fit models were calculated with phyML 3.3 (Guindon and Gascuel, 2003). For each taxonomically fully sampled orthologous group, tree topologies from Ophrys and Gymnadenia single-copy gene branches were extracted. In addition, we also extracted topologies where one Ophrys species was missing. The extraction of tree topologies was automated with an in-house R script. Moreover, for both Ophrys and Gymnadenia, we extracted topologies where gene duplications happened only within a monophyletic group of a given species. In the latter case, all but one of the duplicate tips was dropped from the phylogeny (keep.tip function from the package ape for R v3.5.0) (R Core Team, 2001). After retrieving (rooted) topologies of target groups, we compared these topologies with Robinson-Foulds distances, where a distance of 0 indicates that topologies are in full agreement with each other (Robinson and Foulds, 1981), using the package phytools (Revell, 2012) for R. Tree visualization was performed using the Bioconductor package Ggtree (Yu et al., 2017) for R. Finally, we compared the annotation, particularly the GO Plant Slim terms, of the different topologies observed for Ophrys and Gymnadenia.
Results
Transcriptome Assemblies and Functional Annotation
All Ophrys individuals were diploid (Figure S2 for O. iricolor s.l.), consistent with previous studies (Xu et al., 2011; Sedeek et al., 2014). After sequencing, a total of 493.5 million paired-end (PE) reads from O. iricolor and 191.9 million from Gymnadenia were produced (Table 1). All the raw sequencing data (totalling 431.8 Gbp from 2111 million PE reads) are available in the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) under the accession numbers in Table 1. We successfully produced 131,528 and 589,218 contigs (Table 1) for O. iricolor and for the Gymnadenia cross-species assembly, respectively, corresponding to 88,664 and 174,633 Coding Sequences (CDSes) (Table 2). The remaining sequences did not match any known gene from the databases queried. The annotation tables can be downloaded from figshare (links in Table 2). Based on the three main Gene Ontology categories (biological process, cellular component, and molecular function), we compared the 14 most common GO Plant Slim terms (Clark et al., 2005) in the Gymnadenia spp. cross-species, O. iricolor and the updated O. sphegodes transcriptomes (Table 1, Figure 2). To avoid overrepresentation of general terms such as “metabolic” or “cellular” processes, we omitted the first 7, 3, and 3 terms for Biological Process, Cellular Component and Molecular Function, respectively. Overall, the three transcriptomes are very similar in GO terms. The main differences between O. sphegodes and O. iricolor are the lack of terms related to “response to endogenous stimulus” in O. iricolor, and the presence of terms related to “vacuole” in O. sphegodes (Figures 2A, B). On the other hand, the Gymnadenia transcriptome differs from the Ophrys transcriptome by showing a high number of genes related to “nucleus” processes and an absence of those related to "membrane" processes (Figure 2B). Finally, BUSCO assessments with the embryophyta lineage database indicated that the completeness of the transcriptomes was 93.4, 91.9, and 94.1% for O. iricolor, O. sphegodes v.2, and cross-species Gymnadenia transcriptomes, respectively (Figure 1A). These results therefore suggest a reasonably high assembly quality of our floral transcriptomes, especially when compared with fully sequenced orchid genomes (encoding the transcripts of all tissues), i.e. the Apostasia genome with a 93.62% completeness, 94.45% in Phalaenopsis equestris and 95.49% in Dendrobium catenatum (all using the embryophyta database) (Zhang et al., 2017). Also, with 87.6, 85.5, and 87% for the larger BUSCO liliopsida lineage database (Figure 1B), for O. iricolor, O. sphegodes v.2 and cross-species Gymnadenia transcriptomes, respectively, our transcriptomes appear relatively complete with respect to monocot-specific genes.
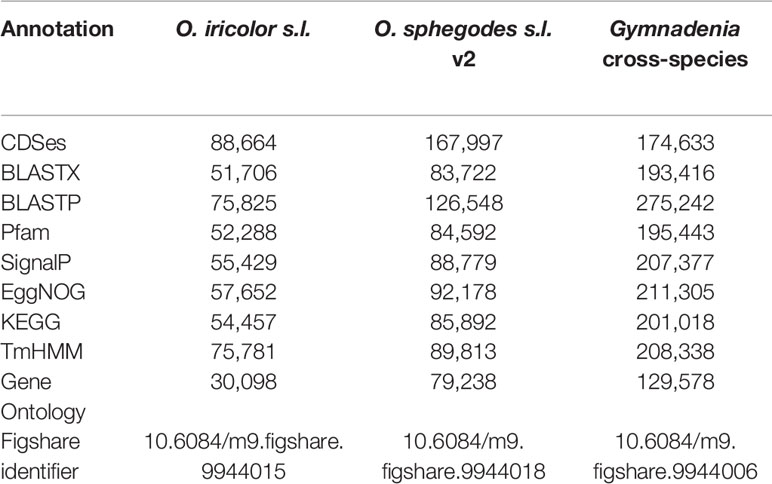
Table 2 Annotation statistics.
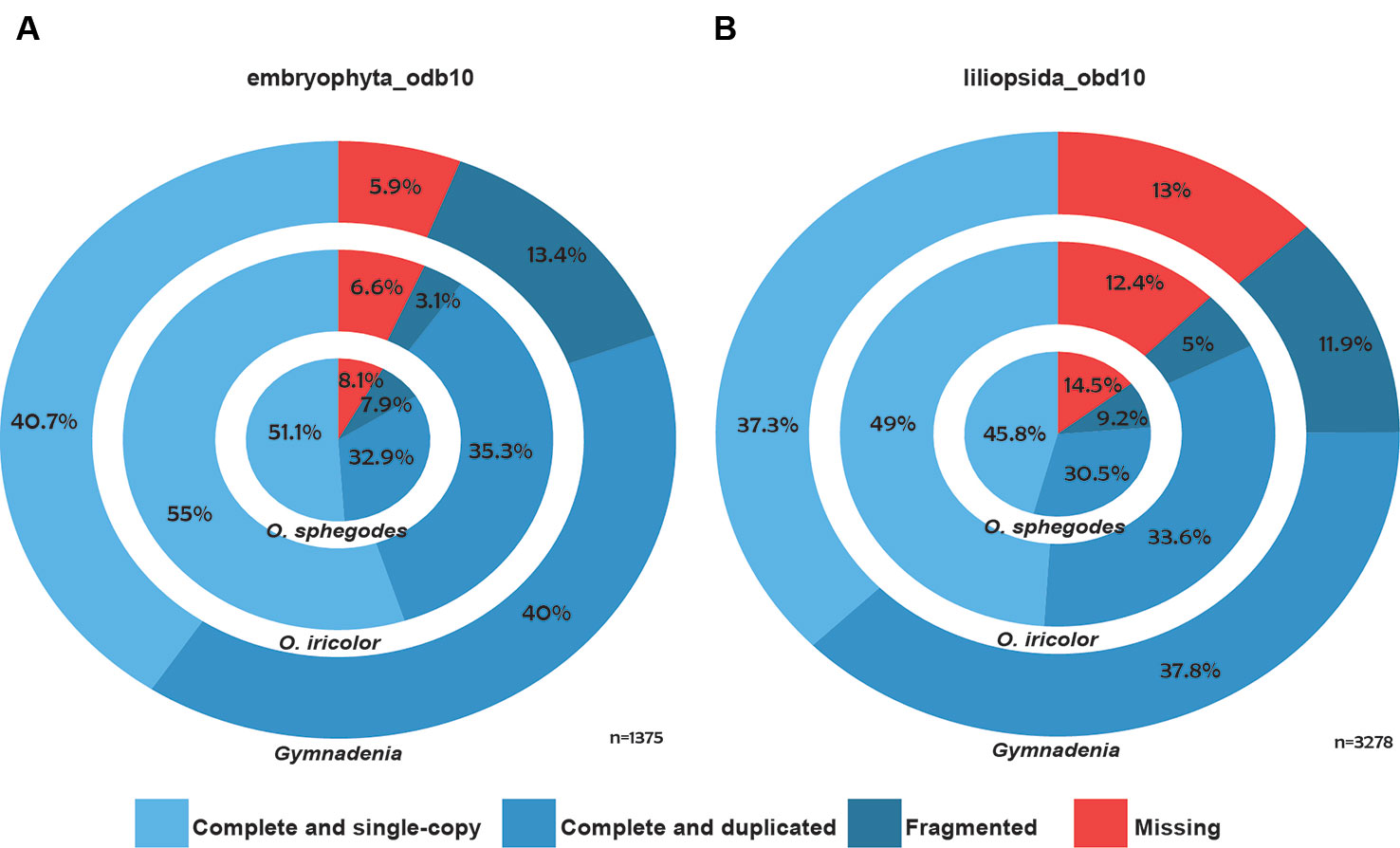
Figure 1 BUSCO assessment. Concentric circles show the BUSCO assessment of O. sphegodes v.2, O. iricolor and cross-species Gymnadenia spp. transcriptomes (from inside to outside), where the first three (blue) categories together are taken as an estimation of transcriptome “completeness”. (A) BUSCO results with embryophyta_odb10 and (B) the larger liliopsida_odb10 databases.
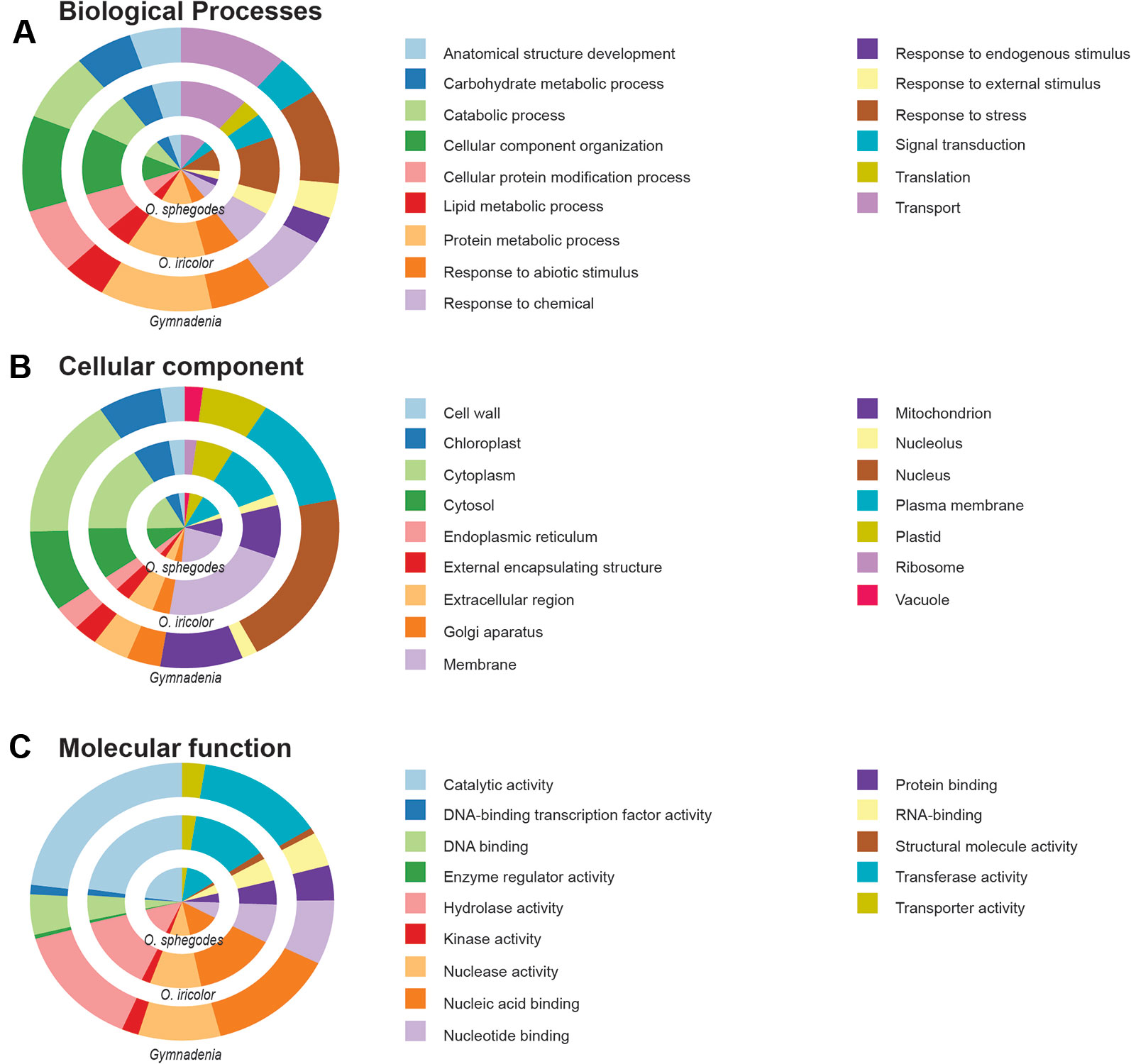
Figure 2 GO Plant Slim functional annotation. GO Plant Slim annotation of the three transcriptomes for the most common (A) Biological Process, (B) Cellular Component and (C) Molecular Function terms.
Phylogenomic Analysis
Overall, we found the 15 orchid species included in this study to share a total of 1,749 gene families. From these gene family phylogenies, 226 contain Ophrys monophyletic groups with no gene duplications sampled from all Ophrys species and 160 contain Gymnadenia monophyletic groups with no gene duplications sampled from all Gymnadenia species separately. In addition, 116 contain informative topologies with one Ophrys species missing; and 153 and 318 topologies contain gene duplications (or alleles) within single-species monophyletic groups for Ophrys and Gymnadenia, respectively. For Ophrys and Gymnadenia, we found 5 and 6 of the 15 possible rooted topologies for four taxa, respectively. In Ophrys, the most common topology (77% of the trees) suggests that O. insectifera s.l. (with O. aymoninii) is not the basalmost clade, but instead places it in a clade with O. sphegodes, whereas O. iricolor takes the basal position (Figures 3A, B). This is also evident from the consensus tree over all orthologous gene groups (Figure 3C) and from all (100%) of the trees missing one Ophrys species (Figure 3B). In the case of Gymnadenia, the distribution of topologies is more even. Yet, the most common topology, supported by 33% of the trees, places G. rhellicani at the basal position in the Gymnadenia tree (Figure 4). Overall, a total of 48% of evaluated Gymnadenia genes show a topology that places G. rhellicani as a sister to all other species. Also, strikingly, only 33% of gene topologies support a sister-species relationship among G. conopsea and G. odoratissima. We compared the GO annotations of each topology in Ophrys and Gymnadenia, but despite some annotation differences between topologies, there is no clear pattern with respect to putatively pollinator-relevant features (Figures S3 and S4). Although not significant, the two most common Ophrys topologies also show the highest average branch length (Figure S5A), whereas two less common topologies have the longest branch lengths in Gymnadenia (Figure S5B, non-significant); these are not united by a common phylogenetic theme (e.g. with respect to G. rhellicani).
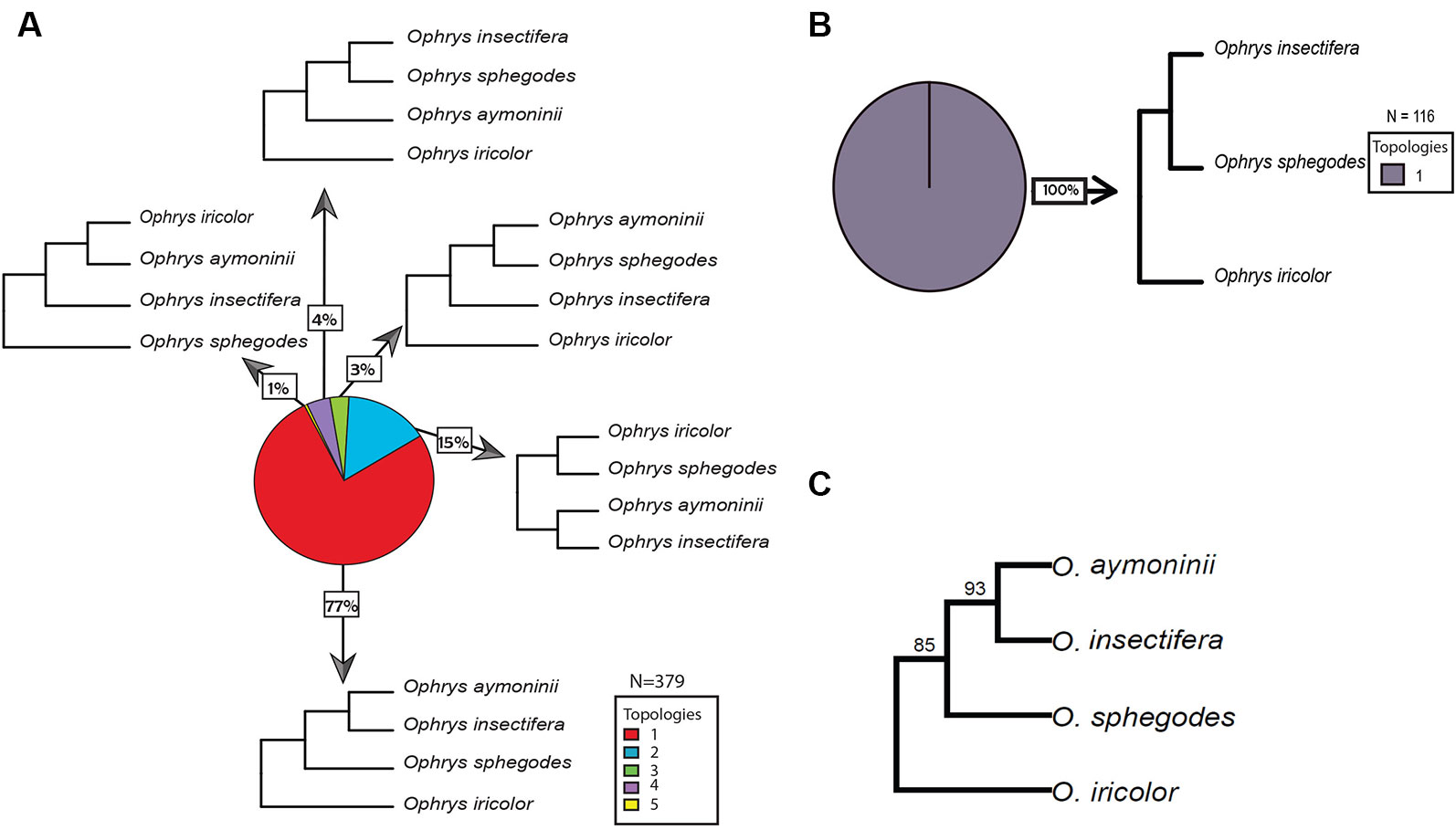
Figure 3 Distribution of gene tree topologies in Ophrys. Proportions of rooted orthologous gene tree topologies (shown without branch lengths) for Ophrys. (A) overview of four-species Ophrys topologies; (B) three-species Ophrys topology; (C) majority-rule consensus tree from all individual 4-species gene trees, where numbers above branches indicate the percentage of individual gene trees supporting a group.
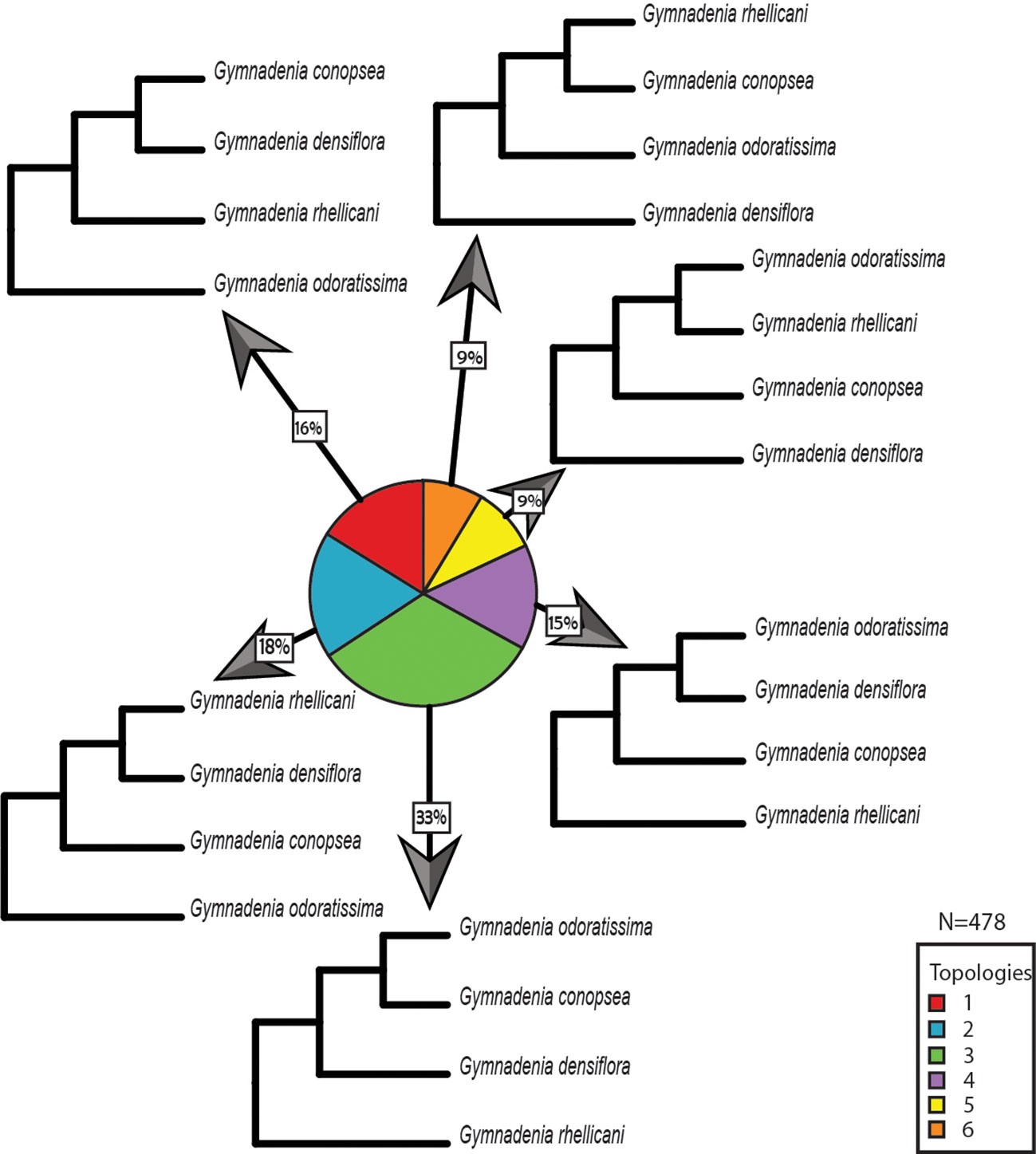
Figure 4 Distribution of gene tree topologies in Gymnadenia. Proportions of rooted orthologous gene tree topologies (shown without branch lengths) for Gymnadenia.
Discussion
This study provides significant new transcriptome sequence resources aimed to improve our knowledge about the highly complex Orchidaceae family. Specifically, we present novel floral transcriptomes of several members of the subtribe Orchidinae of the Orchidoideae subfamily, covering both sexually deceptive and rewarding orchids. Overall, there were no striking differences between sexually deceptive and rewarding orchids when comparing the most common annotation terms based on Gene Ontology categories. This is not a surprise, because the GO Slim categories approach, although providing a large vocabulary to describe the functional categories, also suffers from a lack of clarity and too broad definitions, resulting in only a vague overview of molecular biology (Smith et al., 2003). At the same time, the phylogenetic proximity of Ophrys and Gymnadenia provides a plausible explanation for the lack of strong differentiation in terms of GO categories and suggests that differences in pollination strategy do not require fundamental changes in the genome-wide repertoire of florally expressed genes. This is in line with the phylogenetic lability of pollination strategies reported within the Orchidinae (Inda et al., 2012).
However, clear differences between Ophrys and Gymnadenia are apparent in terms of the transcriptome-wide distribution of gene tree topologies. For phylogeny reconstructions, rather than concatenating sequences, we evaluated multiple gene family trees separately. Trees derived from concatenated sequences do not reveal discrepancies between individual genes that are expected under a standard coalescent process, i.e., the more recent a species split is, the more tree topologies are expected due to incomplete lineage sorting (Takahata, 1989). Disagreement between gene trees and species trees has been observed in an increasing number of studies suggesting that the combination of a large amount of ancestral polymorphism and post-speciation gene flow between taxa can lead to large systematic differences between gene and species trees (Green et al., 2010; Novikova et al., 2016; Filiault et al., 2018; Malinsky et al., 2018).
Interestingly, the two most common gene tree topologies recovered for Ophrys reflect previous published phylogenetic reconstructions, our topologies 1 and 2 (Figure 3) corresponding to the phylogenies published most recently by Bateman et al. (2018b) and Breitkopf et al. (2015), respectively. Breitkopf et al.'s reconstruction suggested the O. insectifera group (clade α, including O. aymoninii) as the basal clade on the tree. By contrast, the phylogenetic reconstruction by Bateman et al. places O. insectifera closer to the O. sphegodes group, whereas a lineage containing the O. fusca complex (clade β, here represented by O. iricolor) is the earliest diverged. Our results, with a consensus of 85% of gene topologies, overwhelmingly support the inner placement of O. insectifera, rather than a basal position (Figure 3C). However, with the wasp-pollinated O. insectifera sister to the clade containing O. sphegodes and the wasp-pollinated O. speculum sister to the clade containing O. iricolor/O. fusca, the phylogeny's implication for the ancestral mode of pollination remains unchanged; the inference of ancestral wasp pollination in the genus Ophrys (Breitkopf et al., 2015) therefore seems unaffected by our findings. Nevertheless, it is striking that we found no strong evidence for discordant phylogenies throughout the genome.
Since the O. insectifera-group member O. aymoninii, a narrow endemic in southern France, is Andrena-pollinated (Paulus and Gack, 1990; Gervasi et al., 2017), phylogenies placing O. aymoninii together with the other Andrena-pollinated linages, O. sphegodes and/or O. iricolor could be (but need not be) an indication of hybridization and introgression via Andrena pollinators. Although our analysis recovers phylogenies (Figure 3A, topologies 3 and 5) consistent with this hypothesis, with only 4% of the gene trees overall, support for pollinator-mediated introgression is weak at best.
Unlike Ophrys with a clearly predominant phylogeny across the transcriptome, Gymnadenia presents a much less clear picture of species relationships. The sister relationship between G. conopsea and G. odoratissima has been supported in several previous studies (e.g. Bateman et al., 2003; Sun et al., 2015) including by a recent genome-wide RAD-Seq (concatenated) SNP data set (Brandrud et al., 2019). This relationship is here supported by the most common topology in the transcriptome (Figure 4, topology 3). Yet this is also the only topology that supports this relationship, accounting for only 33% of orthologous gene groups evaluated. We must therefore conclude that, from a genomic perspective, the sister relationship of G. conopsea and G. odoratissima is not beyond doubt.
The genus Gymnadenia now typically includes its former sister genus Nigritella as a subgenus. Initial hypotheses built on morphological data (Wucherpfennig, 2002), anthocyanin pigments (Strack et al., 1989), or AFLP markers (Ståhlberg, 1999) suggested the separation of the two genera. Early molecular phylogenies (usually based solely on ITS) typically sampled only G. conopsea, G. odoratissima, and a single member of Nigritella, which was generally the outgroup to the sister Gymnadenia species (Hedrén et al., 2000). When additional species were sampled and added to this basic phylogeny, G. densiflora (or, depending on the sampling, G. borealis) was shown to be the sister taxon to members of Nigritella, arguing for combining the genera (Pridgeon et al., 1997; Bateman et al., 1997; Bateman et al., 2003; Stark et al., 2011; Efimov, 2013). Addition of three nuclear genes did not change this topology (Rey, 2011; Sun et al., 2015). Interestingly, where authors considered multiple phylogenetic methods, conflict seems to arise in tree construction, with parsimony showing Nigritella as the outgroup to G. conopsea/G. densiflora/G. odoratissima/G. borealis, while Bayesian and maximum likelihood analyses demonstrate a sister relationship between Nigritella and either G. borealis or G. densiflora (Rey, 2011; Inda et al., 2012). In a major upgrade to the generic phylogeny, Brandrud et al. (2019) performed RAD-Seq, with contrasting results to the ITS-based phylogenies. Their phylogeny shows four Nigritella species as the outgroup to five Gymnadenia species, with no sister relation between G. densiflora and Nigritella, and the relevant nodes show high support.
Given the often contradictory results of earlier circumscription attempts, it is perhaps not too surprising that the different Gymnadenia gene topologies are relatively evenly distributed and that we see no single Gymnadenia phylogeny standing out as the best supported tree. However, the most common gene tree topology shows G. rhellicani as the outgroup to the other three sampled species (Figure 4, topology 3), in agreement with the recent RAD-Seq-based concatenated SNP analysis by Brandrud et al. (2019). Nonetheless, overall support for versus against a basal position of G. rhellicani is equivocal, at 48% of gene trees for (topologies 3 and 4) versus 52% against a basal position. The prevalence of other supported topologies (generally with G. odoratissima rather than G. rhellicani as the outgroup) suggests a complex population genetic history within the genus, perhaps partially due to gene exchange and incomplete lineage sorting. Neither gene annotation (Figure S4) nor average gene tree branch lengths (Figure S5) for topologies with basal G. rhellicani placement stand out as an indication of adaptive processes. Although Gymnadenia and Nigritella have produced one stable hybrid offspring, the apomict G. runei (Teppner and Klein, 1989) and other hybrids may be found, some dispute about their frequency exists (Claessens and Kleynen, 2011; Brandrud et al., 2019). Taken together, our analysis of Gymnadenia hints at a complex relationship among species that we are only beginning to understand. Whether this apparently more complicated pattern of genome-wide relationships in Gymnadenia as compared to Ophrys is due to the difference in pollination systems is currently unclear, although Gymnadenia's less specialized pollination strategy would certainly present more opportunities for hybridization.
Using multiple gene family trees instead of one concatenated tree has proven to be a useful approach (Boussau et al., 2013; One Thousand Plant Transcriptomes Initiative, 2019). Concatenation of sequences implies that loci with a larger number of phylogenetically informative sites can bias the inference such that it may not be representative of patterns of unlinked genes throughout the genome. Also, such an approach holds no explicit information about the specific other topologies that may be useful for disentangling more complex evolutionary patterns of relationships throughout the genome, as would clearly be of interest in cases such as Gymnadenia. This problem is likely to be more severe in phylogenies of closely related species where excessive incomplete linage sorting may be expected and where a more sophisticated coalescent-based analysis may be valuable. Additionally, a consensus tree of individual gene trees (e.g. Figure 3C) is informative of the proportions of those genes in the genome that support a certain species relationship. Moreover, it is important to note that unlike a bootstrap pseudoreplicate approach, this allows for real quantification of proportions of independently segregating loci and/or functional genes and is thus more biologically meaningful. So far, our analysis only covers a small part of the genome. However, given a high-quality genome reference, future integration of this approach along chromosomes may be able to reconstruct the ancestry of individual chromosomal fragments and thereby shed light on the detailed evolutionary patterns and the role of selection (see Filiault et al., 2018) in shaping lineage divergence. The significant new sequence resources provided in this study may be a first step towards realizing this goal for European orchids in the future.
Data Availability Statement
The datasets analysed for this study can be found in the NCBI accessions PRJNA574279 and PRJNA504609, and as indicated in Table 1 and 2.
Author Contributions
Designed the project: PS. Drafted the manuscript: LP, with assistance from PS, KB, and RK. Revised the manuscript: all authors. Extracted material and prepared the libraries: JC, KS, and RK. Sequenced and processed the raw data: WQ, CA. Assembled and annotated the transcriptomes: LP, KB, JC, AR, and WQ. Conducted phylogenomic analysis: LP. Interpreted the results: LP, KB, RK, and PS. Acquired funding: KB, RK, and PS.
Funding
This work was financially supported by the Swiss National Science Foundation (SNF) (31003A_155943 to PS). Additional support came from the University of Zurich Research Priority Programme “Evolution in Action” (PS/AR and F. Schiestl/RK), a University of Zurich Forschungskredit grant to RK, a PLANT FELLOWS Postdoctoral Fellowship grant to KB (European Union: FP7-PEOPLE-2010-COFUND Proposal 267243), and several travel grants from the Georges & Antoine Claraz Foundation Zurich (to LP, RK, KB, and PS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Kriton Kalantidis, University of Crete, for access to his lab and support during sampling. We thank Karin Gross, Danae Laina and Daniel Gervasi for help with field work and sample collection, Shuqing Xu for sharing his scripts, and Florian Schiestl for support and sharing data. We also thank our funding sources as well as members of the Schlüter and Schiestl labs for assistance and helpful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01553/full#supplementary-material
References
Abascal, F., Zardoya, R., Telford, M. J. (2010). TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38, 7–13. doi: 10.1093/nar/gkq291
Ackerman, J. D. (1986). Mechanisms and evolution of food-deceptive pollination system in orchids. Lindleyana 1, 108–113.
Antonelli, A., Dahlberg, C. J., Carlgren, K. H. I., Appelqvist, T. (2009). Pollination of the lady's slipper orchid (Cypripedium calceolus) in Scandinavia - Taxonomic and conservational aspects. Nord. J. Bot. 27, 266–273. doi: 10.1111/j.1756-1051.2009.00263.x
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25. doi: 10.1038/75556
Ayasse, M., Schiestl, F. P., Paulus, H. F., Löfstedt, C., Hansson, B., Ibarra, F., et al. (2000). Evolution of reproductive strategies in the sexually deceptive orchid Ophrys sphegodes: how does flower-specific variation of odor signals influence reproductive success? Evolution 54, 1995–2006. doi: 10.1111/j.0014-3820.2000.tb01243.x
Balao, F., Trucchi, E., Wolfe, T. M., Hao, B. H., Lorenzo, M. T., Baar, J., et al. (2017). Adaptive sequence evolution is driven by biotic stress in a pair of orchid species (Dactylorhiza) with distinct ecological optima. Mol. Ecol. 26, 3649–3662. doi: 10.1111/mec.14123
Bateman, R. M., Pridgeon, A. M., Chase, M. W. (1997). Phylogenetics of subtribe Orchidinae (Orchidoideae, Orchidaceae) based on nuclear ITS sequences. 2. Infrageneric relationships and reclassification to achieve monophyly of Orchis sensu stricto. Lindleyana 12, 113–141.
Bateman, R. M., Hollingsworth, P. M., Preston, J., Yi-Bo, L., Pridgeon, A. M., Chase, M. W. (2003). Molecular phylogenetics and evolution of Orchidinae and selected Habenariinae (Orchidaceae). Bot. J. Linn. Soc. 142, 1–40. doi: 10.1046/j.1095-8339.2003.00157.x
Bateman, R. M., Murphy, A. R. M., Hollingsworth, P. M., Hart, M., Denholm, I., Rudall, P. J. (2018a). Molecular and morphological phylogenetics of the digitate-tubered clade within subtribe Orchidinae s.s. (Orchidaceae: Orchideae). Kew Bull. 73, 54. doi: 10.1007/s12225-018-9782-1
Bateman, R. M., Sramkó, G., Paun, O. (2018b). Integrating restriction site-associated DNA sequencing (RAD-seq) with morphological cladistic analysis clarifies evolutionary relationships among major species groups of bee orchids. Ann. Bot. 121, 85–105. doi: 10.1093/aob/mcx129
Bell, A. K., Roberts, D. L., Hawkins, J. A., Rudall, P. J., Box, M. S., Bateman, R. M. (2009). Comparative micromorphology of nectariferous and nectarless labellar spurs in selected clades of subtribe Orchidinae (Orchidaceae). Bot. J. Linn. Soc. 160, 369–387. doi: 10.1111/j.1095-8339.2009.00985.x
Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M.-C., Estreicher, A., Gasteiger, E., et al. (2003). The SWISS-PROT protein knowledge base and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Boussau, B., Szöllosi, G. J., Duret, L., Gouy, M., Tannier, E., Daubin, V. (2013). Genome-scale coestimation of species and gene trees. Genome Res. 23, 323–330. doi: 10.1101/gr.141978.112
Brandrud, M. K., Paun, O., Lorenz, R., Baar, J., Hedrén, M. (2019). Restriction-site associated DNA sequencing supports a sister group relationship of Nigritella and Gymnadenia (Orchidaceae). Mol. Phylogenet. Evol. 136, 21–28. doi: 10.1016/j.ympev.2019.03.018
Brantjes, N. B. M. (1981). Ant, bee and fly pollination in Epipactis palustris (L.) Crantz (Orchidaceae). Acta Botanica Neerl. 30, 59–68. doi: 10.1111/j.1438-8677.1981.tb00387.x
Braunschmid, H., Mükisch, B., Rupp, T., Schäffler, I., Zito, P., Birtele, D., et al. (2017). Interpopulation variation in pollinators and floral scent of the lady's-slipper orchid Cypripedium calceolus L. Arthropod-Plant Interact. 11, 363–379. doi: 10.1007/s11829-017-9512-x
Breitkopf, H., Onstein, R. E., Cafasso, D., Schlüter, P. M., Cozzolino, S. (2015). Multiple shifts to different pollinators fuelled rapid diversification in sexually deceptive Ophrys orchids. New Phytol. 1, 377–389. doi: 10.1111/nph.13219
Cameron, K. M., Chase, M. W., Whitten, W. M., Kores, P. J., Jarrell, D. C., Albert, V. A., et al. (1999). A phylogenetic analysis of the Orchidaceae: Evidence from rbcL nucleotide sequences. Am. J. Bot. 86, 208–224. doi: 10.2307/2656938
Capella-Gutiérrez, S., Silla-Martínez, J. M., Gabaldón, T. (2009). TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Capella-Gutiérrez, S., Kauff, F., Gabaldón, T. (2014). A phylogenomics approach for selecting robust sets of phylogenetic markers. Nucleic Acids Res. 42, e54. doi: 10.1093/nar/gku071
Chase, M. W., Cameron, K. M., Freudenstein, J. V., Pridgeon, A. M., Salazar, G., Berg, C., et al. (2015). An updated classification of Orchidaceae. Bot. J. Linn. Soc. 177, 151–174. doi: 10.1111/boj.12234
Chittka, L., Menzel, R. (1992). The evolutionary adaptation of flower colours and the insect pollinators' colour vision. J. Comp. Physiol. A Neuroethology Sens. Neural Behav. Physiol. 171, 171–181. doi: 10.1007/BF00188925
Claessens, J., Kleynen, J. (2011). The Flower of the European Orchid: Form and Function, Claessens and Kleynen.
Claessens, J., Kleynen, J. (2017). The Pollination of European Orchids Part 6: nectar as attractant: Gymnadenia conopsea and Neottia ovata. J. Hardy Orchid Soc. 14, 110–136.
Clark, J. I., Brooksbank, C., Lomax, J. (2005). It's all GO for plant scientists. Plant Physiol. 138, 1268–1279. doi: 10.1104/pp.104.058529
Cozzolino, S., Widmer, A. (2005). Orchid diversity: An evolutionary consequence of deception? Trends Ecol. Evol. 20, 487–494. doi: 10.1016/j.tree.2005.06.004
Cozzolino, S., Roma, L., Schlüter, P. M., Scopece, G. (2019). Different filtering strategies of Genotyping-By-Sequencing data provide complementary resolutions of species boundaries and relationships in a clade of sexually deceptive orchids. J. Syst. Evol. in press. doi: 10.1111/jse.12493
Cribb, P. J., Kell, S. P., Dixon, K. W., Barrett, R. L. (2003). “Orchid conservation: a global perspective,” in Dixon, K. W., Kell, S. P., Barrett, R. L., Cribb, P. J. (eds.) Orchid Conservation, (Kinabalu, Malaysia: Natural History Publications, Kota). 1–24.
Dafni, A., Ivri, Y. (1979). Pollination ecology of, and hybridization between, Orchis coriophora L. and O. collina Sol. ex Russ. (Orchidaceae) in Israel. New Phytol. 83, 181–187. doi: 10.1111/j.1469-8137.1979.tb00740.x
Deng, H., Zhang, G. Q., Lin, M., Wang, Y., Liu, Z. J. (2015). Mining from transcriptomes: 315 single-copy orthologous genes concatenated for the phylogenetic analyses of Orchidaceae. Ecol. Evol. 5, 3800–3807. doi: 10.1002/ece3.1642
R Core Team. (2018). R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing: Vienna, Austria. https://www.R-project.org/.
Domínguez, E., Bahamonde, N. (2013). Gavilea araucana (Phil.) M. N. Correa: first record of an orchid for Chile on Sphagnum peatland in Magallanes. Biodiv. J. 4, 125–128.
Dressler, R. L. (1993). Phylogeny and Classification of the Orchid Family. Cambridge, UK: Cambridge University Press.
Efimov, P. G. (2013). Sibling species of fragrant orchids (Gymnadenia: Orchidaceae, Magnoliophyta) in Russia. Russian J. Genet. 49, 299–309. doi: 10.1134/S102279541302004X
Filiault, D. L., Ballerini, E. S., Mandáková, T., Aköz, G., Derieg, N. J., Schmutz, J., et al. (2018). The Aquilegia genome provides insight into adaptive radiation and reveals an extraordinarily polymorphic chromosome with a unique history. eLife 7, e36426. doi: 10.7554/eLife.36426
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: The protein families database. Nucleic Acids Res. 42, 138–141. doi: 10.1093/nar/gkt1223
Fitch, W. M. (1970). Distinguishing homologous from analogous proteins. Syst. Zool. 19, 99. doi: 10.2307/2412448
Fu, L., Niu, B., Zhu, Z., Wu, S., Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gaskett, A. C. (2011). Orchid pollination by sexual deception: Pollinator perspectives. Biol. Rev. 86, 33–75. doi: 10.1111/j.1469-185X.2010.00134.x
Gervasi, D. D. L., Selosse, M. A., Sauve, M., Francke, W., Vereecken, N. J., Cozzolino, S., et al. (2017). Floral scent and species divergence in a pair of sexually deceptive orchids. Ecol. Evol. 7, 6023–6034. doi: 10.1002/ece3.3147
Givnish, T. J., Spalink, D., Ames, M., Lyon, S. P., Hunter, S. J., Zuluaga, A., et al. (2015). Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proc. Roy. Soc. B. Biol. Sci. 282, 20151553. doi: 10.1098/rspb.2015.1553
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gravendeel, B., Smithson, A., Slik, F. J. W., Schuiteman, A. (2004). Epiphytism and pollinator specialization: Drivers for orchid diversity? Phil. Trans. Roy. Soc. B. Biol. Sci. 359, 1523–1535. doi: 10.1098/rstb.2004.1529
Green, R. E., Krause, J., Briggs, A. W., Maricic, T., Stenzel, U., Kircher, M., et al. (2010). A draft sequence of the Neandertal genome. Science 328, 710–722. doi: 10.1126/science.1188021
Guindon, S., Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hedrén, M., Klein, E., Teppner, H. (2000). Evolution of polyploids in the European orchid genus Nigritella: evidence from allozyme data. Phyton 40, 239–275.
Huber, F. K., Kaiser, R., Sauter, W., Schiestl, F. P. (2005). Floral scent emission and pollinator attraction in two species of Gymnadenia (Orchidaceae). Oecologia 142, 564–575. doi: 10.1007/s00442-004-1750-9
Inda, L. A., Pimentel, M., Chase, M. W. (2012). Phylogenetics of tribe Orchideae (Orchidaceae: Orchidoideae) based on combined DNA matrices: inferences regarding timing of diversification and evolution of pollination syndromes. Ann. Bot. 110, 71–90. doi: 10.1093/aob/mcs083
Jensen, R. A. (2001). Orthologs and paralogs - we need to get it right. Genome Biol. 2, interactions 1002.1–1002.3. doi: 10.1186/gb-2001-2-8-interactions1002
Jersáková, J., Johnson, S. D., Kindlmann, P. (2006). Mechanisms and evolution of deceptive pollination in orchids. Biol. Rev. 81, 219–235. doi: 10.1017/S1464793105006986
Johnson, S. D., Hobbhahn, N., Bytebier, B. (2013). Ancestral deceit and labile evolution of nectar production in the African orchid genus Disa. Biol. Lett. 9, 20130500. doi: 10.1098/rsbl.2013.0500
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kellenberger, R. T., Byers, K. J. R. P., De Brito Francisco, R. M., Staedler, Y. M., LaFountain, A. M., Schönenberger, J., et al. (2019). Emergence of a floral colour polymorphism by pollinator-mediated overdominance. Nat. Commun. 10, 63. doi: 10.1038/s41467-018-07936-x
Krogh, A., Larsson, È., Heijne, G. V., Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kullenberg, B., Bergström, G. (1976). The pollination of Ophrys orchids. Botaniska Notiser 29, 11–19.
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, W., Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, L., Stoeckert, C. J., Jr., Roos, D. S. (2003). OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Malinsky, M., Svardal, H., Tyers, A. M., Miska, E. A., Genner, M. J., Turner, G. F., et al. (2018). Whole-genome sequences of Malawi cichlids reveal multiple radiations interconnected by gene flow. Nat. Ecol. Evol. 2, 1940–1955. doi: 10.1038/s41559-018-0717-x
Mant, J. G., Schiestl, F. P., Peakall, R., Weston, P. H. (2002). A phylogenetic study of pollinator conservatism among sexually deceptive orchids. Evolution 56, 888–898. doi: 10.1554/0014-3820(2002)056[0888:apsopc]2.0.co;2
Novikova, P. Y., Hohmann, N., Nizhynska, V., Tsuchimatsu, T., Ali, J., Muir, G., et al. (2016). Sequencing of the genus Arabidopsis identifies a complex history of nonbifurcating speciation and abundant trans-specific polymorphism. Nat. Genet. 48, 1077–1082. doi: 10.1038/ng.3617
One Thousand Plant Transcriptomes Initiative. (2019). One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685. doi: 10.1038/s41586-019-1693-2
Pamilo, P., Nei, M. (1988). Relationships between gene trees and species trees. Mol. Biol. Evol. 5, 568–583. doi: 10.1093/oxfordjournals.molbev.a040517
Paulus, H. F., Gack, C. (1990). Pollinators as prepollinating isolation factors: Evolution and speciation in Ophrys (Orchidaceae). Isr. J. Bot. 39, 43–79.
Paulus, H. F. (2018). Pollinators as isolation mechanisms: field observations and field experiments regarding specificity of pollinator attraction in the genus Ophrys (Orchidaceae und Insecta, Hymenoptera, Apoidea). Entomol. Generalis 37, 261–316. doi: 10.1127/entomologia/2018/0650
Pease, J. B., Hahn, M. W. (2015). Detection and polarization of introgression in a five-taxon phylogeny. Syst. Biol. 64, 651–662. doi: 10.1093/sysbio/syv023
Pease, J. B., Haak, D. C., Hahn, M. W., Moyle, L. C. (2016). Phylogenomics reveals three sources of adaptive variation during a rapid radiation. PloS Biol. 14, e1002379. doi: 10.1371/journal.pbio.1002379
Petersen, T. N., Brunak, S., Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785. doi: 10.1038/nmeth.1701
Phillips, R. D., Peakall, R. (2018). Breaking the rules: discovery of sexual deception in Caladenia abbreviata (Orchidaceae), a species with brightly coloured flowers and a non-insectiform labellum. Aust. J. Bot. 66, 95–100. doi: 10.1071/BT17151
Pridgeon, A. M., Bateman, R. M., Cox, A. V., Hapeman, J. R., Chase, M. W. (1997). Phylogenetics of subtribe Orchidinae (Orchidoideae, Orchidaceae) based on nuclear ITS sequences. 1. Intergeneric relationships and polyphyly of Orchis sensu lato. Lindleyana 12, 89–109.
Revell, L. J. (2012). Phytools: an R package for phylogenetic comparative biology (and other things). Meth. Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Rey, M. (2011). Speciation in Gymnadenia (Orchidaceae): a phylogenetic reconstruction and underlying ecological processes. (master thesis) University of Zürich, Supervisor Florian P. Schiestl.
Robinson, D. F., Foulds, L. R. (1981). Comparison of phylogenetic trees. Math. Biosci. 53, 131–147. doi: 10.1016/0025-5564(81)90043-2
Roma, L., Cozzolino, S., Schlüter, P. M., Scopece, G., Cafasso, D. (2018). The complete plastid genomes of Ophrys iricolor and O. sphegodes (Orchidaceae) and comparative analyses with other orchids. PloS One 13, e0204174. doi: 10.1371/journal.pone.0204174
Salzmann, C. C., Nardella, A. M., Cozzolino, S., Schiestl, F. P. (2007). Variability in floral scent in rewarding and deceptive orchids: the signature of pollinator-imposed selection? Ann. Bot. 100, 757–765. doi: 10.1093/aob/mcm161
Santorum, J. M., Darriba, D., Taboada, G. L., Posada, D. (2014). Jmodeltest.org: selection of nucleotide substitution models on the cloud. Bioinformatics 30, 1310–1311. doi: 10.1093/bioinformatics/btu032
Schiestl, F. P., Schlüter, P. M. (2009). Floral isolation, specialized pollination, and pollinator behavior in orchids. Annu. Rev. Entomol. 54, 425–446. doi: 10.1146/annurev.ento.54.110807.090603
Schiestl, F. P., Ayasse, M., Paulus, H. F., Löfstedt, C., Hansson, B. S., Ibarra, F., et al. (1999). Orchid pollination by sexual swindle. Nature 399, 421–421. doi: 10.1038/20829
Schiestl, F. P., Ayasse, M., Paulus, H. F., Löfstedt, C., Hansson, B. S., Ibarra, F., et al. (2000). Sex pheromone mimicry in the early spider orchid (Ophrys sphegodes): patterns of hydrocarbons as the key mechanism for pollination by sexual deception. J. Comp. Physiol. 186, 567–574. doi: 10.1007/s003590000112
Schiestl, F. P., Peakall, R., Mant, J. G., Ibarra, F., Schulz, C., Franke, S., et al. (2003). The chemistry of sexual deception in an orchid-wasp pollination system. Science 302, 437–438. doi: 10.1126/science.1087835
Schlüter, P. M., Schiestl, F. P. (2008). Molecular mechanisms of floral mimicry in orchids. Trends Plant Sci. 13, 228–235. doi: 10.1016/j.tplants.2008.02.008
Schlüter, P. M., Ruas, P. M., Kohl, G., Ruas, C. F., Stuessy, T. F., Paulus, H. F. (2009). Genetic patterns and pollination in Ophrys iricolor and O. mesaritica (Orchidaceae): Sympatric evolution by pollinator shift. Bot. J. Linn. Soc. 159, 583–598. doi: 10.1111/j.1095-8339.2009.00957.x
Schlüter, P. M., Xu, S., Gagliardini, V., Whittle, E., Shanklin, J., Grossniklaus, U., et al. (2011). Stearoyl-acyl carrier protein desaturases are associated with floral isolation in sexually deceptive orchids. Proc. Natl. Acad. Sci. 108, 5696–5701. doi: 10.1073/pnas.1013313108
Schlüter, P. M. (2018). The magic of flowers or: speciation genes and where to find them. Am. J. Bot. 105, 1957–1961. doi: 10.1002/ajb2.1193
Sedeek, K. E. M., Qi, W., Schauer, M. A., Gupta, A. K., Poveda, L., Xu, S., et al. (2013). Transcriptome and proteome data reveal candidate genes for pollinator attraction in sexually deceptive orchids. PloS One 8, e64621. doi: 10.1371/journal.pone.0064621
Sedeek, K. E. M., Scopece, G., Staedler, Y. M., Schönenberger, J., Cozzolino, S., Schiestl, F. P., et al. (2014). Genic rather than genome-wide differences between sexually deceptive Ophrys orchids with different pollinators. Mol. Ecol. 23, 6192–6205. doi: 10.1111/mec.12992
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Smith, B., Williams, J., Steffen, S. (2003). The ontology of the Gene Ontology. AMIA Annu. Symp. Proc. 2003, 609.
Ståhlberg, D. (1999). Polyploid evolution in the European orchid genus Nigritella: evidence from DNA fingerprinting, (master thesis) Lund University, Supervisor Mikael Hedrén.
Stark, C., Michalski, S. G., Babik, W., Winterfeld, G., Durka, W. (2011). Strong genetic differentiation between Gymnadenia conopsea, and G. densiflora despite morphological similarity. Plant Syst. Evol. 293, 213–226. doi: 10.1007/s00606-011-0439-x
Strack, D., Busch, E., Klein, E. (1989). Anthocyanin patterns in european orchids and their taxonomic and phylogenetic relevance. Phytochemistry 28, 2127–2139. doi: 10.1016/S0031-9422(00)97931-7
Sun, M., Schlüter, P. M., Gross, K., Schiestl, F. P. (2015). Floral isolation is the major reproductive barrier between a pair of rewarding orchid sister species. J. Evol. Biol. 28, 117–129. doi: 10.1111/jeb.12544
Takahata, N. (1989). Gene genealogy in three related populations: consistency probability between gene and population trees. Genetics 122, 957–966.
Teppner, H., Klein, E. (1989). Gymnigritella runei spec. nova (Orchidaceae-Orchideae) aus Schweden. Phyton 29, 161–173.
Wong, D. C. J., Amarasinghe, R., Rodriguez-Delgado, C., Eyles, R., Pichersky, E., Peakall, R. (2017). Tissue-specific floral transcriptome analysis of the sexually deceptive orchid Chiloglottis trapeziformis provides insights into the biosynthesis and regulation of its unique UV-B dependent floral volatile, chiloglottone 1. Front. Plant Sci. 8, 1260. doi: 10.3389/fpls.2017.01260
Wucherpfennig, W. (2002). Nigritella: Gattung oder Untergattung? Jber. Naturwiss. Ver. Wuppertal 55, 46–61.
Xu, S., Schlüter, P. M., Scopece, G., Breitkopf, H., Gross, K., Cozzolino, S., et al. (2011). Floral isolation is the main reproductive barrier among closely related sexually deceptive orchids. Evolution 65, 2606–2620. doi: 10.1111/j.1558-5646.2011.01323.x
Xu, S., Schlüter, P. M., Schiestl, F. P. (2012). Pollinator-driven speciation in sexually deceptive orchids. Int. J. Ecol. 2012, 285081. doi: 10.1155/2012/285081
Xu, S., Brockmöller, T., Navarro-Quezada, A., Kuhl, H., Gase, K., Ling, Z., et al. (2017). Wild tobacco genomes reveal the evolution of nicotine biosynthesis. Proc. Natl. Acad. Sci. 114, 6133–6138. doi: 10.1073/pnas.1700073114
Yu, G., Smith, D. K., Zhu, H., Guan, Y., Lam, T. T. (2017). GGTREE: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Meth. Ecol. Evol. 8, 28–36. doi: 10.1111/2041-210X.12628
Zhang, G. Q., Xu, Q., Bian, C., Tsai, W., Yeh, C. (2016). The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep. 6, 19029. doi: 10.1038/srep19029
Keywords: phylogenomics, orchids, Ophrys, Gymnadenia, transcriptome, pollination strategy
Citation: Piñeiro Fernández L, Byers KJRP, Cai J, Sedeek KEM, Kellenberger RT, Russo A, Qi W, Aquino Fournier C and Schlüter PM (2019) A Phylogenomic Analysis of the Floral Transcriptomes of Sexually Deceptive and Rewarding European Orchids, Ophrys and Gymnadenia. Front. Plant Sci. 10:1553. doi: 10.3389/fpls.2019.01553
Received: 20 July 2019; Accepted: 07 November 2019;
Published: 29 November 2019.
Edited by:
Jen-Tsung Chen, National University of Kaohsiung, TaiwanReviewed by:
Ashley N. Egan, Aarhus University, DenmarkAlejandra Vázquez-Lobo, Universidad Autónoma del Estado de Morelos, Mexico
Copyright © 2019 Piñeiro Fernández, Byers, Cai, Sedeek, Kellenberger, Russo, Qi, Aquino Fournier and Schlüter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Piñeiro Fernández, bGF1cmEucGluZWlyb0BzeXN0Ym90LnV6aC5jaA==; Philipp M. Schlüter, cGhpbGlwcC5zY2hsdWV0ZXJAdW5pLWhvaGVuaGVpbS5kZQ==