 Maguta Kibe1,2
Maguta Kibe1,2 Sudha K. Nair3
Sudha K. Nair3 Biswanath Das1
Biswanath Das1 Jumbo M. Bright1
Jumbo M. Bright1 Dan Makumbi1Johnson Kinyua2
Dan Makumbi1Johnson Kinyua2 L. M. Suresh1
L. M. Suresh1 Yoseph Beyene1
Yoseph Beyene1 Michael S. Olsen1
Michael S. Olsen1 Boddupalli M. Prasanna1
Boddupalli M. Prasanna1 Manje Gowda1*
Manje Gowda1*- 1International Maize and Wheat Improvement Center, Nairobi, Kenya
- 2Jomo Kenyatta University of Agriculture and Technology, Nairobi, Kenya
- 3International Maize and Wheat Improvement Center, Hyderabad, India
Gray leaf spot (GLS) is one of the major maize foliar diseases in sub-Saharan Africa. Resistance to GLS is controlled by multiple genes with additive effect and is influenced by both genotype and environment. The objectives of the study were to dissect the genetic architecture of GLS resistance through linkage mapping and genome-wide association study (GWAS) and assessing the potential of genomic prediction (GP). We used both biparental populations and an association mapping panel of 410 diverse tropical/subtropical inbred lines that were genotyped using genotype by sequencing. Phenotypic evaluation in two to four environments revealed significant genotypic variation and moderate to high heritability estimates ranging from 0.43 to 0.69. GLS was negatively and significantly correlated with grain yield, anthesis date, and plant height. Linkage mapping in five populations revealed 22 quantitative trait loci (QTLs) for GLS resistance. A QTL on chromosome 7 (qGLS7-105) is a major-effect QTL that explained 28.2% of phenotypic variance. Together, all the detected QTLs explained 10.50, 49.70, 23.67, 18.05, and 28.71% of phenotypic variance in doubled haploid (DH) populations 1, 2, 3, and F3 populations 4 and 5, respectively. Joint linkage association mapping across three DH populations detected 14 QTLs that individually explained 0.10–15.7% of phenotypic variance. GWAS revealed 10 significantly (p < 9.5 × 10–6) associated SNPs distributed on chromosomes 1, 2, 6, 7, and 8, which individually explained 6–8% of phenotypic variance. A set of nine candidate genes co-located or in physical proximity to the significant SNPs with roles in plant defense against pathogens were identified. GP revealed low to moderate prediction correlations of 0.39, 0.37, 0.56, 0.30, 0.29, and 0.38 for within IMAS association panel, DH pop1, DH pop2, DH pop3, F3 pop4, and F3 po5, respectively, and accuracy was increased substantially to 0.84 for prediction across three DH populations. When the diversity panel was used as training set to predict the accuracy of GLS resistance in biparental population, there was 20–50% reduction compared to prediction within populations. Overall, the study revealed that resistance to GLS is quantitative in nature and is controlled by many loci with a few major and many minor effects. The SNPs/QTLs identified by GWAS and linkage mapping can be potential targets in improving GLS resistance in breeding programs, while GP further consolidates the development of high GLS-resistant lines by incorporating most of the major- and minor-effect genes.
Introduction
Maize is the most important cereal crop in sub-Saharan Africa (SSA), where more than 80% of the population rely on it as a source of food, income, and livelihood (Prasanna et al., 2020). Gray leaf spot (GLS) is a one of the major foliar diseases of maize caused by the polycyclic pathogens Cercospora zeae-maydis and Cercospora zeina (Crous et al., 2006; He et al., 2018). In eastern Africa, C. zeae-maydis is more prevalent. GLS poses a serious problem to maize production with estimated yield losses of more than 70% (Liu et al., 2016) under favorable conditions. The disease caused severe economic losses in SSA (Ward, 1996; Vivek et al., 2010; Kibata et al., 2011; Bekeko et al., 2018; Yigrem and Yohannes, 2019). Therefore, maize breeding programs in SSA typically incorporate GLS resistance in product pipelines.
Diagnostic symptoms of GLS include necrotic and chlorotic spots that run parallel to the leaf veins, rectangular fleck-type lesions; later-stage infection leads to severe blighting of leaves, stalk rotting, severe lodging, and premature death (Latterell and Rossi, 1983). Several factors contribute to the prevalence of GLS including conducive environment for disease development, monoculture of maize, and adoption of conservation tillage, which allows fungal inoculum to build up on crop residues. Moderate to high temperatures coupled with prolonged periods of high relative humidity also favor the development of disease symptoms (Ward, 1996). Chemical control has been recommended to combat GLS, but the application of fungicides is not economical especially for small and marginal farmers, and is also hazardous to human health with negative impacts on environment (Dhami et al., 2015). Breeding for resistant germplasm through conventional methods and by integrating advanced molecular tools is the most effective method to control diseases and to ensure maize-based food security in SSA.
Quantitative trait loci (QTLs) underlying resistance to several diseases in maize have been identified in the last two decades (Welz and Geiger, 2000; Shi et al., 2014; Xu et al., 2014; de Jong et al., 2018; He et al., 2018; Sitonik et al., 2019; Du et al., 2020; Lv et al., 2020). Despite the substantial number of QTLs reported, majority of them had huge confidence intervals, which represented large segments of chromosomes. In many cases flanking markers are very far from the causative mutations that can be easily lost during meiotic recombination and consequently limit their usefulness in breeding applications. Use of small mapping populations with low mapping resolution is also another major reason for the failure to identify robust and reliable markers associated with disease resistance. Although a biparental population-based genetic mapping approach offers high QTL detection power, the resolution remains low (Holland, 2007). In recent QTL mapping studies, the size of the mapping populations usually ranged between 100 and 300 individuals (Lehmensiek et al., 2001; Zwonitzer et al., 2010; Berger et al., 2014; He et al., 2018).
Genome-wide association studies (GWAS) have shown great potential by detecting QTL with high resolution, besides faster and accurate determination of recombination breakpoints (Zhu et al., 2008), but the detection power is fairly low and the false-positive rate is often high (Mammadov et al., 2015). Nevertheless, GWAS has been used successfully to identify QTL or genomic regions for some major diseases in maize at the whole-genome level, including maize lethal necrosis (MLN) (Gowda et al., 2015; Sitonik et al., 2019; Nyaga et al., 2020) and tar spot complex (Mahuku et al., 2015). GWAS of GLS was reported earlier in temperate maize germplasm (Benson et al., 2015; Mammadov et al., 2015) where significant SNPs associated with GLS resistance have been identified using diverse panels. However, GWAS for GLS resistance in tropical maize germplasm adapted to SSA agroecology was not reported so far.
Molecular marker-assisted breeding for improving disease resistance in maize is implemented in a few cases where QTLs with major effects were identified and validated; minor-effect QTLs are not part of the selection process, as these are often not consistent across different genetic backgrounds (Kuki et al., 2018). Genomic prediction (GP) is a newer approach that estimates the effects of all markers simultaneously, while omitting the stringent significance testing needed to identify QTL (Meuwissen et al., 2001). For GP, genetic markers spanning the whole genome are used with the assumption that all QTLs will be in linkage disequilibrium (LD) with at least one of these markers (Heffner et al., 2010). GP combines phenotypic and genotypic data of the training population to obtain genomic estimated breeding values (GEBVs) of the testing population that has been genotyped (Crossa et al., 2017; Wang et al., 2018). GP captures even the small effect QTLs/genes that are not detected by marker-assisted selection (Hayes et al., 2009).
In GP, the number of markers tend to be higher than the number of phenotypic observations; to account for this, GP applies various algorithms and models including genomic best linear unbiased prediction (GBLUP), rr-BLUP (Ridge regression), and Bayes (Crossa et al., 2017). GP reduces the time required for variety development and the cost per cycle as compared to phenotypic selection (Crossa et al., 2017; Beyene et al., 2019). However, applying GP in an association mapping population shows more effectiveness for traits possessing high heritability (Combs and Bernardo, 2013).
With this background, the objectives of the study were as follows: (1) to phenotypically characterize a genetically diverse association mapping panel and biparental populations for their responses to GLS, including correlation with other agronomic traits; (2) to conduct population-based QTL mapping and joint linkage association mapping (JLAM); (3) to identify marker-trait associations for GLS resistance through GWAS; and (4) to assess the usefulness of GP in breeding for GLS resistance in tropical maize.
Materials and Methods
Plant Materials and Field Trials
An association mapping panel developed at the international Maize and Wheat Improvement Center (CIMMYT), called the Improved Maize for African soils (IMAS) panel (Ertiro et al., 2020a), three doubled haploid (DH) populations [pop1 CML 550 x CML494 (107 lines); pop2 CML550 x CML504 (211 lines); pop3 CML550 x CML511 (107 lines)] (Ertiro et al., 2020b), and two F3 populations [pop4 CZL0618 x LaPostaSeqC7-F71-1-2-1-1B (183 lines); and pop5 CZL074 x LaPostaSeqC7-F103-1-2-1-1B (172 lines)] (Semagn et al., 2012; Zhang et al., 2015) were used in this study (Supplementary Table S1). The IMAS panel, comprising 410 CIMMYT maize (sub)tropical inbred lines, was used to evaluate the genetic architecture of resistance to several major diseases through GWAS (Gowda et al., 2015; Sitonik et al., 2019). The IMAS panel included lines adapted to tropical lowlands, African mid-elevation/subtropical, and the tropical highlands. All three DH populations used in this study were also used in earlier studies on MLN and low N stress conditions (Sitonik et al., 2019; Ertiro et al., 2020b); the two F3 populations were used to study the grain yield under optimum and drought stress conditions (Semagn et al., 2012; Zhang et al., 2015). The IMAS panel was evaluated in four location–year combinations [Kakamega (0°17′3.19″ N 34°45′8.24″ E, 1535 masl) and Kitale (1.0191° N 35.0023° E, 1900 masl) in 2013 and 2014]. DHpop2 and DH pop3 were evaluated in Kakamega and Kitale for 2 years in 2014 and 2015. DH pop1 was evaluated only in 2015 in the same two locations. F3 pop4 and F3 pop5 were evaluated in two locations in Kakamega and Embu (0°31′52″ S 37°27′02″ E, 1406 masl) at 2011 in Kenya (Supplementary Table S1). The list of the inbred lines, DH lines and F3 populations, and their phenotypic and marker data is available for all in CIMMYT depository1.
All the genotypes from IMAS panel, DH, and F3 populations were planted in 4-m-long single row plots in an alpha lattice design with two replications in independent trials at locations mentioned above. Two seeds were planted per hill and thinned to a single plant per hill, 3 weeks after emergence to ensure uniform plant density. Standard agronomic practices were followed. The chosen locations were natural hotspots for GLS; uniform disease infection across the trials at each location was observed. The IMAS panel, and the DH and F3 populations were evaluated for their responses to GLS in two to four environments (Supplementary Table S1). GLS disease severity is typically at its peak between the growth stages of tasseling and physiological maturity; therefore, disease severity data were recorded at the mid-silking and hard dough stages, and scored plot-wise on an ordinal scale of 1 (highly resistant, without disease symptoms) to 9 (highly susceptible, leading to necrosis). On the IMAS panel, in addition to GLS severity scoring, data were also collected for several other agronomic traits, including anthesis date (AD), anthesis-silking interval (ASI), plant height (PH), ear height (EH), ear position (EPO), ears per plant (EPP), husk cover (HC), ear rot (ER), corn rust (PS), Turcicum leaf blight (TLB), grain yield (GY), and grain moisture (MOI).
Phenotypic Data Analyses
Since the phenotypic data for GLS was recorded based on an ordinal scale, we evaluated whether the data met the assumptions of the applied statistical model, i.e., normally distributed, constant variance, and independent (Rawlings et al., 1998). The analysis revealed that the GLS data met all the assumptions. For every population, each location–year combination was treated as an independent environment that resulted in four environments for the IMAS panel, DH pop2, and DH pop3, whereas the number of environments was two for DH pop1, F3 pop4, and F3 pop5. Analysis of variance for individual and across environments was undertaken using the ASREML-R (Gilmour et al., 2009) for each biparental population and the IMAS panel. The following statistical model was used to estimate variance components:
where Yijko is the phenotypic performance of the ith genotype at the jth environment in the kth replication of the oth incomplete block, μ is an intercept term, Gi is the genetic effect of the ith genotype, Ej is the effect of the jth environment, (GE)ij is the interaction effect between genotype and environment, R(E)kj is the effect of the kth replication at the jth environment, B(R.E)ojk is the effect of the oth incomplete block in the kth replication at the jth environment, and eijko is the residual. The genotypic effect (Gi), genotype by environment interaction, and effect of incomplete blocks were treated as random effects in order to estimate their variances and residual error variance. Environments and replications were treated as fixed effects. Assuming fixed genotypic effects, a mixed linear model (MLM) was fitted to obtain the best linear unbiased estimates (BLUEs). With ASREML-R, the significance of variance components were tested by model comparison (full model vs. half model) with likelihood ratio tests in which the halved P values were used as an approximation. Heritability (H2) was estimated as the ratio of genotypic to phenotypic variance components.
Broad-sense heritability (H2) was calculated for all the traits using the following equation:
where σ2g is the genotype variance, σ2gl is the genotype × environment interaction variance, and σ2ε is the error variance, with l representing number of environments and r denoting number of replications. META-R software (Alvarado et al., 2015) was used to obtain best linear unbiased prediction (BLUP) for each genotype across environments.
Genotypic Data and Linkage Mapping
More detailed explanation on the molecular markers used and the linkage map construction for DH and F3 populations are available in earlier studies (Semagn et al., 2012; Sitonik et al., 2019; Ertiro et al., 2020b). In brief, the DNA of all inbred lines of the IMAS AM panel and the biparental populations was extracted from seedlings at 3–4 leaf stages and genotyped using the GBS platform at the Institute for Genomic Diversity, Cornell University, Ithaca, United States, using high-density markers, as per the procedure described by Elshire et al. (2011). For all three DH populations, TASSEL ver5.2 (Bradbury et al., 2007) was used to exclude SNPs with a heterozygosity of >5%. Whereas for all the five biparental populations, a minor allele frequency (MAF) of <0.05 and a minimum count of 90% were excluded by filtering from raw GBS SNP markers. Further, for each population, markers that are homozygous for both the parents and polymorphic between the parents were retained. Finally, SNPs were further filtered based on minimum distance between the markers. We used the criteria of minimum distance between adjacent SNPs as ≥200 kilobase pairs (kbps) to ensure uniform distribution of markers throughout the genome. For JLAM, markers from all three DH pops were combined, and markers with <1% missing value and >5% MAF and heterozygosity of <5% were retained. Finally, a set of 7490 SNPs that are uniformly distributed across the genome was used for JLAM analyses.
QTL IciMapping version 4.1 (Meng et al., 2015) was used to construct the linkage map based on data from all five biparental populations. QTL IciMapping was used to remove the highly correlated SNPs that do not provide any additional information by using an inbuilt tool BIN. This resulted in retention of 2105, 2699, 1962, 1130, and 1047 high-quality SNPs in DHpop1, pop2, pop3, F3pop4, and F3pop5, respectively. These SNPs were used to construct linkage maps using the MAP function, by selecting the most significant markers using stepwise regression. A likelihood ratio test was used to calculate the logarithm of odds (LOD) for each marker at a score of >3 with a 30-cM maximum distance between two loci. Three steps involved in constructing the linkage were grouping, ordering, and rippling. The Recombination Counting and ORDering (RECORD) algorithm was used to order markers. Grouping was done at LOD score > 3.0, and Sum of adjacent criterion (SAD) ripple was used to confirm marker order. The Kosambi mapping function (Kosambi, 1944) was used to transform the recombination frequencies between two linked loci. BLUPs across environments were used to detect QTLs based on Inclusive interval mapping (ICIM) for each population. Phenotypic variation explained by individual QTL and total variation explained by QTLs were estimated. QTL naming was done with letter “q” indicating QTL, followed by abbreviation of trait name, the chromosome, and marker position, respectively.
Joint Linkage Association Mapping
For JLAM, high-quality, uniformly distributed 7490 SNPs across three DH populations were selected. The SNPs were then used to construct a linkage map based on their physical positions. A biometric model (Würschum et al., 2012) was used to perform JLAM, with BLUPs across environments and population being applied for analysis. After testing several biometric models, one that performed well for association studies in multiple biparental populations (Würschum et al., 2012) was used to conduct the JLAM. This model controls the differences in population means by incorporating population effect and the genetic background by using cofactors and marker effects across populations. This model was explained in detail by Liu et al. (2011) and Würschum et al. (2012). In brief, with this model, as a first step, cofactors were selected based on the Schwarz Bayesian Criterion (SBC, Schwarz, 1978) by including a population effect, and in the second step, P-values were calculated for the F-test by using a full model (including SNP effect) versus a reduced model (without SNP effect). Cofactors were selected by using PROC GLM SELECT from SAS 9.4 (SAS Institute Inc, 2015) and genome-wide scans for QTLs were applied in R version 3.2.5 (R Core Team, 2015).
Genome-Wide Association Analyses
TASSEL ver5.2 (Bradbury et al., 2007) was used for GWAS. SNPs with a heterozygosity of <5%, a MAF of >0.05, and a minimum count of 90% were included by filtering from raw GBS data sets, and 337,110 high-quality SNPs were retained for further analysis in the IMAS AM panel. Distribution of these 337,110 GBS markers in the maize genome is presented with different color keys in the Supplementary Figure S1. BLUPs across environments were used as phenotypes in association mapping scans. Principal component analysis (PCA) was performed using TASSEL ver5.2. The principal components were used to correct for population structure and to create a two-dimensional plot to enable visualization of the probable population structure. A MLM that computes both PCs and kinship matrix (K) was applied for GWAS to correct for population structure (Yu and Buckler, 2006). The kinship matrix was calculated in Tassel ver 5.2 using a normalized Identity by State (IBS) option. The extent of LD of the genome was based on physical distances between the SNPs and the adjacent pairwise r2 values between high-quality SNPs from GBS (Remington et al., 2001). The “nlin” function in R was used to fit non-linear models into the genome-wide LD data by incorporating r2 as responses (y axis) and pairwise distances (x axis) as predictors. The average estimator for LD decay was calculated at a “significance” threshold of r2 = 0.1 and r2 = 0.2 cutoff points in relation to distance (Hill and Weir, 1988), and a representative scatter plot was drawn as LD between adjacent markers versus chromosome distance (kb). In this study, we used a MAF of 0.05 to assess LD as was applied in other studies (Li et al., 2014; Vos et al., 2017).
Scans for genome-wide marker-trait associations were carried out to detect main effect QTLs. R2 statistics was used to assess the amount of phenotypic variation explained by the model by simultaneously fitting all significant SNPs in a linear model. Multiple testing correction was performed to determine the significance threshold, where instead of 337,110 independent tests, the total number of tests was estimated based on the average extent of LD at r2 = 0.1 (Cui et al., 2016). With respect to the above, significant associations were declared when P-values in independent tests were less than 9.5 × 10–6. Candidate genes containing or being adjacent to the significant SNPs were obtained from the B73 gene set (version 2.0) in Maize GDB. BLAST searches were performed with 50-bp source sequences of the significantly associated SNPs against the “B73” RefGen_v22.
Genomic Prediction
RR-BLUP was used to carry out GP using a fivefold cross-validation (Zhao et al., 2011). BLUEs across environments for each of the biparental populations and across three DH populations were used for the analysis. For all biparental populations and IMAS panel, the same set of high-quality uniformly distributed 4000 SNPs with no missing values and MAF > 0.05 were used. Approaches used for GP include the “within population” approach in which individual biparental population and IMAS set were sampled to form a training and prediction set, a “joint population” or combined population prediction approach where data from three DH populations were combined and sampled randomly to form a testing and training set, and “across population” prediction in which IMAS association panel was used as a training set and each DH population was used as a testing set. For each approach, 100 iterations were done for sampling of the training and validation sets. RR-BLUP was implemented using the statistical software R (R Core Team, 2015).
Results
The IMAS panel with a set of 410 lines and five biparental populations were evaluated against GLS in Kenya in two to four environments. GLS disease severity scores indicated comparable disease pressures across the tested environments as indicated by significant genotypic variance at each environment for each population (Supplementary Table S1). Further, significant (p < 0.05) Pearson correlations were also observed among phenotypic values determined at different environments for each population (Supplementary Table S2). This suggested that there was enough GLS disease pressure in each environment and the combined analysis across environments was not biased. The significant environmental variations (data not shown) observed for GLS indicated that the environments were distinct and provided unique information on the individual lines in the linkage and association mapping population.
The disease severity was high in each of the environments as well as across environments, with the susceptible checks CLYN265 and DTPYC9-F13-2-3-1-2-B showing a score of 7 on the 1–9 scale. The frequency of the phenotypic values followed a near-normal distribution for individual populations and combined DH populations as well as IMAS panel (Figure 1). The IMAS panel, on average, was moderately resistant to GLS with a mean of 3.98, assessed on the 1–9 scale. Four parental lines of DH populations, CML494, CML504, CML511, and CML550, recorded GLS scores of 3.77, 3.39, 3.31, and 4.94, respectively. The DH populations developed from the four parents were moderately resistant with mean GLS scores of 5.24, 3.90, and 3.29 in DH pop1, 2, and 3, respectively, while the GLS score in the combined DH populations across environments was 4.52. For the F3 populations, the parental lines LapostaSeq-C7-F71 and CZL074 were GLS-resistant with mean scores of 2.60 and 3.00, respectively, whereas the other two parents LapostaSeq-C7-F108 and CZL618 had mean scores of 3.58 and 4.01, respectively. F3 pop4 and 5 recorded mean GLS scores of 3.24 and 3.84, respectively. Thus, significant variation for GLS disease severity was observed in all populations.
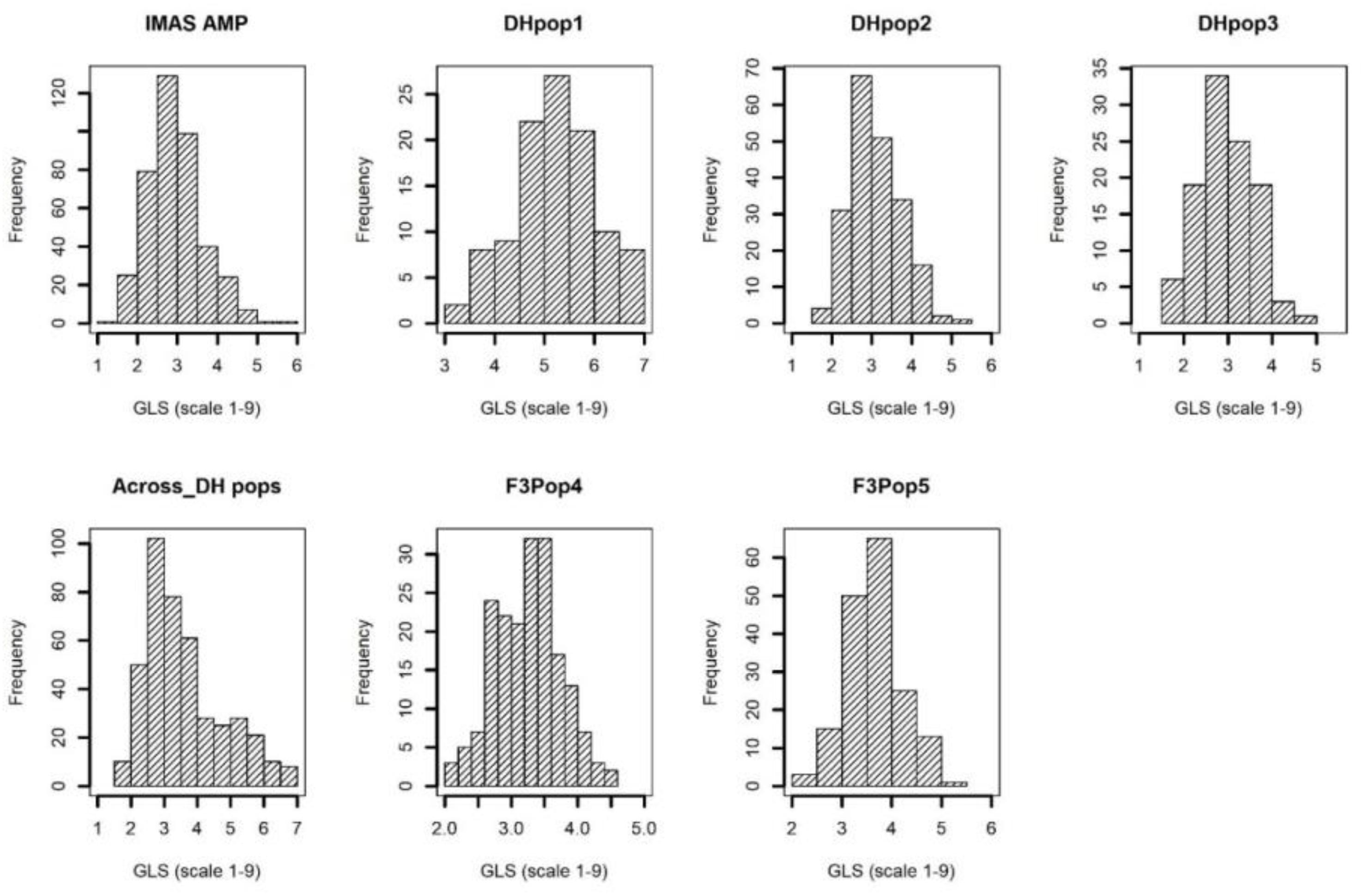
Figure 1. Phenotypic distribution of GLS disease severity in IMAS association mapping panel, three DH populations, across DH populations, and two F3 populations evaluated in two to four environments.
ANOVA, calculated across environments, revealed significant genotypic and genotype by environment (G × E) interaction variance in the IMAS panel (Table 1). Individual biparental populations and the combined DH populations had significant genotypic and G × E interaction variance except for F3 pop5 where the G × E interaction variance was not significant. Heritability estimates were moderate, ranging from 0.43 to 0.68. F3 pop4 had the lowest heritability estimates while DH pop2 had the highest estimates of heritability. The IMAS panel and combined DH population had heritability estimates of 0.56. Each population displayed adequate disease expression for both susceptible and resistant lines for response to GLS. The IMAS panel used in this study showed significant variations for all other agronomic traits including plant height, grain yield, ear height, and ear position (data not shown). GLS is negatively and significantly correlated with grain yield, anthesis date, ear position, grain moisture, plant height, and ear height (Figure 2). Correlation among GLS and anthesis date was significant, but low magnitude (-0.27), showing that flowering time is not a cofound effect in GLS resistance. Of the traits surveyed, the largest positive correlation was estimated between ear height and ear position (r = 0.84, p < 0.01) and between ear height and plant height (r = 0.82, p < 0.01).
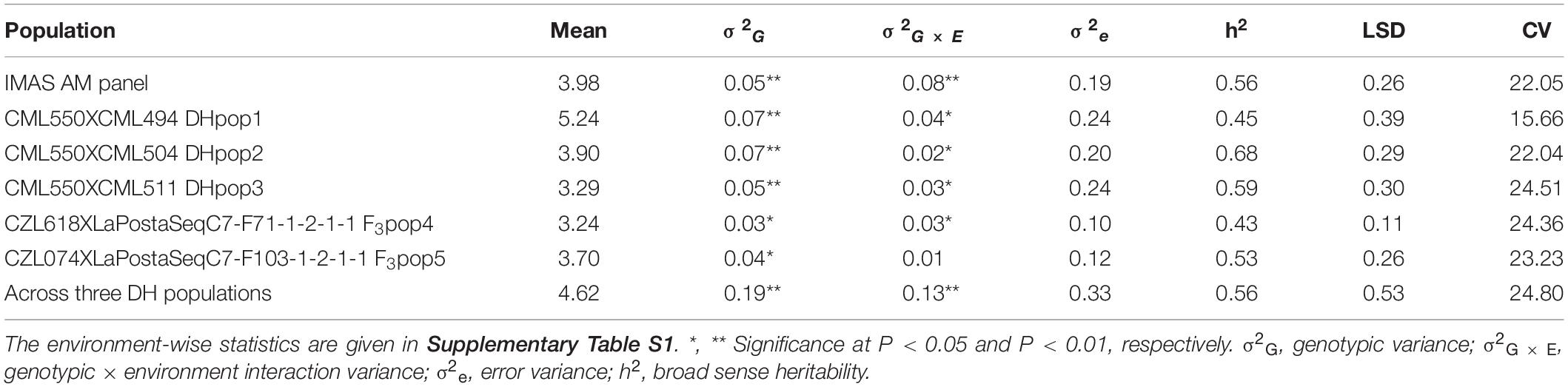
Table 1. Means and components of variance for gray leaf spot disease severity for maize inbred lines from IMAS association panel, three DH and two F3 populations, as well as across DH populations evaluated in two to four environments.
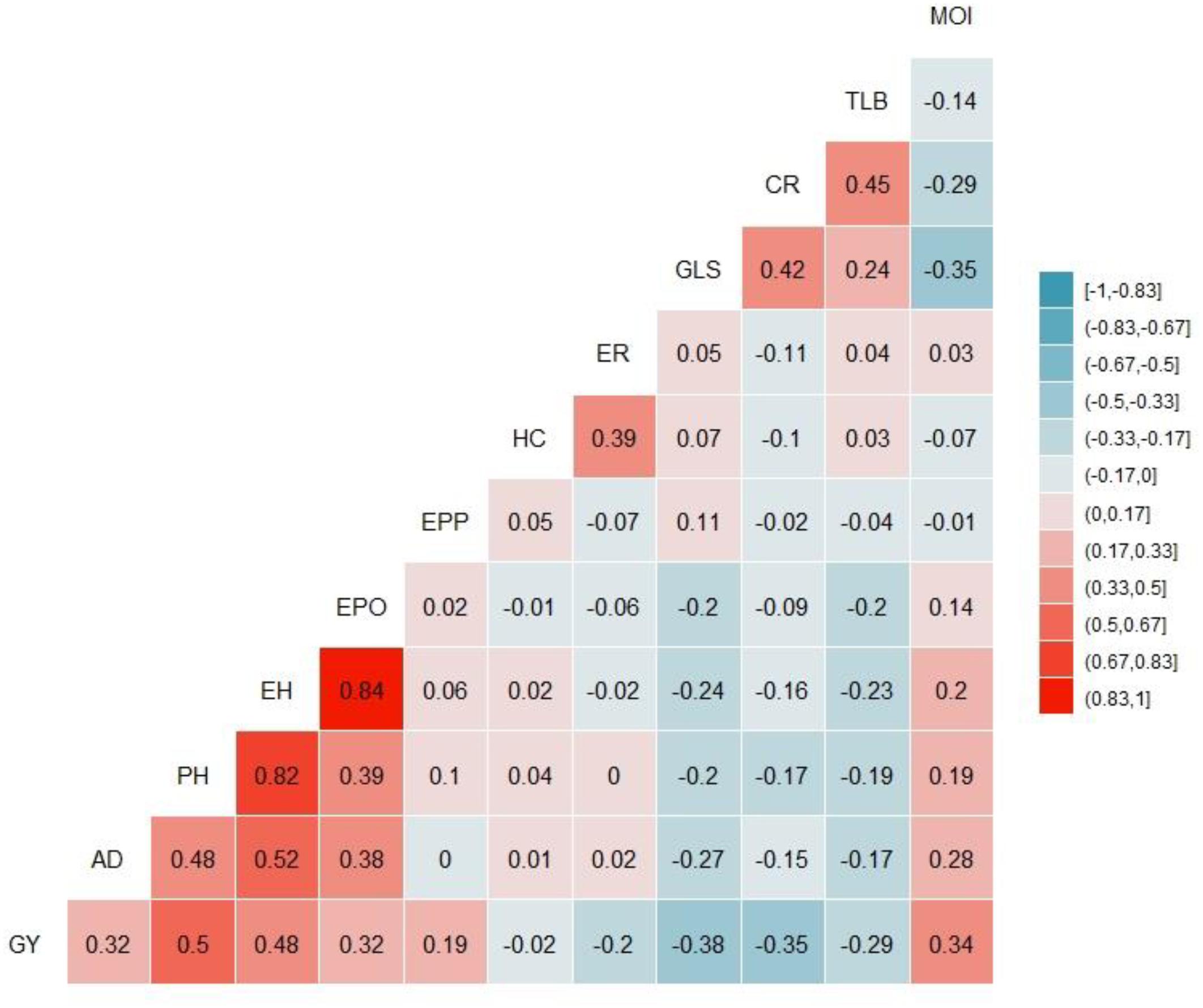
Figure 2. Phenotypic correlations among 12 traits evaluated in four environments. The correlation level is color-coded according to the color key plotted on the extreme right. Correlations with >0.12, and >0.15 were significant at 0.05 and 0.01 levels, respectively. GY, grain yield; AD, days to anthesis; PH, plant height; EH, ear height; EPO, ear position; EPP, number of ears per plant; HC, husk cover; GLS, gray leaf spot; TLB, Turcicum leaf blight; CR, corn rust; MOI, grain moisture content.
Population structure was diagnosed using STRUCTURE software for K (number of clusters/fixed subgroups). Most of the significant change was observed when K was increased from one to two and one to four. Structure results of K = 4 was the best probable partition, as they showed high consistency with significant delta K values, geographical origin, and pedigree history (Supplementary Figure S2). PCA indicated a clear diverse population structure within the panel. The first two PCs, PC1 and PC2, explained a variation of 35.99 and 19.56%, respectively. Further, LD for the entire genome was calculated using 11,035 SNPs. LD decay of the panel was rapid with increase in physical distance. Physical distance at a cutoff value of r2 = 0.1 was found to be 14.97 kb and that at r2 = 0.2 was 5.23 kb (Supplementary Figure S3).
Linkage map for each of the populations was constructed. The number of progenies or lines, markers, map lengths, and average genetic distances between the markers for each biparental population are presented in Supplementary Table S3. Linkage mapping detected one QTL (qGLS5-217) for GLS in DH pop1 on chromosome 5, which explained 11% of the phenotypic variance (Table 2). Five QTLs were identified for DH pop2 on chromosomes 1, 5, 7, and two on chromosome 8, which individually explained phenotypic variance ranging from 1.5 to 28.2%, and together explained 49.7% of the total phenotypic variance. In DH pop3, only two QTLs were detected on chromosomes 1 and 10, which individually explained 16.6 and 8.7% of phenotypic variance, and together explained 23.7% total phenotypic variance. Seven QTLs were detected in F3 pop4, which individually explained 3.21–14.36% of phenotypic variance and together contributed for 18.05% of total phenotypic variance. In the F3 Pop5, seven QTLs were detected, distributed on chromosomes 1, 2, 4, 5, and 6, individually explaining 2.53–12.79% of phenotypic variance. The QTL on chromosome 7 (qGLS7-105) in DH pop2 was found to explain the largest proportion of phenotypic variance (28.2%). Overlapping of QTLs was compared based on the physical distance between the flanking markers, as the linkage maps were not comparable due to different sets of markers used. The QTL qGLS1-104 on F3 pop5 overlapped with the qGLS1-123 on F3 pop4, qGLS1-155 in DH pop3, and qGLS1-158 in DH pop2 (Table 2). Another QTL on chromosome 4 qGLS4-190 in F3 pop4 overlapped with qGLS4-157 detected in F3 pop5. QTL qGLS5-217 on DH pop1 overlapped with qGLS5-217 QTL detected on F3 pop5. The QTLs detected between 155 and 159 Mbp on chromosome 1 in DH pop2 and DH pop3 were in close physical proximity and could possibly represent the same QTL.
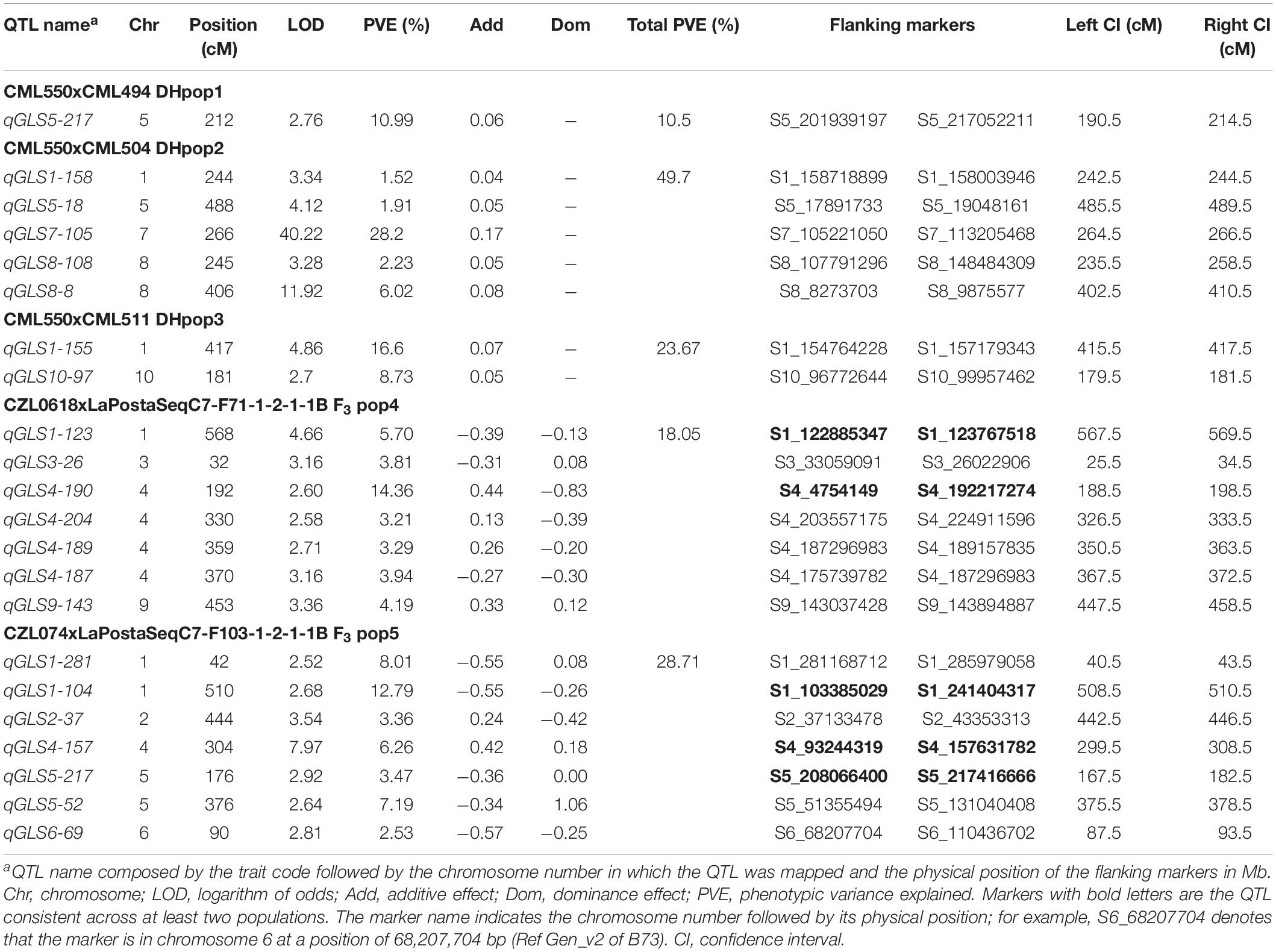
Table 2. Detection of QTL associated with resistance to GLS, their physical positions, and genetic effects in three DH populations and two F3 populations.
Joint linkage association mapping analyses revealed 14 QTLs distributed across six chromosomes, 3 QTLs on chromosome 1, 5 QTLs on chromosome 5, 2 QTLs each on chromosomes 4 and 8, and 1 QTL each on chromosomes 6 and 9. These QTLs individually explained 0.1–15.7% of the phenotypic variance (Table 3). The QTL on chromosome 4 (qGLS4-163) had the largest effect at 15.7% of phenotypic variation and was found overlapping with the QTL qGLS4-190 detected on F3 pop4 (Tables 2, 3). In addition, qGLS1-158 overlapped with the QTL qGLS1-104 detected in F3 pop5 and qGLS1-158 in DH pop2. Another QTL, qGLS4-154, detected in JLAM also overlapped with QTL qGLS4-190 in F3 pop4 and qGLS4-157 in F3 pop5. A QTL on chromosome 5, qGLS5-19, overlapped with QTL qGLS5-18 detected on DH pop2. Two QTLs detected in JLAM qGLS5-213 and qGLS5-216 were falling within the confidence interval of the QTL qGLS5-217 detected in DH pop1 and F3 pop5 (Tables 2, 3). Large variation was observed for both allele substitution effects (α effect) even with changing in signs and QTL effects (Table 3).
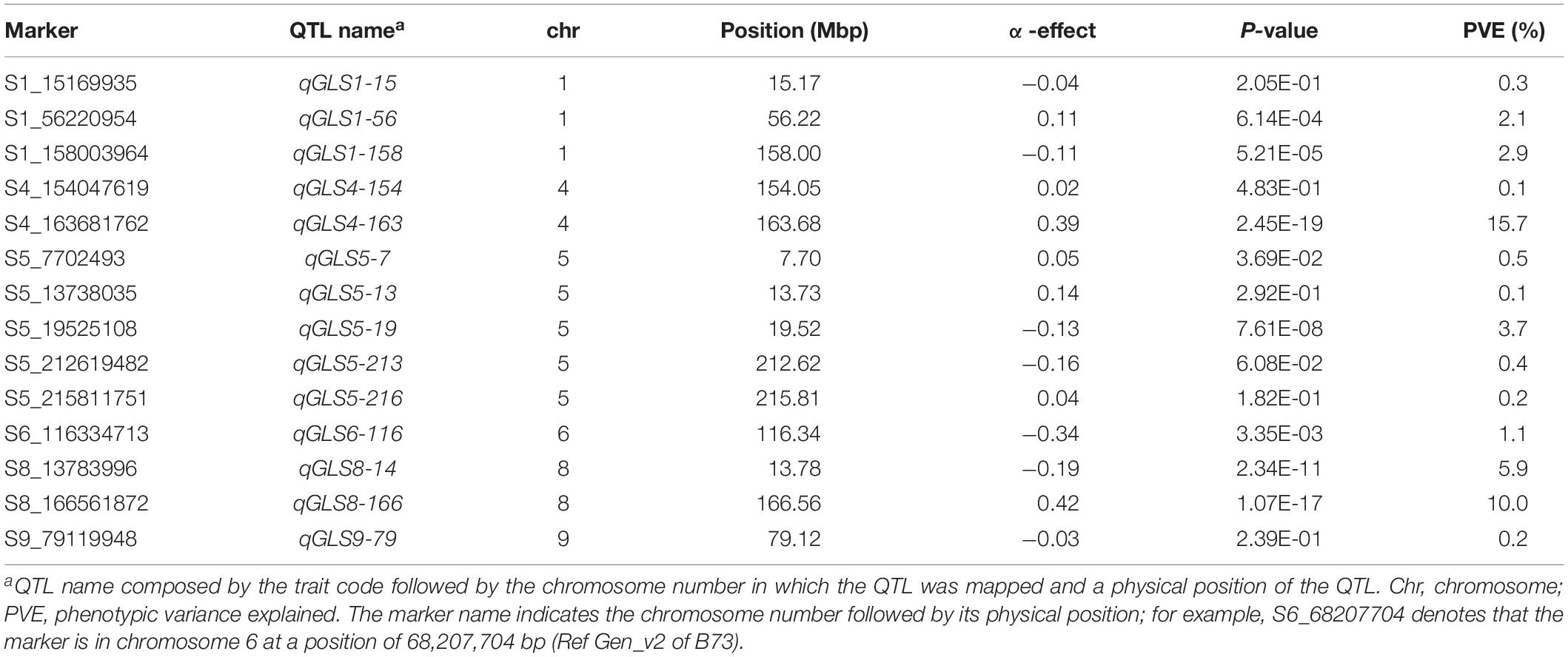
Table 3. Analysis of GLS trait-associated markers, allele substitution (α) effects, and the total phenotypic variance (R2) of the joint linkage association mapping based on combined data from three DH populations.
We performed GWAS based on MLM, which corrects for both the population structure and familial relatedness to avoid type I errors. Further, type II errors was avoided by observing the distribution of null versus alternative hypotheses in quantile–quantile (QQ) plots (Figure 3). GWAS analyses revealed the quantitative genetic nature of the GLS resistance and identified 10 marker-trait associations (MTA) in five chromosomes (Table 4) based on a significant threshold p value of 9.5 × 10–6. SNPs identified for GLS severity explained moderate phenotypic variation ranging from 6 to 9%. The SNP S1_215340710 is co-located with QTL qGLS1-104 detected in biparental F3 pop5 (Tables 2, 4). The B73 maize genome reference V2.0 sequence was used to identify putative candidate genes based on significant SNP associations. Sets of putative candidates were identified together with their predicted functions (Table 4).
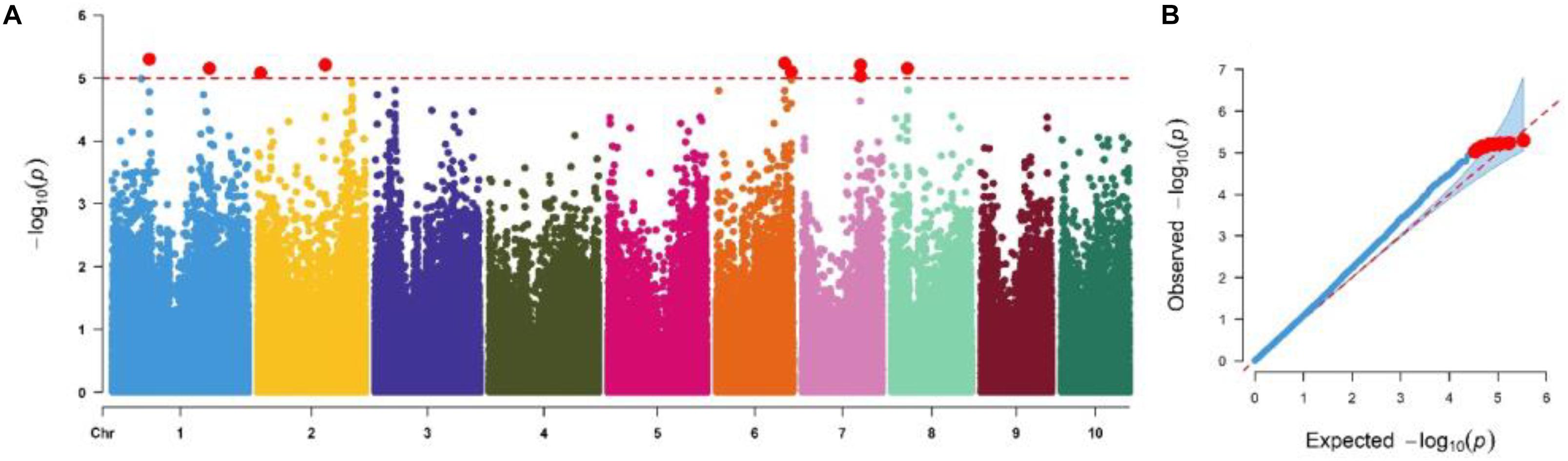
Figure 3. (A) Manhattan and quantile–quantile plots of a mixed linear model for GLS resistance in the IMAS association mapping panel evaluated in four environments. The dashed horizontal line depicts the significance threshold (P = 9.5 × 10– 6). The X-axis indicates the SNP location along the 10 chromosomes, with chromosomes separated by different colors; Y-axis is the -log10(P observed) for each analysis. (B) Quantile–quantile plots.
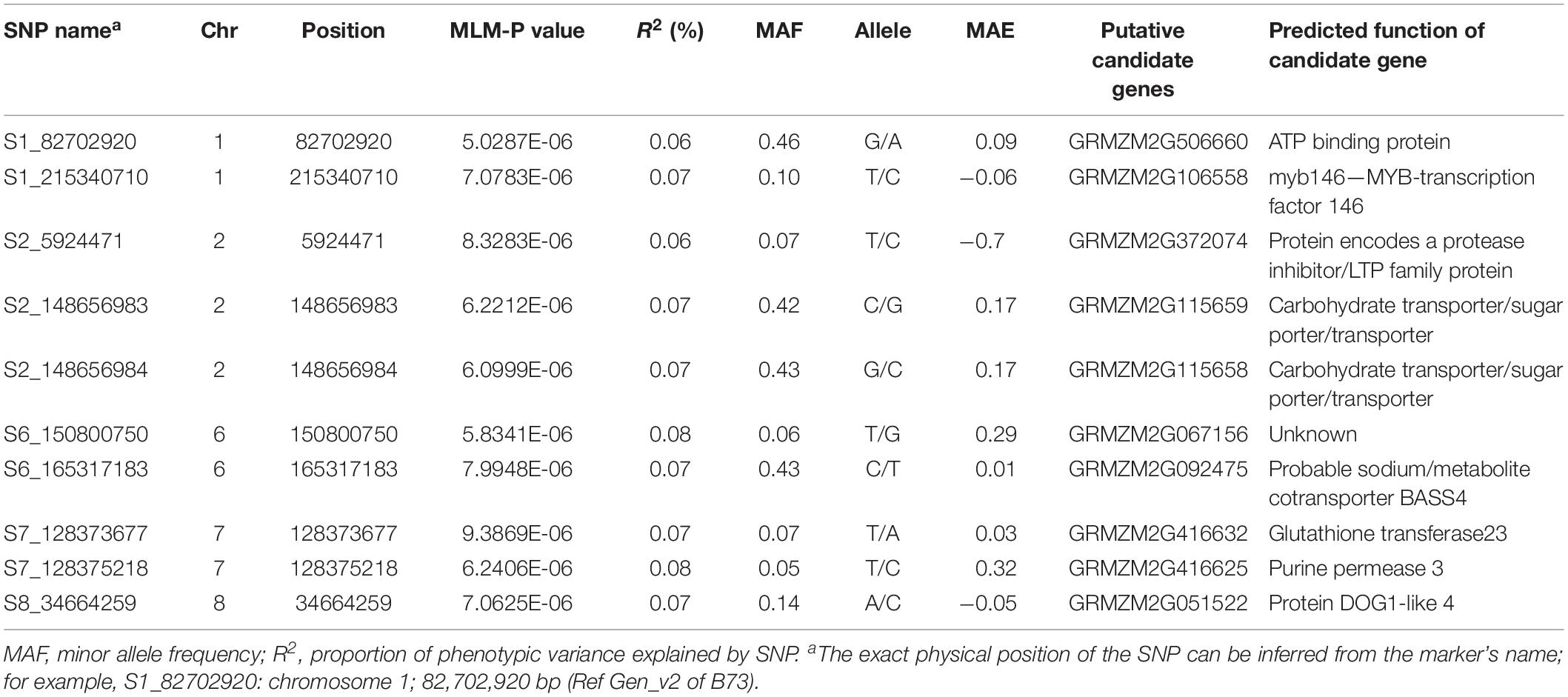
Table 4. List of significant SNPs and candidate genes associated with GLS resistance in the IMAS association panel evaluated in four environments.
The predictive ability of the GP model was determined as the correlation between GEBVs and observed phenotypes. The fivefold cross-validation revealed low to moderate prediction correlations of 0.39, 0.37, 0.56, 0.30, 0.29, and 0.38 for within IMAS association panel, DH pop1, DH pop2, DH pop3, F3 pop4, and F3 po5, respectively (Figure 4). The prediction correlations were increased to 0.84 when the prediction was based on combined three DH populations. Combining both IMAS association panel and all DH populations increased the diversity of the training set, but the correlation was still high at 0.77. In breeding, prediction of new population using one diverse panel as common training set is desired. However, this could affect the prediction accuracy, particularly for complex traits. Nevertheless, for GLS, using IMAS association panel as the training set, we predicted the response of each DH population and observed cross-validated correlations of 0.29, 0.36, and 0.14 for DH pop1, DH pop2, and DH pop3, respectively (Figure 4).
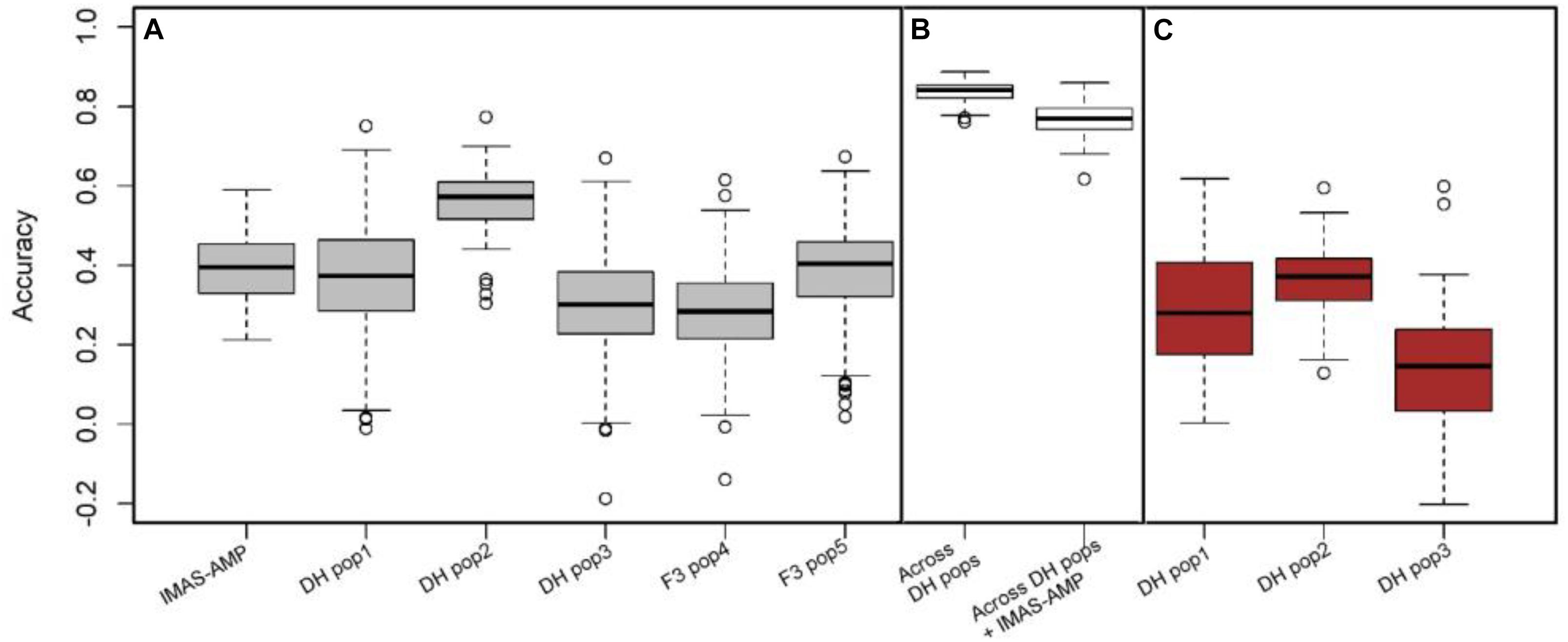
Figure 4. Genome-wide prediction accuracies for GLS resistance in biparental and IMAS AM panel based on three different scenarios. (A) Estimation and prediction within IMAS AM panel and biparental populations; (B) estimation and prediction combined DH populations and combined all DH populations and IMAS AM panel; (C) prediction of the DH population using IMAS AM panel as a training set with fivefold cross-validation.
Discussion
Gray leaf spot is an economically important foliar disease of maize in SSA. With the changing climates and ever-increasing maize monoculture in SSA, GLS becomes a more serious threat to maize production in the future. Understanding the genetic basis of GLS resistance is important to design an effective breeding strategy for developing and deploying GLS-resistant parental lines and hybrids. Breeding for GLS resistance is influenced by several factors including genotype, environment, and their interactions. The present study used CIMMYT’s genetically diverse tropical and subtropical maize breeding lines and populations, with the aim to identify genomic regions for GLS resistance and validate the same across different populations and earlier reports. Five biparental populations were used to detect the QTL for GLS resistance with high detection power, besides an association mapping panel to find marker-trait associations.
The phenotypic data for GLS disease severity score in each of the biparental populations as well as combined three DH populations and association mapping panel supported the quantitative nature for GLS resistance (Figure 1). Several previous studies using either biparental populations (Berger et al., 2014; Liu et al., 2016) or an association mapping panel (Mammadov et al., 2015; Kuki et al., 2018) indicated polygenic control of GLS resistance. We also observed significant genotypic variance and moderate heritabilities indicating good prospects for introgressing GLS resistance in breeding programs, similar to the observations in some earlier studies (Berger et al., 2014; Mammadov et al., 2015; Liu et al., 2016; Kuki et al., 2018).
Tropical and subtropical maize germplasm possess a huge amount of unexplored genetic diversity, which provides insights into resistance for several key traits used in breeding programs (Liu et al., 2016). Moderate to high heritability within the biparental populations for GLS resistance is reported to be controlled by additive gene action (Gordon et al., 2006). QTL analyses in five biparental populations revealed 22 potential QTLs providing resistance to GLS. Several QTLs were consistently detected at least in two populations; for example, QTL qGLS1-104 on F3 pop5 overlapped with the QTLs on F3 pop4, DH pop2, and DH pop3. Another QTL qGLS4-190 in F3 pop4 overlapped with QTL in F3 pop5, and QTL qGLS5-217 in DH pop1 overlapped with QTL detected on F3 pop5 (Table 2). There were five QTLs (qGLS1-104, qGLS1-155, qGLS4-190, qGLS5-217 and qGLS7-105) with major effects (>10% phenotypic variance explained) detected on chromosomes 1, 4, 5, and 7 in five biparental populations. A major QTL (qGLS5-217) identified in DH Pop1, explaining about 11% of total phenotypic variance and located between 201 and 207 Mbp, was consistent with a consensus QTL in bin 5.03 reported by Shi et al. (2007) in a meta-QTL study. A study carried out by Liu et al. (2016) also identified a major QTL on bin 5.04, which coincided with a similar QTL for flowering time and showed linkage between GLS resistance and flowering time. In the findings of QTL mapping within DH pop2, only the QTL on chromosome 7 between 105 and 113 Mbp, qGLS7-105, was found to have a significant effect (R2 of 28.3%). For this major QTL qGLS7-105, the segregation alleles from two tightly linked flanking SNPs reveal that DH lines and inbred lines from the IMAS panel with low DS scores were strongly associated with GLS-tolerant parent CML504 (Supplementary Figure S4). However, the efficiency of these flanking markers should be assessed further through KASP (Kompetitive allele-specific PCR) assays, where we can check these markers’ ability to identify GLS-resistant and susceptible genotypes. In the previous study carried out in South Africa, Berger et al. (2014) identified various QTL hotspots for GLS resistance in chromosomal bins 7.02 and 7.03. “Hotspot,” in this case, is a genomic region comprising multiple genes (clusters) that correspond to a particular trait or a group of related traits. Similar hotspots for GLS have been observed in various studies within chromosome 1 (Clements et al., 2000; Lehmensiek et al., 2001; Pozar et al., 2009; Berger et al., 2014; Lv et al., 2020). Our findings also suggest a possible hotspot within DH pop3 on chromosome 1; QTL qGLS1-155 explained 16.6% phenotypic variation. The same case applies for two QTLs (qGLS8-108 and qGLS8-8) on chromosome 8 within DH pop2, as was also reported by Bubeck et al. (1993) and Chung et al. (2011). Overall, several QTLs were consistent with the previous studies indicating their reliability to be used in applied breeding.
Joint linkage association mapping explores both within- and across-population variations that enabled the detection of novel QTL, which would have been omitted by linkage mapping. In line with this expectation, we found that 8 out of 14 QTLs detected were novel, not observed in linkage mapping. JLAM results indicated the influence of many genes with minor effects on GLS resistance, as shown by the many identified QTLs that explained low phenotypic variation. In general, linkage mapping confines a QTL to a 10–20 cM interval because of limited recombination events during development of mapping population. Identifying QTLs that are consistent across populations, fine mapping of these QTLs, and construction of a high-resolution linkage map could help identify markers for cloning the QTL. On the contrary, six QTLs detected through JLAM overlapped with the QTL detected in linkage mapping, revealing not only stability but also help in reducing the confidence interval of the QTLs and may even be closer to the causal variant responsible for GLS resistance.
Population structure is an essential aspect as it influences detected MTAs including the false positives in an association mapping panel. The present study used CIMMYT’s elite maize lines from multiple breeding programs, including Africa highland, mid-elevation, and lowland tropics. We observed a moderate population structure with PC1 and PC2 explaining 35.99 and 19.56% of variation, respectively (Supplementary Figure S2). The diversity panel lines did not cluster into a group but were scattered among different groups. Similar findings were also observed by George et al. (2004) who studied the diversity of Asian inbred lines including CIMMYT’s tropical and subtropical lines of the region and concluded that the lines had a significant genetic diversity.
The mapping resolution of GWAS and number of SNPs required for desired marker density are dependent on the magnitude of LD and LD decay with genetic distance (Myles et al., 2009). The correlation between alleles in different genomic locations is generally based on the historical recombination between polymorphisms and hence requires a large population for study. The genome-wide average LD decay was 14.97 kb at r2 = 0.1 and 5.23 kb at r2 = 0.2, similar to Rashid et al. (2018) observed in their association panel. LD decayed rapidly with distance between sites but showed substantial variation among loci. Previous studies indicated a rapid decay in tropical maize germplasm as compared to the temperate germplasm; the high LD decay in tropical maize suggests a broader genetic base, resulting from high recombination events (Lu et al., 2009). This provides breeders with an opportunity to select germplasm that integrates high grain yield with disease resistance and abiotic stress tolerance.
The ad hoc statistics ΔK was used to determine the optimum number of subgroups based on the output log likelihood of data [LnP (D)] of STRUCTURE. The peaks of the line plot (Supplementary Figure S2) suggest that the population could be divided into two or four distinct groups in order of possibility, with the K = 4 of delta K intersecting with LnP (D) showing higher possibility. At K = 2, the panel could be grouped into tropical and subtropical groups with group 2 (G2) occupying the bulk. When K = 4, G2 appeared as a mixed group and was further divided into three groups. These three groups could be divided into highland, lowland, and mid-elevation. The clustering of the population using STRUCTURE correlated highly with known pedigree information and origin of the lines.
Multiple studies have been carried out earlier to evaluate the genetic architecture of GLS resistance using both biparental populations and association mapping panels, especially using temperate maize germplasm. Berger et al. (2014), Shi et al. (2014), and Liu et al. (2016), used F2:3 families, association mapping panel, and F7 RILs, respectively, and identified several QTLs for GLS resistance. Detection of QTLs for resistance to GLS was found to be variable between seasons and locations (Bubeck et al., 1993; Lukman et al., 2012). The use of a large tropical association mapping panel and testing across multiple environments in the present study enabled us to identify putatively associated SNPs for GLS resistance. We identified 10 SNPs significantly associated with GLS each explaining between 6 and 8% of total phenotypic variation. This concurred with the polygenic nature of GLS resistance where many genes with minor effects control disease resistance (Asea et al., 2012). Comparing QTL positions between different studies was difficult due to the use of different populations, genetic maps, and statistical tests to define QTL; therefore, we have opted to use the maize core bin regions to compare the QTL (Wisser et al., 2006; Balint-Kurti et al., 2008). Three chromosomal bins were identified in this study: 8.03 with two significantly associated SNPs, and 7.03 and 1.05 were the same as those identified by Shi et al. (2014).
The most significant SNP for GLS was in proximity with the GRMZM2G506660 gene in bin 1.05, which encodes for adenosine-5′-triphosphate (ATP) binding protein (ABP). ABPs contain a binding site for interaction with ATP, an energy molecule. This binding site forms a platform for conversion of ATP to adenosine diphosphate (ADP), realizing energy for use by the protein, or conforms to the protein to change shape and act as a catalytic enzyme. Many ADPs being transmembrane proteins are responsible for the transport of a wide variety of micro- and macromolecules across intra- and extracellular membranes. They have roles in cellular motility, membrane transport, and regulation of various metabolic processes. GRMZM2G115658 on chromosome 2 bin 2.05 is a carbohydrate transporter gene, while GRMZM2G092475 on chromosome 6 bin 6.07 is a sodium/metabolite co-transporter gene within the chloroplast. Chloroplast plays a crucial role in metabolism and energy supply to photosynthetic organisms. They possess transporters and channels within the thylakoid membranes and envelope, hence mediating exchange of metabolites and ions within the chloroplast stroma, cytosol, and different sub-compartments of the chloroplast. GRMZM2G115658 is involved in the regulatory movement of carbohydrate in and out or within a cell through a pore or transporter, and in turn regulating the amount of energy within a cell.
With all these putative candidate genes identified based on associated SNPs, it is evident that transporter genes and channels are somehow involved in plant defense, well demonstrated by the RTM system in Arabidopsis (Chisholm et al., 2000; Gowda et al., 2015), also suggesting that plants can resist pathogens in multiple ways. Since plant pathogens need to move within and between cells through transporter channels for spreading infection, there is high probability that the putative candidate genes identified in this study are involved in disease resistance in maize. However, this warrants further research on the roles of various transport proteins before potential use in strategies for enhancing resistance to GLS.
GRMZM2G372074 is a lipid transfer protein encoding gene on chromosome 2 bin 2.02 characterized by a hydrophobic cavity. It functions in the transfer of lipophilic compounds to cuticular surface of epidermal cells and is directly involved in plant defense against pathogens. Minimal amounts of glycophosphatidylinositol (GPI)-anchored lipid transfer proteins (LPTGs) increase susceptibility to penetration of epidermal cell by pathogens (Fahlberg et al., 2019). GRMZM2G416632 on chromosome 7 bin 7.03 is a gene coding for glutathione transferase23 (GST), which are multifunctional enzymes, highly induced by a wide range of biotic stress factors. This enzyme was found to be up-regulated by early phase of fungal microbial infections. Silencing and overexpression of specific GSTs modify pathogen multiplication rates and subsequent amounts of pathogens in plants (Gullner et al., 2018).
GWAS and GP can be carried out simultaneously since both use the same set of markers and mapping populations, and it captures both minor- and major-effect QTLs (Chaikam et al., 2019; Sitonik et al., 2019). GP is helpful in accelerating the breeding cycle by facilitating the rapid selection of superior genotypes through ease in genotyping and availability of a wide range of markers, which capture maximum favorable alleles. The potential for different GS-based models in identifying lines with favorable traits in maize has been examined in various studies (Zhang et al., 2015; Crossa et al., 2017; Gowda et al., 2018; Beyene et al., 2019). Moderate to high accuracies observed in this study for the biparental populations and diversity panel offer promise in breeding for GLS resistance in tropical maize. Prediction accuracy of the diversity panel was in agreement with various studies on moderately complex traits like MLN (Gowda et al., 2015), maize chlorotic mottle virus (Sitonik et al., 2019), and northern corn leaf blight (Chung et al., 2011). According to Sitonik et al. (2019), significant genetic structure and high LD between adjacent markers of the diversity panel resulted in a moderate prediction accuracy, which could also be attributed to its moderate heritability. Combining the individual DH populations and applying cross-validations to obtain the training set and prediction set from the total DH population resulted in substantial improvement in the prediction accuracy (Figure 4). This was due to the increase in population size of the training set and high relatedness between training and prediction sets. Adding the GWAS panel to the combined DH populations increased the population size but resulted in a slight drop in the accuracy, which was due to the varying degrees of relatedness between the sets and increase in diversity of the training set.
The relative advantage of GP over phenotypic selection determines its routine use in breeding programs. This implementation is dependent on cost-effective and high-throughput genotyping technology like GBS, which provides a platform to genotype many maize lines at relatively low costs (Elshire et al., 2011). This study showed consistent results between GP and phenotypic selection accuracy indicating the possibility to integrate GP with phenotypic selection to improve the efficiency with less resources (Beyene et al., 2019). In terms of gain per year, GP is much more efficient taking into account the likelihood of completing three cycles per year (Lorenzana and Bernardo, 2009). Rapid decline in the cost of genotyping makes it possible to routinely apply GP in breeding. Combining GWAS and the predictive capabilities of GP will also improve the prediction accuracy by using information on major QTLs from GWAS or linkage mapping. Prediction accuracy dropped for the across- and within-population approach where the GWAS population was used as the training population, similar to Liu et al. (2018) and Sitonik et al. (2019) who used diversity panel or natural population to predict biparental populations. The prediction within each DH population revealed an accuracy of 0.37, 0.56, and 0.30 for DH pop1, 2, and 3, respectively, while predicting each biparental population by using the IMAS panel as training set showed 0.29, 0.36, and 0.14 accuracy for DH pop 1, 2 and 3, respectively. These correlations are low compared to prediction within populations. However, when the square root of heritability is compared to the GP correlations, they show that observed gain in phenotypic selection and GP were comparable. This relatively high prediction accuracy by using the IMAS panel as training set could be attributed to the populations’ relatedness to the diversity panel as all parents of the three DH populations were part of the GWAS mapping panel and are related to several lines derived from a (sub)tropical breeding program. The reduction in accuracy is also attributed to the magnitude of trait genotypic variability and heritability in each population. Nevertheless, the predicted accuracies are positive, and under the assumption of three cycles per year possibility, the total selection gain is comparable to phenotypic selection gain. The study thus shows promise for using a common training population or historical data to predict GLS resistance in several connected but independent populations.
Conclusion
In this study, we used one association panel comprising 410 (sub)tropical maize inbred lines for GWAS and GP to understand the genetic basis of resistance to GLS. We also studied five biparental populations using linkage mapping and JLAM to understand the underlying architecture of the trait. Phenotypic correlations of studied traits indicated the potential use of these populations for selection of superior lines. Linkage mapping identified several minor- and major-effect QTLs with a few overlapping across populations, while many QTLs are population-specific for GLS resistance. GWAS scan revealed 10 SNPs associated with GLS resistance. The putative candidate genes identified in the study and their proposed functions require further validation to confirm the involvement of these genes in GLS resistance. Several QTLs identified in this study were found to be overlapping across different analyses, and with QTLs or associated SNPs reported earlier in temperate maize germplasm. These genomic regions can serve as potential selection targets to improve resistance to GLS. GP can be used within populations to predict the response of the germplasm to GLS resistance. Having a common training population comprising lines with diverse representation from a breeding program with good quality phenotypic data and genotyped with high-density markers holds promise in breeding for GLS resistance.
Data Availability Statement
The datasets presented in this study can be found in online repositories at: https://data.cimmyt.org/dataset.xhtml; jsessionid=dc30e94cd6c7318ac8df044c42eb?persistentId=hdl%3 A11529%2F10548467&version=DRAFT. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
MK, BD, DM, BP, SN, and MG conceived the experiments. BD, MG, MK, LS, and YB conducted the field evaluations and phenotyping. MG, SN, and JB coordinated the GBS experiments. MG and MK carried out the GWAS and GS analyses. MG, BD, DM, YB, MO, BP, MK, JB, LS, JK, and SN interpreted the results and drafted the manuscript. All the authors contributed to the article and approved the submitted version.
Funding
The research was supported by the Bill and Melinda Gates Foundation (B&MGF) and the United States Agency for International Development (USAID) through the Stress Tolerant Maize for Africa (STMA, B&MGF Grant # OPP1134248) Project, AG2MW (Accelerating Genetic Gains in Maize and Wheat for Improved Livelihoods, B&MGF Investment ID INV-003439) project, Improved Maize for African Soils (IMAS) Project, and the CGIAR Research Program on Maize (MAIZE). MAIZE receives W1&W2 support from the Governments of Australia, Belgium, Canada, China, France, India, Japan, Korea, Mexico, Netherlands, New Zealand, Norway, Sweden, Switzerland, United Kingdom, and United States and the World Bank.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our sincere thanks to CIMMYT technicians Carolyne Odhiambo and Veronica Ogugo in Kenya. We also thank Dr. Edward S. Buckler and the Institute of Genomic Diversity, Cornell University, for the high-density genotyping (GBS) and imputation service.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.572027/full#supplementary-material
Footnotes
- ^ https://data.cimmyt.org/dataset.xhtml;jsessionid=dc30e94cd6c7318ac8df044c42eb?persistentId=hdl%3A11529%2F10548467&version=DRAFT
- ^ https://www.maizegdb.org/gbrowse/maize_v2
References
Alvarado, G., López, M., Vargas, M., Pacheco, A., Rodríguez, F., Burgueño, J., et al. (2015). META-R (Multi Environment Trail Analysis with R for Windows) Version 5.0 - CIMMYT Research Software Dataverse - CIMMYT Dataverse Network. Available online at: https://data.cimmyt.org/dataset.xhtml?persistentId=hdl:11529/10201 (accessed March 20, 2020).
Asea, G., Vivek, B. S., Lipps, P. E., and Pratt, R. C. (2012). Genetic gain and cost efficiency of marker-assisted selection of maize for improved resistance to multiple foliar pathogens. Mol. Breed. 29, 515–527. doi: 10.1007/s11032-011-9568-8
Balint-Kurti, P. J., Zwonitzer, J. C., Pea, G., Pè, M. E., Lee, M., and Cardinal, A. J. (2008). Identification of quantitative trait loci for resistance to southern leaf blight and days to anthesis in two maize recombinant inbred line populations. Phytopathology 98, 315–320. doi: 10.1094/phyto-98-3-0315
Bekeko, Z., Fininsa, C., Hussien, S., Hussien, T., and Wegari, D. (2018). Resistance of some selected maize genotypes to gray leaf spot disease (Cercospora zeae-maydis) in Ethiopia. J. Gene Environ. Resour. Cons. 6, 1–13.
Benson, J. M., Poland, J. A., Benson, B. M., Stromberg, E. L., and Nelson, R. J. (2015). Resistance to gray leaf spot of maize: genetic architecture and mechanisms elucidated through nested association mapping and near-isogenic line analysis. PLoS Genet. 11:e1005045. doi: 10.1371/journal.pgen.1005045
Berger, D. K., Carstens, M., Korsman, J. N., Middleton, F., Kloppers, F. J., Tongoona, P., et al. (2014). Mapping QTL conferring resistance in maize to gray leaf spot disease caused by Cercospora zeina. BMC Genet 15:60. doi: 10.1186/1471-2156-15-60
Beyene, Y., Gowda, M., Olsen, M., Robbins, K. R., Pérez-Rodríguez, P., Alvarado, G., et al. (2019). Empirical comparison of tropical maize hybrids selected through genomic and phenotypic selections. Front. Plant Sci. 10:1502. doi: 10.3389/fpls.2019.01502
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Bubeck, D. M., Goodman, M. M., Beavis, W. D., and Grant, D. (1993). Quantitative trait loci controlling resistance to gray leaf spot in maize. Cell Biol. Mol. Genet. 33, 838–847. doi: 10.1088/978-0-7503-1094-9ch2
Chaikam, V., Gowda, M., Nair, S. K., Melchinger, A. E., and Boddupalli, P. M. (2019). Genome-wide association study to identify genomic regions influencing spontaneous fertility in maize haploids. Euphytica 215, 1–14. doi: 10.1007/s10681-019-2459-5
Chisholm, S. T., Mahajan, S. K., Whitham, S. A., Yamamoto, M. L., and Carrington, J. C. (2000). Cloning of the Arabidopsis RTM1 gene, which controls restriction of long-distance movement of tobacco etch virus. Proc. Natl. Acad. Sci. U.S.A. 97, 489–494. doi: 10.1073/pnas.97.1.489
Chung, C. L., Poland, J., Kump, K., Benson, J., Longfellow, J., Walsh, E., et al. (2011). Targeted discovery of quantitative trait loci for resistance to northern leaf blight and other diseases of maize. Theor. Appl. Genet. 123, 307–326. doi: 10.1007/s00122-011-1585-9
Clements, M. J., Dudley, J. W., and White, D. G. (2000). Quantitative trait loci associated with resistance to gray leaf spot of corn. Phytopathology 90, 1018–1025. doi: 10.1094/PHYTO.2000.90.9.1018
Combs, E., and Bernardo, R. (2013). Accuracy of genomewide selection for different traits with constant population size, heritability, and number of markers. Plant Genome 6, 1–7. doi: 10.3835/plantgenome2012.11.0030
Crossa, J., Pérez-Rodríguez, P., Cuevas, J., Montesinos-López, O., Jarquín, D., de los Campos, G., et al. (2017). Genomic selection in plant breeding: methods, models, and perspectives. Trends Plant Sci. 22, 961–975. doi: 10.1016/j.tplants.2017.08.011
Crous, P. W., Groenewald, J. Z., Groenewald, M., Caldwell, P., Braun, U., and Harrington, T. C. (2006). Species of Cercospora associated with grey leaf spot of maize. Stud. Mycol. 55, 189–197. doi: 10.3114/sim.55.1.189
Cui, Z., Luo, J., Qi, C., Ruan, Y., Li, J., Zhang, A., et al. (2016). Genome-wide association study (GWAS) reveals the genetic architecture of four husk traits in maize. BMC Genomics 17:946. doi: 10.1186/s12864-016-3229-6
de Jong, G., Pamplona, A. K. A., Von Pinho, R. G., and Balestre, M. (2018). Genome-wide association analysis of ear rot resistance caused by Fusarium verticillioides in maize. Genomics 110, 291–303. doi: 10.1016/j.ygeno.2017.12.001
Dhami, N. B., Kim, S., Paudel, A., Shrestha, J., and Rijal, T. R. (2015). A review on threat of gray leaf spot disease of maize in Asia. J. Maize Res. Dev. 1, 71–85. doi: 10.3126/jmrd.v1i1.14245
Du, L., Yu, F., Zhang, H., Wang, B., Ma, K., Yu, C., et al. (2020). Genetic mapping of quantitative trait loci and a major locus for resistance to grey leaf spot in maize. Theor. Appl. Genet. 133, 2521–2533. doi: 10.1007/s00122-020-03614-z
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:e19379. doi: 10.1371/journal.pone.0019379
Ertiro, B. T., Labuschagne, M., Olsen, M., Das, B., Prasanna, B. M., and Gowda, M. (2020a). Genetic dissection of nitrogen use efficiency in tropical maize through genome-wide association and genomic prediction. Front. Plant Sci. 11:474. doi: 10.3389/fpls.2020.00474
Ertiro, B. T., Olsen, M., Das, B., Gowda, M., and Labuschagne, M. (2020b). Genetic dissection of grain yield and agronomic traits in maize under optimum and low-nitrogen stressed environments. Int. J. Mol. Sci. 21:543. doi: 10.3390/ijms21020543
Fahlberg, P., Buhot, N., Johansson, O. N., and Andersson, M. X. (2019). Involvement of lipid transfer proteins in resistance against a non-host powdery mildew in Arabidopsis thaliana. Mol. Plant Pathol. 20, 69–77. doi: 10.1111/mpp.12740
George, M. L., Regalado, E., Warburton, M., Vasal, S., and Hoisington, D. (2004). Genetic diversity of maize inbred lines in relation to downy mildew. Euphytica 135, 145–155. doi: 10.1023/B:EUPH.0000014904.80451.7f
Gilmour, A. R., Cullis, B. R., and Thompson, R. (2009). ASReml User Guide. Release 3.0. Australia: NSW Department of Primary Industries.
Gordon, S. G., Lipps, P. E., and Pratt, R. C. (2006). Heritability and components of resistance to Cercospora zeae-maydis derived from maize inbred VO613Y. Phytopathology 96, 593–598. doi: 10.1094/PHYTO-96-0593
Gowda, M., Beyene, Y., Makumbi, D., Semagn, K., Olsen, M. S., Bright, J. M., et al. (2018). Discovery and validation of genomic regions associated with resistance to maize lethal necrosis in four biparental populations. Mol. Breed. 2018:66.
Gowda, M., Mahuku, G., Bright, J. M., Gowda, M., Babu, R., Das, B., et al. (2015). Genome-wide association and genomic prediction of resistance to maize lethal necrosis disease in tropical maize germplasm. Theor. Appl. Genet. 128, 1957–1968. doi: 10.1007/s00122-015-2559-0
Gullner, G., Komives, T., Király, L., and Schröder, P. (2018). Glutathione S-transferase enzymes in plant-pathogen interactions. Front. Plant Sci. 871:1836. doi: 10.3389/fpls.2018.01836
Hayes, B. J., Bowman, P. J., Chamberlain, A. J., and Goddard, M. E. (2009). Invited review: genomic selection in dairy cattle: progress and challenges. J. Dairy Sci. 92, 433–443. doi: 10.3168/jds.2008-1646
He, W., Yang, L., Leng, Y., Zhang, B., Yang, J., Li, L., et al. (2018). QTL mapping for resistance of maize to grey leaf spot. J. Phytopathol. 166, 167–176. doi: 10.1111/jph.12673
Heffner, E. L., Lorenz, A. J., Jannink, J. L., and Sorrells, M. E. (2010). Plant breeding with Genomic selection: gain per unit time and cost. Crop Sci. 50, 1681–1690. doi: 10.2135/cropsci2009.11.0662
Hill, W. G., and Weir, B. S. (1988). Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 33, 54–78. doi: 10.1016/0040-5809(88)90004-4
Holland, J. B. (2007). Genetic architecture of complex traits in plants. Curr. Opin. Plant Biol. 10, 156–161. doi: 10.1016/j.pbi.2007.01.003
Kibata, G., Smith, J., Langat, B., Simons, S., and Kinyua, Z. (2011). Status of grey leaf spot disease in Kenyan maize production ecosystems. Afr. Crop Sci. J. 18, 183–194. doi: 10.4314/acsj.v18i4.68647
Kosambi, D. (1944). The estimation of map distances from recombination values. Ann. Eugen. 12, 172–175. doi: 10.1111/j.1469-1809.1943.tb02321.x
Kuki, M. C., Scapim, C. A., Rossi, E. S., Mangolin, C. A., Do Amaral, A. T., and Barth Pinto, R. J. (2018). Genome wide association study for gray leaf spot resistance in tropical maize core. PLoS One 13:e0199539. doi: 10.1371/journal.pone.0199539
Latterell, F. M., and Rossi, A. E. (1983). Gray leaf spot corn: a disease on the move. Plant Sci 67, 842–847. doi: 10.1094/pd-67-842
Lehmensiek, A., Nelson, S. S. W., Esterhuizen, A. M., and Retief, A. E. (2001). Genetic mapping of gray leaf spot (GLS) resistance genes in maize. Theor. Appl. Genet. 103, 797–803. doi: 10.1007/s001220100599
Li, X., Han, Y., Wei, Y., Acharya, A., Farmer, A. D., Ho, J., et al. (2014). Development of an alfalfa SNP array and its use to evaluate patterns of population structure and linkage disequilibrium. PLoS One 9:e0084329. doi: 10.1371/journal.pone.0084329
Liu, L., Zhang, Y. D., Li, H. Y., Bi, Y. Q., Yu, L. J., Fan, X. M., et al. (2016). QTL mapping for gray leaf spot resistance in a tropical maize population. Plant Dis. 100, 304–312. doi: 10.1094/PDIS-08-14-0825-RE
Liu, W., Gowda, M., Steinhoff, J., Peter, H., Horst, F., and Wu, T. (2011). Association mapping in an elite maize breeding population. Theor. Appl. Genet. 123, 847–858. doi: 10.1007/s00122-011-1631-7
Liu, X., Wang, H., Wang, H., Guo, Z., Xu, X., Liu, J., et al. (2018). Factors affecting genomic selection revealed by empirical evidence in maize. Crop J. 6, 341–352. doi: 10.1016/j.cj.2018.03.005
Lorenzana, R. E., and Bernardo, R. (2009). Accuracy of genotypic value predictions for marker-based selection in biparental plant populations. Theor. Appl. Genet. 120, 151–161. doi: 10.1007/s00122-009-1166-3
Lu, Y., Yan, J., Guimarães, C. T., Taba, S., Hao, Z., Gao, S., et al. (2009). Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor. Appl. Genet. 120, 93–115. doi: 10.1007/s00122-009-1162-7
Lukman, R., Afifuddin, A., and Lubberstedt, T. (2012). Unraveling the Genetic Diversity of Maize Downy Mildew in Indonesia. J. Plant Pathol. Microbiol. 04, 2–9. doi: 10.4172/2157-7471.1000162
Lv, X., Song, M., Cheng, Z., Yang, X., Zhang, X., Zhou, Z., et al. (2020). qGLS1.02, a novel major locus for resistance to gray leaf spot in maize. Mol. Breed. 40:59. doi: 10.1007/s11032-020-01134-8
Mahuku, G., Lockhart, B. E., Wanjala, B., Jones, M. W., Kimunye, J. N., Stewart, L. R., et al. (2015). Maize Lethal Necrosis (MLN), an emerging threat to maize-based food security in sub-saharan africa. Phytopathology 105, 956–965. doi: 10.1094/phyto-12-14-0367-fi
Mammadov, J., Sun, X., Gao, Y., Ochsenfeld, C., Bakker, E., Ren, R., et al. (2015). Combining powers of linkage and association mapping for precise dissection of QTL controlling resistance to gray leaf spot disease in maize (Zea mays L.). BMC Genomics 16:916. doi: 10.1186/s12864-015-2171-3
Meng, L., Li, H., Zhang, L., and Wang, J. (2015). QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 3, 269–283. doi: 10.1016/j.cj.2015.01.001
Meuwissen, T. H., Hayes, B. J., and Goddard, M. E. (2001). Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829.
Myles, S., Peiffer, J., Brown, P. J., Ersoz, E. S., Zhang, Z., Costich, D. E., et al. (2009). Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21, 2194–2202. doi: 10.1105/tpc.109.068437
Nyaga, C., Gowda, M., Beyene, Y., and Muriithi, W. (2020). Genome-wide analyses and prediction of resistance to MLN in large tropical maize germplasm. Genes 11:16. doi: 10.3390/genes11010016
Pozar, G., Butruille, D., Silva, H. D., McCuddin, Z. P., and Penna, J. C. V. (2009). Mapping and validation of quantitative trait loci for resistance to Cercospora zeae-maydis infection in tropical maize (Zea mays L.). Theor. Appl. Genet. 118, 553–564. doi: 10.1007/s00122-008-0920-2
Prasanna, B. M., Nair, S. K., Babu, R., Gowda, M., Zhang, X., Xu, Y., et al. (2020). “Increasing genetic gains in maize in stress-prone environments of the tropics,” in Genomic Designing of Climate-Smart Cereal Crops, ed. C. Kole (Cham: Springer).
R Development Core Team. (2015). R: A Language And Environment For Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rashid, Z., Singh, P. K., Vemuri, H., Zaidi, P. H., Prasanna, B. M., and Nair, S. K. (2018). Genome-wide association study in Asia-adapted tropical maize reveals novel and explored genomic regions for sorghum downy mildew resistance. Sci. Rep. 8, 1–12. doi: 10.1038/s41598-017-18690-3
Rawlings, J. O., Pantula, S. G., and Dickey, D. A. (1998). Applied Regression Analysis - A Research Tool. Berlin: Springer Science & Business Media, doi: 10.1109/mei.2002.981332
Remington, D. L., Thornsberry, J. M., Matsuoka, Y., Wilson, L. M., Whitt, S. R., Doebley, J., et al. (2001). Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. U.S.A. 98, 11479–11484. doi: 10.1073/pnas.201394398
SAS Institute Inc. (2015). SAS 9.4 Intelligence Platform: System Admin- Istration Guide. Cary: SAS Institute Inc.
Schwarz, G. (1978). Estimating the dimension of a model. Ann Stat 6, 461–464. doi: 10.1214/aos/1176344136
Semagn, K., Magorokosho, C., Vivek, B. S., Makumbi, D., Beyene, Y., Mugo, S., et al. (2012). Molecular characterization of diverse CIMMYT maize inbred lines from eastern and southern Africa using single nucleotide polymorphic markers. BMC Genom. 13:113. doi: 10.1186/1471-2164-13-113
Shi, L., Lv, X., Weng, J., Zhu, H., Liu, C., Hao, Z., et al. (2014). Genetic characterization and linkage disequilibrium mapping of resistance to gray leaf spot in maize (Zea mays L.). Crop J. 2, 132–143. doi: 10.1016/j.cj.2014.02.001
Shi, L. Y., Li, X. H., Hao, Z. F., Xie, C. X., Ji, H. L., Lian, H., et al. (2007). Comparative QTL mapping of resistance to gray leaf spot in maize based on bioinformatics. Agric. Sci. China 6, 1411–1419. doi: 10.1016/S1671-2927(08)60002-4
Sitonik, C., Suresh, L. M., Beyene, Y., Olsen, M. S., Makumbi, D., Oliver, K., et al. (2019). Genetic architecture of maize chlorotic mottle virus and maize lethal necrosis through GWAS, linkage analysis and genomic prediction in tropical maize germplasm. Theor. Appl. Genet. 132, 2381–2399. doi: 10.1007/s00122-019-03360-x
Vivek, B. S., Odongo, O., Njuguna, J., Imanywoha, J., Bigirwa, G., Diallo, A., et al. (2010). Diallel analysis of grain yield and resistance to seven diseases of 12 African maize (Zea mays L.) inbred lines. Euphytica 172, 329–340. doi: 10.1007/s10681-009-9993-5
Vos, P. G., Paulo, M. J., Voorrips, R. E., Visser, R. G. F., van Eck, H. J., and van Eeuwijk, F. A. (2017). Evaluation of LD decay and various LD-decay estimators in simulated and SNP-array data of tetraploid potato. Theor. Appl. Genet. 130, 123–135. doi: 10.1007/s00122-016-2798-8
Wang, X., Xu, Y., Hu, Z., and Xu, C. (2018). Genomic selection methods for crop improvement: current status and prospects. Crop J. 6, 330–340. doi: 10.1016/j.cj.2018.03.001
Ward, J. M. J. (1996). Epidemiology and Management of Gray Leaf Spot: A New Disease of Maize in South Africa. South Africa: University of Natal Pietermar. PhD Thesis.
Welz, H. G., and Geiger, H. H. (2000). Genes for resistance to northern corn leaf blight in diverse maize populations. Plant Breed. 119, 1–14. doi: 10.1046/j.1439-0523.2000.00462.x
Wisser, R. J., Balint-Kurti, P. J., and Nelson, R. J. (2006). The genetic architecture of disease resistance in maize: a synthesis of published studies. Phytopathology 96, 120–129. doi: 10.1094/PHYTO-96-0120
Würschum, T., Liu, W., Gowda, M., Maurer, H. P., Fischer, S., Schechert, A., et al. (2012). Comparison of biometrical models for joint linkage association mapping. Heredity 108, 332–340. doi: 10.1038/hdy.2011.78
Xu, L., Zhang, Y., Shao, S., Chen, W., Tan, J., Zhu, M., et al. (2014). High-resolution mapping and characterization of qRgls2, a major quantitative trait locus involved in maize resistance to gray leaf spot. BMC Plant Biol. 14:230.
Yigrem, M., and Yohannes, M. (2019). Participatory evaluation of maize varieties for the management of maize gray leaf spot disease (Cercospora zeae maydis) at Gondar Zuria district, northwest Ethiopia. J. Dryl. Agric. 5, 1–6. doi: 10.5897/joda2018.0001
Yu, J., and Buckler, E. S. (2006). Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 17, 155–160. doi: 10.1016/j.copbio.2006.02.003
Zhang, X., Pérez-Rodríguez, P., Semagn, K., Beyene, Y., Babu, R., López-Cruz, M. A., et al. (2015). Genomic prediction in biparental tropical maize populations in water-stressed and well-watered environments using low-density and GBS SNPs. Heredity 114, 291–299. doi: 10.1038/hdy.2014.99
Zhao, Y., Gowda, M., Liu, W., Würschum, T., Maurer, H. P., Longin, F. H., et al. (2011). Accuracy of genomic selection in European maize elite breeding populations. Theor. Appl. Genet. 124, 769–776. doi: 10.1007/s00122-011-1745-y
Zhu, C., Gore, M., Buckler, E. S., and Yu, J. (2008). Status and prospects of association mapping in plants. Plant Genome. 1:5. doi: 10.3835/plantgenome2008.02.0089
Zwonitzer, J. C., Coles, N. D., Krakowsky, M. D., Arellano, C., Holland, J. B., McMullen, M. D., et al. (2010). Mapping resistance quantitative trait loci for three foliar diseases in a maize recombinant inbred line population—evidence for multiple disease resistance? Phytopathology 100, 72–79. doi: 10.1094/phyto-100-1-0072
Keywords: GLS, GP, GWAS, SNP, disease resistance, JLAM
Citation: Kibe M, Nair SK, Das B, Bright JM, Makumbi D, Kinyua J, Suresh LM, Beyene Y, Olsen MS, Prasanna BM and Gowda M (2020) Genetic Dissection of Resistance to Gray Leaf Spot by Combining Genome-Wide Association, Linkage Mapping, and Genomic Prediction in Tropical Maize Germplasm. Front. Plant Sci. 11:572027. doi: 10.3389/fpls.2020.572027
Received: 12 June 2020; Accepted: 29 September 2020;
Published: 02 November 2020.
Edited by:
Nicolas Rispail, Institute for Sustainable Agriculture, Spanish National Research Council, SpainReviewed by:
Ana Butron, Misión Biológica de Galicia, Spanish Council for Scientific Research (MBG-CSIC), SpainMauricio Carlos Kuki, Instituto GDM, Brazil
Copyright © 2020 Kibe, Nair, Das, Bright, Makumbi, Kinyua, Suresh, Beyene, Olsen, Prasanna and Gowda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manje Gowda, TS5Hb3dkYUBjZ2lhci5vcmc=