 Karolina Heyduk1,2*
Karolina Heyduk1,2* Edward V. McAssey1,3,4
Edward V. McAssey1,3,4 Jane Grimwood5Shengqiang Shu6
Jane Grimwood5Shengqiang Shu6 Jeremy Schmutz5,6
Jeremy Schmutz5,6 Michael R. McKain7
Michael R. McKain7 Jim Leebens-Mack8
Jim Leebens-Mack8- 1School of Life Sciences, University of Hawai’i at Mānoa, Honolulu, HI, United States
- 2Department of Ecology and Evolutionary Biology, Yale University, New Haven, CT, United States
- 3Department of Biological Sciences, Quinnipiac University, Hamden, CT, United States
- 4Department of Biology and Environmental Science, University of New Haven, West Haven, CT, United States
- 5HudsonAlpha Institute for Biotechnology, Huntsville, AL, United States
- 6Lawrence Berkeley National Laboratory, US Department of Energy Joint Genome Institute, Berkeley, CA, United States
- 7Department of Biological Sciences, University of Alabama, Tuscaloosa, AL, United States
- 8Department of Plant Biology, University of Georgia, Athens, GA, United States
Hybridization in plants results in phenotypic and genotypic perturbations that can have dramatic effects on hybrid physiology, ecology, and overall fitness. Hybridization can also perturb epigenetic control of transposable elements, resulting in their proliferation. Understanding the mechanisms that maintain genomic integrity after hybridization is often confounded by changes in ploidy that occur in hybrid plant species. Homoploid hybrid species, which have no change in chromosome number relative to their parents, offer an opportunity to study the genomic consequences of hybridization in the absence of change in ploidy. Yucca gloriosa (Asparagaceae) is a young homoploid hybrid species, resulting from a cross between Yucca aloifolia and Yucca filamentosa. Previous analyses of ∼11 kb of the chloroplast genome and nuclear-encoded microsatellites implicated a single Y. aloifolia genotype as the maternal parent of Y. gloriosa. Using whole genome resequencing, we assembled chloroplast genomes from 41 accessions of all three species to re-assess the hybrid origins of Y. gloriosa. We further used re-sequencing data to annotate transposon abundance in the three species and mRNA-seq to analyze transcription of transposons. The chloroplast phylogeny and haplotype analysis suggest multiple hybridization events contributing to the origin of Y. gloriosa, with both parental species acting as the maternal donor. Transposon abundance at the superfamily level was significantly different between the three species; the hybrid was frequently intermediate to the parental species in TE superfamily abundance or appeared more similar to one or the other parent. In only one case—Copia LTR transposons—did Y. gloriosa have a significantly higher abundance relative to either parent. Expression patterns across the three species showed little increased transcriptional activity of transposons, suggesting that either no transposon release occurred in Y. gloriosa upon hybridization, or that any transposons that were activated via hybridization were rapidly silenced. The identification and quantification of transposon families paired with expression evidence paves the way for additional work seeking to link epigenetics with the important trait variation seen in this homoploid hybrid system.
Introduction
Hybridization between related species has the potential to generate novel genotypic and phenotypic combinations, sometimes resulting in the origin of new species. Understanding the factors that promote the process of hybridization, as well as the maintenance of newly created hybrids, has been of considerable interest to both the fields of ecology and evolution (Gross and Rieseberg, 2005). As the generation of biodiversity is of primary importance to evolutionary biology, many studies have sought to determine whether or not newly created hybrids are reproductively isolated from parental species and are capable of persisting in a hybrid state for many generations. The tools aimed at studying plant hybridization include observational studies of plants and their pollinators in the wild (Leebens-Mack and Milligan, 1998; Hersch and Roy, 2007), reciprocal transplant studies across multiple environments (Wang et al., 1997), manual pollinations between related species (Sun et al., 2018), cytogenetics (Thórsson et al., 2001), and population genomics (Bredeson et al., 2016). Hybridization can result in allopolyploid individuals, in which hybridization occurs at the same time as chromosome doubling, as well as homoploids, in which there is no change in chromosome number (for a review, see Rieseberg, 1997; Soltis and Soltis, 2009). Transposable element content and abundance has been hypothesized to contribute to genome dominance in allopolyploid species (Edger et al., 2017; Bird et al., 2018), but change in ploidy makes it difficult to assess its importance relative to hybridization in the genesis of a new species. Homoploid hybrid species provide an opportunity to focus on the effects of hybridization while controlling for ploidy level (Ungerer et al., 2009; Staton et al., 2012).
Investigation of hybridization almost always begins with a detailed understanding of the genetics and life history of the putative parental and hybrid species. In the case of wild sunflowers, numerous studies have focused on how Helianthus annuus and H. petiolaris have hybridized multiple independent times to form three homoploid hybrid species: H. anomalus, H. deserticola, and H. paradoxus (Rieseberg, 1991; Rieseberg et al., 2003). These hybrid species are morphologically distinct from their parents and each other (Rieseberg et al., 2003), display varying levels of salt tolerance (Welch and Rieseberg, 2002; Karrenberg et al., 2006), show gene expression differences (Lai et al., 2006), and exhibit population genetic patterns consistent with selective sweeps (Sapir et al., 2007). The repeated formation of homoploid hybrids in Helianthus has increased our understanding of hybrid speciation from both ecological and genomic perspectives, yet it is only one example of homoploid hybridization in flowering plants. Another well-studied example of homoploid hybridization is in Iris nelsonii, a hybrid suspected to have genetic contributions from more than two species based on patterns of both nuclear and plastid genetic variation (Arnold, 1993). The fitness of the hybrid species relatives to the parental species varies depending on the moisture of the environments, implying that genotype-by-environment interactions differentially affect parental and hybrid genotypes, a phenomenon that can lead to hybrid speciation (Johnston et al., 2001).
While hybridization’s effect on the generation of biodiversity and the movement of adaptive traits between species has been well established, the effect on the genome is only recently being fully understood. McClintock (1984), described hybridization as a “challenge” or “shock” for the genome; the merger of two separate genomes in a single nucleus results in a completely novel genomic environment. Post hybridization, alleles once restricted to separate species now interact in a new cellular setting, allowing for the formation of novel phenotypes, epistatic interactions, and potentially significant and rapid evolutionary change. Possible outcomes of hybridization and subsequent genome shock include: alteration of gene expression (Hegarty et al., 2009; Xu et al., 2009); chromosomal rearrangements (Rieseberg et al., 1995; Lai et al., 2005; Danilova et al., 2017); genome dominance, in which one progenitor genome expresses and/or retains more genes (Rapp et al., 2009; Bardil et al., 2011; Schnable et al., 2011; Yoo et al., 2013; Edger et al., 2017; Bird et al., 2018); epigenetic perturbation (Salmon et al., 2005), which in turn can lead to a release of silencing of repetitive elements and allows for subsequent repeat proliferation (Ungerer et al., 2006; Parisod et al., 2009).
Repetitive elements in particular have been implicated in the divergence of hybrid species from their progenitors. For example, RNA-seq suggests that established homoploid hybrid sunflowers, as opposed to newly synthesized hybrids, have elevated transposon expression levels (Renaut et al., 2014). In two of these hybrid sunflower species fluorescent in situ hybridization studies identified expansions of Gypsy retrotransposons relative to the progenitor species (Staton and Ungerer, 2009). Gypsy and Copia elements are typically the most abundant superfamilies in plant genomes, and are both Class I retrotransposons that replicate via a “copy and paste” mechanism (Wessler et al., 1995), in contrast to the variety of Class II DNA transposons that replicate via a “cut and paste” mechanism (Feschotte and Pritham, 2007). Transposons can affect traits by disrupting genes, duplicating or re-organizing genes (Xiao et al., 2008), or they can land upstream and create new patterns of gene expression (Studer et al., 2011). The accumulation of transposons contributes to a large proportion of genome size variation seen in plants (Tenaillon et al., 2011), and ectopic recombination between transposable elements can result in genomic deletions and are a major force in genome evolution (Devos et al., 2002).
While homoploid hybrid systems are relatively rare, recent efforts to sequence the genomes of Yucca (Asparagaceae) species allows us to investigate the effects of hybridization on a homoploid genome. Yucca aloifolia L. and Yucca filamentosa L. are emergent models in understanding the evolution of CAM photosynthesis, as the species use CAM and C3, respectively (Heyduk et al., 2016). The two species also hybridize to form Y. gloriosa L. (Rentsch and Leebens-Mack, 2012), which is photosynthetically intermediate and a relatively recently derived homoploid hybrid species (Trelease, 1902). All three species have genome sizes of ∼2.8 Gb (Heyduk unpublished) and are sympatric in the Southeastern United States, with Y. filamentosa found across a broader range of the eastern seaboard, including into New England and the Midwest; Y. aloifolia is restricted largely to the Southeastern United States and reaches only as far north as North Carolina. Yucca gloriosa is even more restricted than either parent in its range, found only in the coastal dune systems of the Atlantic seaboard and, based on herbarium records, along the coast of the Gulf of Mexico. It is thought that Y. aloifolia was introduced into the Southeastern United States from Mexico or the Caribbean by Spanish colonists (Trelease, 1902; Groman and Pellmyr, 2000). Perhaps as a result of the human-involved introduction, Y. aloifolia has escaped the dependence on the obligate Yucca-yucca moth pollination mutualism and can be pollinated by the yucca moth Tegeticula yuccasella (Leebens-Mack and Pellmyr, 2004) or introduced generalist honeybees (Apis mellifera) (Rentsch and Leebens-Mack, 2014). Yucca filamentosa still retains its obligate pollination mutualism with the yucca moths (Tegeticula yuccasella and T. cassandra) (Pellmyr, 1999), and overlaps in flowering time with Y. aloifolia briefly and only in some years, suggesting that hybridization between the two species may be rare.
Previous work suggested no variation in chloroplast or microsatellite repeats in a small sampling of Y. aloifolia genotypes, and further indicated that Y. aloifolia is the maternal parent in any hybridization events that led to Y. gloriosa (Rentsch and Leebens-Mack, 2012). Through a whole genome sequencing project that aims to assemble the genomes of Y. aloifolia and Y. filamentosa, resequencing was performed on individuals of all three Yucca species. Using the resequencing data, we sought to re-test hypotheses on the number and direction of hybridization events in Y. gloriosa. Specifically, we assembled maternally inherited chloroplast genomes, which can inform not only the evolutionary history, but also the direction of hybridization. We assessed whether all hybrid Y. gloriosa individuals were nested within Y. aloifolia on a phylogenetic tree and haplotype network, consistent with the hypothesis of a single hybridization event with Y. aloifolia as the maternal parent. We further examined the repeat landscape of all three species to determine if repeat content in the hybrid is intermediate between the two parents, or if transgressive repeat abundance exists, suggesting a degree of post-hybridization genomic shock. Finally, using existing RNA-sequencing datasets in the three species of Yucca, we examined the activity of repeats using mRNA reads as a proxy. Through the use of high throughput genomic data, we find that Y. gloriosa is the result of repeated and bi-directional hybridization events that evidently led to minimal repeat proliferation. Our findings further suggest that there is little evidence of repetitive element release in Y. gloriosa as a result of hybridization.
Materials and Methods
DNA Sampling, Library Preparation, and Sequencing
Clones of 41 individuals (5 from Y. aloifolia, 24 from Y. gloriosa, and 12 form Y. filamentosa) were collected throughout the Southeastern United States from 2013 to 2015 and planted in the University of Georgia greenhouse (Figure 1 and Supplementary Table 1). In 2018, approximately 100 mg of fresh tissue was harvested from fully expanded leaves and kept on ice until DNA extraction, using a CTAB protocol with sorbitol addition that removes secondary compounds before DNA purification (Doyle, 1987; Štorchová et al., 2000). DNA was visualized on a 1.5% agarose gel to measure integrity and quantified via Qubit. Samples were shipped to the HudsonAlpha Institute for Biotechnology, where Illumina 350 basepair PCRfree fragment libraries were constructed using standard protocols. Each library was uniquely barcoded and sequenced on a NovaSeq 6000 with paired end 150bp reads. Data is available on the NCBI Sequence Read Archive (for a full list of SRA accessions, see Supplementary Table 1).
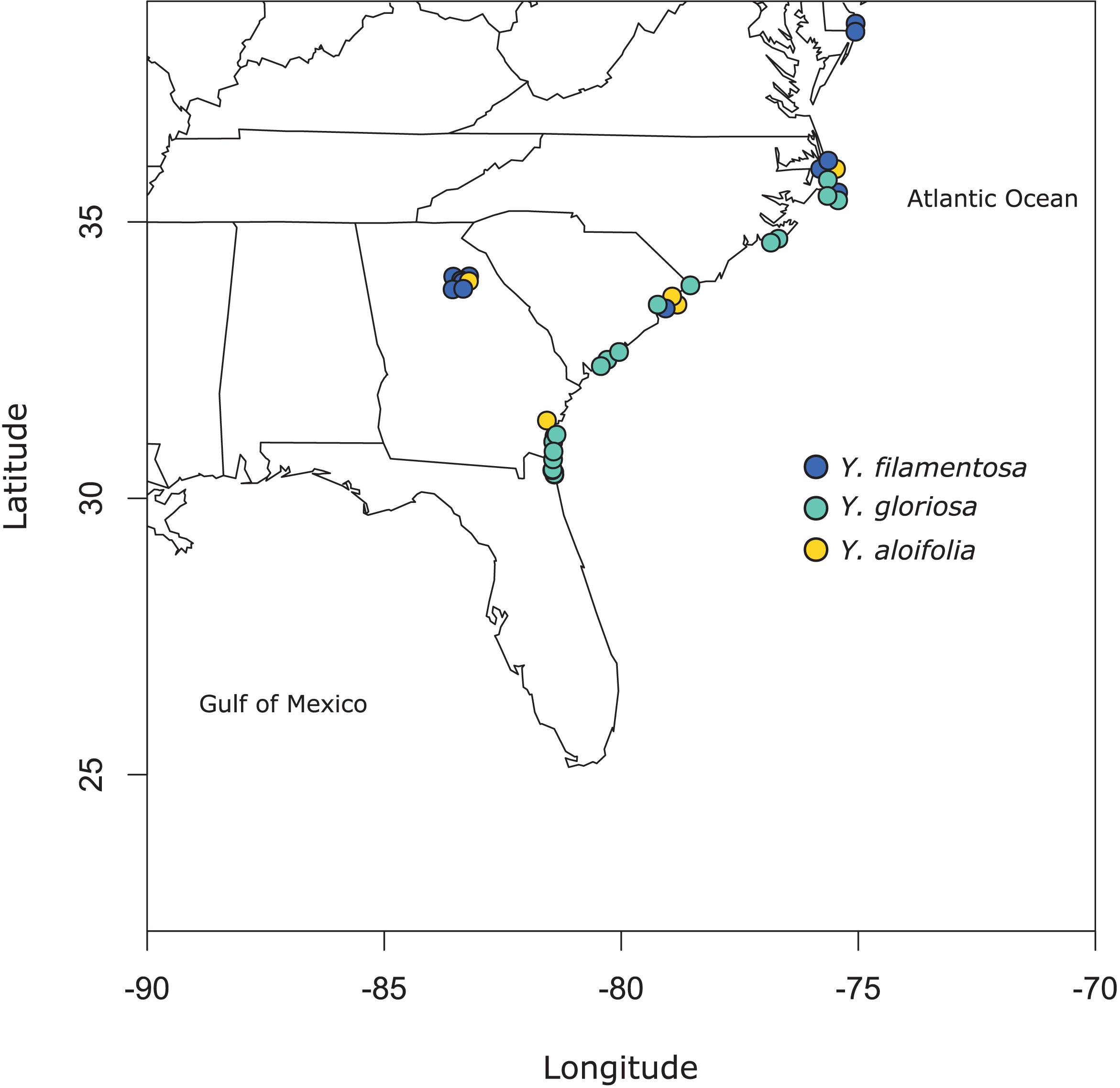
Figure 1. Map of populations sampled for this study. See Supplementary Table 1 for full geographic locality information.
Chloroplast Genome Assembly and Analysis
Raw reads were first quality trimmed using Trimmomatic v 0.36 (Bolger et al., 2014). Due to the sheer size of the sequence data per individual—roughly 400–800 million reads—a subset of four million paired-end reads was randomly sub-sampled from each library’s trimmed dataset in order to speed up computational analyses. The sub-sampled data were used as input into the program Fast-Plast1, which assembles plastid genomes by first mapping reads to a reference plastid genome (here we used a previously assembled Y. filamentosa chloroplast genome; McKain et al., 2016).
Chloroplast genomes of Agave americana (NCBI accession: KX519714.1, Abraham et al., 2016) and Nolina atopocarpa (NCBI accession: NC_032708.1) were used as outgroups for phylogenetic analyses. All Yucca chloroplast assemblies as well as Agave and Nolina were aligned using MAFFT (Katoh and Standley, 2013). The alignment was manually inspected for misaligned regions, and as a result three Yucca genotypes (Y. aloifolia YA7, and Y. gloriosa YG13 and YG61) containing considerable misalignments indicative of a sub-optimal genome assembly were not included in further analyses. The second inverted repeat (IR) region was removed before tree estimation: an aligned Y. aloifolia chloroplast genome sequence (YA23) was annotated for the IR by conducting a BLASTn (Altschul et al., 1990) against itself. The position of an inverted self-hit in YA23 was used to remove the second IR from the multi-species alignment. The optimal model of molecular evolution (GTR + Gamma) was determined using JModelTest v2 and BIC penalized-likelihood (Darriba et al., 2012) on the CIPRES gateway (Miller et al., 2010). The multiple sequence alignment was then used to estimate a chloroplast phylogeny using RAxML v8.2.11, with 500 bootstrap replicates (Stamatakis, 2006). The entire chloroplast alignment (with both IR) of the Yucca species without outgroups was also used to construct a median joining chloroplast haplotype network using PopArt (epsilon = 0) (Leigh and Bryant, 2015). Chloroplast genome assemblies were annotated in Geneious Prime 2019.2.3, using the built-in annotation tool with the previously published Y. filamentosa annotation as a reference (NCBI accession: KX931467, McKain et al., 2016). Chloroplast genome assemblies are available on NCBI’s GenBank (Supplementary Table 1), and the plastid alignment and newick files can be found on github2.
Repetitive Content Classification and Analysis
In a similar fashion to the chloroplast sequence processing, one million trimmed paired-end reads were randomly sub-sampled for an analysis of transposon content. In order to ensure that only nuclear repetitive sequences were being analyzed, reads were first mapped to Yucca chloroplast and mitochondrial genome sequences (reference files are available at JGI Genome Portal3) using Bowtie v2 with default settings (Langmead and Salzberg, 2012) to be flagged for removal. The nuclear data were retained and further processed in preparation for downstream steps, including: converting bam mapping files to fastq files using SAMTools v1.9 (Li et al., 2009) and BEDTools v2.26 (Quinlan and Hall, 2010), interleaving fastq files so that pairs are found sequentially in a single file (script4, from Boisvert et al., 2010), and converting fastq files to fasta files with the FASTX-Toolkit v 0.145.
Transposome (Staton and Burke, 2015) was used to cluster and identify repetitive DNA sequences in all 41 Yucca genotypes using a Yucca-specific reference. Briefly, RepeatModeler (Smit and Hubley, 2008) was used to predict repeat families de novo on the assembled Yucca genomes; RepeatModeler uses both RECON (Bao and Eddy, 2002) and RepeatScout (Price et al., 2005) to identify repeat family consensus sequences. To remove false positives (e.g., repetitive domains within genes), the predicted RepeatModeler consensus sequences were searched for functional PFAM and Panther domains. If no domains—or only known transposable element domains—were found in a given putative repeat family, it was retained as a true repeat; if only false positive domains were identified, the family was removed from further analysis. Putative repeat families that had a combination of transposable element and false positive domains, or had otherwise unknown domain classes, underwent manual curation.
For annotating Y. aloifolia and Y. filamentosa repeats via Transposome, we used the species-specific RepeatModeler families (repetitive element reference files are available at JGI Genome Portal; see footnote). For Y. gloriosa hybrid individuals, we concatenated the two parental repeat databases. Finally, we used the following parameters in our usage of Transposome: percent identity = 90%, a required fraction of overlap between pairwise matches of 0.55, a minimum cluster size of 100, a merge threshold of 1,000, and a BLAST e-value of 1. Cross-species comparisons of transposons included the average amount of total repetitive DNA as well as the relative amounts (genomic proportion) of transposon superfamilies. In R v. 3.6.1 and v. 4.0.2 (R Core Team, 2019), we used ANOVA to determine whether there were significant differences between species in the relative amount of repetitive DNA in each of the 10 transposon superfamilies and abundance of individual family lineages within a superfamily. Post hoc tests were conducted with the emmeans package in R6. Additionally, a data matrix containing each individual’s relative amount of repetitive DNA for each of the 10 superfamilies served as the input for a principal components analysis, using the prcomp() function in R. Throughout, we use the transposable element classification system described in Wicker et al. (2007).
Repetitive Element Activity via mRNAseq
Many repetitive elements contain sequences that are involved in their replication and therefore are transcribed into mRNA; transcripts produced from these repeats can be detected by mRNA sequencing (Hollister et al., 2011; Dion-Côté et al., 2014). While read counts from mRNA sequencing are a proxy for transcription of a repeat, no assumptions can be made as to the successful integration of a repeat copy into the genome post transcription; a variety of genomic mechanisms exist to silence and degrade repetitive element-derived transcripts (Lisch, 2009; Fultz et al., 2015). Nevertheless, as a first approximation of repeat activity, we used previously published mRNA-seq data on the three species of Yucca analyzed here (Heyduk et al., 2019). Briefly, RNA from leaf tissue was collected from all three species of Yucca growing in growth chambers set to 30°C/18°C day/night temperatures, with ∼400 μmol m–2 s–1 of light at leaf level, and 40% humidity in a 12 h day/night light regime. While the previous study further assessed gene expression under drought, here only libraries from well-watered plants taken during the daytime were analyzed. The original study used 2–3 genotypes per species, each of which had 2–3 replicates that were taken from different time points during the day. Because replication within a genotype is confounded with time, we limited our analyses to considering only species-specific differences rather than examining genotypic differences within species. Final species-level replication varied from 6 in Y. aloifolia to 9 in Y. gloriosa and Y. filamentosa.
RNA reads were mapped to the same repeat databases used in Transposome; Y. aloifolia and Y. filamentosa reads were mapped to each species’ specific repeat reference, while Y. gloriosa reads were mapped to a merged parental reference. RNA reads were mapped via Kallisto v 0.43 using default parameters (Bray et al., 2016). For Y. gloriosa, counts were summed in cases where both parental species had a consensus sequence for a given repeat family. Libraries were first normalized by the Trimmed Mean of M-values (TMM) (Robinson and Oshlack, 2010) as implemented in EdgeR (Robinson et al., 2010), then scaled by overall abundance of that repeat family as estimated by Transposome. To scale, a matrix consisting of all repeat abundances across all genotypes from the three Yucca species was scaled by the maximum abundance of all families identified by Transposome. These scaled abundance values were then used as a multiplier of the TMM normalized read counts. By normalizing by genomic abundance, expression of repeats could then be compared across genotypes and species that have varying genomic fraction of the repeat families. Once normalized and scaled, we tested for significant expression within species using a glm intercept model in the glm.nb() function in the R package MASS (Venables and Ripley, 2013), which employs a negative binomial model appropriate for count data that exhibits a degree of over dispersion. Differentially expressed repeats between species were also tested with a negative binomial model, and post hoc tests were done using the emmeans() function from the R package emmeans.
Results
Plastid Phylogenetic and Haplotype Analyses
Despite the relatedness between the three Yucca species studied here, there was enough divergence between the species’ chloroplast genomes to identify highly supported clades of chloroplast haplotypes (Figure 2). Y. gloriosa genotypes were found nested within three separate clades (Figure 2). A single Y. gloriosa genotype, YG16, was within a clade that otherwise contained all of the Y. filamentosa individuals that were analyzed. Three Y. gloriosa genotypes (YG12, YG55, and YG56) were placed in a clade with two Y. aloifolia genotypes (YA23 and YA11). The remaining 18 Y. gloriosa genotypes were grouped with the remaining two Y. aloifolia individuals (YA3 and YA32).
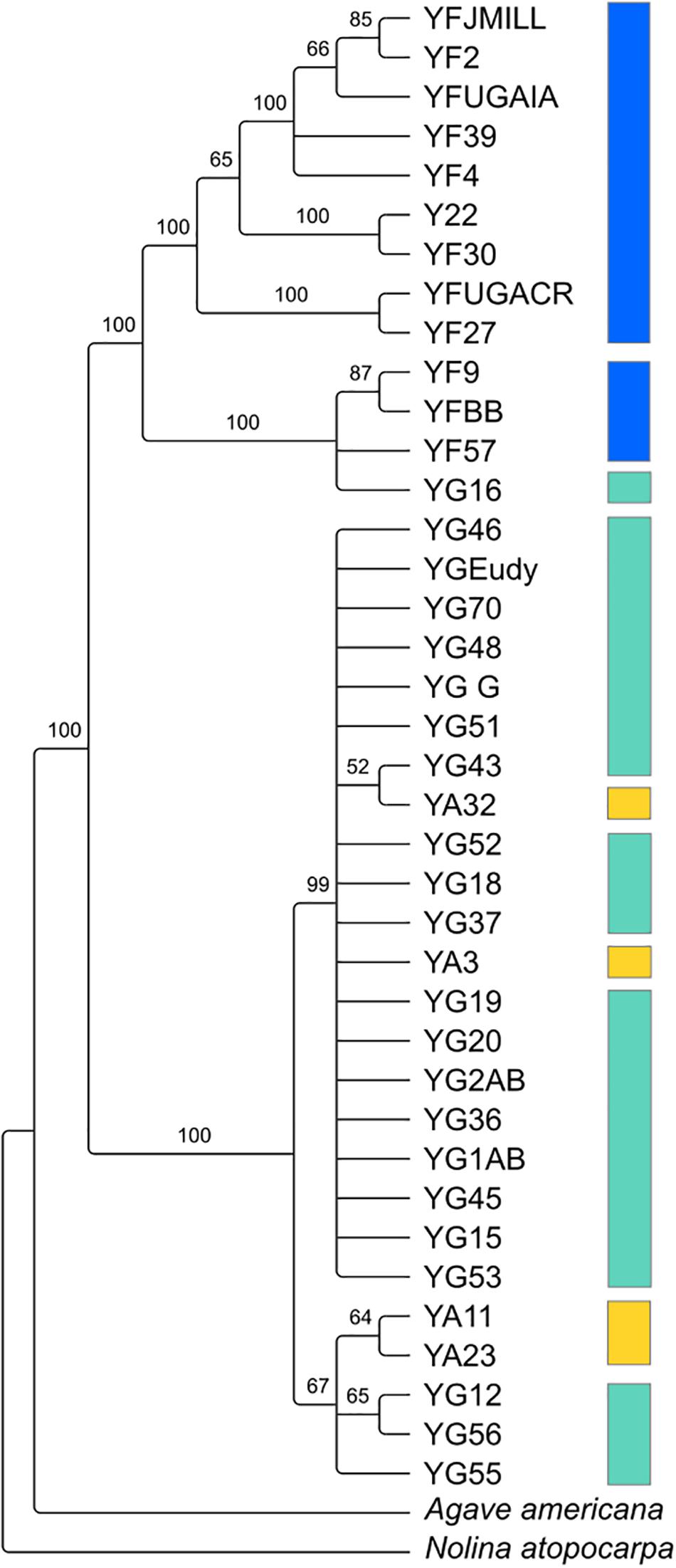
Figure 2. RAxML estimated phylogeny of the plastome using only one inverted repeat copy in the alignment. Bootstrap support indicated on the branches, with nodes that had less than 50 bootstrap support collapsed. Colors indicate the three species: Y. aloifolia (yellow), Y. filamentosa (blue), and Y. gloriosa (teal).
PopArt haplotype analysis (Leigh and Bryant, 2015) identified the same patterns found in the maximum likelihood-based phylogeny. Over 350 substitutions differentiated the two major groupings of genotypes (Y. aloifolia and Y. filamentosa-like chloroplast genomes; Figure 3). Yucca filamentosa had considerably more chloroplast haplotypes compared to Y. aloifolia (7 vs. 2, respectively; Figure 3). In contrast to previous analysis of nuclear simple repeats (Rentsch and Leebens-Mack, 2012), genetic diversity was seen not only in the Y. aloifolia chloroplast genomes but also for Y. gloriosa, which had four substitutions separating the different Y. aloifolia-like haplotypes, and over 400 substitutions separating the single Y. filamentosa-like haplotype from other individuals of Y. gloriosa.
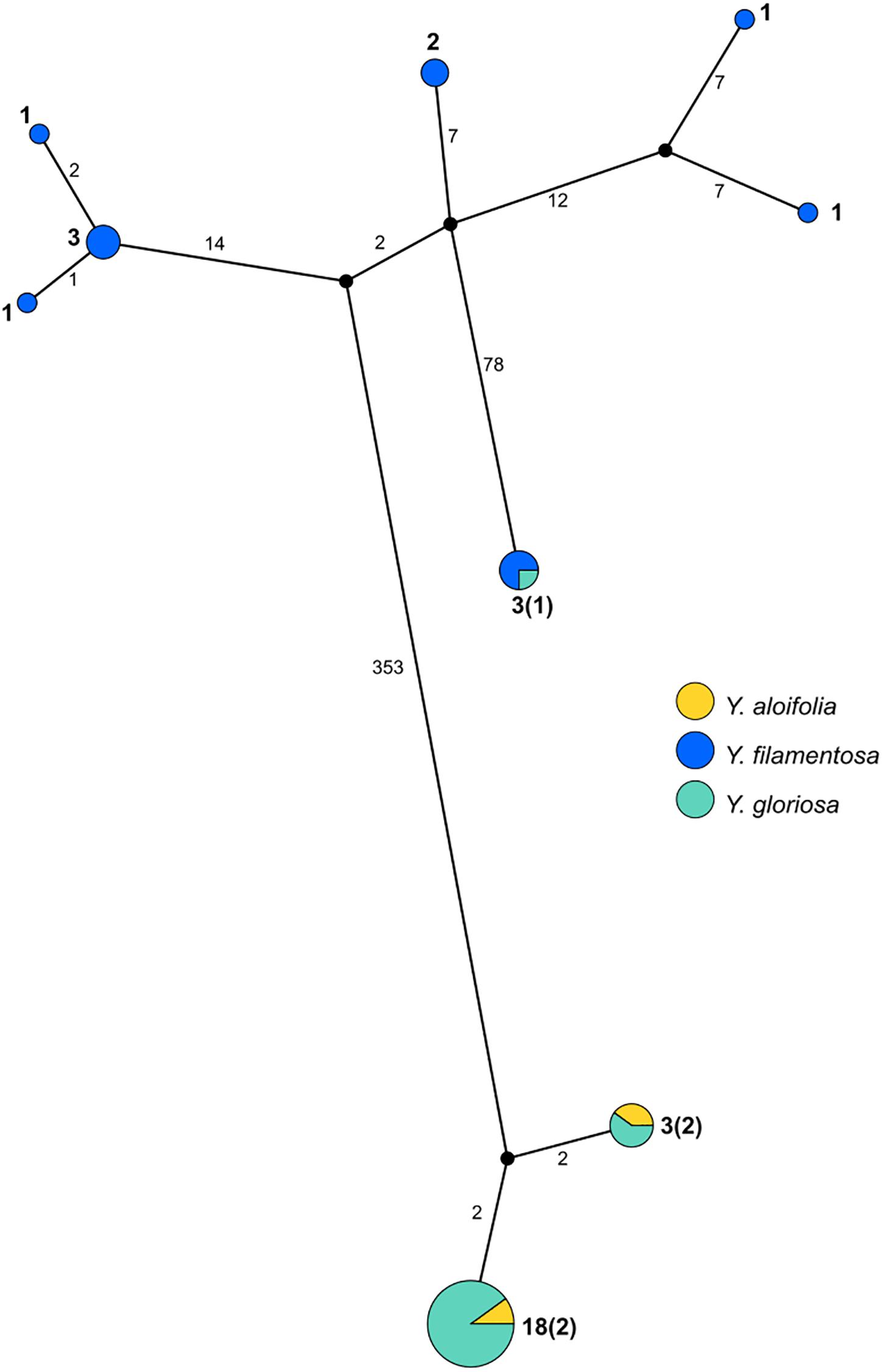
Figure 3. Haplotype network estimated from the entire plastome alignment across all three species, excluding outgroup accessions. Haplotype estimated via PopArt, with number of substitutions separating haplotypes on branches and size (number of individuals) indicated in boldface numbers next to haplotypes, with majority number and, in parentheses, minority number.
Repetitive Fraction of Yucca Genomes
The fraction of the genome containing repetitive DNA significantly differed between the three species [p < 0.001, F(2, 38) = 17.853]. While Y. aloifolia (mean repetitive genome fraction = 0.658; SD = 0.0138) and Y. gloriosa (mean = 0.662; SD = 0.0215) had statistically indistinguishable amount of repetitive DNA, Y. filamentosa was significantly lower than both species (mean = 0.621; SD = 0.0167; p < 0.01 for both post hoc comparisons). Moreover, the fraction of the genome composed of the various repeat families varied across the three species. The most abundant type of repeat in all three genomes were members of the Gypsy superfamily (Figure 4A), comprising ∼39% of the total genome, although species did not significantly differ in overall Gypsy abundance. The second most abundant superfamily in the Yucca genomes, at about ∼16.5%, was Copia (Figure 4A). Yucca gloriosa had significantly more Copia elements than either parent (post hoc comparison of Y. gloriosa to either parent p < 0.001). The third most abundant repeat superfamily was DNA Helitrons, at ∼3.5%, which had significantly different abundances between all three species (post hoc comparison p < 0.01). In general, the variation in repeat superfamily abundance between the three species was large enough to distinguish each species (Supplementary Figure 1), though intraspecific variation in repeat abundance was apparent as well. Certain transposable element superfamilies not typically seen in plant genomes—including non-LTR Zisupton, Novosib, and Line-2 (L2) elements—were found at non-zero abundance levels in the Yucca species (Supplementary Table 2), but are not considered further here, as they may be the result of contamination in DNA isolation (e.g., from fungi).
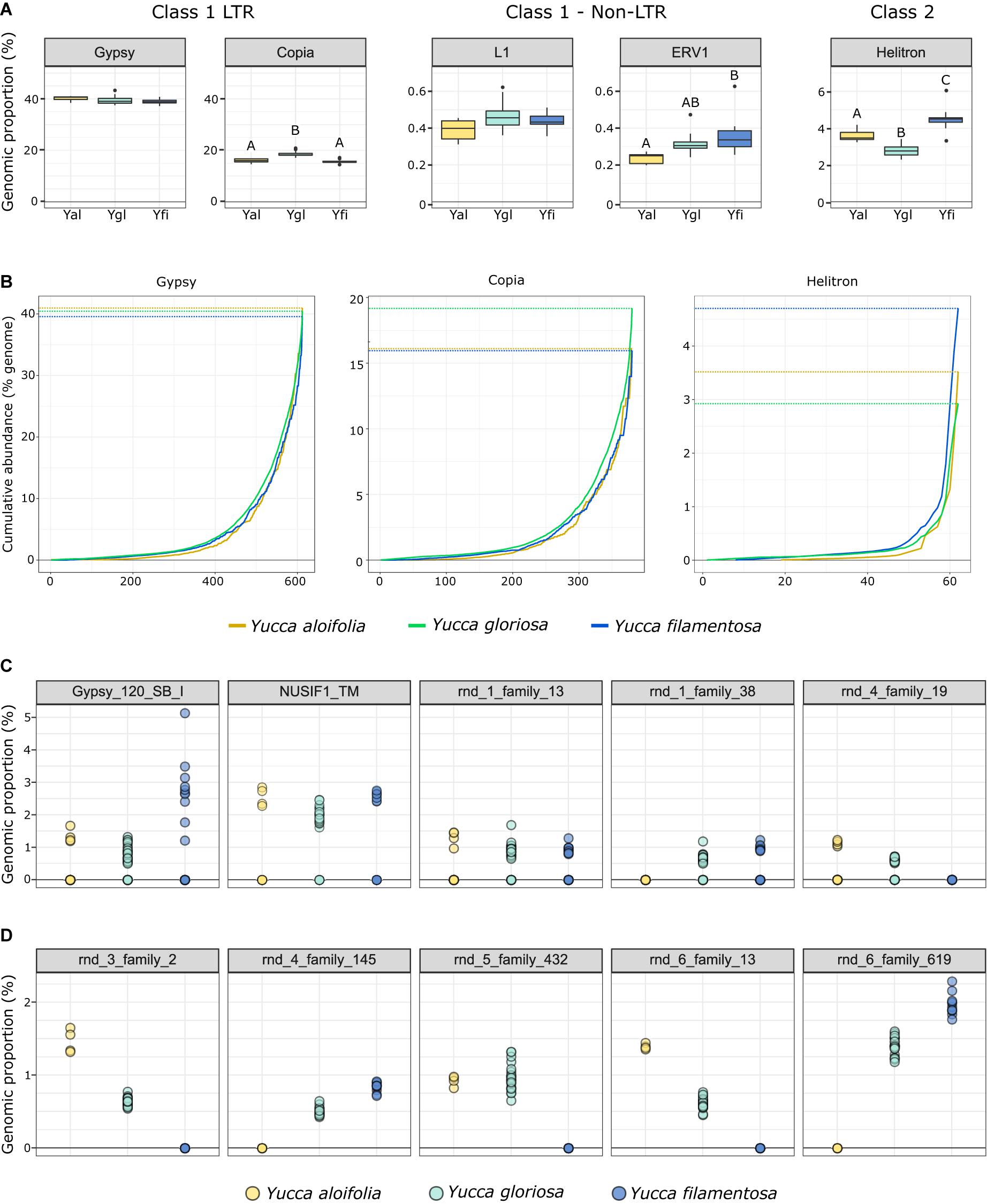
Figure 4. (A) The genomic proportion (as a%) of the genome of a subset of transposable element families in Y. aloifolia (yellow), Y. gloriosa (teal), and Y. filamentosa (blue). Letters indicate significant differences based on Tukey post hoc tests from an ANOVA (abundance ∼ species) per repeat superfamily; shared letters indicate no significant difference at p < 0.01. y-axes are scaled by category (LTR, non-LTR, Class II). (B) Cumulative abundance of individual families per superfamily, ordered from lowest average abundance to highest average abundance. (C,D) Individual genotype abundance values for the five most abundant families in Gypsy (C) and Copia (D).
At the family level (repeat lineages within a superfamily), while variation across the three species existed (Supplementary Table 3), it was rare that a single family contributed to the differences seen at the superfamily level (Figure 4A). In particular, high abundance families in Copia showed a pattern of parental-specific ancestry, in that they were found in only one of the two parental species. Furthermore, these high abundance, parental-specific repeat families also had high abundance inherited in the hybrid, resulting in an overall higher abundance of Copia elements in Y. gloriosa relative to either parent (Figures 4B,C). In contrast, high abundance Gypsy elements were not parental-specific, and the hybrid therefore had equivalent abundance values as the parental abundance (Figures 4B,C). These high abundance, parental-specific families in the Copia superfamily drive the overall species difference at the superfamily level.
Repeat mRNA Expression
Transposome abundance analysis of Y. aloifolia and Y. filamentosa identified 504 and 726 repeat families in at least one genotype of either species, respectively; only 231 repeat families were present in both parental species. Of the 231 families present in both species, only 118 and 119 repeat families had significantly non-zero expression in Y. aloifolia and Y. filamentosa, respectively (Benjamini-Hochberg adjusted p < 0.01) (Supplementary Table 4), and only 92 families were both present in all three species and had detectable expression in any one library (Figure 5). Only 27 families were significantly expressed in both parental species (Table 1). Repeat families with significant expression were typically from Gypsy (64 and 61% of total families expressed in Y. aloifolia and Y. filamentosa, respectively) and Copia (25, 27%) superfamilies. Yucca gloriosa had largely overlapping expression with its parental species; the hybrid shared significant expression of 74 families with Y. aloifolia and 70 families with Y. filamentosa. Yucca gloriosa had only two families that were not also significantly expressed in either parent: one a member of the Gypsy superfamily, the other belonging to the Copia superfamily, and both had genomic abundance at less than 1%. Overall, normalized and scaled expression values were positively correlated with the genomic abundance of a family across the three species [r = 0.23, t(5, 214) = 4.9, p < 0.001 for full data, r = 0.41, t(5,204) = 31.83, p < 0.001 for families with TPM < 2,000].
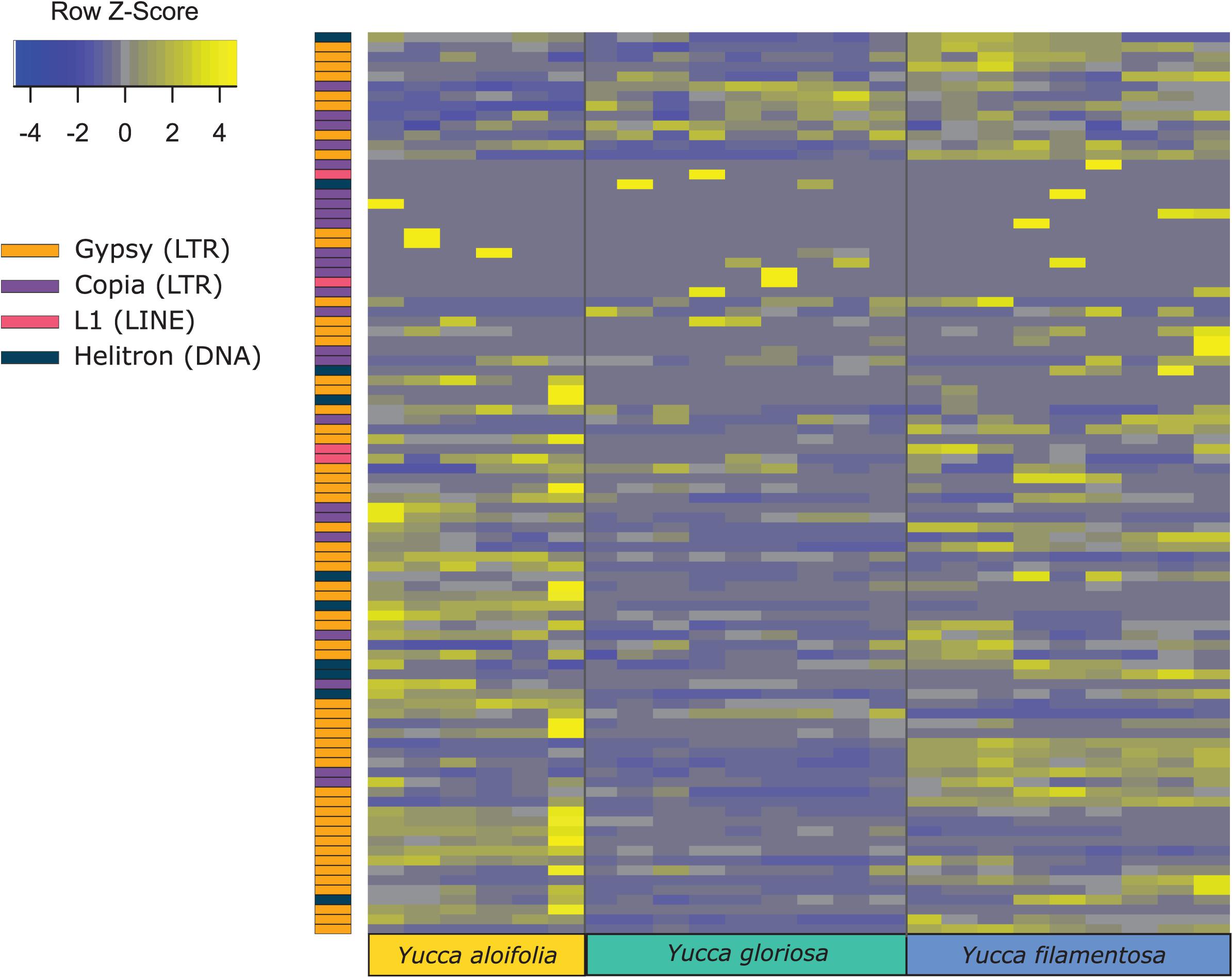
Figure 5. Heatmap of normalized and scaled expression of 92 repeat families that were both present in all three species and had any detectable expression in any library. Individual rows are repeat clusters, and columns represent individual RNA-seq library replicates for each genotype.
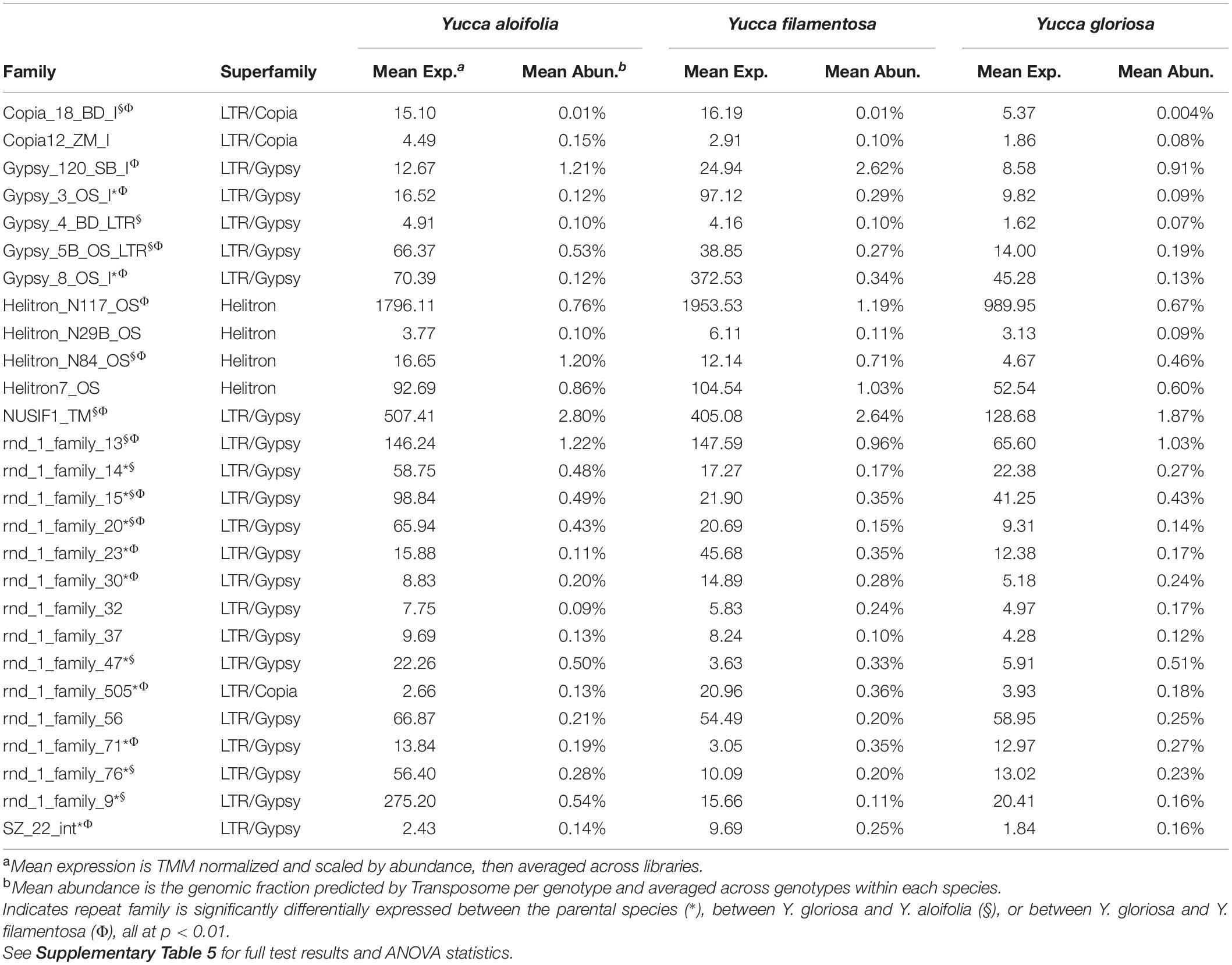
Table 1. Mean expression and abundance of repeat families in Y. aloifolia and Y. filamentosa significantly expressed above zero (p < 0.01).
In comparing the repeat families that are significantly expressed in any of the three species, Y. gloriosa showed little transgressive expression patterns; in most of the 178 repeat families that had significant post hoc comparisons, Y. gloriosa was not statistically different than one of its parental species. There were only three repeat families where expression differed significantly in all three species (post hoc p < 0.01) (Supplementary Table 5), and in 5 families, Y. gloriosa exhibited an expression level that was significantly different than the pattern shared in the two parental species (post hoc p < 0.01) (Figure 6). In all five cases, Y. gloriosa expression was significantly lower than the parental species’ expression, though notably not zero. In general, however, the expression levels of repetitive elements in Y. gloriosa were shared with one or both parental species. Nine transposons families showed shared expression in Y. gloriosa and Y. filamentosa that differed significantly from Y. aloifolia, and seven transposons had shared expression between Y. gloriosa and Y. aloifolia that differed significantly from Y. filamentosa. The majority of transposons had shared expression between the two parents, but significantly different expression between Y. gloriosa and either Y. aloifolia (n = 76) or Y. filamentosa (n = 77). There was a single transposon family where the parental species had significantly different expression from each other and Y. gloriosa’s expression was not significantly different than either parent.
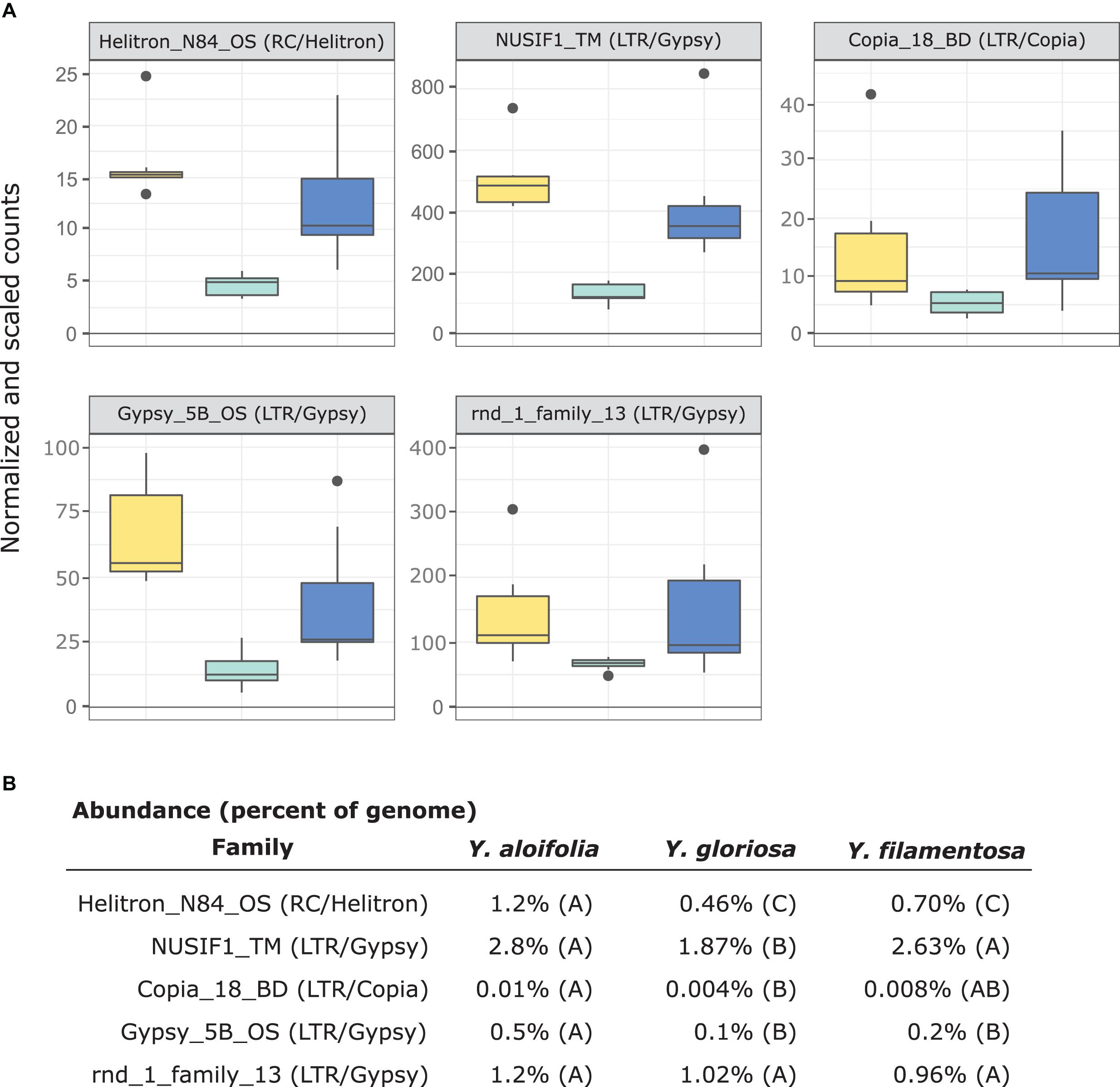
Figure 6. (A) Expression plot of the 5 TE families that were significantly differentially expressed between Y. gloriosa (teal) and both of its parental species (Y. aloifolia = yellow, Y. filamentosa = blue). TMM-normalized count data that is further scaled by abundance is plotted. (B) Mean percent abundance per species, as estimated by Transposome, and the result of post hoc test using emmeans() in R on the results of a negative binomial generalized linear model. Shared letters indicate no significant difference at a p < 0.01.
Discussion
By increasing both the number of Yucca genotypes and assessing the whole chloroplast genome we have greatly improved resolution of the history of homoploid hybridization in Yucca relative to previous analyses of simple sequence repeats and short fragments of the chloroplast (Rentsch and Leebens-Mack, 2012). Whereas the previous work inferred a single, shared plastid haplotype in Y. aloifolia and Y. gloriosa, our findings implicate multiple origins of Y. gloriosa with both Y. aloifolia and Y. filamentosa acting as maternal parents. Moreover, analyses of nuclear TE abundances document overall quite similar TE landscapes across the three species, but certain families showed species-specific shifts in abundance. Using mRNA to assess current transposon activity, we find little evidence for ongoing release of transposons in the hybrid genome.
Reciprocal Parentage and Multiple Origins
Using 15–40x whole genome resequencing data, chloroplast assemblies for 38 individuals of Yucca across three species provided robust re-assessment of the history of this hybrid system. The presence of three separate clades containing Y. gloriosa (Figure 2) strongly suggests that not only can Y. aloifolia act as the maternal parent in the cross, as previously suggested, but that a reciprocal cross with Y. filamentosa as the maternal parent was viable enough to produce at least one extant lineage in Y. gloriosa. While Y. filamentosa acting as the maternal parent in at least one cross is a parsimonious explanation for the data, the presence of a Y. filamentosa chloroplast in Y. gloriosa could also be due to a backcrossing event in which a Y. gloriosa pollen grain sired a seed on a Y. filamentosa individual. Such a backcross is unlikely to have happened recently. All of the genotypes of Y. gloriosa in this study (n = 24), as well as 2 and 6 of the genotypes for Y. aloifolia and Y. filamentosa from this study, respectively, have been phenotyped extensively for photosynthesis related traits (Heyduk et al., 2020), and a recent backcrossed hybrid would be expected to have photosynthetic physiology more similar to Y. filamentosa than Y. aloifolia, as the parents are strongly divergent in whether they use C3 photosynthesis or CAM, respectively. However, the genotype of Y. gloriosa with the Y. filamentosa chloroplast haplotype (YG16) has strong signatures of CAM, including nocturnal CO2 uptake as well as acid accumulation, traits which are diagnostic of the CAM phenotype displayed by Y. aloifolia (Heyduk et al., 2020). Additionally, the three species are very easy to distinguish in the field by leaf morphology: Y. filamentosa has filamentous leaf margins, Y. aloifolia has serrated leaf margins, and Y. gloriosa has smooth leaf margins. However these observations cannot rule out a more ancient backcrossing event, in which an original Y. filamentosa x Y. gloriosa cross’s progeny thereafter crossed only within Y. gloriosa, which over time would largely dampen the addition of the Y. filamenotsa nuclear genome but the chloroplast haplotype would remain.
The two clades of Y. gloriosa individuals that group with Y. aloifolia further support the inference that Y. gloriosa is derived from multiple hybridization events. However, as with the one instance of a Y. filamentosa chloroplast in Y. gloriosa, it is difficult to rule out recent backcrossing as the source of this observation (though leaf margins of all Y. gloriosa individuals sampled here had smooth margins that are diagnostic of this species in the wild). Earlier literature suggests that Y. aloifolia was introduced into the Southeastern United States ∼500 years ago by European colonists, who likely brought the plant from the Caribbean or Central America (Trelease, 1893; Groman and Pellmyr, 2000; Pellmyr, 2003). Within that time frame, however, the age of the hybridization events remains unknown. Additional analysis of re-resequencing data will assist in determining the number and timing of putative hybridization events. For example, the length of parental haplotype segments in a hybrid genome is related to the degree of recombination across the hybrid genome; short haplotype blocks would indicate a greater degree of recombination and, therefore, an older hybridization event. On the other hand, longer intact parental haplotype blocks in the hybrid may point to more recent hybridization. Moreover, the length of these haplotype blocks will vary between individuals, and may point to a mixture of both older and younger hybridization events within Y. gloriosa.
Previous work on the three Yucca species suggested that all Y. aloifolia and Y. gloriosa individuals shared a single chloroplast haplotype (Rentsch and Leebens-Mack, 2012). Comparisons across the entire chloroplast genome show that four nucleotide differences separated the two clades of Y. aloifolia and Y. gloriosa individuals. Over 400 genetic changes separate the Y. filamentosa and YG16 haplotypes from all Y. aloifolia and the remaining Y. gloriosa haplotypes. In agreement with the previous work, this study documents low plastid genetic diversity within Y. aloifolia and most Y. gloriosa samples. Yucca aloifolia is introduced into the Southeastern United States and likely suffered a bottleneck, resulting in lower overall diversity. The current sample of Y. gloriosa individuals identified one individual with a Y. filamentosa-derived haplotype. Additionally, this analysis identified seven discrete haplotypes within Y. filamentosa, which parallels the greater number of alleles per locus in Y. filamentosa suggested by previous work (Rentsch and Leebens-Mack, 2012).
Any attempt at describing the frequency of hybrid formation will be largely affected by the number of individuals in the germplasm collection. The original collection area spanned a large portion of the Southeastern United States in order to capture a significant amount of genetic diversity within the genus. Collections of Y. gloriosa likely represent many of the extant populations, but the ranges of both Y. aloifolia and Y. filamentosa are much larger than sampled here. As a result, any interpretation of geographic patterns to the chloroplast phylogeny or haplotype network are hampered by relatively low sampling of the parental genetic diversity. For example, the single Y. gloriosa individual found with a Y. filamentosa chloroplast (YG16) was collected in South Carolina, while Y. filamentosa individuals with the most similar haplotypes were collected in Delaware, North Carolina, and South Carolina. This haplotype grouping is clearly not geographically localized to one portion of the Atlantic coast and could be the result of missing genetic diversity in our analysis. Additionally, the Southeastern United States coastline experiences hurricanes and/or tropical storms on nearly an annual basis. Such storms have the potential to both disperse genets as well as eradicate entire populations and could make geographic interpretation of extant diversity difficult.
Transposable Element Abundance and Amplification
Genome resequencing provides a relatively unbiased sampling of the genome, allowing us to estimate the genomic fraction composed of transposable elements. Among sequenced plant genomes, transposable element contribution to genome size ranges from 14% in Eragrostis tef to 85% in Zea mays (Wendel et al., 2016). While all three Yucca species described in this work fall within the described range, the three species varied in the total amount of repetitive DNA with Y. filamentosa having significantly less repetitive DNA that Y. aloifolia and Y. gloriosa (62% vs. 65/66%). However, variation in abundance of particular repeat superfamilies does suggest superfamily-specific changes between the three species. Copia elements, the second most abundant superfamily of repeat in all three species, were more abundant in Y. gloriosa relative to both parents, suggesting an amplification of this superfamily post-hybridization. While Class 2 elements represent a relatively small proportion of Yucca genomes, Helitrons were found more often in Y. filamentosa compared to either Y. aloifolia or Y. gloriosa. Helitrons are capable of generating a tremendous amount of structural novelty, including the ability to capture and re-distribute pieces of genes (Yang and Bennetzen, 2009). As genomes become available for these species, it will be possible to analyze the extent to which all types of transposable elements have facilitated structural rearrangements and have affected expression of neighboring genes.
Previous work in various hybrid systems has shown incredible changes to the genomes post-hybridization. In a wallaby x kangaroo cross, reduced methylation of the genome resulted in the proliferation of a novel transposable element that caused significant structural changes to the chromosomes (O’Neill et al., 1998). Interspecific hybrids in Drosophila had an increase in transposable element mobilization relative to parental species (Vela et al., 2014). Three independent homoploid hybrids in Helianthus all show increased genome size due to expansion of repetitive elements, particularly in Ty3/gypsy-like LTR elements (Ungerer et al., 2006, 2009). In Yucca, however, there seems to be little indication that transposable elements were released from silencing mechanisms and proliferated in the hybrid Y. gloriosa. Instead, Y. gloriosa shows similar abundance of transposable elements relative to its progenitor species, though with a notable increase in Copia elements in the hybrid (Figure 4). Extant genotypes of Y. gloriosa have little in the way of increased repeat expression (Figure 6); whether this means no genomic shock initially happened upon hybridization, or that the genome has had sufficient time to stabilize repetitive elements, remains unclear.
Finally, the three Yucca species provide an excellent system within which to describe the role of repetitive content on novel phenotypic evolution and adaptation. Yucca gloriosa has been studied extensively for its intermediate photosynthetic phenotype (Heyduk et al., 2016, 2019). When well-watered, the majority of carbon fixation happens during the day through the C3 cycle, although low levels of CAM activity are present. When drought stress, Y. gloriosa can switch to predominantly CAM photosynthesis, but the degree to which individual genotypes do so varies. The hybrid’s photosynthetic phenotype is novel, in that neither parent displays CAM induction upon drought stress, nor the ability to switch from primarily C3 carbon fixation to primarily CAM. On first glance, negligible differences in repeat content and activity in Y. gloriosa relative to its parents suggest that repetitive content is unlikely to underlie the novel photosynthetic phenotype in the hybrid. However, here we only assessed overall abundance and activity in extant individuals; location of repeats in the hybrid relative to the parental species, as well as older repetitive content bursts, still have the potential to create transgressive and novel phenotypes in the hybrid. Repetitive elements can alter gene expression and gene networks by inserting into regulatory regions (Kunarso et al., 2010; Wang et al., 2013), can interfere with alternative splicing (Leprince et al., 2001; Li et al., 2014), and can be a general source of genomic variation and rapid evolution (González et al., 2010; Schrader et al., 2014). Moreover, transposable element activity can increase in response to environmental stressors (Makarevitch et al., 2015) and can play a role in forming stress-induced regulatory networks (Naito et al., 2009). Whether transposable elements are responsible for Y. gloriosa’s ability to upregulate CAM photosynthesis under drought stress remains to be tested.
Conclusion
Since the chloroplast phylogeny and haplotype network imply multiple hybridization events contributing to the origin of Y. gloriosa, new hypotheses regarding the repeatability of transposon accumulation can now be tested. For example, since YG16 appears to most likely be derived from a distinct hybridization event relative to other Y. gloriosa genotypes, we can assess whether the genomic organization of its transposable elements is vastly different from the major clade of Y. gloriosa genotypes grouping with Y. aloifolia (Figure 2). Integrating transposable element abundance and expression with other types of genomic data, including RNA-seq and bisulfite sequencing, may help us understand the potential for insertions to differentially regulate genes. The Yucca system is particularly powerful, in that the parental species are strongly divergent in photosynthetic pathway and the hybrid segregates for many of the same traits; this provides a framework in which to understand the role of repeats in regulating these genes in Y. gloriosa.
Given the massively expanding availability of whole genome sequence data, hypothesis-driven comparative analyses of genome content and structure are becoming more tractable. In this work, reads that normally would have been filtered out were instead analyzed to address whether a hybrid species had multiple and/or reciprocal origins. Furthermore, these reads helped provide a first glance into the repetitive landscape of 40 genotypes across three related species. While whole genomes will ultimately have the greatest ability to answer many of the questions brought up in this work, the approaches used here are quicker, less expensive, and generate many hypotheses for testing at the genome level in the future.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in Supplementary Table 1.
Author Contributions
JG prepared libraries and sequenced samples. SS annotated repetitive content in the parental genomes. EM and KH conducted all analyses and wrote the manuscript. MM optimized plastome assembly and assisted with the manuscript. JL-M and JS were integral to overall project planning and management and assisted with the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a DOE Joint Genome Institute Community Science Project award to KH. The work conducted by the US DOE Joint Genome Institute is supported by the Office of Science of the US Department of Energy under Contract no. DE-AC02-05CH11231.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge Amanda L. Cummings for assistance with DNA preparation, the Georgia Advanced Computing Resource Center, and the staff at the University of Georgia greenhouses, in particular Michael Boyd and Gregory Cousins. We thank the DOE Joint Genome Institute and collaborators for pre-publication access to the WGS data from Yucca accessions herein and to repeat databases from the genome sequences of Yucca aloifolia and Yucca filamentosa. This manuscript has been released as a pre-print at bioRxiv, https://doi.org/10.1101/2020.06.14.150078. This is publication #95 from the School of Life Sciences, University of Hawai’i at Mānoa.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.573767/full#supplementary-material
Footnotes
- ^ https://github.com/mrmckain/Fast-Plast
- ^ https://github.com/kheyduk/Yucca_plastome
- ^ https://genome.jgi.doe.gov
- ^ https://github.com/sebhtml/ray/blob/master/scripts/interleave-fastq.py
- ^ http://hannonlab.cshl.edu/fastx_toolkit/
- ^ https://github.com/rvlenth/emmeans
References
Abraham, P. E., Yin, H., Borland, A. M., Weighill, D., Lim, S. D., De Paoli, H. C., et al. (2016). Transcript, protein and metabolite temporal dynamics in the CAM plant Agave. Nat. Plants 2:16178.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Arnold, M. L. (1993). Iris nelsonii (Iridaceae): origins and genetic composition of a homoploid hybrid species. Am. J. Bot. 80, 577–583. doi: 10.1002/j.1537-2197.1993.tb13843.x
Bao, Z., and Eddy, S. R. (2002). Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276. doi: 10.1101/gr.88502
Bardil, A., de Almeida, J. D., Combes, M. C., Lashermes, P., and Bertrand, B. (2011). Genomic expression dominance in the natural allopolyploid Coffea arabica is massively affected by growth temperature. New Phytol. 192, 760–774. doi: 10.1111/j.1469-8137.2011.03833.x
Bird, K. A., VanBuren, R., Puzey, J. R., and Edger, P. P. (2018). The causes and consequences of subgenome dominance in hybrids and recent polyploids. New Phytol. 220, 87–93. doi: 10.1111/nph.15256
Boisvert, S., Laviolette, F., and Corbeil, J. (2010). Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J. Comput. Biol. 17, 1519–1533. doi: 10.1089/cmb.2009.0238
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bray, N. L., Pimentel, H., Melsted, P., and Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi: 10.1038/nbt.3519
Bredeson, J. V., Lyons, J. B., Prochnik, S. E., Wu, G. A., Ha, C. M., Edsinger-Gonzales, E., et al. (2016). Sequencing wild and cultivated cassava and related species reveals extensive interspecific hybridization and genetic diversity. Nat. Biotechnol. 34, 562–570. doi: 10.1038/nbt.3535
Danilova, T. V., Akhunova, A. R., Akhunov, E. D., Friebe, B., and Gill, B. S. (2017). Major structural genomic alterations can be associated with hybrid speciation in Aegilops markgrafii (Triticeae). Plant J. 92, 317–330. doi: 10.1111/tpj.13657
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Devos, K. M., Brown, J. K. M., and Bennetzen, J. L. (2002). Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 12, 1075–1079. doi: 10.1101/gr.132102
Dion-Côté, A.-M., Renaut, S., Normandeau, E., and Bernatchez, L. (2014). RNA-seq reveals transcriptomic shock involving transposable elements reactivation in hybrids of young lake whitefish species. Mol. Biol. Evol. 31, 1188–1199. doi: 10.1093/molbev/msu069
Doyle, J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Edger, P. P., Smith, R., McKain, M. R., Cooley, A. M., Vallejo-Marin, M., Yuan, Y., et al. (2017). Subgenome dominance in an interspecific hybrid, synthetic allopolyploid, and a 140-year-old naturally established neo-allopolyploid monkeyflower. Plant Cell 29, 2150–2167. doi: 10.1105/tpc.17.00010
Feschotte, C., and Pritham, E. J. (2007). DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 41, 331–368. doi: 10.1146/annurev.genet.40.110405.090448
Fultz, D., Choudury, S. G., and Slotkin, R. K. (2015). Silencing of active transposable elements in plants. Curr. Opin. Plant Biol. 27, 67–76. doi: 10.1016/j.pbi.2015.05.027
González, J., Karasov, T. L., Messer, P. W., and Petrov, D. A. (2010). Genome-wide patterns of adaptation to temperate environments associated with transposable elements in Drosophila. PLoS Genet. 6:e1000905. doi: 10.1371/journal.pgen.1000905
Groman, J. D., and Pellmyr, O. (2000). Rapid evolution and specialization following host colonization in a yucca moth. J. Evol. Biol. 13, 223–236. doi: 10.1046/j.1420-9101.2000.00159.x
Gross, B. L., and Rieseberg, L. H. (2005). The ecological genetics of homoploid hybrid speciation. J. Hered. 96, 241–252. doi: 10.1093/jhered/esi026
Hegarty, M. J., Barker, G. L., Brennan, A. C., Edwards, K. J., Abbott, R. J., and Hiscock, S. J. (2009). Extreme changes to gene expression associated with homoploid hybrid speciation. Mol. Ecol. 18, 877–889. doi: 10.1111/j.1365-294x.2008.04054.x
Hersch, E. I., and Roy, B. A. (2007). Context-dependent pollinator behavior: an explanation for patterns of hybridization among three species of Indian paintbrush. Evolution 61, 111–124. doi: 10.1111/j.1558-5646.2007.00009.x
Heyduk, K., Burrell, N., Lalani, F., and Leebens-Mack, J. (2016). Gas exchange and leaf anatomy of a C3-CAM hybrid, Yucca gloriosa (Asparagaceae). J. Exp. Bot. 67, 1369–1379. doi: 10.1093/jxb/erv536
Heyduk, K., Ray, J. N., Ayyampalayam, S., Moledina, N., Borland, A., Harding, S. A., et al. (2019). Shared expression of Crassulacean acid metabolism (CAM) genes predates the origin of CAM in the genus Yucca. J. Exp. Bot. 70, 6597–6609. doi: 10.1093/jxb/erz105
Heyduk, K., Ray, J. N., and Leebens-Mack, J. (2020). Leaf anatomy is not correlated to CAM function in a C3+CAM hybrid species, Yucca gloriosa. Ann. Bot. 2020:mcaa036. doi: 10.1093/aob/mcaa036
Hollister, J. D., Smith, L. M., Guo, Y.-L., Ott, F., Weigel, D., and Gaut, B. S. (2011). Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. U.S.A. 108, 2322–2327. doi: 10.1073/pnas.1018222108
Johnston, J. A., Grise, D. J., Donovan, L. A., and Arnold, M. L. (2001). Environment-dependent performance and fitness of Iris brevicaulis, I. fulva (Iridaceae), and hybrids. Am. J. Bot. 88, 933–938. doi: 10.2307/2657046
Karrenberg, S., Edelist, C., Lexer, C., and Rieseberg, L. (2006). Response to salinity in the homoploid hybrid species Helianthus paradoxus and its progenitors H. annuus and H. petiolaris. New Phytol. 170, 615–629. doi: 10.1111/j.1469-8137.2006.01687.x
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kunarso, G., Chia, N.-Y., Jeyakani, J., Hwang, C., Lu, X., Chan, Y.-S., et al. (2010). Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 42, 631–634. doi: 10.1038/ng.600
Lai, Z., Gross, B. L., Zou, Y. I., Andrews, J., and Rieseberg, L. H. (2006). Microarray analysis reveals differential gene expression in hybrid sunflower species. Mol. Ecol. 15, 1213–1227. doi: 10.1111/j.1365-294x.2006.02775.x
Lai, Z., Nakazato, T., Salmaso, M., Burke, J. M., Tang, S., Knapp, S. J., et al. (2005). Extensive chromosomal repatterning and the evolution of sterility barriers in hybrid sunflower species. Genetics 171, 291–303. doi: 10.1534/genetics.105.042242
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leebens-Mack, J., and Milligan, B. (1998). Pollination biology in hybridizing Baptisia (Fabaceae) populations. Am. J. Bot. 85:500. doi: 10.2307/2446433
Leebens-Mack, J., and Pellmyr, O. (2004). Patterns of genetic structure among populations of an oligophagous pollinating yucca moth (Tegeticula yuccasella). J. Hered. 95, 127–135. doi: 10.1093/jhered/esh025
Leigh, J. W., and Bryant, D. (2015). popart : full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116. doi: 10.1111/2041-210x.12410
Leprince, A. S., Grandbastien, M. A., and Meyer, C. (2001). Retrotransposons of the Tnt1B family are mobile in Nicotiana plumbaginifolia and can induce alternative splicing of the host gene upon insertion. Plant Mol. Biol. 47, 533–541.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, Q., Xiao, G., and Zhu, Y.-X. (2014). Single-nucleotide resolution mapping of the Gossypium raimondii transcriptome reveals a new mechanism for alternative splicing of introns. Mol. Plant 7, 829–840. doi: 10.1093/mp/sst175
Lisch, D. (2009). Epigenetic regulation of transposable elements in plants. Annu. Rev. Plant Biol. 60, 43–66. doi: 10.1146/annurev.arplant.59.032607.092744
Makarevitch, I., Waters, A. J., West, P. T., Stitzer, M., Hirsch, C. N., Ross-Ibarra, J., et al. (2015). Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet. 11:e1004915. doi: 10.1371/journal.pgen.1004915
McClintock, B. (1984). The significance of responses of the genome to challenge. Science 226, 792–801. doi: 10.1126/science.15739260
McKain, M. R., McNeal, J. R., Kellar, P. R., Eguiarte, L. E., Pires, J. C., and Leebens-Mack, J. (2016). Timing of rapid diversification and convergent origins of active pollination within Agavoideae (Asparagaceae). Am. J. Bot. 103, 1717–1729. doi: 10.3732/ajb.1600198
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, 1–8.
Naito, K., Zhang, F., Tsukiyama, T., Saito, H., Hancock, C. N., Richardson, A. O., et al. (2009). Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 461, 1130–1134. doi: 10.1038/nature08479
O’Neill, R. J., O’Neill, M. J., and Graves, J. A. (1998). Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature 393, 68–72. doi: 10.1038/29985
Parisod, C., Salmon, A., Zerjal, T., Tenaillon, M., Grandbastien, M.-A., and Ainouche, M. (2009). Rapid structural and epigenetic reorganization near transposable elements in hybrid and allopolyploid genomes in Spartina. New Phytol. 184, 1003–1015. doi: 10.1111/j.1469-8137.2009.03029.x
Pellmyr, O. (1999). Systematic revision of the yucca moths in the Tegeticula yuccasella complex (Lepidoptera: Prodoxidae) north of Mexico. Syst. Entomol. 24, 243–271. doi: 10.1046/j.1365-3113.1999.00079.x
Pellmyr, O. (2003). Yuccas, Yucca moths, and coevolution: a review. Ann. Mo. Bot. Gard. 90, 35–55. doi: 10.2307/3298524
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl. 1), i351–i358.
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
R Core Team (2019). R: A Language and Environment for Statistical Computing. Vienna: R foundation for statistical computing.
Rapp, R. A., Udall, J. A., and Wendel, J. F. (2009). Genomic expression dominance in allopolyploids. BMC Biol. 7:18. doi: 10.1186/1741-7007-7-18
Renaut, S., Rowe, H. C., Ungerer, M. C., and Rieseberg, L. H. (2014). Genomics of homoploid hybrid speciation: diversity and transcriptional activity of long terminal repeat retrotransposons in hybrid sunflowers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130345. doi: 10.1098/rstb.2013.0345
Rentsch, J. D., and Leebens-Mack, J. (2012). Homoploid hybrid origin of Yucca gloriosa: intersectional hybrid speciation in Yucca (Agavoideae, Asparagaceae). Ecol. Evol. 2, 2213–2222. doi: 10.1002/ece3.328
Rentsch, J. D., and Leebens-Mack, J. (2014). Yucca aloifolia (Asparagaceae) opts out of an obligate pollination mutualism. Am. J. Bot. 101, 2062–2067. doi: 10.3732/ajb.1400351
Rieseberg, L. H. (1991). Homoploid reticulate evolution in helianthus (asteraceae): evidence from ribosomal genes. Am. J. Bot. 78, 1218–1237. doi: 10.1002/j.1537-2197.1991.tb11415.x
Rieseberg, L. H. (1997). Hybrid origins of plant species. Annu. Rev. Ecol. Syst. 28, 359–389. doi: 10.1146/annurev.ecolsys.28.1.359
Rieseberg, L. H., Raymond, O., Rosenthal, D. M., Lai, Z., Livingstone, K., Nakazato, T., et al. (2003). Major ecological transitions in wild sunflowers facilitated by hybridization. Science 301, 1211–1216. doi: 10.1126/science.1086949
Rieseberg, L. H., Van Fossen, C., and Desrochers, A. M. (1995). Hybrid speciation accompanied by genomic reorganization in wild sunflowers. Nature 375, 313–316. doi: 10.1038/375313a0
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Robinson, M. D., and Oshlack, A. (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11:R25.
Salmon, A., Ainouche, M. L., and Wendel, J. F. (2005). Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Mol. Ecol. 14, 1163–1175. doi: 10.1111/j.1365-294x.2005.02488.x
Sapir, Y., Moody, M. L., Brouillette, L. C., Donovan, L. A., and Rieseberg, L. H. (2007). Patterns of genetic diversity and candidate genes for ecological divergence in a homoploid hybrid sunflower, Helianthus anomalus. Mol. Ecol. 16, 5017–5029. doi: 10.1111/j.1365-294x.2007.03557.x
Schnable, J. C., Springer, N. M., and Freeling, M. (2011). Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. U.S.A. 108, 4069–4074. doi: 10.1073/pnas.1101368108
Schrader, L., Kim, J. W., Ence, D., Zimin, A., Klein, A., Wyschetzki, K., et al. (2014). Transposable element islands facilitate adaptation to novel environments in an invasive species. Nat. Commun. 5:5495.
Soltis, P. S., and Soltis, D. E. (2009). The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 60, 561–588. doi: 10.1146/annurev.arplant.043008.092039
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Staton, S. E., Bakken, B. H., Blackman, B. K., Chapman, M. A., Kane, N. C., Tang, S., et al. (2012). The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 72, 142–153. doi: 10.1111/j.1365-313x.2012.05072.x
Staton, S. E., and Burke, J. M. (2015). Transposome: a toolkit for annotation of transposable element families from unassembled sequence reads. Bioinformatics 31, 1827–1829. doi: 10.1093/bioinformatics/btv059
Staton, S. E., and Ungerer, M. C. (2009). The genomic organization of Ty3/gypsy-like retrotransposons in Helianthus (Asteraceae) homoploid hybrid species. Am. J. Bot. 96, 1646–1655. doi: 10.3732/ajb.0800337
Štorchová, H., Hrdlièková, R., Chrtek, J., Tetera, M., Fitze, D., and Fehrer, J. (2000). An improved method of DNA isolation from plants collected in the field and conserved in saturated NaCl/CTAB solution. Taxon 49, 79–84. doi: 10.2307/1223934
Studer, A., Zhao, Q., Ross-Ibarra, J., and Doebley, J. (2011). Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 43, 1160–1163. doi: 10.1038/ng.942
Sun, C., Ma, Z., Zhang, Z., Sun, G., and Dai, Z. (2018). Factors Influencing Cross Barriers in Interspecific Hybridizations of Water Lily. J. Am. Soc. Hortic. Sci. 143, 130–135. doi: 10.21273/jashs04302-17
Tenaillon, M. I., Hufford, M. B., Gaut, B. S., and Ross-Ibarra, J. (2011). Genome size and transposable element content as determined by high-throughput sequencing in maize and Zea luxurians. Genome Biol. Evol. 3, 219–229. doi: 10.1093/gbe/evr008
Thórsson, A. T., Salmela, E., and Anamthawat-Jónsson, K. (2001). Morphological, cytogenetic, and molecular evidence for introgressive hybridization in birch. J. Hered. 92, 404–408. doi: 10.1093/jhered/92.5.404
Trelease, W. (1893). Further studies of yuccas and their pollination. Missouri Bot. Garden Annu. Rep. 1893, 181–226. doi: 10.2307/2992178
Trelease, W. (1902). The yucceae. Missouri Bot. Garden Annu. Rep. 1902, 27–133. doi: 10.2307/2400121
Ungerer, M. C., Strakosh, S. C., and Stimpson, K. M. (2009). Proliferation of Ty3/gypsy-like retrotransposons in hybrid sunflower taxa inferred from phylogenetic data. BMC Biol. 7:40. doi: 10.1186/1741-7007-7-40
Ungerer, M. C., Strakosh, S. C., and Zhen, Y. (2006). Genome expansion in three hybrid sunflower species is associated with retrotransposon proliferation. Curr. Biol. 16, R872–R873.
Vela, D., Fontdevila, A., Vieira, C., and García Guerreiro, M. P. (2014). A genome-wide survey of genetic instability by transposition in Drosophila hybrids. PLoS One 9:e88992. doi: 10.1371/journal.pone.0088992
Wang, H., McArthur, E. D., Sanderson, S. C., Graham, J. H., and Freeman, D. C. (1997). Narrow hybrid zone between two species of Big Sagebrush (Artemisia tridentata: Asteraceae). IV. Reciprocal transplant experiments. Evolution 51, 95–102. doi: 10.2307/2410963
Wang, X., Weigel, D., and Smith, L. M. (2013). Transposon variants and their effects on gene expression in Arabidopsis. PLoS Genet. 9:e1003255. doi: 10.1371/journal.pgen.1003255
Welch, M. E., and Rieseberg, L. H. (2002). Habitat divergence between a homoploid hybrid sunflower species, Helianthus paradoxus (Asteraceae), and its progenitors. Am. J. Bot. 89, 472–478. doi: 10.3732/ajb.89.3.472
Wendel, J. F., Jackson, S. A., Meyers, B. C., and Wing, R. A. (2016). Evolution of plant genome architecture. Genome Biol. 17:37.
Wessler, S. R., Bureau, T. E., and White, S. E. (1995). LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr. Opin. Genet. Dev. 5, 814–821. doi: 10.1016/0959-437x(95)80016-x
Wicker, T., Sabot, F., Hua-Van, A., Bennetzen, J. L., Capy, P., Chalhoub, B., et al. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982. doi: 10.1038/nrg2165
Xiao, H., Jiang, N., Schaffner, E., Stockinger, E. J., and van der Knaap, E. (2008). A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 319, 1527–1530. doi: 10.1126/science.1153040
Xu, Y., Zhong, L., Wu, X., Fang, X., and Wang, J. (2009). Rapid alterations of gene expression and cytosine methylation in newly synthesized Brassica napus allopolyploids. Planta 229, 471–483. doi: 10.1007/s00425-008-0844-8
Yang, L., and Bennetzen, J. L. (2009). Distribution, diversity, evolution, and survival of Helitrons in the maize genome. Proc. Natl. Acad. Sci. U.S.A. 106, 19922–19927. doi: 10.1073/pnas.0908008106
Keywords: homoploid, hybrid, Yucca, chloroplast, transposable element, genomic shock
Citation: Heyduk K, McAssey EV, Grimwood J, Shu S, Schmutz J, McKain MR and Leebens-Mack J (2021) Hybridization History and Repetitive Element Content in the Genome of a Homoploid Hybrid, Yucca gloriosa (Asparagaceae). Front. Plant Sci. 11:573767. doi: 10.3389/fpls.2020.573767
Received: 17 June 2020; Accepted: 17 December 2020;
Published: 15 January 2021.
Edited by:
Hanna Weiss-Schneeweiss, University of Vienna, AustriaReviewed by:
Tony Heitkam, Technische Universität Dresden, GermanySteven Dodsworth, University of Bedfordshire, United Kingdom
Copyright © 2021 Heyduk, McAssey, Grimwood, Shu, Schmutz, McKain and Leebens-Mack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karolina Heyduk, aGV5ZHVrQGhhd2FpaS5lZHU=