 Darshan T. Dharajiya
Darshan T. Dharajiya Nitin Shukla
Nitin Shukla Maharshi Pandya
Maharshi Pandya Amrutlal K. Patel
Amrutlal K. Patel- Gujarat Biotechnology Research Centre (GBRC), Department of Science and Technology, Government of Gujarat, Gandhinagar, Gujarat, India
Cumin (Cuminum cyminum L.), an important spice crop belonging to the Apiaceae family is infected by Fusarium oxysporum f. sp. cumini (Foc) to cause wilt disease, one of the most devastating diseases of cumin adversely affects its production. As immune responses of cumin plants against the infection of Foc are not well studied, this research aimed to identify the genes and pathways involved in responses of cumin (cv. GC-2, GC-3, GC-4, and GC-5) to the wilt pathogen. Differential gene expression analysis revealed a total of 2048, 1576, 1987, and 1174 differentially expressed genes (DEGs) in GC-2, GC-3, GC-4, and GC-5, respectively. In the resistant cultivar GC-4 (resistant against Foc), several important transcripts were identified. These included receptors, transcription factors, reactive oxygen species (ROS) generating and scavenging enzymes, non-enzymatic compounds, calcium ion (Ca2+) transporters and receptors, R-proteins, and PR-proteins. The expression of these genes is believed to play crucial roles in conferring resistance against Foc. Gene ontology (GO) analysis of the up-regulated DEGs showed significant enrichment of 19, 91, 227, and 55 biological processes in GC-2, GC-3, GC-4, and GC-5, respectively. Notably, the resistant cultivar GC-4 exhibited enrichment in key GO terms such as ‘secondary metabolic process’, ‘response to reactive oxygen species’, ‘phenylpropanoid metabolic process’, and ‘hormone-mediated signaling pathway’. Furthermore, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed the enrichment of 28, 57, 65, and 30 pathways in GC-2, GC-3, GC-4, and GC-5, respectively, focusing on the up-regulated DEGs. The cultivar GC-4 showed enrichment in pathways related to steroid biosynthesis, starch and sucrose metabolism, fatty acid biosynthesis, butanoate metabolism, limonene and pinene degradation, and carotenoid biosynthesis. The activation or up-regulation of various genes and pathways associated with stress resistance demonstrated that the resistant cultivar GC-4 displayed enhanced defense mechanisms against Foc. These findings provide valuable insights into the defense responses of cumin that could contribute to the development of cumin cultivars with improved resistance against Foc.
1 Introduction
Cumin (Cuminum cyminum L.) from the Apiaceae family is a small annual and herbaceous plant (Mnif and Aifa, 2015). Cumin seeds have several applications in industries like spice, pharmaceutical, and food, and are also used in traditional Ayurvedic medicine to treat several diseases (Arun et al., 2016; Sharma et al., 2016; Srinivasan, 2018; Choudhary et al., 2021). India is the leading country in the world in cumin cultivation area, production, consumption, and supplier/exporter since 2010 (Meena et al., 2018; Kumar et al., 2021). In India, Gujarat has been the leading state of cumin production and productivity followed by Rajasthan (Kumar et al., 2021). However, cumin production is adversely affected by biotic factors, mainly fungal diseases such as wilt (Fusarium oxysporum f. sp. cumini (Foc)), blight (Alternaria burnsii), and powdery mildew (Erysiphe polygoni) (Dange, 1995). Among the fungal diseases, wilt, caused by F. oxysporum f. sp. cumini, is considered a severe disease that hinders worldwide cumin production. It causes high yield losses, up to 100% in severely damaged areas with disease-promoting environments (Abo-Elyousr et al., 2022). Because of the prolonged existence and accumulation of Foc in soil, there is no effective control strategy available at present. Therefore, understanding of Foc resistance mechanism, effective chemical-free and environmentally-safe strategies to reduce the substantial loss caused by Foc in cumin production are important and highly required.
The wilt pathogen can infect plants in all the stages of growth, but the severity of wilt increases with the age of the plant. In the severe infection, the plants and leaves wilt and die. Seeds formed by infected plants are thin, small, and shriveled. The seeds are often contaminated during harvesting and the pathogen spreads to new areas (Lodha and Mawar, 2014). Wilt disease in cumin can be managed by cultural, biological, and chemical approaches. While various fungicides may provide effective control of Foc, the excessive use of these chemicals poses significant hazards to the environment and human health, as well as the issue of resistance development (Abo-Elyousr et al., 2022). With the absence of an effective wilt management approach, research efforts have primarily focused on identifying resistant sources and utilizing them in resistance breeding programs. There are limited resistant sources against wilt pathogens in the available germplasm throughout the world (Lodha and Mawar, 2014).
In recent years, transcriptome analysis in different crops like flax (Boba et al., 2021), cotton (Hou et al., 2021), banana (Dong et al., 2020), mung bean (Chang et al., 2021), and common bean (Leitão et al., 2021) have been performed to elucidate the genes and pathways for fusarium wilt (caused by different subspecies or special forms of F. oxysporum in different crops) resistance. Response of plants towards F. oxysporum infection varied in different crops. The defense responses of cumin to Foc infection are not well known even though this information is very important to plan effective approaches against destructive diseases. The identification of genes responsible for resistance against Foc can be of great importance for understanding the detailed molecular mechanism of resistance in cumin to fusarium wilt disease and ultimately utilizing the genes related to Foc resistance for cultivar improvement in cumin. Hence, the main objective of this study was to reveal the genes and pathways responsible for Foc resistance in cumin cultivars by transcriptome analysis and to understand the mechanism of resistance.
2 Materials and methods
2.1 Plant sample collection
Plant samples of four cumin cultivars namely, Gujarat cumin-2 (GC-2) (susceptible to wilt), GC-3 (resistant to wilt), GC-4 (resistant to wilt), and GC-5 (moderately resistant to wilt) (Meena et al., 2010) were collected from Seed Spices Research Station, Sardarkrushinagar Dantiwada Agricultural University (SDAU), Jagudan (23.5177° N Latitude, 72.4125° E Longitude), Mehsana, Gujarat in the year of 2020. The samples of healthy and wilt diseased plants of all the cultivars were collected at their reproductive stage from the control plot and wilt sick plot, respectively. The plants showing wilt disease symptoms were collected and used for the isolation of pathogens. The pure culture of isolated fungus was observed under microscope and identified by PCR amplification using ITS1 (5’-TCCGTAGGTGAACCTGCGG-3’) and ITS4 (5’-TCCTCCGCTTATTGATATGC-3’) primers and sequencing of ITS region of ribosomal DNA by automatic capillary sequencer ABI 3500XL (Applied Biosystems, CA, USA). The sequence data were used for BLAST analysis on NCBI.
2.2 RNA extraction, library preparation, and transcriptome sequencing
Plant samples (aerial parts of the plant) were powdered using liquid nitrogen in a mortar and pestle. Total RNA was extracted using RNeasy® Plant Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions. Extracted RNA samples were checked on denatured agarose gel. The purity of RNA samples was checked using a QIAxpert spectrophotometer (Qiagen, Germany). The quantity and integrity of the RNA were assessed by the Qubit™ 4 Fluorometer (Thermo Fisher Scientific, MA, USA) and Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA), respectively. A total of eight cDNA libraries (GC-2-H, GC-2-I, GC-3-H, GC-3-I, GC-4-H, GC-4-I, GC-5-H, and GC-5-I) from RNA samples were prepared using Ion Total RNA-Seq Kit v2 (Thermo Fisher Scientific, MA, USA). Template preparation was done by Ion Chef™ Instrument (Thermo Fisher Scientific, MA, USA) and sequencing was performed on Ion GeneStudio™ S5 Plus System (Thermo Fisher Scientific, MA, USA). The raw sequences were submitted to sequence read archive (SRA) – NCBI (Supplementary Table S1).
2.3 Transcript assembly and differential gene expression analysis
The quality assessment of reads was performed by FastQC (v.0.11.9). After a quality check of reads, PRINSEQ++ (v.1.2.4) was used to remove low-quality sequences (Q value <20 and read length <100 bp). The high-quality reads were reconstructed and assembled into unigenes by Trinity (v.2.8.5) with default parameters. The assembled sequences were annotated with Transdecoder (v.5.5.0) for prediction of the open reading frame. The individual samples were mapped and quantified against the assembled transcriptome using salmon (v.1.9.0). The gene expression levels of individual transcripts were estimated by transcripts per million (TPM). The counts data was simulated using the Seqgendiff package (v.1.2.3) (Gerard, 2020) and analysis of differentially expressed genes (DEGs) between infected and healthy plants of each cultivar was performed with the DeSeq2 package (v.1.34.0) in R. The genes with threshold p-value ≤ 0.05 | log2FoldChange (log2FC) ≥ 2 and ≤ −2 were considered significant DEGs. The Venn diagrams for DEGs were drawn by using InteractiVenn (Heberle et al., 2015), an online tool.
2.4 Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis
The transcripts from individual samples were annotated using the KOBAS-i (v.3.0) tool (Bu et al., 2021). The biological significance of up- and down-regulated genes was assessed by the KEGG pathway and GO enrichment which provided three ontologies, viz., biological process, cellular component, and molecular function using ShinyGO (v.0.76.3) (Ge et al., 2020). The enrichment analysis was performed by hypergeometric distribution followed by false discovery rate (FDR) correction. The data of annotated DEGs were used in R Studio for graphical visualization with the ggplot2 package (3.4.0) and pheatmap package (1.0.12) from the bioconductor. The illustration for the proposed defense response in cumin against Foc was prepared by Microsoft PowerPoint 2016.
3 Results
3.1 RNA-seq data processing and de novo assembly
To study the variation in gene expression in GC-2, GC-3, GC-4, and GC-5, healthy and infected plants of these cultivars were collected. The fungal culture isolated from infected plants was confirmed as F. oxysporum by microscopic observation and sequence analysis (Supplementary File 1). Transcriptomes of healthy and infected plants of cumin cultivars were analyzed. De novo assembly of cumin was constructed using Trinity and individual transcriptome samples were mapped against the de novo assembly of cumin. The assembly contained a median contig length of 344 bp with a total assembled 29,836,958 bases. It represented 62,845 ‘genes’ and 65,389 transcripts with GC content of 44.35% and N50 value of 501 bp (Supplementary Table S2). A total of 169,992,277 reads were generated from eight libraries with a mean of 21,249,035 reads (Supplementary Table S3). The mapping of eight samples with the de novo assembly of cumin ranged from 82.19% to 95.42% with an average of 92.2% (Supplementary Table S3).
3.2 Identification of differentially expressed genes (DEGs)
The healthy and infected samples of each cultivar were used to identify DEGs. The total DEGs were identified with a threshold of p-value ≤ 0.05 and with log2FC ≥ 2.0 and ≤ −2.0 for up- and down-regulated DEGs, respectively. Among all the annotated DEGs, duplicates were removed for further analysis. Among these DEGs, 807, 1239, 1567, and 837 were up-regulated and 1337, 466, 574, and 406 were down-regulated in GC-2, GC-3, GC-4, and GC-5, respectively (Figure 1).
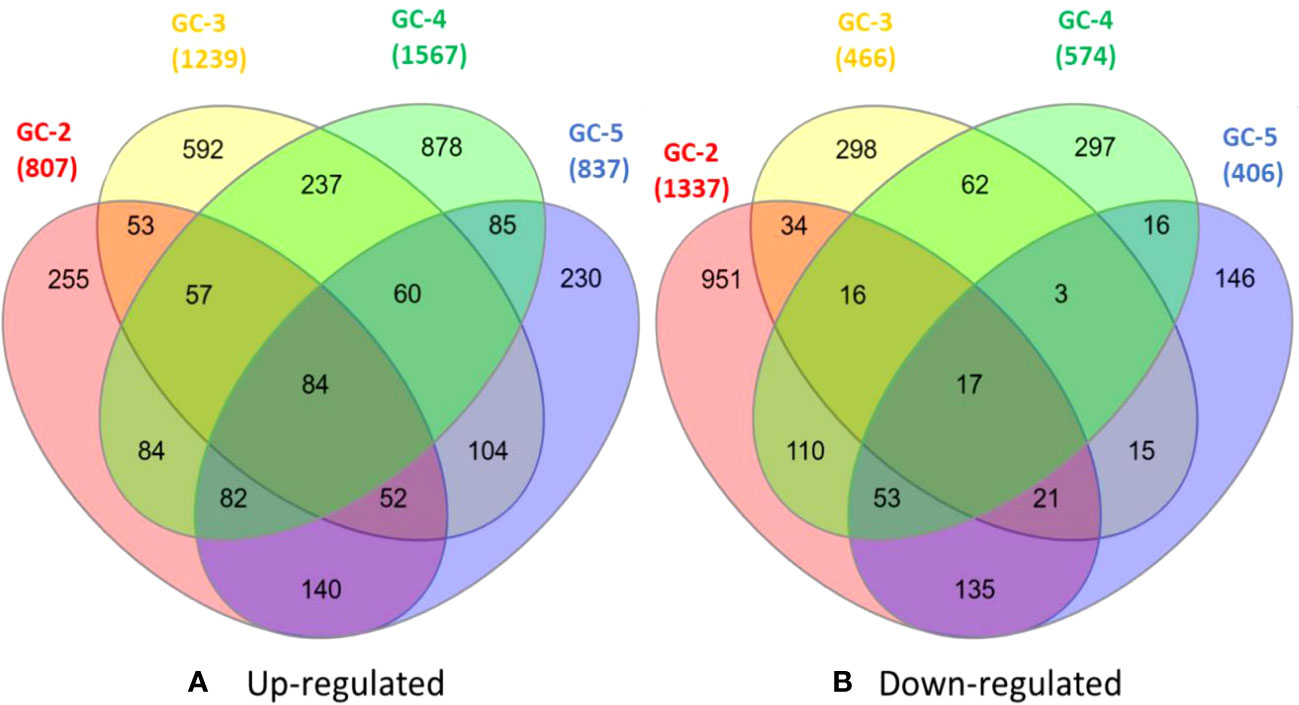
Figure 1 Venn diagrams of DEGs (A) up-regulated and (B) down-regulated healthy and diseased plants of cumin cultivars (GC-2, GC-3, GC-4 and GC-5). The cut-off set for DEGs was, log2FC ≥ 2 for up-regulated and ≤ −2 for down-regulated. The cut-off for p-value ≤ 0.05 was set for all DEGs.
The list of DEGs of all the cultivars is given in Supplementary File 2. Inter-cultivar comparison of up-regulated DEGs of GC-2 (susceptible) and GC-3 (resistant) showed that 561 and 993 were unique in GC-2 and GC-3, respectively while comparing down-regulated DEGs, 1249 and 378 were unique in GC-2 and GC-3, respectively. By comparing up-regulated DEGs of GC-2 (susceptible) and GC-4 (resistant), 500 and 1260 DEGs were unique in GC-2 and GC-4, respectively while comparing down-regulated DEGs, 1141 and 378 were unique in GC-2 and GC-4, respectively. The unique and common DEGs for susceptible and resistant/moderately resistant cultivars have been shown in Figure 2.
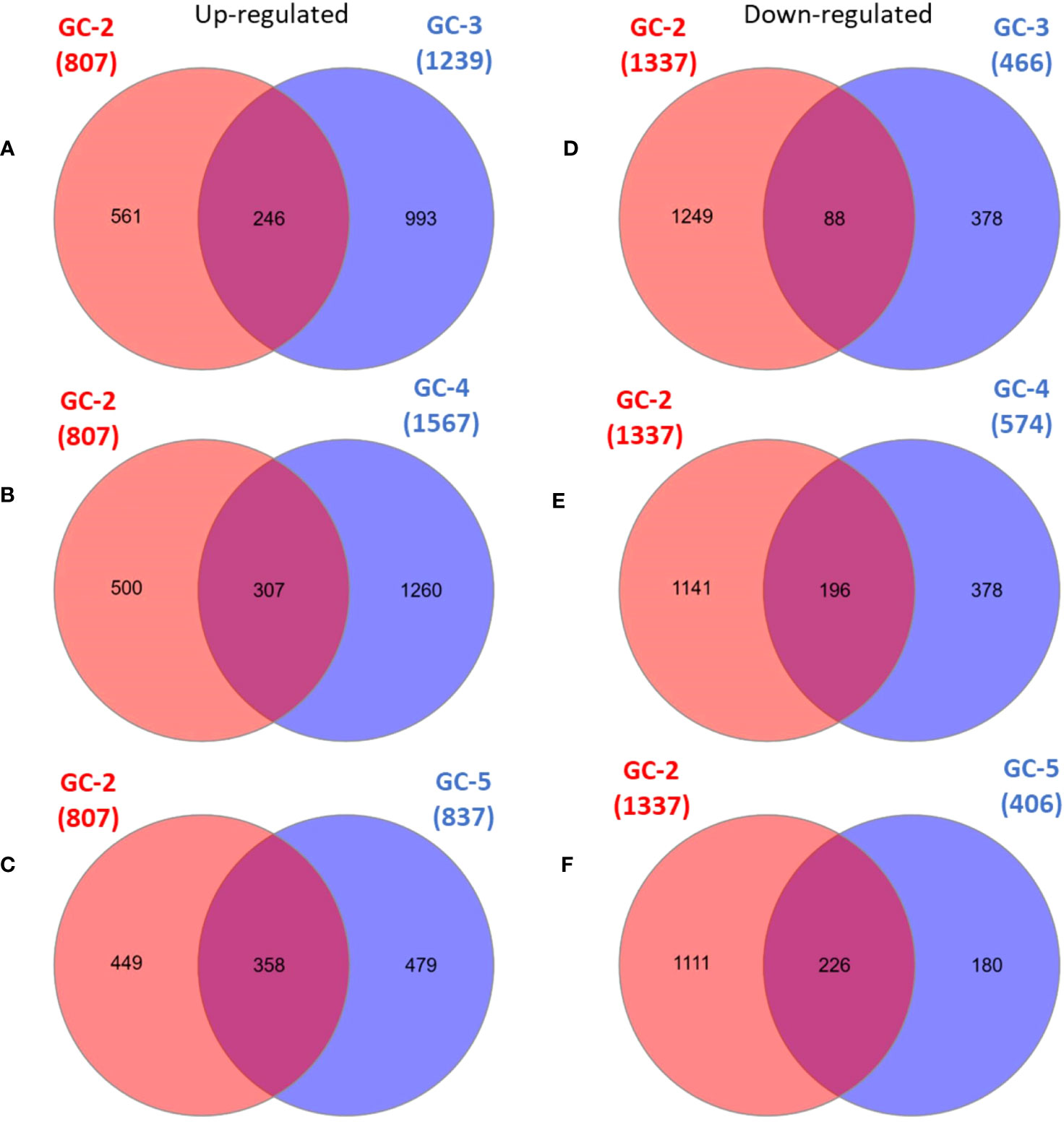
Figure 2 Venn diagrams of DEGs in susceptible cultivar (GC-2) vs resistant (GC-3 and GC-4)/moderately resistant cultivar (GC-5). (A) up-regulated DEGs: GC-2 vs GC-3, (B) up-regulated DEGs: GC-2 vs GC-4, (C) up-regulated DEGs: GC-2 vs GC-5, (D) down-regulated DEGs: GC-2 vs GC-3, (E) down-regulated DEGs: GC-2 vs GC-4, (F) down-regulated DEGs: GC-2 vs GC-5. The cut-off for the log2FC was set ≥ 2 for up-regulated DEGs and ≤ −2 for down-regulated DEGs. The cut-off for p-value was ≤ 0.05 for all DEGs.
The volcano plots representing the distribution of DEGs in healthy and infected plants of GC-2, GC-3, GC-4, and GC-5 are presented in Figure 3. By considering the cut-off p-value ≤ 0.05 and log2FC ≥ 2 for up-regulated DEGs and ≤ −2 for down-regulated DEGs a total of 2048, 1576, 1987, and 1174 DEGs were considered for GC-2, GC-3, GC-4, and GC-5, respectively. More DEGs with high Log2FC were plotted for GC-4 compared to the susceptible cultivar (GC-2). The hierarchical clustering properly divided the healthy plant samples from the infected plant samples representing the differential regulation of genes based on normalized counts for the top fifty DEGs in all four cultivars (Supplementary Figures S1–S4).
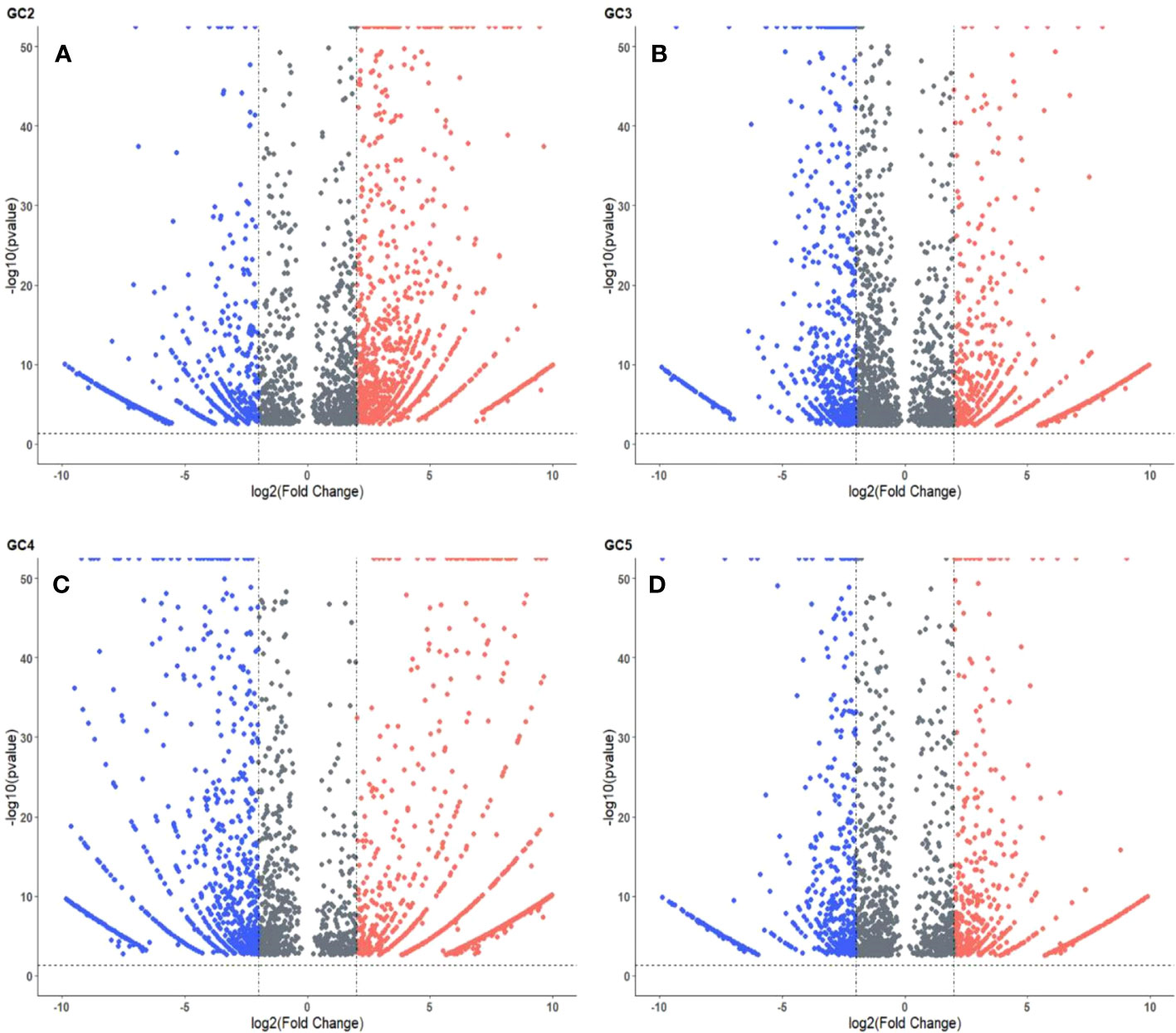
Figure 3 Volcano plot of DEGs in cumin cultivars (A) GC2, (B) GC3, (C) GC4 and (D) GC5. The cut-off for the log2FC was set ≥ 2 for up-regulated DEGs and ≤ −2 for down-regulated DEGs. The cut-off for p-value was ≤ 0.05 for all DEGs. Red and blue colors indicate up-regulated and down-regulated DEGs, respectively.
3.3 Functional classification of DEGs
Test sets for GO analysis with up-regulated DEGs from all the cultivars resulted in 44, 41, 130, and 22 enriched significant GO terms for molecular function in GC-2, GC-3, GC-4, and GC-5, respectively. The details on no. of genes involved in the pathway and enriched genes in individual pathways in each cultivar are given in Supplementary File 3. In GC-4, enriched unique key GO terms for molecular function were ‘transmembrane transporter activity’, ‘structural constituent of cytoskeleton’, ‘proteasome-activating activity’, ‘peptidase activity’, ‘isoprenoid binding’, ‘hormone binding’, ‘endopeptidase activity’, ‘catalase activity’ and ‘abscisic acid binding’ (Figure 4). In GC-3, unique GO terms for molecular function were ‘enriched channel activity’ and ‘ammonia-lyase activity’. The GO analysis for down-regulated DEGs, resulted in 67, 37, and 22 enriched GO terms for molecular function in GC-2, GC-4, and GC-5, respectively (Supplementary File 3). No significant enrichment in GO terms for molecular function was observed in GC-3. Many of the GO terms for molecular function enriched for down-regulated in the susceptible cultivar (GC-2) were found to be up-regulated DEGs in resistant cultivars. The results of GO for molecular function by DEGs in different cultivars are represented in Figure 4.
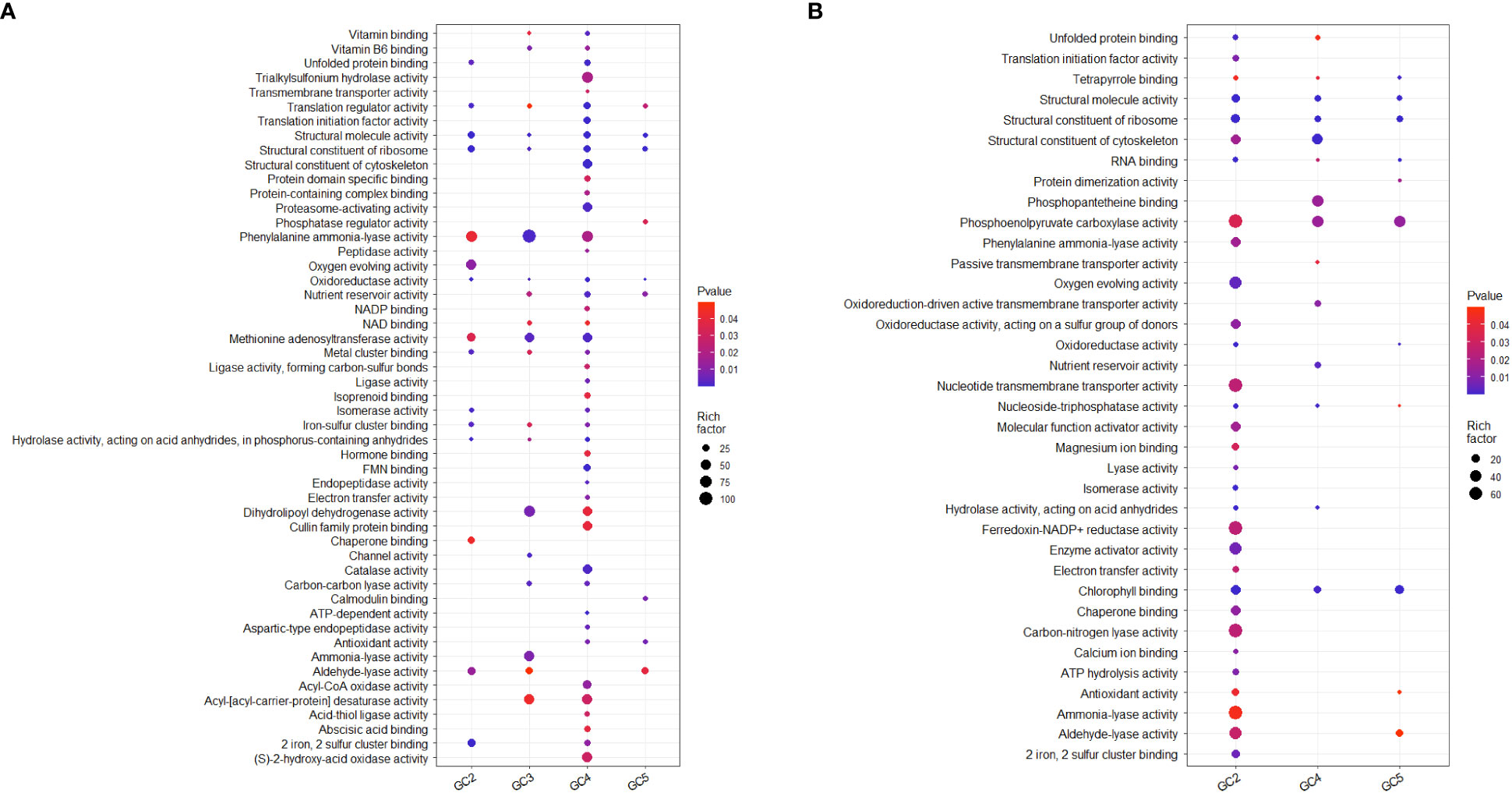
Figure 4 Overview of selected enriched GO terms for molecular function (A) up-regulated and (B) down-regulated in transcriptome of cumin cultivars in response to Foc infection. The color shows p-value (Bejamini Hochberg) and size of the dots represents the rich factor of significant genes involved in each corresponding process. Rich factor is the ratio of the number of DEGs to the total number of genes in a given GO term. In GC-3 cultivar, no significant enrichment was observed for down-regulated DEGs hence not considered in the plot.
The results of GO analysis showed that total up-regulated significantly enriched terms for biological processes in GC-2, GC-3, GC-4, and GC-5 were 19, 91, 227, and 55, respectively and their details are given in Supplementary File 4. The enriched GO terms for biological processes in GC-4 were ‘secondary metabolic process’, ‘response to reactive oxygen species’, ‘response to organic substance’, ‘response to endogenous stimulus’, ‘phenylpropanoid metabolic process’, ‘hydrogen peroxide metabolic process’ and ‘hormone-mediated signaling pathway’. Compared to GC-4, GC-3 had less no. of uniquely up-regulated GO terms for biological processes i.e. ‘terpenoid synthetic process’, ‘response to abiotic stimulus’, ‘protein refolding’, ‘olefinic compound biosynthetic process’, and ‘isoprenoid biosynthetic process’. There were some commonly enriched GO terms for biological processes in GC-3 and GC-4, e.g. ‘translation’, ‘small molecule biosynthetic process’, ‘protein folding’, ‘oxidative phosphorylation’, ‘organic acid metabolic process’, and ‘cinnamic acid biosynthetic process’ (Figure 5). GO analysis for down-regulated DEGs, resulted in 118, 74, and 55 enriched GO terms for biological processes in GC-2, GC-4, and GC-5, respectively (Supplementary File 4). No significant enrichment in GO terms for biological processes was observed in GC-3. Some GO terms for biological processes like, ‘response to inorganic substance’, ‘response to chemical’, ‘response to abiotic stimulus’, ‘protein repair’, ‘pigment biosynthetic process’, ‘cinnamic acid biosynthetic process’ etc. were enriched for down-regulated DEGs in susceptible cultivar (GC-2) and most of these terms were up-regulated in resistant cultivars (Figure 5).
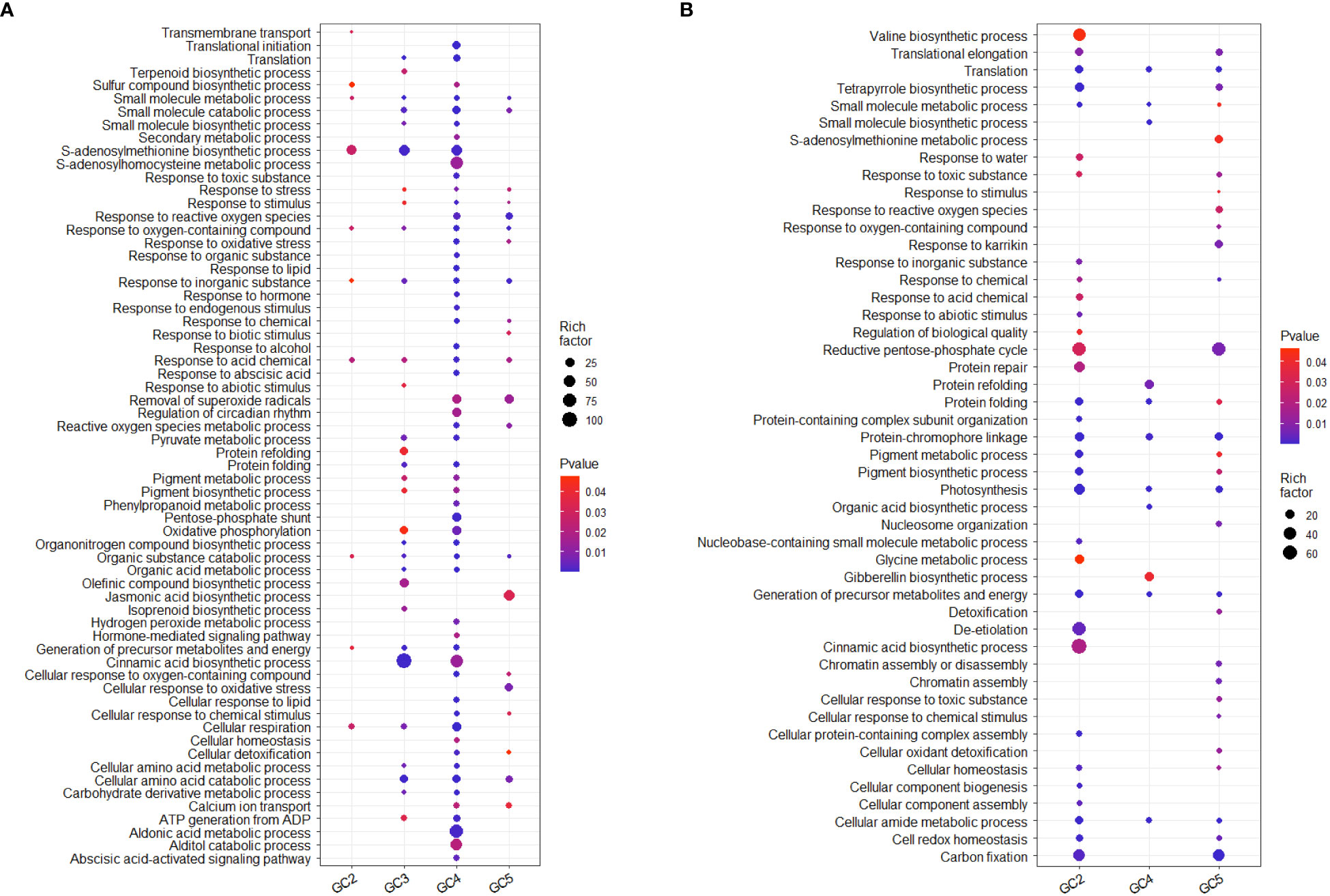
Figure 5 Overview of selected enriched GO terms for biological process (A) up-regulated and (B) down-regulated in transcriptome of cumin cultivars in response to Foc infection. The color shows p-value (Bejamini Hochberg) and size of the dots represents the rich factor of significant genes involved in each corresponding process. Rich factor is the ratio of the number of DEGs to the total number of genes in a given GO term. In GC-3 cultivar, no significant enrichment was observed for down-regulated DEGs hence not considered in the plot.
Considering all the up-regulated DEGs in different cultivars, 29 and 44 GO terms for cellular components were significantly enriched in resistant cultivars, GC-3 and GC-4, respectively. Considering up-regulated DEGs, no GO term for cellular components was significantly enriched in GC-2 and GC-5. In the case of down-regulated DEGs, 71, 4, 51, and 34 GO terms for cellular components were significantly enriched in GC-2, GC-3, GC-4, and GC-5, respectively (Supplementary File 5).
3.4 KEGG pathway enrichment analysis
KEGG pathway analysis based on all the DEGs showed that 76 and 51 pathways were significantly up- and down-regulated, respectively in one or more cultivar(s) (Supplementary Files 6, 7). Many of them were probably directly or indirectly involved in plant defense against biotic or abiotic stresses in at least one of the cultivars. Considering the up-regulated DEGs, 28, 57, 65, and 30 KEGG pathways were enriched in GC-2, GC-3, GC-4, and GC-5, respectively. Among those enriched KEGG pathways, some metabolic pathways i.e. steroid biosynthesis, limonene and pinene degradation, butanoate metabolism, RNA degradation, starch and sucrose metabolism, aminoacyl-tRNA biosynthesis, fatty acid biosynthesis, inositol phosphate metabolism, biotin metabolism, and carotenoid biosynthesis were only enriched in GC-4 (resistant) and photosynthesis, tropane, piperidine, and pyridine alkaloid biosynthesis, SNARE interactions in vesicular transport, tyrosine metabolism, folate biosynthesis were only enriched in GC-3 (resistant) (Figure 6). Some pathways like phenylpropanoid pathway, phenylalanine metabolism, ubiquitin-mediated proteolysis, circadian rhythm, alanine, aspartate and glutamate metabolism, arginine biosynthesis, proteasome, phenylalanine, tyrosine and tryptophan biosynthesis, ascorbate and aldarate metabolism, propanoate metabolism, endocytosis, nucleocytoplasmic transport were enriched in GC-3 and GC-4 (Figure 6). The detailed results of KEGG pathway analysis for up-regulated DEGs of all the cultivars is shown in Supplementary File 6. The number of genes enriched in most of the individual pathways mentioned above was highest in GC-4, followed by GC-3, GC-5, and GC-2 (Supplementary File 6).
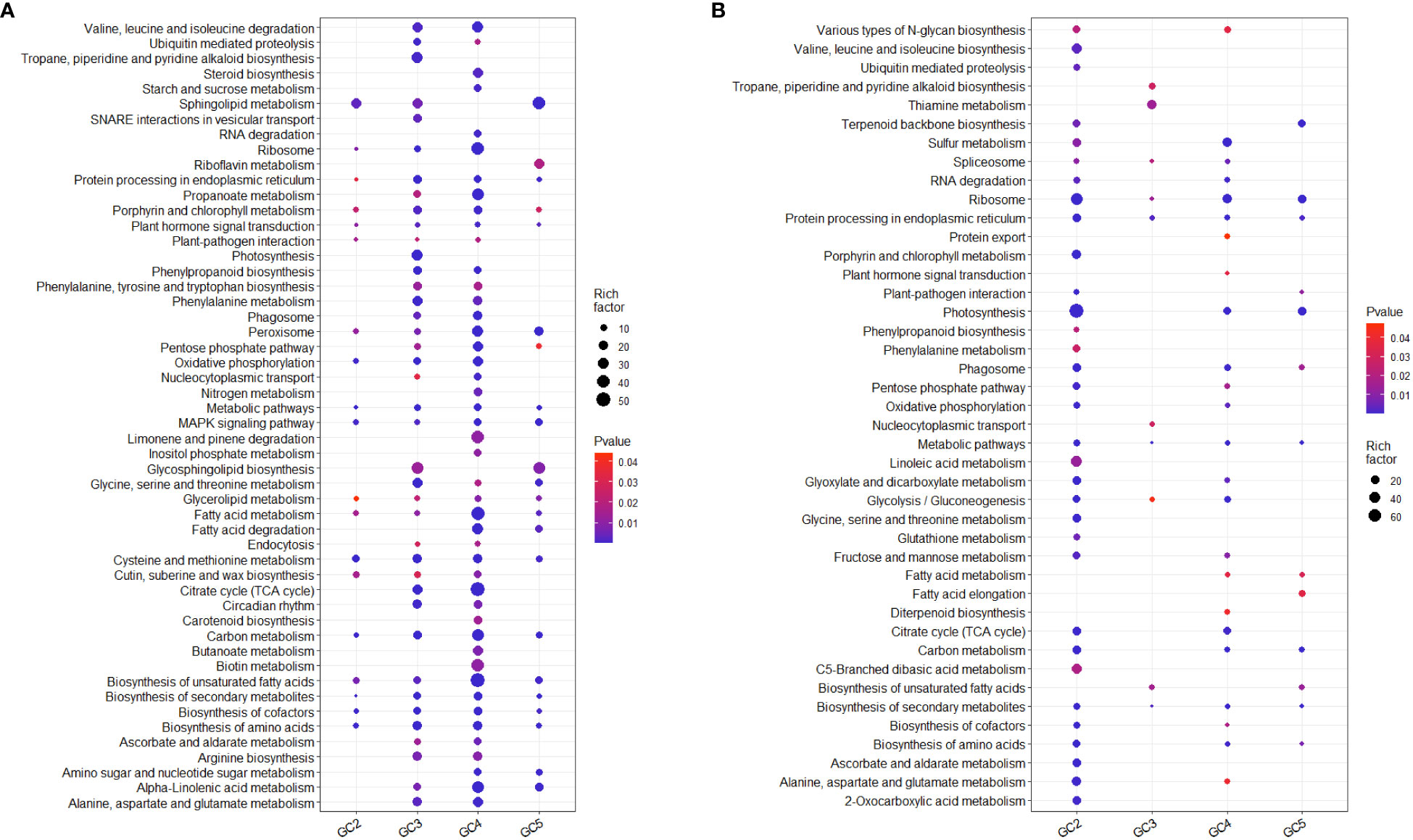
Figure 6 Enriched KEGG pathways of significant differentially expressed genes in cumin cultivars in response to Foc infection. (A) Up-regulated KEGG pathways and (B) Down-regulated KEGG pathways. The color of the dots represents the range of P-value and size represents the rich factor of significant genes involved in a pathway. Rich factor is the ratio of the number of DEGs to the total number of genes in a given KEGG pathway.
While considering down-regulated DEGs, 39, 11, 32, and 15 KEGG pathways were enriched in GC-2, GC-3, GC-4, and GC-5, respectively. Pathways enriched for down-regulated DEGs in GC-2 (susceptible) only were mostly enriched for up-regulated DEGs in resistant cultivars. The number of genes in enriched pathways was higher in susceptible cultivar (GC-2) compared to resistant or moderately resistant cultivars (Supplementary File 7).
3.5 Important DEGs involved in pathways related to disease-resistance
DEGs in different cultivars were used to identify some important genes that might be responsible for the response to the cumin wilt pathogen. Some of those genes from various important pathways have been given in Table 1.
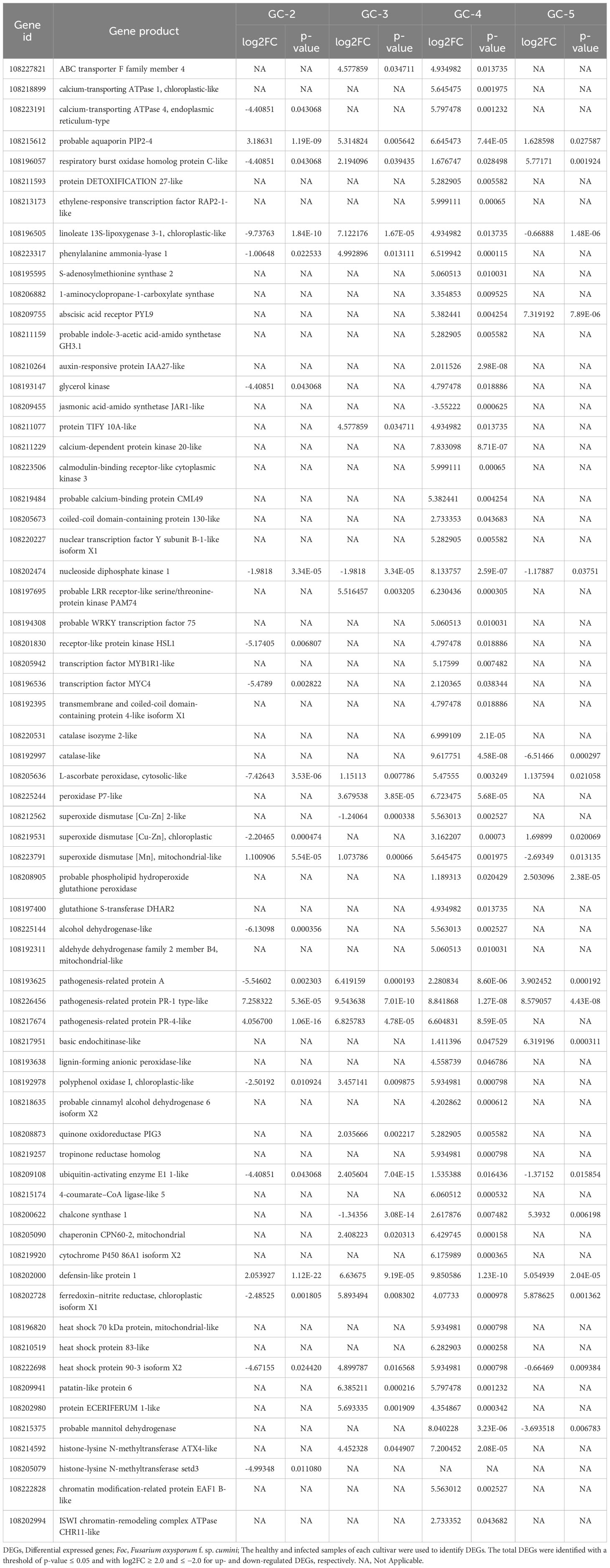
Table 1 Important DEGs encoding transporters, receptors, enzymes and other proteins involved in immune response of cumin cultivars during Foc infection.
During the infection of Foc in cumin cultivars, some signal receptors and transporters for the transport of signals or molecules might have been involved in an immune response. Surface signal receptors or cytoplasmic receptors like ‘calcium-dependent protein kinase 20-like’ (108211229), ‘calmodulin-binding receptor-like cytoplasmic kinase’ (3108223506), ‘coiled-coil domain-containing protein 130-like’ (108205673), ‘nucleoside diphosphate kinase 1’ (108202474), ‘probable LRR receptor-like serine/threonine-protein kinase PAM74’ (108197695), ‘transmembrane and coiled-coil domain-containing protein 4-like isoform X1’ (108192395), and ‘receptor-like protein kinase HSL1’ (108201830) were up-regulated in GC-4. The DEGs for transporters were ‘ABC transporter F family member 4’ (108227821), ‘calcium-transporting ATPase 1, chloroplastic-like’ (108218899), ‘calcium-transporting ATPase 4, endoplasmic reticulum-type’ (108223191), ‘respiratory burst oxidase homolog protein C-like’ (108196057), and ‘protein DETOXIFICATION 27-like’ (108211593) which showed up-regulation in one or both of the resistant cultivars (GC-3 and GC-4). ‘Probable aquaporin PIP2-4’ (108215612) was up-regulated in all the cultivars but Log2FC was higher in GC-3 and GC-4 compared to the susceptible cultivar (Table 1). The activation of such signaling pathways altered gene expression which mainly depended on different transcription factors. Such differentially expressed transcription factors included ‘probable WRKY transcription factor 75’ (108194308), ‘transcription factor MYB1R1-like’ (108205942), ‘transcription factor MYC4’ (108196536), and ‘nuclear transcription factor Y subunit B-1-like isoform X1’ (108220227) which were up-regulated in GC-4 only.
As a result of the immune response towards pathogen infection, ROS and H2O2 flux increased which was scavenged by different antioxidant enzymes in the cell. The DEGs for antioxidant enzymes i.e. ‘catalase isozyme 2-like’ (108220531), ‘catalase-like’ (108192997), ‘superoxide dismutase [Cu-Zn] 2-like’ (108212562), ‘superoxide dismutase [Cu-Zn], chloroplastic’ (108219531), ‘glutathione S-transferase DHAR2’ (108197400), and ‘probable phospholipid hydroperoxide glutathione peroxidase’ (108208905) showed up-regulation in GC-4 only. Expression of the other two ROS scavenging enzymes ‘L-ascorbate peroxidase, cytosolic-like’ (108205636) and ‘peroxidase P7-like’ (108225244) showed up-regulation in GC-3 and GC-4. ‘Superoxide dismutase [Mn], mitochondrial-like’ (108223791) showed up-regulation in all the cultivars.
The expression profiles of different genes involved in pathways of stress-related hormones like jasmonic acid (JA), salicylic acid (SA), ethylene, and abscisic acid (ABA) showed up-regulation of ‘ethylene-responsive transcription factor RAP2-1-like’ (108213173), ‘linoleate 13S-lipoxygenase 3-1, chloroplastic-like’ (108196505), ‘phenylalanine ammonia-lyase 1’ (108223317), ‘S-adenosylmethionine synthase 2’ (108195595), ‘1-aminocyclopropane-1-carboxylate synthase’ (108206882), ‘abscisic acid receptor PYL9’ (108209755), ‘glycerol kinase’ (108193147), ‘jasmonic acid-amido synthetase JAR1-like’ (108209455), and ‘protein TIFY 10A-like’ (108211077) in one or both of the resistant cultivars (Table 1).
Some DEGs involved in defense response including ‘glutathione S-transferase DHAR2’ (108197400), ‘alcohol dehydrogenase-like’ (108225144), ‘endochitinase-like’ (108217951), ‘lignin-forming anionic peroxidase-like’ (108193638), ‘polyphenol oxidase I, chloroplastic-like’ (108192978), ‘probable cinnamyl alcohol dehydrogenase 6 isoform X2’ (108218635), ‘quinone oxidoreductase PIG3’ (108208873), ‘tropinone reductase homolog’ (108219257), ‘ubiquitin-activating enzyme E1 1-like’ (108209108), ‘4-coumarate–CoA ligase-like 5’ (108215174), ‘chalcone synthase 1’ (108200622), ‘chaperonin CPN60-2, mitochondrial’ (108205090), ‘cytochrome P450 86A1 isoform X2’ (108219920), ‘ferredoxin–nitrite reductase, chloroplastic isoform X1’ (108202728), ‘heat shock 70 kDa protein, mitochondrial-like’ (108196820), ‘heat shock protein 83-like’ (108210519), ‘patatin-like protein 6’ (108209941), ‘protein ECERIFERUM 1-like’ (108202980), probable mannitol dehydrogenase (108215375) etc. were up-regulated in GC-4 and might have involved in different pathways for secondary metabolite synthesis, cell wall strengthening and pathways for enhancing pathogen resistance in plant. Among those DEGs, defensin-like protein 1 (108202000) was up-regulated in all the cultivars but the Log2FC was higher in resistant cultivars compared to the susceptible cultivar (Table 1).
The change in the expression of genes providing defense against pathogens might be due to epigenetic changes occurring in GC-4. Some enzymes involved in histone modification e.g. ‘histone-lysine N-methyltransferase ATX4-like’ (108214592) (catalyzes H3K4me3) was up-regulated in GC-4 whereas ‘histone-lysine N-methyltransferase setd3’ (108205079) (catalyzes H3K4me3) was down-regulated in susceptible cultivar (GC-2). Moreover, enzymes involved in chromatin modification e.g. ‘chromatin modification-related protein EAF1 B-like’ (108222828) (involvement in histone acetyltransferase complex) and ‘ISWI chromatin-remodeling complex ATPase CHR11-like’ (108202994) (possesses intrinsic ATP-dependent nucleosome-remodeling activity) were up-regulated in GC-4 only. The methylation, acetylation, and chromatin remodeling pattern might have altered the expression of genes involved in defense against the pathogen.
4 Discussion
Fusarium wilt caused by many forms of soil-borne pathogen F. oxysporum is a widespread plant disease. Several hundred plant species are susceptible to F. oxysporum, including economically important food crops like cumin, coriander, legumes, vegetables, and melons. In the recent years, transcriptome analysis has been performed in different crops like sesame (Wei et al., 2016), banana (Sun et al., 2019; Dong et al., 2020), flax (Boba et al., 2021), cotton (Hou et al., 2021), some pulse crops like mung bean (Chang et al., 2021) and common bean (Leitão et al., 2021) to elucidate the enrichment of pathways or genes for fusarium wilt resistance. Since the cumin defense mechanism against wilt pathogens has not yet been revealed, we attempted to recognize the genes and pathways associated with fusarium wilt resistance in different cultivars by comparing their transcriptome.
Mechanisms of resistance to F. oxysporum are very complex and a network of phytohormone signaling (Boba et al., 2021). In the present study, some defense-related genes and pathways have been enriched during Foc infection. Among the enriched GO terms for biological processes in GC-4, some of them have been reported to be enriched in plants during Fusarium infection such as ‘secondary metabolic process’ (Kaushal et al., 2021), ‘response to reactive oxygen species’ (Chang et al., 2021), ‘response to organic substance’ (Toueni et al., 2016), ‘response to endogenous stimulus’ (Xiong et al., 2021), ‘phenylpropanoid metabolic process’ (Zhu et al., 2021), ‘hydrogen peroxide metabolic process’ (Zhang et al., 2019) and ‘hormone-mediated signaling pathway’ (Zhang et al., 2018) considering up-regulated DEGs.
Considering up-regulated DEGs, more KEGG pathways were enriched in GC-4 followed by GC-3, GC-5, and GC-2. The steroid biosynthesis pathway was enriched in GC-4. In this pathway, genes up-regulated in one or both of the resistant cultivars in the present study, have an important role in biotic stress resistance i.e. fungal infection in plants (Kong et al., 2005; Lu et al., 2020; Pandian et al., 2020). Butanoate metabolism pathway enriched in only GC-4 possessed genes that were reported to be up-regulated during F. oxysporum f. sp. vasinfectum infection in cotton (Yao et al., 2019). ‘Glutamate decarboxylase’ increases gamma-aminobutyric acid (GABA) accumulation which has been observed during various plant-pathogen interactions and has been associated with disease resistance response against tomato pathogen Ralstonia solanacearum (Wang et al., 2019a). ‘Probable enoyl-CoA hydratase 1, peroxisomal’ is involved in β-oxidation of fatty acids which produces cytotoxic ROS as byproducts (Yu et al., 2019). The fatty acid biosynthesis pathway was significantly enriched in resistant cultivars which is in agreement with its enrichment observed in pepper during fusarium wilt (Zhu et al., 2021). Genes up-regulated in resistant cultivars were involved in enhancing resistance against fungal diseases in plants (Lee et al., 2021; Peng et al., 2022). Up-regulated DEGs in the inositol phosphate metabolism pathway in GC-4 were also found to be up-regulated in the previous studies e.g. in pepper during Fusarium infection (Zhu et al., 2021).
In GC-4, some genes involved in disease resistance were up-regulated which might have provided resistance against Foc. Up-regulation of these genes during fungal infection have been supported by some previous reports like, LRR-RLK in resistant rubber tree against Corynespora cassiicola (Roy et al., 2019), transcription factors WRKY, and MYB in apple plant showing resistance against Fusarium proliferatum f. sp. malus domestica (Duan et al., 2022), superoxide dismutase, catalase, ascorbate peroxidase and monodehydroascorbate reductase in resistant plant of cucumber against the infection of Alternaria cucumerina (Sa et al., 2022), ascorbate and carotenoids in pepper against Fusarium infection (Zhu et al., 2021), calcium-dependent protein kinase (CDPK) and R-proteins in banana resistant plant against wilt pathogen (Sun et al., 2019), calmodulin (CaM) and calmodulin-like protein (CML) in wild cabbage resistant to Plasmodiophora brassicae during infection (Zhang et al., 2016), pathogenesis related protein-1 (PR-1) and PR-2 in rose in response to powdery mildew (Chandran et al., 2021), in tomato plants infected with F. oxysporum (Slezina et al., 2021), chitinase and glucanase in apple plant resistant to Fusarium infection (Duan et al., 2022), defensin-like protein 1 in tomato plants infected with F. oxysporum (Slezina et al., 2021) and lignin biosynthesis in cotton during Fusarium wilt infection (Hou et al., 2021). Several ROS scavenging enzymes were more expressed in GC-4 during Foc infection. Our findings were in agreement with the findings of the previous study related to Alternaria leaf spot resistance in cucumber (Sa et al., 2022). Catalase as a major H2O2-scavenging enzyme is also widely involved in plant immunity (Yan et al., 2021). Over-accumulation of ROS, a result of pathogen infection, probably leads to chlorotic and membrane lipid peroxidation (Sa et al., 2022).
During Foc infection in GC-4, interactions among different defense hormones might have a crucial role in resistance against the pathogen. ‘Probable indole-3-acetic acid-amido synthetase GH3.1’ is an auxin-responsive gene that acts in the auxin-dependent development of plants for activating biotic stress resistance pathways independent of salicylic acid signaling and jasmonic acid signaling which enhances resistance to both fungal and bacterial pathogens (Ding et al., 2008). ‘Auxin-responsive protein IAA27-like’ regulates the auxin and ethylene signaling pathways and it is also involved in the regulation of strigolactone biosynthesis. Strigolactones are plant hormones and root-derived signals that regulate shoot branching and respond against parasitic and symbiotic interactions ultimately enhancing plant growth and diminishing the effects of different stresses (Iqbal and Peng, 2022). ‘1-aminocyclopropane-1-carboxylate (ACC) synthase’ which is involved in ethylene biosynthesis by catalyzing the conversion of S-adenosyl-L-methionine (SAM) into ACC was up-regulated in GC-4 only. In a past study, its involvement in ethylene biosynthesis and signaling for disease resistance probably by activating the production of ROS and phytoalexins in rice during M. oryzae infection was reported (Yang et al., 2017). Overexpression of ‘abscisic acid receptor PYL9’ induced the elongation of lateral roots in the presence of abscisic acid (ABA) and recovery of lateral roots from ABA inhibition via MYB transcription factors (Xing et al., 2016). In the present study, ‘abscisic acid receptor PYL9’ was significantly up-regulated in GC-4 only that indicated the possible mechanism of resistance in GC-4. ‘Jasmonic acid-amido synthetase JAR1-like’ was down-regulated and ‘linoleate 13S-lipoxygenase 3-1, chloroplastic-like’ were up-regulated in GC-4 in the present study and involved in jasmonic acid pathway. These genes might have been associated with the downstream JA-responsive resistance genes (Luo et al., 2019). ‘Phenylalanine ammonia-lyase 1’ gene is involved in salicylic acid pathway and was up-regulated in GC-4. SA synthesis and action have been induced according to pathogen-associated molecular patterns (PAMPs) and play an important role in immune responses in plants against fungal pathogens (Luo et al., 2019).
In the present study, we also found the up-regulation of several defense-related genes. ‘Pathogenesis-related protein PR-1 type-like’ was significantly up-regulated in all the cultivars which are among the most abundantly produced proteins in plants during pathogen infection. Its expression has been considered a sign of salicylic acid-mediated disease resistance. PR-1 has broad antimicrobial activity and its overexpression in plants results in increased resistance to fungi and bacteria (Breen et al., 2017). ‘Basic endochitinase-like’ protein was up-regulated in GC-4. Interaction between plant and pathogen directs the rapidity of chitinase (a type of PR-proteins) induction in plant tissues (Vaghela et al., 2022). The expression of ‘lignin-forming anionic peroxidase-like’ was increased in GC-4. It is an important enzyme in lignin biosynthesis which results in secondary cell wall synthesis ultimately providing a physical barrier for pathogens to enhance plant resistance. It was in agreement with the results found in the past in cotton during Fusarium infection (Yao et al., 2019). The probable mannitol dehydrogenase gene was up-regulated in GC-4 only which might be involved in pathogen-secreted mannitol catabolism (Meena et al., 2015). Some genes i.e. ‘glycerol kinase’ and ‘heat shock protein 90-2-like’ were up-regulated in GC-4 and down-regulated in GC-2. These genes were reported to be up-regulated in resistant plants to provide resistance against pathogens (Li et al., 2021; Xiao et al., 2022). The gene for ‘respiratory burst oxidase homolog protein C-like’ (RBOHC) was up-regulated in GC-3, GC-4, and GC-5 and down-regulated in GC-2. Similarly, it was reported that in Arabidopsis it was up-regulated for resistance against Botrytis cinerea (van Rensburg et al., 2020).
Cutin, suberine, and wax biosynthesis pathway was significantly enriched in all the cultivars except GC-5. In the recent past, it was observed that it was significantly involved in resistance against Fusarium verticillioides infection in sugarcane by the KEGG pathway enrichment analysis (Wang et al., 2019b). ‘Protein ECERIFERUM 1-like’ promotes long-chain alkane wax biosynthesis to enhance plant response to biotic and abiotic stresses (Bourdenx et al., 2011). ‘Omega-hydroxypalmitate O-feruloyl transferase-like’ is possibly involved in cutin and suberin biosynthesis and the increase in its transcripts might be an indication of a strengthening of the barrier against the fungal pathogen (Balestrini et al., 2020; Shi et al., 2021). Hence, the up-regulation of some of the genes involved in immune response in resistant cultivars indicated that they might be crucial in providing resistance against wilt pathogens in cumin.
The variation in epigenetic changes in resistant and susceptible cultivars might have played a crucial role in the defense response against pathogens due to the variable expression of different genes. Enzymes involved in histone modification e.g. ‘histone-lysine N-methyltransferase ATX4-like’ (catalyzes histone H3 lysine 4 trimethylation (H3K4me3)) were up-regulated in GC-4 whereas ‘histone-lysine N-methyltransferase setd3’ (catalyzes H3K4me3) was down-regulated in susceptible cultivar (GC-2) only. The enrichment of H3K4me3 at transcription start sites (TSSs) promotes transcription by recruiting PHD-domain-containing proteins (e.g. TATA-box-binding protein associated factor 3 (TAF3)) involved in transcription initiation (Wang et al., 2023). H3K4me is catalyzed by a conserved protein complex (COMPASS-like complex) and is mainly located in euchromatin (Xie and Duan, 2023). Generally, H3K4me3 is associated with transcriptionally active regions hence, the methylation pattern might have activated the genes involved in defense against the wilt pathogen in GC-4. Enzymes involved in chromatin modification e.g. ‘chromatin modification-related protein EAF1 B-like’ (involvement in histone acetyltransferase complex) and ‘ISWI chromatin-remodeling complex ATPase CHR11-like’ (108202994) (possesses intrinsic ATP-dependent nucleosome-remodeling activity) were up-regulated in GC-4 only in the present study. ‘Chromatin modification-related protein EAF1 B-like’ is a component of the NuA4 histone acetyltransferase complex. This complex acetylates nucleosomal histones H2A and H4 for the transcriptional activation of related genes. The acetylation of histones controlled the expression of different developmental stages in plants (Bieluszewski et al., 2015). ‘ISWI chromatin-remodeling complex ATPase CHR11-like’ exhibits intrinsic ATP-dependent nucleosome-remodeling activity. Chromatin-remodeling factors regulate the transcription initiation at many developmental stages of the plant life cycle (Huanca-Mamani et al., 2005). The epigenetic changes like methylation, acetylation, and chromatin remodeling patterns in GC-4 might have altered the expression of genes involved in controlling developmental stages and stress responses for defense against the wilt pathogen.
From the results of DEGs, KEGG, and GO enrichment analysis, the probable immune response to the wilt pathogen, Foc in the resistant cultivar (GC-4) has been proposed (Figure 7). The PAMPs might be detected by the plant receptors. These receptors possessing a kinase domain ultimately activated different signaling pathways like the mitogen-activated protein kinase (MAPK) signaling pathway in the cytoplasm (Nag et al., 2022). It activated some transcription factors (e.g. WRKY75 and MYB1R1) which in turn, up-regulated defense-related genes and pathways in the nucleus. The detection of PAMPs by receptors might have elicited ROS in the apoplastic region by the induction of NADPH oxidases, polyamine oxidases, and peroxidases. The elevated level of ROS and H2O2 might be scavenged by the up-regulation of ROS scavenging enzymes. The production of H2O2 and ROS from mitochondria, chloroplast, and peroxisomes in response to the pathogen attack initiated different signaling pathways toward the resistance against the pathogen. The ROS production in the apoplastic region might have a role in cell wall loosening and cross-linking. Those ROS might also enter into the cytoplasm via aquaporins (e.g. plasma membrane intrinsic protein (PIP2-4)) where it also induces ROS-dependent activation of the MAPK cascade. It also activated calcium channels on the plasma membrane which increased the Ca+2 concentration in the cytoplasm activating CDPKs and calmodulin-like protein. These CDPKs might have relayed the signals to the nucleus of the cell which leads to altering the gene expression of important defense-related genes and pathways. The effector molecules secreted by pathogens might have been recognized by the receptors from R-proteins and they might have blocked the effectors’ activity to block the induction of signaling pathways by kinases after pathogen-associated molecular pattern molecules (PAMPs) detection. Effectors might activate immune responses in plants via different signaling pathways (Jose et al., 2020; Nag et al., 2022). Pattern-triggered immunity (PTI) and effector-triggered immunity (ETI) activated defense signaling pathways inducing expression of PR-proteins (e.g. chitinase, peroxidases, etc.), phytoalexins, R-proteins, and some defense-related pathways as mentioned in results. The phenylpropanoid pathway is involved in the biosynthesis of lignin and ultimately secondary cell wall (SCW) synthesis, hence providing a physical barrier to pathogen entry in the plant cell (Yao et al., 2019). Phytoalexins and some PR-proteins attack the pathogen and reduce the infection. The activation of signaling pathways enhanced the expression of stress-responsive phytohormones (e.g. SA, JA, and ABA) which regulated the expression of other stress-responsive genes (Ali et al., 2018). Some of the PR-proteins might have been involved in programmed cell death (PCD) (Shao et al., 2021). The overall initiation for expression of various genes in response to the infection might be due to epigenetic changes like histone methylation, histone acetylation, and chromatin remodeling. The probable mechanism of defense in wilt GC-4 against F. oxysporum f. sp. cumini is shown in Figure 7. The activation or up-regulation of different genes and pathways related to defense possibly provided resistance in the GC-3 and/or GC-4 cultivars.
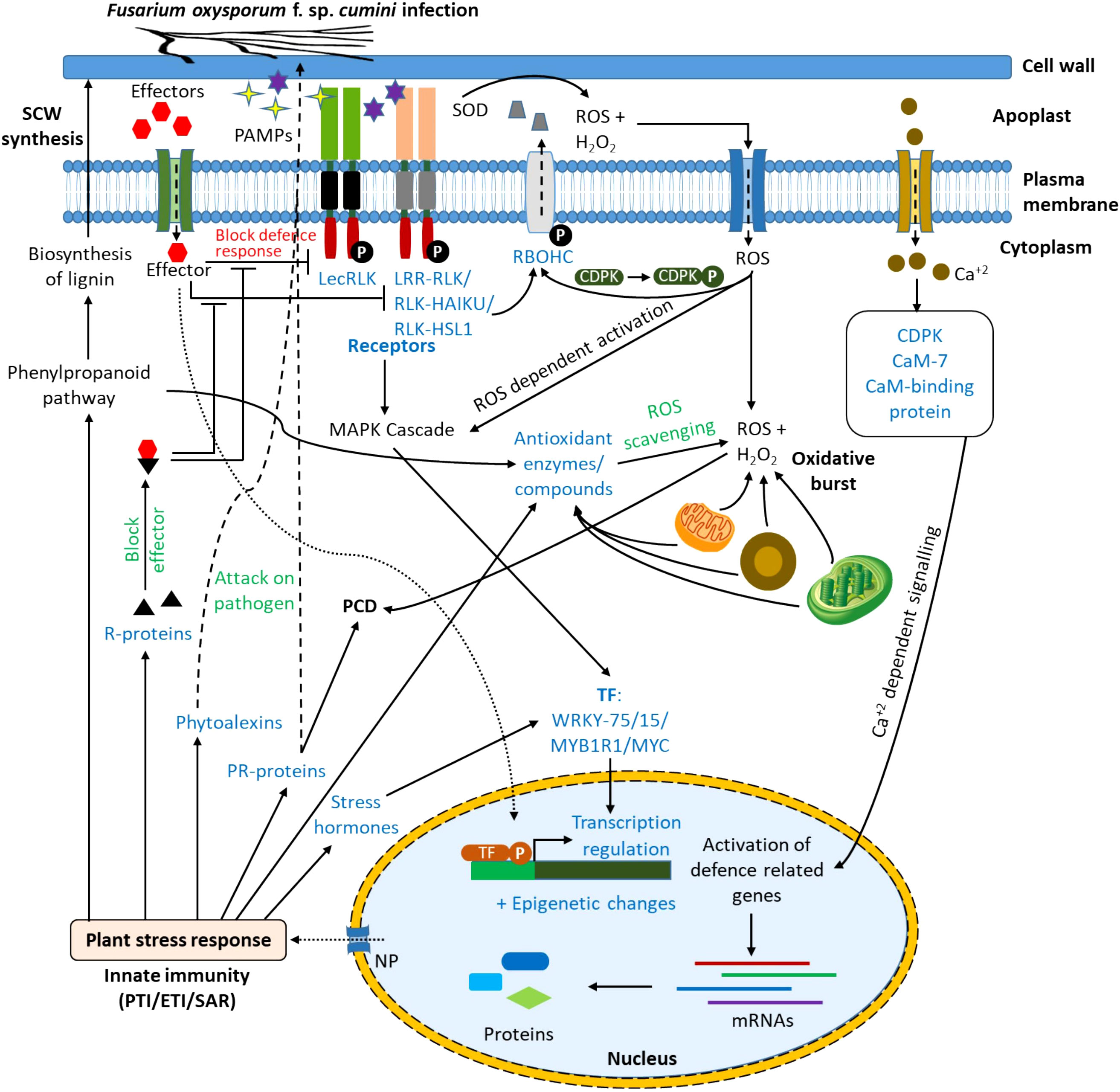
Figure 7 Response in resistant cultivars of cumin to F. oxysporum f. sp. cumini infection (CaM, Calmodulin; CDPK, Calcium dependent protein kinase; CML, Calmodulin-like protein; ETI, Effector triggered immunity; LecRLK, Lectin receptor-like kinases; LRR-RLK, Leucine-rich repeat receptor-like kinases; NP, Nuclear pore; PAMPs, Pathogen-associated molecular patterns; PCD, Programmed cell death; PR-protein, Pathogenesis-related protein; PTI, Pattern-triggered immunity; RBOHC, Respiratory burst oxidase homolog protein C (Calcium-dependent NADPH oxidase); RLK-HAIKU2, Receptor-like kinase- HAIKU2 domain; RLK-HLS1, Receptor-like kinase- HLS1 domain; ROS, Reactive oxygen species; R-protein, Resistance protein; SAR, Systemic acquired resistance; SCW, Secondary cell wall; SOD: Superoxide dismutase; TF, Transcription factor).
5 Conclusion
The transcriptome analysis of wilt-resistant and susceptible cultivars of cumin indicated the variation in the regulation of different pathways and genes by altering signaling pathways and immune system-related pathways. The up-regulation of receptors (e.g. RLKs) for recognition of PAMPs, transcription factors, ROS generating and ROS scavenging enzymes, Ca+2 transporters and receptors, R-proteins, PR-proteins and, phytoalexins in GC-4 might have provided resistance against Foc. Some important pathways enriched in GC-4 and GC-3 were the phenylpropanoid pathway, TCA cycle, phenylalanine metabolism, ubiquitin-mediated proteolysis, ascorbate and aldarate metabolism, and propanoate metabolism. Pathways like steroid biosynthesis, starch and sucrose metabolism, fatty acid biosynthesis, butanoate metabolism, inositol phosphate metabolism, limonene, and pinene degradation, and carotenoid biosynthesis were enriched for up-regulated DEGs in GC-4. The activation or up-regulation of different genes and pathways related to the defense provided resistance in the GC-4 cultivar. These results provide insights to develop cumin cultivars resistant to wilt, in addition to allowing the investigation of the detailed mechanisms underlying cumin defense responses against Foc.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
DTD: wrote the first draft of the manuscript, literature review, and transcriptome data analysis; NS: drafted the bioinformatics pipeline for data analysis and transcriptome data analysis; MP: sample collection, library preparation, and sequencing; MJ: guidance in the data analysis and manuscript proof-reading; AKP: guidance in the data analysis and manuscript proof-reading; CGJ: conception and design of the study, guidance in the data analysis and manuscript proof-reading. All authors contributed to the article and approved the submitted version.
Funding
The project was funded by the Department of Science and Technology, Government of Gujarat (DST- GoG), Gandhinagar, India with project code no. GBRC/GoG-DST/JD1/AGR/2017-18/11.
Acknowledgments
The authors are also thankful to Dr. A. U. Amin, Research Scientist, Seed Spices Research Station, Sardarkrushinagar Dantiwada Agricultural University (SDAU), Jagudan, Mehsana, Gujarat, India for his guidance and support in the collection of plant samples.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1204828/full#supplementary-material
References
Abo-Elyousr, K. A. M., Saad, M. M., Al-Qurashi, A. D., Ibrahim, O. H. M., Mousa, M. A. A. (2022). Management of cumin wilt caused by fusarium oxysporum using native endophytic bacteria. Agronomy 12, 2510. doi: 10.3390/agronomy12102510
Ali, S., Ganai, B. A., Kamili, A. N., Bhat, A. A., Mir, Z. A., Bhat, J. A., et al. (2018). Pathogenesis-related proteins and peptides as promising tools for engineering plants with multiple stress tolerance. Microbiol. Res. 212, 29–37. doi: 10.1016/j.micres.2018.04.008
Arun, K. B., Aswathi, U., Venugopal, V. V., Madhavankutty, T. S., Nisha, P. (2016). Nutraceutical properties of cumin residue generated from ayurvedic industries using cell line models. J. Food Sci. Technol. 53, 3814–3824. doi: 10.1007/s13197-016-2372-z
Balestrini, R., Ghignone, S., Quiroga, G., Fiorilli, V., Romano, I., Gambino, G. (2020). Long-term impact of chemical and alternative fungicides applied to Grapevine cv Nebbiolo on Berry Transcriptome. Int. J. Mol. Sci. 21, 6067. doi: 10.3390/ijms21176067
Bieluszewski, T., Galganski, L., Sura, W., Bieluszewska, A., Abram, M., Ludwikow, A., et al. (2015). AtEAF1 is a potential platform protein for Arabidopsis NuA4 acetyltransferase complex. BMC Plant Biol. 15 (1), 1–15. doi: 10.1186/s12870-015-0461-1
Boba, A., Kostyn, K., Kozak, B., Zalewski, I., Szopa, J., Kulma, A. (2021). Transcriptomic profiling of susceptible and resistant flax seedlings after Fusarium oxysporum lini infection. PloS One 16, e0246052. doi: 10.1371/journal.pone.0246052
Bourdenx, B., Bernard, A., Domergue, F., Pascal, S., Léger, A., Roby, D., et al. (2011). Overexpression of Arabidopsis ECERIFERUM1 promotes wax very-long-chain alkane biosynthesis and influences plant response to biotic and abiotic stresses. Plant Physiol. 156, 29–45. doi: 10.1104/pp.111.172320
Breen, S., Williams, S. J., Outram, M., Kobe, B., Solomon, P. S. (2017). Emerging insights into the functions of pathogenesis-related protein 1. Trends Plant Sci. 22, 871–879. doi: 10.1016/j.tplants.2017.06.013
Bu, D., Luo, H., Huo, P., Wang, Z., Zhang, S., He, Z., et al. (2021). KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 49, W317–W325. doi: 10.1093/nar/gkab447
Chandran, N. K., Sriram, S., Prakash, T., Budhwar, R. (2021). Transcriptome changes in resistant and susceptible rose in response to powdery mildew. J. Phytopathol. 169, 556–569. doi: 10.1111/jph.13028
Chang, Y., Sun, F., Sun, S., Wang, L., Wu, J., Zhu, Z. (2021). Transcriptome analysis of resistance to Fusarium Wilt in mung bean (Vigna radiata L.). Front. Plant Sci. 12, 679629. doi: 10.3389/fpls.2021.679629
Choudhary, S., Mishra, B. K., Singh, R., Sharma, R. (2021). Bacterial diversity and bio-chemical properties in the rhizosphere soils of Cumin and Coriander. Trop. Ecol. 62, 368–376. doi: 10.1007/s42965-021-00155-4
Dange, S. R. S. (1995). Diseases of cumin (Cuminum cyminum L.) and their management. J. Spices Aromat. Crop 4, 57–60.
Ding, X., Cao, Y., Huang, L., Zhao, J., Xu, C., Li, X., et al. (2008). Activation of the indole-3-acetic acid–amido synthetase GH3-8 suppresses expansin expression and promotes salicylate-and jasmonate-independent basal immunity in rice. Plant Cell 20, 228–240. doi: 10.1105/tpc.107.055657
Dong, H., Ye, Y., Guo, Y., Li, H. (2020). Comparative transcriptome analysis revealed resistance differences of Cavendish bananas to Fusarium oxysporum f. sp. cubense race1 and race4. BMC Genet. 21, 1–17.
Duan, Y., Ma, S., Chen, X., Shen, X., Yin, C., Mao, Z. (2022). Transcriptome changes associated with apple (Malus domestica) root defense response after Fusarium proliferatum f. sp. malus domestica infection. BMC Genomics 23, 484. doi: 10.1186/s12864-022-08721-3
Ge, S. X., Jung, D., Yao, R. (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. doi: 10.1093/bioinformatics/btz931
Gerard, D. (2020). Data-based RNA-seq simulations by binomial thinning. BMC Bioinf. 21, 1–14. doi: 10.1186/s12859-020-3450-9
Heberle, H., Meirelles, G. V., da Silva, F. R., Telles, G. P., Minghim, R. (2015). InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinf. 16, 1–7. doi: 10.1186/s12859-015-0611-3
Hou, J., Zhao, F., Yang, X., Li, W., Xie, D., Tang, Z., et al. (2021). Lignin synthesis related genes with potential significance in the response of upland cotton to fusarium wilt identified by transcriptome profiling. Trop. Plant Biol. 14, 106–119. doi: 10.1007/s12042-020-09278-9
Huanca-Mamani, W., Garcia-Aguilar, M., León-Martínez, G., Grossniklaus, U., Vielle-Calzada, J. P. (2005). CHR11, a chromatin-remodeling factor essential for nuclear proliferation during female gametogenesis in Arabidopsis thaliana. Proc. Natl. Acad. Sci. 102 (47), 17231–17236. doi: 10.1073/pnas.0508186102
Iqbal, M. Z., Peng, Y. (2022). Indole acetic acid/auxin-responsive gene TrIAA27 of white clover improves growth and enhances drought and salt tolerance in Arabidopsis. doi: 10.21203/rs.3.rs-2292011/v1
Jose, J., Ghantasala, S., Roy Choudhury, S. (2020). Arabidopsis transmembrane receptor-like kinases (RLKs): a bridge between extracellular signal and intracellular regulatory machinery. Int. J. Mol. Sci. 21, 4000. doi: 10.3390/ijms21114000
Kaushal, M., Mahuku, G., Swennen, R. (2021). Comparative transcriptome and expression profiling of resistant and susceptible banana cultivars during infection by Fusarium oxysporum. Int. J. Mol. Sci. 22, 3002. doi: 10.3390/ijms22063002
Kong, L., Anderson, J. M., Ohm, H. W. (2005). Induction of wheat defense and stress-related genes in response to Fusarium graminearum. Genome 48, 29–40. doi: 10.1139/g04-097
Kumar, M., Sahoo, P. K., Kushwaha, D. K., Saxena, S. N., Mani, I., Singh, G., et al. (2021). Cumin cultivation: Present status and future prospects. Pharma Innov. J. 10, 1121–1123.
Lee, K., Lee, J. G., Min, K., Choi, J. H., Lim, S., Lee, E. J. (2021). Transcriptome analysis of the fruit of Two strawberry cultivars “Sunnyberry” and “Kingsberry” that show different susceptibility to Botrytis cinerea after harvest. Int. J. Mol. Sci. 22, 1518. doi: 10.3390/ijms22041518
Leitão, S. T., Santos, C., Araújo, S., de, S., Rubiales, D., Vaz Patto, M. C. (2021). Shared and tailored common bean transcriptomic responses to combined fusarium wilt and water deficit. Hortic. Res. 8:1–18. doi: 10.1038/s41438-021-00583-2
Li, Q., Shah, N., Zhou, X., Wang, H., Yu, W., Luo, J., et al. (2021). Identification of micro ribonucleic acids and their targets in response to Plasmodiophora brassicae infection in Brassica napus. Front. Plant Sci. 12, 734419. doi: 10.3389/fpls.2021.734419
Lu, J., Xu, Y., Wang, J., Singer, S. D., Chen, G. (2020). The role of triacylglycerol in plant stress response. Plants 9, 472. doi: 10.3390/plants9040472
Luo, J., Xia, W., Cao, P., Xiao, Z., Zhang, Y., Liu, M., et al. (2019). Integrated transcriptome analysis reveals plant hormones jasmonic acid and salicylic acid coordinate growth and defense responses upon fungal infection in poplar. Biomolecules 9, 12. doi: 10.3390/biom9010012
Meena, M., Prasad, V., Zehra, A., Gupta, V. K., Upadhyay, R. S. (2015). Mannitol metabolism during pathogenic fungal–host interactions under stressed conditions. Front. Microbiol. 6, 1019. doi: 10.3389/fmicb.2015.01019
Meena, M. D., Lal, G., Meena, S. S., Meena, N. K. (2018). Production and export performances of major seed spices in India during pre and post-WTO period. Int. J. Seed Spices 8 (1), 21–30.
Meena, R. S., Anwer, M. M., Lal, G., Kant, K., Mehta, R. S. (2010). Advanced production technology of cumin. ICAR-National research centre on seed spices tabiji. Ajmer (Rajasthan), 6.
Mnif, S., Aifa, S. (2015). Cumin (Cuminum cyminum L.) from traditional uses to potential biomedical applications. Chem. Biodivers. 12, 733–742. doi: 10.1002/cbdv.201400305
Nag, P., Paul, S., Shriti, S., Das, S. (2022). Defence response in plants and animals against a common fungal pathogen, Fusarium oxysporum. Curr. Res. Microb. Sci., 3:100135. doi: 10.1016/j.crmicr.2022.100135
Pandian, B. A., Sathishraj, R., Djanaguiraman, M., Prasad, P. V. V., Jugulam, M. (2020). Role of cytochrome P450 enzymes in plant stress response. Antioxidants 9, 454. doi: 10.3390/antiox9050454
Peng, J., Aluthmuhandiram, J. V. S., Chethana, K. W. T., Zhang, Q., Xing, Q., Wang, H., et al. (2022). An nmrA-like protein, lws1, is important for pathogenesis in the woody plant pathogen lasiodiplodia theobromae. Plants 11, 2197. doi: 10.3390/plants11172197
Roy, C. B., Liu, H., Rajamani, A., Saha, T. (2019). Transcriptome profiling reveals genetic basis of disease resistance against Corynespora cassiicola in rubber tree (Hevea brasiliensis). Curr. Plant Biol. 17, 2–16. doi: 10.1016/j.cpb.2019.02.002
Sa, R., Tao, L., Zhang, X., Liu, D., Chen, L., Wang, J., et al. (2022). The effect of Alternaria leaf spot on the antioxidant system of cucumber seedlings. Eur. J. Plant Pathol. 164, 125–138. doi: 10.1007/s10658-022-02545-3
Shao, D., Smith, D. L., Kabbage, M., Roth, M. G. (2021). Effectors of plant necrotrophic fungi. Front. Plant Sci. 12, 687713. doi: 10.3389/fpls.2021.687713
Sharma, L. K., Agarwal, D., Rathore, S. S., Malhotra, S. K., Saxena, S. N. (2016). Effect of cryogenic grinding on volatile and fatty oil constituents of cumin (Cuminum cyminum L.) genotypes. J. Food Sci. Technol. 53, 2827–2834. doi: 10.1007/s13197-016-2258-0
Shi, C.-H., Wang, X.-Q., Xu, J.-F., Zhang, Y.-X., Qi, B., Jun, L. (2021). Dissecting the molecular mechanism of russeting in sand pear (Pyrus pyrifolia Nakai) by metabolomics, transcriptomics, and proteomics. Plant J. 108, 1644–1661. doi: 10.1111/tpj.15532
Slezina, M. P., Istomina, E. A., Korostyleva, T. V., Kovtun, A. S., Kasianov, A. S., Konopkin, A. A., et al. (2021). Molecular insights into the role of cysteine-rich peptides in induced resistance to Fusarium oxysporum infection in tomato based on transcriptome profiling. Int. J. Mol. Sci. 22, 5741. doi: 10.3390/ijms22115741
Srinivasan, K. (2018). Cumin (Cuminum cyminum) and black cumin (Nigella sativa) seeds: traditional uses, chemical constituents, and nutraceutical effects. Food Qual. Saf. 2, 1–16. doi: 10.1093/fqsafe/fyx031
Sun, J., Zhang, J., Fang, H., Peng, L., Wei, S., Li, C., et al. (2019). Comparative transcriptome analysis reveals resistance-related genes and pathways in Musa acuminata banana’Guijiao 9’in response to Fusarium wilt. Plant Physiol. Biochem. 141, 83–94. doi: 10.1016/j.plaphy.2019.05.022
Toueni, M., Ben, C., Le Ru, A., Gentzbittel, L., Rickauer, M. (2016). Quantitative resistance to Verticillium wilt in Medicago truncatula involves eradication of the fungus from roots and is associated with transcriptional responses related to innate immunity. Front. Plant Sci. 7, 1431. doi: 10.3389/fpls.2016.01431
Vaghela, B., Vashi, R., Rajput, K., Joshi, R. (2022). Plant chitinases and their role in plant defense–a comprehensive review. Enzyme Microb. Technol., 159:110055. doi: 10.1016/j.enzmictec.2022.110055
van Rensburg, H. C., Takács, Z., Freynschlag, F., Toksoy Öner, E., Jonak, C., den Ende, W. (2020). Fructans prime ROS dynamics and Botrytis cinerea resistance in Arabidopsis. Antioxidants 9, 805.
Wang, H., Fan, Z., Shliaha, P. V., Miele, M., Hendrickson, R. C., Jiang, X., et al. (2023). H3K4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature 615 (7951), 339–348. doi: 10.1038/s41586-023-05780-8
Wang, G., Kong, J., Cui, D., Zhao, H., Niu, Y., Xu, M., et al. (2019a). Resistance against Ralstonia solanacearum in tomato depends on the methionine cycle and the γ-aminobutyric acid metabolic pathway. Plant J. 97, 1032–1047. doi: 10.1111/tpj.14175
Wang, Z., Li, Y., Li, C., Song, X., Lei, J., Gao, Y., et al. (2019b). Comparative transcriptome profiling of resistant and susceptible sugarcane genotypes in response to the airborne pathogen Fusarium verticillioides. Mol. Biol. Rep. 46, 3777–3789. doi: 10.1007/s11033-019-04820-9
Wei, L., Zhang, H., Duan, Y., Li, C., Chang, S., Miao, H. (2016). Transcriptome comparison of resistant and susceptible sesame (Sesamum indicum L.) varieties inoculated with Fusarium oxysporum f. sp. sesami. Plant Breed. 135, 627–635. doi: 10.1111/pbr.12393
Xiao, X., Wang, R., Khaskhali, S., Gao, Z., Guo, W., Wang, H., et al. (2022). A novel glycerol kinase gene osNHO1 regulates resistance to bacterial blight and blast diseases in rice. Front. Plant Sci., 12:3032. doi: 10.3389/fpls.2021.800625
Xie, S. S., Duan, C. G. (2023). Epigenetic regulation of plant immunity: from chromatin codes to plant disease resistance. aBIOTECH, 4:1–16. doi: 10.1007/s42994-023-00101-z
Xing, L., Zhao, Y., Gao, J., Xiang, C., Zhu, J.-K. (2016). The ABA receptor PYL9 together with PYL8 plays an important role in regulating lateral root growth. Sci. Rep. 6, 27177. doi: 10.1038/srep27177
Xiong, X.-P., Sun, S.-C., Zhu, Q.-H., Zhang, X.-Y., Liu, F., Li, Y.-J., et al. (2021). Transcriptome analysis and RNA interference reveal GhGDH2 regulating cotton resistance to verticillium wilt by JA and SA signaling pathways. Front. Plant Sci. 12, 654676. doi: 10.3389/fpls.2021.654676
Yan, Y., Wang, P., Wei, Y., Bai, Y., Lu, Y., Zeng, H., et al. (2021). The dual interplay of RAV5 in activating nitrate reductases and repressing catalase activity to improve disease resistance in cassava. Plant Biotechnol. J. 19, 785–800. doi: 10.1111/pbi.13505
Yang, C., Li, W., Cao, J., Meng, F., Yu, Y., Huang, J., et al. (2017). Activation of ethylene signaling pathways enhances disease resistance by regulating ROS and phytoalexin production in rice. Plant J. 89, 338–353. doi: 10.1111/tpj.13388
Yao, Z., Chen, Q., Chen, D., Zhan, L., Zeng, K., Gu, A., et al. (2019). The susceptibility of sea-island cotton recombinant inbred lines to Fusarium oxysporum f. sp. vasinfectum infection is characterized by altered expression of long noncoding RNAs. Sci. Rep. 9, 2894. doi: 10.1038/s41598-019-39051-2
Yu, L., Fan, J., Xu, C. (2019). Peroxisomal fatty acid β-oxidation negatively impacts plant survival under salt stress. Plant Signal. Behav. 14, 1561121. doi: 10.1080/15592324.2018.1561121
Zhang, Q., Gao, M., Wu, L., Wu, H., Chen, Y., Wang, Y. (2018). Expression network of transcription factors in resistant and susceptible tung trees responding to Fusarium wilt disease. Ind. Crops Prod. 122, 716–725. doi: 10.1016/j.indcrop.2018.05.041
Zhang, X., Liu, Y., Fang, Z., Li, Z., Yang, L., Zhuang, M., et al. (2016). Comparative transcriptome analysis between broccoli (Brassica oleracea var. italica) and wild cabbage (Brassica macrocarpa Guss.) in response to Plasmodiophora brassicae during different infection stages. Front. Plant Sci. 7, 1929. doi: 10.3389/fpls.2016.01929
Zhang, Y., Yang, N., Zhao, L., Zhu, H., Tang, C. (2019). Transcriptome analysis reveals the mechanism by which the biocontrol fungus Chaetomium globosum CEF-082 controls Verticillium wilt in cotton. doi: 10.21203/rs.2.15513/v1
Keywords: Cuminum cyminum L., fusarium wilt, disease resistance, induced systemic resistance, transcriptome, Fusarium oxysporum f. sp. cumini
Citation: Dharajiya DT, Shukla N, Pandya M, Joshi M, Patel AK and Joshi CG (2023) Resistant cumin cultivar, GC-4 counters Fusarium oxysporum f. sp. cumini infection through up-regulation of steroid biosynthesis, limonene and pinene degradation and butanoate metabolism pathways. Front. Plant Sci. 14:1204828. doi: 10.3389/fpls.2023.1204828
Received: 12 April 2023; Accepted: 21 September 2023;
Published: 17 October 2023.
Edited by:
Magdi A. A. Mousa, King Abdulaziz University, Saudi ArabiaReviewed by:
Taras P. Pasternak, Miguel Hernández University of Elche, SpainKarthikeyan Adhimoolam, Jeju National University, Republic of Korea
Kamal A. M. Abo-Elyousr, Assiut University, Egypt
Copyright © 2023 Dharajiya, Shukla, Pandya, Joshi, Patel and Joshi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Madhvi Joshi, amQxQGdicmMucmVzLmlu; Amrutlal K. Patel, amQyQGdicmMucmVzLmlu; Chaitanya G. Joshi, ZGlyZWN0b3JAZ2JyYy5yZXMuaW4=