 Ting He
Ting He Zhaoyu Ren
Zhaoyu Ren Ghulam Muhae-Ud-Din
Ghulam Muhae-Ud-Din Qingyun Guo2
Qingyun Guo2 Taiguo Liu
Taiguo Liu Wanquan Chen
Wanquan Chen Li Gao
Li Gao- 1State Key Laboratory for Biology of Plant Disease and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China
- 2Key Laboratory of Agricultural Integrated Pest Management, Qinghai University, Xining, China
Tilletia laevis Kühn [synonym T. foetida (Wallr.) Liro] can lead to a wheat common bunt, which is one of the most serious diseases affecting kernels, a serious reduction in grain yield, and losses can reach up to 80% in favorable environments. To understand how wheat tassels respond to T. laevis, based on an RNA-Seq technology, we analyzed a host transcript accumulation on healthy wheat tassels and on tassels infected by the pathogen. Our results showed that 7,767 out of 15,658 genes were upregulated and 7,891 out of 15,658 genes were downregulated in wheat tassels. Subsequent gene ontology (GO) showed that differentially expressed genes (DEGs) are predominantly involved in biological processes, cellular components, and molecular functions. Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis showed that 20 pathways were expressed significantly during the infection of wheat with T. laevis, while biosynthesis of amino acids, carbon metabolism, and starch and sucrose metabolism pathways were more highly expressed. Our findings also demonstrated that genes involved in defense mechanisms and myeloblastosis (MYB) transcription factor families were mostly upregulated, and the RNA-seq results were validated by quantitative real-time polymerase chain reaction (qRT-PCR). This is the first report on transcriptomics analysis of wheat tassels in response to T. laevis, which will contribute to understanding the interaction of T. laevis and wheat, and may provide higher efficiency control strategies, including developing new methods to increase the resistance of wheat crops to T. laevis-caused wheat common bunt.
Introduction
Common bunt of wheat, which may have been caused by Tilletia laevis Kühn (synonym T. foetida (Wallr.) Liro), which is one of the most serious fungal diseases affecting the wheat crops globally (Goates, 2012; Bokore et al., 2019). This disease affects wheat crop growth and production via the infection of roots, vascular bundles of stems, leaves, tassels, and grains by replacing the grain materials and leads to a marked decline in yield and quality (Goates and Peterson, 1999; Lu et al., 2005; Goates, 2012). Common bunt can be controlled with different fungicides coated with seeds, and incorporating resistance into cultivars is still important in many wheat breeder programs worldwide and increases interest in production (Matanguihan et al., 2011; Din et al., 2021). Therefore, an extensive breeding research has been carried out to understand the genetics of disease resistance, as well as the underlying mechanisms by which new cultivars resistant to the pathogen can be produced (Dumalasová and Bartoš, 2010). Additionally, extensive and continuous use of fungicides potentially pollutes the environment and generates resistance in pathogens (Zhu et al., 2017). Therefore, an improved understanding of the defense mechanisms used by wheat crops in response to common bunt will contribute to the design of new and safer control strategies and aid in the development of resistant cultivars.
Plants have evolved in a number of strategies to effectively fight against pathogens, involving a series of morph-physiological responses, including callose deposition, cell wall modifications, hypersensitive reactions, and the production of defense-related proteins, antimicrobial metabolites, and proteins (Cohn et al., 2001; Muhae-Ud-Din et al., 2020a; Ren et al., 2020; Xu et al., 2021). These morph-physiological responses are linked with numerous pathogenesis-related genes and transcription factors. Thus, with advancement in technology, approaches in comparative “omics” have effectively donated to the effort of defining gene functions and our understanding of their expression and alterations in accumulation during plant pathogen interactions (Mathioni et al., 2011; Zhu et al., 2017; Ren et al., 2020). Transcriptome responses provide new insights into the molecular mechanisms of plant resistance during the interaction of plants and pathogens. An RNA-Seq has been evaluated for a series of host-pathogen interactions, including wheat and T. controversa (Ren et al., 2020), mango and Fusarium mangiferae (Liu et al., 2016), tomato and Xanthomonas perforans race T3 (Du et al., 2015), wheat and Puccinia striiformis f. sp. tritici (Poretti et al., 2021), banana and F. oxysporum f. sp. cubense (Li et al., 2013), avocado and Rosellinia necatrix (Zumaquero et al., 2019), wheat and Heterodera avenae (Kong et al., 2015), pea and Phytophthora pisi (Hosseini et al., 2015), and soybean and F. oxysporum (Lanubile et al., 2015). Based on comparative transcriptomic analysis, many mycoblastosis (MYB) transcription factors and defense-related genes (Thilmony et al., 2006; Mukherjee et al., 2010; Proietti et al., 2013) were found to be involved in defense mechanisms and in primary and secondary resistance-associated signal transduction paths of plants. For example, defense-related genes were found to be significantly upregulated in wheat after T. controversa inoculation (Ren et al., 2020), phytohormone-related genes were upregulated in potato after Ralstonia solanacearum (Zuluaga et al., 2015), and MYB transcription factor families were upregulated in wheat after T. controversa (Chen et al., 2021) and R. cerealis infections (Shan et al., 2016).
Wheat crops are staple food crops in many countries (Shewry, 2009). Although extensive research has been done to date to analyze wheat-pathogen interactions (Zhang et al., 2012; Haueisen et al., 2019; Ren et al., 2020), very little literature is known about transcriptome responses between wheat and T. laevis, which lead to wheat common bunt being an emerging and serious fungal disease that causes dramatic quality and quantity losses in wheat. This study is the first global transcription analysis of wheat tassels in response to T. laevis. to develop resistance by contributing to the control of common bunt disease.
Materials and Methods
Plant and Fungal Material
Healthy wheat kernels (Cultivar: Dongxuan 3) and T. laevis were collected from the Institute of Plant Protection, Chinese Academy of Agricultural Sciences, China. The grains were sterilized and vernalized, and ten wheat seedlings were transplanted into every pot. Based on morphological characterization of teliospores of T. laevis (Supplementary Figure 1) and specific band and sequence of SCAR marker of T. laevis (Supplementary Figure 2), we identified the pathogen of T. laevis based on the previous reports (Xu et al., 2020; Qin et al., 2021). Then, the identified T. laevis was use in this study. The teliospores of T. laevis were cultured following the methods of previously published reports (Muhae-Ud-Din et al., 2020a). Briefly, two to three kernels were crushed by a centrifuge machine, disinfected with 2 ml of 0.25% NaClO and cultured at 16°C and 60% relative humidity for 14 days in an incubator (LT-36VLC8, Percival, United States). After 16 days of incubation, hyphal growth was checked under an inverted microscope (IX83, Olympus, Japan). Hyphae were collected and mixed with ddH2O and used to inoculate wheat plants at a concentration of 106 spores/ml with an OD600 of 0.15. One milliliter of obtained mycelium was injected into tassels, and injection was repeated for 3 times with a 1-day interval. The tassels injected with ddH2O were used as a control in this study. Ten pots were used for T. laevis inoculation and for control separately. The tassels (6 ± 5 cm in length) were detached from T. laevis-inoculated and control plants, with three biological replicates for every treatment.
RNA Extraction From Wheat Tassels
Wheat tassel RNA was extracted from each sample using a total plant RNA extraction kit (Ambion, TX, United States), according to the manufacturer’s instructions. The quality and integrity of RNA were determined by using agarose gel electrophoresis, while the concentration was determined through a NanoDrop 2000 (Denovix, United States), and RNA was stored at −80°C for further use.
Transcriptome Sequencing of Purified RNA
The raw reads were filtered by using the high-throughput sequencing Illumina HiSeq 4,000 platform. Clean reads were generated by removing reads with adaptors, reads where the number of unknown bases was more than 10%, low-quality reads (those in which more than 50% of bases presented a quality of ≤ 10) and poly-N (unrecognized bases). Clean reads were then aligned to the reference sequence1 using the software hisat2 (Kim et al., 2015) with the parameters set by the system.
Kyoto Encyclopedia of Genes and Genomes and Gene Ontology Enrichment Analysis
The Kyoto Encyclopedia of Genes and Genomes (KEGG)2 pathway analysis was performed by using GPSeq, and gene ontology (GO) enrichment analysis was performed with FDR < 0.05, representing the significantly expressed genes (Kanehisa et al., 2008; Young et al., 2010), NR, EggNOG database, NT, KOG/COG (Tatusov et al., 2000), Pfam (Bateman et al., 2002), and SWISS-Prot (Bairoch and Apweiler, 1997).
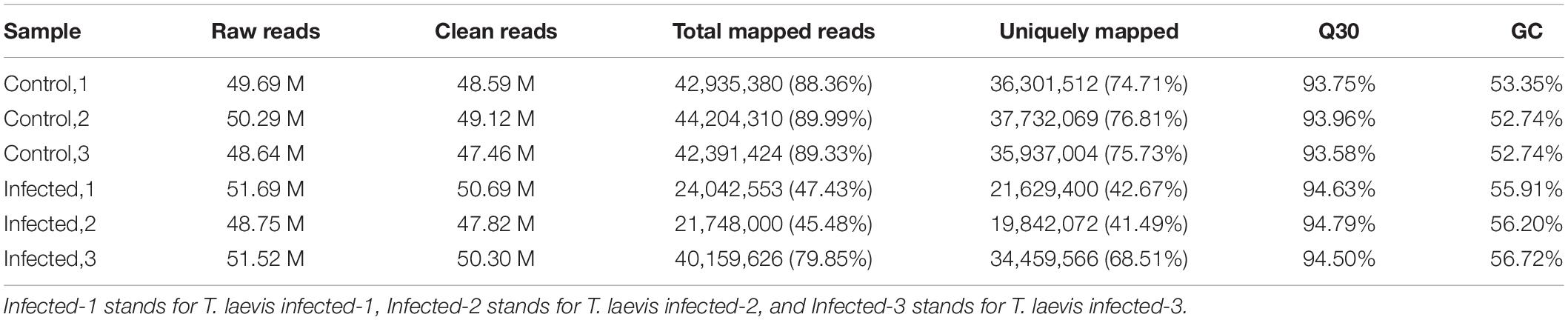
Table 1. Transcriptome analysis of RNA-Seq data.
Verification of Differentially Expressed Genes by Quantitative Real-Time PCR
First-strand cDNA was synthesized by using 1.5 μg purified RNA by using the Trans Script One-Step gDNA Removal and cDNA Synthesis SuperMix Kit (Beijing Quanshijin Biotechnology Co., Ltd., China) and stored at -20°C for further use. A PerfectStart Green qPCR SuperMix (TransGen Biotech, Beijing, China) kit was used for quantitative real-time polymerase chain reaction (qRT-PCR). The 20-μl reaction mixtures contained 1 μl template, 0.4 of each primer (10 μM), 10 μl SuperMix (Beijing Quanshijin Biotechnology Co., Ltd., China) and 8.2 μl ddH2O. The amplification protocol was 94°C for 30 s, followed by 40 cycles of 94°C for 5 s, 60°C for 30 s, and 72°C for 30 s. All cDNA and qRT-PCRs were carried out in triplicate. The 2–ΔΔCt method was used to calculate the relative expression of each gene (Pfaffl, 2001). The primers used for validation are listed in Supplementary Table 1.
Results
Transcriptomic Analysis of RNA-Seq Data
Based on RNA-Seq, we identified the alterations in wheat genes when the spike was infected by T. laevis. Six cDNA libraries (three T. laevis-infected and three control) were sequenced. Approximately 49.69, 50.29, and 48.64 million raw reads were produced from the control samples control-1, control-2, and control-3, respectively, and raw reads in T. laevis samples were 51.69, 48.75, and 51.52 million in infected samples, respectively. Similarly, 48.59, 49.12, and 47.46 million clean reads were produced from control samples control-1, control-2, and control-3, respectively, and clean reads in T. laevis samples were 50.69, 47.82, and 50.30 million in infected samples, respectively. Total mapped reads and uniquely mapped reads of the above-mentioned transcripts ranged from 45.48 to 89.99% and 41.49 to 76.81%, respectively. Additionally, the Q30 and GC of these transcripts ranged from 93.58 to 94.79% and 52.74 to 56.72%, respectively (Supplementary Table 1). Next, the differentially expressed genes (DEGs) were recognized by comparing the fragments per kilobase of transcript per million mapped reads (FPKM) value of every gene between control and T. laevis-infected samples. For control-1 DEGs, 5,402 (FPKM ≥ 10), 28,967 (FPKM 1–10), 9,525 (FPKM 0.5–1), and 64,011 (FPKM 0–0.5) genes were differentially expressed. For T. laevis-infected DEGs, 6,732 (FPKM ≥ 10), 18,573 (FPKM 1–10), 6,006 (FPKM 0.5–1), and 76,234 (FPKM 0–0.5) genes were differentially expressed. The DEGs of T. laevis-infected were showed (Figure 1). In addition, the sample-to-sample cluster analysis showed that the repeatability between each group of sequencing samples was high, indicating that the repeatability and reliability of the transcriptome sequencing data meet the needs of further analysis (Figure 2). Furthermore, principal component analysis (PCA) was performed for both control- and T. laevis-infected samples. The PCA of the samples of the infection group and the control group found that the control group was concentrated in the first and third quadrants, and the infection group was concentrated mainly in the second and fourth quadrants, indicating that there were differences and good repeatability within each group (Figure 3).
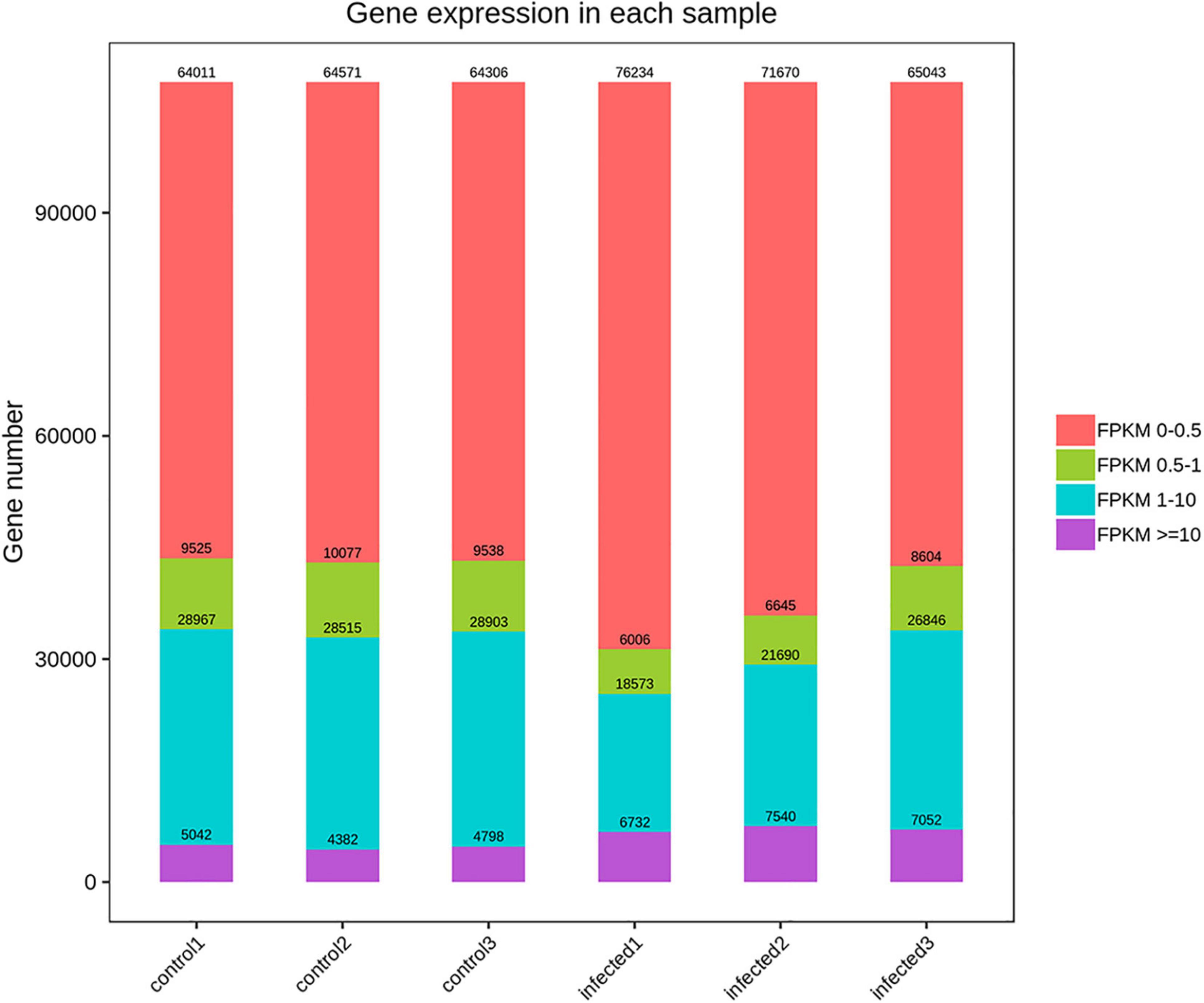
Figure 1. Summary of differentially expressed genes (DEGs). Numbers of DEGs between the control and T. laevis infection groups.
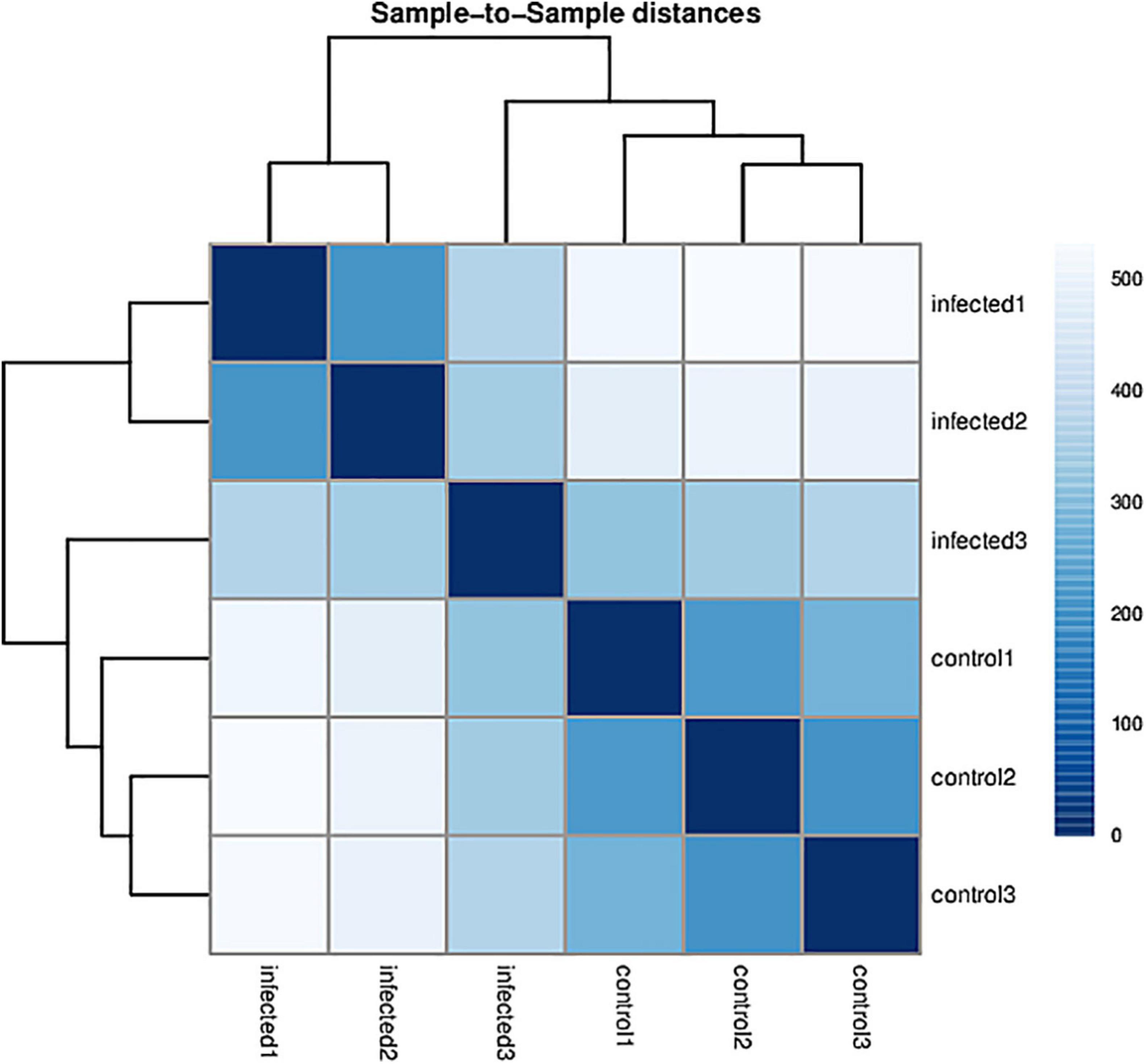
Figure 2. Sample-to-sample clustering analysis for checking batch effects and their similarity.
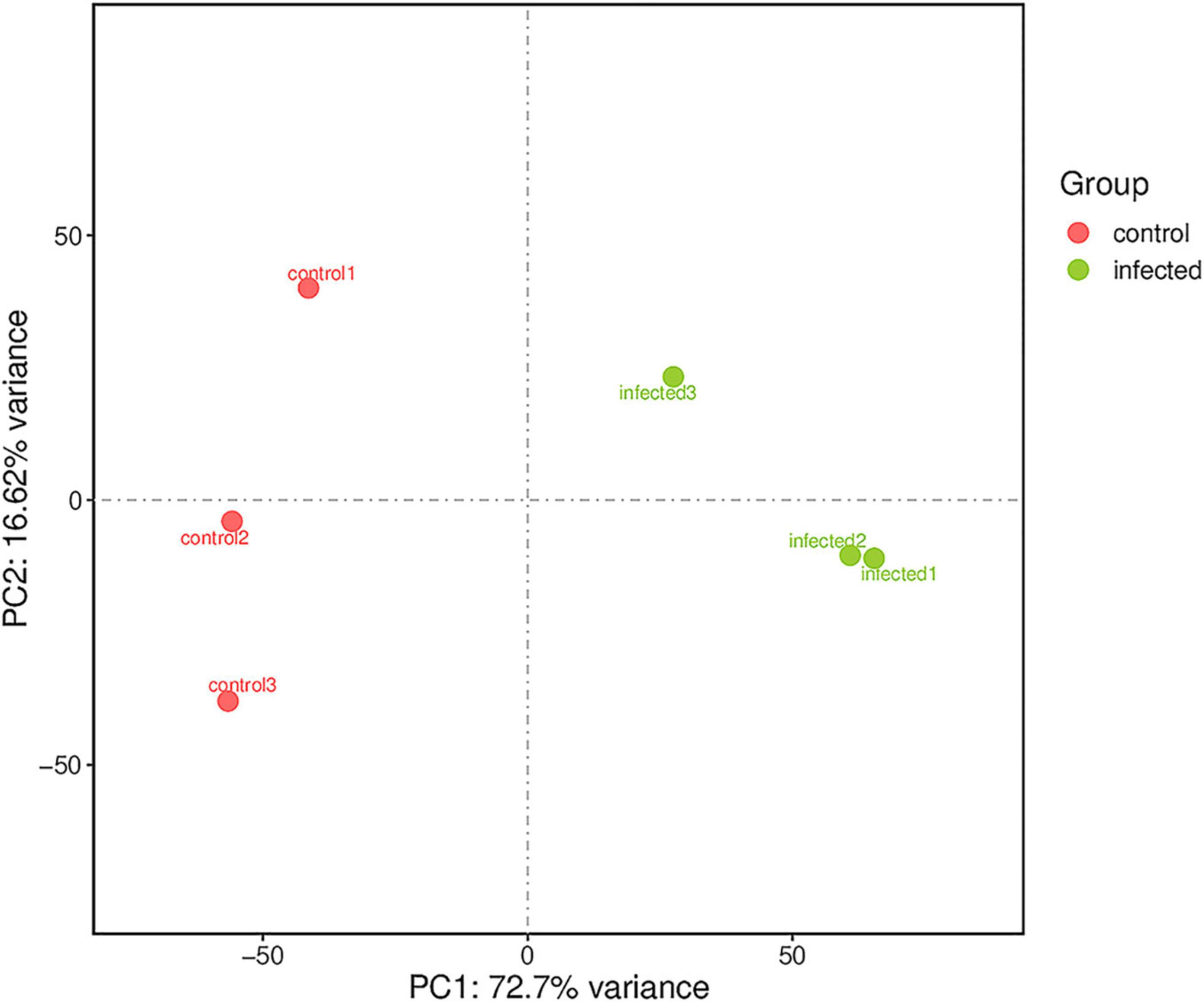
Figure 3. Principal component analysis (PCA) for gene expression patterns. The first and second PCAs explained 72.7 and 16.62% of the variance, respectively.
Analysis of Differential Gene Expression Patterns
The differentially expressed genes were recognized in T. laevis-infected and control libraries. By comparing the tassels infected with T. laevis and the control, 49.6% genes (7,767 genes out of 15,658 genes) were found to be upregulated and 50.4% genes (7,891 genes out of 15,658 genes) were found to be downregulated (Figure 4 and Supplementary Table 2). In further analysis of the differential expression of T. laevis-infected and control samples by using cluster expression analysis, the results showed that there were 0.8% more downregulated genes than upregulated genes in T. foetida plants (Figure 5).
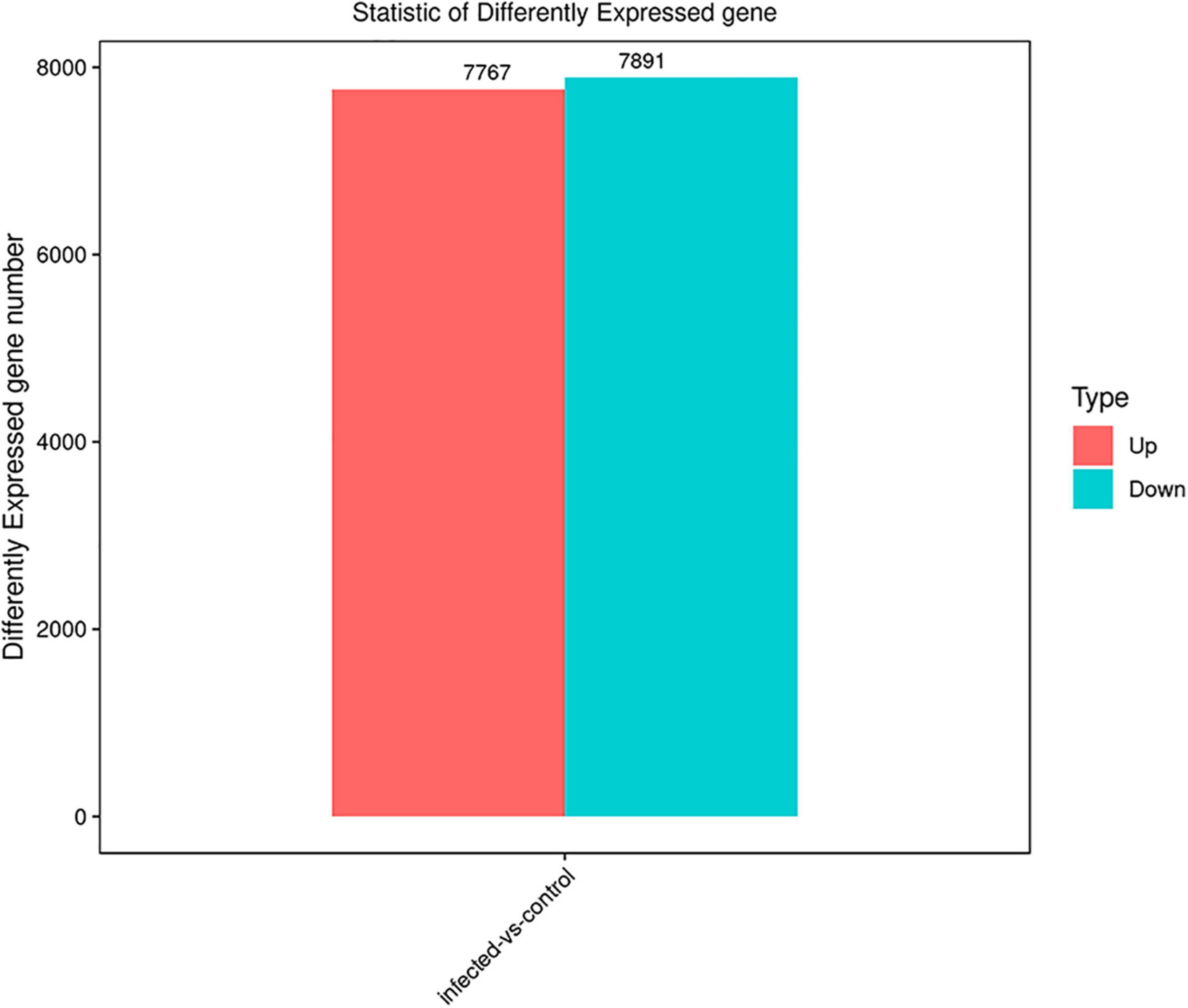
Figure 4. Significant differentially expressed genes (DEGs) in T. laevis-infected vs. control samples. Up- or downregulated DEGs in response to T. laevis infection.
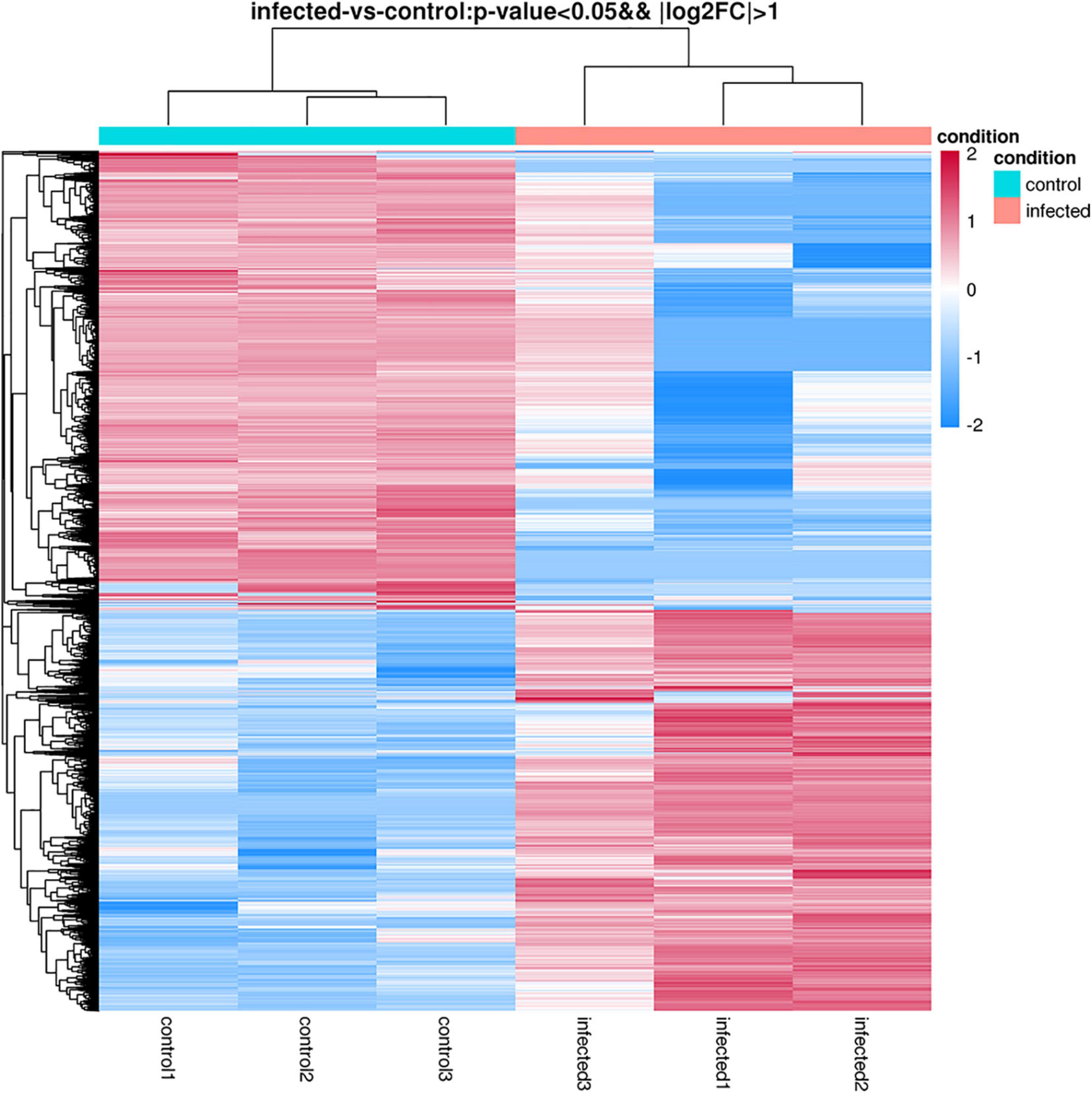
Figure 5. Hierarchical clustering heatmap of DEGs according to changes in expression in response to T. laevis infection. Each column shows a library, and each row shows a DEG expression. The colors blue, white, and red indicate low, medium, and high expression patterns of genes, respectively.
Gene Ontology Enrichment Analysis of Differentially Expressed Genes
The GO categories were developed to evaluate potential DEG functions. The DEGs were classified into 48 functional categories, including biological process (21), cellular component (13), and molecular function (14). The results showed that in the biological process category, GO was associated mainly with cellular component organization, establishment of localization, localization, metabolic process, regulation of biological process, response to stimulus, and single organism process. Meanwhile, in the cellular component category, DEGs were primarily associated with cells, cell parts, membranes, membrane parts, and organelles. Similarly, in the molecular function category, DEGs were associated primarily with binding, catalytic activity, nucleic acid binding transcription factor activity, and transporter activity (Figure 6). In addition, KEGG pathway mapping was also carried out, and the results showed that 200 different pathways were identified (Supplementary Table 3). Of these pathways, 20 were peroxisome, regulation of autophagy, plant hormone signal transduction, FoxO signaling pathway, etc. (Figure 7).
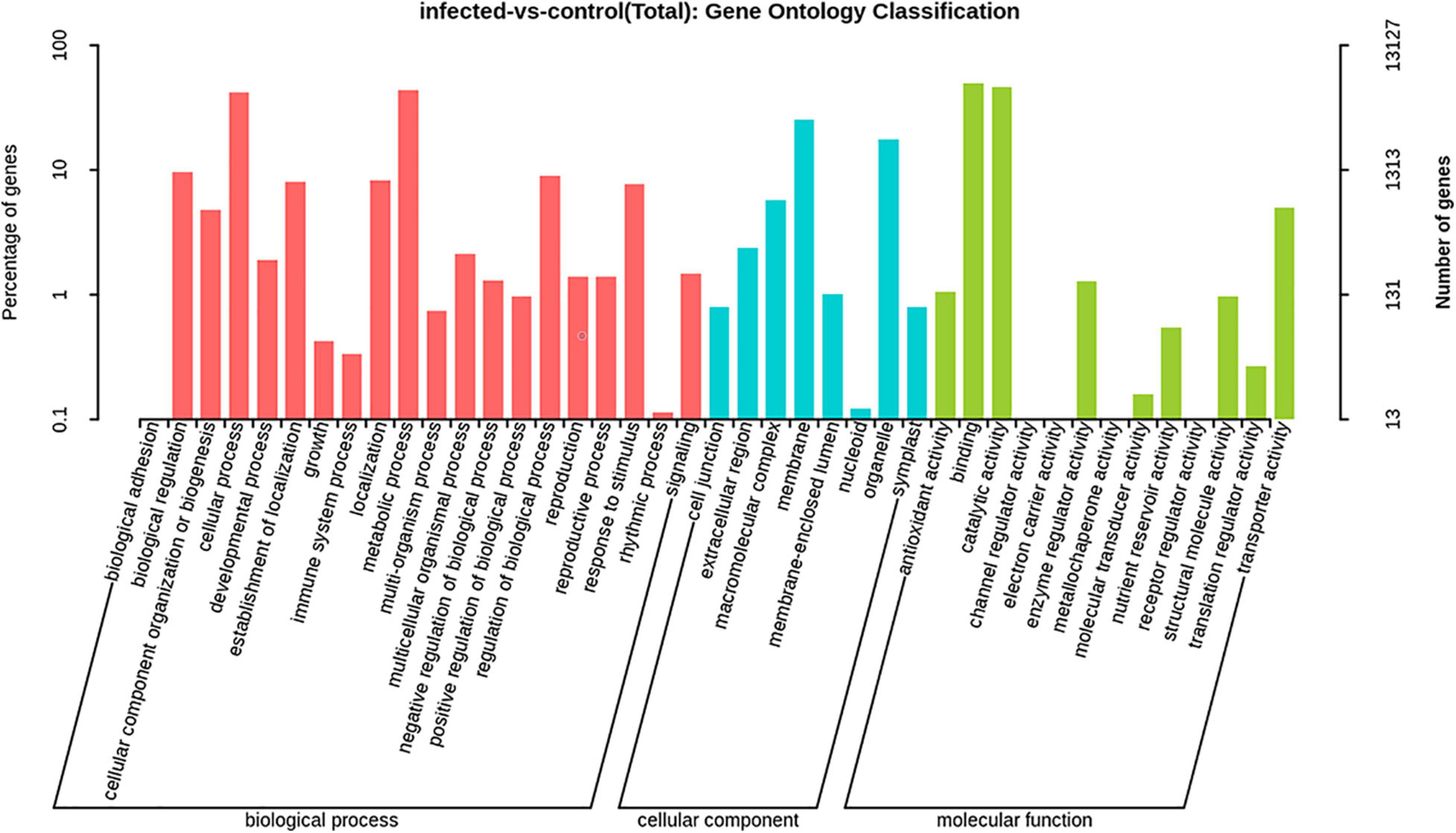
Figure 6. Gene ontology (GO) enrichment analysis of significant DEGs of T. laevis-infected and control samples. Annotations are grouped by biological process, cellular component, and molecular function.
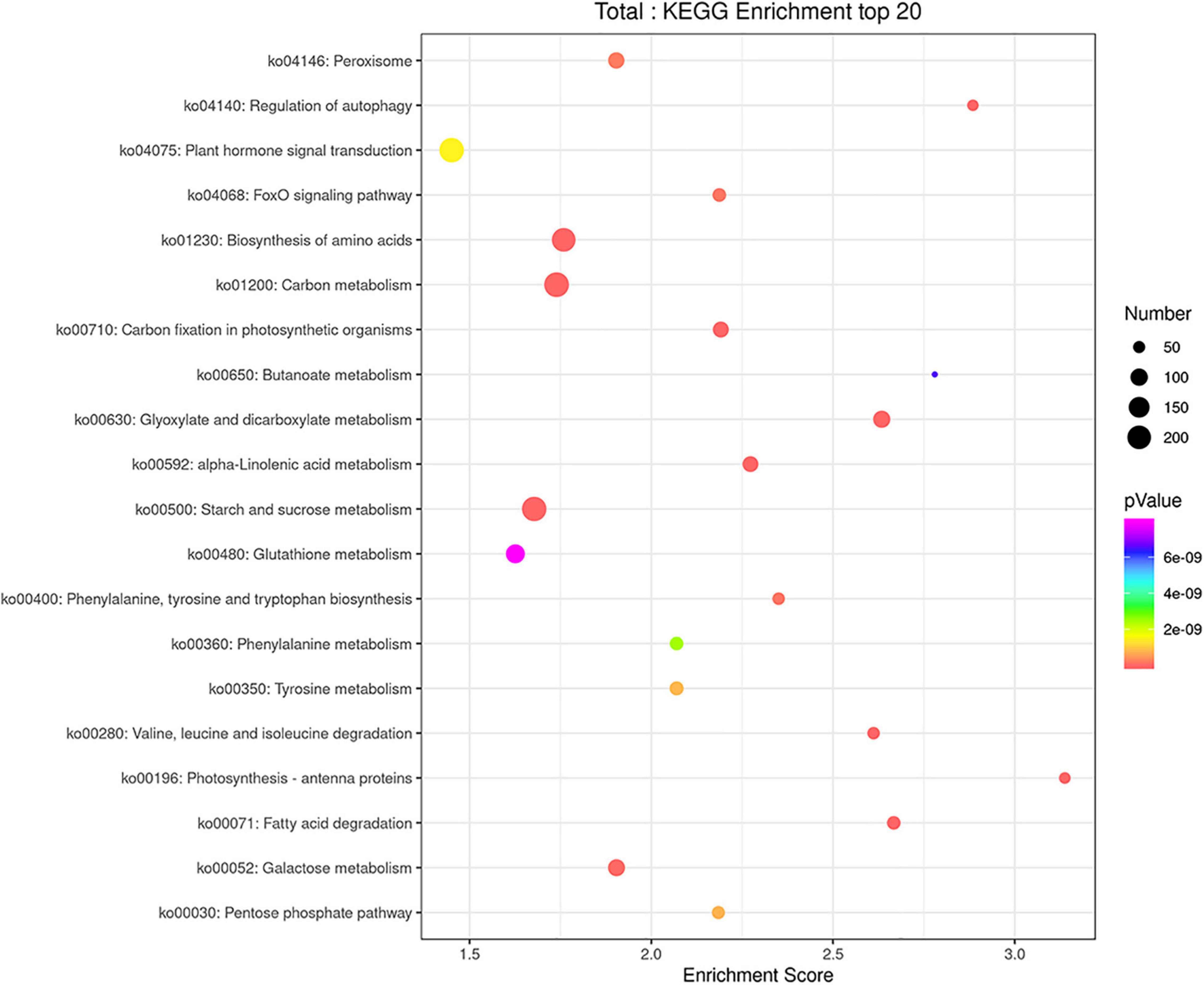
Figure 7. KEGG enrichment analysis scatter plot representing pathways of DEGs in response to T. laevis infection. The colors blue, white and red indicate low, medium, and high expression patterns of genes, respectively.
Differential Expression of Defense-Related Myeloblastosis Transcription Factor After Tilletia laevis Infection
Our results showed that after T. laevis infection, the expression of pathogenesis-related (PR) genes changed. The results showed that 6 pathogenesis-related genes, 3 thaumatin-like protein, 3 chitinase, 86 peroxidase, and 15 glucanase genes, changed during T. laevis infection, and their expression is shown in Supplementary Table 4. The expression of MYB transcription factors was also changed after T. laevis infection. The results showed that 10 of 14 and 4 of 14 MYB transcription factors were upregulated and downregulated, respectively (Supplementary Table 5).
Validation of RNA Sequencing Results by Quantitative Real-Time PCR
To verify the reliability of the obtained transcriptome data, we screened out some of the differentially expressed genes for qRT-PCR verification. Taking the stress resistance of tassels infected by T. laevis as the screening standard, 9 differential genes were selected for quantitative verification. The expression trends in the qRT-PCR results validated the transcription sequencing results, which indicated a high degree of reproducibility between transcript abundances assayed using RNA-Seq and the expression profiles revealed by qRT-PCR data (Figure 8 and Supplementary Table 6).
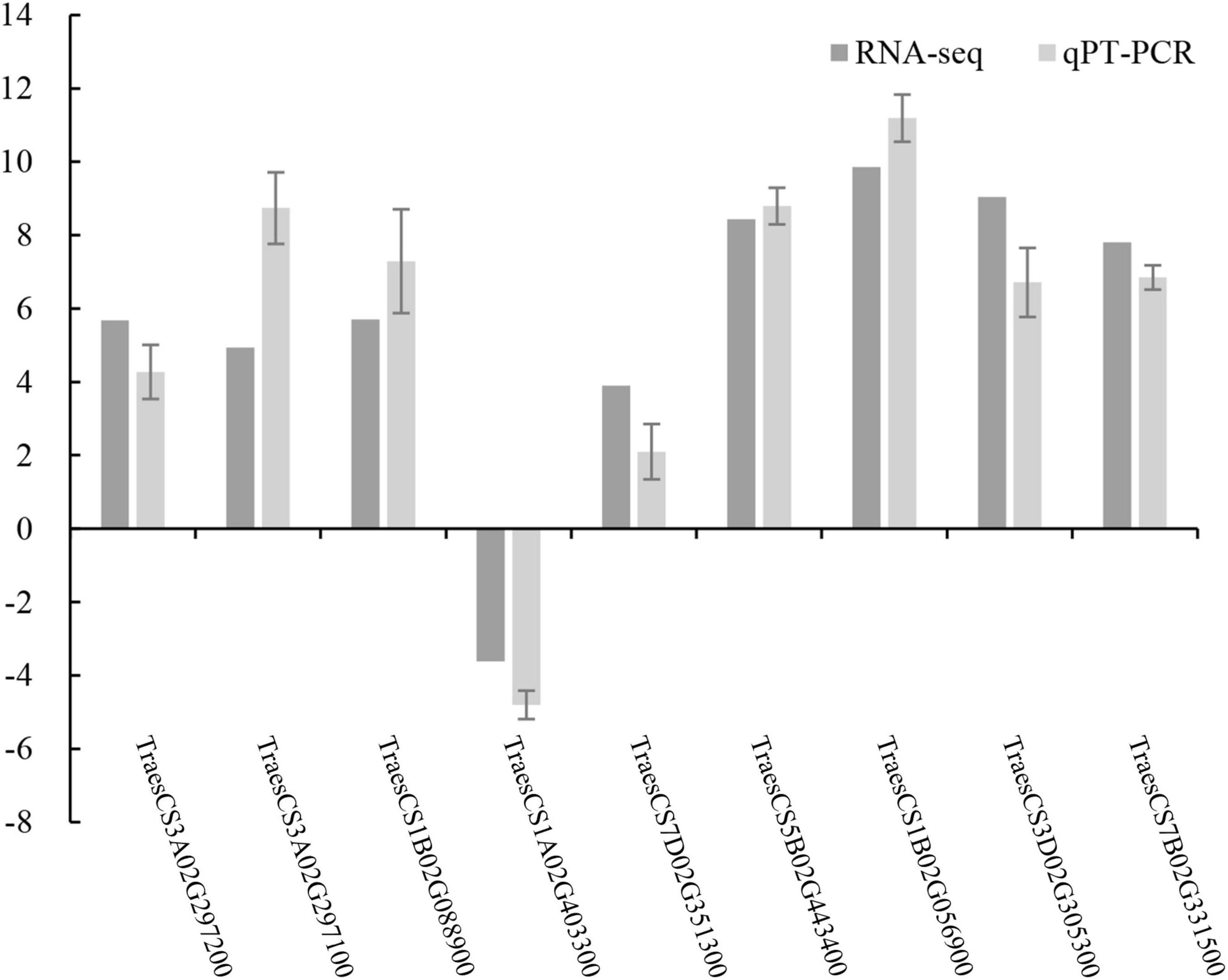
Figure 8. Validation of RNA-Seq data by quantitative real-time PCR (qRT-PCR).
Discussion
Common bunt is one of the most serious diseases of wheat caused by T. laevis, and common grains are partly or totally replaced by fugus gulls, leading to 80% of wheat yield loss. To date, due to a lack of information on interactions between wheat tassels and T. laevis at the transcript level, therefore, it is essential to explore plant-pathogen interactions in the reproductive organs (tassels) of wheat. To determine plant responses to abiotic and biotic stresses, a large-scale gene expression analysis was performed (Desmond et al., 2008). Gene expression levels upon infection by pathogens have been broadly investigated in various crops using RNA-Seq, such as in wheat crops against T. controversa (Ren et al., 2020), F. graminearum (Erayman et al., 2015) and obligate pathogens (Poretti et al., 2021). In this study, based on RNA-Seq, we used the highly susceptible cultivar “Dongxuan 3” with T. laevis to analyze the response of the gene expression levels in fungal infected plants. Our findings showed that a different set of defense genes was differentially and specifically expressed and that a series of signaling molecules in wheat tassels were triggered by T. laevis.
Pathogenesis-related genes have key roles in the plant defense system against fungal pathogen infection (Sels et al., 2008). Based on the literature, thaumatin, chitinase, peroxidase, glucanase, and PR-10 enhance resistance to T. controversa and F. pseudograminearum in wheat (Desmond et al., 2008; Muhae-Ud-Din et al., 2020b; Chen et al., 2021), and Ustilaginoidea virens in rice (Han et al., 2015). Consistent with our findings, PR genes in wheat have been induced by T. controversa (Ren et al., 2020). We compared the transcript levels between T. laevis-inoculated and control plants in the tassels. The expression of 113 defense-related genes was changed after T. laevis inoculation (Supplementary Table 5), including 6 PRs, 3 thaumatin, 3 chitinase, 86 peroxidase, and 15 glucanase. Myeloblastosis (MYB) transcription factors form one of the largest protein superfamilies in plants and are involved in diverse biological processes, including cell wall biosynthesis, cell cycle regulation, reproduction and development, and play a significant role in controlling the transcription of defense-related genes (Millar and Gubler, 2005; Dubos et al., 2010; Zhang et al., 2015; Shan et al., 2016). For example, the R2R3-MYB transcription factor has increased the resistance in Arabidopsis against Alternaria brassicicola and Botrytis cinerea and was also involved in the tolerance of oxidative and osmotic stresses (Mengiste et al., 2003). The HvMYB6 improves the immunity level in barley against Blumeria graminis (Chang et al., 2013). Overexpression of the MYB gene TaPIMP1 induced a resistance in wheat against Bipolaris sorokiniana (Zhang et al., 2012). Our results were consistent with the above studies and showed that 14 MYB transcription factors were altered after T. laevis inoculation: 10 were upregulated and 4 were downregulated (Supplementary Table 5). The above changes in the transcription levels of different gene families might play a role in disease suppression against T. laevis.
Upon plant pathogen infection, plant defense responses contain transcriptional regulation of a large number of plant host genes. An RNA-Seq profiling suggests that plants activate different sets of defense-related genes to overcome severity of the disease in various crops (Conrath, 2011; Ren et al., 2020). Some defense-related proteins, such as thaumatin, PR proteins, chitinase, peroxidase, and glucanase, are capable of destroying the cell wall components of fungal pathogens and boosting the defense response of plants (Chen and Chen, 2002). In Tilletia species-infected wheat crops, the genes encoding chitinase, lipase, PR1.1, PR1.2, defensins and PR-10 were previously reported to be highly expressed in fungi-infected plants compared to control plants at different time intervals (Muhae-Ud-Din et al., 2020a). Desmond et al. (2008) reported that the expression of PR-1, β-1,3-glucanase, thaumatin-like proteins, and PR-10 was induced significantly in wheat crops after F. pseudograminearum infection. Similarly, the expression of peroxidase involved in reactive oxygen species (ROS) was induced after F. pseudograminearum infection (Kawano, 2003; Desmond et al., 2008). Transcription factors play roles in activating primary response genes after pathogen infection (Herschman, 1991; Li et al., 2016). Therefore, transcriptional regulation of plant genes is a part of the plant defense mechanism and plays an important role in inducing plant disease resistance (Chen and Chen, 2002). Similarly, in plant–pathogen interactions, pathogens take nutrients from plants, and plant cells try to stop nutrient movements by altering carbon metabolism and transport (Chen et al., 2011; Kretschmer et al., 2017; Kanwar and Jha, 2019). Some genes regulated the phytosynthetic activity repressed by affecting the photosynthetic activity (Rolfe and Scholes, 2010; Windram et al., 2012; Smith et al., 2014). In the present study, the transcription levels of 113 defense-related genes were changed after the T. laevis infection, including 6 PR genes, 3 thaumatin-like proteins, 3 chitinases, 86 peroxidases, and 15 glucanases (Supplementary Table 4). The 44 PRS genes were upregulated, and 69 PRs were downregulated. The above changes in the transcription levels of different gene families might play a role in the suppression of disease against T. laevis.
The GO enrichment analysis revealed that in the “biological process” category, cellular process, metabolic process, and single-organism process; in the “cellular component” category, cell, cell part, membrane, and membrane part; and in the “molecular function” category, binding and catalytic activity had the highest number of DEGs during plant pathogen interactions (Figure 6). These results indicated that the pathogens mobilized the primary and secondary metabolisms and finally regulated the expression of related genes through signal transduction and ion transport, which clearly induced the immune defense responses and may have a role in disease suppression. After pathogen infection, plants spend more energy in signaling and transportation processes to defend themselves than morph physiological and reproductive processes (Siemens et al., 2006; Berger et al., 2007). Furthermore, KEGG pathway analysis showed that most DEGs were characterized by biosynthesis of amino acids, carbon metabolism, and starch and sucrose metabolism (Figure 7). Amino acids have key roles in plant species, such as nitrogen sources, stress-reducing agents, hormone precursors (Zhao, 2010; Maeda and Dudareva, 2012) and as signaling factors of various physiological processes in plant species (Teixeira et al., 2018). Sucrose is an important element of assimilated carbon, which is a by-product during photosynthesis and then transported from source to sink tissues via the vascular system (Tauzin and Giardina, 2014). Overall, our findings showed that defense-related genes, including PR genes and MYB transcription, were mostly upregulated after T. laevis infection, suggesting that these gene families play important roles in common bunt resistance in wheat and may contribute to the control of wheat common bunt by regulating the over-expression defense genes.
Data Availability Statement
The data presented in the study and deposited in the Supplementary Material.
Author Contributions
LG designed the experiments. TH and ZR performed the experiments. GM and LG analyzed the data. TH, GM-U-D, and LG wrote the manuscript. WC, TL, and QG provided some materials. All authors reviewed the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31761143011) and the Ministry of Agriculture (CARS-03).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.823907/full#supplementary-material
Supplementary Figure 1 | Morphological characterization of teliospores of T. laevis.
Supplementary Figure 2 | Specific band and sequence of SCAR marker of T. laevis.
Footnotes
References
Bairoch, A., and Apweiler, R. (1997). The SWISS-PROT protein sequence data bank and its supplement TrEMBL. Nucleic Acids Res. 25, 31–36. doi: 10.1093/nar/25.1.31
Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S. R., et al. (2002). The Pfam protein families database. Nucleic Acids Res. 30, D138–D141. doi: 10.1093/nar/30.1.276
Berger, S., Sinha, A. K., and Roitsch, T. (2007). Plant physiology meets phytopathology: plant primary metabolism and plant–pathogen interactions. J. Exp. Bot. 58, 4019–4026. doi: 10.1093/jxb/erm298
Bokore, F. E., Cuthbert, R. D., Knox, R. E., Singh, A., Campbell, H. L., Pozniak, C. J., et al. (2019). Mapping quantitative trait loci associated with common bunt resistance in a spring wheat (Triticum aestivum L.) variety Lillian. Theor. Appl. Genet. 132, 3023–3033. doi: 10.1007/s00122-019-03403-3
Chang, C., Yu, D., Jiao, J., Jing, S., Schulze-Lefert, P., and Shen, Q. H. (2013). Barley MLA immune receptors directly interfere with antagonistically acting transcription factors to initiate disease resistance signaling. Plant Cell 25, 1158–1173. doi: 10.1105/tpc.113.109942
Chen, C., and Chen, Z. (2002). Potentiation of developmentally regulated plant defense response by AtWRKY18, a pathogen-induced arabidopsis transcription factor. Plant Physiol. 129, 706–716. doi: 10.1104/pp.001057
Chen, D., Muhae-Ud-din, G., Liu, T., Chen, W., Liu, C., and Gao, L. (2021). Wheat varietal response to Tilletia controversa J. G. Kühn using qRT-PCR and laser confocal microscopy. Genes 12:425. doi: 10.3390/genes12030425
Chen, L. Q., Hou, B. H., Lalonde, S., Takanaga, H., Hartung, M. L., Qu, X. Q., et al. (2011). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532. doi: 10.1038/nature09606.Sugar
Cohn, J., Sessa, G., and Martin, G. B. (2001). Innate immunity in plants. Curr. Opin. Immunol. 13, 55–62. doi: 10.1126/science.1171647
Conrath, U. (2011). Molecular aspects of defence priming. Trends Plant Sci. 16, 524–531. doi: 10.1016/j.tplants.2011.06.004
Desmond, O. J., Manners, J. M., Schenk, P. M., Maclean, D. J., and Kazan, K. (2008). Gene expression analysis of the wheat response to infection by Fusarium pseudograminearum. Physiol. Mol. Plant Pathol. 73, 40–47. doi: 10.1016/j.pmpp.2008.12.001
Din, G. M. U., Du, Z., Zhang, H., Zhao, S., Liu, T., Chen, W., et al. (2021). Effects of Tilletia foetida on microbial communities in the rhizosphere soil of wheat seeds coated with different concentrations of Jianzhuang. Microb. Ecol. 82, 736–745. doi: 10.1007/s00248-021-01696-w
Du, H., Wang, Y., Yang, J., and Yang, W. (2015). Comparative transcriptome analysis of resistant and susceptible tomato lines in response to infection by Xanthomonas perforans Race T3. Front. Plant Sci. 6:1173. doi: 10.3389/fpls.2015.01173
Dubos, C., Stracke, R., Grotewold, E., Weisshaar, B., Martin, C., and Lepiniec, L. (2010). MYB transcription factors in Arabidopsis. Trends Plant Sci. 15, 573–581. doi: 10.1016/j.tplants.2010.06.005
Dumalasová, V., and Bartoš, P. (2010). Reaction of wheat, alternative wheat and triticale cultivars to common bunt, Czech. J. Genet. Plant Breed. 46, 14–20. doi: 10.17221/73/2009-cjgpb
Erayman, M., Turktas, M., Akdogan, G., Gurkok, T., Inal, B., Ishakoglu, E., et al. (2015). Transcriptome analysis of wheat inoculated with Fusarium graminearum. Front. Plant Sci. 6:867. doi: 10.3389/fpls.2015.00867
Goates, B. J. (2012). Identification of new pathogenic races of common bunt and dwarf bunt fungi, and evaluation of known races using an expanded set of differential wheat lines. Plant Dis. 96, 361–369.
Goates, B. J., and Peterson, G. L. (1999). Relationship between soilborne and seedborne inoculum density and the incidence of dwarf bunt of wheat. Plant Dis. 83, 819–824.
Han, Y., Zhang, K., Yang, J., Zhang, N., Fang, A., Zhang, Y., et al. (2015). Differential expression profiling of the early response to Ustilaginoidea virens between false smut resistant and susceptible rice varieties. BMC Genome 16:955. doi: 10.1186/s12864-015-2193-x
Haueisen, J., Möller, M., Eschenbrenner, C. J., Grandaubert, J., Seybold, H., Adamiak, H., et al. (2019). Highly flexible infection programs in a specialized wheat pathogen. Ecol. Evol. 9, 275–294. doi: 10.1002/ece3.4724
Herschman, H. R. (1991). Primiry response genes induced by growth factors and tumor promoters. Annu. Rev. Biochem. 60, 281–319.
Hosseini, S., Elfstrand, M., Heyman, F., Jensen, D. F., and Karlsson, M. (2015). Deciphering common and specific transcriptional immune responses in pea towards the oomycete pathogens Aphanomyces euteiches and Phytophthora pisi. BMC Genomics 16:627. doi: 10.1186/s12864-015-1829-1
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. doi: 10.1093/nar/gkm882
Kanwar, P., and Jha, G. (2019). Alterations in plant sugar metabolism: signatory of pathogen attack. Planta 249, 305–318. doi: 10.1007/s00425-018-3018-3
Kawano, T. (2003). Roles of the reactive oxygen species-generating peroxidase reactions in plant defense and growth induction. Plant Cell Rep. 21, 829–837. doi: 10.1007/s00299-003-0591-z
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kong, L., Wu, D., Huang, W., Peng, H., Wang, G., Cui, J., et al. (2015). Large-scale identification of wheat genes resistant to cereal cyst nematode Heterodera avenae using comparative transcriptomic analysis. BMC Genomics 16:801. doi: 10.1186/s12864-015-2037-8
Kretschmer, M., Croll, D., and Kronstad, J. W. (2017). Maize susceptibility to Ustilago maydis is influenced by genetic and chemical perturbation of carbohydrate allocation. Mol. Plant Pathol. 18, 1222–1237. doi: 10.1111/mpp.12486
Lanubile, A., Muppirala, U. K., Severin, A. J., Marocco, A., and Munkvold, G. P. (2015). Transcriptome profiling of soybean (Glycine max) roots challenged with pathogenic and non-pathogenic isolates of Fusarium oxysporum. BMC Genomics 16:1089. doi: 10.1186/s12864-015-2318-2
Li, B., Meng, X., He, L. S., and He, P. (2016). Transcriptional regulation of pattern-triggered immunity in plants. Physiol. Behav. 176, 100–106. doi: 10.1016/j.chom.2016.04.011
Li, C., Shao, J., Wang, Y., Li, W., Guo, D., Yan, B., et al. (2013). Analysis of banana transcriptome and global gene expression profiles in banana roots in response to infection by race 1 and tropical race 4 of Fusarium oxysporum f. sp. cubense. BMC Genomics 14:851. doi: 10.1186/1471-2164-14-851
Liu, F., Wu, J. B., Zhan, R. L., and Ou, X. C. (2016). Transcription profiling analysis of mango–Fusarium mangiferae interaction. Front. Microbiol. 7:1443. doi: 10.3389/fmicb.2016.01443
Lu, Z. X., Gaudet, D. A., Frick, M., Puchalski, B., Genswein, B., and Laroche, A. (2005). Identification and characterization of genes differentially expressed in the resistance reaction in wheat infected with Tilletia tritici, the common bunt pathogen. J. Biochem. Mol. Biol. 38, 420–431. doi: 10.5483/bmbrep.2005.38.4.420
Maeda, H., and Dudareva, N. (2012). The shikimate pathway and aromatic amino acid biosynthesis in plants. Annu. Rev. Plant Biol. 63, 73–105. doi: 10.1146/annurev-arplant-042811-105439
Matanguihan, J. B., Murphy, K. M., and Jones, S. S. (2011). Control of common bunt in organic wheat. Plant Dis. 95, 92–103.
Mathioni, S. M., Beló, A., Rizzo, C. J., Dean, R. A., and Donofrio, N. M. (2011). Transcriptome profiling of the rice blast fungus during invasive plant infection and in vitro stresses. BMC Genomics 12:49. doi: 10.1186/1471-2164-12-49
Mengiste, T., Chen, X., Salmeron, J., and Dietrich, R. (2003). The BOTRYTIS SUSCEPTIBLE1 gene encodes an R2R3MYB transcription factor protein that is required for biotic and abiotic stress responses in Arabidopsis. Plant Cell 15, 2551–2565.
Millar, A. A., and Gubler, F. (2005). The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell 17, 705–721. doi: 10.1105/tpc.104.027920
Muhae-Ud-Din, G., Chen, D., Liu, T., Chen, W., and Gao, L. (2020a). Characterization of the wheat cultivars against Tilletia controversa Kühn, causal agent of wheat dwarf bunt. Sci. Rep. 10:9029. doi: 10.1038/s41598-020-65748-w
Muhae-Ud-Din, G., Chen, D., Liu, T., Chen, W., and Li, G. (2020b). Methyljasmonate and salicylic acid contribute to the control of Tilletia controversa Kühn, causal agent of wheat dwarf bunt. Sci. Rep. 10:19175. doi: 10.1038/s41598-020-76210-2
Mukherjee, A. K., Carp, M. J., Zuchman, R., Ziv, T., Horwitz, B. A., and Gepstein, S. (2010). Proteomics of the response of Arabidopsis thaliana to infection with Alternaria brassicicola. J. Proteomics 73, 709–720. doi: 10.1016/j.jprot.2009.10.005
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 29, e45–e45.
Poretti, M., Sotiropoulos, A. G., Graf, J., Jung, E., Bourras, S., Krattinger, S. G., et al. (2021). Comparative transcriptome analysis of wheat lines in the field reveals multiple essential biochemical pathways suppressed by obligate pathogens. Front. Plant Sci. 12:1981. doi: 10.3389/fpls.2021.720462
Proietti, S., Bertini, L., Timperio, A. M., Zolla, L., Caporale, C., and Caruso, C. (2013). Crosstalk between salicylic acid and jasmonate in Arabidopsis investigated by an integrated proteomic and transcriptomic approach. Mol. Biosyst. 9, 1169–1187.
Qin, D. D., Xu, T. S., Liu, T. G., Chen, W. Q., and Gao, L. (2021). First report of wheat common bunt caused by Tilletia laevis in Henan Province, China. Plant Dis. 105:215.
Ren, Z., Liu, J., Din, G. M. U., Zhang, H., Du, Z., Chen, W., et al. (2020). Transcriptome analysis of wheat spikes in response to Tilletia controversa Kühn which cause wheat dwarf bunt. Sci. Rep. 10:21567. doi: 10.1038/s41598-020-78628-0
Rolfe, S. A., and Scholes, J. D. (2010). Chlorophyll fluorescence imaging of plant–pathogen interactions. Protoplasma 247, 163–175. doi: 10.1007/s00709-010-0203-z
Sels, J., Mathys, J., De Coninck, B. M. A., Cammue, B. P. A., and De Bolle, M. F. C. (2008). Plant pathogenesis-related (PR) proteins: a focus on PR peptides. Plant Physiol. Biochem. 46, 941–950. doi: 10.1016/j.plaphy.2008.06.011
Shan, T., Rong, W., Xu, H., Du, L., Liu, X., and Zhang, Z. (2016). The wheat R2R3-MYB transcription factor TaRIM1 participates in resistance response against the pathogen Rhizoctonia cerealis infection through regulating defense genes. Sci. Rep. 6:28777. doi: 10.1038/srep28777
Siemens, J., Keller, I., Sarx, J., Kunz, S., Schuller, A., Nagel, W., et al. (2006). Transcriptome analysis of Arabidopsis clubroots indicate a key role for cytokinins in disease development. Mol. Plant Microbe Interact. 19, 480–494. doi: 10.1094/MPMI-19-0480
Smith, J. E., Mengesha, B., Tang, H., Mengiste, T., and Bluhm, B. H. (2014). Resistance to Botrytis cinerea in Solanum lycopersicoides involves widespread transcriptional reprogramming. BMC Genomics 15:334. doi: 10.1186/1471-2164-15-334
Tatusov, R. L., Galperin, M. Y., Natale, D. A., and Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. doi: 10.1093/nar/28.1.33
Tauzin, A. S., and Giardina, T. (2014). Sucrose and invertases, a part of the plant defense response to the biotic stresses. Front. Plant Sci. 5:293. doi: 10.3389/fpls.2014.00293
Teixeira, W. F., Fagan, E. B., Soares, L. H., Soares, J. N., Reichardt, K., and Neto, D. D. (2018). Seed and foliar application of amino acids improve variables of nitrogen metabolism and productivity in soybean crop. Front. Plant Sci. 9:396. doi: 10.3389/fpls.2018.00396
Thilmony, R., Underwood, W., and He, S. Y. (2006). Genome-wide transcriptional analysis of the Arabidopsis thaliana interaction with the plant pathogen Pseudomonas syringae pv. tomato DC3000 and the human pathogen Escherichia coli O157:H7. Plant J. 46, 34–53. doi: 10.1111/j.1365-313X.2006.02725.x
Windram, O., Madhou, P., Mchattie, S., Hill, C., Cooke, E., Jenkins, D. J., et al. (2012). Arabidopsis defense against Botrytis cinerea: chronology and regulation deciphered by high-resolution temporal transcriptomic analysis. Plant Cell 24, 3530–3557.
Xu, T. S., Qin, D. D. Muhae-Ud-Din, G., Liu, T. G., Chen, W. Q., and Gao, L. (2021). Characterization of histological changes at the tillering stage (Z21) in resistant and susceptible wheat plants infected by Tilletia controversa Kühn. BMC Plant Biol. 21:49.
Xu, T. S., Yao, Z. Q., Liu, J. J., Zhang, H., Muhae-Ud-Din, G., Zhao, S. F., et al. (2020). Development of droplet digital PCR for the detection of Tilletia laevis, which causes common bunt of wheat, based on the SCAR marker derived from ISSR and real-time PCR. Sci. Rep. 10:16106.
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, 1–12.
Zhang, Z., Chen, J., Su, Y., Liu, H., Chen, Y., Luo, P., et al. (2015). TaLHY, a 1R-MYB transcription factor, plays an important role in disease resistance against stripe rust fungus and ear heading in wheat. PLoS One 10:e0127723. doi: 10.1371/journal.pone.0127723
Zhang, Z., Liu, X., Wang, X., Zhou, M., Zhou, X., Ye, X., et al. (2012). An R2R3 MYB transcription factor in wheat, Ta PIMP 1, mediates host resistance to Bipolaris sorokiniana and drought stresses through regulation of defense-and stress-related genes. New Phytol. 196, 1155–1170.
Zhao, Y. (2010). Auxin biosynthesis and its role in plant development. Annu. Rev. Plant Biol. 61, 49–64. doi: 10.1146/annurev-arplant-042809-112308
Zhu, L., Ni, W., Liu, S., Cai, B., Xing, H., and Wang, S. (2017). Transcriptomics analysis of apple leaves in response to Alternaria alternata apple pathotype infection. Front. Plant Sci. 8:22. doi: 10.3389/fpls.2017.00022
Zuluaga, A. P., Lu, H., Góngora-castillo, E., Vaillancourt, B., Coll, N., Buell, C. R., et al. (2015). Transcriptome responses to Ralstonia solanacearum infection in the roots of the wild potato Solanum commersonii. BMC Genomics 16:246. doi: 10.1186/s12864-015-1460-1
Zumaquero, A., Kanematsu, S., Nakayashiki, H., Matas, A., Martínez-Ferri, E., Barceló-Muñóz, A., et al. (2019). Transcriptome analysis of the fungal pathogen Rosellinia necatrix during infection of a susceptible avocado rootstock identifies potential mechanisms of pathogenesis. BMC Genomics 20:1016. doi: 10.1186/s12864-019-6387-5
Keywords: transcriptomic, wheat tassel, Tilletia foetida, defense response, wheat common bunt
Citation: He T, Ren Z, Muhae-Ud-Din G, Guo Q, Liu T, Chen W and Gao L (2022) Transcriptomics Analysis of Wheat Tassel Response to Tilletia laevis Kühn, Which Causes Common Bunt of Wheat. Front. Plant Sci. 13:823907. doi: 10.3389/fpls.2022.823907
Received: 28 November 2021; Accepted: 10 January 2022;
Published: 22 February 2022.
Edited by:
Tika Adhikari, North Carolina State University, United StatesReviewed by:
Malkhan Singh Gurjar, Indian Agricultural Research Institute (ICAR), IndiaBasharat Ali, University of Agriculture, Faisalabad, Pakistan
Copyright © 2022 He, Ren, Muhae-Ud-Din, Guo, Liu, Chen and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Gao, eGlhb2dhb3N4QGhvdG1haWwuY29t
†These authors have contributed equally to this study