 Inkyu Park
Inkyu Park Jun-Ho Song
Jun-Ho Song Sungyu Yang
Sungyu Yang Sungwook Chae
Sungwook Chae Byeong Cheol Moon
Byeong Cheol Moon- 1Herbal Medicine Resources Research Center, Korea Institute of Oriental Medicine, Naju, South Korea
- 2Herbal Medicine Research Division, Korea Institute of Oriental Medicine, Daejeon, South Korea
Trichosanthes is a genus in Cucurbitaceae comprising 90–100 species. Trichosanthes species are valuable as herbaceous medicinal ingredients. The fruits, seeds, and roots of species such as T. kirilowii and T. rosthornii are used in Korean traditional herbal medicines. T. rosthornii is only found in China, whereas in South Korea two varieties, T. kirilowii var. kirilowii and T. kirilowii var. japonica, are distributed. T. kirilowii var. kirilowii and T. kirilowii var. japonica have different fruit and leaf shapes but are recognized as belonging to the same species. Furthermore, although its members have herbal medicine applications, genomic information of the genus is still limited. The broad goals of this study were (i) to evaluate the taxonomy of Trichosanthes using plastid phylogenomic data and (ii) provide molecular markers specific for T. kirilowii var. kirilowii and T. kirilowii var. japonica, as these have differences in their pharmacological effectiveness and thus should not be confused and adulterated. Comparison of five Trichosanthes plastid genomes revealed locally divergent regions, mainly within intergenic spacer regions (trnT-UGU–trnL-UAA: marker name Tri, rrn4.5–rrn5: TRr, trnE-UUC–trnT-GGU: TRtt). Using these three markers as DNA-barcodes for important herbal medicine species in Trichosanthes, the identity of Trichosanthes material in commercial medicinal products in South Korea could be successfully determined. Phylogenetic analysis of the five Trichosanthes species revealed that the species are clustered within tribe Sicyoeae. T. kirilowii var. kirilowii and T. rosthornii formed a clade with T. kirilowii var. japonica as their sister group. As T. kirilowii in its current circumscription is paraphyletic and as the two varieties can be readily distinguished morphologically (e.g., in leaf shape), T. kirilowii var. japonica should be treated (again) as an independent species, T. japonica.
Introduction
Trichosanthes L. is a large genus of the tribe Sicyoeae Schrad. (Cucurbitaceae Juss.) comprising 90–100 species (de Wilde and Duyfjes, 2010; de Boer and Thulin, 2012). The members of the genus are diecious, rarely monecious, perennial climbing herbs, characterized by unlobed or palmately lobed, rarely compound leaves with branched tendrils, a usually distinct fimbriate corolla, and ovoid to globose or elongated fusiform pepos (Huang et al., 2011; de Boer and Thulin, 2012). The center of diversity for the genus is Southeast Asia, and its range is India to East Asia and southeast Australia (de Wilde and Duyfjes, 2010; Huang et al., 2011; de Boer and Thulin, 2012). Trichosanthes is of broader interest for several reasons, including being a model plant for sex determination in plants and for its medicinal properties (de Boer et al., 2012; Yu et al., 2018; Lu et al., 2021). South Korea hosts two varieties of a Trichosanthes species: T. kirilowii var. kirilowii and T. kirilowii var. japonica (Kim and Choi, 2018). T. kirilowii var. japonica is distinguished from T. kirilowii var. kirilowii based on the color of its fruits. In addition, the two T. kirilowii varieties are distinguished based on leaf shape as well as seed color (Ohba, 1999).
Sequence data from the plastid genome has made it possible to identify medicinal plants (Park et al., 2018b, 2019a,b, 2020). T. kirilowii and T. rosthornii are important in Korean and Chinese traditional herbal medicine, as their roots and seeds are used as Trichosanthis Radix and Trichosanthis Semen (herbal medicinal names), respectively. Such medicines are used for their immunomodulatory, anti-tumor, and anti-HIV properties (Yu et al., 2018). Furthermore, T. kirilowii and T. rosthornii fruits are used in traditional Chinese medicine as Trichosanthis Fructus and Trichosanthis Pericarpium, respectively. T. kirilowii var. japonica has been reported to prevent proliferation of leukemia cell lines in vitro (Kim et al., 2003). For quality control and to ensure the safety and effectiveness of their ingredients, only the Korean Ministry of Food and Drug Safety designates and regulates Trichosanthis Radix and Trichosanthis Semen and the roots and seeds of T. kirilowii and T. rosthornii as medicine (Korea Institute of Oriental Medicine [KIOM], 2020). Since T. kirilowii var. japonica is not regulated by law, the use of T. kirilowii var. japonica as a medicine in South Korea poses a problem. There are some efficacy reports, but there are insufficient reports related to pharmacological studies, component analysis, or physicochemical composition. Trichosanthis Radix and Trichosanthis Semen are obtained in the form of slices and powders in Korean traditional markets. In general, distinguishing authentic from inauthentic herbal products is challenging for the untrained eye. Therefore, appropriate methods are required to discriminate good quality herbal products from adulterated preparations. Adulterants may cause negative side effects and quality problems. The purity of herbal medicine ingredients can be tested using species-specific molecular markers. As mentioned above, T. kirilowii var. kirilowii and T. kirilowii var. japonica are distributed in South Korea. Owing to a lack of major morphological differences in their floral characteristics and their sympatric distribution in South Korea, distinguishing them is often challenging. To protect the original herbal medicine–T. kirilowii var. kirilowii and T. rosthornii–from adulterants, molecular markers could be used for testing and identification.
Plastid genomes are useful for species classification, identification, population genetics studies, diversity and evolutionary analysis, and can infer well-resolved phylogenetic relationships, even at the species level (Jansen et al., 2007; Parks et al., 2009). Furthermore, plastid genomes are useful for DNA barcoding approaches. Insertion/deletion (indel) genetic variants from the plastid genome are useful markers for species identification and discrimination (Dong et al., 2012; Li et al., 2015; Park et al., 2019b). Several studies have developed indel markers to identify the correct herbal medicinal plants among related species. Kim et al. (2015) reported the plastid genome of an important herbal medicinal plant, Panax ginseng, and indel markers from sequence variants of plastid genomes that could discriminate 14 Panax ginseng accessions. Aconitum species, which have toxic components such as aconitine, are extensively used in herbal medicine (Park et al., 2017). The complete plastid genomes of Aconitum coreanum and A. carmichaelii have revealed indel sequence variation among the Aconitum species. An A. coreanum species-specific marker was developed and fully distinguished nine other Aconitum accessions. Furthermore, indel markers were developed to distinguish the important herbal medicine Pharbitidis Semen (seeds of Ipomoea nil and I. purpurea) from closely related Ipomoea species (Park et al., 2018b). Such cases involving traditional herbal medicine attest to the value of plastid genomes. However, chloroplast capture, which refers to the introgression of one plastid genome into another species due to hybridization, has also been reported (Acosta and Premoli, 2010; Stegemann et al., 2012; Kawabe et al., 2018). Therefore, it is recommended that both nuclear and plastid DNA be used together for accurate species identification.
In this study, our aim was to explore the taxonomic identities of T. kirilowii var. kirilowii and T. kirilowii var. japonica and to facilitate their distinction. We also aimed to authenticate the use of Trichosanthes in herbal medicine. To this end, the plastid genomes of five Trichosanthes accessions were sequenced and compared based on nucleotide variation and genome structure. We used the data to test the phylogenetic relationships among five Trichosanthes taxa (four species, one with two varieties). Finally, to authenticate Trichosanthes species-based herbal medicine, we developed and tested novel molecular marker sets based on genetically variable plastid regions.
Materials and Methods
Plant Materials and Morphological Analysis
Fresh T. kirilowii var. kirilowii and T. kirilowii var. japonica leaves were collected from five natural habitats in South Korea (Supplementary Figure 1), and one T. rosthornii individual was collected from its natural habitat in China (Supplementary Table 1). All specimens were registered with the Korean Herbarium of Standard Herbal Resources (Index Herbariorum Code KIOM) at the Korea Institute of Oriental Medicine (KIOM). Two accessions each of T. kirilowii var. kirilowii and T. kirilowii var. japonica and one accession of T. rosthornii were also used for plastid genome analysis (Supplementary Table 1).
To investigate the morphology of T. kirilowii var. kirilowii and T. kirilowii var. japonica, mature leaves, fruits, and seeds from all 25 samples of each variety (five individuals × five collection sites) were selected (Supplementary Table 1). The general shape, degree of division, and hairiness were observed in detail under a stereomicroscope (Olympus SZX16, Olympus, Tokyo, Japan). The Royal Horticultural Society Color Chart® (Royal Horticultural Society, 5th edition) was used to determine the color of the seeds.
Genome Sequencing and Assembly
DNA was extracted using a DNeasy Plant Maxi Kit (Qiagen, Valencia, CA, United States) according to the manufacturer’s instructions. Illumina short-insert paired-end sequencing libraries (TruSeq DNA Nano kit) were constructed and sequenced using the NextSeq500 platform (Illumina, San Diego, CA, United States). De novo assembly was used to construct plastid genomes from the resulting whole-genome shotgun sequencing reads. CLC quality trim v 4.2.1 (CLC Inc., Aarhus, Denmark) was used to trim and check the quality of the reads. Trimmed paired-end reads (Phred score ≥ 20) were assembled using the CLC genome assembler v 4.2.1 (CLC Inc.) using default parameters. Principal contigs representing the plastid genome were retrieved from the total contigs using Nucmer (Delcher et al., 2003), and aligned contigs were ordered using the plastid genome sequence of Hodgsonia macrocarpa (NC_039628) as a reference (Zeng et al., 2018). The representative plastid contigs were arranged in order based on a previously reported plastid genome sequence and connected into a single draft sequence by joining the overlapping terminal sequences. Assembly errors were identified in the initial assembly contigs and manually corrected by the mapping of raw reads to assembled sequences. SOAP de novo gap closer v 0.99 was used to fill gaps based on the alignments of paired-end reads (Luo et al., 2012). LSC/IR, IR/SSC, SSC/IR, and IR/LSC regions of completed plastid genomes were validated using PCR-based sequencing. IR boundaries were amplified using 20 ng of genomic DNA in a 20-μL PCR mixture (SolgTM 2X Taq PCR smart mix 1, Solgent, Daegeon, South Korea) with 10 pmol of each primer (Bioneer, Daejeon, South Korea). Amplification was performed using a Pro Flex PCR system (Applied Biosystems, Waltham, MA, United States) according to the following program: initial denaturation at 95°C for 2 min; 35 cycles at 95°C for 1 min, 60°C for 1 min, 72°C for 1.5 min; and final extension at 72°C for 5 min. PCR products were separated on 2% agarose gels at 150 V for 40 min. To validate IR boundary sequences, each PCR product was rescued from the agarose gel, subcloned into the pGEM-T Easy vector (Promega, Madison, WI, United States), and sequenced using a DNA sequence analyzer (ABI 3730, Applied Biosystems Inc., Foster City, CA, United States) to determine sizes and verify the sequences of amplicons. Primer information and sequence alignment results are listed in Supplementary Tables 2, 3.
Genome Annotation and Comparative Analysis
Gene annotation of the five Trichosanthes plastid genomes was performed using GeSeq v. 1.76 (Tillich et al., 2017). Protein-coding sequences were manually curated and confirmed using Artemis v. 1.8 (Carver et al., 2008), then checked against the National Center for Biotechnology Information (NCBI) protein database. The tRNAs were confirmed with tRNAscan-SE 1.21 (Lowe and Eddy, 1997). IR region sequences were confirmed using IR finder and RepEx v. 1.0 (Michael et al., 2019). Circular maps of the five Trichosanthes plastid genomes were obtained using OGDRAW v. 1.3.1 (Greiner et al., 2019). GC content and relative synonymous codon usage (RSCU) of all five Trichosanthes plastid genomes were analyzed using MEGA6 (Tamura et al., 2013). The codon usage distribution of 33 Cucurbitaceae plastid genomes was visualized using the Heatmapper program utilizing an average linkage clustering method and the Euclidean distance measurement method (Babicki et al., 2016). The mVISTA program v. 2.0 was used in Shuffle-LAGAN mode to compare the five Trichosanthes plastid genomes with the T. kirilowii var. kirilowii 1 plastid genome as a reference. DnaSP v. 6.1 was used to calculate nucleotide variability (Pi) among the five Trichosanthes plastid genomes (Rozas et al., 2017). Pi value represented nucleotide variability as a measure of genetic variation at the nucleotide level for five Trichosanthes accessions. Each plastid genome was divided into genes, introns, and intergenic regions.
Repeat Analysis
Tandemly arranged repeats of short DNA motifs, 1–6 bp in length, called simple sequence repeats (SSRs), were detected in the five Trichosanthes plastid genomes using MISA-web1 (Beier et al., 2017). The following criteria were used for detecting SSRs: SSR motif length between one and six nucleotides with the minimum number of repeat parameters set to 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. Tandem repeats (>20 bp) were identified using the Tandem Repeats Finder v. 4.07 (Benson, 1999) using a minimum alignment score and maximum period size of 50 and 500, respectively, and the identity of repeats was set to ≥90%.
Indel Marker Development and Trichosanthes Validation
To detect species-specific variants, we aligned the five Trichosanthes plastid genomes using MAFFT v. 7 (Katoh et al., 2002). Three regions with indels (hereafter named Tri, TRr, and TRtt), located in the intergenic spacers trnT-UGU–trnL-UAA (Tri), rrn4.5–rrn5 (TRr), and trnE-UUC–trnT-GGU (TRtt), were selected as candidate marker regions, and the sequences of the whole intergenic spacers were extracted from the aligned plastid genomes of all species using Bioedit v. 7.2 (Hall, 1999). Primers flanking the three variable indel regions were designed using Primer-BLAST2. The specificity of the indel markers was confirmed using PCR amplification with 20 ng of genomic DNA extracted from 23 samples of Trichosanthes species in a 20-μl PCR mixture (SolgTM 2 × Taq PCR smart mix 1, Solgent, Daejeon, South Korea) with 10 pmol of each of the Tri, TRr, and TRtt primers. Tri, TRr, and TRtt were amplified on a Pro Flex PCR system (Applied Biosystems, Waltham, MA, United States) with the following amplification parameters: initial denaturation at 95°C for 2 min; 35 cycles at 95°C for 50 s, 62°C for 50 s, 72°C for 50 s, and a final extension at 72°C for 5 min. PCR products were separated on 2% agarose gel for 40 min at 150 V. Each PCR product was isolated using a gel extraction kit (Qiagen), subcloned into a pGEM-T Easy vector (Promega, Madison, WI, United States), and sequenced using a DNA sequence analyzer (ABI 3730, Applied Biosystems Inc., Foster City, CA, United States). The Trichosanthes accessions used are listed in Supplementary Table 4. The commercial products are listed in Supplementary Table 5. Tri, TRr, and TRtt primer sequences are listed in Supplementary Table 6.
Phylogenetic Analysis
A total of 19 plastid genomes were used for phylogenetic analyses: 17 from Trichosanthes species and one each from Cucumis melo var. makuwa (MF536700) and Cucumis melo var. momordica (MF536701) as the outgroup. Of these, 12 plastid genome sequences were downloaded from NCBI GenBank (Supplementary Table 7). MAFFT (Katoh et al., 2002) was used to align the plastid genomes, and alignments were manually adjusted using Bioedit (Hall, 1999). Subsequently, each aligned gene (CDS) was extracted using Geneious prime v. 2021.13, yielding 58 single CDS alignments. The alignment files were filtered to remove ambiguously aligned regions using GBLOCKS v. 0.91b (Castresana, 2000), and concatenated using Geneious. The best-fitting model of nucleotide substitutions was determined based on Akaike Information Criterion in JModeltest v. 2.1.10 (Darriba et al., 2012). Maximum likelihood (ML) analysis was performed using RaxML v. 8.0.5 (Stamatakis, 2014) with 1,000 bootstrap replicates based on the GTR + I + G model. Bayesian Inference (BI) analysis was carried out using MrBayes v. 3.2.2 (Ronquist et al., 2012), with two independent runs and four chains run simultaneously for 5,000,000 generations. Trees were sampled every 100,000 generations, with the first 25% discarded as burn-in. The 50% majority-rule consensus tree was visualized using Figtree v. 1.4.2 (Rambaut, 2014), with posterior probabilities (PP) estimated from the sampled trees after the burn-in fraction was discarded. Nuclear ITS sequences were obtained using the above method (Indel marker development and Trichosanthes validation) using KIOM specimens (Supplementary Table 1). The ITS region was amplified using ITS1 (TCC GTA GGT GAA CCT GCG G) and ITS4 (TCC GCT TAT TGA TAT GC) primers, as described previously (White et al., 1990). Phylogenetic analysis was carried out using the method applied for the plastid genomes.
Results
Morphological Characteristics of Trichosanthes kirilowii vars. kirilowii and japonica
The floral parts (female and male flowers) were remarkably similar (Figures 1A,B). However, leaf lobation patterns were clearly distinct. The leaves of T. kirilowii var. kirilowii were deeply (about half to two-thirds) 5-lobed (rarely 3- or 7-lobed) with a rhombic-obovate to oblong-shaped middle lobe (Figure 1C). In contrast, the leaves of T. kirilowii var. japonica were shallowly (up to the middle) 3-lobed, rarely almost unlobed or 5-lobed, with an unlobed and triangular to triangular-ovate middle lobe (Figure 1D). In the case of the fruit, pepos were globose to oblong in T. kirilowii var. kirilowii (Figure 1E), and oblong to ellipsoidal in T. kirilowii var. japonica (Figure 1F). Both had oblong- to oblong-ovate-shaped seeds, but seed color was somewhat different. Based on the RHS Color Chart, T. kirilowii var. kirilowii seeds were bright orange/brown (RHS Color Chart 164A, B to 165A, B; Figure 1G), whereas T. kirilowii var. japonica seeds were a dark orange/brown (RHS Color Chart 174A, B to 175A, B; Figure 1H).

Figure 1. Characteristics of Trichosanthes (kirilowii var.) kirilowii and T. (kirilowii var.) japonica in Korea (A) Inflorescence of T. kirilowii. (B) Inflorescence of T. japonica. (C) Leaf shape of T. kirilowii. (D) Leaf shape of T. japonica. (E) Fruit of T. kirilowii. (F) Fruit of T. japonica. (G) Seeds of T. kirilowii. (H) Seeds of T. japonica.
Trichosanthes Plastid Genome Organization
Sequencing generated 1.5–1.8 Gb raw paired-end read data and 1.1–1.5 Gb trimmed reads (Supplementary Tables 8, 9). We obtained two to four initial plastid contigs from all the assembled contigs (Supplementary Figure 2). The complete plastid genomes of the Trichosanthes accessions had mean read depths of 570×, 547×, 852×, 303×, and 84×. They varied in length from 156,790 to 157,155 bp, and all had a typical quadripartite structure with an LSC region of 86,047–86,242 bp, SSC region of 18,219–18,341 bp, and IR regions of 86,048–86,243 bp (Figure 2, Table 1 and Supplementary Figure 3). The GC content was 37.1% in all five accessions. In general, the GC content of the IR (42.8%) was higher than those of the LSC (34.8–34.9%) and the SSC (31.0–31.2%) (Table 1). All Trichosanthes plastid genomes had 113 genes, including 79 protein-coding, four rRNA, and 30 tRNA genes (Table 2). They had 18 intron-containing genes, with three duplicate genes (ndhB, trnI-GAU, and trnA-UGC) in the IR regions, 16 of which had a single intron and two of which (ycf3 and clpP) had three introns (Supplementary Table 10). The infA gene was probably a pseudogene, with an early stop codon in all five plastid genomes. To identify IR contractions and extensions, we analyzed the IR boundaries of five Trichosanthes accessions in comparison to H. macrocarpa (Supplementary Figure 4). Rps19 was confined to the LSC in H. macrocarpa but extended into the IR regions in Trichosanthes. The ycf1 and ndhF genes were located at the IRa/SSC junction. The rpl2 genes of all species were located in the IRa region, with part of the gene duplicated in IRb. Analysis of codon usage and anticodon recognition patterns indicated that the five Trichosanthes accession plastid genomes contained 26,737–26,769 codons, and leucine, isoleucine, and serine were the most abundant (Supplementary Figure 5). The relative synonymous codon usage (RSCU) values indicated synonymous codon usage bias with high proportions of A or T in the third position. To identify codon patterns in Cucurbitaceae plastid genomes, we analyzed codon distribution in 33 plastid genomes using the hierarchical clustering method (Supplementary Figure 6). RSCU values below 1.00 indicated cases where codons were used less frequently than expected, whereas RSCUs values above 1.00 indicated cases were codons were used more frequently than expected. In Supplementary Figure 6, the colors green and red indicate strong (RSCU value > 1) and weak (RSCU value < 1) codon usage bias, respectively. The codons with an A or T in the third position had a strong codon bias. Most RSCU values had similar patterns in Cucurbitaceae. AGA (arginine) usually had high RSCU values. However, Gynostemma had genus-specific patterns–e.g., AGG (arginine) yielded particularly low RSCU values.
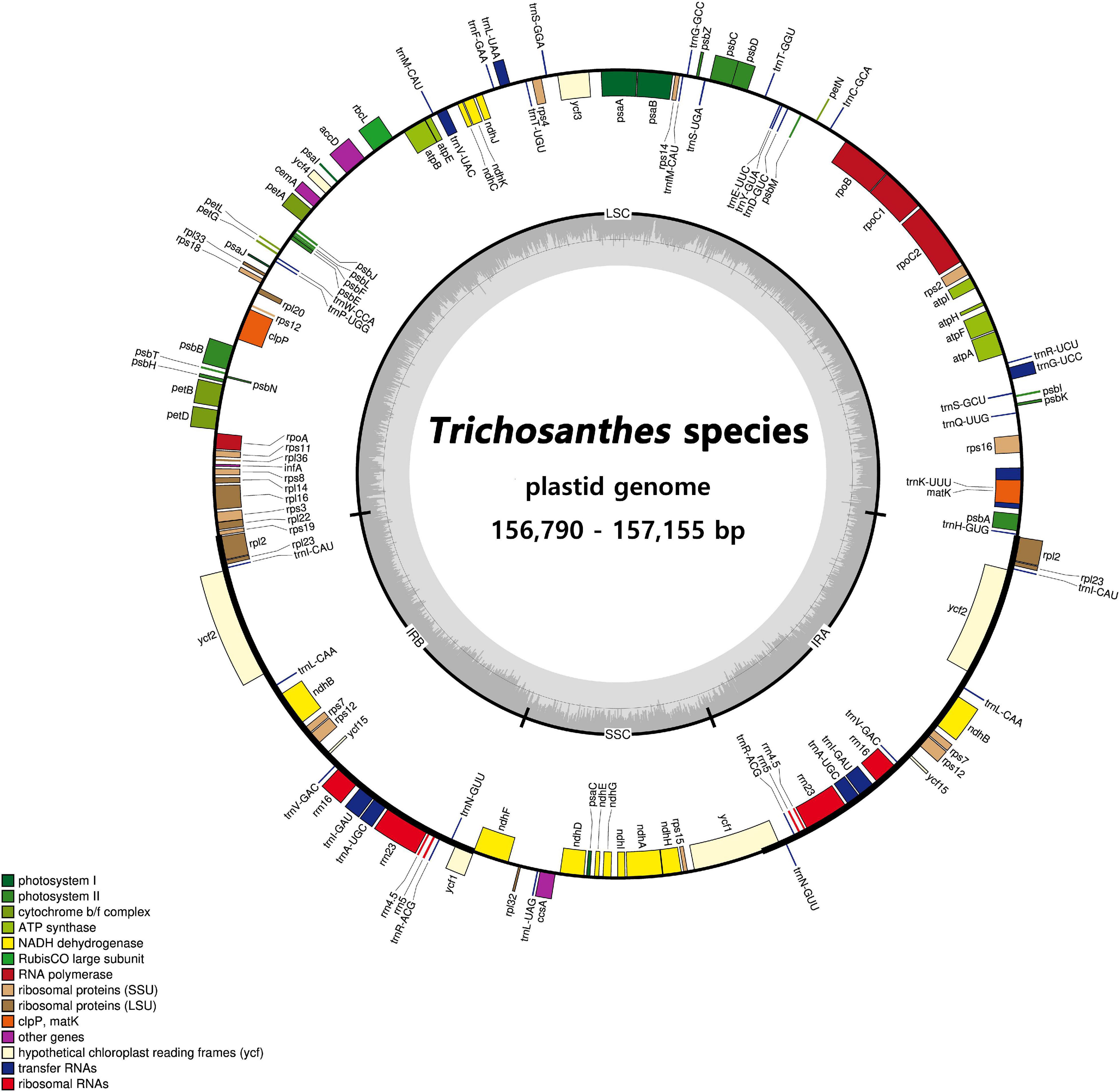
Figure 2. Circular gene map of plastid genomes from Trichosanthes species. Genes drawn inside the circle are transcribed clockwise, and those outside the circle are transcribed counterclockwise. The darker gray in the inner circle represents GC content. The gene map corresponds to the T. kirilowii chloroplast genome.
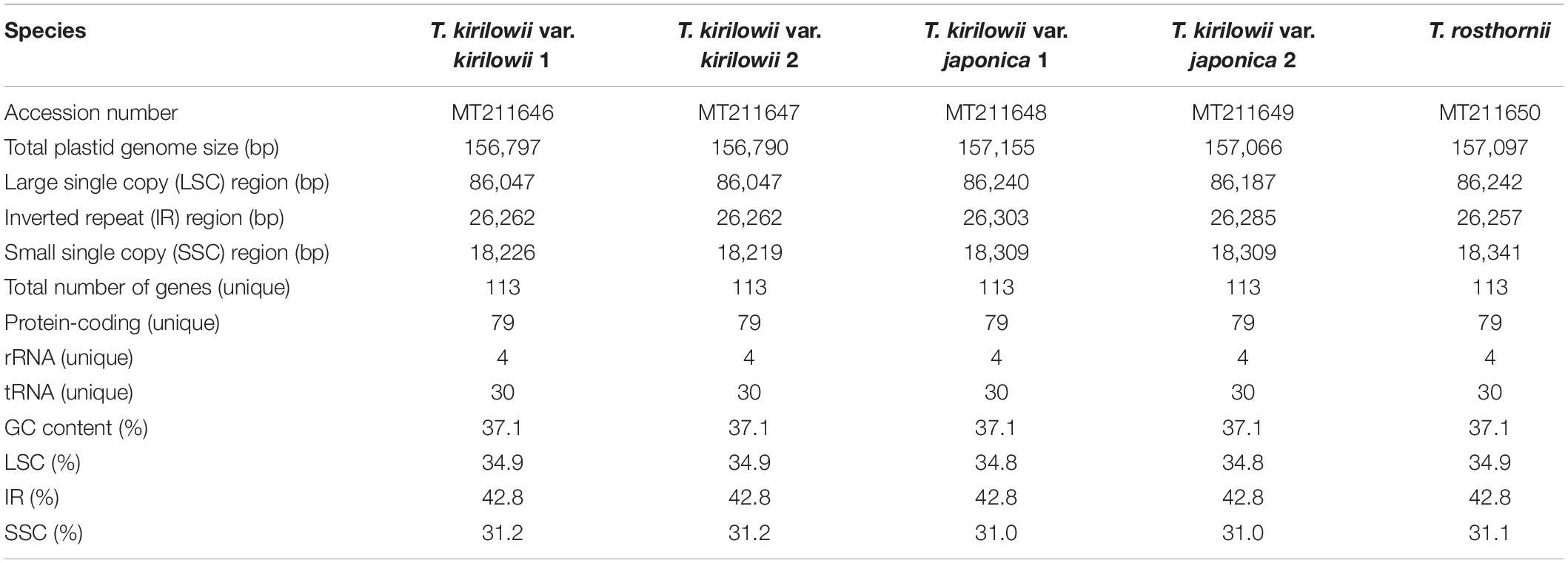
Table 1. Features of Trichosanthes plastid genomes.

Table 2. Genes in the plastid genomes of Trichosanthes species.
SSR and Tandem Repeat Analysis in Trichosanthes Plastid Genomes
The five Trichosanthes accessions had 64–75 SSRs in total (Figure 3A). Most SSRs were in the LSC and SSC regions within intergenic spacers (Figures 3A,B). The mononucleotide motif was the most abundant in all the accessions (Figure 3C). No hexanucleotide motif repeats were found. Most tandem repeats were located in the LSC (Figure 3D). T. kirilowii var. kirilowii and T. kirilowii var. japonica had tandem repeats in different genomic regions (Figure 3E). T. rosthornii had fewer tandem repeats than other Trichosanthes taxa. There were 7–16 tandem repeats of >20 bp identified in the five Trichosanthes accessions, and most were 21–40 bp long (Figure 3F).
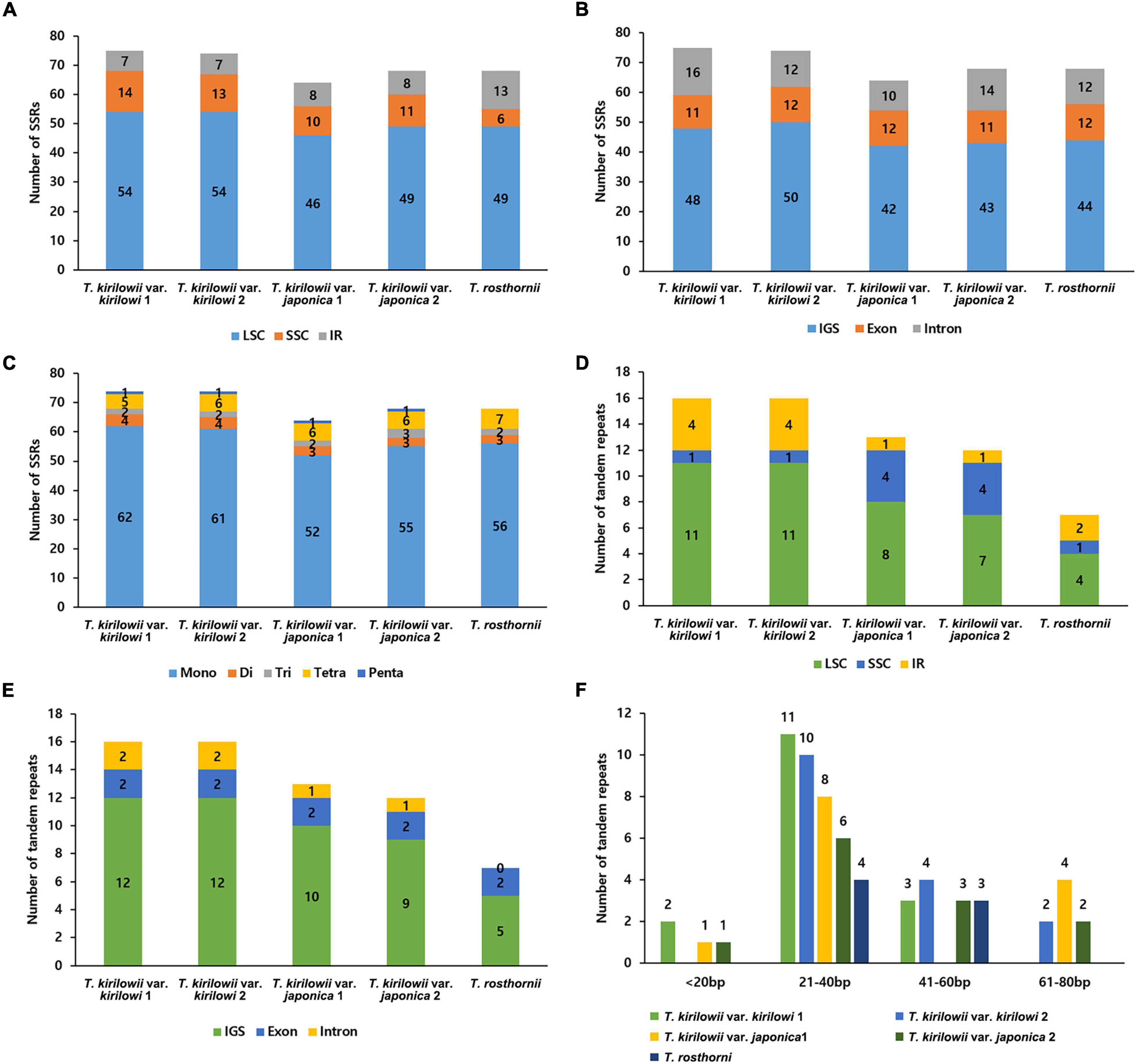
Figure 3. Distribution of repeat sequences in the five Trichosanthes plastid genomes. (A) Number of SSRs in genomic regions. (B) Distribution of SSRs in intergenic spacer (IGS), exon, and intron regions. (C) Distribution of SSR types. (D) Distribution of tandem repeats in genomic regions. (E) Distribution of tandem repeats in intergenic spacer (IGS), exon, and intron regions. (F) Distribution of tandem repeat lengths.
Comparative Analysis of Trichosanthes Plastid Genomes
To identify divergent regions, we analyzed the plastid genomes of all five Trichosanthes accessions using the mVISTA program with T. kirilowii var. kirilowii as a reference (Figure 4). Overall, the alignment revealed that IR regions were better conserved than the single-copy regions, and intergenic regions were more divergent than genic regions, except for ycf1, ycf2, rps3, and rpl22. The major regions of divergence (including less than 50% similarity) were identified in the intergenic regions (IGS) trnT-UGU–trnL-UAA, trnE-UUC–trnT-GGU, rrn4.5–rrn5. The differences observed between T. kirilowii var. kirilowii and T. kirilowii var. japonica in the regions could indicate that they are two different species. To determine sequence divergence in the five Trichosanthes accessions, we calculated the nucleotide diversity as a Pi value (Figure 5). The IGS region in atpF-atpH exhibited high divergence, with a Pi of 0.0198. In the genic region, the value of Pi for ndhC was 0.00496, indicating that the genic region was more highly conserved than the IGS region, as expected. In the present study, although the plastid genomes of Trichosanthes exhibited a highly conserved structure, highly local sequence variability was detected in IGS.
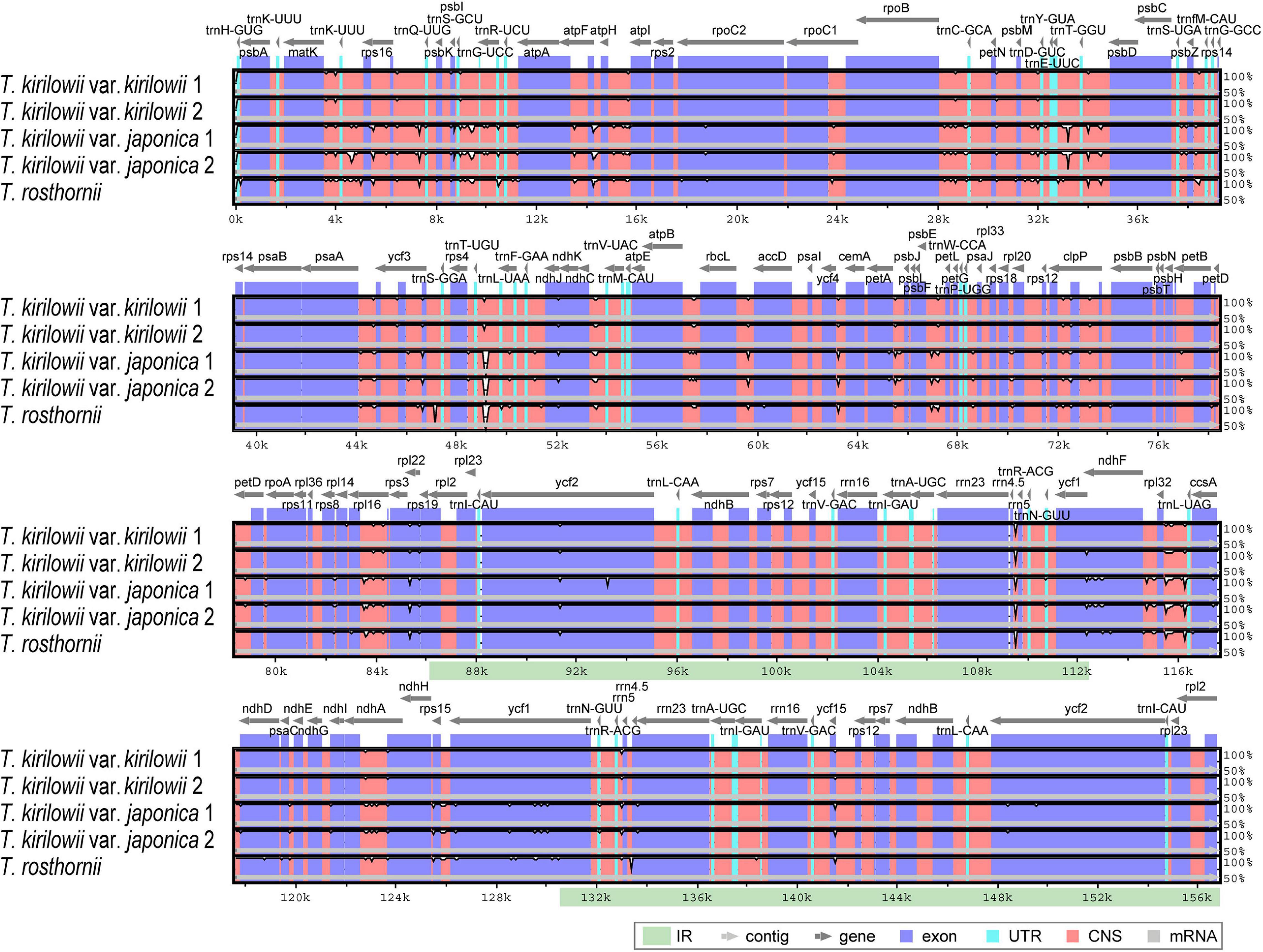
Figure 4. Comparison of Trichosanthes plastid genomes using mVISTA. Complete plastid genomes of five Trichosanthes species were compared, with T. kirilowii var. kirilowii 1 as the reference. The gray arrows above the alignment indicate genes. Different colors represent different regions (coding and non-coding). The horizontal axis indicates the coordinates within the chloroplast genome. The vertical scale represents the percentage of identity, ranging from 50 to 100%.
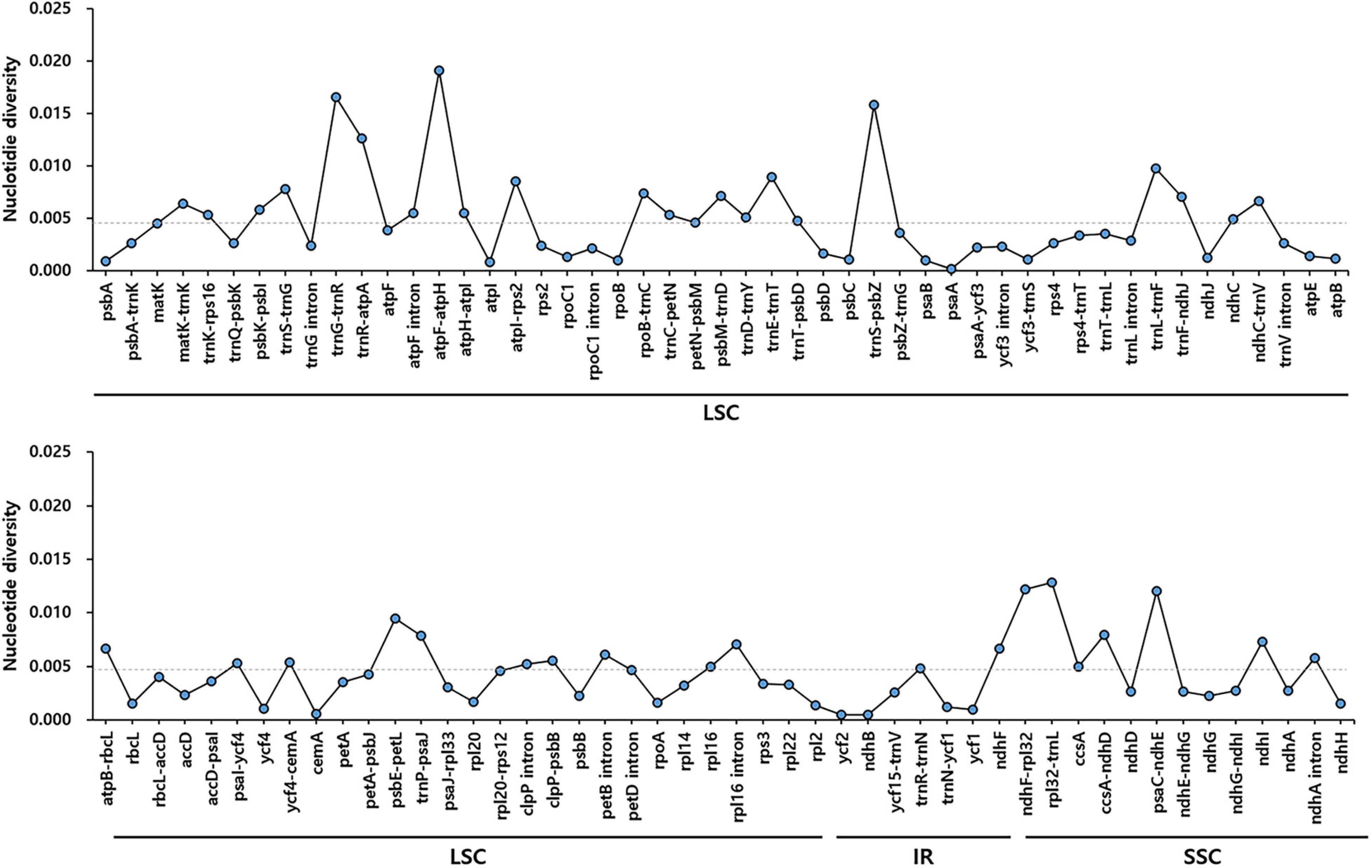
Figure 5. Comparison of nucleotide diversity (Pi) values among the five Trichosanthes species. The dotted line indicates the average Pi value by gene and intergenic region in five Trichosanthes plastid genomes. Regions with Pi = 0 were excluded.
New Molecular Markers for Distinguishing Trichosanthes Species
We compared the nuclear ITS sequences of T. kirilowii var. kirilowii, T. kirilowii var. japonica, T. rosthornii, and T. rubriflos. We did not find any variation among T. kirilowii and T. rosthornii (Supplementary Figure 7). Only T. kirilowii var. japonica and T. rubriflos had a few differences compared to the other species, showing the limitation of this universal DNA barcode in distinguishing Trichosanthes at the species level and below. To address the problem, we developed a marker set to distinguish T. kirilowii var. kirilowii, T. kirilowii var. japonica, and T. rosthornii using indels. Three intergenic regions (trnT-UGU–trnL-UAA, rrn4.5–rrn5, and trnE-UUG–trnT-GGU) were revealed to be considerably helpful by containing species-specific indels (Supplementary Figure 8).
Tri, TRr, and Trtt markers were successfully amplified for all samples (Figure 6). The Tri marker (located in the trnT-UGU–trnL-UAA region) differentiated T. kirilowii var. kirilowii from T. kirilowii var. japonica, T. rosthornii, and T. rubriflos. The Tri marker was 640, 512, and 502 bp long in T. kirilowii var. kirilowii, T. kirilowii var. japonica, and T. rosthornii, respectively. The TRr marker (located in the rrn4.5–rrn5 region) contained a 32-bp deletion in T. rosthornii, discriminating this species from the others. The Trtt marker (located in the trnE-UUG–trnT-GGU region) contained an 88-bp deletion in T. kirilowii var. japonica compared to the other Trichosanthes (Figure 6 and Supplementary Figure 8). Therefore, the four Trichosanthes taxa could be unambiguously discriminated using the three markers developed in the present study (Figure 6). Furthermore, we surveyed 15 commercial drugs made of crude Trichosanthes tissues from South Korea (Supplementary Table 5). Only four products were identified as T. kirilowii var. kirilowii using the Tri marker; no products were identified as T. rosthornii using the TRr marker. Nine products were identified as T. kirilowii var. japonica using Trtt. The two products without marked asterisks on Figure 6 were inferred to be T. rubriflos through the simultaneous use of all three markers (Tri, TRr, and Trtt). All fruit products were identified to be T. kirilowii var. japonica, while root and seed products were identified to be T. kirilowii var. japonica (four root and two seed products), T. rubriflos (one root and one seed product) or T. kirilowii var. kirilowii (one root and three seed products).
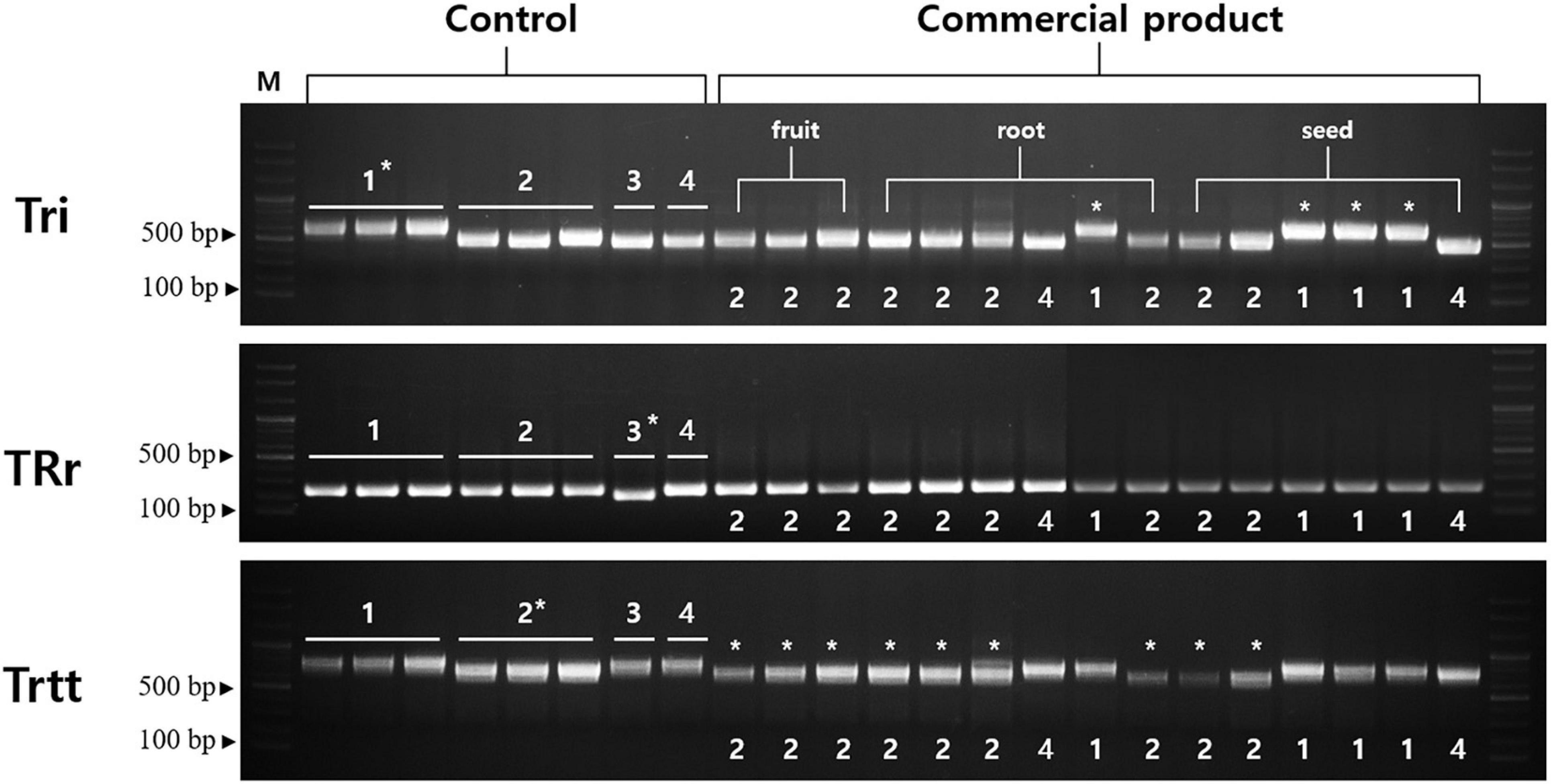
Figure 6. Gel images of indel markers for Trichosanthes. Primers for Tri, TRr, and Trtt were tested with 15 commercial products on the Korean market. Control samples were used in a marker test (Supplementary Table 4). Information regarding the commercial products is provided in Supplementary Table 5. An asterisk indicates matching between band patterns of control samples and commercial products. 1, Trichosanthes kirilowii var. kirilowii; 2, Trichosanthes kirilowii var. japonica; 3, Trichosanthes rosthornii; 4, Trichosanthes rubriflos.
Phylogenetic Relationships Among Trichosanthes Species Within Cucurbitaceae
To verify the phylogenetic relationships among Trichosanthes, we identified 73 protein-coding sequences (60,681 bp in total) shared by our five Trichosanthes accessions and 14 other Trichosanthes accessions, with Cucumis melo subsp. melo and Cucumis melo var. momordica as the outgroup (Figure 7 and Supplementary Figure 9). Phylogenetic relationships inferred from BI and ML were essentially identical (Figure 7 and Supplementary Figure 9). All phylogenetic relationships were strongly supported (BI PP = 1.0). Trichosanthes formed a monophyletic group with the exception of, T. nervifolia that is grouped with T. sect. Involucraria instead of with the other included species of sect. Trichosanthes. T. kirilowii var. kirilowii and T. kirilowii var. japonica were clustered with T. rosthornii and T. homophylla into T. sect. Foliobracteola. Whereas T. kirilowii var. kirilowii and T. kirilowii var. japonica were monophyletic, T. kirilowii as a whole was paraphyletic due to the nested positions of T. rosthornii and T. homophylla.

Figure 7. Phylogenetic relationships of Trichosanthes. Bayesian Inference (BI) topology is shown. The numbers above branches represent BI posterior probability, and Maximum Likelihood (ML) bootstrap support; branches without support values have maximum support (PP 1, BS 100).
Discussion
Features of Trichosanthes Plastid Genomes
In the present study, we newly determined the complete plastid genomes of five Trichosanthes accessions. Of those, T. kirilowii var. japonica and T. rosthornii were sequenced for the first time. Plastid genomes in Trichosanthes have 113 unique genes and their gene order, GC contents, genomic structure, and overall length (156,790–157,155 bp) are within the ranges described previously for angiosperm plastid genomes (Millen et al., 2001). Trichosanthes plastid genomes have one pseudogene, infA, which is the result of an early stop codon. The gene infA frequently contains deletions and early stop codons in angiosperm plastid genomes (Wicke et al., 2011). It has been transferred from plastid genomes to the nucleus in many plants. The infA encoded translation initiation factor was independently lost during land plant evolution (Wicke et al., 2011; Menezes et al., 2018). Codon usage is an essential factor for the expression of genetic information correctly, and it plays an important role in shaping plastid genome evolution (Wang et al., 2016; Yi et al., 2018). High relative synonymous codon usage (RSCU) values correspond to more highly conserved plastid genes (Wang et al., 2016; Ivanova et al., 2017; Zuo et al., 2017). The five Trichosanthes accessions have nearly the same codons, which are similar to those of other plastid genomes (Wang et al., 2016; Raman et al., 2019). We also surveyed the RSCU values of 28 other Cucurbitaceae plastid genomes. Half of the codons had a high codon bias (Supplementary Figure 6) and are denoted in green in the figure (RSCU > 1). This result is similar to those of other plastid genomes, and most codons with high RSCU values had an A or T in the third position of their amino acid. The plastid genomes of Gynostemma species have codon bias patterns that are slightly different from those of other Cucurbitaceae. In particular, AGG (arginine) codons had relatively low RSCU values. The RSCU values of the Trichosanthes species and other Cucurbitaceae plastid genomes were similar to those of other higher plants.
Repeat Sequences in Trichosanthes Plastid Genomes
Simple sequence repeats or microsatellites of 1–6 nucleotides are widely distributed throughout genomes (Curci et al., 2015). They are useful in population genetics studies and for discriminating species, and facilitate phylogenetic studies due to their high polymorphism at the intra- and interspecific levels (Powell et al., 1995; Zalapa et al., 2012). In the present study, mononucleotide SSRs comprised approximately 83% of all SSRs (55–62 SSRs), which were mostly detected in IGS regions (Figure 3). The finding is similar to those of previous reports, in which most of the mononucleotide repeats were A and T repeats due to an abundance of polyamines and polythymines in the plastid genome (Qian et al., 2013; Yang et al., 2016; Wang et al., 2018). SSRs identified in the Trichosanthes plastid genome could provide useful genetic resources for Trichosanthes species identification and population genetics studies. Repeat sequences facilitate phylogenetic research and species identification (Asaf et al., 2016). Tandem repeats, 7–16 nucleotides long, were detected in the five Trichosanthes plastid genomes. Among the five Trichosanthes taxa, T. rosthornii had the fewest tandem repeat sequences. T. kirilowii var. kirilowii and T. kirilowii var. japonica exhibited differences in SSRs and tandem repeats. In general, repeats are very similar between individuals of a similar species (Park et al., 2018b), but the two T. kirilowii varieties are completely different in terms of repeats. Furthermore, we did not observe a clear difference in the tandem repeat copy number, as was seen in previous studies for distinguishing species (Hong et al., 2017; Park et al., 2018b); however, a general repeat sequence variation was observed (Park et al., 2017). Such repeat sequences serve as molecular markers in population genetics and phylogenetic studies of Cucurbitaceae, including Trichosanthes.
Genetic Variation in the Trichosanthes Accessions
According to the mVISTA results, the plastid genome of Trichosanthes has low diversity, and its genic regions are more conserved than its IGS regions, the latter of which are consistent with angiosperm plastid genomes in general (Shaw et al., 2007; Huo et al., 2019; Song et al., 2019). The trnT–trnL, rrn4.5–rrn5, and trnE–trnT regions were observed to be hotspot regions for genetic variation (Figure 4). The hotspot regions in plant species are caused by mutation events (Morton and Clegg, 1993; Maier et al., 1995; Liu et al., 2018, 2019), and can be used as DNA barcodes to distinguish species or genera, depending on the variability of the regions. Such regions have been successfully used for the development of molecular markers to efficiently distinguish species (Cho et al., 2015; Hong et al., 2017; Park et al., 2018a, b). In terms of nucleotide diversity (Pi), most divergent regions were non-coding, and this is consistent with other plastid genomes, which have been reported to have highly variable non-coding regions at trnG–trnR, trnR–atpA, atpF–atpH, trnS–psbZ, trnL–trnF, ndhC–trnV, psbE–petL, ndhF–rpl32, and rpl32–trnL (Wang et al., 2010; Schroeder et al., 2016; Zhitao et al., 2017; Liu et al., 2019; Smidt et al., 2020).
Inverted repeat (IR) contraction and expansion in angiosperm plastid genomes cause plastid genome size to vary (Raubeson et al., 2007). Previous studies have identified extremely short IRs, or the loss of IR regions and genes (Wang et al., 2008; Zhu et al., 2016). Compared to H. macrocarpa, Trichosanthes exhibits a highly conserved IR length and gene positions. However, The IR region length ranged from 26,257 to 26,303, meaning that there was some contraction/expansion in the IR region (Supplementary Figure 4). This phenomenon has also been observed in other Cucurbitaceae plastid genomes (Zhang et al., 2018; Bellot et al., 2020).
Molecular Marker Development and Commercial Products Screening in Korean Herbal Markets
Authentication of herbal materials is essential for quality control, safety, and herbal medicine efficacy. Medicinal plants are extensively used for disease prevention, side effects management, and for their pharmacological effects (Yu et al., 2018). Adulterants in herbal medicine could have similar morphology, uncertain origins, and often bear names that are similar to those of the original ingredient (Han et al., 2016; Ichim, 2019). Their use may bring about negative side effects and quality problems. In Korean herbal medicine, the roots and seeds of T. kirilowii and T. rosthornii are considered authentic Trichosanthis Radix and Trichosanthis Semen, respectively (Korea Institute of Oriental Medicine [KIOM], 2020). However, T. kirilowii var. kirilowii and T. kirilowii var. japonica, distributed in South Korea, appear highly morphologically similar to the naked eye, and therefore, are often misused or used interchangeably.
In the present study, we developed an indel marker set to facilitate the distinction of authentic and adulterated Trichosanthes materials. Three novel markers–Tri, TRr, and Trtt–can completely discriminate T. kirilowii var. kirilowii from T. kirilowii var. japonica, T. rosthornii, and T. rubriflos (Figure 6). We also tested commercial products in South Korea. Among the 15 product samples tested, only four were composed of T. kirilowii, and no T. rosthornii was found, indicating that only the four were authentic. Most commercial products are prepared from T. kirilowii var. japonica, the adulterant. In the present study, we encountered problems in the context of quality control for authentic herbal medicines. The novel indel markers developed in the present study will facilitate rapid and accurate authentication of Trichosanthes herbal medicines, as well as the determination of whether they have been adulterated. Consequently, further studies are required to test the quality of the numerous commercial products available in markets.
Phylogenetic Relationships Among Trichosanthes and Taxonomic Identity of the T. kirilowii Complex
Our reconstructed phylogeny of the genus Trichosanthes is generally consistent with a recent infrageneric classification by de Boer and Thulin (2012). However, T. nervifolia, which currently is placed in sect. Trichosanthes together with T. cucumerina (de Boer et al., 2012), was instead closely related to taxa in sect. Involucraria. This topological conflict between plastome and nrDNA phylogenies may be due to chloroplast capture but additional morphological, micromorphological, anatomical, and palynological characteristics are needed to specify the exact sectional position of T. nervifolia. Considering the results of previous studies and those of the present study, the two infra-specific taxa of T. kirilowii have clear genetic differences that are large enough to distinguish them as separate species based on the phylogenetic species concept, as also concluded by de Boer et al. (2012) and Liu et al. (2021). Moreover, their habitats are different. T. kirilowii var. kirilowii is widely distributed across the temperate and subtropical regions of East Asia in inland habitats, such as forests, shrublands, and grasslands (Huang et al., 2011), whereas T. kirilowii var. japonica is rather narrowly distributed in Korea and Japan in islands and coastal regions (Ohba, 1999; Kim and Choi, 2018; Supplementary Figure 7). Finally, the two infra-specific taxa were clearly identified based on combined morphological characters such as leaf and fruit shape, and seed color according to the taxonomic concept of Regel (1868). We therefore propose that T. kirilowii var. japonica (≡T. kirilowii subsp. japonica) be recognized again as a separate species considering its distinct genetics, morphology, and geographical distribution (Huang et al., 2009; de Boer et al., 2012; Liu et al., 2021). Further molecular identification and in-depth phylogenetic studies, as well as morphological studies, using various and abundant samples of Trichosanthes are required to identify the unique traits among the species to facilitate their identification and classification.
From an herbal medicine perspective, T. kirilowii and T. rosthornii form a monophyletic clade, and T. japonica was separated from them. Therefore, the use of T. japonica as a medicinal herb is not recommended because its effects are unknown. The results of the present study offer data on the authenticity of Korean herbal medicine resources, particularly Trichosanthes, which could enhance the quality and safety of Trichosanthes herbal medicines.
Conclusion
The present study identified the distinct morphological traits between T. kirilowii s. str (=T. kirilowii var. kirilowii) and T. japonica (=T. kirilowii var. japonica). The plastid genomes of the five Trichosanthes accessions studied were highly conserved with respect to gene content, gene orientation, GC content, and local variations. Most divergences were detected in non-coding regions (trnT–UGU–trnL–UAA, rrn4.5–rrn5, trnE–UUC–trnT–GGU). Such hotspot regions were used to create a novel marker set that successfully discriminated T. kirilowii from T. japonica and T. rosthornii in commercial herbal medicine products. Overall, our results distinguish T. kirilowii and T. japonica based on complete plastid genomes, novel marker sets, and phylogenetic relationships. Furthermore, our results could facilitate herbal medicine quality control by enabling the authentication of herbal medicines containing T. kirilowii and T. rosthornii.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
IP designed the experimental framework, drafted and revised the manuscript, performed experiments, and carried out genome analysis. J-HS, SY, SC, and BM collected and identified plant materials. IP and BM revised the manuscript. All authors contributed to the experiments and approved the final manuscript.
Funding
This research was funded by a grant from the Development of Sustainable Application for Standard Herbal Resources (KSN2012320) from the Korea Institute of Oriental Medicine (KIOM), South Korea.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.559511/full#supplementary-material
Footnotes
- ^ http://misaweb.ipk-gatersleben.de/
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi
- ^ https://www.geneious.com
References
Acosta, M. C., and Premoli, A. C. (2010). Evidence of chloroplast capture in South American Nothofagus (subgenus Nothofagus, Nothofagaceae). Mol. Phylogenet. Evol. 54, 235–242. doi: 10.1016/j.ympev.2009.08.008
Asaf, S., Khan, A. L., Khan, A. R., Waqas, M., Kang, S. M., Khan, M. A., et al. (2016). Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant Sci. 7:843. doi: 10.3389/fpls.2016.00843
Babicki, S., Arndt, D., Marcu, A., Liang, Y., Grant, J. R., Maciejewski, A., et al. (2016). Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 44, W147–W153.
Beier, S., Thiel, T., Munch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bellot, S., Mitchell, T. C., and Schaefer, H. (2020). Phylogenetic informativeness analyzes to clarify past diversification processes in Cucurbitaceae. Sci. Rep. 10:488.
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Carver, T., Berriman, M., Tivey, A., Patel, C., Bohme, U., Barrell, B. G., et al. (2008). Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 24, 2672–2676. doi: 10.1093/bioinformatics/btn529
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Cho, K. S., Yun, B. K., Yoon, Y. H., Hong, S. Y., Mekapogu, M., Kim, K. H., et al. (2015). Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS One 10:e0125332. doi: 10.1371/journal.pone.0125332
Curci, P. L., De Paola, D., Danzi, D., Vendramin, G. G., and Sonnante, G. (2015). Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other Asteraceae. PLoS One 10:e0120589. doi: 10.1371/journal.pone.0120589
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
de Boer, H. J., Schaefer, H., Thulin, M., and Renner, S. S. (2012). Evolution and loss of long-fringed petals: a case study using a dated phylogeny of the snake gourds, Trichosanthes (Cucurbitaceae). BMC Evol. Biol. 12:108. doi: 10.1186/1471-2148-12-108
de Boer, H. J., and Thulin, M. (2012). Synopsis of Trichosanthes (Cucurbitaceae) based on recent molecular phylogenetic data. PhytoKeys 12, 23–33. doi: 10.3897/phytokeys.12.2952
de Wilde, W. J., and Duyfjes, B. E. (2010). “Cucurbitaceae,” in Flora Malesiana, Series I, Seed Plants, ed. H. P. Nooteboom (Leiden: Netherlands Centre for Biodiversity Naturalis), 1–333.
Delcher, A. L., Salzberg, S. L., and Phillippy, A. M. (2003). Using MUMmer to identify similar regions in large sequence sets. Curr. protoc. Bioinform. 10:10.3.
Dong, W., Liu, J., Yu, J., Wang, L., and Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One 7:e35071. doi: 10.1371/journal.pone.0035071
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64.
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Han, J., Pang, X., Liao, B., Yao, H., Song, J., and Chen, S. (2016). An authenticity survey of herbal medicines from markets in China using DNA barcoding. Sci. Rep. 6:18723.
Hong, S. Y., Cheon, K. S., Yoo, K. O., Lee, H. O., Cho, K. S., Suh, J. T., et al. (2017). Complete chloroplast genome sequences and comparative analysis of Chenopodium quinoa and C. album. Front. Plant Sci. 8:1696. doi: 10.3389/fpls.2017.01696
Huang, L. Q., Le, C. X., Cheng, J. R., and Lou, Z. C. (2009). Taxonomic Study of Medicinal Plants of Trichosanthes L. Volume 2. Shanghai: Shanghai Science and Technology Press, 27–33.
Huang, L. Q., Lu, A. M., and Jeffrey, C. (2011). “Trichosanthes,” in Flora of China, Volume 19, eds A. M. Lu and C. Jeffrey (St. Louis: Missouri Botanical Gardens Press), 36–45.
Huo, Y., Gao, L., Liu, B., Yang, Y., Kong, S., Sun, Y., et al. (2019). Complete chloroplast genome sequences of four Allium species: comparative and phylogenetic analyzes. Sci. Rep. 9:12250.
Ichim, M. C. (2019). The DNA-based authentication of commercial herbal products reveals their globally widespread adulteration. Front. Pharmacol. 10:1227. doi: 10.3389/fphar.2019.01227
Ivanova, Z., Sablok, G., Daskalova, E., Zahmanova, G., Apostolova, E., Yahubyan, G., et al. (2017). Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Front. Plant Sci. 8:204. doi: 10.3389/fpls.2017.00204
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Depamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U.S.A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kawabe, A., Nukii, H., and Furihata, H. Y. (2018). Exploring the history of chloroplast capture in arabis using whole chloroplast genome sequencing. Int. J. Mol. Sci. 19:602. doi: 10.3390/ijms19020602
Kim, K., Lee, S. C., Lee, J., Lee, H. O., Joh, H. J., Kim, N. H., et al. (2015). Comprehensive survey of genetic diversity in chloroplast genomes and 45S nrDNAs within Panax ginseng species. PLoS One 10:e0117159. doi: 10.1371/journal.pone.0117159
Kim, S. C., Park, S. Y., Her, K. H., Kim, S. J., and Kang, H. K. (2003). HL-60 effect of extracts of T. kirilowii var. japonica on the growth of HL-60 leukemia cells. Yakhak Hoeji 47, 319–324.
Kim, Y. D., and Choi, B. H. (2018). “Trichosanthes,” in Flora of Korea Editorial Committee. The Genera of Vascular Plants of Korea, ed. C.-W. Park (Seoul: Hongreung Publishing Co.), 536–537.
Korea Institute of Oriental Medicine [KIOM] (2020). Defining Dictionary for Medicinal. (Herbs). Available online at: https://oasis.kiom.re.kr/herblib/ (accessed on 4 March, 2020).
Li, X., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y., and Chen, S. (2015). Plant DNA barcoding: from gene to genome. Biol. Rev. Camb. Philos. Soc. 90, 157–166. doi: 10.1111/brv.12104
Liu, H., Su, Z., Yu, S., Liu, J., Yin, X., Zhang, G., et al. (2019). Genome comparison reveals mutation hotspots in the chloroplast genome and phylogenetic relationships of Ormosia species. Biomed. Res. Int. 2019:7265030.
Liu, L., Wang, Y., He, P., Li, P., Lee, J., Soltis, D. E., et al. (2018). Chloroplast genome analyzes and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genomics 19:235. doi: 10.1186/s12864-018-4633-x
Liu, Q., Lin, N., Zhang, D. G., Huangm, X. H., Wang, H. C., Yang, J., et al. (2021). Trichosanthes sunhangii (Cucurbitaceae), a new species from Hubei, China. Phytotaxa 479, 87–294.
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Lu, Q. B., Liu, C. Q., and Huang, S. X. (2021). Moths pollinate four crops of Cucurbitaceae in Asia. J. Appl. Entomol. 1, 1–9. doi: 10.1002/9780470015902.a0003723.pub2
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18.
Maier, R. M., Neckermann, K., Igloi, G. L., and Kossel, H. (1995). Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 251, 614–628. doi: 10.1006/jmbi.1995.0460
Menezes, A. P. A., Resende-Moreira, L. C., Buzatti, R. S. O., Nazareno, A. G., Carlsen, M., Lobo, F. P., et al. (2018). Chloroplast genomes of Byrsonima species (Malpighiaceae): comparative analysis and screening of high divergence sequences. Sci. Rep. 8:2210.
Michael, D., Gurusaran, M., Santhosh, R., Hussain, M. K., Satheesh, S. N., Suhan, S., et al. (2019). RepEx: A web server to extract sequence repeats from protein and DNA sequences. Comput. Biol. Chem. 78, 424–430. doi: 10.1016/j.compbiolchem.2018.12.015
Millen, R. S., Olmstead, R. G., Adams, K. L., Palmer, J. D., Lao, N. T., Heggie, L., et al. (2001). Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13, 645–658. doi: 10.2307/3871412
Morton, B. R., and Clegg, M. T. (1993). A chloroplast DNA mutational hotspot and gene conversion in a noncoding region near rbcL in the grass family (Poaceae). Curr. Genet. 24, 357–365. doi: 10.1007/bf00336789
Ohba, H. (1999). “Trichosanthes,” in Flora of Japan, Vol. 2c, eds K. Iwatsuki, D. E. Boufford, and H. Ohba (Tokyo: Kodansha), 196–199.
Park, I., Kim, W. J., Yang, S., Yeo, S. M., Li, H., and Moon, B. C. (2017). The complete chloroplast genome sequence of Aconitum coreanum and Aconitum carmichaelii and comparative analysis with other Aconitum species. PLoS One 12:e0184257. doi: 10.1371/journal.pone.0184257
Park, I., Song, J. H., Yang, S., Kim, W. J., Choi, G., and Moon, B. C. (2019a). Cuscuta species identification based on the morphology of reproductive organs and complete chloroplast genome sequences. Int. J. Mol. Sci. 20:2726. doi: 10.3390/ijms20112726
Park, I., Song, J. H., Yang, S., and Moon, B. C. (2020). Comparative analysis of Actaea chloroplast genomes and molecular marker development for the identification of authentic Cimicifugae Rhizoma. Plants 9:157. doi: 10.3390/plants9020157
Park, I., Yang, S., Kim, W. J., Noh, P., Lee, H. O., and Moon, B. C. (2018a). Authentication of herbal medicines Dipsacus asper and Phlomoides umbrosa using DNA barcodes, chloroplast genome, and sequence characterized amplified region (SCAR) marker. Molecules 23:1748. doi: 10.3390/molecules23071748
Park, I., Yang, S., Kim, W. J., Noh, P., Lee, H. O., and Moon, B. C. (2018b). The complete chloroplast genomes of six Ipomoea species and indel marker development for the discrimination of authentic Pharbitidis Semen (Seeds of I. nil or I. purpurea). Front. Plant Sci. 9:965. doi: 10.3389/fpls.2018.00965
Park, I., Yang, S., Kim, W. J., Song, J. H., Lee, H. S., Lee, H. O., et al. (2019b). Sequencing and comparative analysis of the chloroplast genome of Angelica polymorpha and the development of a novel indel marker for species identification. Molecules 24:1038. doi: 10.3390/molecules24061038
Parks, M., Cronn, R., and Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84. doi: 10.1186/1741-7007-7-84
Powell, W., Morgante, M., Mcdevitt, R., Vendramin, G. G., and Rafalski, J. A. (1995). Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proc. Natl. Acad. Sci. U.S.A. 92, 7759–7763. doi: 10.1073/pnas.92.17.7759
Qian, J., Song, J., Gao, H., Zhu, Y., Xu, J., Pang, X., et al. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS One 8:e57607. doi: 10.1080/23802359.2020.1778574
Raman, G., Park, S., Lee, E. M., and Park, S. (2019). Evidence of mitochondrial DNA in the chloroplast genome of Convallaria keiskei and its subsequent evolution in the Asparagales. Sci. Rep. 9:5028.
Rambaut, A. (2014). FigTree v 1.4. 2 Molecular Evolution, Phylogenetics and Epidemiology. Edinburgh: Univ. Edinburgh, Inst. Evol. Biol.
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: analyzes including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8:174. doi: 10.1186/1471-2164-8-174
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sanchez-Delbarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Schroeder, H., Cronn, R., Yanbaev, Y., Jennings, T., Mader, M., Degen, B., et al. (2016). Development of molecular markers for determining continental origin of wood from white oaks (Quercus L. sect. Quercus). PLoS One 11:e0158221. doi: 10.1371/journal.pone.0158221
Shaw, J., Lickey, E. B., Schilling, E. E., and Small, R. L. (2007). Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am. J. Bot. 94, 275–288. doi: 10.3732/ajb.94.3.275
Smidt, E. C., Paez, M. Z., Vieira, L. D. N., Viruel, J., De Baura, V. A., Balsanelli, E., et al. (2020). Characterization of sequence variability hotspots in Cranichideae plastomes (Orchidaceae, Orchidoideae). PLoS One 15:e0227991. doi: 10.1371/journal.pone.0227991
Song, Y., Zhang, Y., Xu, J., Li, W., and Li, M. (2019). Characterization of the complete chloroplast genome sequence of Dalbergia species and its phylogenetic implications. Sci. Rep. 9:20401.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stegemann, S., Keuthe, M., Greiner, S., and Bock, R. (2012). Horizontal transfer of chloroplast genomes between plant species. Proc. Natl. Acad. Sci. U.S.A. 109, 2434–2438. doi: 10.1073/pnas.1114076109
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11.
Wang, R. J., Cheng, C. L., Chang, C. C., Wu, C. L., Su, T. M., and Chaw, S. M. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8:36. doi: 10.1186/1471-2148-8-36
Wang, W., Wu, Y., Yan, Y., Ermakova, M., Kerstetter, R., and Messing, J. (2010). DNA barcoding of the Lemnaceae, a family of aquatic monocots. BMC Plant. Biol. 10:205. doi: 10.1186/1471-2229-10-205
Wang, X., Zhou, T., Bai, G., and Zhao, Y. (2018). Complete chloroplast genome sequence of Fagopyrum dibotrys: genome features, comparative analysis and phylogenetic relationships. Sci. Rep. 8:12379.
Wang, Y., Zhan, D. F., Jia, X., Mei, W. L., Dai, H. F., Chen, X. T., et al. (2016). Complete chloroplast genome sequence of Aquilaria sinensis (Lour.) Gilg and evolution analysis within the malvales order. Front. Plant Sci. 7:280. doi: 10.3389/fpls.2016.00280
White, T. J., Bruns, T., Lee, S., and Taylor, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. Guide Methods Appl. 18, 315–322. doi: 10.1016/b978-0-12-372180-8.50042-1
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Muller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Yang, Y., Zhou, T., Duan, D., Yang, J., Feng, L., and Zhao, G. (2016). Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 7:959. doi: 10.3389/fpls.2016.00959
Yi, S., Li, Y., and Wang, W. (2018). Selection shapes the patterns of codon usage in three closely related species of genus Misgurnus. Genomics 110, 134–142. doi: 10.1016/j.ygeno.2017.09.004
Yu, X., Tang, L., Wu, H., Zhang, X., Luo, H., Guo, R., et al. (2018). Trichosanthis fructus: botany, traditional uses, phytochemistry and pharmacology. J. Ethnopharmacol. 224, 177–194. doi: 10.1016/j.jep.2018.05.034
Zalapa, J. E., Cuevas, H., Zhu, H., Steffan, S., Senalik, D., Zeldin, E., et al. (2012). Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 99, 193–208. doi: 10.3732/ajb.1100394
Zeng, C. X., Hollingsworth, P. M., Yang, J., He, Z. S., Zhang, Z. R., Li, D. Z., et al. (2018). Genome skimming herbarium specimens for DNA barcoding and phylogenomics. Plant Methods 14:43.
Zhang, X., Zhou, T., Yang, J., Sun, J., Ju, M., Zhao, Y., et al. (2018). Comparative analyzes of chloroplast genomes of cucurbitaceae species: lights into selective pressures and phylogenetic relationships. Molecules 23:2165. doi: 10.3390/molecules23092165
Zhitao, N., Shuying, Z., Jiajia, P., Ludan, L., Jing, S., and Xiaoyu, D. (2017). Comparative analysis of Dendrobium plastomes and utility of plastomic mutational hotspots. Sci. Rep. 7:2073.
Zhu, A., Guo, W., Gupta, S., Fan, W., and Mower, J. P. (2016). Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 209, 1747–1756. doi: 10.1111/nph.13743
Keywords: Trichosanthes, plastid genome, divergent region, phylogenetic relationship, indel marker, herbal medicine
Citation: Park I, Song J-H, Yang S, Chae S and Moon BC (2021) Plastid Phylogenomic Data Offers Novel Insights Into the Taxonomic Status of the Trichosanthes kirilowii Complex (Cucurbitaceae) in South Korea. Front. Plant Sci. 12:559511. doi: 10.3389/fpls.2021.559511
Received: 06 May 2020; Accepted: 08 July 2021;
Published: 27 July 2021.
Edited by:
Gerald Matthias Schneeweiss, University of Vienna, AustriaReviewed by:
Sidonie Bellot, Royal Botanic Gardens, Kew, United KingdomHanno Schaefer, Technical University of Munich, Germany
Copyright © 2021 Park, Song, Yang, Chae and Moon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Byeong Cheol Moon, YmNtb29uQGtpb20ucmUua3I=