 Kai Ma1,2,3†
Kai Ma1,2,3† Ruiqiang Xu1,2†Yu Zhao1,2,3Liqun Han1,2,3Yuhui Xu1,2Lili Li1,2,3Juan Wang1,2*Ning Li1,2*
Ruiqiang Xu1,2†Yu Zhao1,2,3Liqun Han1,2,3Yuhui Xu1,2Lili Li1,2,3Juan Wang1,2*Ning Li1,2*- 1Institute of Horticultural Crops, Xinjiang Academy of Agricultural Sciences, Urumqi, China
- 2Key Laboratory of Horticulture Crop Genomics and Genetic Improvement in Xinjiang, Urumqi, China
- 3Xinjiang Fruit Science Experiment Station, Ministry of Agriculture and Rural Affairs, Urumqi, China
Melatonin widely mediates multiple developmental dynamics in plants as a vital growth stimulator, stress protector, and developmental regulator. N-acetylserotonin methyltransferase (ASMT) is the key enzyme that catalyzes the final step of melatonin biosynthesis in plants and plays an essential role in the plant melatonin regulatory network. Studies of ASMT have contributed to understanding the mechanism of melatonin biosynthesis in plants. However, AMST gene is currently uncharacterized in most plants. In this study, we characterized the JrASMT gene family using bioinformatics in a melatonin-rich plant, walnut. Phylogenetic, gene structure, conserved motifs, promoter elements, interacting proteins and miRNA analyses were also performed. The expansion and differentiation of the ASMT family occurred before the onset of the plant terrestrialization. ASMT genes were more differentiated in dicotyledonous plants. Forty-six ASMT genes were distributed in clusters on 10 chromosomes of walnut. Four JrASMT genes had homologous relationships both within walnut and between species. Cis-regulatory elements showed that JrASMT was mainly induced by light and hormones, and targeted cleavage of miRNA172 and miR399 may be an important pathway to suppress JrASMT expression. Transcriptome data showed that 13 JrASMT were differentially expressed at different periods of walnut bud development. WGCNA showed that JrASMT1/10/13/23 were coexpressed with genes regulating cell fate and epigenetic modifications during early physiological differentiation of walnut female flower buds. JrASMT12/28/37/40 were highly expressed during morphological differentiation of flower buds, associated with altered stress capacity of walnut flower buds, and predicted to be involved in the regulatory network of abscisic acid, salicylic acid, and cytokinin in walnut. The qRT-PCR validated the results of differential expression analysis and further provided three JrASMT genes with different expression profiles in walnut flower bud development. Our study explored the evolutionary relationships of the plant ASMT gene family and the functional characteristics of walnut JrASMT. It provides a valuable perspective for further understanding the complex melatonin mechanisms in plant developmental regulation.
Introduction
Melatonin (N-acetyl-5-methoxytryptamine), a widely known tryptophan derivative that regulates reproductive physiology and circadian rhythms, was initially identified in bovine pineal tissue and subsequently demonstrated to be common in various plant tissues (Dubbels et al., 1995; Hattori et al., 1995; Lerner et al., 2002). Melatonin participates in the growth and development of plants in different tissues and periods (Wang et al., 2016; Zhang et al., 2017), especially as an antioxidant regulating the resistance of plants to various adverse environments (Zhang et al., 2014; Wang et al., 2015), and is considered a potential phytohormone (Arnao and Hernández-Ruiz, 2019). Similar to that in animals, melatonin synthesis in higher plants requires four enzymatic reactions catalyzed by at least six enzymes, including tryptophan decarboxylase (TDC), tryptophan hydroxylase (TPH), tryptamine 5-hydroxylase (T5H), serotonin N-acetyltransferase (SNAT), and N-acetylserotonin methyltransferase (ASMT; Back et al., 2016). ASMT is involved in the O-methylation of N-acetylserotonin in the last melatonin biosynthesis step and is regarded as the vital terminal enzyme for melatonin production (Tan and Reiter, 2020).
ASMT genes have been identified in more than 10 species, including Arabidopsis (Arabidopsis thaliana), rice (Oryza sativa), tomato (Solanum lycopersicum), pepper (Capsicum annuum), apple (Malus domestica), and wheat (Triticum aestivum; Liu et al., 2017; Pan et al., 2019; Zhan et al., 2019; Bhowal et al., 2021; Wang et al., 2022). Functions of ASMT in plant biotic and abiotic stress responses have been extensively characterized. Rice OsASMT1 was the first ASMT gene analyzed and cloned from recombinant Escherichia coli, and its expression level was induced by the aging process and highly correlated with melatonin content (Kang et al., 2011). Further studies demonstrated the presence of multiple ASMT genes in rice, and independent overexpression of OsASMT1, OsASMT2, and OsASMT3 increased rice ASMT enzyme activity and drought stress tolerance (Park et al., 2013a). Increased AtASMT expression and enzyme sulfhydryl modification promoted stomatal closure and osmotic stress tolerance in Arabidopsis under exogenous H2S treatment (Wang et al., 2021). Cadmium infiltration enhanced ATASMT1 transcript expression levels and melatonin biosynthesis in mature Arabidopsis leaves (Byeon et al., 2016). Four SlASMTs in tomato were thought to respond to multiple pathogens (Liu et al., 2017). The expression of SlASMTs was induced by selenium and can alleviate cadmium stress (Li et al., 2016). The apple genome contains at least 37 MdASMT genes. Elevated expression of MdASMT1 increased apple stomatal performance and water content under drought stress. MdASMT11 and MdASMT14 are assumed to play essential roles in the response of apple rootstocks to abiotic stresses (Wang et al., 2022). ASMT also participated in multiple growth and developmental dynamics in plants. The melatonin-deficient ASMT rice exhibited accelerated senescence in detached flag leaves and significantly reduced yield (Byeon and Back, 2016). Overexpression of AtASMT caused massive melatonin accumulation and synergized with the phytohormone abscisic acid (ABA) to inhibit seed germination in Arabidopsis (Lv et al., 2021). At least 16 CaASMT genes were identified in pepper. They were differentially expressed during pepper pericarp formation and development (Pan et al., 2019). Exogenous melatonin induced the strawberry ASMT expression and accelerated the ripening of strawberry fruits through the ABA pathway (Mansouri et al., 2021). Overexpression of SlASMT genes increased the heat shock protein (HSP) profile and expression of autophagy-related genes in tomatoes (Xu et al., 2016). MeASMT2 and MeASMT3 in cassava simultaneously promote melatonin bioactivation and similarly interact with autophagy-related genes to positively stimulate dynamic changes in cassava autophagic activity (Wei et al., 2020). Although decisive molecular genetic data are not yet available, the possible role of AMST in plant flower development has been implied by several researches. Melatonin levels and OsASMT expression were induced in parallel during rice flower development (Park et al., 2013b). Three SlASMT genes were tissue-specific expressed in flowers and buds in tomatoes (Liu et al., 2017). Seasonal light signal repressed MdASMT9 expression in apple, thereby promoting flowering (Zhang et al., 2019a).QTL analysis of cowpea [Vigna unguiculata (L.) Walp.] RIL population showed that five Arabidopsis ASMT orthologs were highly correlated with floral scent (Lo et al., 2020).
Walnuts (Juglans regia) are an important nut crop that nutritionists and consumers generally favor due to the high nutritional value of their seeds and their effectiveness in preventing many diseases (Ros et al., 2018). Compared to other nuts, walnuts are richer in melatonin (Verde et al., 2022). The available studies have mainly focused on pathological experiments or nutritional value evaluation of melatonin in walnuts (Bonomini et al., 2018; Cheng et al., 2021; Kamoun et al., 2021; Steffen et al., 2021). However, systematic reports on the mechanism of melatonin synthesis and inheritance in walnut are still lacking. The publication of the whole walnut genome has made it possible to investigate this important fruit crop using bioinformatics (Marrano et al., 2020). Genome-wide isolation and identification of the walnut ASMT gene family have not been reported. In this study, 46 walnut ASMT gene family members were identified, and their physicochemical properties, gene characteristics, phylogeny, evolutionary status, promoter cis-regulatory elements (CRE), and potential interaction relationships were analyzed by bioinformatics methods. Transcriptome and qRT–PCR data were also used to screen the expression dynamics and differential coexpression network of JrASMTs during the development of walnut flower buds. This research provides a global reference for the functional study of the ASMT gene family in walnut and a theoretical basis for exploring the function and molecular mechanism of melatonin during walnut flower bud development.
Materials and Methods
Plant Materials
Conventionally cultivated walnut (J. regia, “Xinxin 2”) was used as research material in this study. Walnuts were grown under natural conditions at Xinjiang Fruit Science Experiment Station of Ministry of Agriculture and Rural Affairs (Yecheng, China). Female flower buds from the terminal buds’ lower 2–3 positions were collected on April 15 (qP1), May 15 (qP2), May 30 (qS1), June 15 (qS2), and July 15 (qS3) in 2021, respectively. All floral bud samples were collected after removing the outer skin and fixed by flavonoid acetic acid (FAA) for subsequent studies.
ASMT Gene Family Identification
Complete genomic data for all species used in this study were downloaded from the EnsemblPlants1 and Phytozome2 public databases; Markov model files of the dimerization domain (PF16864) and O-methyltransferase domain (PF00891) were downloaded from the Pfam3 public database (El-Gebali et al., 2019). The amino acid sequences of OsASMT1/2/3 downloaded from the Rice Genome Annotation Project4 and AtASMT1 downloaded from The Arabidopsis Information Resource5 were used as query sequences for local BLAST (E-value: 1e−5); HMMER software was simultaneously used to search for walnut protein sequences (E-value: 1e−5; Wheeler and Eddy, 2013). Amino acid sequences of all subsequent genes were verified by SMART,6 Pfam,7 and CDD8 databases for complete conserved structural domains (Schultz et al., 2000; Marchler-Bauer et al., 2007). ASMT genes of Arabidopsis (A. thaliana), tomato (S. lycopersicum), rice (O. sativa), sorghum (Sorghum bicolor) and Apple (M. domestica) were collected from published studies (Zhan et al., 2019; Bhowal et al., 2021; Wang et al., 2022). Confirmed walnut ASMT genes were submitted to the ExPASy9 and WoLF PSORT10 online sites to calculate physicochemical properties and predict subcellular localization.
Gene Structure, Conserved Motifs, and Phylogenetic Analysis
Annotation information of ASMT genes was extracted from walnut whole genome files using Perl scripts. MEME software was used to analyze the conserved motifs of JrASMTs (motif number: 20, min-width: 6; Bailey et al., 2009). TBtools was used to visualize the gene structure and conserved motifs (Chen et al., 2020). Multiplex sequence alignment was performed using Clustal X 2.1 (Larkin et al., 2007). Phylogenetic trees were constructed by IQ-TREE software (automatic calculation of the optimal model, bootstrap ≥1,000; Minh et al., 2020). The ASMT amino acid sequences of all species were used to construct species phylogenetic trees based on OrthoFinder (STAG algorithm; Emms and Kelly, 2019). The evolutionary tree was embellished using the iTOL11 online website.
Homologous Genes, cis-Regulatory Elements, and Interaction Network Analysis
Inter- and intraspecies synteny and collinearity analysis was performed using MCScanX software (Wang et al., 2012). Synonymous and nonsynonymous substitution rates between paralogous homologous pairs were calculated by KaKs Calculator 2.0 (Wang et al., 2010). The CREs in the ASMT promoter sequences (2,000 bp sequence upstream of each gene) were predicted with PlantCARE12 (Lescot et al., 2002). Visualization of homologous relationships and CREs was also performed using TBtools. All JrASMT proteins were submitted to STRING13 to predict interactions (Szklarczyk et al., 2019). The miRNAs targeting ASMT were predicted through the PmiREN14 online website (Expectation: 5, Gaps: 0, Identity: 12; Guo et al., 2020). The interaction network was visualized by Cytoscape software (Su et al., 2014).
Analysis of JrASMT Gene Expression Profiles in Flower Bud Development
RNA-seq data for early physiological differentiation (P1), late physiological differentiation (P2), critical morphological differentiation (PS), early morphological differentiation (S1), and late morphological differentiation (S2) of female walnut buds were obtained from published studies (PRJNA673588).
All transcriptome data were mapped to the walnut reference genome by HISAT2 (Kim et al., 2019). Gene expression was normalized by fragments per kilobase of exon model per million mapped reads (FPKM). Differentially expressed genes were filtered using DESeq2 (Love et al., 2014), with differential levels set to absolute values of the FoldChange greater than 2 and FDR values less than 0.01. The differentially expressed genes were plotted and hierarchically clustered based on TBtools.
Weighted Gene Coexpression Network Analysis
Weighted gene coexpression network analysis (WGCNA) of all differentially expressed genes was performed based on the publicly available R package (“WGCNA”) in the R language (v 4.1.2) environment (Langfelder and Horvath, 2008). Genes with a mean FPKM of less than 0.1 and those with more than 50% deletions in a set of replicates were removed. Soft thresholds were calculated by the scale-free topological fit index and mean connectivity. Hierarchical clustering of genes was performed based on the topological overlap measure (TOM). The minimum number of genes in the module was 60, and high similarity modules were merged based on a threshold of 0.2.
Gene Function Annotation and Core Network Extraction
De novo annotation of transcriptome data was completed via eggNOG-Mapper (v 5.0; Huerta-Cepas et al., 2017). Enrichment analysis (p-value < 0.01) and visualization of annotation results from Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) were performed using R. The cytoHubba plugin in Cytoscape was used to extract specific modules with JrASMTs with high connectivity (Chin et al., 2014). Cytoscape was used to visualize core coexpression networks.
Paraffin Sectioning and Real-Time Fluorescence Quantification
Walnut female bud samples fixed in FAA for 24 h and softened by glycerol immersion were treated with a continuous gradient of ethanol (70, 85, 95, and 100%) and a mixture of anhydrous ethanol and xylene (v:v = 1:1) for dehydration, and then were immersed in 60°C liquid paraffin. Slicing was done using a Leica Microtome (Leica, Weztlar, Germany) with a thickness of 8 μM, sections were stained using Fast Green FCF (Sangon Biotech, Shanghai, China), Nikon Eclipse Ts2 microscope (Nikon, Shanghai, China) was used for observation of the sections, and images were processed using NIS-Elements.
Total RNA was extracted from different tissues by RNA extraction kits (Tiangen, DP441, Beijing, China). First-strand cDNA was synthesized using Vazyme HiScript II 1st Strand cDNA Synthesis Kit (Vazyme Biotech Co., Ltd., China). Specific primers were designed based on 18 selected coding sequences of JrASMTs (Supplementary Table 1), and Sangon Biotech Co., Ltd. (Shanghai, China) synthesized the primers. The Actin gene was used as an internal reference and real-time fluorescence quantification by Maxima SYBR Green/ROX qPCR Master Mix. Three biological replicates were included in this experiment, and gene expression was calculated by 2−ΔΔCt (Livak and Schmittgen, 2001). qP1 period samples were set as the control.
Results
Genome-Wide Identification of the Walnut ASMT Family
The whole-genome sequence of walnut was used to identify ASMT proteins. Candidate genes were initially screened by a local BLAST search of query sequences and an hmmsearch of Markov model files. A total of 46 genes were finally identified as members of the walnut ASMT family by validation of their conserved structural domains and were named in order of their position on the walnut genome (JrASMT1-46). The physical and chemical properties showed that most of the walnut ASMT proteins (71.74%) had amino acid lengths ranging from 335 aa to 385 aa (Supplementary Table 2). Forty-one (89.13%) JrASMTs were acidic proteins, and the isoelectric points (pI) of all JrASMT proteins ranged from 4.91 to 8.91, with an aliphatic index of 71.7 to 105.02. Nineteen (41.30%) JrASMTs were hydrophobic proteins, with GRAVY values ranging from 0 to 0.175. Twenty-six (56.52%) JrASMTs were hydrophilic proteins, with GRAVY values ranging from −0.017 to −0.247. The prediction of subcellular localization indicated that most of the JrASMT proteins were distributed in the cytoplasm (29), and the remaining JrASMT proteins were distributed in the cytoskeleton (12), nucleus (2), chloroplast (2), and peroxisome (1).
Phylogeny, Gene Structure, and Conserved Motif Analysis of JrASMTs
The phylogenetic tree was constructed with the amino acid sequences of all JrASMTs to explore the relationships among walnut ASMTs. Forty-six JrASMTs were distributed in five evolutionary distinct groups (Figure 1A). Group I contained only two members, JrASMT40 and JrASMT37. Group II contained 14 JrASMTs. Group III, Group IV, and Group V were located in a common internal node. Among them, Group III contained only one JrASMT38, which was considered to be an outgroup. Group IV contained 12 JrASMTs. Group V contained the largest number of JrASMTs at 17. Gene structure analysis showed that JrASMT genes contained 1–4 exons and 1–3 introns, and only 11 JrASMT genes contained complete 3′ and 5’ UTRs. JrASMT13 and JrASMT41 contained only one exon and one intron (Figure 1B). Conserved motif analysis showed that some JrASMT genes have more conserved motif sequences than others (Figure 1C), four JrASMT (JrASMT28, JrASMT20, JrASMT46, JrASMT19) genes had only partial motifs due to their short protein length. The different groups of JrASMT proteins contained different motifs, mainly at the 5′ terminus (motif13, motif26) or 3′ terminus (motif14, motif17).
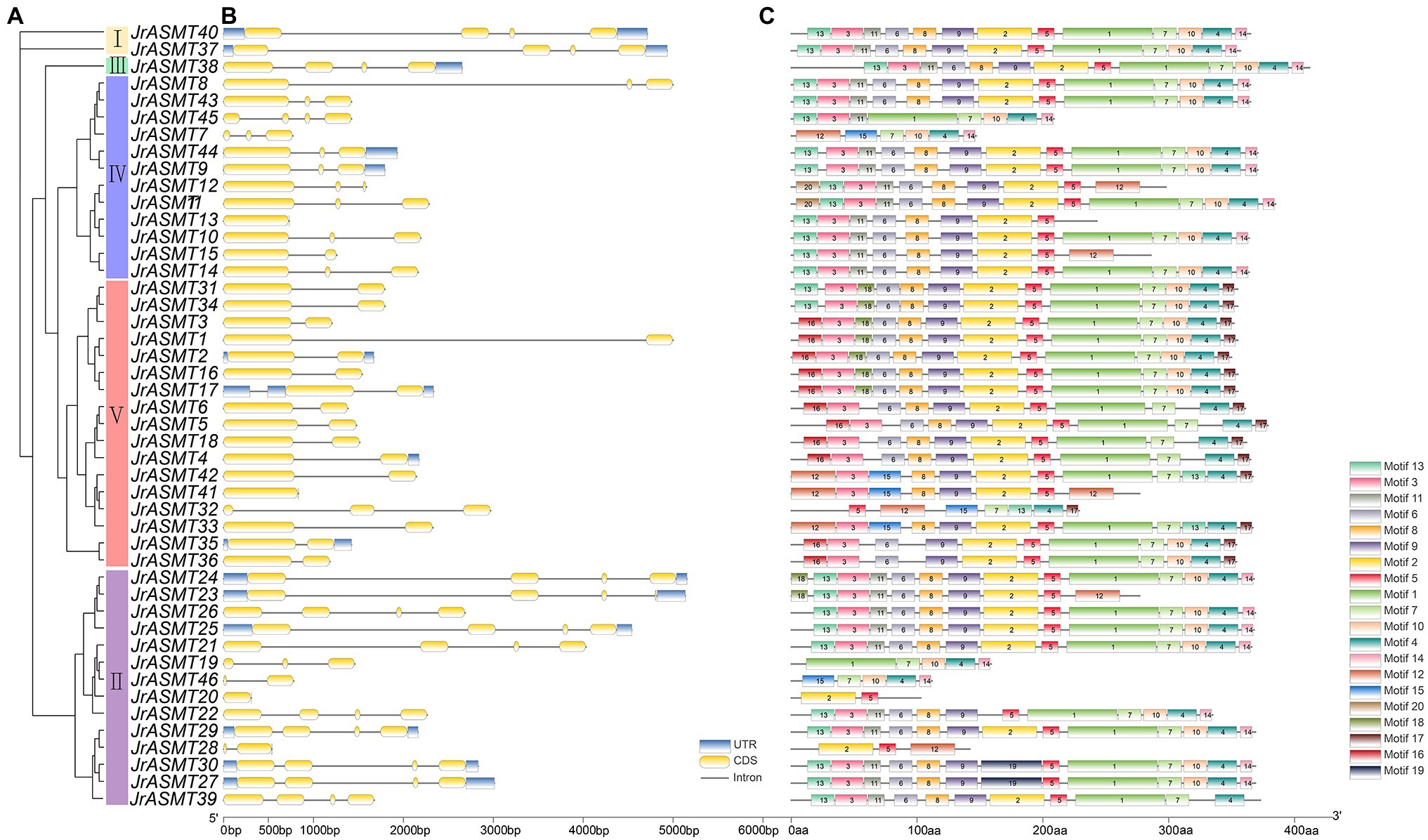
Figure 1. Phylogenetic, gene structure, and motif analysis of walnut JrASMT genes. (A) The phylogenetic tree was constructed by IQ-TREE. Forty-six ASMT genes were divided into five subgroups (I–V). (B) TBtools were used to show the gene structure, with UTRs in blue, exons in yellow, and introns in black solid lines. (C) The motifs of JrASMT. Rectangles with different colors and numbers represented different motifs.
Evolutionary Relationship and Phylogenetic Analysis of JrASMTs in Multiple Plants
To assess the evolutionary status of JrASMT in major plant species, 584 ASMT proteins were identified from 27 plant species by multiple methods (Supplementary File 1). These plants included four algae, two mosses, one pteridophyte, one gymnosperm, four monocotyledons, and 15 dicotyledons. All protein sequences were manually validated for structural domains by the CDD, Pfam, and SMART databases to ensure accurate identification. The species tree constructed base on OrthoFinder showed that ASMT genes were present in all selected species (Figure 2A). Only a single copy of the ASMT gene was present in the cyanobacteria D. salina and C. stagnale. Two green algae, C. reinhardtii and C. zofingiensis, contained 3 ASMT genes each. All selected embryophytes contained at least nine ASMT genes, with A. thaliana containing 19 ASMT genes and J. regia containing the most ASMT genes at 46. A clear differentiation among ASMT genes occurred at different taxonomic levels. Dicotyledonous and monocotyledonous plants were strictly assigned to different branches on the species tree. The number of ASMT genes was not strongly correlated with species genome size. The larger genomes of H. vulgare, Z. mays, and G. hirsutum contained only 24, 18, and 22 ASMT genes. Woody plants seemed to possess more ASMT genes. In addition to walnuts, P. persica, Morella rubra, M. alba, and M. domestica contained 44, 34, 37, and 37 ASMT genes, respectively. However, A. chinensis contained only nine ASMTs.
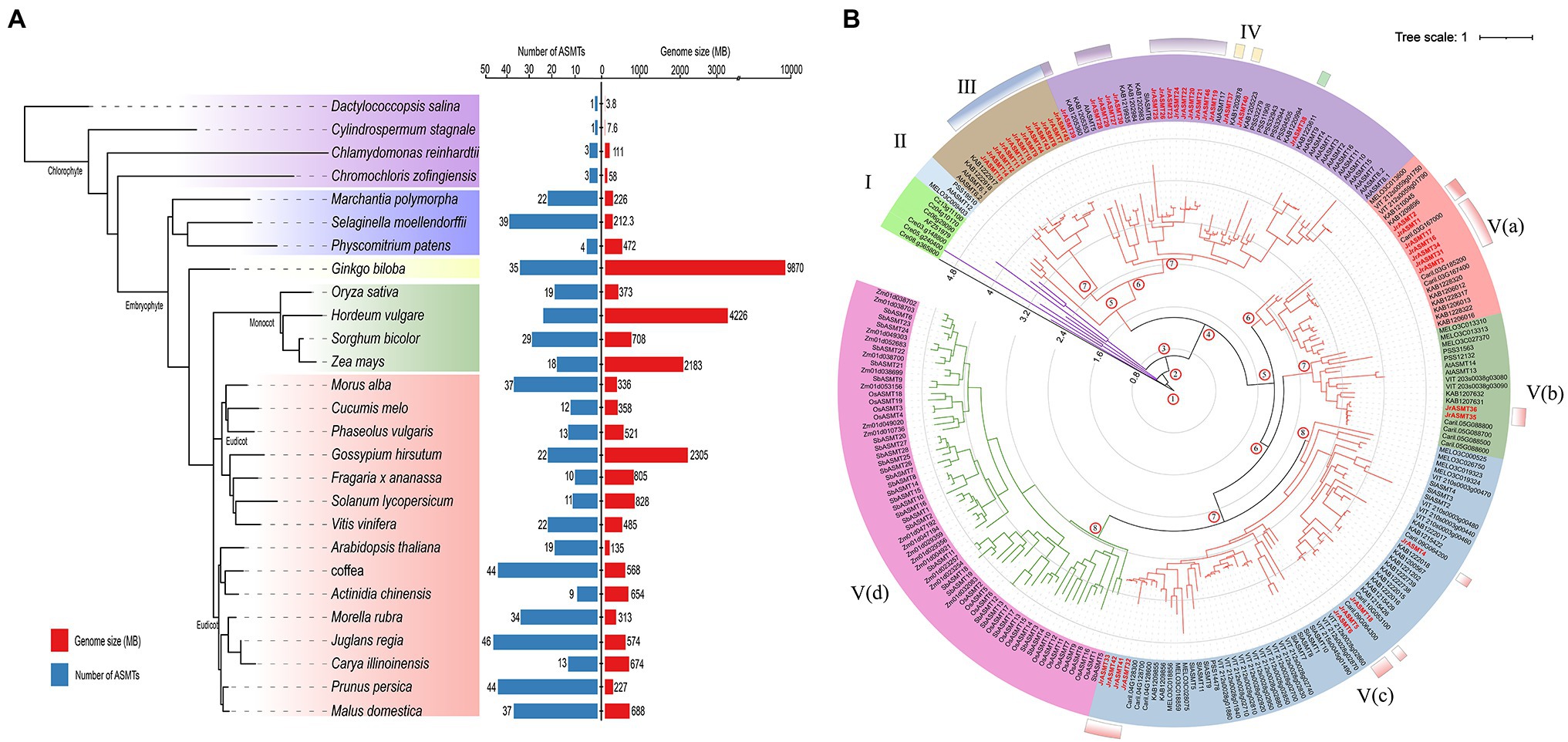
Figure 2. Numerical statistics and taxonomic status analysis of the ASMT family in multiple plants. (A) The species tree of the ASMT family was constructed based on OrthoFinder. All selected plants were divided into four subgroups, distinguished by different colored branches. The statistics of the respective genome size and number of ASMT family members for each species were shown on the right. (B) The phylogenetic tree was constructed with IQ-TREE. The ASMT of algae was used as the midpoint root. Different colored branches represented different taxonomic levels, purple for algae, red for dicotyledons and green for monocotyledons. The length of the branches represented the evolutionary distance. Two hundred and thirty-seven ASMT genes were divided into five subgroups (I–V) and displayed in different background colors. The black axes inside the evolutionary tree displayed the tree scale. The numbers at the internal nodes represented the level of clades. The classification results of JrASMT in Figure 1 were displayed in the outer circle of the evolutionary tree as identical gradient color rectangles.
The 237 protein sequences from 14 species, which contained one cyanobacteria, two green algae, three monocotyledons, and eight dicotyledons, were used to construct a phylogenetic tree with the D. salina ASMT protein as the midpoint root (Figure 2B). All ASMT proteins were distributed within five main different subgroups (I–V), the subgroup V were further elaborated into Va, Vb, Vc, and Vd. With subgroup I containing only algae distributed in four clades. Subgroup V(d) contained only monocotyledonous plants, and the ASMT genes in this subgroup shared similar branch lengths and close evolutionary distances. Interestingly, a general divergence of the ASMT family in eudicot occurred, as they were distributed in subgroups II-V. Subgroups IV and V(c) contained 46 and 63 ASMT genes, respectively, which are relatively high numbers. Subgroup II contained the fewest genes, with only three ASMTs from A. thaliana, Cucumis melo, and A. chinensis. Subgroup V(a) contained genes from five species: C. melo, Vitis vinifera, M. rubra, J. regia and C. illinoinensis. Specifically, Arabidopsis ASMTs were absent from this group. Walnut ASMT genes were distributed in five of the four subgroups of Dictyostelium, but each subgroup member did not match the grouping results of the previous phylogenetic tree (Figure 1A). The introduction of many outgroups allowed for a more precise delineation of the JrASMT divergence relationships.
Analysis of Paralogous and Direct Homologous Genes
Gene doubling and duplication are important pathways for the adaptive evolution of organisms and the acquisition of diverse phenotypes. The 46 putative JrASMT genes were unevenly distributed on the 10 chromosomes of walnut (Figure 3A). Twelve (26.09%) of these JrASMT genes were concentrated on walnut chromosome 4 (Chr4), which had the highest number. In contrast, only 1 (2.17%) JrASMT was found on chromosome 12 (Chr12). The results of the walnut ASMT collinearity gene analysis showed that only 7 (15.22%) JrASMTs were found to have four pairs of paralogous homologs (JrASMT3/JrASMT31, JrASMT4/JrASMT18, JrASMT21/JrASMT37 and JrASMT37/JrASMT40). JrASMT3/JrASMT31 was classified in subgroup V(a) of the phylogenetic tree (Figure 2B). JrASMT4/JrASMT18 was classified in subgroup V(c). JrASMT21/JrASMT37 and JrASMT37/JrASMT40 were both classified in subgroup IV. All paralogous homologous genes were derived from segmental duplication, and no tandem duplication events occurred. Synonymous and nonsynonymous mutation rates were calculated for all paralogous homologous genes (Supplementary Table 3). The Ka/Ks values for both homologous pairs JrASMT37/JrASMT21 and JrASMT4/JrASMT18 were close to 1, and these genes were almost free from environmental selection and showed neutral evolution, while gene pairs JrASMT3/JrASMT31 and JrASMT37/JrASMT40 had Ka/Ks values of 0.296 and 0.092, respectively, suggesting that these two gene pairs were subject to strong purifying selection and were functionally stable.
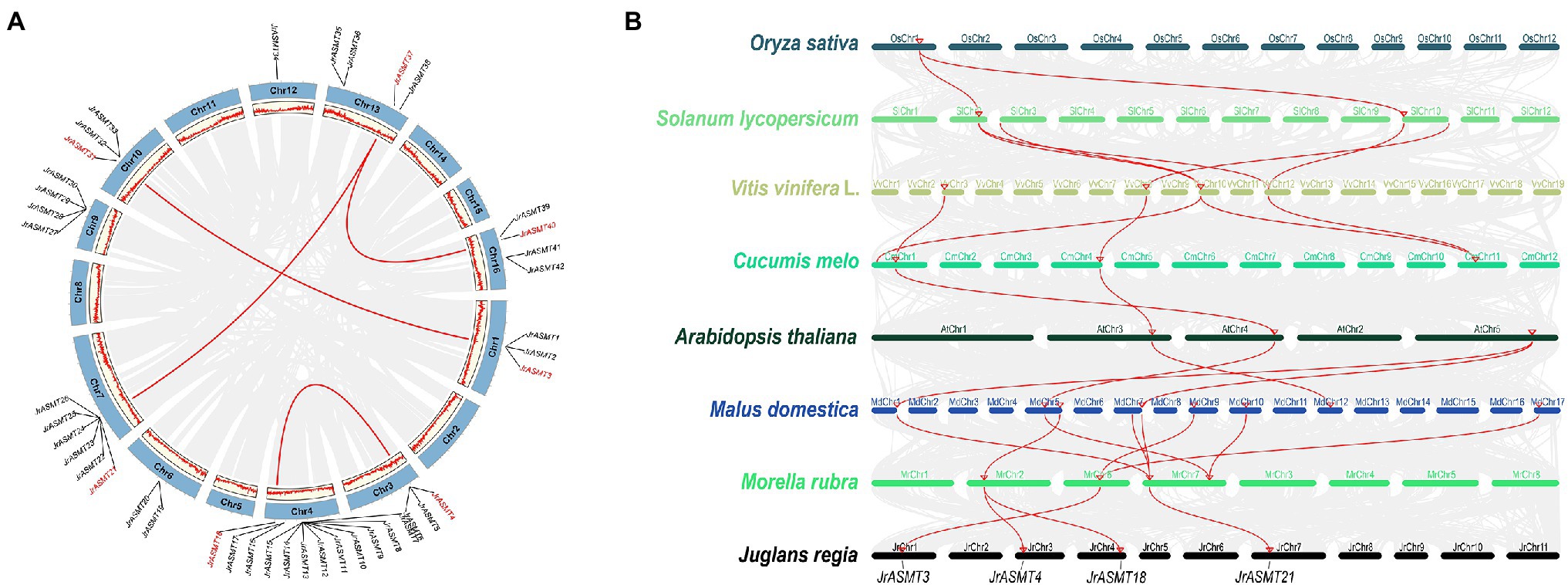
Figure 3. Synteny and collinearity analysis of the ASMT family. (A) Distribution of JrASMTs on walnut chromosomes and analyzed of paralogous genes. The outer circle indicated the 16 chromosomes of walnut, and the inner circle showed the density of genes on the chromosomes. The gray lines represented genes and blocks with paralogous relationship, the red lines represented paralogous JrASMT genes, and the corresponding JrASMTs name were marked in red. (B) Collinearity analysis of ASMT in multiple species. Latin names of species were labeled on the left. The ranking order was according to the proximity of the relatives in the species tree. Chromosomes of different species were shown in different colors. The gray lines represented genes or blocks with collinearity, and the red lines were ASMT genes with collinearity. Four JrASMTs name were labeled below.
The ASMT genes from eight plant species were chosen for collinearity analysis according to their distant relationships on the species phylogenetic tree (Figure 2A), including one monocotyledon (O. sativa) and seven dicotyledons (S. lycopersicum, V. vinifera L., C. melo, A. thaliana, M. domestica, M. rubra, J. regia; Figure 3B). Rice contained only one gene that has orthologous relationship with two genes in the dicotyledonous tomato plant, while the number of orthologous genes increased significantly inside the dicotyledonous clade. There were five pairs of orthologous relationships formed by four ASMT genes, with two each between tomato and grape, grape and melon, although the origin of these orthologous relationships was not the same. Only two ASMT gene pairs had four orthologous relationship between melon and Arabidopsis. In direct contrast, there were eight direct orthologous gene pairs between the nine apple ASMT genes and the four poppy ASMT genes. Three poplar ASMT genes were orthology to JrASMT1, JrASMT4, JrASMT18, and JrASMT21, and these same JrASMTs were located in subgroups IV, V(a), and V(c). JrASMT4 and JrASMT18 shared the same orthologous ASMT gene with poplar. Collectively, there are four conserved direct homologous relationships within the ASMT gene family among dicotyledonous plants.
Analysis of cis-Regulatory Elements in JrASMTs
Transcription factors regulate gene expression by binding to CRE sites in promoter regions. To explore the possible biological functions and expression patterns of walnut ASMT genes, the 2,000 bp upstream sequences of all JrASMT genes were subjected to CRE analysis (Figure 4A). Except for core response elements and functionally unknown elements, a total of 1,076 CREs were characterized in all JrASMT promoter regions. These CREs could be broadly classified into four categories: light-response elements (34.94%), growth- and development-related elements (21.84%), stress-response elements (10.41%) and hormone-response elements (32.81%). Among them, there were 21 types of light-response elements, mainly G-boxes (13.38%), GATA motifs (3.07%), GT1 motifs (3.25%), JrASMT expression was widely regulated by multiple modes of light-mediated. There were 11 types of growth- and development-related elements, including hyphal tissue-specific expression element CAT-box (2.23%) and circadian rhythm element circadian (1.49%). There were six types of stress-response elements, mainly the LTR (3.25%) and MBS (2.70%). It suggested that JrASMT was sensitive to low temperature and drought environment. JrASMT was extensively involved in walnut hormonal regulatory networks, 11 hormone-response elements were observed, including the ABA response element ABRE (12.17%); growth hormone response elements AuxRR-core (1.30%); jasmonic acid (JA) response elements CGTCA-motif (6.13%) and TGACG-motif (6.13%); gibberellin response elements P-box (1.30%); and salicylic acid (SA) response elements TCA-element (2.23%). More than 30 CREs were found in the promoter regions of JrASMT8, JrASMT43, JrASMT45, JrASMT7, JrASMT32, and JrASMT20, and multiple pathways may widely regulate these genes in walnut. JrASMT13, JrASMT5, and JrASMT22 each had fewer than 15 CREs in the promoter region, implying that these genes expression may be mediated in fewer ways. The promoter regions shared a similar distribution of CREs among the more closely related JrASMT genes. The JrASMT8, JrASMT43, JrASMT45, and JrASMT7 genes in subgroup IV, which were distributed in the same clade, all contained a large number of G-box elements and hormone-related elements. The promoter regions of the JrASMT31 and JrASMT34 genes in subgroup V(a) contained only O2-site elements. JrASMT23, JrASMT24, JrASMT25, and JrASMT26 in subgroup II also showed similar transcription factor binding sites, and they all contained high amounts of AREs, G-boxes, and ABREs. Overall, these results provided evidence for the expression patterns of walnut ASMT genes.
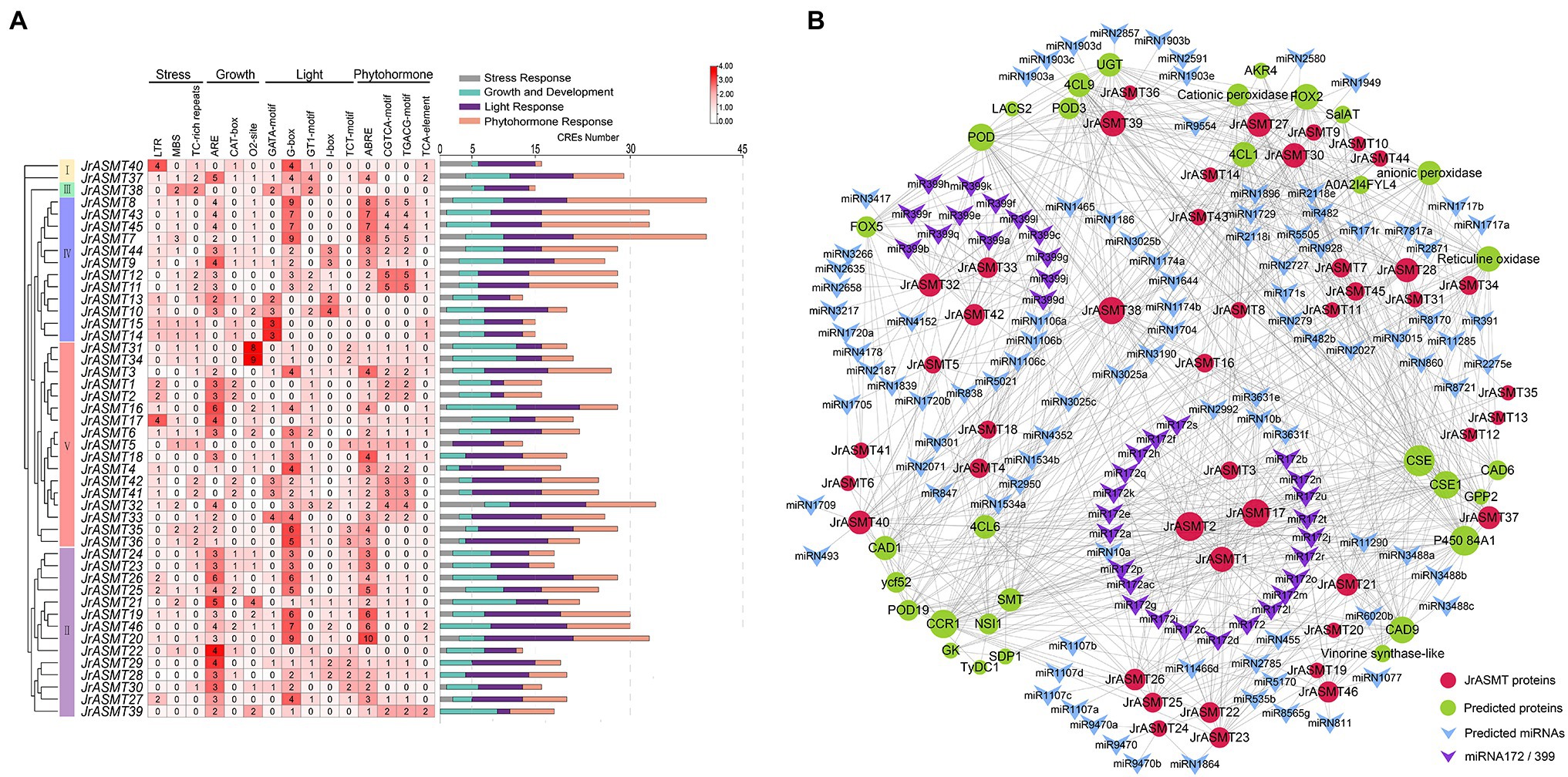
Figure 4. CRE analysis of the promoter region of JrASMT, and interacting proteins and miRNAs prediction. (A) The CRE statistics of JrASMT promoter regions. CREs were classified into four categories: stress-response, growth and development, light-response and phytohormone-response. JrASMT was arranged in the order of phylogenetic tree. The names of CREs were marked on the top. The heat map showed the number of 15 main CREs corresponding to each JrASMT. The bar chart on the right side counted all the number of four types of CREs in each JrASMT, and different types of CREs were indicated by different colors. (B) Network diagram of proteins and miRNAs interacted with JrASMT. Gray connecting lines indicated the presence of interactions. JrASMT was represented by red circles, and interacting proteins were represented by light green circles, with the size of the circles represented the number of interaction relationships. miRNAs were represented by V-shaped rectangles, where miRNA172 and miRNA399 were shown in purple.
Network Analysis of JrASMT Proteins and miRNAs
Plants possess complex and indirect gene expression networks. Molecular interactions are an important component of plant complicated expression patterns. The interrelationship between miRNA and mRNA or between proteins is essential for plants to display different expression patterns. Based on online databases, potential miRNAs and proteins that target or interact with JrASMT were predicted (Supplementary Table 4). Only six JrASMT proteins (JrASMT27/28/30 and JrASMT37/39/40) were predicted to exhibit nine pairs of direct interactions with each other (Supplementary Figure 1). A total of 27 JrASMT proteins were predicted to directly or indirectly interact with 31 proteins (Figure 4B), the main ones being CAFFEOYL SHIKIMATE ESTERASE (CSE), cytochrome P450 84A1, cinnamoyl-CoA reductase (CCR), peroxidase (POD) and cinnamyl alcohol dehydrogenase (CAD), which interacted with 40, 37, 34, 31, and 30 proteins, respectively. Forty-two JrASMT genes were predicted to have potential targeting relationships with 132 miRNAs from 75 families (Supplementary Table 3). miR2118, miR482, miRN2785, miRN1729, and miR5505 interacted with the most genes, targeting 9, 9, 7, 6, and 5 JrASMTs, respectively. The miR172 and miR399 families were the most abundant, with 23 types miR172 and 13 types miR399 targeting the JrASMT1/2/3/17 and JrASMT32/33/42 genes, respectively. JrASMTs distributed in the same clade on the phylogenetic tree had similar protein interaction relationships. The JrASMT27/28/30/37/38/39/40 genes classified in subgroup IV had an almost identical number of interactions (mean degree = 87), and the other six genes (JrASMT21/22/23/25/26/46) in this subgroup had a degree of 59. Five genes (JrASMT2/3/16/17/34) in subgroup V(a) all had a degree equal to 18, while the degrees for two genes (JrASMT35/36) in subgroup V(b) and the seven genes (JrASMT4/5/6/18/32/41/42) in subgroup V(c) were all equal to 2. There may be complex synergistic expression or functional redundancy of these genes.
Expression Dynamics of JrASMTs During Walnut Bud Development
We retrieved the expression patterns of all JrASMT family genes during physiological differentiation (P1, P2), the physiological differentiation and morphological differentiation critical period (PS), and morphological differentiation (S1, S2) of walnut buds from published transcriptome data. Significant differential expression occurred in 13 of 46 JrASMT genes during walnut flower bud development. The heatmap shows that differentially expressed ASMT genes were hierarchically clustered into two groups based on their expression during the P and S periods (Figure 5A). JrASMT1, JrASMT10-14, JrASMT23, and JrASMT24 showed similar expression patterns; all eight genes had the highest expression in the P1 and P2 periods and decreased expression in the PS-S2 period. The expression of JrASMT28/29/30, JrASMT37, and JrASMT40 increased during later floral bud development: JrASMT28/29/30 were highly expressed during S2, and both JrASMT37 and JrASMT40 were highly expressed during PS-S2. Among all the differentially expressed JrASMTs, only JrASMT40 was relatively highly expressed in all periods, reaching a significant difference only under the P1 vs. S1 comparison, indicating that this gene may play an essential role in walnut flower bud development.
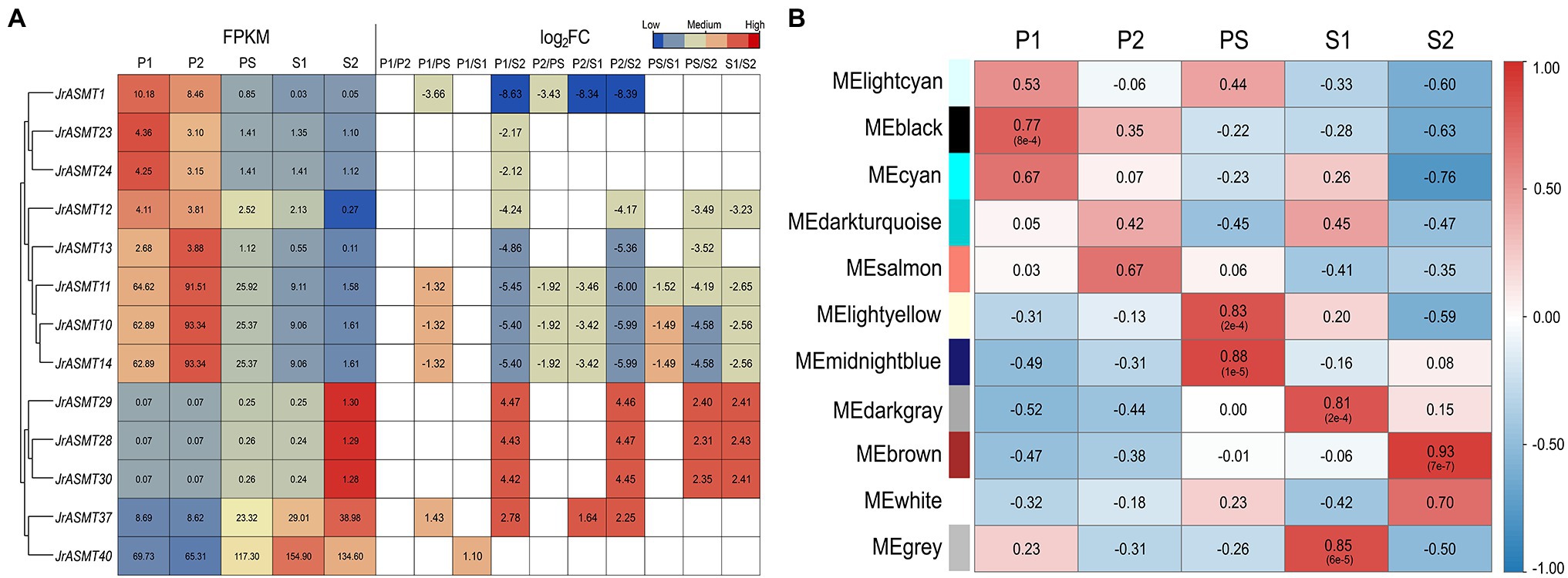
Figure 5. The expression analysis of JrASMTs in walnut flower bud development. (A) The DEG heat map of JrASMT. Genes were hierarchically clustered based on expressions. The normalized expressions were shown on the left, and the fold change under each difference grouping were shown on the right, in log2FC. The colors of the heat map represent the magnitude of expressions and fold change. (B) Correlation of each module with the developmental period. The module names were on the left and the different periods were on top. The color of the heat map represented the magnitude and properties of the correlation. The numbers in the heat map were the correlation values and the numbers in parentheses were the p-values.
To further determine the underlying functions of JrASMT genes in walnut bud development, all differentially expressed genes (DEGs) were used to conduct weighted gene coexpression network analysis after data filtering. The soft threshold for constructing the adjacency matrix was 27 (correlation over 78%). Except for those at PS and S1, walnut buds at different developmental stages showed significant differences in their expression patterns (Supplementary Figure 2). Total of 12,664 DEGs were assigned in 11 merged modules (Supplementary Figure 3). The connectivity of eigengenes was analyzed for differences between modules and the magnitude of association between module genes and traits, and the results showed significant differences between modules (Figure 5B, Supplementary Figure 4). The black module containing 4,316 genes was highly correlated with the P1 period (p = 8e−4, R = 0.77). The light-yellow module containing 263 genes (p = 2e−4, R = 0.83) and the midnight-blue module containing 334 genes (p = 1e−5, R = 0.88) were highly correlated with the PS period. The dark-gray module containing 220 genes (p = 2e−4, R = 0.81) and the gray module containing 1,181 genes (p = 6e−5, R = 0.85) were highly correlated with the S1 period, and the brown module containing 4,528 genes was highly correlated with the S2 period (p = 7e−7, R = 0.93).
Association Module and Functional Prediction of JrASMTs
JrASMT1/10/13/23 were specifically classified in the black module, JrASMT40 was specifically classified in the dark-gray module, and JrASMT12/28/37 were specifically classified in the brown module. GO, and KEGG annotation and enrichment analysis were performed for genes in these three modules (Figure 6). In the black module, 182 (8.90%) genes were enriched in the cell cycle term, 115 (5.62%) genes were enriched in the mitotic cell cycle term, 206 (12.96%) genes were enriched in the chromosome and associated protein pathway, and 187 (11.76%) genes were enriched in the carbohydrate metabolism pathway. These results suggested that genes in the black module played roles in cell cycle regulation, DNA replication and repair, and nuclear chromosome segregation. In the dark-gray module, 44 (12.61%) genes were enriched in the catalytic activity term, 17 (4.87%) genes in the endomembrane system term, 17 (4.87%) genes in the cell wall organization or biogenesis term, and 18 (1.48%) genes in protein families: metabolism pathway. A large number of DEGs were also enriched in functions related to cell wall synthesis and metabolism, glycolic acid, lignin, cellulose, and other metabolic processes in plants. This result indicated that genes in the dark-gray module were involved in cell wall formation during morphological differentiation of walnut flower buds and mediated various secondary metabolic pathways. In the brown module, 485 (23.00%) genes were enriched in response to chemical terms, 292 (13.85%) genes in response to oxygen-containing compound terms, 206 (9.77%) genes in transcription regulator activity terms, 92 (13.53%) genes in environmental information processing pathways, and 51 (7.50%) genes in plant hormone signals. The genes of the brown module were significantly associated with the response of walnut buds to various environmental factors and extensively involved in multiple signaling and hormone regulatory networks in the late stage of walnut bud morphological differentiation.
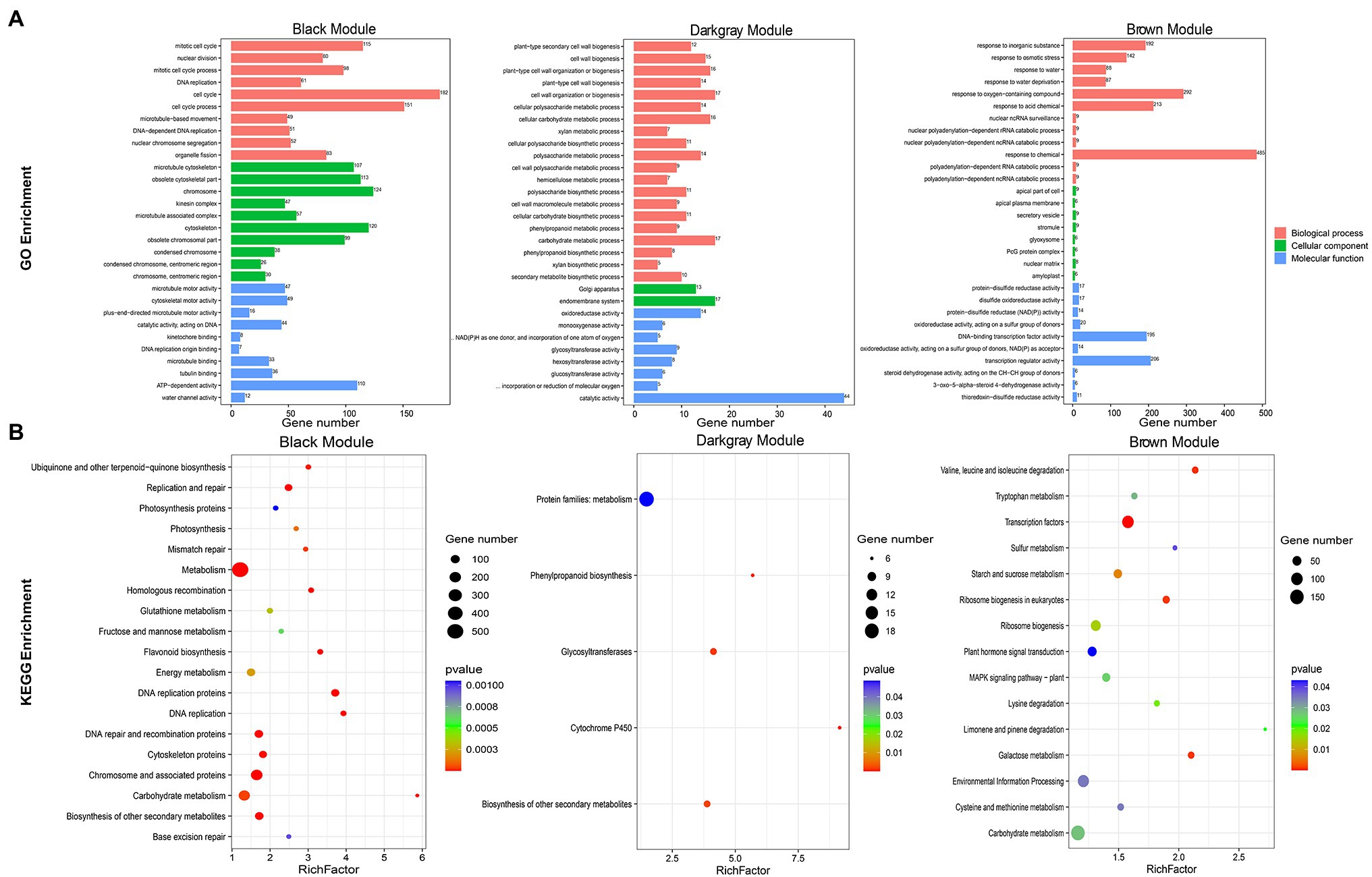
Figure 6. Functional annotation of black, dark-gray, and brown modules genes. (A) Bar chart of GO enrichment for the three modular genes. GO enrichment terms were shown on the left. Module names were labeled above the bar graph. (B) Bubble plots of KEGG enrichment for the three modular genes. KEGG enrichment pathway were shown on the left. The RichFactor represented the level of enrichment. The size of dots represented the number of genes. The color of the dots represented the q-value of the enrichment. The module names were labeled above the bubble diagram.
The Coexpression Network of JrASMTs in Walnut Flower Bud Development
Genes coexpressed with JrASMTs in the three modules were screened. The black, dark-gray and brown modules contained 500 (11.58%), 91 (41.36%) and 582 (12.85%) genes coexpressed with JrASMT genes, respectively. The core subnetworks of the three modules were extracted by cytoHubba. The interactions and expressions were visualized (Figure 7). Except for genes with unknown functions, in the black module, JrASMT1/10/13/23 were coexpressed with several cell process-related genes, such as the mitotic checkpoint protein coding gene Budding uninhibited by benzimidazole (BUB), which is involved in the cell cycle; Protein arginine methyltransferase (PRMT); Cyclin B2 (CYCB2), which belongs to the cyclin family; and NPK1-activating kinesin (NACK), which belongs to the TRAFAC class of myosins. In addition, the JrASMT genes in the black module were also associated with the Fasciclin I family protein coding gene (FAS1), glutamyl-tRNA (Gln) amidotransferase subunit coding gene (Amidase), Casparian strip membrane domain protein (CASP), and other coexpressed genes. Almost all functionally known coexpressed genes were highly expressed during P1 and P2, except for LRR receptor-like serine threonine-protein kinase (LRR-RLKs), PRMT, and CASP, which had elevated expression during morphological differentiation. In contrast, the genes coexpressed with JrASMT40 in the dark-gray module were more functionally diverse, containing Lonely guy (LOG), which activates the cytokinin-activating enzyme Constitutive disease resistance (CDR) and Responsive to desiccation 22 (RD22). Two transporter protein family members ATP-binding cassette (ABCG) and Hypothetical protein (HHP) involved in the AMPK signaling pathway; Adenosine-5′-phosphosulfate (APS) involved in the purine metabolism pathway; and Glycerol-3-phosphate acyltransferase (GPAT) involved in the glycerolipid metabolism pathway, were found in the coexpression network that also included Chitinase-like protein (CTL), Trichome birefringence-like (TBL), and Glycosyltransferase 2 (GT2) involved in cellulose synthase, encoding Isochorismatase family protein (ICS). All genes expression levels in the dark-gray module increased during the PS period; most reached maximum expression at the S1 period and remained unchanged or decreased at the S2 period. In the brown module, JrASMT12/28/37 was coexpressed with the gene encoding DNA-directed RNA polymerase III subunit (RNAPIII), protein translation factor SUI1 homolog coding gene (SUI1), Poly(A) binding protein (PABP), and other genes. These genes are involved in transcriptional regulation or translational modifications. The network likewise contained multiple Receptor-like serine–threonine protein kinases (SRKs) and LRR-RLKs. JrASMT12/28/37 was also coexpressed with Beta carbonic anhydrase (BCA), Proline/serine-rich protein (PRP), and a Tetratricopeptide repeat protein coding gene (TPR). All coexpressed genes were most highly expressed during the S2 period. However, JrASMT12 was more highly expressed during the P1 and P2 periods.
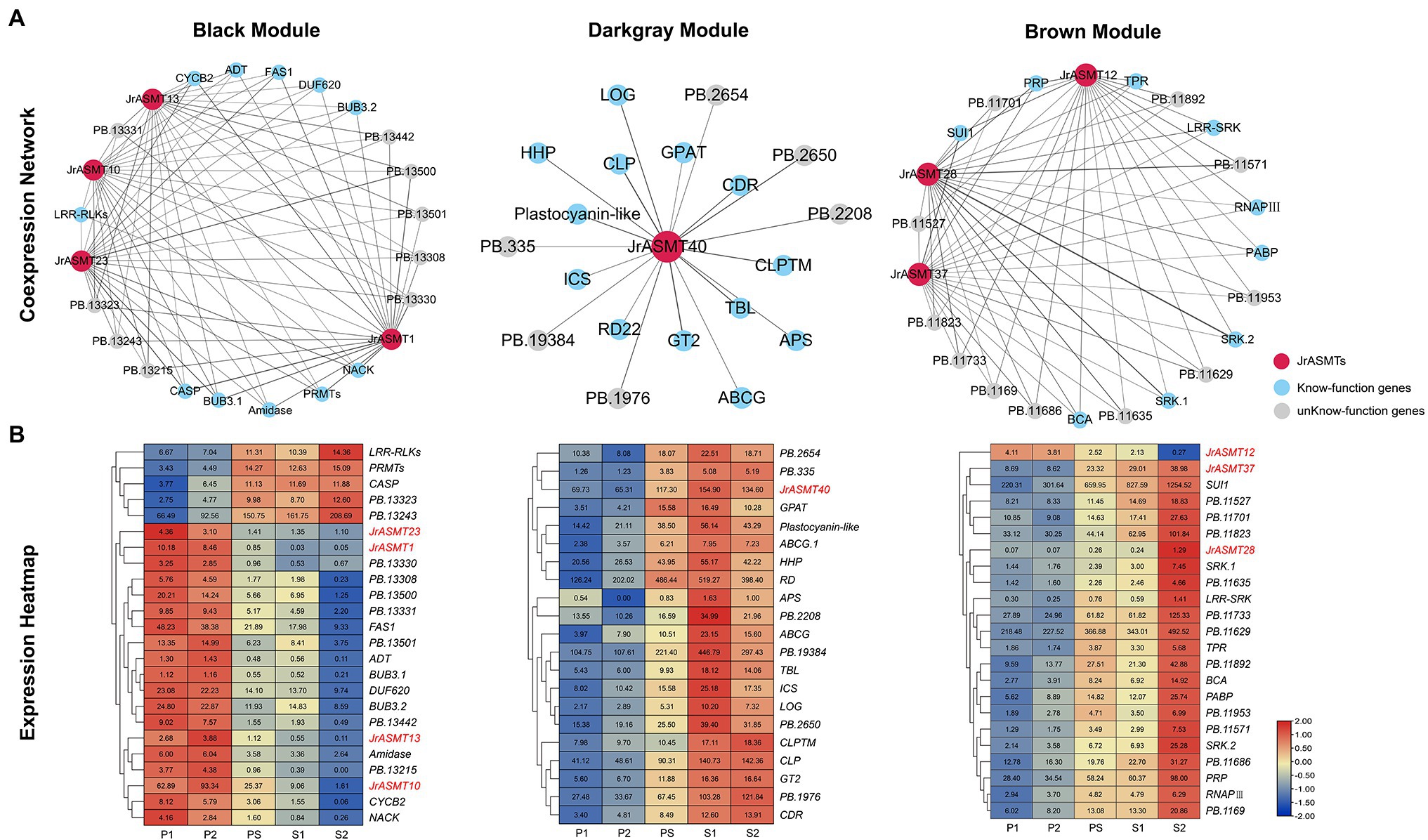
Figure 7. Construction and expression analysis of core coexpression networks. (A) The core coexpression networks with JrASMT in the three modules. JrASMT was represented by red circles, known-function genes were represented by light blue circles, and unknown-function genes were represented by gray circles. Gray connecting lines indicated the presence of coexpression relationships, the thickness of the line indicated the confidence level of coexpression. The names of the modules were labeled above the network diagram. (B) Heat map of gene expression in the three coexpression networks. The horizontal axis was samples from different periods. The vertical axis was the genes name. The different colors represent the high or low expression levels. All genes were clustered hierarchically based on expression patterns. JrASMT was labeled in red.
Tissue Sectioning and Real-Time Fluorescence Quantification
To verify the expression pattern of JrASMT during the development of walnut buds, flower bud tissues from walnuts at different periods were collected. The Paraffin section results showed that the walnut buds in the qP1 and qP2 periods were undergoing physiological differentiation (Figure 8A), and those in qS1, qS2, and qS3 were undergoing bud morphological differentiation. Eighteen JrASMTs, including 11 DEGs in the transcriptome and seven non-DEGs, were used for real-time fluorescence quantification in these samples (Figure 8B). Similar to several previous results, multiple JrASMTs were significantly differentially expressed and clustered at different periods of walnut bud development. JrASMT1/10/12/13/14 were significantly higher in qP1 and qP2 but barely expressed during morphological differentiation. JrASMT23 had no significant differential expression in the qP1, qP2, and qS1 periods but was slightly decreased during the qS2 and qS3 periods. JrASMT3/28/29/30/37/40 were commonly expressed at different stages. However, they were only highly and specifically expressed at the later stages of flower bud development. The expression profiles of JrASMT7/9/18/31/34/45 were also relatively similar, with all of these genes being significantly highly expressed at qS2 and weakly expressed at qS1 and qS3. Most of the differentially expressed JrASMT genes except for JrASMT23 showed similar expression trends with RNA-seq data. JrASMT7, JrASMT9, and JrASMT45 were highly expressed at qS2; these genes, which were not considered DEGs in the transcriptome, apparently played important roles during the morphological differentiation of the walnut bud.
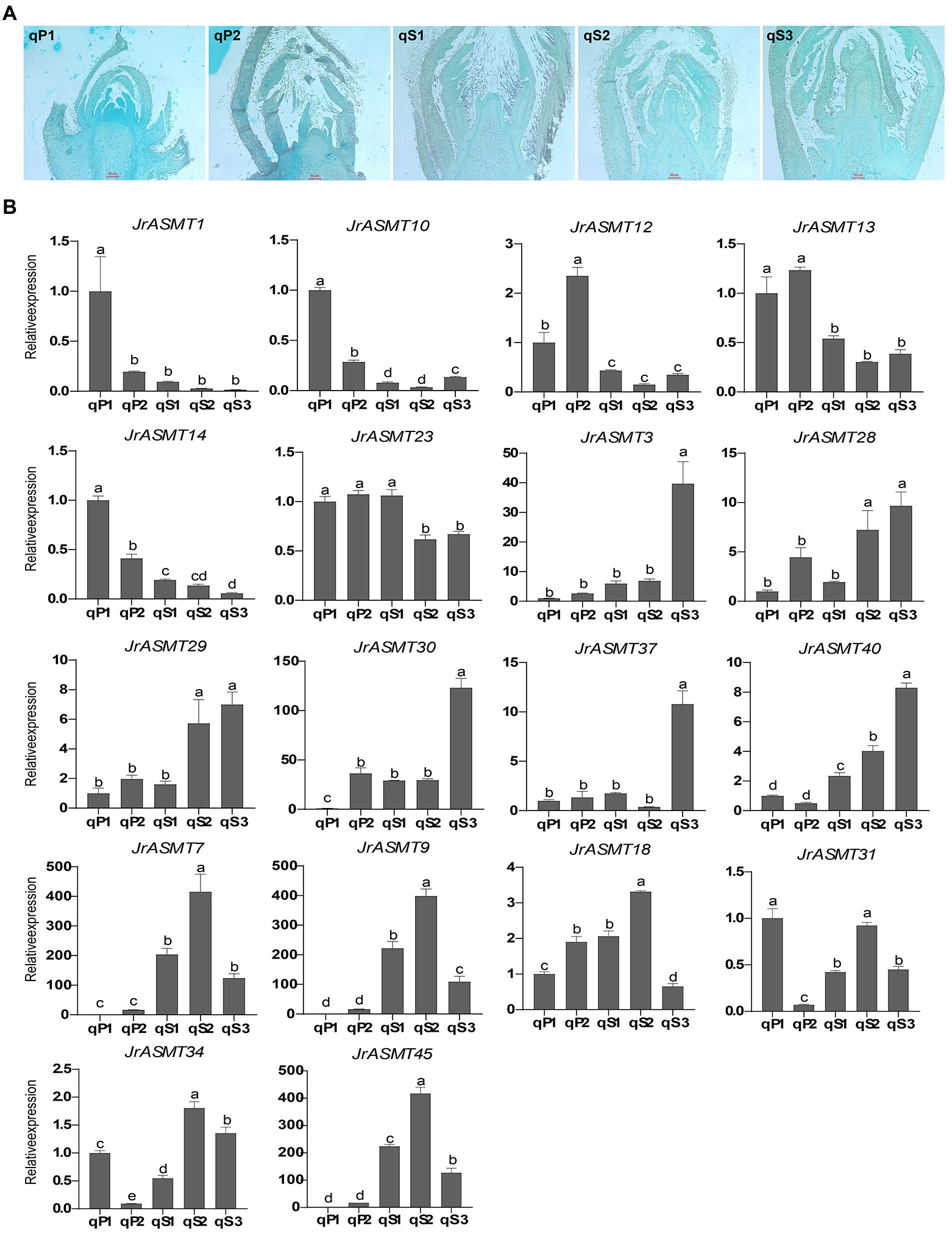
Figure 8. Paraffin sectioning and Real-time quantitative PCR. (A) Results of paraffin sections of female flower buds of walnut at different periods, corresponding sample numbers were marked in the upper left corner. (B) Bar chart of the 18 JrASMTs Real-time quantitative PCR. The horizontal axis showed the names of samples from different periods. The vertical axis showed the real-time quantitative result. The corresponding JrASMTs name was labeled above the bar chart. The standard deviations were shown with error bars. Letters indicated the significance of differences between expressions. The expression at qP1 was used for normalization.
Discussion
Identification of ASMT Gene Family Members in Walnut
As an indole-like molecular substance, melatonin is most well known for its ubiquitous regulation of circadian rhythms, immune systems, and antioxidant activities in living organisms (Zhao et al., 2019). With the study of melatonin synthesis, metabolism, and catabolism in higher plants, melatonin is considered a potentially pleiotropic phytohormone that regulates plant growth and development, life processes, redox dynamics, biotic and abiotic stresses, and fruit preservation (Back, 2021). Several studies have demonstrated that many melatonin synthases in plants interact directly with various proteins, including transcription factors (Wei et al., 2018). The formation of these protein complexes implies that melatonin synthases play an essential role in plant melatonin-mediated physiological and biochemical processes. ASMT is considered to be the key enzyme for melatonin synthesis.
This study identified 46 possible ASMT genes from walnuts based on published genomic data. Even though the selection criteria of hmmsearch and local BLAST were stringent and all genes were similarly validated for structural domains, the numbers of ASMTs in walnuts were still obviously higher than those in all currently identified species, including rice (19), Arabidopsis (19), tomato (13), pepper (16), wild mulberry (20), and apple (37). Some of the JrASMTs may have lost their functions. SlASMT13, which encodes only 103 amino acids in tomatoes, was determined to be a pseudogene (Liu et al., 2017). We observed the same presence of JrASMT20 (103 aa) and JrASMT46 (112 aa) genes encoding short peptides in walnuts. Despite our retention of these two members, whether they were pseudogenes similar to SlASMT13 needs further confirmation. Another possible reason for such a large number of JrASMTs is the high degree of redundancy within the family; for example, JrASMT31 on chromosome 10 and JrASMT34 on chromosome 12 encode proteins with identical sequences, and JrASMT8 and JrASMT43 likewise share identical amino acid sequences. These functionally redundant genes increased the number of ASMT family members identified.
JrASMT genes contained relatively few exons; the simple gene structure confers a faster expression response of ASMT to ensure the realization of dynamic changes in melatonin content in plants. Most JrASMTs shared similar conserved motifs, except for members limited by the encoded protein length. The ASMT enzymes were more conserved among plants and localized in the cytoplasm, cytoskeleton, and chloroplast. Overall, these results were consistent with previous reports (Wang et al., 2022). Interestingly, an X-ray crystal structure analysis showed that the Homo sapiens ASMT enzyme only had a helical domain at the N-terminal end that interacted with multiple monomers (Botros et al., 2013). However, in terms of conserved motifs only, both the N-terminal and C-terminal motifs of JrASMT were significantly different, which provided a possibility for differences in the three-dimensional structure and substrate affinity between plant and animal ASMT enzymes. Although the crystal structure of the rice melatonin synthase OsTDC has been studied (Zhou et al., 2020), data from high-resolution X-ray or Cryo-transmission electron microscopy of plant ASMT enzymes are still lacking. CREs is an important regulator of gene expression, identification of CREs in the promoter region of JrASMT revealed that many elements are associated with light-response and hormone-response, consistent with the manner in which melatonin is predominantly induced in plants (Arnao and Hernández-Ruiz, 2021; Li et al., 2021; Tiwari et al., 2021; Yao et al., 2021). The promoter of JrASMT20 was distributed with many ABA response elements ABRE, melatonin and ABA are both essential components of the phytohormone-mediated network (Zhang et al., 2014, 2017; Hu et al., 2021), JrASMT20 was probably involved in the synergistic response of melatonin and ABA in walnuts. JA response elements were enriched in the promoter region of JrASMT11 and JrASMT12. Melatonin and JA were considered to regulate together important plant physiological changes in biotic and abiotic stresses (Imran et al., 2021; Tiwari et al., 2021), these two genes may mediate the interaction between melatonin and JA. Two groups of JrASMTs were predicted to be separately targeted by a large number of miR172 and miR399 family members. miR172 regulation of meristem size, trichome initiation, stem elongation, branch meristem, and flowering capacity has been extensively characterized (Lian et al., 2021). miR399 plays an important role in plant resistance to various abiotic stresses (Li et al., 2020; Pegler et al., 2021). These results implied that miRNA172 and miR399 may antagonize the melatonin biosynthesis in walnuts under certain developmental stages or growth environments. This regulation is not mentioned in other studies (Liu et al., 2017; Wang et al., 2022).
Expansion and Differentiation of ASMT Genes During the Evolution of Land Plants
Evolution is the source of all biological functions. Exploring the evolutionary trajectories of species at the molecular (DNA, RNA, protein) scale is a common approach in modern evolutionary biology. Previous research on the evolution of 62 ASMT genes in 13 species during plant terrestrialization showed that ASMT genes first appeared in primitive bacteria and expanded to embryophytes (Zhao et al., 2021). Our study increased the number of species and ASMT proteins identified. ASMT genes were found in both cyanobacteria and green algae at early evolutionary stages, with the difference that only one single-copy ASMT gene was found in cyanobacteria, whereas this number was apparently increased in green algae. Considering the more complex life system and significantly greater photosynthetic capacity of green algae (Khan et al., 2020), we infer that the ASMT gene family had already expanded before plants began terrestrialization to better regulate melatonin biosynthesis and better accommodate new environmental and biological functional requirements.
The number of ASMT gene copies in higher plants does not appear to be strongly correlated with evolutionary status or genome size, and the evolution of ASMT genes may have been influenced by a variety of complex factors, thereby perpetuating events such as gene duplication, loss, or mutation.
Significant divergence of ASMT genes has occurred within higher plants. A phylogenetic tree of 237 ASMT proteins provides direct evidence. Monocotyledonous and dicotyledonous clades are distributed on completely different evolutionary branches. All monocotyledonous phyla are in the same clade V(d), and dicotyledonous phyla are distributed in II–V(c) six different clades. This coincides with previous results for rice, Arabidopsis, tomato, and sorghum (Bhowal et al., 2021). The difference is that the introduction of a large number of ASMT proteins in our study classified dicotyledonous plant ASMTs into four subgroups more reliably, which was not suggested by other phylogenetic analyses of ASMT families. Considering that the functions of most ASMT proteins, including those in Arabidopsis, have not been characterized in detail, these ASMT proteins were crudely classified into subgroups II-V(c) based on their distant relationship with algal ASMTs. There are obvious evolutionary distance differences between different subgroup clades. Subgroups II–IV share the same internal node, and they maintain a very close relationship with algal ASMTs. Subgroup V(a) contains only perennial woody plant ASMT members. Taken together, these results undoubtedly show a high degree of sequence and functional differentiation of ASMT genes in Dictyostelium. Three algal ASMT proteins are in separate clades and at different evolutionary distances, further demonstrating that the differentiation of ASMT genes occurred early in biological evolution.
Plant evolution is usually accompanied by mutations and the loss of large segments of genes. Molecular evolution encounters immense background noises; identifying the evolutionary trajectory of genes among species is difficult (Williams et al., 2021). The orthology of ASMTs between different species was searched according to the distances between ASMTs. Although more or fewer orthologous genes existed between some species depending on the degree of species differentiation, in general, there were roughly four pairs orthologous ASMTs among dicots. The results of walnut collinearity analysis showed that these genes were also paralogous homologs, suggesting that these genes have maintained a high degree of collinearity among and within species in the evolution of the ASMT family. In contrast, only one ASMT gene is orthologous to these other genes in rice. Considering that the number of ASMT genes in both primitive bacteria and cyanobacteria was one, we believed that there was a single copy of ASMT genes in the ancient ancestor, which began to replicate and diverge in late algal evolution. Multiple whole-genome duplication events in higher plants caused chromosome duplication, rearrangements, and losses that expanded the number of ASMT genes, and the ancestral ASMT gene finally reached four copies in dicotyledons. These genes may represent the most conserved and typical functions of ASMTs. Both evolutionary trees and homology relationships suggest that monocot ASMT genes had fewer copies and were more conserved. This may be related to the evolution of the ASMT toward caffeic acid O-methyltransferase (COMT) during plant terrestrialization (Zhao et al., 2021). The reasons for this evolutionary drive playing a prominent role in monocotyledonous plants still need further investigation.
Diversified Functions of JrASMTs in Walnut Flower Bud Development
Flowering in plants requires a complex series of physiological and biochemical processes, and several flowering pathways, such as photoperiod, vernalization, autonomous, and gibberellin, have been identified in Arabidopsis. The floral regulator gene Flowering locus T (FT) and the repressor of FT, Flowering locus C (FLC), are vital genes (Izawa, 2021). It has been demonstrated that melatonin participates in the flowering process in plants. High melatonin concentrations can inhibit flower opening in Chenopodium rubrum through the Photoinductive cycle (Kolář et al., 2003). The differential expression of TDC in herbaceous peony (Paeonia lactiflora) flowers during different developmental periods caused significant changes in melatonin accumulation (Zhao et al., 2018). TDC, T5H, and COMT were highly expressed in rice before and after flowering, and the melatonin content was significantly higher than that in other tissues (Park et al., 2013b). Exogenous melatonin administration (500 μM) delayed flowering time and increased the stability of two flowering repressors of the DELLA protein in Arabidopsis (Shi et al., 2016). Nevertheless, the mechanisms involved in melatonin regulation of floral developmental processes and flowering time are still poorly understood. On the one hand, melatonin plays multiple roles in plant floral organs, often responding to stress and pressure (Sharif et al., 2018). On the other hand, melatonin exhibits clear dose-dependent and interspecies differences during flower development. Different amounts and concentrations of exogenous hormones may lead to conflicting effects (Arnao and Hernández-Ruiz, 2020).
Similar to the results of other melatonin synthases in plant flowers (Zhao et al., 2018; Lee et al., 2019; Pan et al., 2021; Tsunoda et al., 2021), JrASMT genes were also participated in walnut flower development in this study. Coexpression analysis divided these genes into three modules. Four ASMT genes in the black module participated in early physiological differentiation. Among their coexpressed genes, BUB3 is a core protein forming the spindle assembly checkpoint (SAC), which is associated with chromosome segregation in mitosis and meiosis (Komaki and Schnittger, 2017). Some components of the SAC promote flowering in Arabidopsis and have indirect interactions with FLC (Bao et al., 2014). CYCB2 is highly expressed during the floral transition of the shoot apical meristem (SAM) in Arabidopsis (Klepikova et al., 2015). NACK regulates and controls cytoplasmic division within the mitogen-activated protein kinase (MAPK) pathway (Liang and Yang, 2019). These genes are evidently involved in floral developmental processes and cell fate, which coincides with melatonin-regulated rhythms. In addition, epistatic modifications may also be important for the involvement of ASMT in flower bud differentiation in walnut. ADT acts as a possible methylation target induced by vernalization to promote beet priming and flowering (Hébrard et al., 2013). PRMT promotes growth and flowering in cauliflower through asymmetric arginine methylation (Niu et al., 2007). Recent studies have revealed that Arabidopsis PRTM promotes FT by positively regulating flowering nuclear factor Ycs via physical interactions to promote FT transcript levels (Zhang et al., 2021).
The four ASMT genes in the dark-gray and brown modules are mainly engaged in the morphological differentiation of walnut flower buds. Among the coexpressed genes, RD22 is a molecular marker of ABA signaling that is mediated by drought, salt, and other abiotic stresses (Harshavardhan et al., 2014; Matus et al., 2014). HHP1 acts as a negative regulator of ABA in the crosstalk between cold stress and osmoregulation (Chen et al., 2010). CDR plays a role in mediating the expression of plant defense genes and enhancing disease resistance through salicylic acid (Ying et al., 2020). LOG converts nucleotide precursors of cytokinins into biologically active forms through the direct activation pathway. The content and location of cytokinins (CK) significantly impact plant flower development and sex differentiation (Kuroha et al., 2009; Ming et al., 2020). These results indicate that melatonin synergizes multiple hormonal regulatory networks during walnut flower development to influence and alter late developmental processes and resistance to multiple biotic-abiotic stresses in walnut flowers. The coexpression of several Receptor-like kinases (RLKs), generally considered receptor proteins for phytohormones and related to multiple stresses (Cui et al., 2022), provides further evidence for this. Some research have suggested interaction between melatonin and NO, GA, and SL (Mukherjee, 2019; Zhang et al., 2019b; Jahan et al., 2021). However, there is no similar direct evidence in our study. TBL is a component of plant cell wall polysaccharide acetylation (Gao et al., 2017). A recent genetic map identified a JrTBL13 gene on chromosome 4 associated with the flowering date in male walnut flowers (Bernard et al., 2020). Interestingly, TBL33 was coexpressed with JrASMT40 in our study. Most of the genes coexpressed with JrASMTs still lack precise functional annotation, and no reliable homologs exist in Arabidopsis.
qRT–PCR results verified the expression of the core network of JrASMT genes during the development of walnut flower buds. Moreover, some JrASMT genes, which were found to not be differentially expressed, exhibited significantly high expression at specific floral developmental stages, JrASMT18 and JrASMT33 were, respectively, homologous with SlASMT2 and SlASMT12, which were specifically expressed in tomato buds and flowers. In our results, JrASMT18 was significantly highly expressed in qS2 period. In addition, Vigun11g097000 was significantly associated with secondary metabolism changes during the V. unguiculata flowering period, and this gene was orthologs with JrASMT42. All these results certainly implying once again the complexity of the mechanisms involved in the regulation of plant flowering by melatonin. Overall, our study on the changes in melatonin synthase expression during germination and differentiation in a long-day flowering plant, walnut, provides a new perspective on the involvement of melatonin in plant flowering.
Conclusion
In this study, 46 members of the ASMT family were identified in walnut. The genes were distributed heterogeneously and in clusters on 10 chromosomes of walnut and were named sequentially. The gene structure, conserved motifs, and phylogeny of all JrASMT proteins were analyzed. The evolution of the ASMT family was examined among different lineages of plants, and ASMT divergence began before the terrestrialization of plants. The presence of four homologous pairs of genes derived from a single copy of the ASMT gene in a distant ancestor is conserved within and among plants of the Dictyostelium phylum. A large number of light- and hormone-responsive elements are present in the promoter region of the JrASMT genes. Members with similar taxonomic status share a similar composition of CREs. RNA-seq data were also used in this study to explore the role of JrASMT in female flower bud development in walnut, and 13 JrASMT showed differential expression. WGCNA further suggested that JrASMT1/10/13/23 were expressed in concert with cell cycle and epistatic modification genes at the stage of physiological differentiation of female flower buds. Similarly, JrASMT12/28/37/40 were involved in phytohormone regulatory networks such as ABA, SA, and CK during flower bud morphological differentiation, regulating flower development and providing stress tolerance in walnut. Paraffin sections and qRT-PCR verified the expression of core network genes and provided other JrASMT members that may have potential functions. In conclusion, this analysis of the ASMT family, a key enzyme for melatonin synthesis, and its study in walnut flowering provides a theoretical basis for further understanding the mechanism of melatonin synthesis and action in higher plants.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
RX, LH, and LL performed the experiment and data analysis. RX, YZ, and KM drafted the manuscript. NL and JW designed the research and revised the manuscript. YX participated in the writing and revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by National Natural Science Foundation of China (31960584), Xinjiang Uygur Autonomous Region “Tianshan Youth” talent program (2020Q031), and The central government guided local scientific and technological development special funds in 2022.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
In particular, we would like to gratefully acknowledge the help of Jisen Shi, Jinhui Chen, and Weihuang Wu of Nanjing Forestry University in this study. They gave valuable advice on many analytical methods early in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.861043/full#supplementary-material
Supplementary File 1 | Amino acid sequence of ASMT gene family in 25 plants.
Supplementary Figure 1 | Sample clustering tree and trait correlation heat map.
Supplementary Figure 2 | Direct interaction network diagram of JrASMT.
Supplementary Figure 3 | Merged dynamic gene modules and genes tree.
Supplementary Figure 4 | Scatter plot of correlation between module membership and gene significance in black, brown and dark grey.
Footnotes
1. ^https://plants.ensembl.org/
2. ^https://phytozome-next.jgi.doe.gov/
5. ^https://www.arabidopsis.org/
7. ^http://pfam.xfam.org/search
8. ^https://www.ncbi.nlm.nih.gov/cdd
9. ^https://web.expasy.org/protparam/
10. ^https://wolfpsort.hgc.jp/
12. ^http://bioinformatics.psb.ugent.be/webtools/plantcare/html/
References
Arnao, M. B., and Hernández-Ruiz, J. (2019). Melatonin: a new plant hormone and/or a plant master regulator? Trends Plant Sci. 24, 38–48. doi: 10.1016/j.tplants.2018.10.010
Arnao, M. B., and Hernández-Ruiz, J. (2020). Melatonin in flowering, fruit set and fruit ripening. Plant Reprod. 33, 77–87. doi: 10.1007/s00497-020-00388-8
Arnao, M. B., and Hernández-Ruiz, J. (2021). Melatonin as a regulatory hub of plant hormone levels and action in stress situations. Plant Biol. 23, 7–19. doi: 10.1111/plb.13202
Back, K. (2021). Melatonin metabolism, signaling and possible roles in plants. Plant J. 105, 376–391. doi: 10.1111/tpj.14915
Back, K., Tan, D.-X., and Reiter, R. J. (2016). Melatonin biosynthesis in plants: multiple pathways catalyze tryptophan to melatonin in the cytoplasm or chloroplasts. J. Pineal Res. 61, 426–437. doi: 10.1111/jpi.12364
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Bao, Z., Zhang, N., and Hua, J. (2014). Endopolyploidization and flowering time are antagonistically regulated by checkpoint component MAD1 and immunity modulator MOS1. Nat. Commun. 5:5628. doi: 10.1038/ncomms6628
Bernard, A., Marrano, A., Donkpegan, A., Brown, P. J., Leslie, C. A., Neale, D. B., et al. (2020). Association and linkage mapping to unravel genetic architecture of phenological traits and lateral bearing in Persian walnut (Juglans regia L.). BMC Genomics 21:203. doi: 10.1186/s12864-020-6616-y
Bhowal, B., Bhattacharjee, A., Goswami, K., Sanan-Mishra, N., Singla-Pareek, S. L., Kaur, C., et al. (2021). Serotonin and melatonin biosynthesis in plants: genome-wide identification of the genes and their expression reveal a conserved role in stress and development. Int. J. Mol. Sci. 22:11034. doi: 10.3390/ijms222011034
Bonomini, F., Borsani, E., Favero, G., Rodella, L. F., and Rezzani, R. (2018). Dietary melatonin supplementation could be a promising preventing/therapeutic approach for a variety of liver diseases. Nutrients 10:1135. doi: 10.3390/nu10091135
Botros, H. G., Legrand, P., Pagan, C., Bondet, V., Weber, P., Ben-Abdallah, M., et al. (2013). Crystal structure and functional mapping of human ASMT, the last enzyme of the melatonin synthesis pathway. J. Pineal Res. 54, 46–57. doi: 10.1111/j.1600-079X.2012.01020.x
Byeon, Y., and Back, K. (2016). Low melatonin production by suppression of either serotonin N-acetyltransferase or N-acetylserotonin methyltransferase in rice causes seedling growth retardation with yield penalty, abiotic stress susceptibility, and enhanced coleoptile growth under anoxic conditions. J. Pineal Res. 60, 348–359. doi: 10.1111/jpi.12317
Byeon, Y., Lee, H.-J., Lee, H. Y., and Back, K. (2016). Cloning and functional characterization of the Arabidopsis N-acetylserotonin O-methyltransferase responsible for melatonin synthesis. J. Pineal Res. 60, 65–73. doi: 10.1111/jpi.12289
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, C.-C., Liang, C.-S., Kao, A.-L., and Yang, C.-C. (2010). HHP1, a novel signalling component in the cross-talk between the cold and osmotic signalling pathways in Arabidopsis. J. Exp. Bot. 61, 3305–3320. doi: 10.1093/jxb/erq162
Cheng, G., Ma, T., Deng, Z., Gutiérrez-Gamboa, G., Ge, Q., Xu, P., et al. (2021). Plant-derived melatonin from food: a gift of nature. Food Funct. 12, 2829–2849. doi: 10.1039/D0FO03213A
Chin, C.-H., Chen, S.-H., Wu, H.-H., Ho, C.-W., Ko, M.-T., and Lin, C.-Y. (2014). cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8:S11. doi: 10.1186/1752-0509-8-S4-S11
Cui, Y., Lu, X., and Gou, X. (2022). Receptor-like protein kinases in plant reproduction: current understanding and future perspectives. Plant Commun. 3:100273. doi: 10.1016/j.xplc.2021.100273
Dubbels, R., Reiter, R. J., Klenke, E., Goebel, A., Schnakenberg, E., Ehlers, C., et al. (1995). Melatonin in edible plants identified by radioimmunoassay and by high performance liquid chromatography-mass spectrometry. J. Pineal Res. 18, 28–31. doi: 10.1111/j.1600-079X.1995.tb00136.x
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Gao, Y., He, C., Zhang, D., Liu, X., Xu, Z., Tian, Y., et al. (2017). Two trichome birefringence-like proteins mediate xylan acetylation, which is essential for leaf blight resistance in rice. Plant Physiol. 173, 470–481. doi: 10.1104/pp.16.01618
Guo, Z., Kuang, Z., Wang, Y., Zhao, Y., Tao, Y., Cheng, C., et al. (2020). PmiREN: a comprehensive encyclopedia of plant miRNAs. Nucleic Acids Res. 48, D1114–D1121. doi: 10.1093/nar/gkz894
Harshavardhan, V. T., Son, L. V., Seiler, C., Junker, A., Weigelt-Fischer, K., Klukas, C., et al. (2014). AtRD22 and AtUSPL1, members of the plant-specific BURP domain family involved in Arabidopsis thaliana drought tolerance. PLoS One 9:e110065. doi: 10.1371/journal.pone.0110065
Hattori, A., Migitaka, H., Iigo, M., Itoh, M., Yamamoto, K., Ohtani-Kaneko, R., et al. (1995). Identification of melatonin in plants and its effects on plasma melatonin levels and binding to melatonin receptors in vertebrates. Biochem. Mol. Biol. Int. 35, 627–634.
Hébrard, C., Trap-Gentil, M.-V., Lafon-Placette, C., Delaunay, A., Joseph, C., Lefèbvre, M., et al. (2013). Identification of differentially methylated regions during vernalization revealed a role for RNA methyltransferases in bolting. J. Exp. Bot. 64, 651–663. doi: 10.1093/jxb/ers363
Hu, E., Liu, M., Zhou, R., Jiang, F., Sun, M., Wen, J., et al. (2021). Relationship between melatonin and abscisic acid in response to salt stress of tomato. Sci. Hortic. 285:110176. doi: 10.1016/j.scienta.2021.110176
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Imran, M., Latif Khan, A., Shahzad, R., Aaqil Khan, M., Bilal, S., Khan, A., et al. (2021). Exogenous melatonin induces drought stress tolerance by promoting plant growth and antioxidant defence system of soybean plants. AoB Plants 13:plab026. doi: 10.1093/aobpla/plab026
Izawa, T. (2021). What is going on with the hormonal control of flowering in plants? Plant J. 105, 431–445. doi: 10.1111/tpj.15036
Jahan, M. S., Shu, S., Wang, Y., Hasan, M. M., El-Yazied, A. A., Alabdallah, N. M., et al. (2021). Melatonin pretreatment confers heat tolerance and repression of heat-induced senescence in tomato through the modulation of ABA- and GA-mediated pathways. Front. Plant Sci. 12:650955. doi: 10.3389/fpls.2021.650955
Kamoun, A., Hammouda, O., Turki, M., Maaloul, R., Chtourou, M., Bouaziz, M., et al. (2021). Moderate walnut consumption improved lipid profile, steroid hormones and inflammation in trained elderly men: a pilot study with a randomized controlled trial. Biol. Sport 38, 245–252. doi: 10.5114/biolsport.2020.97676
Kang, K., Kong, K., Park, S., Natsagdorj, U., Kim, Y. S., and Back, K. (2011). Molecular cloning of a plant N-acetylserotonin methyltransferase and its expression characteristics in rice. J. Pineal Res. 50, 304–309. doi: 10.1111/j.1600-079X.2010.00841.x
Khan, A. K., Kausar, H., Jaferi, S. S., Drouet, S., Hano, C., Abbasi, B. H., et al. (2020). An insight into the algal evolution and genomics. Biomol. Ther. 10:1524. doi: 10.3390/biom10111524
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Klepikova, A. V., Logacheva, M. D., Dmitriev, S. E., and Penin, A. A. (2015). RNA-seq analysis of an apical meristem time series reveals a critical point in Arabidopsis thaliana flower initiation. BMC Genomics 16:466. doi: 10.1186/s12864-015-1688-9
Kolář, J., Johnson, C. H., and Macháčková, I. (2003). Exogenously applied melatonin (N-acetyl-5-methoxytryptamine) affects flowering of the short-day plant Chenopodium rubrum. Physiol. Plant. 118, 605–612. doi: 10.1034/j.1399-3054.2003.00114.x
Komaki, S., and Schnittger, A. (2017). The spindle assembly checkpoint in arabidopsis is rapidly shut off during severe stress. Dev. Cell 43, 172.e5–185.e5. doi: 10.1016/j.devcel.2017.09.017
Kuroha, T., Tokunaga, H., Kojima, M., Ueda, N., Ishida, T., Nagawa, S., et al. (2009). Functional analyses of LONELY GUY cytokinin-activating enzymes reveal the importance of the direct activation pathway in Arabidopsis. Plant Cell 21, 3152–3169. doi: 10.1105/tpc.109.068676
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Lee, H. Y., Lee, K., and Back, K. (2019). Knockout of Arabidopsis serotonin N-Acetyltransferase-2 reduces melatonin levels and delays flowering. Biomol. Ther. 9:712. doi: 10.3390/biom9110712
Lerner, A. B., Case, J. D., Takahashi, Y., Lee, T. H., and Mori, W. (2002). Isolation of melatonin, the pineal gland factor that lightens melanocytes. J. Am. Chem. Soc. 80:2587. doi: 10.1021/ja01543a060
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Li, X., Chen, P., Xie, Y., Yan, Y., Wang, L., Dang, H., et al. (2020). Apple SERRATE negatively mediates drought resistance by regulating MdMYB88 and MdMYB124 and microRNA biogenesis. Hortic. Res. 7:98. doi: 10.1038/s41438-020-0320-6
Li, M.-Q., Hasan, M. K., Li, C.-X., Ahammed, G. J., Xia, X.-J., Shi, K., et al. (2016). Melatonin mediates selenium-induced tolerance to cadmium stress in tomato plants. J. Pineal Res. 61, 291–302. doi: 10.1111/jpi.12346
Li, Y., Liu, C., Shi, Q., Yang, F., and Wei, M. (2021). Mixed red and blue light promotes ripening and improves quality of tomato fruit by influencing melatonin content. Environ. Exp. Bot. 185:104407. doi: 10.1016/j.envexpbot.2021.104407
Lian, H., Wang, L., Ma, N., Zhou, C.-M., Han, L., Zhang, T.-Q., et al. (2021). Redundant and specific roles of individual MIR172 genes in plant development. PLoS Biol. 19:e3001044. doi: 10.1371/journal.pbio.3001044
Liang, Y.-J., and Yang, W.-X. (2019). Kinesins in MAPK cascade: how kinesin motors are involved in the MAPK pathway? Gene 684, 1–9. doi: 10.1016/j.gene.2018.10.042
Liu, W., Zhao, D., Zheng, C., Chen, C., Peng, X., Cheng, Y., et al. (2017). Genomic analysis of the ASMT gene family in Solanum lycopersicum. Molecules 22:1984. doi: 10.3390/molecules22111984
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lo, S., Fatokun, C., Boukar, O., Gepts, P., Close, T. J., and Muñoz-Amatriaín, M. (2020). Identification of QTL for perenniality and floral scent in cowpea (Vigna unguiculata [L.] Walp.). PLoS One 15:e0229167. doi: 10.1371/journal.pone.0229167
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lv, Y., Pan, J., Wang, H., Reiter, R. J., Li, X., Mou, Z., et al. (2021). Melatonin inhibits seed germination by crosstalk with abscisic acid, gibberellin, and auxin in Arabidopsis. J. Pineal Res. 70:e12736. doi: 10.1111/jpi.12736
Mansouri, S., Sarikhani, H., Sayyari, M., and Soleimani Aghdam, M. (2021). Melatonin accelerates strawberry fruit ripening by triggering GAMYB gene expression and promoting ABA accumulation. Sci. Hortic. 281:109919. doi: 10.1016/j.scienta.2021.109919
Marchler-Bauer, A., Anderson, J. B., Derbyshire, M. K., DeWeese-Scott, C., Gonzales, N. R., Gwadz, M., et al. (2007). CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 35, D237–D240. doi: 10.1093/nar/gkl951
Marrano, A., Britton, M., Zaini, P. A., Zimin, A. V., Workman, R. E., Puiu, D., et al. (2020). High-quality chromosome-scale assembly of the walnut (Juglans regia L.) reference genome. GigaScience 9:giaa050. doi: 10.1093/gigascience/giaa050
Matus, J. T., Aquea, F., Espinoza, C., Vega, A., Cavallini, E., Santo, S. D., et al. (2014). Inspection of the grapevine BURP superfamily highlights an expansion of RD22 genes with distinctive expression features in berry development and ABA-mediated stress responses. PLoS One 9:e110372. doi: 10.1371/journal.pone.0110372
Ming, X., Tao, Y.-B., Fu, Q., Tang, M., He, H., Chen, M.-S., et al. (2020). Flower-specific overproduction of cytokinins altered flower development and sex expression in the perennial woody plant Jatropha curcas L. Int. J. Mol. Sci. 21:640. doi: 10.3390/ijms21020640
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Mukherjee, S. (2019). Insights into nitric oxide–melatonin crosstalk and N-nitrosomelatonin functioning in plants. J. Exp. Bot. 70, 6035–6047. doi: 10.1093/jxb/erz375
Niu, L., Lu, F., Pei, Y., Liu, C., and Cao, X. (2007). Regulation of flowering time by the protein arginine methyltransferase AtPRMT10. EMBO Rep. 8, 1190–1195. doi: 10.1038/sj.embor.7401111
Pan, Y., Zhao, X., and Chen, D.-X. (2021). Different development phase of transcription proteomics and metabolomics of flower of Lonicera macranthoides. Zhongguo Zhong Yao Za Zhi 46, 2798–2805. doi: 10.19540/j.cnki.cjcmm.20210227.101
Pan, L., Zheng, J., Liu, J., Guo, J., Liu, F., Liu, L., et al. (2019). Analysis of the ASMT gene family in pepper (Capsicum annuum L.): identification, phylogeny, and expression profiles. Int. J. Genom. 2019:e7241096. doi: 10.1155/2019/7241096
Park, S., Byeon, Y., and Back, K. (2013a). Functional analyses of three ASMT gene family members in rice plants. J. Pineal Res. 55, 409–415. doi: 10.1111/jpi.12088
Park, S., Le, T.-N. N., Byeon, Y., Kim, Y. S., and Back, K. (2013b). Transient induction of melatonin biosynthesis in rice (Oryza sativa L.) during the reproductive stage. J. Pineal Res. 55, 40–45. doi: 10.1111/jpi.12021
Pegler, J. L., Nguyen, D. Q., Oultram, J. M. J., Grof, C. P. L., and Eamens, A. L. (2021). Molecular manipulation of the mir396 and mir399 expression modules alters the response of Arabidopsis thaliana to phosphate stress. Plan. Theory 10:2570. doi: 10.3390/plants10122570
Ros, E., Izquierdo-Pulido, M., and Sala-Vila, A. (2018). Beneficial effects of walnut consumption on human health: role of micronutrients. Curr. Opin. Clin. Nutr. Metab. Care 21, 498–504. doi: 10.1097/MCO.0000000000000508
Schultz, J., Copley, R. R., Doerks, T., Ponting, C. P., and Bork, P. (2000). SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 28, 231–234. doi: 10.1093/nar/28.1.231
Sharif, R., Xie, C., Zhang, H., Arnao, M. B., Ali, M., Ali, Q., et al. (2018). Melatonin and its effects on plant systems. Molecules 23:2352. doi: 10.3390/molecules23092352
Shi, H., Wei, Y., Wang, Q., Reiter, R. J., and He, C. (2016). Melatonin mediates the stabilization of DELLA proteins to repress the floral transition in Arabidopsis. J. Pineal Res. 60, 373–379. doi: 10.1111/jpi.12320
Steffen, L. M., Yi, S. Y., Duprez, D., Zhou, X., Shikany, J. M., and Jacobs, D. R. (2021). Walnut consumption and cardiac phenotypes: the coronary artery risk development in young adults (CARDIA) study. Nutr. Metab. Cardiovasc. Dis. 31, 95–101. doi: 10.1016/j.numecd.2020.09.001
Su, G., Morris, J. H., Demchak, B., and Bader, G. D. (2014). Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinformatics 47, 8.13.1–8.13.24. doi: 10.1002/0471250953.bi0813s47
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi: 10.1093/nar/gky1131
Tan, D.-X., and Reiter, R. J. (2020). An evolutionary view of melatonin synthesis and metabolism related to its biological functions in plants. J. Exp. Bot. 71, 4677–4689. doi: 10.1093/jxb/eraa235
Tiwari, R. K., Lal, M. K., Kumar, R., Mangal, V., Altaf, M. A., Sharma, S., et al. (2021). Insight into melatonin-mediated response and signaling in the regulation of plant defense under biotic stress. Plant Mol. Biol. doi: 10.1007/s11103-021-01202-3
Tsunoda, Y., Hano, S., Imoto, N., Shibuya, T., Ikeda, H., Amagaya, K., et al. (2021). Physiological roles of tryptophan decarboxylase revealed by overexpression of SlTDC1 in tomato. Sci. Hortic. 275:109672. doi: 10.1016/j.scienta.2020.109672
Verde, A., Míguez, J. M., Leao-Martins, J. M., Gago-Martínez, A., and Gallardo, M. (2022). Melatonin content in walnuts and other commercial nuts. Influence of cultivar, ripening and processing (roasting). J. Food Compos. Anal. 105:104180. doi: 10.1016/j.jfca.2021.104180
Wang, Q., An, B., Wei, Y., Reiter, R. J., Shi, H., Luo, H., et al. (2016). Melatonin regulates root meristem by repressing auxin synthesis and polar auxin transport in Arabidopsis. Front. Plant Sci. 7:1882. doi: 10.3389/fpls.2016.01882
Wang, Z., Mu, Y., Hao, X., Yang, J., Zhang, D., Jin, Z., et al. (2021). H2S aids osmotic stress resistance by S-sulfhydration of melatonin production-related enzymes in Arabidopsis thaliana. Plant Cell Rep. 41, 365–376. doi: 10.1007/s00299-021-02813-2
Wang, H., Song, C., Fang, S., Wang, Z., Song, S., Jiao, J., et al. (2022). Genome-wide identification and expression analysis of the ASMT gene family reveals their role in abiotic stress tolerance in apple. Sci. Hortic. 293:110683. doi: 10.1016/j.scienta.2021.110683
Wang, P., Sun, X., Wang, N., Tan, D.-X., and Ma, F. (2015). Melatonin enhances the occurrence of autophagy induced by oxidative stress in Arabidopsis seedlings. J. Pineal Res. 58, 479–489. doi: 10.1111/jpi.12233
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., and Yu, J. (2010). KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinform. 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Wei, Y., Bai, Y., Cheng, X., Zhu, B., Reiter, R. J., and Shi, H. (2020). The dual roles of melatonin biosynthesis enzymes in the coordination of melatonin biosynthesis and autophagy in cassava. J. Pineal Res. 69:e12652. doi: 10.1111/jpi.12652
Wei, Y., Liu, G., Chang, Y., Lin, D., Reiter, R. J., He, C., et al. (2018). Melatonin biosynthesis enzymes recruit WRKY transcription factors to regulate melatonin accumulation and transcriptional activity on W-box in cassava. J. Pineal Res. 65:e12487. doi: 10.1111/jpi.12487
Wheeler, T. J., and Eddy, S. R. (2013). nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489. doi: 10.1093/bioinformatics/btt403
Williams, T. A., Schrempf, D., Szöllősi, G. J., Cox, C. J., Foster, P. G., and Embley, T. M. (2021). Inferring the deep past from molecular data. Genome Biol. Evol. 13:evab067. doi: 10.1093/gbe/evab067
Xu, W., Cai, S.-Y., Zhang, Y., Wang, Y., Ahammed, G. J., Xia, X.-J., et al. (2016). Melatonin enhances thermotolerance by promoting cellular protein protection in tomato plants. J. Pineal Res. 61, 457–469. doi: 10.1111/jpi.12359
Yao, J.-W., Ma, Z., Ma, Y.-Q., Zhu, Y., Lei, M.-Q., Hao, C.-Y., et al. (2021). Role of melatonin in UV-B signaling pathway and UV-B stress resistance in Arabidopsis thaliana. Plant Cell Environ. 44, 114–129. doi: 10.1111/pce.13879
Ying, X., Redfern, B., Gmitter, F. G., and Deng, Z. (2020). Heterologous expression of the Constitutive Disease Resistance 2 and 8 Genes from Poncirus trifoliata restored the hypersensitive response and resistance of Arabidopsis cdr1 mutant to bacterial pathogen pseudomonas syringae. Plan. Theory 9:821. doi: 10.3390/plants9070821
Zhan, H., Nie, X., Zhang, T., Li, S., Wang, X., Du, X., et al. (2019). Melatonin: a small molecule but important for salt stress tolerance in plants. Int. J. Mol. Sci. 20:709. doi: 10.3390/ijms20030709
Zhang, Z., Hu, Q., Liu, Y., Cheng, P., Cheng, H., Liu, W., et al. (2019b). Strigolactone represses the synthesis of melatonin, thereby inducing floral transition in Arabidopsis thaliana in an FLC-dependent manner. J. Pineal Res. 67:e12582. doi: 10.1111/jpi.12582
Zhang, P., Li, X., Wang, Y., Guo, W., Chachar, S., Riaz, A., et al. (2021). PRMT6 physically associates with nuclear factor Y to regulate photoperiodic flowering in Arabidopsis. aBIOTECH 2, 403–414. doi: 10.1007/s42994-021-00065-y
Zhang, J., Shi, Y., Zhang, X., Du, H., Xu, B., and Huang, B. (2017). Melatonin suppression of heat-induced leaf senescence involves changes in abscisic acid and cytokinin biosynthesis and signaling pathways in perennial ryegrass (Lolium perenne L.). Environ. Exp. Bot. 138, 36–45. doi: 10.1016/j.envexpbot.2017.02.012
Zhang, H., Wang, L., Shi, K., Shan, D., Zhu, Y., Wang, C., et al. (2019a). Apple tree flowering is mediated by low level of melatonin under the regulation of seasonal light signal. J. Pineal Res. 66:e12551. doi: 10.1111/jpi.12551
Zhang, H.-J., Zhang, N., Yang, R.-C., Wang, L., Sun, Q.-Q., Li, D.-B., et al. (2014). Melatonin promotes seed germination under high salinity by regulating antioxidant systems, ABA and GA4 interaction in cucumber (Cucumis sativus L.). J. Pineal Res. 57, 269–279. doi: 10.1111/jpi.12167
Zhao, D., Wang, R., Liu, D., Wu, Y., Sun, J., and Tao, J. (2018). Melatonin and expression of tryptophan decarboxylase gene (TDC) in herbaceous peony (Paeonia lactiflora Pall.) flowers. Molecules 23:1164. doi: 10.3390/molecules23051164
Zhao, D., Yao, Z., Zhang, J., Zhang, R., Mou, Z., Zhang, X., et al. (2021). Melatonin synthesis genes N-acetylserotonin methyltransferases evolved into caffeic acid O-methyltransferases and both assisted in plant terrestrialization. J. Pineal Res. 71:e12737. doi: 10.1111/jpi.12737
Zhao, D., Yu, Y., Shen, Y., Liu, Q., Zhao, Z., Sharma, R., et al. (2019). Melatonin synthesis and function: evolutionary history in animals and plants. Front. Endocrinol. 10:249. doi: 10.3389/fendo.2019.00249
Keywords: melatonin, ASMT (N-acetylserotonin methyltransferase), walnut, bioinformatics, floral bud development
Citation: Ma K, Xu R, Zhao Y, Han L, Xu Y, Li L, Wang J and Li N (2022) Walnut N-Acetylserotonin Methyltransferase Gene Family Genome-Wide Identification and Diverse Functions Characterization During Flower Bud Development. Front. Plant Sci. 13:861043. doi: 10.3389/fpls.2022.861043
Edited by:
Qing-Yong Yang, Huazhong Agricultural University, ChinaReviewed by:
Dong Fang Ma, Yangtze University, ChinaJunliang Yin, Yangtze University, China
Chen Zhu, Fujian Agriculture and Forestry University, China
Copyright © 2022 Ma, Xu, Zhao, Han, Xu, Li, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Wang, d2FuZ2p1YW5AeGFhcy5hYy5jbg==; Ning Li, bGluaW5nQHhhYXMuYWMuY24=
†These authors share first authorship